

Abstract
Infection of human cells with poliovirus induces the proliferation of double-membraned cytoplasmic vesicles whose surfaces are used as the sites of viral RNA replication and whose origin is unknown. Here, we show that several hallmarks of cellular autophagosomes can be identified in poliovirus-induced vesicles, including colocalization of LAMP1 and LC3, the human homolog of Saccharomyces cerevisiae Atg8p, and staining with the fluorophore monodansylcadaverine followed by fixation. Colocalization of LC3 and LAMP1 was observed early in the poliovirus replicative cycle, in cells infected with rhinoviruses 2 and 14, and in cells that express poliovirus proteins 2BC and 3A, known to be sufficient to induce double-membraned vesicles. Stimulation of autophagy increased poliovirus yield, and inhibition of the autophagosomal pathway by 3-methyladenine or by RNA interference against mRNAs that encode two different proteins known to be required for autophagy decreased poliovirus yield. We propose that, for poliovirus and rhinovirus, components of the cellular machinery of autophagosome formation are subverted to promote viral replication. Although autophagy can serve in the innate immune response to microorganisms, our findings are inconsistent with a role for the induced autophagosome-like structures in clearance of poliovirus. Instead, we argue that these double-membraned structures provide membranous supports for viral RNA replication complexes, possibly enabling the nonlytic release of cytoplasmic contents, including progeny virions, from infected cells.
Poliovirus is a well-studied positive-strand RNA virus, and here it is shown to subvert the machinery of the cellular autophagy pathway at several stages in the viral life cycle
Introduction
Upon infection by poliovirus and many other positive-strand RNA viruses, dramatic changes are rapidly induced in the cellular environment. Poliovirus infection causes the massive rearrangement of intracellular membranes, with double-membraned vesicles 200–400 nm in diameter accumulating in the cytoplasm [1,2]. Immunoelectron microscopy has revealed that the cytoplasmic surfaces of these membranous vesicles are the sites of viral RNA replication [3,4,5] and, indeed, all known positive-strand RNA viruses of eukaryotic cells replicate their RNA on cytoplasmic membranes. It is thought that one function for membrane localization of viral RNA replication proteins is to promote their oligomerization [6,7].
It has been noted previously that several of the features displayed by the vesicles induced during poliovirus infection [1,2] are known to be shared with cellular membranous structures termed autophagosomes. During cellular autophagy, cells break down cytoplasmic proteins and organelles within autophagosomes, double-membraned structures that become degradative upon maturation. Originally identified as a process induced by cellular starvation, autophagy is now appreciated as a cellular response to a variety of stimuli, including hormone treatment, and is a feature of normal development in several organisms (reviewed in [8]). In mammalian cells, two chemical inducers of autophagy are tamoxifen [9,10] and rapamycin [11,12,13]. Cells that express the estrogen receptor respond to tamoxifen treatment by accumulating large amounts of autophagosomes [9,10]. Rapamycin inhibits the function of the mammalian target of rapamycin, a known repressor of the autophagic pathway (reviewed in [14]). In yeast, formation and maturation of autophagosomes requires the functions of many genes (reviewed in [15]). Human homologs of several of the yeast autophagy genes have been recently identified, including LC3, the human homolog of Atg8p, and it is likely that much of the autophagic pathway is conserved [16,17,18].
In mammalian cells, the formation and maturation of autophagosomes involves the stepwise acquisition of proteins from disparate cellular compartments (reviewed in [19,20,21]). Nascent autophagosomes form either de novo [22,23] or from the endoplasmic reticulum (ER) [24,25] and comprise cellular cytoplasm surrounded by two lipid bilayers that fuse from a C-shaped intermediate. Although relatively protein-poor [22,26], they have been shown to contain a modified, lipidated form of LC3 [16]. As maturation proceeds, the late autophagosome acquires LAMP1 before lysosomal fusion [16,25,27,28], which defines the “mature autolysosome.” Mature autolysosomes are no longer surrounded by double membranes and, having degraded the inner membrane and much of the cytosolic contents, become electron-dense [27,29]. Various intermediates among these stages have also been visualized, including incompletely fused double membranes and incompletely degraded inner membranes [23,24,25].
Although poliovirus-induced vesicles display several hallmarks of autophagosomes, other origins for the poliovirus-induced vesicles have been suggested [30,31,32]. Poliovirus RNA replication is known to be inhibited by brefeldin A, which has led to the suggestion that the virally induced vesicles might derive from the COPI pathway, a known target of brefeldin A [32,33,34]. However, this would be inconsistent with the apparent origin of the poliovirus-induced vesicles from the ER: many images consistent with the budding of the poliovirus-induced vesicles directly from the ER have been reported [3,31]. Furthermore, poliovirus proteins 2BC and 3A induce the formation of vesicles that are biochemically and ultrastructurally similar to those formed in poliovirus-infected cells [35]. The 3A protein and 2C, a proteolytic product of 2BC, localize to the ER when expressed in isolation [35,36]. Therefore, it is likely that poliovirus proteins 3A and 2BC, or larger precursors, localize to the ER early in infection and subsequently promote vesiculation from ER membranes.
Poliovirus-induced vesicles have been shown to contain human COPII proteins Sec13p and Sec31p early in their formation, leading to the hypothesis that they are modified COPII vesicles, subverted from the anterograde transport pathway [31]. Although this is an attractive suggestion, it is incongruent with other findings concerning the poliovirus-induced vesicles: their frequently observed double-membraned morphology, their cytosolic contents, and the abundance of components from throughout the secretory pathway, including LAMP1, a marker of late endosomes and lysosomes [1,2,35]. Furthermore, in Saccharomyces cerevisiae, components of the COPII pathway, encoded by the SEC12, 16, 23, and 24 genes, are known to be required for autophagy [37,38,39], making it possible that the Sec13p and Sec31p proteins could have other functions in addition to their known roles in anterograde traffic. The size of the poliovirus-induced vesicles, at 200–400 nm in diameter, does not immediately suggest whether they are related to the COPII or autophagosomal pathways: typical diameters of COPII vesicles and autophagosomes are 50–100 nm [40,41] and 500–1,000 nm, respectively [24,25].
Finally, the idea that poliovirus infection induces autophagosome formation to facilitate viral growth is surprising in view of existing data that autophagy is an effective antimicrobial host response in many cases (reviewed in [42,43]). During herpes virus infection, there is good correlation between the induction of autophagy and the presence of effective antiviral responses. The process of autophagy is thought to be destructive to herpes virus because mutant viruses that fail to inhibit autophagy and other antiviral responses showed large decreases in yield [44]. During Streptococcus infection, genetic disruption of the autophagy pathway in the infected host cells resulted in increased bacterial yield, consistent with a role for autophagy in bacterial clearance [45].
Specific markers for autophagosome formation have become available, facilitating the identification of membranes derived from the autophagic pathway (reviewed in [43,46]). Here, we explore the role of several constituents of autophagosome machinery in poliovirus- and rhinovirus-infected cells by monitoring: the presence of autophagosomal protein LC3 in virally induced vesicles, the acquisition of colocalization of LC3 and LAMP1 in virally infected cells, the viral induction of punctate structures that stain with monodansylcadaverine (MDC), and the effects of perturbing the autophagosomal pathway pharmacologically and via RNA interference on intracellular and extracellular virus yield. Our data support the hypothesis that poliovirus, and likely the closely related rhinovirus, induce the formation of autophagosome-like structures to serve as the membrane scaffolds for RNA replication. We further suggest that double-membraned vesicles, by forming a luminal cytoplasmic compartment, may facilitate prelytic viral exit from infected cells.
