

Abstract
Infection with Borrelia burgdorferi (Bb) causes Lyme disease in humans. In small rodents, the natural reservoir species of this spirochete, infections lead to only modest disease manifestations, despite causing persistence infection. While B cell responses are central for controlling bacterial tissue burden and disease manifestations, they lack classical aspects of T-dependent responses, such as sustained IgG affinity maturation and longevity, corresponding with a rapid collapse of germinal centers. Instead, the antibody response is characterized by strong and ongoing secretion of IgM, whose origins and impact on protective immunity to B. burgdorferi remains unknown. Here we demonstrate that Bb infection-induced IgM in mice was produced continuously, mainly by conventional B, not B-1 cells, in a T-independent manner. While IgM was passively protective and restricted early bacteremia, its production had no effects on bacterial dissemination into solid tissues, nor did it affect Borrelia tissue-burden. The latter was controlled by the induction of bactericidal IgG, as shown comparing infections in wild type mice with those of mice lacking exclusively secreted (s)IgM−/−, all class-switched antibodies via deletion of aicda (AID−/−), and all secreted antibodies (sIgM−/− × AID−/−). Consistent with the notion that B. burgdorferi infection drives production of IgM over more tissue-penetrable IgG, we demonstrated increased short- and long-term IgM antibody responses also to a co-administered, unrelated antigen. Thus, the continued production of IgM may explain the absence of Bb in the blood.
Introduction
Borrelia burgdorferi sensu lato (Bb) is the causative agent of Lyme disease, a chronic, non-resolving, and often debilitating systemic emerging tick-transmitted infectious disease in humans, dogs and horses, among other species (1, 2). In mice, major natural reservoir hosts, Bb causes persistent infections with minimal clinical manifestations of disease, indicating that the bacteria have evolved complex strategies to evade mammalian immune responses. Indeed, our previous work showed that Bb rapidly enters draining LNs at the site of infection altering tissue structures of experimentally-infected mice (3, 4). While strong antibody production ensues and germinal centers initially form, a rapid collapse of germinal centers is noted around one month of infection, despite ongoing infection (3–5). This collapse in T-dependent germinal center responses correlated with the loss of high-affinity Borrelia-specific serum IgG antibodies (6). Together, these data demonstrate that Bb targets the inductive site of adaptive immunity and suggests that the antibody responses, while strongly induced and measurable, may be functionally compromised.
LNs are sites of early, strong and Borrelia-specific extrafollicular antibody production in infected mice (3). Nearly half of this response consisted of IgM (3). Beginning around three months after Bb infection, specific IgM-secreting plasma cells also begin to accumulate in the bone marrow (5). Bb-specific IgM has been found previously in the sera of 28 day Bb-infected mice (7). This is consistent with clinical studies, which have shown the presence of IgM responses to multiple Bb antigens even as far out as 10 years after the appearance of erythema migrans, an early manifestation of the disease in some humans, including individuals treated with antibiotics (8, 9). A remarkable observation, given the short half-life of IgM, considered to be under 24h.
It is unknown whether the ongoing, long-term production of Bb-specific IgM ultimately benefits the host or the pathogen. IgM has numerous functional characteristics that may benefit the host. IgM’s multiple binding sites enable high-avidity binding, particularly to repeat antigens on bacterial or viral surfaces and to bacterial lipoproteins, which are abundant on the surface of Bb (10). Indeed, while class-switch recombination to IgG is considered a hallmark of protective immunity, studies with various pathogens have shown a necessary role for antigen-specific IgM in clearing infections and protecting the host from future challenge (11–13). IgM responses were shown also to be highly protective against infections with the blood-resident B. hermsii, the relapsing fever spirochete. In that infection the presence of IgM strongly reduced bacteremia, and therefore disease (14, 15). On the other hand, pentameric IgM, because of its large size, does not usually extravasate into connective tissues where Bb resides largely in the apparent absence of inflammation, thus perhaps precluding it from interactions once Bb has disseminated.
To define the function of IgM in immunity to Bb we studied its origins, protective capacity, and effects on the course of Bb infection. We demonstrate the strong and persistent production of passively protective Bb-specific IgM mainly by conventional B cells in a T-independent manner. IgM was shown non-redundant for controlling bacteremia, but not Bb dissemination and tissue burden, the latter was shown to require IgG. Immunization with a non-related antigen during Bb infection enhanced IgM responses compared to responses in non-infected mice, suggesting that Bb may drive IgM responses to reduce antibody-mediated tissue clearance.
Material and Methods
Borrelia burgdorferi
A low-passage clonal strain of sensu stricto Borrelia burgdorferi (cN40) was grown in modified Barbour-Stoenner-Kelley II medium at 33°C and enumerated with a Petroff-Hauser bacterial counting chamber (Baxter Scientific, McGaw Park, IL) for inoculation of SCID mice. Borrelia burgdorferi lysate was obtained by growing N40 to log-phase (8–10 days) followed by pelleting by centrifugation, resuspended in cold PBS with MgCl2 and centrifuged repeatedly at 17,500 g for 5 min at 4°C. Protein concentrations were determined by Bradford assay (Bio-Rad). Aliquots were stored at −20°C.
Mice
Eight- to twelve-week old female C57BL/6J, B6.Cg-IghaThy1aGpi1a/J (Igha), B6.CB17-Prkdcscid/SzJ (SCID), B6.129P2-Tcrbtm1MomTcrdtm1Mom/J (ß/∂ T cell KO), B6.129S2-Igh6tm1Cgn/J (B cell KO) BALB/cByJ, B6.129S2-Cd40lgtm1Imx/J (CD40L−/−), CBySmn.CB17-Prkdcscid/J (BALB/c-SCID) female or male mice were purchased (The Jackson Laboratory). Late term pregnant Hsd:ICR (CD-1) mice (Harlan) and their 21-days old pups were used for passive protection assays. Breeders of mice that lack secreted IgM but not membrane bound IgM (sIgM−/−) originally generated by Jianzhu Chen (16), AID (AID−/−) (17), and mice knocked out for both sIgM and AID (sIgM × AID−/−) (18) on a C57BL/6 background, were kindly provided by Frances Lund (University of Alabama at Birmingham). All mice were kept under SPF housing conditions in ventilated microisolator cages. Mice were euthanized by over-exposure to carbon dioxide.
Ig allotype chimeras were generated as previously described (19, 20) to distinguish B-1 and B-2 cells and the IgM they secrete, using allotype and isotype specific reagents. In brief, newborn B6 pups (Igh-b) were treated with 0.1 mg of anti-IgM-b (AF6–78.25) i.p. to deplete all host B cells. On day 2 after birth, pups received 5×106 peritoneal cavity (PerC) cells from 8–12 weeks old congenic Igh-a donors as a source for B-1 cells. Pups were treated with 0.2 mg of AF6–78.25 i. p. biweekly for an additional six weeks. At that time de novo B-1 cell development from host bone marrow is strongly abrogated (21, 22). Mice were used two months after cessation of antibody treatment to allow full reconstitution of conventional B cells (Igh-b). The B-1 cell pool remains largely Igh-a (23, 24). Reverse chimeras were made also, in which newborn Igh-a mice were treated with anti-IgM-a (DS-1.1) and received cells from B6 (Igh-b) donors.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols involving animals were approved by the Animal Use and Care Committee at UC Davis.
Bactericidal assay
Bb was grown for 5 −7 days to mid-log phase at 33°C and adjusted to a density of 2.6 × 107 (Fig. 3B); 7.28 × 105 (Fig. 3C) and 8 × 105 (Fig. 3D) cells/ml, respectively. Sera pooled from day 31-infected C57BL/6 Igh-a or Igh-b expressing mice, sIgM−/− and SCID mice or graded doses purified IgG from indicated sources were diluted in modified Barbour-Stoenner-Kelley II (BSK) medium. To some cultures, as indicated, 1% serum from non-infected RAG−/− mice was added as a source of complement. Antibody/complement mixtures were added together with Bb to 0.5ml tubes containing BSK medium to generate a total volume of 0.2ml. As indicated, graded doses of antibodies were added to achieve concentrations of 0.0125, 0.025, 0.0375 and 0.05 mg IgG/ml, respectively. Tubes were briefly vortexed and then cultured for 48 hours at 33°C. Live Bb were counted using a hemocytometer and a darkfield microscope.
Figure 3. Similar bactericidal activity of immune sera from sIgM−/− and wildtype Bb-infected mice and their purified IgG.
