

Abstract
When lineages of hosts and microbial symbionts engage in intimate interactions over evolutionary timescales, they can diversify in parallel (i.e., co-diversify), producing associations between the lineages’ phylogenetic histories. Tests for co-diversification of individual microbial lineages and their hosts have been developed previously, and these have been applied to discover ancient symbioses in diverse branches of the tree of life. However, most host-microbe relationships are not binary but multipartite, in that a single host-associated microbiota can contain thousands of microbial lineages, generating numerous challenges for assessing co-diversification. Here, we review recent evidence for co-diversification in complex microbiota, highlight the limitations of prior studies, and outline a hypothesis testing approach designed to overcome some of these limitations. We advocate for the use of microbiota-wide scans for co-diversifying symbiont lineages and discuss tools developed for this purpose. Tests for co-diversification for simple host symbiont systems can be extended to entire phylogenies of microbial lineages (e.g., metagenome-assembled or isolate genomes, amplicon sequence variants, etc.) sampled from host clades, thereby providing a means for identifying co-diversifying symbionts present within complex microbiota. The relative ages of symbiont clades can corroborate co-diversification, and multi-level permutation tests can account for multiple comparisons and phylogenetic non-independence introduced by repeated sampling of host species. Discovering co-diversifying lineages will generate powerful opportunities for interrogating the molecular evolution and lineage turnover of ancestral, host-species specific symbionts within host-associated microbiota.
Keywords: Cospeciation, coevolution, metagenomics, mutualism, parasitism
Graphical Abstract
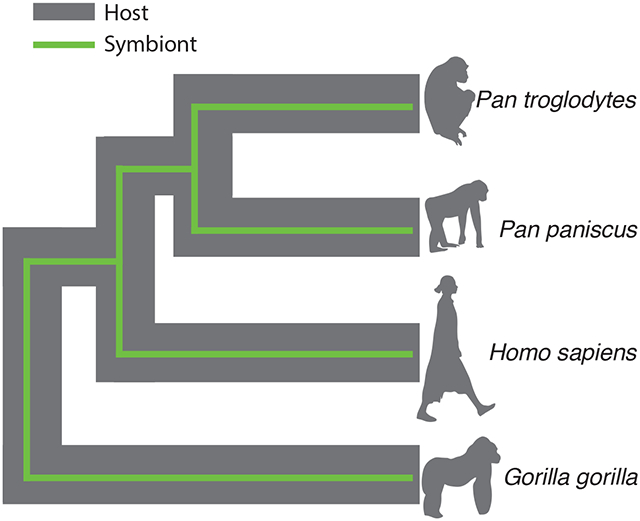
How can we identify the symbionts in complex microbiomes? In this study we evaluate recent evidence that certain lineages within animal gut microbial communities have co-diversified with their host species and populations, and we present statistical approaches for identifying co-diversifying microbial lineages while accounting for multiple testing and phylogenetic non-independence. Discovering the co-diversifying lineages in microbiomes enables discrimination between transient microbial lineages and ancestral, host-species specific symbionts that have been maintained over host evolutionary timescales.
Introduction
Co-diversification—the synchronized bifurcation of two or more lineages of organism—is a canonical consequence of intimate and long-term symbiosis. Testing for co-diversification can reveal ancient symbioses and generate phylogenetic frameworks for the study of their evolutionary histories. The expectation that closely interacting lineages should co-diversify was articulated as early as 1913 by Heinrich Fahrenholz, a German parasitologist studying the ectoparasites of vertebrates, who noted that ectoparasites (e.g., lice) of closely related host species tended to display morphological similarities not seen in those from more distantly related hosts (Farenholtz, 1913). Over 75 years later, the predicted association between phylogenetic trees was tested and validated with protein electrophoretic and DNA sequence data from rodents and their ectoparasites, revealing the concurrent diversification of host and symbiont clades (Hafner and Nadler, 1988; Hafner et al., 1994).
Since the earliest studies, numerous examples of co-diversification between hosts and symbionts have been detected. Discoveries of co-diversification in simple host-symbiont systems have been reviewed elsewhere (De Vienne et al., 2013; Hernández-Hernández et al., 2021) and will not be discussed in detail here. These examples span the symbiosis continuum, from parasites to commensals and mutualists, as well as diverse branches of the eukaryotic tree of life, including plants and their herbivores (Maron et al., 2019), animals and their microbial symbionts (Perreau and Moran, 2022), and fungi and their plant hosts (Otero et al., 2011).
Historically, tests for co-diversification have been applied to individual pairs of interacting lineages (i.e., a host clade and a single symbiont clade), but most symbioses between hosts and microbial lineages take place in the context of complex microbial consortia consisting of dozens, hundreds, or even thousands of linages. For example, humans as a species (Homo sapiens) harbor upwards of one thousand distinct bacterial species across oral, urogenital, lung, and gut microbiomes (Almeida et al., 2019; Almeida et al., 2021). Testing for co-diversification in such multipartite communities of symbionts raises several challenges not faced in the analysis of binary host-symbiont systems. Identifying co-diversified symbionts within microbiota can be complicated by issues such as where on the microbial phylogeny to test for co-diversification events, how to correct for the multiple testing inherent in the assessment of co-diversification across many symbiont lineages within a microbiota, and how to account properly for phylogenetic non-independence.
Here, we review recent progress in the study of co-diversification within complex, host-associated microbiota and outline a hypothesis testing framework that overcomes several of the limitations of prior studies. We discuss the history of discoveries for co-diversification in complex microbiota, including the rapid advancement in this area afforded by genome-resolve metagenomics approaches. We conclude that tests for co-diversification originally designed for simple host-symbiont systems can be readily extended to analyses of entire microbiota, and we consider strategies regarding sampling design for future studies of microbiota co-diversification.
