

Abstract
Celiac disease (CeD) is a widespread, gluten-induced autoimmune disorder that lacks any medicinal therapy. Towards the goal of developing non-dietary treatments for CeD, research has focused on elucidating its molecular and cellular etiology. A model of pathogenesis has emerged centered on interactions between three molecular families: certain class II major histocompatibility complex proteins on antigen presenting cells, deamidated gluten-derived peptides, and T cell receptors on inflammatory CD4+ T cells. Growing evidence suggests that this pathogenic axis can be pharmacologically targeted to protect a patient from some of the adverse effects of dietary gluten. Further studies have revealed the existence of additional host and environmental contributors to disease initiation and tissue destruction. This review summarizes our current understanding of CeD pathogenesis and how it is being harnessed for therapeutic design and development.
Keywords: celiac disease, gluten, antigen presenting cells, major histocompatibility complex, T cells, transglutaminase, autoimmunity, clinical trials, therapeutics
Celiac disease: from bench to bedside
Celiac disease (CeD) is a systemic inflammatory disorder caused by dietary gluten that affects around 0.5–1% of the world’s population [1]. This number is growing, especially among women and children, as disease awareness and the availability of screening become more widespread [2]. In recent years, effective diagnostic tools have been developed and a strong clinical understanding of the disease has emerged (Box 1). Despite these advances, there are, as of now, no approved non-dietary therapies for CeD, and complications of disease persist even in patients who initiate dietary therapy (Box 2).
Box 1. Clinical presentation and diagnosis of celiac disease.
The clinical presentation of CeD is diverse [92]. Pathology occurs only in response to the consumption of cereal grains and, as a result, typically begins following introduction of wheat, barley, or rye into a child’s diet. However, symptoms can begin and disease can be diagnosed at any age [93]. The typical symptoms associated with CeD are gastrointestinal (e.g., abdominal pain, bloating, nausea, diarrhea) but there can be extra-intestinal manifestations of disease as well (e.g., fatigue, blistering skin rashes) [94]. In more severe cases, patients may present with iron-deficiency anemia, osteopenia, weight loss, and failure to meet growth milestones (especially among children) [93]. As diagnostic tools have improved, a growing proportion of CeD cases are diagnosed in the absence of overt symptoms [94].
The formal diagnosis of CeD is multi-pronged and varies around the world. When a physician suspects CeD, the most useful (and least invasive) initial test is a serum IgA assay for the presence of autoantibodies against tissue transglutaminase (tTG or TG2). Notwithstanding high specificity (typically >90%), the positive predictive value of this test is insufficient for definitively diagnosing a lifelong condition such as CeD [95]. In cases of IgA deficiency or equivocal test results, alternative serologies can be assessed (e.g., anti-tTG IgG, antibodies against deamidated gliadin peptides, anti-endomysial IgA). In the United States, definitive diagnosis of CeD requires an upper-gastrointestinal endoscopy and duodenal biopsy that reveals evidence of villous atrophy, as quantified through a decreased villous height to crypt depth ratio (Vh:Cd) [96]. Other histologic changes observed in the duodenal biopsy are also informative, such as an increased number of intraepithelial lymphocytes (IELs) [36]. The guidelines for a formal diagnosis of CeD are less strict for children in Europe. There, the combination of serum anti-tTG IgA titers at least 10-fold higher than the upper limit of normal and a positive anti-endomysial IgA titer on a second sampling override the need for confirmatory biopsy [97]. Importantly, the entire diagnostic protocol for CeD hinges on a patient retaining gluten in their diet, as its removal promotes mucosal healing [98] and normalization of anti-tTG serology [99]. As such, there remains a need for clinically useful diagnostic protocols in cases where patients have self-initiated a gluten-free diet (GFD).
Box 2. Clinical preseCurrent treatment and complications of celiac disease.
Because enteropathy in CeD depends on the presence of the causative antigen (i.e., gluten proteins from wheat, barley, or rye), the therapy for disease is lifelong gluten exclusion. Currently, gluten-free diet (GFD) is the only approved treatment for CeD. While theoretically simple, maintaining a GFD is challenging for many patients. This is because small doses of gluten (~50 mg/day) can induce intestinal inflammation and symptom exacerbation in some patients [100]. Low levels of gluten exposure can result from inadvertent cross-contamination of gluten-free meals at home or in a restaurant or through ingestion of a product containing an unspecified source of gluten. Moreover, even when implemented perfectly, a GFD carries with it a risk of nutrient deficiencies, especially in fiber and micronutrients such as iron and zinc, because of the cooccurrence of these substances with gluten-containing foods or supplementation of these nutrients in flours [101]. There are additional psychosocial implications of the GFD including food-anxiety and the increased financial burden of purchasing gluten-free foods [102]. Despite these shortcomings, GFD can be an effective treatment that results in symptomatic resolution and mucosal healing in many patients [98]. In rare cases (estimated 1–2%) termed refractory CeD, GFD does not palliate symptoms and mucosal inflammation persists [103,104].
Inability to maintain a GFD by CeD patients leads to a spectrum of complications primarily due to chronic small intestinal inflammation. Beyond the symptoms resulting from ongoing gluten exposure, the most common complications are nutrient deficiencies from decreased absorptive surface area in the gut leading to anemia and osteopenia [94]. Inflammation carries with it a low but especially problematic risk of malignant transformation of normal epithelium or immune cells into adenocarcinoma or intestinal lymphoma, respectively [105].
These clinical characteristics have inspired significant efforts in basic research and tool development for the study of CeD (Fig. 1). From this work has come an integrated model of celiac disease pathogenesis based on a prototypical interaction in the adaptive immune system between CeD-associated class II major histocompatibility complex (MHC) molecules, gluten-derived antigens, and CD4+ T cell receptors. Additionally, much has been uncovered about essential components of disease pathogenesis outside of this axis, including the role of certain microbes, the innate immune system, and B cells. These advances in our understanding of pathogenesis have inspired the development of many putative therapeutic agents whose modes of action are founded in recent biochemical and immunological insights. In this review, we discuss fundamental advancements in the study of CeD pathogenesis from a mechanistic perspective and how these discoveries have inspired clinical development towards the goal of identifying non-dietary CeD therapies.
Figure 1. Abriged history of celiac disease insights and tool development.
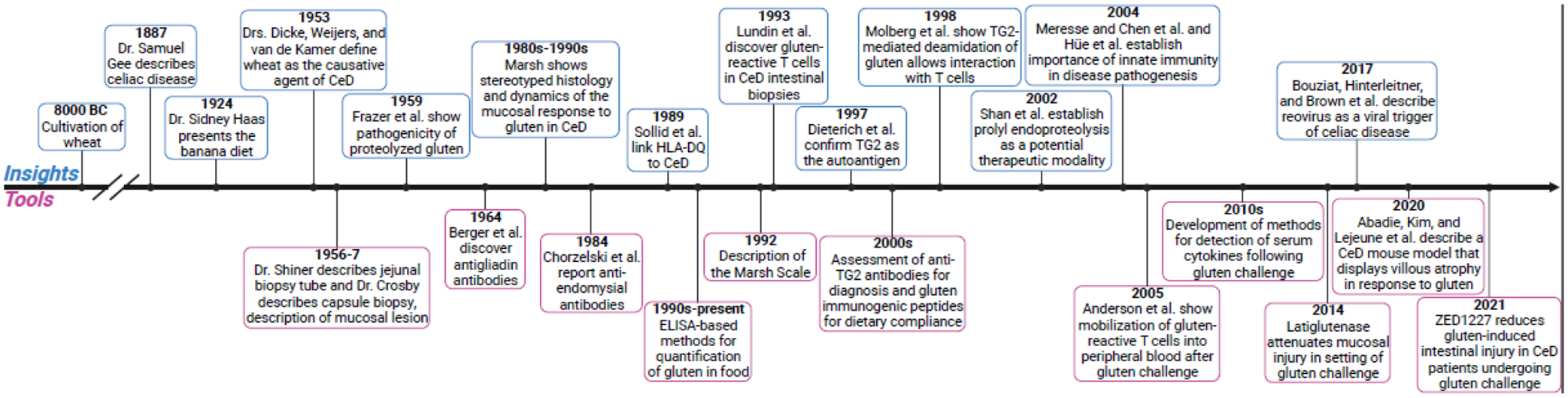
Brief timeline of fundamental advancements in our understanding of celiac disease pathogenesis and tool development for the study, diagnosis, and/or treatment of celiac disease [6,25,37,42,43,61,71,106–120].