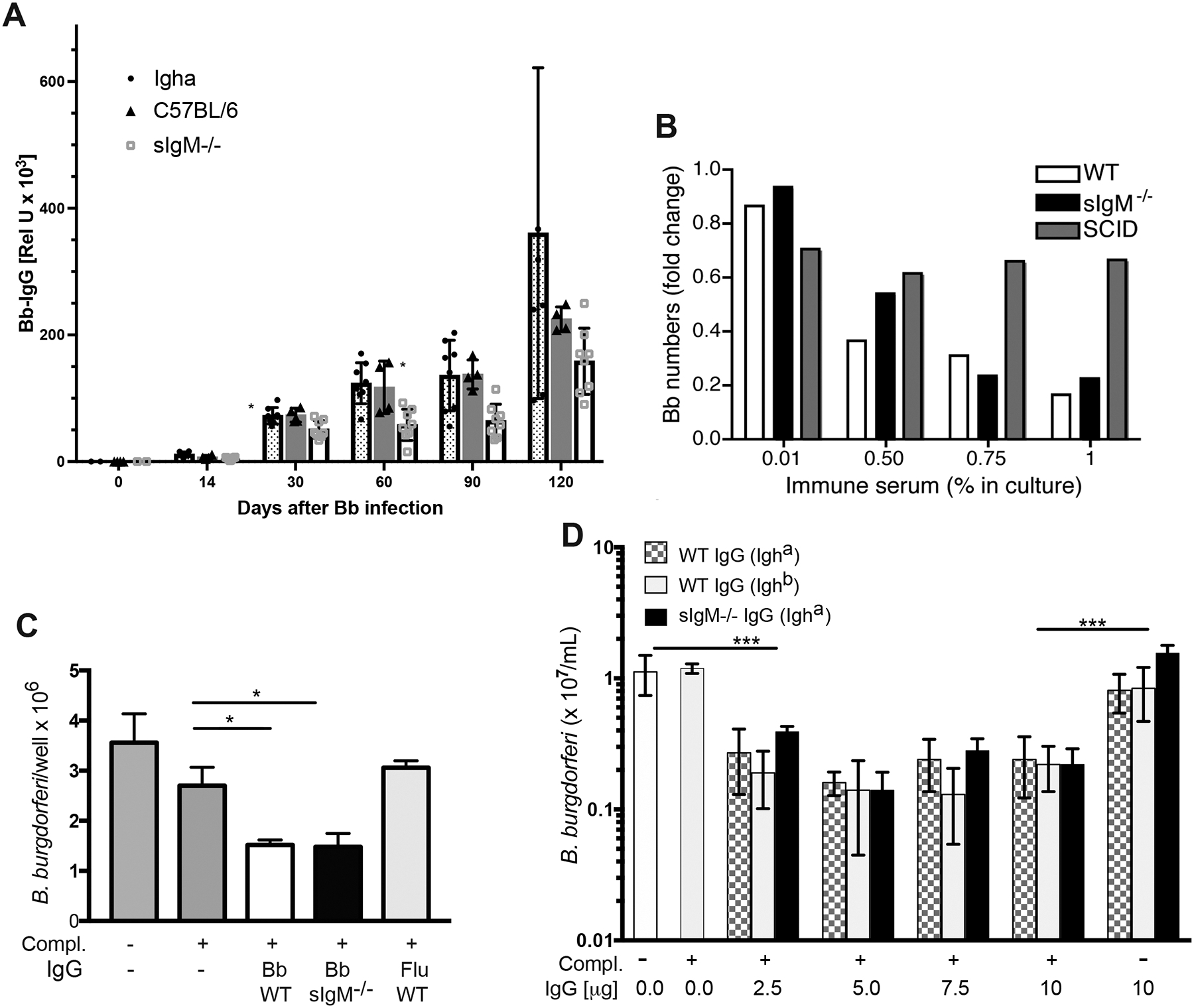
(A) Shown are mean serum Bb-specific IgG levels measured by ELISA at indicated times of Bb infection, expressed as relative units compared to a standard immune serum. Compared were sera from C57BL/6 congenic Igh-a mice and sIgM−/− mice (n = 8/group) and C57BL/6 mice (n=4). (B) Shown are mean fold-changes of Bb numbers in BSK cultures grown for 48h in the presence of indicated concentration serum from 31-days Borrelia-infected B6 (open bars), sIgM−/− (black) or SCID (grey) mice compared to cultures with medium alone. Shown is one of four experiments done using duplicate cultures that gave similar results. (C) Shown are mean ± SD Bb counts in cultures with BSK medium plus addition of 1% RAG−/− serum from non-infected mice as source of complement, where indicated, and 5μg purified IgG from sera of C57BL/6 or sIgM−/− mice infected for 31 days with Bb, or from C57BL/6 mice infected with influenza virus as a negative control. Results are pooled from 3 experiments done with cultures in triplicate. (D) Cultures were set-up as in (C) with indicated graded concentrations of purified IgG from C57BL/6 mice (checkered), B6 congenic but Igh-a expressing mice (light grey) or sIgM−/− mice (black) infected for 31 days with Bb. Cultures were done in quadruplicate and data represent one of 3 experiments done. Significance was tested for (A) by two-way ANOVA (p < 0.0015); for (C) and (D) using unpaired, one-tailed Student’s t-test comparing each culture to the BSK only control. *p < 0.05; ***p < 0.001.
Infections and immunizations
Mice were infected with tissue-adapted Bb cN40 by transferring small pieces of ear tissue from SCID mice, infected for at least 14 days, into the right hind leg under the skin of recipients, as described previously (3). To generate Bb-infected ear tissue, B6 SCID mice were infected s.c. with 104 culture-grown Bb or 1×106 in BALB/c-SCID. Infected SCID mice were euthanized and ears were cleaned with 70% ethanol and diluted Nolvasan (Pfizer) before removal. Lateral tail vein blood collections were performed to obtain serum. For influenza immunization, mice were injected subcutaneously on the right side of the tail base with 500 hemagglutinating units (HAU) of A/PR8 (H1N1) in alum (Imject Alum, Thermo Fisher). Virus was propagated in embryonated hen eggs, and HAU determined as previously described (25).
Antibiotic treatment
Groups of mice infected with Bb were treated i.p. twice daily for 5 days followed by treatment once daily for 25 days with 16 mg/kg ceftriaxone in 100 μL PBS. Controls received PBS only.
Antibody purification
IgM was purified from sera of wildtype and β/δ T cell KO mice by column affinity chromatography, using immobilized anti-IgM mAb (clone 331, in-house generated with Thermo Scientific Pierce AminoLink Plus Immobilization Kit, pH 7.2 coupling buffer). IgM concentrations were determined by ELISA in comparison to a purified mouse IgM standard (Southern Biotech).
ELISA
Borrelia burgdorferi-specific ELISAs were performed by coating 96-well plates (Maxisorb, Thermo Fisher Scientific) with 1 μg/mL of N40 Bb lysate overnight at room temperature. Alternatively, coating was done with four immunodominant recombinant Bb proteins, decorin binding protein A (DbpA), outer surface protein C (OspC), arthritis related protein (Arp) and Borrelia membrane protein A (BmpA), as previously described (3). ELISAs were performed as previously described (3, 5). In brief, after blocking for 1 hour, diluted sera were added to the plates and incubated for 2 hours. Antibody binding was revealed using biotin conjugated anti-IgM, anti-IgG, or anti-IgA (Southern Biotech) followed by an incubation with SA-HRP (Vector Laboratories) for 1 hour and then substrate for 20 min. Reactions were stopped with 1 N sulfuric acid and absorbance was read at 450 nm and reference wavelength of 595 nm using a SpectraMax M5 (Molecular Devices). Relative units antibody levels were calculated by comparison to a day-120 immune serum titrated on the same plates from mice infected with host-adapted spirochetes. To determine total concentrations of IgM, IgA or total Ig, ELISA was done as above but plates were coated with anti-IgM, -A or IgG (Southern Biotech) and binding was revealed with biotinylated antibodies to the matching Ig-isotype. Standards used were purified mouse IgM, -A or -G (Southern Biotech).
ELISPOT
To probe for Bb-specific antibody producing cells, 96-well plates (Multiscreen HA filtration; Millipore) were coated overnight as for ELISA plates. For influenza specific antibody producing cells, plates were coated with influenza A/PR8 (1000 HAU/mL). IgM-secreting cells were identified on plates coated with anti-IgM (clone 331, in-house generated by purification from hybridoma supernatants via protein G column affinity chromatography). After blocking with PBS/4% BSA for 1 h,our 5×105 cells in medium (RPMI 1640, 292 μg/mL L- glutamine, 100 μg/mL of penicillin and streptomycin, 10% heat inactivated FCS, and 0.03 M 2-ME) were placed in the starting well and 2-fold serially diluted. Cells were incubated at 37°C overnight then lysed with water. Antibody binding by B-1 and B-2 cells was revealed by adding biotin conjugated anti-IgM-a (DS-1.1) or anti-IgM-b (AF6–78.25), respectively, (both in house generated) in PBS/2% BSA for 2 hours, SA-HRP (Vector Laboratories) in PBS/2% BSA for 1 hour and revealing with 3-amino-9-ethylcarbazole (Sigma-Aldrich) for 10 min. Plates were dried and spots were counted in all wells with dilutions resulting in visible and countable spots using a stereomicroscope. Mean numbers ± SD were calculated from cell counts of all wells with countable spots.
Passive protection assay
To test for passive protective capacity of serum IgM from wildtype and ß/∂ T cell KO mice, purified serum IgM from day 60 infected mice was injected i. p. at various concentrations into 3-weeks old CD-1 pups prior to challenge with culture grown Bb. Injection of PBS or pre-immune sera served as negative controls. For all Bb challenge infections, s.c. inoculation of 104 N40 Bb between the shoulder blades was done 18 hours post antibody transfer. Mice were euthanized two weeks post infection and urinary bladder, heart base, and inoculation site were collected for qPCR and/or for culture in BSK II medium.