Tests for co-diversification in bipartite host-microbe symbioses
Tests for co-diversification in binary host-symbiont systems can be divided into two general classes of methods: topology- or distance-based methods and event-based methods (De Vienne et al., 2013; Dismukes et al., 2022). For both classes of methods, host and symbiont phylogenies (or phylogenetic distance matrices) are required as input, and the validity of downstream conclusions about co-diversification reflects the quality of the phylogenies used for the analysis. Exceptions to this general limitation of tests for co-diversification are event-based Bayesian methods or distance-based methods that account for tree uncertainty (Huelsenbeck et al., 2000; Pérez-Escobar et al., 2016; Balbuena et al., 2020). We will not comprehensively review software packages and specific implementations of tests for co-diversification in simple host-symbiont systems here, as this is provided by previous work (De Vienne et al., 2013; Dismukes et al., 2022; Groussin et al., 2020). Below, we briefly summarize the rationales and methodologies underlying distance- and event-based methods as context for discussion about how these approaches can be extended to test for co-diversification within complex, host-associated microbiota.
Distance-based tests for co-diversification assess the association between host and symbiont relative divergence times (e.g., Legendre et al., 2002; Hommola et al., 2009; Balbuena et al., 2013; Mramba et al., 2013; Hutchinson et al., 2017). In general, these tests attempt to estimate the probability of observing by chance a degree of association between symbiont and host phylogenies equal to or greater than that observed between the true symbiont and host phylogenies. These tests require generation of a distribution of degrees of association (e.g., correlation coefficients) between symbiont and host phylogenetic distances under the null hypothesis of no history of co-diversification. In practice, these null distributions can be generated through random permutations of the host and symbiont tip labels or the incidence table indicating the presence and absence of symbiont lineages within host lineages.
Distance-based methods have the advantage of not requiring estimations of the relative probability of co-diversification, extinction, or host switching events, i.e., these methods provide non-parametric tests of association between symbiont and host phylogenies. However, if multiple symbiont lineages from individual host species are sampled, then the p-values derived from such methods may indicate either co-diversification of symbionts with hosts or host-species specificity of symbionts in the absence of co-diversification, as the null hypothesis being tested is that of no association between host and symbiont phylogenies. For example, significant p-values can be obtained from permutation tests if symbionts from individual host species form monophyletic clades on the symbiont phylogeny, even in the absence of co-diversification of symbiont lineages and host species (Nishida and Ochman, 2021). Therefore, significant results must be interpreted with caution, and evidence for co-diversification carefully examined through approaches that account for pseudoreplication, such as subsampling symbiont phylogenies to a single lineage from each host species.
In contrast to distance-based tests, event-based tests (e.g., Merkle and Middendorf, 2005) explicitly model co-diversification, host-switching, within-host diversification, and extinction of symbiont lineages along a host phylogeny. These methods afford the ability to calculate the relative likelihoods of different sets of events (e.g., strict co-diversification or rampant host switching) given the observed symbiont phylogenies and probabilities for the occurrence of each type of event per unit of time or branch length. However, the probabilities of co-diversification, host-switching, within-host diversification, and extinction events are rarely known for the host and symbiont lineages of interest, and event-based tests can be prone to over- or under-parametrization (De Vienne et al., 2013). Nevertheless, with accurate parameters event-based methods provide exceptional power for inferring specific instances of co-diversification between symbionts and hosts.
Alternative explanations for apparent co-diversification
Significant associations between symbiont and host phylogenetic trees provides strong evidence for co-diversification, but the lack of such associations does not necessarily preclude a history of co-diversification. For instance, the ability to accurately identify a true history of co-diversification events from phylogenetic analyses can be obscured by more recent histories of symbiont host switching or extinction, or by missing data. Conversely, detection of significant associations between symbiont and host phylogenies does not by itself conclusively demonstrate a history co-diversification, because such associations can also be produced by alternative processes. One prominent alternative explanation for congruence between symbiont and host phylogenies is the successive colonization of hosts by symbionts in combination with ‘ecological fitting’ (Janzen, 1985). For instance, if hosts display phylogenetic signal in traits that affect symbiont colonization, the pattern in which host and symbiont phylogenies mirror one another can be produced in the absence of a history of co-diversification by the successive colonization by symbionts of host lineages along the host phylogeny. One approach for differentiating co-diversification and ecological fitting hypotheses is to incorporate information about the timing of host and symbiont divergence events (discussed further below). For instance, if host and symbionts have co-diversified, the ages of nodes supporting co-diversification in host and symbiont trees should be associated with one another—a prediction that is not necessarily expected in the case of ecological fitting in the absence of co-diversification.