The TCR-gluten peptide-MHC axis
At its core, CeD pathogenesis is driven by CD4+ T cells. More specifically, gluten-induced inflammation in a patient depends on the interaction of three key molecules: a gluten-derived antigen, a T cell receptor, and a class II MHC heterodimer capable of presenting the antigenic peptide to the T cell receptor (Fig. 2). Among autoimmune disorders, CeD is unique because the causative antigen and genetic background (i.e., the necessary MHC haplotype) have been extensively characterized.
Figure 2. Central pathogenic axis of celiac disease.
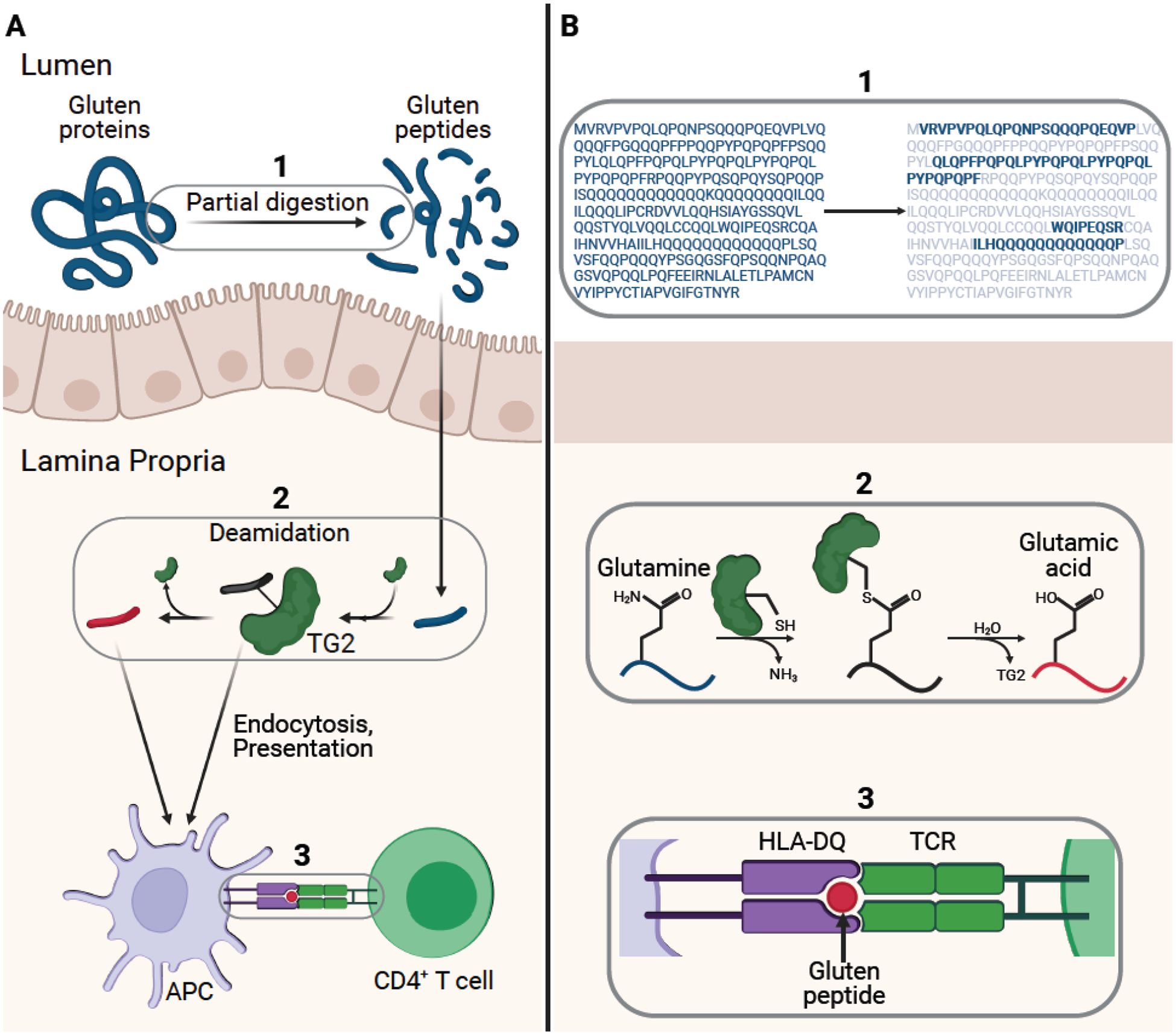
(A) Gluten proteins are partially digested by gastrointestinal proteases to yield gluten peptides (blue). Specific glutamine residues within these liberated peptides are deamidated by transglutaminase 2 (TG2). The deamidated peptides (red) or TG2- gluten peptide complexes undergo endocytosis by professional antigen presenting cells and the peptides are subsequently presented on disease-associated HLA-DQ receptors. Upon recognition by T cell receptors on disease-specific CD4+ T cells, these MHC-peptide complexes trigger the hallmark inflammatory response. (B) (1) shows the digestion of α2-gliadin by gastric and intestinal proteases into long peptides (bold) [6]. (2) depicts the two-step process of TG2 catalyzed gluten peptide deamidation. (3) zooms in on the central HLA-peptide-TCR interaction.
Structurally related peptides derived from gluten proteins in many common cereal grains (e.g., gliadins and glutenins in wheat, hordeins in barley, and secalins in rye) are the causative antigens in CeD [3–5]. Glutens belong to a class of proteins known as prolamins, so called because of the abundance of proline and glutamine residues in their primary sequences. The over-representation of proline is important as the unique structure imparted by this amino acid renders gluten proteins resistant to complete digestion by the proteases and peptidases of the human gastrointestinal tract [6]. Because gluten proteins are only partially broken down, they persist in the upper intestine as long oligomers and can ultimately bind to the clefts of disease-relevant MHC proteins.
A genetic prerequisite of CeD is the possession of a specific set of HLA alleles encoding class II MHC heterodimers. Specifically, HLA-DQ2 (comprised of DQA1*05 and DQB1*02 monomers) or HLA-DQ8 (DQA1*03 and DQB1*0302) are required for CeD [7]. Class II MHC receptors are expressed by professional antigen presenting cells (APCs) such as B cells, macrophages, and dendritic cells (DCs). They have evolved to hold antigens derived from foreign extracellular proteins and present them to CD4+ T cells. In the case of CeD, the structures of the HLA-DQ2 and HLA-DQ8 clefts are such that they can accommodate certain proline- and glutamine-rich motifs found in gluten peptides [8]. Previous work has demonstrated expansion of mucosal DCs and macrophages in the CeD small intestine following gluten exposure, suggesting a role for these cells in pathogenesis [9].