Flow cytometry
Single cell suspensions from inguinal LNs, spleens, peritoneal cavity, and bone marrow were stained as previously described (3). After Fc receptor blocking (10 μg anti-CD16/32, clone 2.4G2, in-house generated), the following antibody conjugations were used at predetermined optimal concentrations to determine frequencies of B cell subsets: CD19-Alexa700, CD8-Cy5PE (eBiosciences), CD24-FITC, CD23-FITC, CD43-PE, CD4-Cy5PE, F4/80-Cy5PE, CD5-biotin, IgM-allophycocyanin, IgM-a-allophycocyanin, IgM-b-allophycocyanin, IgD-Cy7PE, IgD-a-Cy7PE, IgD-b-Cy7PE (all in-house generated), Streptavidin-Qdot605 (Invitrogen). Staining was performed on ice for 20 min. For dead cell discrimination, cells were stained with live/dead violet stain (Invitrogen by Life Technologies) on ice for 30 min. Data acquisition was performed on a 13-color FACSAria instrument (BD Biosciences) (26). Data were analyzed using FlowJo software.
qPCR for detection of Bb DNA
Quantitative real-time PCR was utilized to detect Bb flagellin (flaB) DNA in LNs, urinary bladder, injection sites and heart base, using primers and probe for flaB as described (27). Samples were amplified with 40 cycles of denaturation at 95°C for 15 sec, annealing at 50°C for 2 min and extension at 60°C for 1 min. Tissue samples were weighed and DNA was extracted from tissues with DNeasy kits (Qiagen, Valencia, CA) according to manufacturer’s instructions. Purified N40 DNA served as positive control and DNA from uninfected mice and water as negative controls. Data were calculated as the copy number of flaB DNA per mg of tissue based on DNA standard.
Western blotting
Whole cell Bb N40 proteins, loaded at 8μg/lane, were fractionated on 10–20% Tris-Glycine gel (Novex) before transfer to a nitrocellulose membrane (Pharmacia Biotech). Blots were blocked with 1% BSA in Tris-buffered-saline solution followed by incubation with immune sera from wild-type and sIgM−/− mice 31 days after infection or serum from uninfected wild-type mice (negative control), diluted at 1:100 for 2 hours. Blots were probed with goat anti-mouse IgG conjugated to alkaline phosphatase (Sigma). Bands were revealed with a substrate solution containing 5-bromo-4-chloro-3-indolylphosphate and Nitro Blue Tetrazolium.
Statistical analysis
Statistical analysis was performed using the Student’s t-test, and the Holm-Sidak method for multiple pairwise comparisons with help from Prism 8 software (GraphPad Software) as well as ANOVA for multi-group comparisons. A p-value of 0.05 was considered statistically significant.
Results
Continued production of Bb-specific IgM after infection
The presence of pathogen-specific serum IgM (with or without IgG) is often used as a diagnostic criterion of early infections. However, infection with Bb has been reported to induce the long-term elaboration of Borrelia-specific IgM in humans (8, 9) and mice (7). We previously showed that during acute infection, IgM antibodies binding to both, lysates of culture-grown Bb as well as to a pool of four immunodominant recombinant Borrelia proteins: Arthritis-resolving protein (Arp, Genbank AF050212), Outer surface protein C (OspC, AY275221), BmpA (AF288609.1) and Decorin-binding protein A (DbpA, AF069252.1) (3, 5), which we also used to assess anti-Borrelia antibodies here.
Borrelia-specific serum IgM increased during the first 30 days of infection of mice with host-adapted spirochetes and persisted for at least 24 weeks (Fig. 1A). 120-day infected mice lacking CD40L, a critical co-stimulator of T-dependent B cell activation, showed similar levels of anti-Borrelia IgM compared to controls, indicating that IgM production did not require cognate T cell help (Fig. 1B). 30-day treatment of mice with antibiotics (ceftriaxone) starting from 45 day after infection let to a significant drop in serum IgM levels against both, Arp and DbpA, compared to Bb-infected and sham-treated mice (Fig. 1C), similar to what we had observed previously for anti-Borrelia IgG responses (28). However, even 5 months after infection serum IgM levels were significantly higher than prior to infection in both the treated and non-treated groups (p = 0.024 and p = 0.029, respectively). Thus, persistent infection with Bb leads to long-term production of T-independent IgM such that IgM and IgG co-production occurred throughout infection (Fig. 1A). Despite extensive antibiotic treatment, Bb-IgM production was retained for months after infection to levels above those prior to infection.
Figure 1. Continuous production of T-independent IgM following Bb infection.
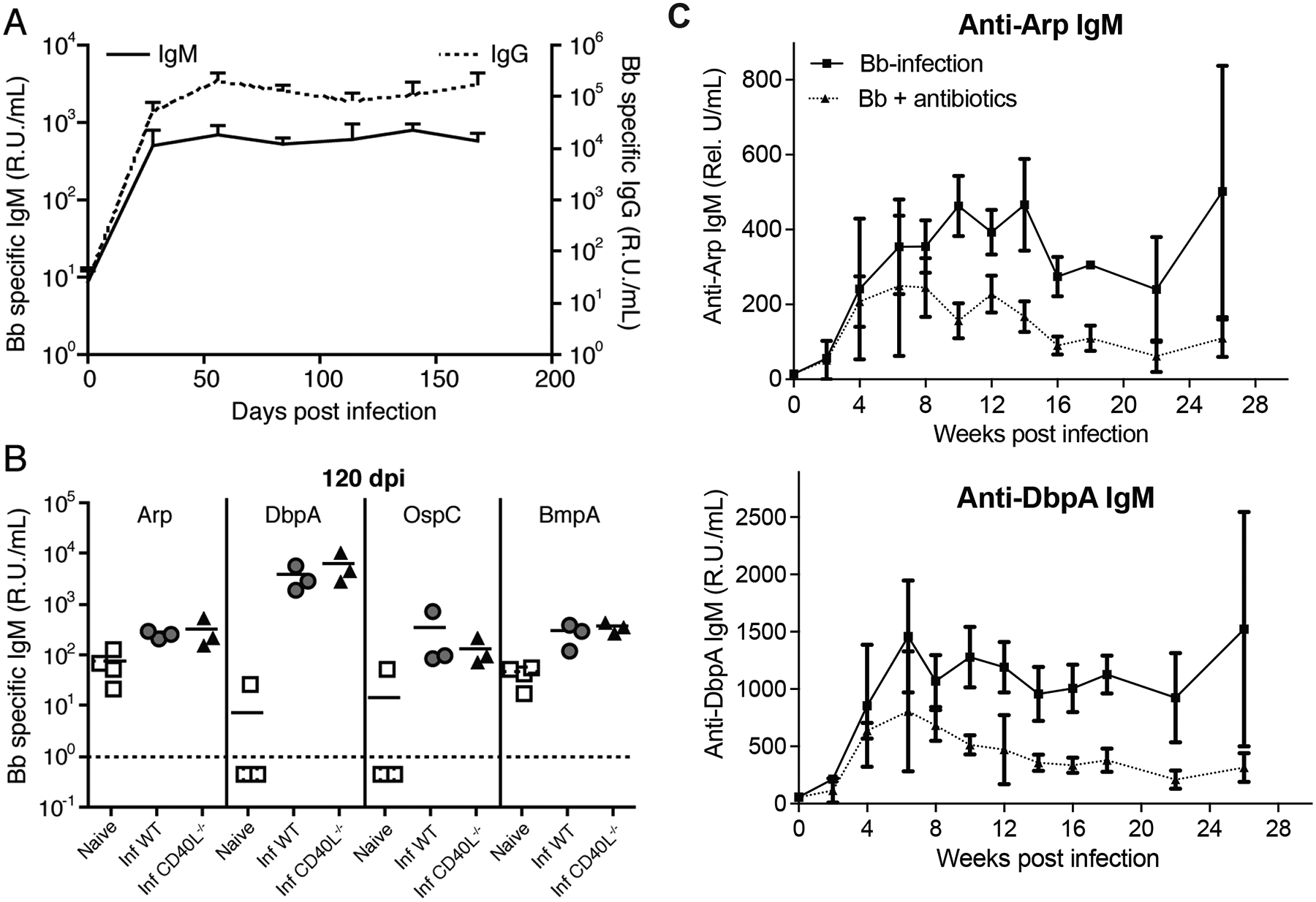
(A) Shown are relative units Bb-specific serum IgM (stippled line) and IgG (solid line) of mice at indicated times after infection with host-adapted spirochetes, as assessed by ELISA with a pool of recombinant Borrelia-proteins (DbpA, Arp, OspC, and BmpA). Each data point represents the mean of the group, + SD (n = 5). Data are from one experiment. Serum IgG titers are shown for comparison and have been reported previously (5). (B) Shown are relative units Bb-specific serum IgM to Borrelia proteins Arp, DbpA, OspC, BmpA in non-infected “naïve” mice, as well as B6 (WT) and congenic CD40L−/− mice day 120 post infection (inf) as assessed by ELISA. Symbols indicate results from individual mice and horizontal line indicates the mean for the group. (C) Relative units Arp- (top) and DbpA- (bottom) specific serum IgM of mice (n = 4) at indicated times after infection treated or not with ceftriaxone for 30 days starting on day 45.