Distinctions between co-diversification, co-evolution, and phylosymbiosis
Co-diversification between hosts and symbionts is indicative of long-term associations, which could provide opportunities for co-evolution, in which interacting lineages act as reciprocal selective forces on and adapt to one another. As other authors have noted, co-evolution and co-diversification are distinct processes, and co-diversification between hosts and symbionts can occur in the absence of co-evolution (Althoff et al., 2014; Moran and Sloan 2015; Russo et al., 2018). For instance, host diversification events can generate barriers to gene flow between microbiota of host lineages, leading to phylogenetic tracking of host lineages by symbionts (Russo et al., 2018; Blasco-Costa et al., 2021). Phylogenetic tracking by symbionts refers specifically to cases in which symbiont diversification depends on host diversification while host diversification is relatively independent of symbiont diversification. In extreme cases, symbionts can be vertically transmitted (i.e., parent to offspring) with high fidelity throughout the course of host diversification, leading to confinement of symbiont lineages within host lineages. These dispersal-based processes do not require ongoing adaptation of symbionts to hosts (or of hosts to symbionts), but increased symbiont dispersal among conspecific hosts relative to among heterospecific hosts can generate patterns of co-diversification. Under this view, symbiont diversification driven by phylogenetic tracking of host lineages is analogous to the effects of vicariance events that have occurred for terrestrial macroorganisms due to continental drift or other large-scale barriers to gene flow (Cowman and Bellwod, 2013; Althoff et al., 2014; Groussin et al., 2020).
An alternative, but not mutually exclusive, process that could maintain host-species specificity of symbionts over host evolutionary timescales and drive co-diversification is ongoing symbiont adaptation to hosts, to other members of the microbiota, or to other environmental factors associated with the host phylogeny. In complex microbiota, there is emerging experimental evidence that host-species specific symbionts adapt to their host species. In the tetrapod gut microbiota, for instance, Lactobacillus strains display competitive advantages within their native hosts relative to strains from other tetrapod host species (Oh et al., 2010; Frese et al., 2011; Duar et al., 2017). Recent work in which whole microbiota from closely related rodent species (genus Mus) were competed in pairwise mixtures within germ-free house mice (Mus musculus domesticus) found that house-mouse microbiota consistently out-competed non-native microbiota from other host species (Sprockett et al., 2023). Further assessing the degree and mechanistic bases of symbiont adaptation to host species within complex microbiota will require develop additional germ-free model systems within individual host clades. These experimental resources will enable reciprocal transplant and competition experiments—the gold standards of tests for local adaptation—of microbiota between closely related hosts.
It is also useful to differentiate between co-diversification of microbiota lineages with their hosts and associations between the taxonomic or functional composition of the microbiota and the host phylogeny. The latter—termed phylosymbiosis—can occur in the absence of co-diversification between individual symbiont lineages and hosts (Moran and Sloan, 2015; Kohl, 2020). For example, more closely related hosts may select for more similar sets of microorganisms from common environmental pools of microorganisms (i.e., habitat filtering), leading to congruence between microbiota-composition dendrograms and host phylogeny without co-diversification between symbiont and host lineages.
Signals of co-diversification in complex microbiota
The prediction that host-restricted symbionts should co-diversify with their hosts can be extended to entire communities of host-associated microorganisms, i.e., microbiota. Compared to tests for phylosymbiosis, tests for co-diversification of individual lineages within complex microbiota and their hosts are relatively rare. This disparity results largely from the methodological difficulties inherent in testing for co-diversification in the microbiota. Testing for phylosymbiosis relies on microbiota-composition dendrograms, which can be readily generated by amplicon or metagenomic sequencing of host species, whereas testing for co-diversification requires a well-resolved symbiont phylogeny, often at the strain-level. Overcoming the methodological barriers that preclude tests for co-diversification is a critical priority for comparative research of host-associated microbiota, because these tests can provide insights into processes that produce patterns of microbiota diversity among hosts that cannot be revealed by examination of the high-level taxonomic or functional composition of the microbiota alone (Moran et al., 2019).
To date, tests for co-diversification in complex, host-associated microbiota have relied primarily on symbiont phylogenies inferred from amplicon-based data, such as 16S rRNA gene or protein-coding amplicon datasets (Table 1). Early evidence for co-diversification of symbionts with hosts in the context of complex microbiota came from amplicon-based studies of 16S rRNA gene regions. Sanders et al. analyzed 16S rRNA gene datasets from ants and apes, finding that many of the differences between the microbiota of closely related host species could be explained by recent bacterial evolution, consistent with a history of co-diversification, rather than taxonomically broad shifts in microbiota composition due to, for example, host dietary changes (Sanders et al., 2014). Similar results were observed by Groussin et al. and Youngblut et al., who observed significant evidence of association between gut bacterial phylogenies based on 16S rRNA sequences and their hosts’ phylogenies (Groussin et al., 2017; Youngblut et al., 2019). One limitation of 16S rRNA gene sequences for inferring co-diversification events, particularly recent events, is the slow rate of evolution of this gene. Estimates from insect endosymbionts and enteric bacteria of mammals suggest that 16S rRNA sequences evolve at rates as slow as 1% per 50 million years (Kuo and Ochman, 2009), affording limited phylogenetic signal to assess co-diversification with bacteria over the timescales of the diversification of multicellular eukaryotes. A corollary of this limitation is that 16S rRNA genes will tend to perform better for assessing co-diversification when the host species of interest are relatively distantly related. For instance, recent 16S rRNA gene-amplicon studies of microbiota of sponges and coral-reef invertebrates found significant signals of host-species specificity consistent with co-diversification in multiple bacterial and archaeal families, including the Endozoicomonadaceaea, Spirochaetaceaea and Nitrosopumilaceae (Pollock et al., 2018; O’Brien et al., 2021). Similarly, 16S rRNA gene-amplicon studies have successfully detected co-diversification between gut bacteria and ant lineages (Hu et al., 2023).
Table 1.