While it was known that intestinal pathology in CeD occurs due to T cell-mediated inflammation in the gut, the link that defined the disease as autoimmune in nature was not identified until later. That link is the enzyme transglutaminase 2 (TG2). TG2 is a ubiquitously expressed enzyme in mammals that crosslinks two proteins or alternatively a protein and a biogenic amine [10]. Mechanistically, TG2 functions analogously to a cysteine protease, but acts on the δ-amide sidechains of select glutamine residues in its protein or peptide substrates. In its first half-reaction, TG2 forms a covalent thioester intermediate with a glutamine residue. In turn, the thioester intermediate is resolved through the attack of an incoming amine (such as the sidechain of a lysine residue from another protein or a small molecule amine), resulting in the formation of a new amide bond. In the absence of a suitable amine donor, TG2 can hydrolyze the enzyme-substrate thioester linkage leading to a net glutamine → glutamate conversion. The latter conversion, known as deamidation, is vital to the pathogenesis of CeD [11].
While native gluten peptides have weak affinity for HLA-DQ2 or -DQ8, gluten peptides that have been regiospecifically deamidated by TG2 bind these class II MHCs significantly better owing to a favorable electrostatic interaction with a cationic residue in the ligand-binding cleft [8]. This chemical role for TG2 suggests the origin of the autoantigen in CeD; recognition of TG2-gluten peptide complexes by autoreactive B cells promotes the generation of TG2-specific antibodies. Notwithstanding their diagnostic utility, the precise role of these autoantibodies in CeD pathogenesis remains unclear and is of considerable interest.
Recognition of TG2-gluten peptide complexes is not only vital for the B cell response in CeD but is also important for the delivery of gluten peptides to other APCs such as DCs and macrophages. Recent work has shown that TG2-gluten peptide complexes, even when present extracellularly at very low concentrations, can be efficiently transported to the endo-lysosomal compartment of APCs (the site where class II MHCs are typically loaded with antigens). This occurs via a receptor-mediated endocytic mechanism that leverages the activity of LDL receptor-related protein 1 (LRP-1) [12], a scavenger receptor that is expressed on a variety of cells including macrophages and dendritic cells. In the process, a deamidated gluten peptide is delivered to the lysosome, allowing it to be efficiently presented to CD4+ T cells. Once the APC activates a T cell, CeD pathogenesis begins with the production of inflammatory cytokines such as IFN-γ [13].
Knowledge of these molecular players paints an elegant, albeit simplified, picture of CeD pathogenesis:
Dietary gluten is partially digested in the upper GI tract, resulting in a build-up of metastable gluten peptides that cross the intestinal epithelial barrier;
Upon encountering catalytically active TG2, some peptides undergo simultaneous lysosomal uptake and regioselective glutamine deamidation;
In the lysosomes of APCs, deamidated gluten peptides are loaded onto HLA-DQ2 or -DQ8 for presentation to CD4+ T cells; and
Once activated, CD4+ T cells orchestrate an inflammatory response that forms the basis of CeD pathology.
Celiac disease pathogenesis beyond CD4+ T Cells
Although the T cell-gluten peptide-APC interaction lies at the center of CeD pathogenesis, a complete description of CeD initiation and progression requires discussion of other molecules, cells, and pathways (Fig. 3). One clear illustration of this is the fact that only about 1% of Caucasian individuals will develop CeD despite 30–40% of them harboring a disease-specific HLA haplotype [14]. This raises questions regarding the biological features underlying the loss of oral tolerance to gluten in a genetically susceptible person. Indeed, while genome-wide association studies have unequivocally linked HLA molecules to CeD, more than 40 additional non-HLA genes have been found to be correlated to the disease, confirming there is more to pathogenesis than CD4+ T cell-mediated inflammation [15]. Many of these genes are involved in T and B cell immunobiology, antigen presenting cell function, and cytokine signaling [16,17].
Figure 3. Expanded pathogenesis of celiac disease beyond the core MHC-peptide- TCR interaction.
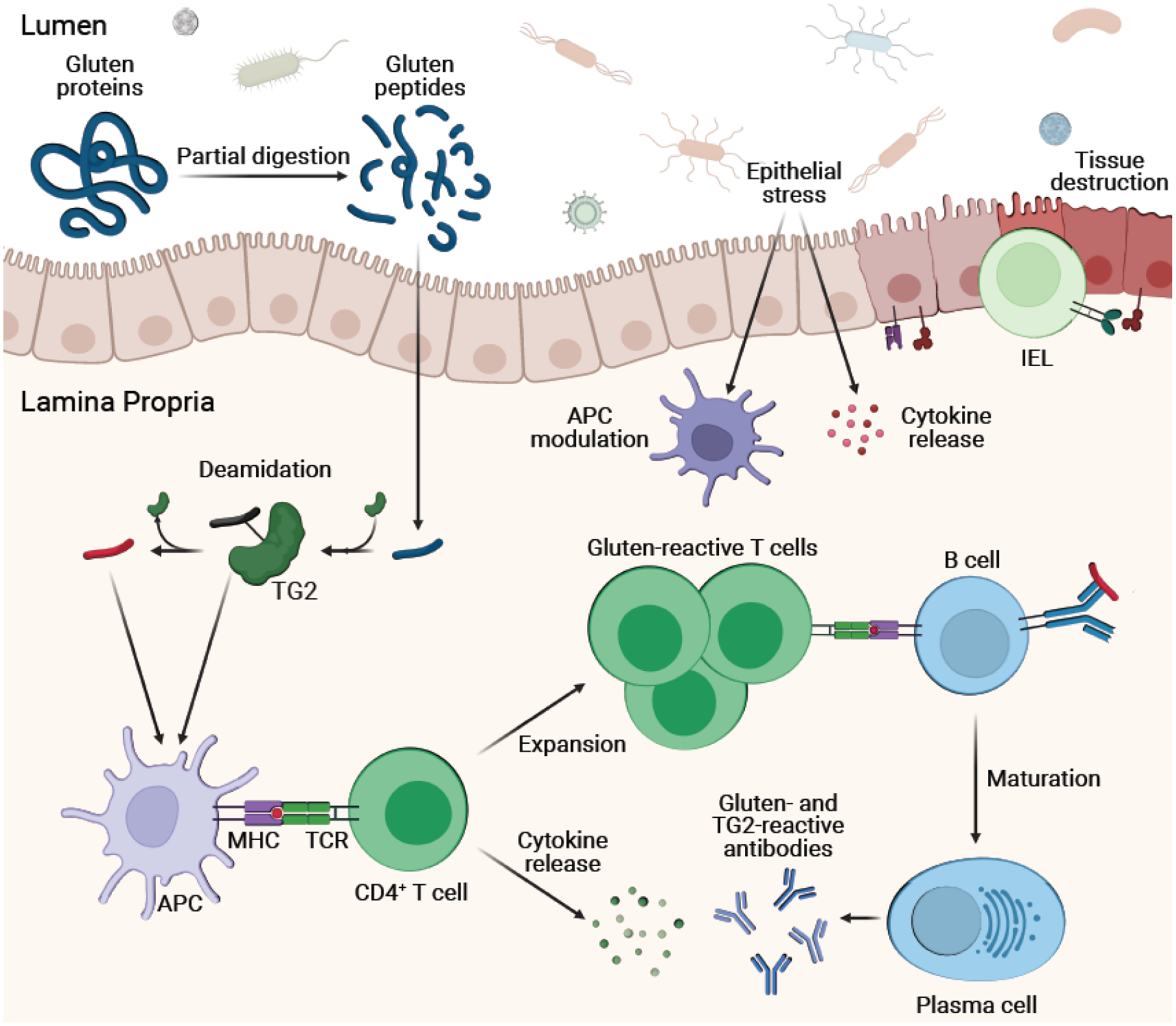
Following activation, CD4+ T cells produce cytokines (e.g., IL-2, IFN-γ, IL-21) and proliferate. Epithelial stress (e.g., from microbes and viruses) induces cytokine release from epithelial cells and upregulation of stress molecules like HLA-E and MICA/B on the intestinal epithelium. Simultaneously, epithelial stress modulates APCs like DCs towards a pro-inflammatory state. This overall inflammatory milieu licenses intraepithelial lymphocytes (IELs) towards a cytotoxic phenotype. Signaling between IELs and gut epithelium (e.g., through NKG2D-MICA interactions) induces cell death, causing tissue destruction. Additionally, B cells recognize deamidated gluten peptides and TG2-gluten peptide complexes through their receptors and subsequently present gluten peptides to CD4+ T cells through class II MHC. Positive signaling induces maturation of B cells to antibody-producing plasma cells.