IgM responses are generated mainly by follicular B cells
Given previous studies reporting that protective serum IgM responses to B. hermsii are derived from innate-like, T-independent CD5neg B-1 cell responses (15), we considered whether the IgM response to Bb response was due to the activation of B-1 cells. To determine the contribution of B-1 cells to Bb-induced immunity, we used an antibody-mediated B cell depletion and adoptive transfer approach to generate chimeric mice in which B-1 and B-2 cells and the IgM they secrete can be distinguished by the Ig-allotype they express (Fig. 2A and Materials and Methods). In the chimeric mice the conventional B cells are derived exclusively from the bone marrow of the host mice (here Igh-b), while the B-1 cells are derived nearly exclusively from the transfer of congenic but allotype-disparate peritoneal cavity B-1 cells (here Igh-a). This approach allows identification of B lineage cells by criteria other than their potentially changing phenotype B cells. We have extensively validated this approach in the past (11, 19, 20, 24, 29). FACS-analysis confirmed that CD19+ IgMa+ IgDa+ cells were CD23- CD43+ CD5+ and CD5- B-1 cells (Fig. 2B), and IgDbhi IgMblo B cells were host bone marrow-derived follicular B cells (Fig. 2C).
Figure 2. B-1 cells do not contribute significantly to IgM production in spleen or LNs after Bb infection.
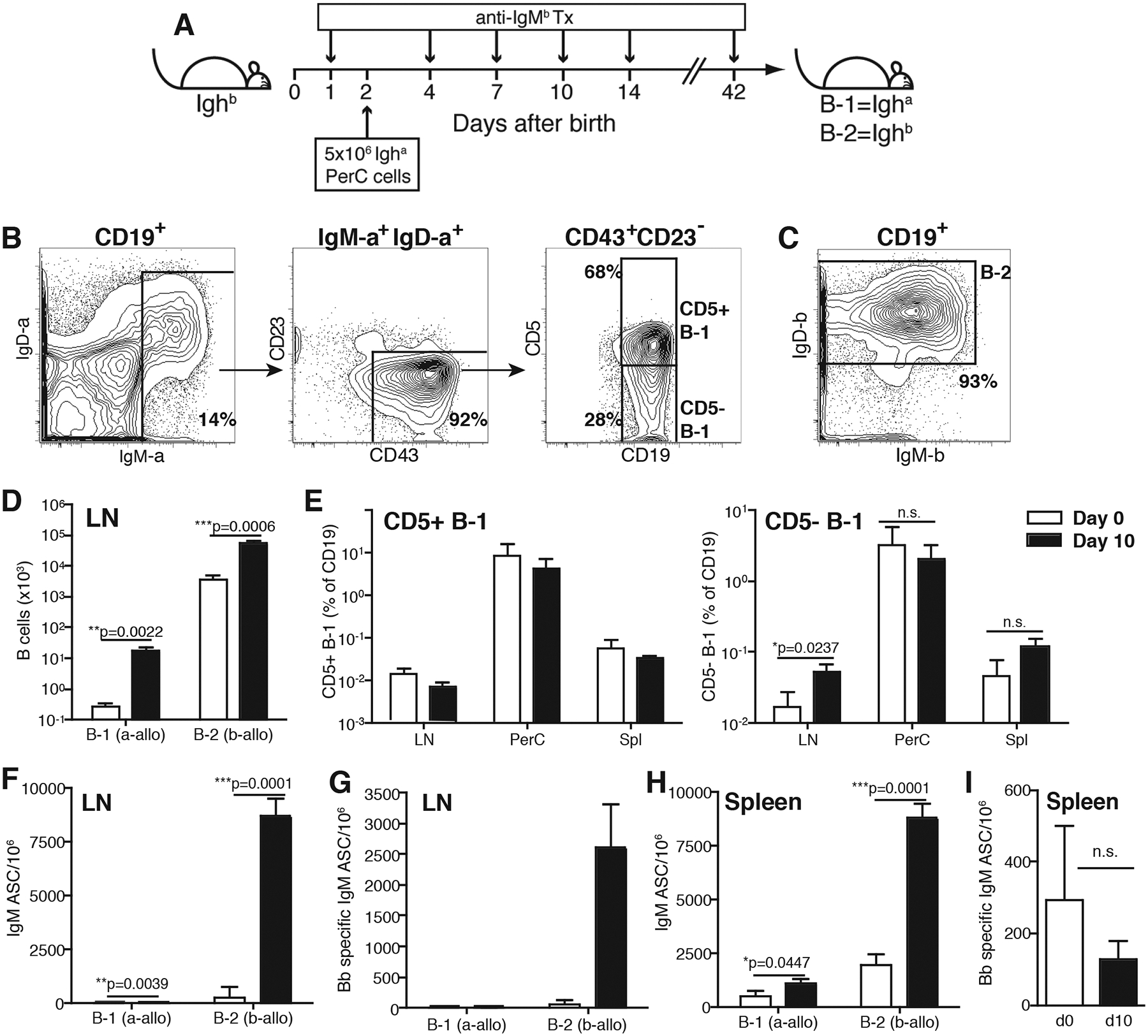
(A) Experimental outline of neonatal Ig-allotype chimera generation. (B) Representative 5% FACS contour plots with outliers of CD19+ B cells from the peritoneal cavity of non-infected mice after gating of live single CD19+ lymphocytes. B-1 cells were identified as IgM-a+ IgD-alo (left panel) CD43+ and CD23− (middle). B-1 cells were further separated based on CD5 expression (right). (C) B-2 cells were identified as host-derived IgM-b+/− IgDhi cells. (D) Total numbers lymph node (LN) CD19+ B cells that were B-1 and B-2, as assessed by Ig-allotype. Bars indicate the mean number + SD before (open) and at 10 days after (filled) infection; (E) relative frequencies CD5+ and CD5- B-1 cell subsets in lymph node (LN), peritoneal cavity wash-out (PerC) and spleen (Spl). (F) Mean numbers + SD of total and (G) Bb-specific IgM-secreting B-1 (Igh-a) and B-2 (Igh-b) cells in LN; (H) Mean numbers + SD of total IgM-secreting B-1 (Igh-a) and B-2 (Igh-b) cells in spleen of chimeras. (I) Total Bb-specific IgM-secreting ASCs in the spleen of B6 mice prior to and at 10 days after Bb infection (n=4/group). Significant statistical values as indicated using unpaired, two-tailed Student’s t-test. Results are from one of two (A-H, n = 3/group) and 4 (I; n=4/group) experiments that gave similar results.
Bb infection of the chimeras resulted in a significant increase of both, B-1 and B-2 cells in the draining lymph nodes (LNs) at day 10 post infection, although the total number of B-1 cells remained very low (Fig. 2D). Only the CD5neg but not CD5+ B-1 cells accumulated to higher levels in the LNs, while their numbers remained unchanged by the infection in peritoneal cavity and spleen (Fig. 2E). This was expected, as our recent studies have identified the CD5− B-1 cells accumulating in draining LNs after infections (influenza and Salmonella) as activated CD5+ B-1 cells that have lost CD5 (30). Total (Fig. 2F), but not Bb-specific (Fig. 2G) IgM secretion by LN B-1 cells increased, when compared to non-infected mice, the vast majority of IgM was produced by host bone marrow-derived, thus mostly B-2 cells, in the LNs (Fig. 2F, G). Nonetheless, the latter may contain a very small amount (<1%) of CD5- B-1 cell-derived IgM, given that previous studies suggested that up to 10% of CD5- B-1 cells to be host-derived in these Ig-allotype chimeras (11, 19, 29). B-1 cells In the spleen of the chimeras, both B-1 and B-2 cell-derived total IgM increased significantly (Fig. 2H), possibly due to the activation of marginal zone B cells (31). Studies in B6 mice showed no significant increases in the frequency of Borrelia-specific IgM-secreting cells in the spleens of day 10 infected B6 mice over a relatively high background, when probed with the cocktail of four recombinant proteins (Fig. 2I). The background presumably represents natural IgM secreting cells (3, 5). Collectively the data demonstrate that conventional LN B cells are major producers of the acute Bb-specific and T-independent IgM response. The data further indicate increased production of non-Bb specific IgM in the spleens in response to Bb infection.
IgM has passive protective capacity
Next, we studied the passive protective capacity of the IgM secreted in response to Bb infection. For this we adoptively transferred graded doses affinity chromatography-purified IgM from 60-day immune serum of either C57BL/6 or T cell-deficient (TCRβ/δ−/−) mice into three-week old mouse pubs, which were challenged with 104 Bb spirochetes 18h later via syringe inoculation. One-week old pups were chosen because, due to their smaller size, they require less serum Ig for protection compared to adult mice. The level of protection was determined by establishing duplicate bacterial cultures of three organs (heart base, urinary bladder and LNs) from each recipient two-weeks after challenge. Mice were scored as positive if at least one culture showed the presence of spirochetes two weeks after culture onset. As little as 18μg purified immune IgM/mouse, derived from either wild type or T-deficient (TCRβ/δ−/−) mice provided full immune protection, while 9μg IgM was partially protective (Table 1). Consistent with the T-independent nature of the IgM (Fig. 1B), there was no difference in the levels of protection with IgM purified from infected wild type or TCRβδ−/− mice. Furthermore, immune serum from TCRβδ−/− also provided full passive protection, while serum from non-infected B cell-deficient mice was non-protective, as expected (Table 1). Thus, infection-induced, T cell-independent IgM can provide passive protection from Bb challenge.
Table 1.