Select examples of studies examining evidence for co-diversification in complex, host-associated microbiota.
| Citation | Bacteria examined | Hosts examined | Evidence for co- diversification |
Methodology |
|---|---|---|---|---|
| Moeller et al. (2016) | Bifidobacteriaceae Lachnospiraceae Bacteroidaceae |
Humans and African apes | Concordance among Bifidobacteriaceae, Bacteroidaceae, and host phylogenies | gyrB-amplicon sequencing |
| Nishida et al. (2021) | Bacteroidaceae | Humans and African apes | Weak association between Bacteroidaceae and host phylogenies | gyrB-amplicon sequencing |
| Powell et al. (2016) | Snodgrassella | Honeybees and bumblebees | Specificity of Snodgrassella alvi strains to bee species | minD-amplicon sequencing |
| Suzuki et al. (2022) | 59 prominent gut bacterial species | Humans | Associations between bacterial and host-population phylogenies | Metagenome assembled genomes |
| Groussin et al. (2017) | Microbiota-wide | Mammals | Associations between bacterial and host phylogenies observed for Alloprevotella, Mitsuokella, Paraprevotella, Allistipes, and other taxa | 16S rRNA gene-amplicon sequencing |
| Sanders et al. (2014) | Microbiota-wide | Cephalotes ants and African apes | Differences among host-species microbiota were driven by recent bacterial evolution, consistent with co-diversification | 16S rRNA gene-amplicon sequencing |
| O’Brien et al. (2021) | Endozoicomonadaceaea, Spirochaetaceaea | Coral-reef invertebrates | Distributions of ASVs across hosts were significantly cluster with host phylogeny, consistent with co-diversification | 16S rRNA gene-amplicon sequencing |
To overcome the limited resolution provided by 16S rRNA genes, other studies have employed marker-gene based approaches focused on protein coding genes. In contrast to 16S rRNA genes, which code for ribosomal RNAs in which nearly every site in the gene has potential to affect the structure and function of the molecular product, protein-coding genes contain numerous silent sites that can mutate without any effect on protein structure. Consequently, compared to 16S rRNA genes, protein-coding genes typically evolve more rapidly and provide greater phylogenetic signal for testing for co-diversification, particularly over recent timescales. For example, Powell et al., showed that 16S rRNA gene sequencing of the bee gut microbiota failed to reveal strain-level symbiont variation, including variation between bee species, but significant host-associated variation was observed when these communities were assayed with amplicon sequencing of minD—a septum site determining protein-coding gene (Powell et al., 2016). A similar approach relying on protein-coding marker genes recently detected evidence of co-speciation between gut bacteria and termite lineages (Arora et al., 2023). Studies using amplicon sequencing of protein coding genes have also been used to test for co-diversification of gut microbiota with humans and African apes, revealing mixed evidence for co-diversification in this host clade (Moeller et al., 2016; Nishida and Ochman, 2021). Moeller et al. sequenced gyrase B (gyrB) genes from three bacterial families—Bifidobacteriaceae, Bacteroidaceae, and Lachnospiraceae, from humans, chimpanzees, bonobos, and gorillas, finding co-phylogenetic signals of co-diversification in two of the three bacterial families (Bifidobacteriaceae and Bacteroidaceae) (Moeller et al., 2016). However, follow-up studies by Nishida and Ochman employed the same methods to survey the Bacteroidaceae present in captive chimpanzees, finding mixed evidence for co-diversification and suggesting that captivity may lead to loss of host-species specific symbionts (Nishida and Ochman, 2021). Similar results were also observed in studies of other captive primates based on 16S rRNA gene amplicon sequencing (Clayton et al., 2016; Houtz et al., 2021).
Though marker-gene approaches based on protein-coding genes provided greater resolution to detect recent co-diversification events compared to 16S rRNA-gene amplicon sequencing, these protein-coding–gene amplicon methods are not without their limitations. A benefit of 16S rRNA gene amplicon sequencing is that the entire microbiota can be surveyed simultaneously with data obtained from a single sequencing library. In contrast, the relatively rapid evolution of protein-coding marker genes requires the design of primers and preparation of libraries for individual bacterial taxa (e.g., families), greatly increasing the up-front effort required to conduct such studies. Moreover, both 16S rRNA-gene and protein-coding–gene amplicon studies are, by definition, limited to interrogations of patterns of co-diversification in individual genes, rather than entire genomes of symbiont lineages.
A recent advance that overcomes both the limitations of 16S rRNA gene and protein-coding gene amplicon sequencing approaches to test for co-diversification is the ability to assemble genomes directly from metagenomic sequence data, i.e., metagenome-assembled genomes (MAGs) (Hugerth et al., 2015; Parks et al., 2017; Bowers et al., 2017) (Table 1). These approaches afford unprecedented resolution for assessing co-diversification in complex host-associated microbiota. For example, Sanders et al. 2023 used a collection of nearly 10,000 MAGs assembled from humans and non-human primates to test for co-diversification in the gut microbiota, finding widespread signals of association between symbiont and host phylogenies across ten gut bacteria phyla, including multiple lineages that appear to have co-diversified with humans and the African apes. Similarly, Suzuki et al. applied MAG-based approaches and co-phylogenetic analyses to identify signals of co-diversification of gut microbiota lineages with populations of Homo sapiens (Suzuki et al., 2022). However, it remains unclear whether the patterns of co-diversification of symbionts with human populations observed by Suzuki et al. reflect fidelity of symbionts to host genealogies, or merely divergence of symbionts between disparate geographic regions (Good, 2022). Nevertheless, results from Suzuki et al. corroborate previous results from studies of Helicobacter pylori showing that this symbiont has diversified in a manner that mirrors human migration routes (Falush et al., 2003). Together with previous work from marker-gene based approaches, these findings provide insights into co-diversification of gut microbiota with human and African-ape hosts at multiple timescales spanning the diversification of host species, populations, and individual genealogies (Figure 1).