The process by which gluten peptides cross the gut epithelium and enter the intestinal lamina propria remains an open question in CeD [18,19]. Some studies have found genetic variation in intestinal barrier and tight junction proteins that differentiate CeD patients from healthy individuals [20,21]. Others have shown the ability for gluten peptides or gluten peptide-antibody complexes to move transcellularly through gut epithelial cells [22,23]. Yet others have suggested a direct role for gluten peptides themselves in modulating barrier integrity [24]. Further work to determine mechanisms underlying gluten peptide translocation and the potential to intercept these pathways for therapy is necessary.
While dietary gluten is undoubtedly the principal environmental driver of CeD, a pathogenic role has also been proposed for viruses and bacteria in inducing the loss of oral tolerance to gluten. One well-described example is reovirus, whereby certain viral strains alter the intestinal immunologic environment and promote polarization of CD4+ T cells toward the inflammatory TH1 subset in the presence of gluten. The resulting inflammatory milieu is thought to license APCs such as CD103+ dendritic cells to aberrantly present dietary antigens such as gluten [25]. Previous cohort studies also showed a correlation between CeD and rotavirus, another double-stranded RNA virus [26]. However, the protective effect of rotavirus vaccination on the development of CeD in children is unclear [27,28]. Analogously, a connection between certain bacterial commensals and pathogens has also been invoked in the CeD literature [29–34]. Interestingly, the gut-colonizing protist Tritrichomonas arnold was shown to protect against the loss of oral tolerance to gluten induced by certain viruses through modulation of the intestinal DC compartment [35]. Future work is needed to assess the role of these organisms and others in the loss and maintenance of oral tolerance to gluten.
Notwithstanding the primary contributions of the adaptive immune response in CeD pathogenesis, innate immunity, orchestrated primarily by intraepithelial lymphocytes (IELs), also plays an important role. Indeed, intraepithelial lymphocytosis has been recognized as a hallmark histological feature of CeD since the pioneering studies of Marsh and coworkers [36,37]. This group of cells is made up largely of CD8+ αβ+ T cells, but also contains some natural killer (NK) cells as well as γδ+ T cells [38,39]. One of the most important cytokines for this compartment is interleukin-15 (IL-15), a key signal for maintenance of homeostasis in DCs, NK cells, and CD8+ T cells [40]. Upregulation of IL-15 in the intestine of CeD patients has been shown to induce a pro-inflammatory environment in multiple ways. Polarization of DCs with IL-15 promotes the generation of an inflammatory phenotype and increases production of pro-inflammatory cytokines like IFN-γ and IL-12-p70 [41]. IL-15 may also interfere with the protective Foxp3+ regulatory T cell response that is involved in maintaining tolerance to gluten [41]. Moreover, IL-15 stimulates the expression of the activating receptor NKG2D on IELs, licensing them for cytotoxic activity. Epithelial stress also leads to the upregulation of NKG2D ligands such as MICA and MICB on the gut epithelium [42]; this is thought to be one of the contributing factors to epithelial cell death in the CeD intestinal mucosa [43,44].
The other arm of the adaptive immune system, B cells and humoral immunity, contributes another essential facet to CeD pathogenesis. CeD is unique for many reasons, including the fact that a dietary antigen induces an autoimmune humoral response. This fact has led many in the field to question just “how autoimmune” celiac disease is [45]. In active disease, plasma cells create both TG2-reactive and gluten-reactive antibodies [46]. While these antibodies have incredible value in disease diagnosis, questions remain about the pathogenic function of these antibodies. One likely role of the B cell receptor is in mediating endocytosis of gluten peptides and TG2-gluten complexes, bringing about efficient delivery of gluten antigens to HLA-DQ2 or -DQ8 for presentation to CD4+ T cells [47]. Indeed, TG2-specific plasma cells are highly abundant in the intestine of celiac disease patients actively consuming gluten. These plasma cells are typically differentiated from naïve B cells and have a less mutated antibody repertoire [48]. Additional work has shown that these plasma cells express HLA-DQ, suggesting that the dual capacity to function as an APC while also secreting soluble immunoglobulins may be vital to the role of cells from the B cell lineage in CeD pathogenesis [49]. Recent reports have also identified gluten-reactive T follicular helper cells in both mice and CeD patients, further supporting a role for B cells in disease [39,50].
Emerging non-dietary celiac disease therapies
The practical, economic, social, and clinical shortcomings of maintaining a strict gluten-free diet have inspired a range of efforts to develop non-dietary therapies for CeD. Many of these potential treatment modalities hinge on the significant recent advancements in our understanding of disease pathogenesis at a molecular and cellular level. They can broadly be divided into two categories; those that act on the gluten-CD4+ T cell axis, and those that modulate off-axis pathways (Fig. 4).
Figure 4. Targets of putative celiac disease therapeutics currently being clinically tested.