Protective capacity of purified IgMa
| Serum Origin | Day after Infection | Passive Transfer | Dose | a Culture Positive/Total |
|---|---|---|---|---|
| B-deficient (μMT) | Day 0 | Serum | 50 μL | 3/3 |
| TCRβ/δ −/− | Day 60 | Serum | 50 μL | 0/3 |
| B6 | Day 60 | IgM | 75 μg | 0/4 |
| 37 μg | 0/4 | |||
| 18 μg | 0/4 | |||
| 9 μg | 1/4 | |||
| βδ T cell KO | Day 60 | IgM | 75 μg | 0/4 |
| 37 μg | 0/4 | |||
| 18 μg | 0/4 | |||
| 9 μg | 2/4 |
Number of Bb-culture positive /total number tested. For each mouse multiple organs were cultured in duplicate for two weeks to grow Bb. Animals were regarded positive if at last one culture contained Bb.
Bactericidal activity of immune sera is independent of sIgM
To explore potential mechanisms of sIgM-mediated immune protection we studied Bb infection in secreted (s)IgM-deficient mice (sIgM−/−). B cells of sIgM−/− mice express surface IgM, and can undergo class-switch recombination, but they cannot secrete IgM due to the deletion of the splice region for sIgM (16). Total serum IgG and IgA concentrations were comparable between C57BL/6 and sIgM−/− mice infected for 31 days with Bb (Suppl. Fig. 1A, C). As seen previously following influenza infection (11, 32), sIgM−/− mice mounted reduced Bb-specific IgG responses, when measured against a cocktail of 4 recombinant Bb proteins compared to wild type C57BL/6 mice (Fig. 3A, p< 0.0015 by two-way ANOVA). Since sIgM−/− mice express the Igh-a allele and C57BL/6 express Igh-b, we also measured Bb-specific responses in Igh-a expressing, C57BL/6 congenic wild type mice. Those mice showed similar serum IgG responses as C57BL/6 mice, but enhanced responses compared to sIgM−/− mice (Fig. 3A). Similarly, sIgM−/− mice had reduced Bb IgG compared to C57BL/6 mice on day 31 after infection when tested on bacterial lysates (Suppl. Fig. 1A). The strongest reductions were noted against DbpA (Suppl. Fig. 1B). As expected, IgM was absent in sIgM−/− mice (Suppl. Fig. 1D). Western-blot analysis did not reveal obvious differences in the pattern of bands from Borrelia lysates recognized by immune serum from either mouse strain (Suppl. Fig. 1E).
We then compared the in vitro borreliacidal activity of sera from day 31-infected C57BL/6 mice and sIgM−/− mice. Serum from infected SCID mice, lacking both sIgM and IgG, served as a negative control (Fig. 3B). Matching the volume of sera from control and sIgM−/−, but not from infected SCID mice, when added to the cultures strongly, and in a dose-dependent manner facilitated killing of spirochetes in vitro, indicating the presence of antibodies with strong borreliacidal activity (Fig. 3B). The activity was lost when sera were heat-inactivated, presumably due to the loss of complement (not shown and see below). In multiple repeats of this assay, we found no consistent statistically significant differences in the bactericidal activity of sIgM−/− and control sera. This was surprising given the relatively large amount Borrelia-IgM in the sera of wild type mice and the reduction in IgG (Fig. 3A).
To determine whether enhanced bactericidal activity of purified IgG in the sera of sIgM−/− mice might offset the lack of sIgM and reduced amounts of IgG, we conducted similar in vitro studies comparing purified IgG from day 31 Bb infected C57BL/6 and sIgM−/− mice. Addition of 5μg IgG from either mouse strain resulted in strong borreliacidal activity when complement was provided to the cultures in form of serum from non-infected RAG1−/− mice. Control IgG from influenza infected mice had no significant effect (Fig. 3C). We repeated the assay also comparing bactericidal activity of graded doses purified IgG from Bb-infected C57BL/6-congenic mice expressing Igh-a. Again, maximal killing of Borrelia was achieved with about 5μg/ml immune-IgG, independent of the Ig-allotype (Fig. 3D). The activity required the presence of complement (Fig. 3D). Together the data suggested that despite the passive immune protective capacity of sIgM, the absence of sIgM does not significantly reduce the antibody-mediated in vitro borreliacidal activity of Bb-immune serum, or its purified IgG.
IgM does not significantly contribute to serum-mediated passive protection
Despite the potential of sIgM to provide immune protection in adaptive transfer experiments (Table 1), the in vitro studies did not support a strong contribution of sIgM in complement-mediated Bb killing. To determine whether the Bb-induced serum IgM contributed significantly to passive protection in the context of whole serum transfer, we compared the passive protective capacity of sera from C57BL/6 mice with that of sIgM-/−. Groups of three-week old mouse pubs (n= 5–7/group) received graded doses (50, 15, and 10 μg/mouse) of serum adjusted to total Ig, as assessed by ELISA. A control group received PBS only. All groups of mice were challenged with 104 Bb spirochetes 18h later via syringe inoculation. Two weeks later, three tissues were taken, and DNA extracted for analysis of Bb copy number by PCR for Bb FlaB (Table 2). All mice that received PBS or sera from non-infected mice were positive for FlaB with high Bb copy numbers. In contrast, all but one mouse in each group were protected from Bb challenge when mice received 50 μg and 15 μg total Ig from either sIgM−/− or control mice (Table 2). The one infected mouse per group had greatly reduced copy numbers compared to the PBS or non-immune serum controls (Table 2). Partial protection was afforded by transfer of 10 μg/mouse immune serum and no significant differences were seen between C57BL/6 and sIgM−/− mice (Table 2). Thus, while IgM can provide passive protection, it did not seem to play a significant role in serum-mediated passive immune protection.
Table 2.
The passive protective capacity of wildtype and sIgM−/− sera
| C57BL/6 Serum | sIgM−/− Serum | PBS (100 μL) | ||
|---|---|---|---|---|
| Inoculation sitea | d0 sera |
5/5b 19,309 (34,580)c |
5/5 2,304 (677) |
7/7 102,841 (241647) |
| 50 μg |
1/4 0.2 (0.4) |
1/4 0.2 (0.5) |
||
| 15 μg |
1/7 6 (16) |
1/7 68 (181) |
||
| 10 μg |
3/7 315 (729) |
2/7 43 (110) |
||
| Urinary bladder | d0 sera |
4/5 6,605 (14,258) |
2/5 639 (908) |
6/7 2,016 (2454) |
| 50 μg |
0/4 0 (0) |
0/4 0 (0) |
||
| 15 μg |
0/7 0 (0) |
0/7 0 (0) |
||
| 10 μg |
3/7 67 (142) |
0/7 0 (0) |
||
| Heart base | d0 sera |
5/5 42,419 (69,133) |
5/5 8,258 (10,787) |
7/7 102,841 (241647) |
| 50 μg |
1/4 1 (2) |
1/4 1 (2) |
||
| 15 μg |
1/7 73 (193) |
1/7 89 (237) |
||
| 10 μg |
4/7 322 (407) |
2/7 89 (154) |
Groups of 3-week-old CD-1 pups (n=7) were treated with 100μl total serum from non-infected (d0 sera) wildtype or sIgM−/− mice, or with sera containing indicated amounts of total Ig from sera of day 31 infected mice as assessed by ELISA. All groups were challenged with Bb 18 hours post serum treatment and inoculation site, urinary bladder and heart base were collected from individual mice and assayed by qPCR for copy number of Bb FlaB.
Number of pups positive by PCR/total number of pups in the group
Mean copy number (SD) of Bb flagellin/mg of tissue of mice positive for B. burgdorferi
Secreted (s)IgM controls bacteremia but not bacterial tissue burden following Bb infection
To further determine the impact of sIgM on the control of Bb infection we compared Borrelia tissue burdens in the LNs, heart base and tibiotarsus joints in C57BL/6 and sIgM−/− mice by qPCR. Despite the significant and continued production of sIgM throughout the infection (Fig. 1A), sIgM did not have a noticeable impact of either Bb tissue load or dissemination kinetics (Fig. 4A). The one exception was a significant increase in Borrelia-burden at day 7 in the heart base of sIgM−/− compared to C57BL/6 control mice (p < 0.028; Fig, 4A, middle panel). The absence of sIgM also did not affect the overall very modest arthritis and carditis manifestations in mice on the B6 background at 121 days after infection (Suppl. Table 1).
Figure 4. IgM controls Bb bacteremia but not tissue burden.