Results
LC3, a Marker of Cellular Autophagosomes, Colocalizes with Proteins of the Poliovirus RNA Replication Complex
To ask whether the membranes on which poliovirus RNA replication complexes assemble contain constituents of autophagosomes, we monitored the localization of both LC3, a specific marker of autophagosomes, and 3A, a critical component of the poliovirus RNA replication complex, in infected cells. LC3 was expressed via DNA transfection as an amino-terminal fusion with green fluorescent protein (GFP). As can be seen in Figure 1, the punctate GFP–LC3 signal colocalized with that of poliovirus RNA replication protein 3A, as visualized by immunofluorescence. No colocalization with poliovirus 3A protein was seen when GFP was not fused to LC3. Therefore, the membranous structures to which 3A and other components of the poliovirus RNA replication complex localize [3] can recruit LC3.
Figure 1. Simultaneous Visualization of GFP-LC3 and Poliovirus Protein 3A (Red).
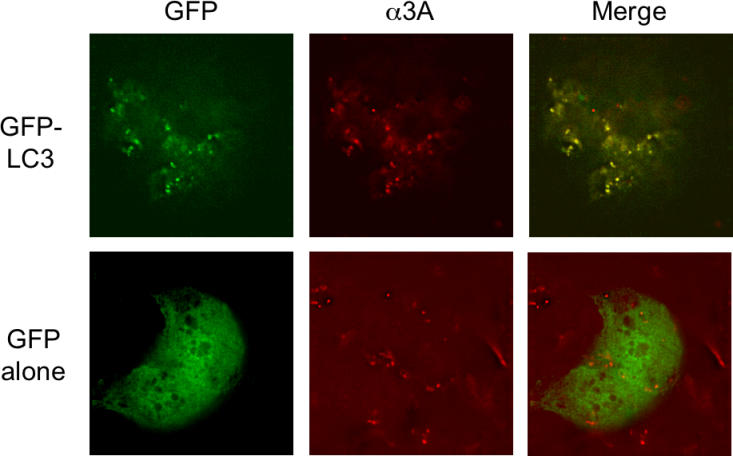
MCF-7 cells were transfected either with a plasmid that expresses a GFP-LC3 fusion protein or GFP alone as indicated. Forty-eight hours posttransfection, cells were infected with poliovirus, then fixed and stained using a monoclonal antibody to poliovirus 3A protein and a rhodamine-conjugated secondary antibody.
LAMP1 and LC3 Colocalize in Both Tamoxifen-Treated, Rapamycin-Treated, and Poliovirus-Infected Cells
In nonautophagic cells, LC3, originally identified as a microtubule-associated protein [47], and LAMP1, a marker of late endosomes and lysosomes, do not colocalize. However, during autophagy, LC3 has been shown to colocalize with lysosomal protein LAMP1, defining the maturation of the nascent autophagosome to the autophagosome [28,48,49]. This process can be seen in Figure 2A, in which LC3 and LAMP1 were shown to colocalize almost completely in MCF-7 cells upon treatment with tamoxifen, a known inducer of autophagy. Similarly, rapamycin treatment of HeLa cells led to the colocalization of GFP–LC3 and LAMP-1, while they remained distinct in untreated cells (Figure 2A). By 3 h postinfection with poliovirus, the LAMP1 and GFP–LC3 signals in HeLa cells also began to colocalize (Figure 2B); this time corresponds to the beginning of viral RNA synthesis in infected HeLa cells [50]. By 4.5 h postinfection, the LAMP1 and GFP–LC3 signals had merged almost completely (Figure 2B). The colocalization of GFP–LC3 and LAMP1 characteristic of autophagosome formation was also seen in cells infected with either of two different serotypes of human rhinovirus (Figure 2C).
Figure 2. Simultaneous Visualization of GFP-LC3 and Resident Lysosomal Protein LAMP1 (Red) during Autophagic Induction, Poliovirus Infection, and Rhinovirus 14 Infection.
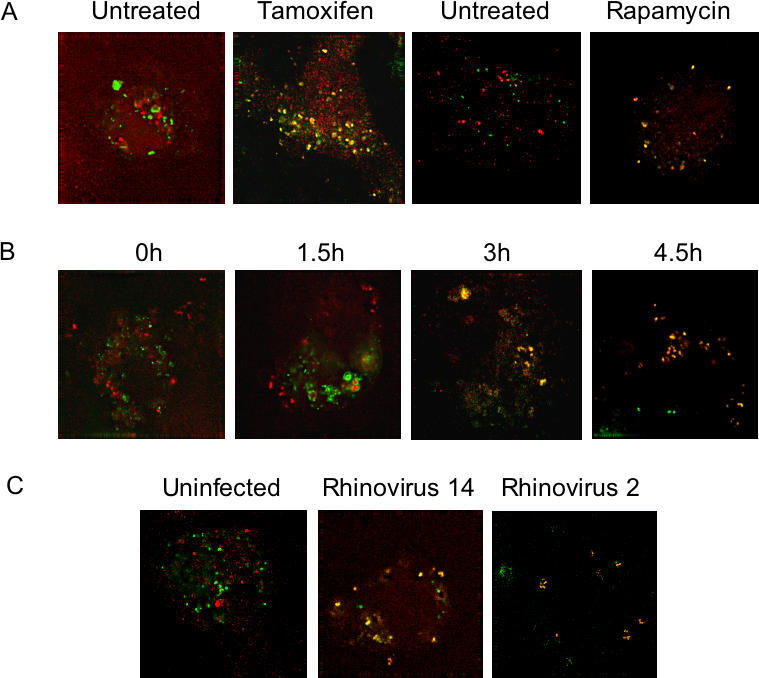
(A) MCF-7 cells were transfected with a plasmid that expresses GFP-LC3 fusion protein and treated with 10 μM tamoxifen in DMSO/EtOH or with DMSO/EtOH alone as indicated. H1-HeLa cells were transfected with the GFP-LC3-expressing plasmid and treated with rapamycin for 5 h or left untreated as indicated.
(B) A time course of poliovirus infection was performed in GFP-LC3-transfected MCF-7 cells, followed by visualization of LAMP1 and GFP-LC3 localization. Infections were with Mahoney type 1 poliovirus at a multiplicity of infection (MOI) of 50 PFU/cell for 5 h at 37 °C.
(C) H1 HeLa cells were transfected with an LC3-GFP-expressing plasmid and either mock-infected or infected with human rhinoviruses as indicated at 50 infectious units/cell for 6 h at 33.5 °C.
Simultaneous Expression of Poliovirus Proteins 2BC and 3A Induces the Colocalization of GFP–LC3 and LAMP1
Expression of poliovirus proteins 2BC and 3A in isolation can induce the formation of double-membraned vesicles that display biochemical and ultrastructural similarity to those formed in poliovirus-infected cells [35]. However, expression of poliovirus protein 2BC alone has been shown to induce the formation of single-membraned vesicles in both mammalian cells and S. cerevisiae [4,35,51,52]. Expression of poliovirus 3A protein in isolation, on the other hand, reduces the rate of ER-to-Golgi traffic and distends ER membranes [36,53]. To test whether 2BC, 3A, or both were sufficient to induce colocalization of GFP–LC3 and LAMP1 as observed in poliovirus-infected cells, proteins 2BC and 3A were expressed singly or in combination in 293T cells in the presence of GFP–LC3. As shown in Figure 3, colocalization of GFP–LC3 and LAMP1 was not observed in control cells or in cells that expressed either 2BC or 3A in isolation. However, coexpression of 2BC and 3A caused GFP–LC3 and LAMP1 to colocalize (Figure 3A), as was seen in cells infected with poliovirus or rhinoviruses treated with tamoxifen or treated with rapamycin (see Figure 2).
Figure 3. Simultaneous Visualization of GFP-LC3 and Resident Lysosomal Protein LAMP1 (Red) in Cells that Express Poliovirus 2BC and 3A Proteins.