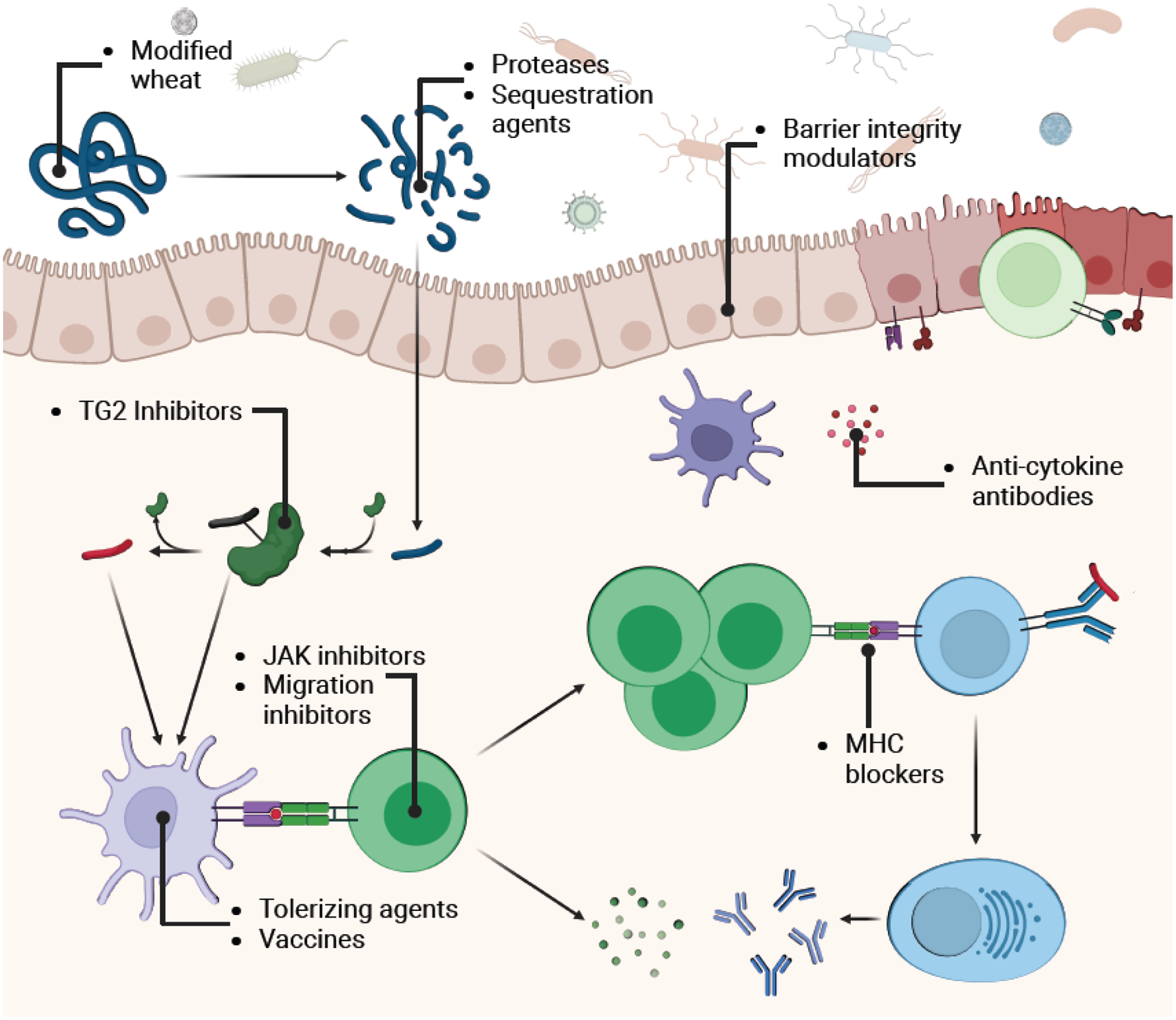
Therapeutic classes and their targets are indicated on the expanded model of pathogenesis. Therapies can operate before, within, or after the gluten-APC-CD4+ T cell interaction (ie., the core pathogenic axis). “On-axis” therapies include genetically engineered non-immunogenic wheat varieties, gluten sequestering agents, gluten degrading proteases or protease mixtures, TG2 inhibitors, and MHC blockers. “Off-axis” therapeutic modalities include anti-cytokine and anti-migration agents, JAK pathway inhibitors, barrier integrity modulators, and gluten tolerizing agents and vaccines.
Therapies that act on the gluten-CD4+ T cell axis
One place for intervention comes even before the ingestion of gluten; the genetic modification of wheat to ablate immunogenic T cell epitopes. The most successful of these non-immunogenic wheat varieties is E82, where RNA interference is used to degrade various mRNAs encoding toxic gliadin and glutenin proteins while sparing nonpathogenic glutenins that contribute to the viscoelastic properties of bread [51]. In a short-term study, celiac disease patients challenged with E82 bread had lower glutendependent IFN-γ responses and lower gluten immunogenic peptides in stool compared to those challenged with regular wheat bread. However, gastrointestinal symptoms between groups were not significantly different, potentially because of the short (1-week) timeline of the challenge [52].
In addition to modifying gluten itself, another gluten-targeting therapy that has been explored is the administration of agents that sequester gluten in the intestinal lumen and prevent peptide translocation into the lamina propria. One such agent is BL-7010, a modified polystyrene that selectively binds gluten in the intestine [53]. Administration of BL-7010 in gluten-sensitized mice mitigated some gluten-induced decrease in Vh:Cd ratio and prevented CD3+ intraepithelial lymphocytosis following gluten exposure [54]. Results from a clinical trial of BL-7010 have not yet been reported (NCT01990885). The polyclonal egg yolk-derived anti-gliadin antibody product AGY has similarly been shown to bind and prevent the absorption of gliadin in the intestine [55]. A phase 1 clinical trial showed safety of AGY in a small cohort of patients (NCT01765647) [56], and a phase 2 clinical trial has recently concluded with results expected soon (NCT03707730).
The best studied class of CeD therapies is recombinant, engineered proteases or protease mixtures designed to degrade gluten peptides in the gastrointestinal tract. Human salivary and gastrointestinal proteases are unable to fully digest proteins derived from cereal grains. This leaves immunogenic peptides intact and free to cross the small intestine epithelial barrier to induce inflammation in CeD patients. Proteases from other organisms, however, have been discovered and optimized to digest these human protease-resistant fragments to short peptides and single amino acids that are unable to elicit immune responses. Within this therapeutic modality, the most comprehensively tested is latiglutenase, a combination enzyme product that contains a glutamine-specific endoprotease and a prolyl endopeptidase that can extensively degrade gluten peptides [57–59]. Initial assessments of this dual enzyme therapy confirmed it was well tolerated and successfully degraded dietary gluten in humans (NCT00626184 and NCT00669825) [60]. Subsequent phase 2 trials of latiglutenase have produced mixed results depending on the specific patient population studied and their diets. Patients who were initially on a GFD but then challenged with a gluten-containing diet and latiglutenase treatment were protected from mucosal damage (NCT00959114 and NCT01255696) [61]. This result was confirmed more recently in a similar gluten-challenge cohort of patients. In addition to preventing mucosal damage, a higher dose of latiglutenase decreased symptoms (NCT03585478) [62]. In a larger cohort of patients with persistent symptoms despite GFD, however, latiglutenase treatment was undifferentiated from placebo with respect to Vh:Cd ratio, intraepithelial lymphocytosis, or anti-tTG serology (likely due to dietary modifications by study participants, a phenomenon known as the Hawthorne effect) [63]. Despite the lack of positive changes in their clinical signs, patients in the high-dose enzyme groups of this study did show symptomatic improvements (NCT01917630) [64]. A phase 2 trial of latiglutenase in symptomatic CeD patients on a GFD is ongoing (NCT04243551). In addition to latiglutenase, two other gluten-digesting agents have been evaluated in the clinic: a prolyl endopeptidase from Aspergillus niger (AN-PEP) [65] and a computationally designed protease originating from Alicyclobacillus sendaiensis kumamolisin-As (TAK-062) [66,67]. Both AN-PEP (NCT00810654 and NCT04788797) [68] and TAK-062 (NCT03701555) [66] were well-tolerated and showed some benefit in short-term gluten challenge. A phase 2 trial of TAK-062 is ongoing (NCT05353985).
Gluten peptides must be deamidated by TG2, either in the intestinal lumen or lamina propria, to be effectively presented to inflammatory CD4+ T cells. As such, inhibiting TG2 activity in the small intestine has been hypothesized to prevent the toxification of native gluten peptides. Significant work has been done towards the development of covalent and non-covalent TG2 inhibitors [69]. The most advanced candidate in this class is ZED1227, a covalent and irreversible TG2 inhibitor with promising potency and selectivity [70]. Phase 1 trials of this compound demonstrated its safety and tolerability in humans (EudraCT 2014–003044-13 and 2015–005283-42). In a phase 2 efficacy study, compared to placebo, ZED1227 attenuated mucosal damage as measured by Vh:Cd and decreased intraepithelial lymphocytosis and patient symptoms significantly in the highest dose group (EudraCT 2017–002241-30) [71].