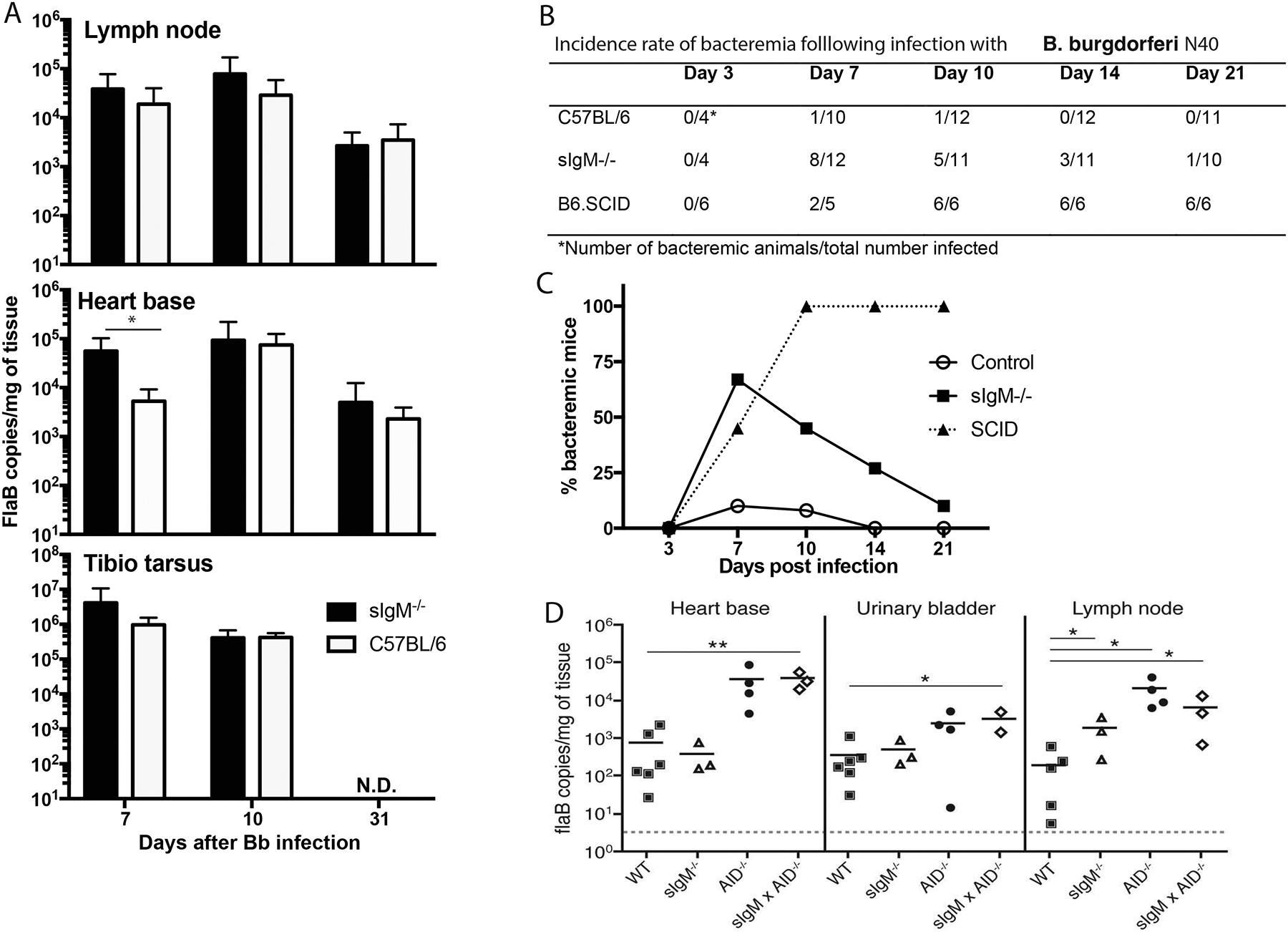
(A) Mean ± SD Bb tissue burden as assessed by qPCR, measuring flaB DNA copy number in indicated tissues of female C57BL/6 controls and sex-matched sIgM−/− mice at indicated days after infection. (B) Incidence of bacteremia in C57BL/6, sIgM−/− and B6.SCID mice at indicated time points after infection with host-adapted Bb N40 (n = 4–12/time point), as assessed by cultures of blood samples. (C) Data from (B) expressed as frequency bacteremic animals per group (culture positive/total infected). (D) Bb tissue burden of indicated tissues of day 121 Bb-infected female B6 (WT), sIgM−/−, AID−/− (mice lacking all class-switched antibodies) and sIgM−/− × AID−/− mice, (mice lacking all secreted antibodies). Each symbol represents one mouse, horizontal line indicates the mean for the group. Adjusted p-values were calculated using the Holm-Sidak method for pairwise multiple comparisons showing differences to WT controls. *p < 0.05; **p < 0.001. N.D., not determined.
Given the rapid dissemination of Bb into various organs, we compared the incidence rate of bacteremia in these mice. In contrast to the lack of effect on Borrelia tissue-loads, the presence of sIgM had a strong and significant impact on the control of bacteremia, particularly during the first two weeks of infection. While bacteremia was observed in at most 10% of infected B6 mice at any time point tested (Fig. 4B, C), over 70% of infected sIgM−/− mice had culturable bacteria in their blood on day 7 of infection, and bacteremia was observed still at day 21 (Fig. 4B, C). As the infection progressed the impact of sIgM became less pronounced, presumably due to the appearance of IgG (Fig. 3A). This is supported by analysis of total Ig-deficient SCID mice, in which all animals became bacteremic by day 10 and remained so throughout the study (Figure 4B, C). Thus, a major role of sIgM was identified in the control of Bb in the blood.
Next we compared the Borrelia tissue loads measured in C57BL/6 and sIgM−/− mice with that of aicda (AID−/−) mice (17), which lack the ability to generate any class-switched Ig but have supra-normal serum IgM concentrations, and with mice lacking all secreted Ig due to a lack of both, sIgM and aicda (sIgM × AID−/−) at day 121 after Bb infection. The data confirmed a requirement for IgG but not IgM in control of Borrelia tissue loads (Fig. 4D). The lack of class-switched antibodies alone in the AID−/− mice enhanced Borrelia-loads to levels similar to that of mice lacking all antibody secretion. And their levels were significantly higher than that of mice that lacked only sIgM or that had normal antibody levels (Fig. 4D). Similar results were obtained when we compared sIgM−/− and Ig-deficient (sIgM−/− × AID−/− mice) on day 30 of infection, the latter showing significantly increased Bb tissue burden (Suppl. Fig. 2A). Since sIgM−/− mice were engineered to express the Igh-a allotype we compared the sIgM−/− to control C57BL/6 mice carrying the Igha allotype. Similar to our previous studies, there was no significant difference in Bb tissue loads between sIgM−/− and wild type Igha-expressing mice, either on day 30 (Suppl. Fig. 2A), or on day 120 (Suppl. Fig. 2B) after Bb infection, suggesting that the Igh-allotype does not significantly affect the quality of the humoral response to Bb.
Together the data demonstrated that IgG but not IgM is critical for the long-term control of Bb tissue-burden or disease induction. Despite that, Borrelia tissue dissemination in mice appeared very little affected by the rate of bacteremia, suggesting Bb main mode of dissemination in mice occurs by means other than via the blood.
Bb infection enhances IgM responses
We had previously noted the relative abundance of IgM secretion in the lymph nodes of Bb infected mice, with Bb-specific antibody production contributing nearly half of antibody-secreting cells even beyond early timepoints of infection (3). Given that IgG but not sIgM controls Bb tissue loads, we aimed to determine whether Borrelia-infection steers immune responses towards exaggerated IgM production. For that we measured antibody-responses to immunization with an unrelated viral antigen in mice simultaneously infected with Bb (Fig. 5A). We chose influenza A/Puerto Rico/8/34 (A/PR8), as it results in strong IgG and only short-lived IgM responses (33). Both groups of mice also received A/PR8-specific transgenic CD4 T cells to provide strong T cell help. Remarkably, the frequencies of A/PR8-specific IgM secreting cells were strongly increased in the LNs of mice that were infected 10 days prior with Bb compared to mice that received A/PR8 immunization alone (Fig. 5B). In addition, 9 weeks after immunization significantly greater numbers of A/PR8-specific IgM-secreting plasma cells were found in the bone marrow of mice infected with Bb (Fig. 5C). Thus, infection with Bb drives B cell responses towards production of IgM, which reduces bacteremia and provides passive protection, but cannot affect Bb once disseminated into solid tissues.
Figure 5. Infection with Bb increases IgM responses to an irrelevant antigen.