Fig. 1. Co-diversification at three levels in humans and African apes.

(A) Studies of protein-coding marker genes and metagenome assembled genomes (MAGs) have provided support for co-diversification between certain gut bacterial symbionts and human and African ape hosts (Moeller et al., 2016; Sanders et al., 2023). However, surveys of captive chimpanzees have found conflicting evidence for co-diversification and host specificity in the gut microbiota (Nishida and Ochman, 2021; Houtz et al., 2021). (B) A recent MAG-based study of human gut microbiota found evidence that multiple lineages of bacterial symbionts have co-diversified with human populations (Suzuki et al., 2022), mirroring previous results observed for Helicobacter pylori (Falush et al., 2003). (C) Suzuki et al. also detected evidence consistent with co-diversification of individual symbionts and human families via vertical transmission through maternal lineages (Suzuki et al., 2022), although these signals are difficult to separate from geographic effects (Good, 2022). The hypothesis that symbiont lineages co-diversify with maternal lineages generates specific predictions about the structure of symbiont phylogenies which have not yet been tested, including the prediction that male lineages may represent evolutionary ‘dead ends’.
Extending tests for co-diversification to multipartite host-microbe systems
A challenge inherent in detecting co-diversified symbionts in complex microbiota is the lack of a priori justification regarding where to test for co-diversification events on the symbiont phylogeny. For a complex microbiota, the phylogeny of symbionts will, by definition, contain many species, typically including species that are distantly related to one another (e.g., distinct phyla). Because diversification of bacterial phyla, classes, orders, families, and even genera can long predate the diversification of eukaryotic host lineages, many bifurcations on phylogenies of microbiota symbionts could not have resulted from co-diversification with hosts. Conversely, divergence of closely related symbiont strains may occur over timescales far shorter than the timescales of host diversification. Because most microorganisms in complex microbiota lack fossil records that could be used for dating the absolute timing of diversification events, it is often not possible to determine at which depth of the symbiont phylogeny to conduct either distance- or event-based tests for co-diversification.
A powerful approach for circumventing this challenge is to conduct unbiased scans of the phylogeny of microbiota symbionts in which each clade is tested individually for co-diversification with hosts. Similar approaches, in which subsets of phylogenies of putatively co-diversifying lineages are tested independently, have been applied previously to assess co-diversification between populations of multicellular organisms, such as mimetic Heliconius butterflies (Hoyal Cuthill and Charelston, 2012; Hoyal Cuthill and Charelston, 2015). Recently, Sanders et al. applied this approach to test for co-diversification between MAGs in the gut microbiota and primate host species (Sanders et al., 2023), but such an approach could in principle be applied to phylogenies of microbiota symbionts based on other data types as well (e.g., 16S rRNA gene or protein-coding amplicon data). In short, the method employed by Sanders et al. traverses the bacterial tree and applies to each node a test for co-diversification (Figure 2). Currently, this workflow, which is available in Python (https://github.com/CUMoellerLab/codiv-tools) and R (https://github.com/DanielSprockett/codiv) implementations, is designed to implement a distance-based test for co-diversification initially developed by Hommola et al. for bipartite host-symbiont systems. This test assesses for individual nodes whether the observed association between symbiont and host phylogenies exceeds the null expectation generated by random permutation of host and symbiont tip labels. However, in principle the workflow could be altered to incorporate any distance- or event-based test for co-diversification. Required input includes a host phylogeny, a symbiont phylogeny, and an incidence table indicating from which host each symbiont lineage was derived. The nodes tested can be restricted to a subset of the symbiont phylogeny, if desired. For instance, for recently diverged host lineages, restricting the scan for co-diversification to the most distal portions of the symbiont phylogeny (e.g., the most distal 10% of the tree) may be desirable to avoid testing symbiont clades that can be assumed to long predate host diversification events. The output is a table containing test statistics and p-values for each clade in the microbial phylogeny indicating the strength of association with the host phylogeny. Thus, the workflow provides a ranking of all nodes in the portion of the bacterial phylogeny tested based on the strength of signal of co-diversification with hosts.
Fig. 2. Multi-level permutation tests enable quantification of co-diversification in complex microbiota.

(A) Phylogeny shows hypothetical relationships among symbionts recovered from a clade of hosts. Scans for co-diversification can be conducted on each node of the symbiont phylogeny (smaller dashed box). Aggregating the statistics of these tests across all or many nodes in the symbiont phylogeny (larger dashed box) can reveal whether the symbiont phylogeny more instances of significant evidence for co-diversification across symbiont clades than expected under the null hypothesis of no co-diversification. (B) Histogram shows the distribution of test statistics for co-diversification derived from permutation tests of host and symbiont tip labels for an individual symbiont clade, corresponding to the smaller dashed box in (A). Vertical dashed lines indicate hypothetical test statistics observed from real data from the clade, with red indicating significant evidence for co-diversification in the clade, blue indicating no evidence for co-diversification, and green indicating negative association between host and symbiont phylogenies. (C) Histogram shows the null distribution of the number of co-diversifying clades detected at a given significance threshold from phylogeny-wide scans for co-diversification conducted after random permutations of the host phylogeny’s tip labels. Vertical dashed lines indicate hypothetical numbers of significantly co-diversifying clades observed in the real dataset, with red indicating more instances of significant co-diversification than expected by chance under the null hypothesis of no co-diversification. As in (B), blue or green dashed lines indicate no evidence for co-diversification across the symbiont phylogeny or significantly fewer instances of co-diversification than expected under the null hypothesis, respectively.