Yet another approach to clinically disrupt the CD4+ T cell axis in CeD pathogenesis is via peptide-MHC blockers, agents that prevent T cell activation by inhibiting the formation of an MHC-peptide-T cell receptor ternary complex. This strategy is particularly well-suited for CeD because of our knowledge of specific deamidated gluten peptide-HLA-DQ pairs [72–74]. Recent efforts have led to the generation of monoclonal antibodies (mAbs) and antibody-like proteins that bind peptide-MHC complexes (so called “TCR-like antibodies”) [75]. One such agent, DONQ52, has recently initiated clinical trials (NCT05425446).
Celiac disease therapies that act off-axis
An alternative approach for the treatment of celiac disease centers not on gluten itself, but instead on the interactions and cellular players involved in initiating and executing the immune response to gluten. Both mAbs and small molecules are being developed for this purpose. For example, because of the role of IL-15 in activating intraepithelial cytotoxic T lymphocytes and exacerbating small intestinal inflammation, mAbs against the cytokine itself or the IL-15 receptor have been clinically tested. AMG714, a monoclonal antibody against IL-15, was investigated in the setting of gluten challenge in CeD patients. While the AMG714 treatment group did not show improvement in Vh:Cd ratios, the cohort did exhibit improved symptom quality indices and the high dose group showed lower intraepithelial lymphocyte counts on biopsy (NCT02637141) [76]. A follow-up phase 2 trial of AMG714 in CeD patients unresponsive to GFD showed no improvement in histopathologic features of CeD, but did show benefit in symptom quality indices (NCT02633020) [77]. A phase 1 trial of a mAb targeting the IL-15Rβ1 subunit, Hu-Mik-β1, was completed in 2019 but the results have not yet been reported (NCT01893775). Separately, tofacitinib, a small molecule pan-JAK inhibitor that inhibits signaling downstream of IL-15, is being tested in patients with type 2 refractory CeD (NCT05636293).
In addition to compounds targeting the IL-15 signaling pathway, agents inhibiting key molecules for cellular migration and trafficking have been employed. For example, α47 integrin is a critical signaling molecule for the homing of T and B cells to the gut [78]. A mAb specific for α4β7 integrin, vedolizumab, has been approved for use in ulcerative colitis and is now being evaluated in CeD patients (NCT02929316). SIRT6 is a sirtuin transcriptional regulator that affects enterocyte biology and is thought to modulate barrier function of the small intestine. IMU-856 is an oral small molecule SIRT6 modulator that is being evaluated in CeD patients for safety and efficacy [79].
While all therapeutic candidates discussed above are intended to block active CeD pathogenesis, a different group of therapies has been designed to test the feasibility of eliciting immune tolerance to gluten in vivo. These desensitizing agents work at a cellular level to inhibit pathogenic immune reactions to gluten. The most clinically advanced of these is TAK-101. TAK-101 is a negatively charged poly(DL-lactide-coglycolide) nanoparticle that encapsulates intact gliadin protein [80]. In nonclinical studies, nanoparticles such as these have been shown to induce a tolerogenic phenotype in APCs such as macrophages and dendritic cells [81,82]. In a phase 2 gluten challenge trial of TAK-101 in CeD patients, the treatment group had a decreased IFN-γ response to gliadin as well as protection from mucosal injury as measured by maintenance of baseline Vh:Cd ratio. Treated patients also showed lower proportions of gut-homing T cells in the peripheral blood, all supporting induction of tolerance to gliadin (NCT03486990 and NCT03738475) [83]. In theory, such drug candidates could also be used as vaccines. Indeed, Nexvax-2 is a mixture of three pre-deamidated gluten peptides that represent important immunogenic epitopes for CD4+ T cell recognition via HLA-DQ2.5 [84,85]. The vaccine is formulated without an adjuvant and administered intradermally. Initial phase 1 studies confirmed the safety and tolerability of the vaccine, despite induction of gastrointestinal symptoms following dosing [84]. The highest dosed group maintained duodenal integrity despite the administration of immunogenic peptides and may have demonstrated blunted IFN-γ release in response to the vaccine peptides, potentially suggesting the viability of this strategy for inducing immune tolerance following repeat injections (NCT02528799) [86]. Unfortunately, a follow-up phase 2 study was discontinued before completion because of futility.
Implications for other autoimmune diseases
Among autoimmune diseases, celiac disease is unique in that the most important causative environmental and genetic factors as well as their mutual interactions have been extensively characterized. Notably, the triggering antigens, gluten peptides, are derived from exogenous non-self proteins, which can be administered to CeD patients in a controlled manner. Furthermore, the principal affected organ in CeD, the small intestine, can be readily sampled via upper gastrointestinal endoscopic procedures. Together, these features have allowed researchers to interrogate the short-term and longer-term molecular, histological, and clinical sequelae of gluten exposure in patients with considerable precision. As just one example, impressive strides have been made in recent years with respect to characterizing the mobilization of gluten-reactive T cells from a patient’s small intestine to the peripheral blood and the concomitant burst of cytokine release in response to an acute gluten challenge [87]. It is possible that peripheral sampling and characterization of the immune compartment may also prove useful in the study of other autoimmune diseases [88], especially those with analogous dependences on HLA molecules (e.g., HLA-B27 in ankylosing spondylitis and HLA-DR2 in multiple sclerosis). Ultimately however, extrapolation of lessons learned from CeD pathogenesis to other autoimmune diseases will hinge on the identification of driver antigens in those disorders.
The role of transglutaminase 2 in CeD is another instructive aspect of the pathogenesis of this autoimmune disorder. Post-translational modification of gluten peptides by TG2 as well as the appearance of anti-TG2 antibodies led to a hypothesis regarding a role for gluten peptide-TG2 complexes. The activity of TG2 in CeD is highly reminiscent of the role of another enzyme, protein arginine deiminase 2 (PAD2), in rheumatoid arthritis (RA). PAD2 carries out the conversion of arginine to citrulline in antigenic peptides in RA, allowing these less positively charged peptide products to be more favorably loaded into HLA-DR4 [89]. The recent demonstration of TG2 inhibition as a potential therapeutic avenue for CeD raises the possibility of PAD2 inhibition for RA treatment. It remains to be seen if other autoimmune diseases share similar dependence on selfproteins for post-translational modification of their causative antigens.
Concluding remarks and future perspectives
The increasing prevalence, risk of significant morbidity, and lack of non-dietary therapy each demand vigorous fundamental mechanistic research and clinical therapeutic development for celiac disease. Over the past 70 years, the field has made significant progress in illuminating the core molecules, cells, and pathways that drive disease. Historical and recent work has unequivocally shown the central role of the class II MHC-deamidated gluten peptide-CD4+ T cell receptor interaction in pathogenesis. More recent efforts have begun to uncover essential players in the initiation of disease as well as the executers of tissue destruction. However, despite this increasingly clear picture of the mechanisms underlying CeD pathogenesis, many open questions remain (see Outstanding Questions). Perhaps the most important is why some people with an HLA genetic predisposition get disease while others do not. The identity of key professional APCs that present deamidated gluten antigens to pathogenic CD4+ T cells at various stages of the disease also remains to be established. Finally, one of the clearest areas for future research is in elucidating the influences of the greater than 40 non-HLA genes implicated in CeD. The development of CeD organoids from patient intestinal tissue appears to be an emerging field for the study of disease as well as for testing potential therapeutics before initiating human trials [90].