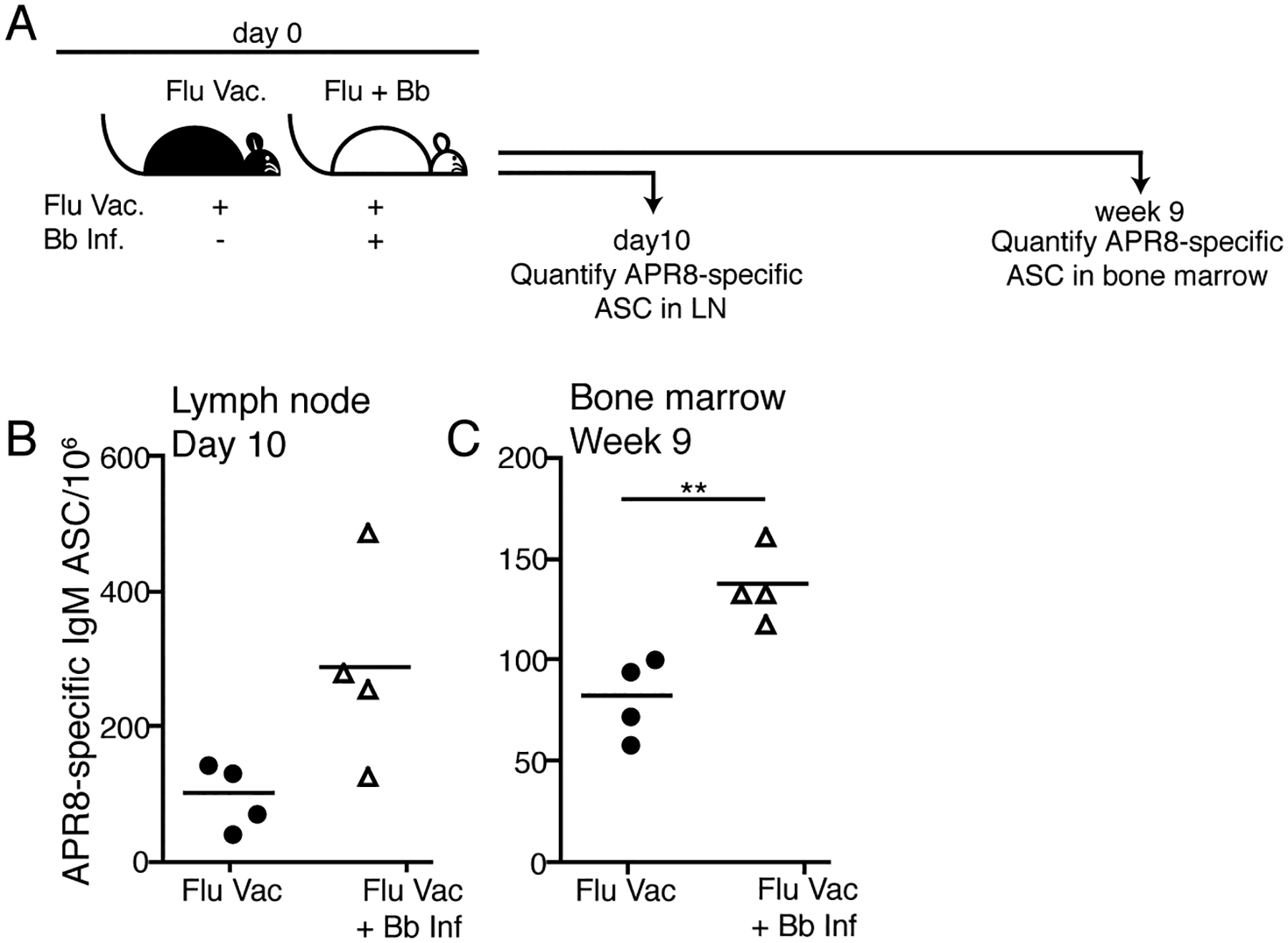
(A) Experimental outline. Two groups of BALB/cByJ mice (n=4 per group) received 1 × 106 CD4 transgenic T cells specific for the hemagglutinin of influenza A/PR8 and were vaccinated s.c. with influenza A/PR8 in alum. One group was also infected with Bb. (B) At day 10 and (C) 9 weeks after infection, respectively, right inguinal LNs and bone marrow were analyzed by ELISPOT to quantify APR8-specific IgM ASC. Each symbol represents one mouse, horizontal line indicates the mean for the group. Data are representative of two independent experiments. Significant statistical values were determined using unpaired, one-tailed Student’s t-test.
Discussion
The complex enzootic life cycle of Bb requires the establishment of infected host reservoirs that enable the continued infection of newly hatched tick larvae and nymphs (34). Mice are one such reservoir species and the investigation into their immune responses can provide important clues about immune evasion strategies the bacteria have developed to establish persistence. Data presented here suggest that the regulation of B cell responses away from class-switched IgG and towards IgM production is one such mechanism. While immune IgM could provide passive immune protection, it did not affect Borrelia-clearance from solid host tissues, presumably due to its large size and the absence of continued tissue-inflammation during infection that otherwise could facilitate IgM extravasation. Interestingly, a heightened induction of IgM was observed not only to Bb, but also to a co-administered vaccine antigen, further strongly suggesting that it is not the quality of the Borrelia antigens, i.e. lipoproteins and carbohydrates, that drives the induction of IgM, but the inflammatory conditions induced by Bb (3, 35). Thus, we propose that the continued production of immune IgM is a manifestation of Bb-mediated B cell response subversion and represents an immune evasion strategy of Bb. It may promote Bb dissemination out of the blood and into the skin, where it can remain until attachment and bite of a tick will induce it to migrate towards the site of the tick-bite.
Strong humoral responses to many infections, both acute and chronic, often induce a serological response pattern characterized by the early and transient appearance of IgM that disappears within a couple weeks following initial infection, followed by an IgG response that is delayed by a few days, but then raises and remains present throughout infection (5). From this, critical serological diagnostic testing and interpretation have been developed that are widely used in the clinics, where the simultaneous measurement of IgM and IgG is used to diagnose acute (IgM alone or IgM plus IgG) from later (IgG only) stages of disease (36). While such a pattern is observed widely with acute resolving infections, it is clear that such as “switch” from IgM to IgG does not occurs in response to all infections. For example, others had previously reported continued IgM production in response to infection of mice with the intracellular bacterium Ehrlichia muris (13). With regards to infection with Bb, our findings are consistent with findings in human patients, in which the continued presence of IgM in patients with positive confirmed diagnosis of Lyme disease was reported previously (8, 9, 37). Thus, classification of Bb infection based on the ratio of IgM : IgG into acute and chronic appears problematic.
Others showed previously that the multiple binding sites of the mostly pentameric IgM, compared to the monomeric IgG, as well as the flexibility of its molecular structure allow for better “coating” of bacterial surface antigens compared to IgG, and thus that IgM might have superior protective capacity (10). However, our in vitro studies did not support a superior bactericidal activity of sIgM at least for complement-mediated killing of Bb, given that the absence of sIgM from serum had no measurable effects on serum bactericidal activity. Interestingly, some previous studies suggested that the presence of sIgM can be detrimental for the host. During malaria infections non-specific IgM was shown to mask IgG binding sites on Plasmodium falciparum (38). During infection with Schistosoma mansoni, specific IgM was shown to bind the outer surface of this parasite and to block eosinophil-mediated killing (39), while IgG of identical specificity supported parasite killing. During Salmonella infection excess antibodies against an erroneous antigen was reported to block the protective capacity of other antigen specific antibodies (40). In that study, HIV infected individuals with non-typhoidal Salmonella infection had an excess of non-bactericidal lipopolysaccharide-specific IgG, which when bound to Salmonella blocked access for bactericidal outer membrane-specific IgG (40).
Of significance, previously studies showed a non-redundant role for IgM in the control of B. hermsii infection, a pathogen closely related to Bb (15). However, the pathogenesis of these two Borrelia-species is distinct, especially with regards to their tissue distribution. B. hermsii causes strong bacteremia and only sporadically infects tissues. In contrast, Bb predominantly resides in connective tissues of multiple organs with only episodic periods of bacteremia (41). Given its size, pentameric IgM may be most effective in binding to pathogens in the blood, as it seems to be able to extravasate into tissues only in the presence of inflammation and other forms of vascular leakage. Continued infection of Bb, in its persistent state, causes very little inflammation. Thus, Bb may evade the immune system by inducing strong IgM responses and residing in tissue where IgM is unable to reach.
It was surprising that the strong increase in the rate of bacteremia in mice lacking sIgM did not significantly affect bacterial burden in the various organs we tested, given that hematogenous dissemination of Bb has been previously indicated (reviewed in (42)). Instead, our data suggest that at least in mice, bacterial dissemination via blood may not contribute greatly to the rapid dissemination of Bb after infection. This is consistent with our previous findings that the dissemination to lymphoid tissues seem to follow lymph, rather than blood flow (3). However, it is conceivable that higher bacterial rates in blood of sIgM−/− mice may induce stronger levels of systemic inflammation and a heightened immune response. However, we did not note any significant differences in clinical or pathological signs of disease between wild type and sIgM−/− mice (Suppl. Table 1).
While much of the Bb infection-induced IgM is generated independent of T cell help (5) and previous studies showed a role for B-1b cell-derived IgM in control of B. hermsii infection (15), we demonstrate here that the vast majority of the IgM response to Bb is produced by conventional B cells (Fig. 2). Only relatively few B-1 cells accumulated in the LNs, and their contribution to overall IgM production was low. The data agree with our findings that the induction of conventional B cell responses to an unrelated vaccine antigen (influenza) shifted towards increased IgM production during infection with Bb. This remarkable effect of the infection on promoting IgM responses rules out the nature of the Bb-antigens alone as the main driver of an IgM-dominated response. Instead, the data suggest that Bb might actively drive IgM over IgG responses.
IgM antibodies are evolutionarily conserved, participating in immune responses as far back as the jawed fish (43). Our findings that Bb might “exploit” this response to its advantage provides an opportunity not only to better understand Bb infections and to identify the unique serological fingerprints that aids their diagnosis, but also to explore the varied functions of this often-overlooked immunoglobulin isotype.
Supplementary Material
Key Points.
B. burgdorferi induces B-2 cells to continuously secrete passively protective IgM
IgM suppresses B. burgdorferi burden in the blood but not in tissues
B. burgdorferi promotes enhanced IgM secretion after vaccination
Acknowledgments
We thank Abigail Spinner for help in operating the BD FACSAria, Dr. Stefan Tunev for IgM purification and passive transfer studies (UC Davis), and Edlin Escobar (UC Davis) for help with PCR analysis.
This work was supported by grants from the Global Lyme Alliance, the Stephen and Alexandra Cohen Foundation, NIH/NIAID R01AI073911 (S.W.B & N.B) R01AI157007 (to N.B), NIH training grants T32-AI060555 (C.J.H) and T-32AI074550 (R.A.E). The work was supported also by the Assistant Secretary of Defense for Health Affairs, through the Tick Borne Disease Research Program under Award No. W81XWH-18-1-0611. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office.