One issue that arises from unbiased scans for co-diversification of symbiont phylogenies from complex microbiota is multiple testing. Because many nodes are tested, it is necessary to establish the Type II error rate for these tests. Moreover, many nodes on the symbiont phylogeny of interest will not be phylogenetically independent from one another, such that efforts must be made to account for pseudoreplication of symbiont lineages (Nishida and Ochman, 2021). Both issues can be addressed by an additional level of permutation testing that goes beyond permutation tests by distance-based tests for co-diversification in bipartite host-symbiont systems. By permuting the host phylogeny’s tip labels a large number of times (e.g., 999 for a significance level of 0.001) and performing for each permutation the scans and permutation tests for co-diversificaiton for individual nodes in the microbiota symbiont phylogeny (e.g., Hommola tests based on correlation coefficients), the distribution of test statistics and p-values observed across nodes in the real dataset can be compared to the distribution generated under the null hypothesis of no co-diversification. This second-order permutation test can reveal whether a microbiota symbiont phylogeny contains a greater number of clades displaying significant evidence for co-diversification than expected by chance (Figure 2C) at a given test-statistic or p-value threshold (e.g., r > 0.75 or p-value < 0.05). When applying distance-based tests in the context of this second-order permutation test, it may be preferrable to consider only test-statistic values (e.g., r > 0.75) instead of p-values, as the former is expected to be more robust to issues pertaining to pseudoreplication and phylogenetic non-independence. This second-order permutation test can provide information about the rate of false discoveries in the scans (which contain multiple comparisons), including the rate of false discoveries due to pseudoreplication and phylogenetic non-independence. For instance, if tests based on permuted labels of the host phylogeny show that 10% of nodes on the microbiota symbiont phylogeny display significant evidence of co-diversification at a given p-value or test-statistic threshold, whereas tests based on the true data identify 50% of the nodes as significantly co-diversifying, a false discovery rate of ~20% can be inferred.
Second order permutation tests can indicate whether there is a stronger signal of co-diversification between microbiota constituents and host lineages than expected by chance under the null hypothesis of no co-diversification, but as noted above in the context of bipartite co-diversifying host-symbiont systems, it is critical to consider that multiple evolutionary processes can generate this pattern. In the context of entire microbiota, one caveat is that constituents of the microbiota are interacting with one another, such that their histories of co-diversification with hosts are non-independent. Under the extreme case, a single highly ecologically connected microbiota constituent that is co-diversifying with host lineages could drive the co-diversification of many other microbiota constituents, even if these latter microbiota constituents display no degree of adaptation to or biased dispersal within their respective host lineages. The hypothesis that focal microbiota constituents may drive co-diversification of other microbiota constituents could be assessed, in principle, by testing for significant overlap (beyond that expected by chance) between the set of microbiota constituents displaying evidence of co-diversification and the set of microbiota constituents displaying positive ecological interactions (e.g., as evidenced by co-abundance patterns or co-culturing experiments).
Use of molecular clocks to corroborate co-diversification
Scans of entire microbiota symbiont phylogenies have the potential to reveal dozens or hundreds of symbiont clades that display signals of co-diversification with host lineages. In cases where evidence of highly parallel co-diversification is observed, the relative ages of putatively co-diversifying symbiont clades can be used to further test the hypothesis that these arose contemporaneously with their host clades. Under the assumption of a uniform or nearly uniform molecular clock for bacterial symbionts, the relative ages of co-diversifying symbiont clades should be positively associated with the relative or absolute ages of their host clades. This cross-symbiont clade prediction can provide another line of evidence that the symbiont clades arose contemporaneously with their hosts.
In cases where absolute host clade ages are known (e.g., through fossil evidence), these ages can further be used to provide absolute dates for divergence events within co-diversifying clades of symbiont. Absolute dating of co-diversifying symbiont clades events promises to provide unprecedented information about the rates of molecular evolution in bacteria. For instance, Sanders et al. employed this approach to co-diversifying bacterial lineages in the primate gut microbiota, revealing significantly different rates of molecular evolution among gut bacterial phyla (Sanders et al., 2023). However, absolute dating of symbiont divergence events using host divergence events first requires strong evidence of co-diversification, such as high levels of topological congruence between symbiont and host phylogenies. In any case, caution is warranted when interpreting ages of apparently co-diversifying symbiont clades dated using host clade ages, as other processes besides co-diversification are in principle capable of generating high degrees of association between symbiont and host phylogenies (as noted above).
Summary
Complex, host-associated microbiota can contain symbionts that co-diversify with host species, symbionts that are readily transmitted among host species lineages as they diversified, as well as symbionts that have been acquired by clades of host species relatively recently. Parsing the constituents of the microbiota into these categories requires phylogenetic approaches that directly compare symbiont and host evolutionary histories. The approaches that have been developed for bipartite symbioses are extendable to studies of entire communities of host-associated symbionts, although doing so requires explicit accounting for multiple testing and pseudoreplication (Figure 2). As methods for profiling microbial communities continue to advance and provide increasingly fine-scale strain-level resolution, such as that afforded by metagenome-assembled genomes, microbiota-wide scans for co-diversification promise to discover evolutionary ancient host-associated symbionts, including those that have been conserved across host generations by natural selection, those that have adapted to their host lineages, and those that have shaped the evolutionary trajectories of host species.
Acknowledgements
This work was funded by award R35 GM138284 from the National Institute of General Medical Sciences to AHM.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Data Availability
No new data were generated for this study.