Outstanding Questions.
What are the roles of non-HLA genes in celiac disease pathogenesis? Can the products of these genes and/or pathways be targeted for CeD therapy?
How is oral tolerance to gluten broken? What are the key molecules, pathways, viruses, organisms, and cells involved in the loss of oral tolerance?
Which antigen presenting cells are responsible for gluten antigen presentation to T cells at different stages of the disease?
Which CeD testing criteria and methods offer the greatest diagnostic accuracy and clinical utility? What are the best methods for assessing dietary compliance and response to treatment?
Will a non-dietary CeD therapy show broad utility for the treatment of CeD in a large trial? Will a non-dietary therapy for CeD allow a patient to resume consumption of gluten, or will medical therapy function only as adjunct to the GFD?
In a clinical setting, while paradigms for CeD diagnosis are generally well established, there remain questions about the optimal strategies for diagnosing and assessing response to gluten-free diet. In this context, the development of sensitive and specific tests for detecting inadvertent gluten exposure and adherence to the gluten-free diet may prove especially useful for the management of CeD patients [91]. Perhaps the most important practical transformation however is likely to come from the emergence of non-dietary medicines for managing this lifelong disorder. Whereas first-generation therapies are likely to be used as adjuncts to a GFD, eventually it may be possible to discover effective disease-modifying treatments that allow patients to consume a normal (or near-normal) diet.
The state of CeD research and clinical trials exemplifies the synergy that exists between fundamental discovery and therapeutic development. A salient example of this is the relatively short 12-year gap between the first demonstration of prolyl endoproteolysis as a gluten detoxification strategy in vitro by Shan and coworkers [6] and the clinical report of latiglutenase offering histological benefit to patients undergoing gluten challenge [61]. Given the rapid pace at which new insights in pathogenesis are translated to putative therapeutic agents in the clinic, it is reasonable to expect that the coming decade will see the first approved non-dietary therapy for celiac disease reach patients.
Highlights.
Celiac disease (CeD) is a chronic inflammatory disorder that occurs in response to gluten-derived peptides in genetically susceptible individuals.
Currently, the only approved treatment for CeD is life-long dietary exclusion of wheat and related cereal grains.
The causative antigens, genetic background, and their interactions have been well-characterized in CeD pathogenesis.
Recent work has begun to elucidate additional genetic and environmental factors important for the initiation of disease and execution of tissue destruction.
This molecular and cellular understanding of CeD pathogenesis has led to the development of numerous putative therapies that are currently being tested in the clinic.
Acknowledgements
This work was supported by a grant from the NIH (R01DK063158 and R01DK126487 to C.K.). H.A.B. was supported by the National Institutes of Health (F30DK132903) and the Stanford University Medical Scientist Training Program (T32GM007365 and T32GM145402). Figures were created with BioRender.com.
Glossary
- Antigen presenting cells (APCs)
a subset of professional immune cells that are capable of loading peptides onto class II major histocompatibility complex (MHC) proteins and presenting them to CD4+ T cells. Classically, this group includes dendritic cells, macrophages, and B cells
- Celiac disease (CeD)
an autoimmune disorder triggered by the ingestion of dietary gluten that occurs in certain genetically susceptible individuals. The primary affected organ in CeD is the small intestine, although extra-intestinal sequelae of gluten-induced inflammation are common
- Gluten
Proline- and glutamine-rich storage proteins in certain cereal grains that harbor the causative antigens of celiac disease. Common gluten sources include wheat (whose grain contains glutenins and gliadins), barley (whose grain contains hordeins) and rye (whose grain contains secalins)
- Gluten-free diet (GFD)
a diet that eliminates wheat, barley, and rye. At present this is the only acceptable form of lifelong therapy for celiac disease. For a dietary product to be gluten-free, it must contain < 20 ppm gluten. As a point of reference, a typical 30 g slice of whole wheat bread contains 3 g gluten
- Human leukocyte antigen DQ2 and human leukocyte antigen DQ8 (HLA-DQ2 and HLA-DQ8)
heterodimeric transmembrane proteins belonging to the family of class II MHC receptors. These isoforms of human class II MHC proteins have the unique ability to bind gluten-derived peptides and present them to CD4+ T cells
- Immunoglobulin G (IgG) and immunoglobulin A (IgA)
different isotypes of antibodies secreted by plasma cells of the B cell lineage
- Intraepithelial lymphocytes (IELs)
immune cells that reside within the epithelial layer of mucosal organs such as the intestine and lungs. They consist primarily of effector (CD8+) T cells harboring an αβ or γδ T cell receptor. In the small intestine of CeD patients, IELs play a role in mediating tissue destruction
- Oral tolerance
the active immunologic state of unresponsiveness to orally delivered antigens (such as gluten). The process by which oral tolerance to gluten is lost (i.e., transformation of gluten from innocuous to immunotoxic) is an open question in CeD
- Transglutaminase 2 (TG2) or tissue transglutaminase (tTG)
a widely expressed enzyme in mammals whose primary physiological function has not yet been established. In CeD, this enzyme post-translationally modifies gluten peptides via deamidation of select glutamine residues (resulting in a net glutamine → glutamate conversion). TG2 is also the major target of autoantibodies in CeD patients
- Villous height to crypt depth ratio (Vh:Cd)
The small intestinal surface is covered with finger-like projections (villi) separated from each other by invaginations (crypts). This morphology can be microscopically visualized in a thin slice of a small intestinal biopsy sample. Because villi appear blunted and crypts become deeper in CeD patients, the average villous height to crypt depth ratio is a definitive and quantitative marker of intestinal tissue damage in CeD
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
C.K. serves as a director of ImmunogenX.