Abbreviations:
- Arp
arthritis-related protein
- Bb
Borrelia burgdorferi
- DbpA
Decorin-binding protein A
- HA
hemagglutinin
- LN
lymph node
- OspC
Outer surface protein C
- sIgM
secreted IgM
References
- 1.Steere AC 2001. Lyme disease. The New England journal of medicine 345: 115–125. [DOI] [PubMed] [Google Scholar]
- 2.Bacon RM, Kugeler KJ, and Mead PS. 2008. Surveillance for Lyme disease--United States, 1992–2006. MMWR. Surveillance summaries : Morbidity and mortality weekly report. Surveillance summaries / CDC 57: 1–9. [PubMed] [Google Scholar]
- 3.Tunev SS, Hastey CJ, Hodzic E, Feng S, Barthold SW, and Baumgarth N. 2011. Lymphoadenopathy during lyme borreliosis is caused by spirochete migration-induced specific B cell activation. PLoS pathogens 7: e1002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hastey CJ, Ochoa J, Olsen KJ, Barthold SW, and Baumgarth N. 2014. MyD88- and TRIF-independent induction of type I interferon drives naive B cell accumulation but not loss of lymph node architecture in Lyme disease. Infect Immun 82: 1548–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hastey CJ, Elsner RA, Barthold SW, and Baumgarth N. 2012. Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J Immunol 188: 5612–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elsner RA, Hastey CJ, and Baumgarth N. 2015. CD4(+) T Cells Promote Antibody Production but Not Sustained Affinity Maturation during Borrelia burgdorferi Infection. Infection and Immunity 83: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu N, Montgomery RR, Barthold SW, and Bockenstedt LK. 2004. Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect Immun 72: 3195–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguero-Rosenfeld ME, Nowakowski J, Bittker S, Cooper D, Nadelman RB, and Wormser GP. 1996. Evolution of the serologic response to Borrelia burgdorferi in treated patients with culture-confirmed erythema migrans. Journal of clinical microbiology 34: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalish RA, McHugh G, Granquist J, Shea B, Ruthazer R, and Steere AC. 2001. Persistence of immunoglobulin M or immunoglobulin G antibody responses to Borrelia burgdorferi 10–20 years after active Lyme disease. Clin Infect Dis 33: 780–785. [DOI] [PubMed] [Google Scholar]
- 10.Tobita T, Oda M, and Azuma T. 2004. Segmental flexibility and avidity of IgM in the interaction of polyvalent antigens. Molecular immunology 40: 803–811. [DOI] [PubMed] [Google Scholar]
- 11.Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, and Chen J. 2000. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med 192: 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, and Engle M. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med 198: 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, and Winslow GM. 2011. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J Immunol 186: 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbour AG, and Bundoc V. 2001. In vitro and in vivo neutralization of the relapsing fever agent Borrelia hermsii with serotype-specific immunoglobulin M antibodies. Infect Immun 69: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alugupalli KR, Gerstein RM, Chen J, Szomolanyi-Tsuda E, Woodland RT, and Leong JM. 2003. The resolution of relapsing fever borreliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J Immunol 170: 3819–3827. [DOI] [PubMed] [Google Scholar]
- 16.Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, and Chen J. 1998. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol 160: 4776–4787. [PubMed] [Google Scholar]
- 17.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563. [DOI] [PubMed] [Google Scholar]
- 18.Kumazaki K, Tirosh B, Maehr R, Boes M, Honjo T, and Ploegh HL. 2007. AID−/−mus−/− mice are agammaglobulinemic and fail to maintain B220-CD138+ plasma cells. J Immunol 178: 2192–2203. [DOI] [PubMed] [Google Scholar]
- 19.Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, and Herzenberg LA. 1999. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proceedings of the National Academy of Sciences of the United States of America 96: 2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savage HP, Yenson VM, Sawhney SS, Mousseau BJ, Lund FE, and Baumgarth N. 2017. Blimp-1-dependent and -independent natural antibody production by B-1 and B-1-derived plasma cells. J Exp Med 214: 2777–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barber CL, Montecino-Rodriguez E, and Dorshkind K. 2011. Reduced production of B-1-specified common lymphoid progenitors results in diminished potential of adult marrow to generate B-1 cells. Proceedings of the National Academy of Sciences of the United States of America 108: 13700–13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lalor PA, Herzenberg LA, Adams S, and Stall AM. 1989. Feedback regulation of murine Ly-1 B cell development. Eur J Immunol 19: 507–513. [DOI] [PubMed] [Google Scholar]
- 23.Forster I, and Rajewsky K. 1987. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic, newborn mice. Eur J Immunol 17: 521–528. [DOI] [PubMed] [Google Scholar]
- 24.Choi YS, and Baumgarth N. 2008. Dual role for B-1a cells in immunity to influenza virus infection. J Exp Med 205: 3053–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doucett VP, Gerhard W, Owler K, Curry D, Brown L, and Baumgarth N. 2005. Enumeration and characterization of virus-specific B cells by multicolor flow cytometry. Journal of immunological methods 303: 40–52. [DOI] [PubMed] [Google Scholar]
- 26.Rothaeusler K, and Baumgarth N. 2006. Evaluation of intranuclear BrdU detection procedures for use in multicolor flow cytometry. Cytometry. Part A : the journal of the International Society for Analytical Cytology 69: 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodzic E, Feng S, Freet KJ, Borjesson DL, and Barthold SW. 2002. Borrelia burgdorferi population kinetics and selected gene expression at the host-vector interface. Infect Immun 70: 3382–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elsner RA, Hastey CJ, Olsen KJ, and Baumgarth N. 2015. Suppression of Long-Lived Humoral Immunity Following Borrelia burgdorferi Infection. PLoS pathogens 11: e1004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith FL, Savage HP, Luo Z, Tipton CM, Lee FE, Apostol AC, Beaudin AE, Lopez DA, Jensen I, Keller S, and Baumgarth N. 2023. B-1 plasma cells require non-cognate CD4 T cell help to generate a unique repertoire of natural IgM. J Exp Med 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savage HP, Kläsener K, Smith FL, Luo Z, Reth M, and Baumgarth N. 2019. TLR induces reorganization of the IgM-BCR complex regulating murine B-1 cell responses to infections. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belperron AA, Dailey CM, Booth CJ, and Bockenstedt LK. 2007. Marginal zone B-cell depletion impairs murine host defense against Borrelia burgdorferi infection. Infect Immun 75: 3354–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen TT, Elsner RA, and Baumgarth N. 2015. Natural IgM prevents autoimmunity by enforcing B cell central tolerance induction. J Immunol 194: 1489–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf AI, Mozdzanowska K, Quinn WJ 3rd, Metzgar M, Williams KL, Caton AJ, Meffre E, Bram RJ, Erickson LD, Allman D, Cancro MP, and Erikson J. 2011. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. The Journal of clinical investigation 121: 3954–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart PE, and Rosa PA. 2018. Physiologic and Genetic Factors Influencing the Zoonotic Cycle of Borrelia burgdorferi. Curr Top Microbiol Immunol 415: 63–82. [DOI] [PubMed] [Google Scholar]
- 35.Tracy KE, and Baumgarth N. 2017. Borrelia burgdorferi Manipulates Innate and Adaptive Immunity to Establish Persistence in Rodent Reservoir Hosts. Front Immunol 8: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dressler F, Whalen JA, Reinhardt BN, and Steere AC. 1993. Western blotting in the serodiagnosis of Lyme disease. The Journal of infectious diseases 167: 392–400. [DOI] [PubMed] [Google Scholar]
- 37.Steere AC, Grodzicki RL, Kornblatt AN, Craft JE, Barbour AG, Burgdorfer W, Schmid GP, Johnson E, and Malawista SE. 1983. The spirochetal etiology of Lyme disease. The New England journal of medicine 308: 733–740. [DOI] [PubMed] [Google Scholar]
- 38.Barfod L, Dalgaard MB, Pleman ST, Ofori MF, Pleass RJ, and Hviid L. 2011. Evasion of immunity to Plasmodium falciparum malaria by IgM masking of protective IgG epitopes in infected erythrocyte surface-exposed PfEMP1. Proceedings of the National Academy of Sciences of the United States of America 108: 12485–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khalife J, Capron M, Capron A, Grzych JM, Butterworth AE, Dunne DW, and Ouma JH. 1986. Immunity in human schistosomiasis mansoni. Regulation of protective immune mechanisms by IgM blocking antibodies. J Exp Med 164: 1626–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacLennan CA, Gilchrist JJ, Gordon MA, Cunningham AF, Cobbold M, Goodall M, Kingsley RA, van Oosterhout JJ, Msefula CL, Mandala WL, Leyton DL, Marshall JL, Gondwe EN, Bobat S, Lopez-Macias C, Doffinger R, Henderson IR, Zijlstra EE, Dougan G, Drayson MT, MacLennan IC, and Molyneux ME. 2010. Dysregulated humoral immunity to nontyphoidal Salmonella in HIV-infected African adults. Science 328: 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barthold SW, de Souza MS, Janotka JL, Smith AL, and Persing DH. 1993. Chronic Lyme borreliosis in the laboratory mouse. The American journal of pathology 143: 959–971. [PMC free article] [PubMed] [Google Scholar]
- 42.Hyde JA 2017. Borrelia burgdorferi Keeps Moving and Carries on: A Review of Borrelial Dissemination and Invasion. Front Immunol 8: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flajnik MF 2002. Comparative analyses of immunoglobulin genes: surprises and portents. Nat Rev Immunol 2: 688–698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.