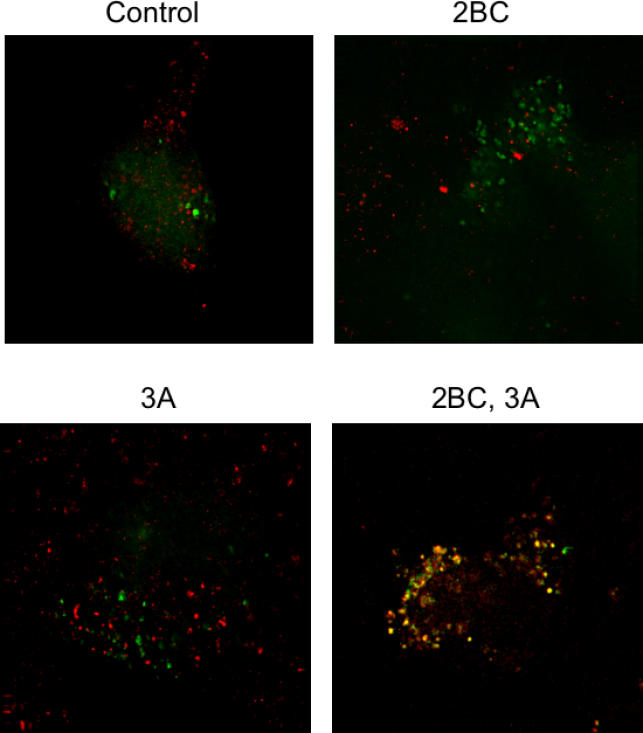
293T cells were transfected with vectors expressing 2BC, 3A, or both and a GFP-LC3 expressing vector for 48 h at 37 °C. Nonexpressing vector DNA was used to ensure that all transfections contained the same amount of DNA.
Poliovirus Infection and Autophagic Induction Induce Punctate MDC Staining
To examine further whether the membranes induced during poliovirus infection display additional characteristics of autophagosomes, we employed fluorescent staining with MDC. Under specific fixation conditions, MDC is retained in autophagosomal membranes [54,55]. As shown in Figure 4, treatment of MCF7 human breast tumor cells with tamoxifen caused the accumulation of punctate structures that retained staining with MDC after fixation. Similar punctate staining was observed in cells infected for 5 h with rhinovirus 14 (Figure 4A) or for 3 h or more with poliovirus (see Figure 4B).
Figure 4. MDC Staining of MCF7 Cells upon Tamoxifen Treatment, Rhinovirus 14 Infection, or Poliovirus Infection.
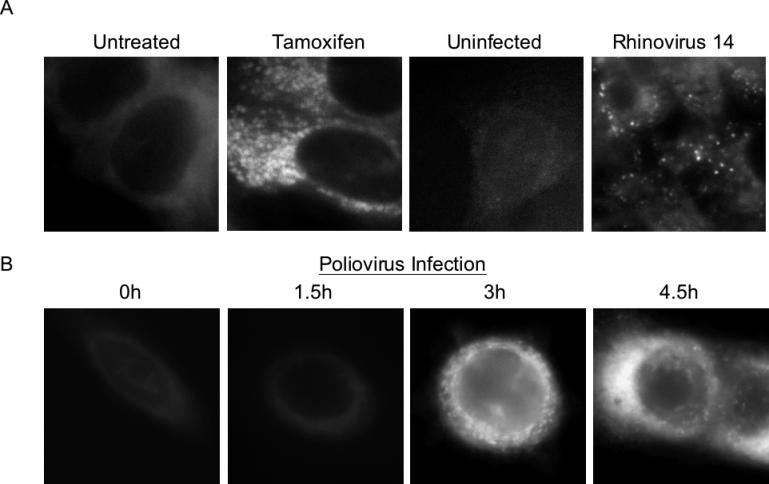
(A) Cells that were treated with 10 μM tamoxifen in DMSO/EtOH or with DMSO/EtOH alone for 48 h at 37 °C, or that were mock-infected or infected with human rhinovirus 14 as in Figure 3, were incubated with 100 μM MDC, fixed, and visualized by deconvolution microscopy in the UV channel as described in Materials and Methods.
(B) Cells were infected with poliovirus at an MOI of 50 PFU/cell for 0, 1.5, 3, or 4.5 h at 37 °C; for each time point, MDC incubation was begun 1 h prior to fixation and visualization.
To test the hypothesis that the MDC-stained structures observed in Figure 4 were the same as the GFP–LC3-containing structures observed in Figure 2, we monitored the localization of GFP–LC3 with MDC. As a live stain, MDC is a poor marker for autophagosomes, because both autophagic membranes and lysosomes are visualized [46]. However, if cells are subjected to a defined fixation protocol after staining with MDC, the dye can be removed from single-membraned, but not double-membraned, vesicles [54,55]. As shown in Figure 5, good colocalization of MDC and LC3 was observed in poliovirus-infected cells, and no evidence of the punctate MDC staining expected of lysosomal membranes could be seen in uninfected cells.
Figure 5. Simultaneous Visualization of GFP-LC3 Localization and MDC Staining (Red) in Uninfected and Poliovirus-Infected Cells.
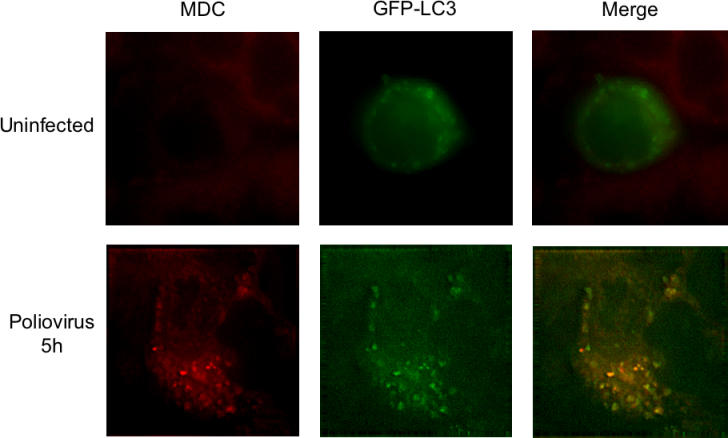
Forty-eight hours posttransfection of MCF-7 cells with a plasmid that expresses a GFP-LC3 fusion protein, cells were mock-infected or infected with poliovirus at an MOI of 50 PFU/cell for 5 h at 37 °C. After fixation, deconvolution microscopy was used to visualize fluorescence from both the MDC and GFP fluorescent molecules. MDC incubation was begun 1 h prior to fixation and visualization.
Compounds That Stimulate and Inhibit Cellular Autophagy Affect Yield of Intracellular Poliovirus
To determine whether the autophagosome-like membranes induced during poliovirus infection perform an antiviral function or facilitate viral replication, we tested the effect on poliovirus yield of pretreating H1–HeLa cells with either tamoxifen or rapamycin, known inducers of autophagy. Yield of intracellular virus increased approximately 4-fold when cells were pretreated with tamoxifen, and 3-fold upon pretreatment with rapamycin (Figure 6A and 6B). Conversely, when cells were treated with 3-methyladenine, a pharmacological inhibitor of autophagy [56], yield of intracellular virus was decreased (Figure 6C). Therefore, during poliovirus infection, the activity of the autophagy pathway correlates with productive viral replication, not with viral destruction.
Figure 6. Intracellular Viral Yields from Poliovirus Infections Performed in the Presence of Pharmacological Inducers and Inhibitors of Autophagy.

(A) H1 HeLa cells were treated with 10 μM tamoxifen in DMSO/EtOH or in DMSO/EtOH alone for 48 h at 37 °C. Cell numbers were determined, and triplicate plates were infected with poliovirus at an MOI of 0.1 PFU/cell for the times indicated. (B) H1 HeLa cells were treated with 50 nM rapamycin in DMSO/EtOH or DMSO/EtOH alone for 3 h before infection with poliovirus as in (A).