References
- 1.Farenholtz H. (1913). Über den einfluss von licht und schatten auf sprosse von holzpflanzen. Heinrich C. [Google Scholar]
- 2.Hafner MS, & Nadler SA (1988). Phylogenetic trees support the coevolution of parasites and their hosts. Nature, 332(6161), 258–259. [DOI] [PubMed] [Google Scholar]
- 3.Hafner MS, Sudman PD, Villablanca FX, Spradling TA, Demastes JW, & Nadler SA (1994). Disparate rates of molecular evolution in cospeciating hosts and parasites. Science, 265(5175), 1087–1090. [DOI] [PubMed] [Google Scholar]
- 4.De Vienne DM, Refrégier G, López-Villavicencio M, Tellier A, Hood ME, & Giraud TJNP (2013). Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytologist, 198(2), 347–385. [DOI] [PubMed] [Google Scholar]
- 5.Hernández-Hernández T, Miller EC, Román-Palacios C, & Wiens JJ (2021). Speciation across the t ree of l ife. Biological Reviews, 96(4), 1205–1242. [DOI] [PubMed] [Google Scholar]
- 6.Maron JL, Agrawal AA, & Schemske DW (2019). Plant–herbivore coevolution and plant speciation. Ecology, 100(7), e02704. [DOI] [PubMed] [Google Scholar]
- 7.Perreau J, & Moran NA (2022). Genetic innovations in animal–microbe symbioses. Nature Reviews Genetics, 23(1), 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otero JT, Thrall PH, Clements M, Burdon JJ, & Miller JT (2011). Codiversification of orchids (Pterostylidinae) and their associated mycorrhizal fungi. Australian Journal of Botany, 59(5), 480–497. [Google Scholar]
- 9.Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, … & Finn RD (2019). A new genomic blueprint of the human gut microbiota. Nature, 568(7753), 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, … & Finn RD (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nature biotechnology, 39(1), 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dismukes W, Braga MP, Hembry DH, Heath TA, & Landis MJ (2022). Cophylogenetic methods to untangle the evolutionary history of ecological interactions. Annual Review of Ecology, Evolution, and Systematics, 53. [Google Scholar]
- 12.Huelsenbeck JP, Rannala B, & Larget B (2000). A Bayesian framework for the analysis of cospeciation. Evolution, 54(2), 352–364. [DOI] [PubMed] [Google Scholar]
- 13.Pérez-Escobar OA, Balbuena JA, & Gottschling M (2016). Rumbling orchids: how to assess divergent evolution between chloroplast endosymbionts and the nuclear host. Systematic Biology, 65(1), 51–65. [DOI] [PubMed] [Google Scholar]
- 14.Balbuena JA, Pérez-Escobar ÓA, Llopis-Belenguer C, & Blasco-Costa I (2020). Random tanglegram partitions (Random TaPas): an Alexandrian approach to the cophylogenetic Gordian knot. Systematic Biology, 69(6), 1212–1230. [DOI] [PubMed] [Google Scholar]
- 15.Groussin M, Mazel F, & Alm EJ (2020). Co-evolution and co-speciation of host-gut bacteria systems. Cell Host & Microbe, 28(1), 12–22. [DOI] [PubMed] [Google Scholar]
- 16.Legendre P, Desdevises Y, & Bazin E (2002). A statistical test for host–parasite coevolution. Systematic biology, 51(2), 217–234. [DOI] [PubMed] [Google Scholar]
- 17.Hommola K, Smith JE, Qiu Y, & Gilks WR (2009). A permutation test of host–parasite cospeciation. Molecular biology and evolution, 26(7), 1457–1468. [DOI] [PubMed] [Google Scholar]
- 18.Balbuena JA, Míguez-Lozano R, & Blasco-Costa I (2013). PACo: a novel procrustes application to cophylogenetic analysis. PloS one, 8(4), e61048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mramba LK, Barber S, Hommola K, Dyer LA, Wilson JS, Forister ML, & Gilks WR (2013). Permutation tests for analyzing cospeciation in multiple phylogenies: applications in tri-trophic ecology. Statistical Applications in Genetics and Molecular Biology, 12(6), 679–701. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson MC, Cagua EF, Balbuena JA, Stouffer DB, & Poisot T (2017). paco: implementing Procrustean Approach to Cophylogeny in R. Methods in Ecology and Evolution, 8(8), 932–940. [Google Scholar]
- 21.Nishida AH, & Ochman H (2021). Captivity and the co-diversification of great ape microbiomes. Nature communications, 12(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clayton JB, Vangay P, Huang HU, Ward T, Hillmann BM, Al-Ghalith GA, … & Knights D (2016). Captivity humanizes the primate microbiome. Proceedings of the National Academy of Sciences, 113(37), 10376–10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houtz JL, Sanders JG, Denice A, & Moeller AH (2021). Predictable and host-species specific humanization of the gut microbiota in captive primates. Molecular Ecology, 30(15), 3677–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merkle D, & Middendorf M (2005). Reconstruction of the cophylogenetic history of related phylogenetic trees with divergence timing information. Theory in Biosciences, 123(4), 277–299. [DOI] [PubMed] [Google Scholar]
- 25.Janzen DH (1985). On ecological fitting. [Google Scholar]
- 26.Althoff DM, Segraves KA, & Johnson MT (2014). Testing for coevolutionary diversification: linking pattern with process. Trends in Ecology & Evolution, 29(2), 82–89. [DOI] [PubMed] [Google Scholar]
- 27.Moran NA, & Sloan DB (2015). The hologenome concept: helpful or hollow?. PLoS biology, 13(12), e1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russo L, Miller AD, Tooker J, Bjornstad ON, & Shea K (2018). Quantitative evolutionary patterns in bipartite networks: Vicariance, phylogenetic tracking or diffuse co-evolution?. Methods in Ecology and Evolution, 9(3), 761–772. [Google Scholar]
- 29.Blasco-Costa I, Hayward A, Poulin R, & Balbuena JA (2021). Next-generation cophylogeny: unravelling eco-evolutionary processes. Trends in Ecology & Evolution, 36(10), 907–918. [DOI] [PubMed] [Google Scholar]