(C) H1 HeLa cells were treated with 10 mM 3-methyladenine in DMSO/EtOH or DMSO/EtOH alone for 3 h before infection with poliovirus as in (A). Viral yields were determined by plaque assay in H1-HeLa cells and expressed as PFU/cell.
A Potential Role for Autophagosome-Like Membranes in Extracellular Delivery of Viruses
To extend the studies with pharmacological agents, we tested the effect of reducing the amount of intracellular autophagy proteins LC3 and Atg12p on poliovirus yield, using double-stranded RNA oligonucleotides designed to target their mRNAs for destruction by RNA interference. Pools of double-stranded oligonucleotides were synthesized to target ATG12 mRNA, and to target both LC3A and LC3B mRNAs (see Materials and Methods). As shown in Figure 7, the intracellular abundance of Atg12p and LC3 proteins could be reduced to 70% and 90%, respectively, of their abundance in cells treated with control double-stranded small interfering RNAs (siRNAs) known to target firefly luciferase mRNA. These reductions in Atg12p and LC3 protein concentration resulted in 3-fold and 4-fold reductions, respectively, in yields of intracellular poliovirus. Even stronger effects of the RNA interference (RNAi)-mediated reduction in autophagy proteins on viral yield were observed, however, when the yields of extracellular virus were examined. The effects of RNAi against ATG12 and LC3 on extracellular virus were 9-fold and 20-fold, respectively (Figure 7). Two potential mechanisms for this preferential reduction in extracellular, as opposed to intracellular virus, seemed possible. The first possibility was that the RNAi-mediated reductions in the intracellular concentrations of Atg12p and LC3 protein reduced cell lysis and therefore lytic release of virus; links between autophagy and apoptosis have been reported (reviewed in [57,58]). The possibility of decreased cellular susceptibility to lysis upon RNAi treatment is difficult to exclude. However, early cell lysis in the presence of reduced Atg12p or LC3 concentrations seemed unlikely because the amounts of virus released were very small, and the observed reductions in extracellular virus release were seen at early time points, before lysis was expected.
Figure 7. Intracellular and Extracellular Viral Yields from Poliovirus Infections of Cells Treated with Small RNA Duplexes to Reduce the Intracellular Concentrations of LC3 and Atg12p Proteins.

(A) H1 HeLa cells (1 × 106) were transfected with 12.5 pm each of eight different RNA duplexes targeted to the LC3A and LC3B mRNAs, or with 100 pm of an RNA duplex targeted to firefly luciferase for 24 h at 37 °C (see Materials and Methods). Cell numbers were determined and triplicate plates were infected with poliovirus at an MOI of 0.1 PFU/cell for the times indicated. Viral yields were determined by plaque assay in H1-HeLa cells and expressed as PFU/cell for the intracellular virus, and as total PFU/plate for the extracellular virus. The relative abundance of LC3 protein in the cells incubated with the control and LC3-targeted RNAi molecules was determined by immunoblot using polyclonal antibodies directed against LC3 and GAPDH.
(B) H1 HeLa cells were transfected with 25 pm each of four RNA duplexes targeted to ATG12 mRNA, or with 100 pm of an RNA duplex targeted to firefly luciferase, for 24 h at 37 °C (see Materials and Methods). Poliovirus infections were performed as in (A). The relative abundance of Atg12p protein was determined by immunoblot using antibodies against human Atg12p and GAPDH.
A second possibility for the greater reduction in extracellular than intracellular virus is that double-membraned structures with the cytoplasm provide a topologically reasonable mechanism for nonlytic release of cytoplasmic contents. Ultrastructural analysis studies of cells infected with poliovirus [1,2] and with rhinovirus 14 (Figure 8A) have revealed the presence of virions and other cytoplasmic material within the lumen of double-membraned vesicles. Presumably, these are double-membraned vesicles that formed relatively late in infection and were therefore able to trap viruses already present in the nearby cytoplasm. The known proximity of RNA replication, protein expression, and virion packaging to the membrane-associated replication complexes [59,60] should facilitate such events. Figure 8B displays an image consistent with the release of the packets of cytosol expected if the outer bilayer of a multilamellar membrane structure were to fuse with the plasma membrane. Figure 8C shows that, in poliovirus-infected cells, such blebs can contain LC3, and Figure 8D shows that they can also contain VP1, a viral capsid protein.
Figure 8. Ultrastructure of H1-HeLa Cells Infected with Human Rhinovirus 14 (HRV14) and Immunoelectron Microscopy of Cells Infected with Poliovirus.
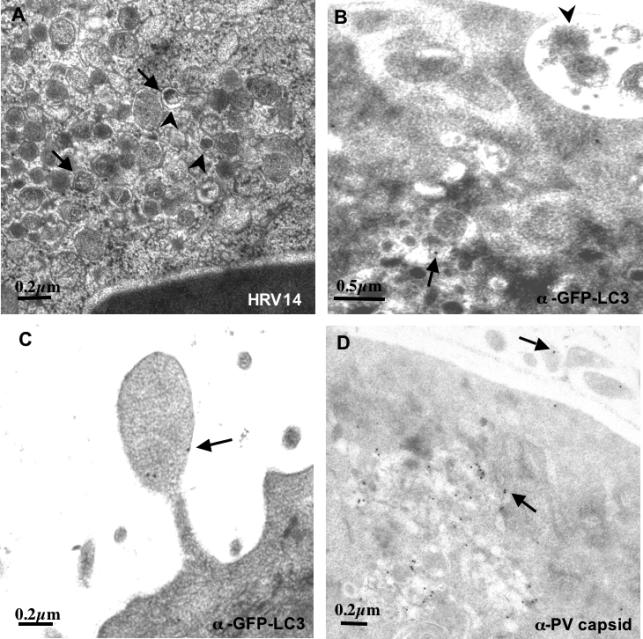
(A) Cells were infected with rhinovirus 14 as in Figure 3 and prepared for electron microscopy by high-pressure freezing. Examples of readily discernable double lipid bilayers are designated with large arrowheads; vesicles that contain intralumenal viral particles are denoted with small arrows.
(B and C) Cells were transfected with a plasmid that expresses GFP-LC3 and subsequently infected with poliovirus for 5 h as in Figure 2. GFP-LC3 was visualized using a secondary antibody coupled to 10-nm gold particles; examples of such particles are denoted with arrows. An arrowhead identifies apparently extracellular packets of cytosol.
(D) Cells transfected with an GFP-LC3-expressing plasmid and infected with poliovirus as in (C) were immunostained using an antibody directed against VP1, a viral capsid protein and visualized using a secondary antibody conjugated to 10-nm gold particles; examples of such particles are identified with arrows.
Discussion
In this work, we have shown that several hallmarks of cellular autophagosomes, including the localization of GFP–LC3 into discrete punctate structures and staining with MDC followed by fixation, can be observed in human cells whether they are treated with tamoxifen or infected with poliovirus or rhinovirus. Furthermore, we have employed a new criterion for autophagy, the colocalization of LC3 and LAMP1, to demonstrate that this intracellular rearrangement was observed in cells treated with tamoxifen, infected with picornaviruses (poliovirus, rhinovirus 2, or rhinovirus 14) or transfected with plasmids that express poliovirus proteins 2BC and 3A (see Figures 2 and 8). Taken together with the previously observed double-membraned morphology, cytoplasmic contents, and complex origin of the membranous structures induced during poliovirus infection [1,2,35], we argue that the viruses utilize components of the autophagosome formation pathway to form the characteristic double-membraned vesicles seen during infection.
Do the autophagosome-like membranous structures induced by poliovirus act as scaffolds for RNA replication, or are they part of the host antiviral response? The numerous positive correlations between the functional presence of autophagosomal pathways and increased viral yield lead us to conclude that the autophagosome-like structures observed during poliovirus infection are not antiviral. Instead, we argue that the double-membraned vesicles induced during poliovirus infection facilitate poliovirus replication, and we hypothesize that poliovirus, rhinovirus 2, and rhinovirus 14 subvert the constituents of the cellular autophagy pathway to form membranous scaffolds on which RNA replication complexes can assemble.
Other positive-strand RNA viruses that have been shown to localize their RNA replication complexes to double-membraned vesicles in the cytoplasm of infected cells are equine artirivirus [61], murine hepatitis virus [62], and SARS virus [63]. The membranes associated with murine hepatitis virus infection have been shown recently to contain LC3 protein [64]. Strikingly, infection of murine ES cell lines deficient for APG5 with murine hepatitis virus was shown to result in a large decrease in the extracellular yield of this enveloped virus; the effect on intracellular viral particles or RNA was not reported. Nevertheless, the large reduction in yield of extracellular virus seen in the absence of Atg5p protein argues that this component of the cellular autophagy pathway is crucial for some step in the formation or egress of infectious virions of this murine coronavirus [64].
For both poliovirus and equine artirivirus, molecular inducers of double-membraned vesicle formation have been identified. Specifically, coexpression of poliovirus proteins 2BC and 3A is required to accumulate double-membraned vesicles [35] and to elicit the colocalization of GFP–LC3 and LAMP1 that correlates with the formation of autophagosomes (see Figure 3A). For equine artirivirus, the coexpression of viral proteins nsp2 and nsp3 is sufficient to induce the formation of double-membraned vesicles [65]. We anticipate that these viral proteins, likely to be capable of mimicking, intercepting, or corrupting the pathway of cellular autophagy, will prove to be useful tools to decipher its mechanism.
Recent work has highlighted an important role for autophagy in the innate immune response of vertebrates to intracellular pathogens. For example, induction of autophagy has been shown to promote clearance of Mycobacterium tuberculosis from infected macrophage [66]. Furthermore, during infection with Shigella flexneri, the wild-type function of the bacterial icsB gene was shown to be required to prevent autophagic degradation, a process that the authors argue is specifically induced by a bacterial protein, the product of the virG gene [67].
Like S. flexneri, successful microorganisms often display strategies to evade potent host defenses. Furthermore, some microorganisms actively subvert otherwise effective host defense responses for their own benefit: for example, the growth of mink focus-forming virus requires apoptotic caspase activity for the maturation of a nonstructural protein [68], and murine cytomegalovirus encodes a chemokine homolog to attract cells of the immune system to the site of infection, which then promote viral dissemination (reviewed in [69]). Similarly, precedents are beginning to be established in which the autophagic pathway or its constituents may be subverted by intracellular pathogens to benefit their own replication (reviewed in [42,43]). As shown in Figure 9, the pathway of autophagosome formation ends with the acquisition of the lysosomal proteases and lipases that render the autophagosome a degradative organelle [27,28]. Mature autolysosomes are no longer bounded by double membranes because the inner membrane and luminal contents are degraded, becoming electron-dense and compact [24]. In Legionella infection, several genes, termed Dot or Icm genes, are required to retard the progression of autophagosome maturation, presumably to benefit bacterial growth within organelles that subvert components of the autophagosome [70,71,72]. We argue that poliovirus, rhinovirus 2, rhinovirus 14, equine encephalitis virus [65], and murine hepatitis virus [64] have similarly evolved a mechanism to accumulate autophagosome-like membranes in the cytoplasm for the duration of the infection. To maintain most of these structures in their double-membraned form, viral infection may both induce their formation and prevent their maturation into degradative organelles.
Figure 9. Pathway of Autophagosome Formation, Autophagic Degradation, and Proposed Steps of Pathway Subversion by Poliovirus and Related Viruses.

Double-membraned autophagosomes form either from ER membrane or de novo, encapsulating cytosol; the action of many gene products, including Atg5p and Atg12p, are required. LC3 protein (the Atg8p homolog) is associated with “sequestration crescents” as well as fully formed double-membraned autophagosomes. LAMP1 acquisition is a hallmark of the maturation of these structures, which eventually fuse with lysosomes to produce mature autophagosomes with single membranes and electron-dense contents. We hypothesize that infection by poliovirus or rhinovirus induces accumulation of autophagosomes to promote viral RNA replication by accelerating the formation of autophagosome-like structures from ER membranes, blocking the maturation of these structures into degradative organelles, or both (upper dotted line). The double-membraned topology makes the extracellular release of virions trapped in the cytosolic lumen topologically plausible, providing a mechanism for viral release in the absence of cell lysis. This could occur either from a double-membraned structure or from one in which only one of the membranes remained (dotted arrows).
Why would a virus choose a double-membraned, autophagosome-like vesicle on which to replicate its RNA? Not all positive-strand RNA viruses utilize such structures. For example, Flock House virus RNA replication complexes assemble on outer mitochondrial membranes [73]. When the RNA replication complexes of a subgenomic Flock House virus RNA were experimentally redirected to the cytoplasmic surface of the ER of S. cerevisiae, the yield of replicated RNA actually increased [73]. Therefore, viral RNA replication displayed no mitochondrion-specific requirement for specific lipids, proteins, or processes. Presumably, however, there are reasons why particular positive-strand RNA viruses target particular intracellular membranes on which to assemble their replication complexes, some of which might not be assayed under single-cycle growth conditions in tissue culture.
A larger effect on extracellular than intracellular virus yield was observed when the abundance of autophagy proteins Atg12p and LC3 was reduced by RNAi (see Figure 7). Possible explanations for the observed preferential decrease in extracellular virions are that the reduction in autophagosome machinery decreased cell lysis early in infection, or that reduced abundance of autophagosomal machinery decreased nonlytic viral escape. Although our data do not yet distinguish between these hypotheses, we will discuss the latter possibility because nonlytic delivery of cytosol to the extracellular milieu could be a unique characteristic of multilamellar vesicles, and we were able to obtain ultrastructural images consistent with this interpretation (see Figure 8).
Nominally lytic viruses are often assumed to spread exclusively via cell lysis. However, the possibility of nonlytic viral release, even for nonenveloped viruses such as poliovirus, has been suggested from numerous reports of persistently infected cell lines that continuously secreted infectious particles [74,75,76,77]. Even more convincingly, when polarized Caco-2 cultures, growing as intact monolayers, as shown by resistance to the passage of dyes and electric current between the apical and basolateral surfaces, were infected with poliovirus, newly synthesized virus was shown to emerge from only the apical surface [78]. That the egress of this virus did not correspond to a detectable breach in the monolayer argued that a nonlytic, and polarized exit route of unknown origin had been utilized.
We suggest a potential mechanism for this nonlytic release of cytosolic viral particles via the formation of double-membraned vesicles throughout the course of infection. Early in infection, the double-membraned structures would entrap cytosol, but this cytosol would be free of virions. However, at later stages of infection, the cytosol trapped by newly generated double-membraned structures would often contain viral particles. Poliovirions and related enteroviruses are relatively resistant to the low pH and active proteolysis that would prevail within the lumen of these vesicles should they mature [79,80]. As depicted in Figure 9, the fusion of the outer membrane of an intact double-membraned structure would result in the release of membrane-bound packets of cytoplasm into the extracellular milieu, whereas fusion of the membrane of a mature autophagosome would result in the direct release of cytosolic contents, and incompletely resolved double-membrane structures would result in the formation of more complex topologies. The presence of both LC3 and poliovirus capsid protein VP1 in extracellular structures adjacent to poliovirus-infected cells (see Figure 8C and 8D) is consistent with the release of at least partially intact packets of cytoplasm during poliovirus infection. We speculate that the formerly intracellular membranes surrounding these packets of cytoplasm would be short-lived outside the cell, with free virus being the eventual result.