- 30.Cowman PF, & Bellwood DR (2013). Vicariance across major marine biogeographic barriers: temporal concordance and the relative intensity of hard versus soft barriers. Proceedings of the Royal Society B: Biological Sciences, 280(1768), 20131541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh PL, Benson AK, Peterson DA, Patil PB, Moriyama EN, Roos S, & Walter J (2010). Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. The ISME Journal, 4(3), 377–387. [DOI] [PubMed] [Google Scholar]
- 32.Frese SA, Benson AK, Tannock GW, Loach DM, Kim J, Zhang M, … & Walter J (2011). The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genetics, 7(2), e1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duar RM, Frese SA, Lin XB, Fernando SC, Burkey TE, Tasseva G, ... & Walter J (2017). Experimental evaluation of host adaptation of Lactobacillus reuteri to different vertebrate species. Applied and Environmental Microbiology, 83(12), e00132–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sprockett DD, Price JD, Juritsch AF, Schmaltz RJ, Real MV, Goldman SL, … & Moeller AH (2023). Home-site advantage for host species–specific gut microbiota. Science Advances, 9(19), eadf5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohl KD (2020). Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Philosophical Transactions of the Royal Society B, 375(1798), 20190251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moran NA, Ochman H, & Hammer TJ (2019). Evolutionary and ecological consequences of gut microbial communities. Annual Review of Ecology, Evolution, and Systematics, 50(1), 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanders JG, Powell S, Kronauer DJ, Vasconcelos HL, Frederickson ME, & Pierce NE (2014). Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Molecular Ecology, 23(6), 1268–1283. [DOI] [PubMed] [Google Scholar]
- 38.Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, & Alm EJ (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nature Communications, 8(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Youngblut ND, Reischer GH, Walters W, Schuster N, Walzer C, Stalder G, … & Farnleitner AH (2019). Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nature communications, 10(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuo CH, & Ochman H (2009). Inferring clocks when lacking rocks: the variable rates of molecular evolution in bacteria. Biology Direct, 4(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollock FJ, McMinds R, Smith S, Bourne DG, Willis BL, Medina M, … & Zaneveld JR (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nature Communications, 9(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Brien PA, Andreakis N, Tan S, Miller DJ, Webster NS, Zhang G, & Bourne DG (2021). Testing cophylogeny between coral reef invertebrates and their bacterial and archaeal symbionts. Molecular Ecology, 30(15), 3768–3782. [DOI] [PubMed] [Google Scholar]
- 43.Hu Y, D'Amelio CL, Béchade B, Cabuslay CS, Łukasik P, Sanders JG, Price S, Fanwick E, Powell S, Moreau CS and Russell JA, 2023. Partner fidelity and environmental filtering preserve stage-specific turtle ant gut symbioses for over 40 million years. Ecological Monographs, 93(1), p.e1560. [Google Scholar]
- 44.Powell E, Ratnayeke N, & Moran NA (2016). Strain diversity and host specificity in a specialized gut symbiont of honeybees and bumblebees. Molecular Ecology, 25(18), 4461–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arora J, Buček A, Hellemans S, Beránková T, Romero Arias J, Fisher BL, … & Bourguignon T (2023). Evidence of cospeciation between termites and their gut bacteria on a geological time scale. Proceedings of the Royal Society B, 290(2001), 20230619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, … & Ochman H (2016). Cospeciation of gut microbiota with hominids. Science, 353(6297), 380–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hugerth LW, Larsson J, Alneberg J, Lindh MV, Legrand C, Pinhassi J, & Andersson AF (2015). Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biology, 16(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, … & Tyson GW (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature Microbiology, 2(11), 1533–1542. [DOI] [PubMed] [Google Scholar]
- 49.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, … & Woyke T (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology, 35(8), 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanders JG, Sprockett DD, Li Y, Mjungu D, Lonsdorf EV, Ndjango JBN, … & Moeller AH (2023). Widespread extinctions of co-diversified primate gut bacterial symbionts from humans. Nature Microbiology, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki TA, Fitzstevens JL, Schmidt VT, Enav H, Huus KE, Mbong Ngwese M, … & Ley RE (2022). Codiversification of gut microbiota with humans. Science, 377(6612), 1328–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Good BH (2022). Limited codiversification of the gut microbiota with humans. bioRxiv. [Google Scholar]
- 53.Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, … & Suerbaum S (2003). Traces of human migrations in Helicobacter pylori populations. Science, 299(5612), 1582–1585. [DOI] [PubMed] [Google Scholar]
- 54.Hoyal Cuthill J, & Charleston M (2012). Phylogenetic codivergence supports coevolution of mimetic Heliconius butterflies. PloS ONE, 7(5), e36464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoyal Cuthill JF, & Charleston M (2015). Wing patterning genes and coevolution of Müllerian mimicry in Heliconius butterflies: Support from phylogeography, cophylogeny, and divergence times. Evolution, 69(12), 3082–3096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated for this study.