Recently, HIV has been shown to exit human macrophages via the fusion of multivesicular bodies with the plasma membrane, rather than by directly budding from the cell surface as in HIV-infected T cells [81,82,83]. HIV gag protein directs the targeting of HIV particles to multivesicular bodies via direct binding to cellular protein Tsg101 [84,85], a component of the ESCRT complex required for the sorting of proteins into endosomes [86,87,88]. Similarly, the intracellular formation of double-membraned vesicles in poliovirus-infected cells provides a topologically reasonable mechanism for the extracellular delivery of cytosolic contents in the absence of cell lysis. We speculate that a mechanism to spread virus within tissues of infected hosts without cell lysis could provide an advantage to those positive-strand viruses that subvert constituents of the cellular pathway of autophagosome formation.
Materials and Methods
Viruses, plasmids, and cells.
Poliovirus Mahoney type 1 was isolated following transfection with an infectious cDNA [89] and propagated as previously described [34]. “Tet-off” MCF7 cells were obtained from Clontech (Palo Alto, California, United States) and propagated in DMEM + 10% FBS; MCF7 cells are highly inducible for autophagosome formation in response to tamoxifen [9]. Poliovirus stocks were titered on both HeLa H1 and MCF7 cells. The multiplicities of infections indicated refer to titers on the appropriate cell lines. H1–HeLa cells were used due to their permissiveness for rhinovirus infection. Rhinovirus stocks were obtained from the American Type Culture Collection ( ATCC, Manassas, Virginia, United States). Virus stocks were prepared in H1–HeLa cells ( ATCC) grown in EMEM supplemented with 0.2 M HEPES and 0.1 M MgCl2, and viral titers were measured by TCID50 as described previously [90].
To construct the GFP–LC3 fusion protein-expressing plasmid, LC3B sequences were amplified by PCR from a Human Lung Library (ResGen, Carlsbad, California, United States) using targeted primers containing EcoRI sites ( ACTGAATTCCCATGCCGTCGGAGAAG and TTTGAATTCTTACACTGACAATTTCA). The LC3A coding region was then inserted into the EcoRI site of pEGFP-C3 (Clontech, Palo Alto, California, United States) to create an EGFP–LC3 fusion protein under the control of the CMV immediate-early promoter. Expression of poliovirus proteins 2BC and 3A was performed as described previously [35]. The 293T cells, which express SV40 T antigen, were used to facilitate expression from the SV40 promoter in these plasmids.
Immunofluorescence
Cells were fixed using freshly made 4% formaldehyde in PBS for 10 min at room temperature. Poliovirus protein 3A and cellular protein LAMP1 were visualized by indirect immunofluorescence. For visualization of poliovirus 3A protein in the presence of GFP–LC3, cells were washed twice in PBS and incubated in a PBS solution that also contained 0.5% saponin, 10 mM sodium azide, 0.125% BSA, 3A monoclonal tissue culture supernatant at a dilution of 1:30, and rhodamine-linked antimouse secondary antibody (SC-2084, Santa Cruz Biotechnologies, Santa Cruz, California, United States) at a dilution of 1:200. Cells were incubated at 4 °C for 45 min, washed twice with PBS, and placed under Vectashield mounting medium (Vector Laboratories, Burlingame, California, United States). Visualization of LAMP1 was performed using a monoclonal LAMP1 antibody (Transduction Laboratories, Lexington, Kentucky, United States) at a dilution of 1:200.
MDC staining and costaining.
MDC (Sigma, St. Louis, United States) was stored at −20 °C under desiccant. A fresh stock solution of 5 mM MDC was made in 1:1 DMSO/EtOH immediately prior to adding to cultures. At 1 h before fixation, fresh medium that contained either 10 μM MDC in DMSO/EtOH or an equivalent volume of DMSO/EtOH was added to the cells. The cells were then fixed using a freshly made 4% formaldehyde solution in PBS for 10 min at room temperature and imaging was performed immediately. To ensure that membrane vesiculation induced by methods other than the induction of autophagy did not show similar MDC staining patterns, Golgi vesiculation was induced with 5 μM ilimaquinone [91] or 5 μM nocodazole [92]; the disappearance of intact Golgi was confirmed using BODIPY-C5 ceramide (Molecular Probes, Eugene, Oregon, United States). Under these conditions, punctate MDC staining was not observed (data not shown).
Microscopy and deconvolution analysis.
Microscopic analysis was carried out on an Olympus IX70 at 100X magnification. Images were captured and deconvolved using SoftWorx 2.50 on an SGI Octane workstation. MDC staining was detected at 360 nm excitation/457 nm emission. GFP–LC3 expression was detected at 490 nm excitation/528nm emission. Rhodamine was detected at 555 nm excitation/617 nm emission. Individual images from each stack were saved as TIFF files and processed in Adobe Photoshop 7.0.
RNA interference to reduce intracellular concentrations of LC3 and ATG12
siRNA SMARTpools, consisting of four RNA duplexes targeting the gene of interest, and a control siRNA targeting firefly luciferase, were purchased from Dharmacon (Lafayette, Colorado, United States). For LC3, both LC3A and LC3B RNAs [93] were targeted, bringing the total number of transfected duplexes to eight. Pools consisted of an equal amount of each duplex. The siRNA sequences are given as sense/antisense pairs. The siRNA sequences for ATG12 were: GGGAAGGACUU ACGGAUGUUU/5′P-ACAUCCGUAAGUCCUUCCCUU; GAACACCAAGUUUCACUGUUU/5′P- ACAGU GAAACUUGGUGUUCUU; GCAGU AGAGCGAACACGAAUU/5′P-UUCGUGUUCGCUCUACUGCUU; and UGUU GCAGCUUCCUACUUCUU/5′P- GAAGU AGGAAGCU GCAACAUU. The siRNA sequences for LC3A were: GGACGGCUUCCUCUAUAUGUU/5′P-CAUAU AGAGGAAGCCGUCCUU; CGGUGAUCAU CGAGCGCUAUU/5′P-U AGCGCUCGAUGAU CACCGUU; ACAU GAGCGAGUUGGUCAAUU/5′P-UU GACCAACUCGCUCAUGUUU; and CGCCCAU CGCGGACAUCUAUU/5′P-UAGAUGU CCGCGAU GGGCGUU. The siRNA sequences for LC3B were: CAAAGUUCCUUGUACCUGAUU/5′P-U CAGGU ACAAGGAACUUUGUU; GAUAAU AGAACGAU ACAAGUU/5′P-CUUGUAUCGUUCUAUUAUCUU; GU AGAAGAUGU CCGACUUAUU/5′P-UAAGU CGGACAUCUUCUACUU; and AGGAGACGUU CGGGAUGAAUU/ 5′P-UUCAU CCCGAACGUCUCCUUU.
Cells were grown to densities of 1—5 × 105 per 6-cm dish in 2.5 ml EMEM without antibiotics, and transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, California, United States) according to manufacturer's instructions. For each 6-cm dish, 100 total pmol of pooled siRNA was diluted in 250 μl of serum-free OptiMEM medium (Invitrogen) and, separately, 5 μl of Lipofectamine 2000 was diluted in 250 μl of OptiMEM. After an incubation of 5 min at room temperature, the diluted RNA and Lipofectamine 2000 were combined and incubated for 20 min at room temperature. The 500-μl mixture was then added to each dish and gently rocked to spread the lipid–RNA complexes. Growth curves and immunoblots were performed 48 h after transfection. Total cell extracts were made using RSB-NP40 extraction buffer (10 mM Tris [pH 7.5], 10 mM NaCl, 1.5 mM MgCl2, 1% NP-40) supplemented with protease inhibitors (Roche, Mannheim, Germany). Extract was separated on a 15% Laemmli gel and transferred to a PVDF membrane for Western blotting. Anti-LC3 immunoblotting was performed using rabbit antibody raised commercially (AnaSpec, San Jose, California, United States) against a peptide comprising the first 16 amino acids of murine LC3 (MPSEKTFKQRRSFEQR). Anti-Atg12p and anti-GAPDH immunoblotting was performed using antibodies from Zymed (South San Francisco, California, United States) and Research Diagnostics (Flanders, New Jersey, United States), respectively. Antibodies were diluted 1:3,000 in a PBS solution that also contained 0.1% Tween-20 and 2% BSA, and detected with alkaline-phosphatase conjugated goat antirabbit antibody, at a dilution of 1:10,000 using the ECF reagent from Amersham Biosciences (Piscataway, New Jersey, United States).
High-pressure freezing, freeze-substitution, and electron microscopy (EM).
For cryofixation and EM analysis, H1–HeLa cells were grown in EMEM supplemented with 0.2 M HEPES and 0.1 M MgCl2 in flasks. Cells were infected with virus at an MOI of 50 PFU/cell for poliovirus or 50 TCID50/cell for rhinovirus, then washed three times with PBS, trypsinized, and collected by centrifugation. The cell pellet was resuspended in 0.15 M mannitol in PBS and collected by centrifugation. Aliquots of the resulting pellet were frozen in a Balzers HPM 10 high-pressure freezing apparatus as described previously [2] and stored in liquid nitrogen. To observe cellular ultrastructure, samples were freeze-substituted in 0.1% tannic acid in acetone at −80 °C, rinsed in acetone, then warmed at −20 °C in the presence of 2% osmium tetroxide in acetone for 16 h, followed by incubation at 4 °C for 4 h. After rinsing in acetone at 4 °C, samples were embedded in Epon-Araldite resin. Thin sections were stained with 2% uranyl acetate and lead citrate and imaged at 80 kV in a JEOL 100C or Philips CM10 EM. For immunostaining, high-pressure frozen samples were freeze-substituted in 0.1% glutaraldehyde-0.05% uranyl acetate in acetone, embedded, stained, and visualized as described previously [35].
Accession Numbers
The SwissProt (http://us.expasy.org/sprot/) accession numbers for the gene products discussed in this article are 2BC and 3A (P03299), Atg12p (O94817), Atg8p (P38182), LAMP1 (P11279), LC3 (Q9GZQ8), mTor (P42345), Sec12p (P11615), Sec13p (P5573), Sec16p (P48415), Sec23p (P15303), Sec24p (P40482), and Sec31p (O94979).
Acknowledgments
We are indebted to Patrice Codogno and Skip Virgin for experimental suggestions and advice and to Peter Sarnow and Katherine Shim for helpful comments on the manuscript. We are grateful to Susan Palmieri and Jon Mulholland with the Stanford Cell Science Imaging Facility for assistance with deconvolution microscopy. This work was supported by the Hutchison Consortium for Translation Research and the National Institutes of Health. WTJ was supported by a National Research Service Award from the National Heart, Lung, and Blood Institute and a Stanford Dean's Fellowship.
Competing interests. The authors have declared that no competing interests exist.
Abbreviations
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- MDC
monodansylcadaverine
- siRNA
small interfering RNA
Author contributions. WTJ, THG, RRK, and KK conceived and designed the experiments. WTJ, THG, and MPT performed the experiments. WTJ and KK analyzed the data. WTJ, MPT, SM, MR, and RRK contributed reagents/materials/analysis tools. KK and WTJ wrote the paper.
Citation: Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, et al. (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3(5): e156.
References
- Dales S, Eggers HJ, Tamm I, Palade GE. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- Schlegel A, Giddings TH, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz K, Egger D, Pasamontes L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology. 1987;60:220–226. doi: 10.1016/0042-6822(87)90063-8. [DOI] [PubMed] [Google Scholar]
- Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology. 1994;202:129–145. doi: 10.1006/viro.1994.1329. [DOI] [PubMed] [Google Scholar]
- Egger D, Teterina N, Ehrenfeld E, Bienz K. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production and viral RNA synthesis. J Virol. 2000;74:6570–6580. doi: 10.1128/jvi.74.14.6570-6580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science. 2002;296:2218–2222. doi: 10.1126/science.1070585. [DOI] [PubMed] [Google Scholar]
- Chen J, Noueiry A, Ahlquist P. An alternate pathway for recruiting template RNA to the brome mosaic virus RNA replication complex. J Virol. 2003;77:2568–2577. doi: 10.1128/JVI.77.4.2568-2577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, et al. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci. 2000;113:1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- Bilir A, Altinoz MA, Erkan M, Ozmen V, Aydiner A. Autophagy and nuclear changes in FM3A breast tumor cells after epirubicin, medroxyprogesterone and tamoxifen treatment in vitro. Pathobiology. 2001;69:120–126. doi: 10.1159/000048766. [DOI] [PubMed] [Google Scholar]
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–2326. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- Shigemitsu K, Tsujishita Y, Hara K, Nanahoshi M, Avruch J, et al. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways: Possible involvement of autophagy in cultured hepatoma cells. J Biol Chem. 1999;274:1058–1065. doi: 10.1074/jbc.274.2.1058. [DOI] [PubMed] [Google Scholar]
- Mordier S, Deval C, Bechet D, Tassa A, Ferrara M. Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J Biol Chem. 2000;275:29900–29906. doi: 10.1074/jbc.M003633200. [DOI] [PubMed] [Google Scholar]
- Meijer AJ. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76. [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Klionsky DJ. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem. 2000;69:303–342. doi: 10.1146/annurev.biochem.69.1.303. [DOI] [PubMed] [Google Scholar]
- Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002;277:13739–13744. doi: 10.1074/jbc.M200385200. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: Many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ. The molecular mechanism of autophagy: Unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- Stromhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J. 1998;335:217–224. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WA Jr. Studies on the mechanisms of autophagy: Formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WA Jr. Studies on the mechanisms of autophagy: Maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fengsrud M, Erichsen ES, Berg T O, Raiborg C, Seglen PO. Ultrastructural characterization of the delimiting membranes of isolated autophagosomes and amphisomes by freeze-fracture electron microscopy. Eur J Cell Biol. 2000;79:871–882. doi: 10.1078/0171-9335-00125. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, et al. Inhibition of autophagy in mitotic animal cells. Traffic. 2002;3:878–893. doi: 10.1034/j.1600-0854.2002.31204.x. [DOI] [PubMed] [Google Scholar]
- Niioka S, Goto M, Ishibashi T, Kadowaki M. Identification of autolysosomes directly associated with proteolysis on the density gradients in isolated rat hepatocytes. J Biochem. 1998;124:1086–1093. doi: 10.1093/oxfordjournals.jbchem.a022225. [DOI] [PubMed] [Google Scholar]
- Tershak DR. Association of poliovirus proteins with the endoplasmic reticulum. J Virol. 1984;52:777–783. doi: 10.1128/jvi.52.3.777-783.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust RC, Landmann L, Gosert R, Tang BL, Hong W, et al. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol. 2001;75:9808–9818. doi: 10.1128/JVI.75.20.9808-9818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina EV, Mackenzie JM, Gorrell RJ, Anderson DA. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae . J Virol. 2002;76:11113–11122. doi: 10.1128/JVI.76.21.11113-11122.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irurzun A, Perez L, Carrasco L. Involvement of membrane traffic in the replication of poliovirus genomes: Effects of brefeldin A. Virology. 1992;191:166–175. doi: 10.1016/0042-6822(92)90178-r. [DOI] [PubMed] [Google Scholar]
- Maynell LA, Kirkegaard K, Klymkowsky MW. Inhibition of poliovirus RNA synthesis by brefeldin A. J Virol. 1992;66:1985–1994. doi: 10.1128/jvi.66.4.1985-1994.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy DA, Giddings TH, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens JR, Giddings TH, Kirkegaard K. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: Genetic and ultrastructural analysis. J Virol. 1997;71:9054–9064. doi: 10.1128/jvi.71.12.9054-9064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, et al. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1:11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Noda T, Ohsumi Y. The early secretory pathway contributes to autophagy in yeast. Cell Struct Funct. 2003;28:49–54. doi: 10.1247/csf.28.49. [DOI] [PubMed] [Google Scholar]
- Rowe T, Aridor M, McCaffery JM, Plutner H, Nuoffer C, et al. COPII vesicles derived from mammalian endoplasmic reticulum microsomes recruit COPI. J Cell Biol. 1996;135:895–911. doi: 10.1083/jcb.135.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Bannykh SI, Rowe T, Balch WE. Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J Biol Chem. 1999;274:4389–4399. doi: 10.1074/jbc.274.7.4389. [DOI] [PubMed] [Google Scholar]
- Dorn BR, Dunn WA, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. 2002;4:1–10. doi: 10.1046/j.1462-5822.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, et al. Autophagy defends cells against invading group A Streptococcus . Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Mann SS, Hammarback JA. Molecular characterization of light chain 3: A microtubule binding subunit of MAP1A and MAP1B. J Biol Chem. 1994;269:11492–11497. [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic. 2002;3:472–482. doi: 10.1034/j.1600-0854.2002.30704.x. [DOI] [PubMed] [Google Scholar]
- Baltimore D, Girard M, Darnell JE. Aspects of the synthesis of poliovirus RNA and the formation of virus particles. J Virol. 1966;29:179–189. doi: 10.1016/0042-6822(66)90024-9. [DOI] [PubMed] [Google Scholar]
- Aldabe R, Carrasco L. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem Biophys Res Commun. 1995;206:64–76. doi: 10.1006/bbrc.1995.1010. [DOI] [PubMed] [Google Scholar]
- Teterina NL, Gorbalenya AE, Egger D, Bienz K, Ehrenfeld E. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol. 1997;71:8962–8972. doi: 10.1128/jvi.71.12.8962-8972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens JR, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995;14:894–907. doi: 10.1002/j.1460-2075.1995.tb07071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- Niemann A, Takatsuki A, Elsasser HP. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem. 2000;48:251–258. doi: 10.1177/002215540004800210. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagic programmed cell death in Drosophila . Cell Death Differ. 2003;10:940–945. doi: 10.1038/sj.cdd.4401280. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Death by design: Apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Pfister T, Egger D, Bienz K. Poliovirus subviral particles associated with progeny RNA in the replication complex. J Gen Virol. 1995;76:63–71. doi: 10.1099/0022-1317-76-1-63. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Johnson KL, Sarnow P, Kirkegaard K. Functional coupling between replication and packaging of poliovirus replicon RNA. J Virol. 1999;73:427–435. doi: 10.1128/jvi.73.1.427-435.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen KW, van der Meer Y, Roos N, Snijder EJ. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol. 1999;73:2016–2026. doi: 10.1128/jvi.73.3.2016-2026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith CS, Tatti KM, Ksiazek TG, Rollin PE, Comer JA, et al. Ultrastructural characterization of SARS coronavirus. Emerg Infect Dis. 2004;10:320–326. doi: 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder EJ, van Tol H, Roos N, Pedersen KW. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol. 2001;82:985–994. doi: 10.1099/0022-1317-82-5-985. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophage. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Best SM, Shelton JF, Pompey JM, Wolfinbarger JB, Bloom ME. Caspase cleavage of the nonstructural protein NS1 mediates replication of Aleutian mink disease virus. J Virol. 2003;77:5305–5312. doi: 10.1128/JVI.77.9.5305-5312.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saederup N, Mocarski ES. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131–129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol. 2002;75:9966–9976. doi: 10.1128/JVI.75.20.9966-9976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Analysis of the intracellular fate of Legionella pneumophila mutants. Ann N Y Acad Sci. 1996;797:8–18. doi: 10.1111/j.1749-6632.1996.tb52944.x. [DOI] [PubMed] [Google Scholar]
- Joshi AD, Sturgill-Koszycki S, Swanson MS. Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell Microbiol. 2001;3:99–114. doi: 10.1046/j.1462-5822.2001.00093.x. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Schwartz MD, Dye BT, Ahlquist P. Engineered retargeting of viral RNA replication complexes to an alternative intracellular membrane. J Virol. 2003;77:12193–12202. doi: 10.1128/JVI.77.22.12193-12202.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbere-Garapin F, Christodoulou C, Crainic R, Pelletier I. Persistent poliovirus infection of human neuroblastoma cells. Proc Natl Acad Sci U S A. 1989;86:7590–7594. doi: 10.1073/pnas.86.19.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE, Bovee M. Persistent infection of human erythroblastoid cells by poliovirus. Virol. 1993;194:200–209. doi: 10.1006/viro.1993.1250. [DOI] [PubMed] [Google Scholar]
- Zhang S, Racaniello VR. Persistent echovirus infection of mouse cells expressing the viral receptor VLA-2. Virol. 1997;235:293–301. doi: 10.1006/viro.1997.8692. [DOI] [PubMed] [Google Scholar]
- Pavio N, Couderc T, Girard S, Sgro J-Y, Blondel B, et al. Expression of mutated poliovirus receptors in human neuroblastoma cells persistently infected with poliovirus. Virology. 2000;274:331–342. doi: 10.1006/viro.2000.0462. [DOI] [PubMed] [Google Scholar]
- Tucker SP, Thronton CL, Wimmer E, Compans RW. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J Virol. 1993;67:4274–4282. doi: 10.1128/jvi.67.7.4274-4282.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JFE, Rowlands DF, Brown F. A physico-chemical sub-grouping of the mammalian picornaviruses. J Gen Virol. 1973;18:171–180. doi: 10.1099/0022-1317-18-2-171. [DOI] [PubMed] [Google Scholar]
- Piirainen L, Hovi T, Roivainen M. Variability in the integrity of human enteroviruses exposed to various simulated in vivo environments. Microb Pathog. 1998;25:131–137. doi: 10.1006/mpat.1998.0221. [DOI] [PubMed] [Google Scholar]
- Pelchin-Matthews A, Kramer B, Marsh M. Infectious HIV-1 assembles in late endosomes in primary macrophage. J Cell Biol. 2003;162:443–455. doi: 10.1083/jcb.200304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. HIV-1 egress is gated through late endosomal membranes. Traffic. 2003;4:902–910. doi: 10.1046/j.1600-0854.2003.00145.x. [DOI] [PubMed] [Google Scholar]
- Ono A, Freed EO. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol. 2004;78:1552–1563. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff A, Ehrlich LS, Cohen SN, Carter CA. Tsg101 control of human immunodeficiency virus type 1 gag trafficking and release. J Virol. 2003;77:9173–9182. doi: 10.1128/JVI.77.17.9173-9182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poornillos O, Higginson DS, Stray KM, Fisher RD, Garrus JE, et al. HIV Gag mimics the Tst101-recruiting activity of the human Hrs protein. J Cell Biol. 2003;162:425–434. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Stefan CJ, Babst M, Emr SD. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol. 2003;160:413–423. doi: 10.1083/jcb.200302136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuchell MD, Garrus JE, Müller B, Stray KM, Ghaffarian S, et al. The human endosomal sorting complex required for transprot (ESCRT-I) and its role in HIV-1 budding. J Biol Chem. 2004;279:36059–36071. doi: 10.1074/jbc.M405226200. [DOI] [PubMed] [Google Scholar]
- Racaniello VR, Baltimore D. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science. 1981;214:916–919. doi: 10.1126/science.6272391. [DOI] [PubMed] [Google Scholar]
- Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest. 1995;96:549–557. doi: 10.1172/JCI118067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa PA, Yucel JK, Veit B, Faulkner DJ, Deerinck T, et al. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell. 1993;73:1079–1090. doi: 10.1016/0092-8674(93)90638-7. [DOI] [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: Regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Dang Y, Dai F, Guo Z, Wu J, et al. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem. 2003;278:29278–29287. doi: 10.1074/jbc.M303800200. [DOI] [PubMed] [Google Scholar]