References
- 1.Singh P, Arora A, Strand TA, Leffler DA, Catassi C, Green PH, et al. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. 2018. Jun;16(6):823–836.e2. [DOI] [PubMed] [Google Scholar]
- 2.King JA, Jeong J, Underwood FE, Quan J, Panaccione N, Windsor JW, et al. Incidence of Celiac Disease Is Increasing Over Time: A Systematic Review and Meta-analysis. Am J Gastroenterol. 2020. Apr;115(4):507–25. [DOI] [PubMed] [Google Scholar]
- 3.Palanski BA, Weng N, Zhang L, Hilmer AJ, Fall LA, Swaminathan K, et al. An efficient urine peptidomics workflow identifies chemically defined dietary gluten peptides from patients with celiac disease. Nat Commun. 2022. Feb 16;13(1):888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biesiekierski JR. What is gluten? Journal of Gastroenterology and Hepatology. 2017;32(S1):78–81. [DOI] [PubMed] [Google Scholar]
- 5.Wieser H Chemistry of gluten proteins. Food Microbiology. 2007. Apr;24(2):115–9. [DOI] [PubMed] [Google Scholar]
- 6.Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural Basis for Gluten Intolerance in Celiac Sprue. Science. 2002. Sep 27;297(5590):2275–9. [DOI] [PubMed] [Google Scholar]
- 7.Sollid LM. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics. 2017. Aug;69(8–9):605–16. [DOI] [PubMed] [Google Scholar]
- 8.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2- mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A. 2004. Mar 23;101(12):4175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ráki M, Tollefsen S, Molberg Ø, Lundin KEA, Sollid LM, Jahnsen FL. A Unique Dendritic Cell Subset Accumulates in the Celiac Lesion and Efficiently Activates Gluten-Reactive T Cells. Gastroenterology. 2006. Aug;131(2):428–38. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang R, Khosla C. Substrates, inhibitors, and probes of mammalian transglutaminase 2. Analytical Biochemistry. 2020. Feb;591:113560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khosla C Celiac Disease: Lessons for and from Chemical Biology. ACS Chem Biol. 2017. Jun 16;12(6):1455–9. [DOI] [PubMed] [Google Scholar]
- 12.Loppinet E, Besser HA, Sewa AS, Yang FC, Jabri B, Khosla C. LRP-1 links post-translational modifications to efficient presentation of celiac disease-specific T cell antigens. Cell Chemical Biology. 2023. Jan;30(1):55–68.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. 1995. Dec 1;37(6):766–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green PHR, Jabri B. Celiac Disease. Annu Rev Med. 2006. Feb 1;57(1):207–21. [DOI] [PubMed] [Google Scholar]
- 15.Spanish Consortium on the Genetics of Coeliac Disease (CEGEC), PreventCD Study Group, Wellcome Trust Case Control Consortium (WTCCC), Trynka G, Hunt KA, Bockett NA, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011. Dec;43(12):1193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007. Jul;39(7):827–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunt KA, Zhernakova A, Turner G, Heap GAR, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008. Apr;40(4):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyman M, Abed J, Lebreton C, Cerf-Bensussan N. Intestinal permeability in coeliac disease: insight into mechanisms and relevance to pathogenesis. Gut. 2012. Sep;61(9):1355–64. [DOI] [PubMed] [Google Scholar]
- 19.Escudero-Hernández C Epithelial cell dysfunction in coeliac disease. In: International Review of Cell and Molecular Biology [Internet]. Elsevier; 2021. [cited 2023 Sep 11]. p. 133–64. Available from: https://linkinghub.elsevier.com/retrieve/pii/S193764482030099X [DOI] [PubMed] [Google Scholar]
- 20.Wapenaar MC, Monsuur AJ, Van Bodegraven AA, Weersma RK, Bevova MR, Linskens RK, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitisAn unusual case of ascites. Gut. 2007. Dec 13;57(4):463–7. [DOI] [PubMed] [Google Scholar]
- 21.Kumar V, Gutierrez-Achury J, Kanduri K, Almeida R, Hrdlickova B, Zhernakova DV, et al. Systematic annotation of celiac disease loci refines pathological pathways and suggests a genetic explanation for increased interferon-gamma levels. Human Molecular Genetics. 2015. Jan 15;24(2):397–409. [DOI] [PubMed] [Google Scholar]
- 22.Schumann M, Richter JF, Wedell I, Moos V, Zimmermann-Kordmann M, Schneider T, et al. Mechanisms of epithelial translocation of the 2-gliadin-33mer in coeliac sprue. Gut. 2008. Feb 13;57(6):747–54. [DOI] [PubMed] [Google Scholar]
- 23.Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, Lebreton C, Ménard S, Candalh C, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. The Journal of Experimental Medicine. 2008. Jan 21;205(1):143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin Induces an Increase in Intestinal Permeability and Zonulin Release by Binding to the Chemokine Receptor CXCR3. Gastroenterology. 2008. Jul;135(1):194–204.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science. 2017. Apr 7;356(6333):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, et al. Rotavirus Infection Frequency and Risk of Celiac Disease Autoimmunity in Early Childhood: A Longitudinal Study. Am J Gastroenterology. 2006. Oct;101(10):2333–40. [DOI] [PubMed] [Google Scholar]
- 27.Vaarala O, Jokinen J, Lahdenkari M, Leino T. Rotavirus Vaccination and the Risk of Celiac Disease or Type 1 Diabetes in Finnish Children at Early Life. Pediatric Infectious Disease Journal. 2017. Jul;36(7):674–5. [DOI] [PubMed] [Google Scholar]
- 28.Hemming-Harlo M, Lähdeaho ML, Mäki M, Vesikari T. Rotavirus Vaccination Does Not Increase Type 1 Diabetes and May Decrease Celiac Disease in Children and Adolescents. Pediatric Infectious Disease Journal. 2019. May;38(5):539–41. [DOI] [PubMed] [Google Scholar]
- 29.Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut. 2015. Mar;64(3):406–17. [DOI] [PubMed] [Google Scholar]
- 30.Petersen J, Ciacchi L, Tran MT, Loh KL, Kooy-Winkelaar Y, Croft NP, et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat Struct Mol Biol. 2020. Jan;27(1):49–61. [DOI] [PubMed] [Google Scholar]
- 31.Sellitto M, Bai G, Serena G, Fricke WF, Sturgeon C, Gajer P, et al. Proof of Concept of Microbiome-Metabolome Analysis and Delayed Gluten Exposure on Celiac Disease Autoimmunity in Genetically At-Risk Infants. Highlander SK, editor. PLoS ONE 2012. Mar 14;7(3):e33387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leonard MM, Valitutti F, Karathia H, Pujolassos M, Kenyon V, Fanelli B, et al. Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proc Natl Acad Sci USA. 2021. Jul 20;118(29):e2020322118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamas B, Hernandez-Galan L, Galipeau HJ, Constante M, Clarizio A, Jury J, et al. Aryl hydrocarbon receptor ligand production by the gut microbiota is decreased in celiac disease leading to intestinal inflammation. Sci Transl Med. 2020. Oct 21;12(566):eaba0624. [DOI] [PubMed] [Google Scholar]
- 34.Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol. 2015. Sep;12(9):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medina Sanchez L, Siller M, Zeng Y, Brigleb PH, Sangani KA, Soto AS, et al. The gut protist Tritrichomonas arnold restrains virus-mediated loss of oral tolerance by modulating dietary antigen-presenting dendritic cells. Immunity. 2023. Aug;56(8):1862–1875.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oberhuber G Histopathology of celiac disease. Biomedicine & Pharmacotherapy. 2000. Aug;54(7):368–72. [DOI] [PubMed] [Google Scholar]
- 37.Marsh MN, Haeney MR. Studies of intestinal lymphoid tissue. VI--Proliferative response of small intestinal epithelial lymphocytes distinguishes gluten- from non-gluten-induced enteropathy. Journal of Clinical Pathology. 1983. Feb 1;36(2):149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayassi T, Ladell K, Gudjonson H, McLaren JE, Shaw DG, Tran MT, et al. Chronic Inflammation Permanently Reshapes Tissue-Resident Immunity in Celiac Disease. Cell. 2019. Feb 21;176(5):967–981.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornberg A, Botella T, Moon CS, Rao S, Gelbs J, Cheng L, et al. Gluten induces rapid reprogramming of natural memory αβ and γδ intraepithelial T cells to induce cytotoxicity in celiac disease. Sci Immunol. 2023. Jul 21;8(85):eadf4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fehniger TA, Caligiuri MA. Interleukin 15: biology and relevance to human disease. Blood. 2001. Jan 1;97(1):14–32. [DOI] [PubMed] [Google Scholar]
- 41.DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011. Mar;471(7337):220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A Direct Role for NKG2D/MICA Interaction in Villous Atrophy during Celiac Disease. Immunity. 2004. Sep;21(3):367–77. [DOI] [PubMed] [Google Scholar]
- 43.Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated Induction by IL15 of a TCR-Independent NKG2D Signaling Pathway Converts CTL into Lymphokine-Activated Killer Cells in Celiac Disease. Immunity. 2004. Sep;21(3):357–66. [DOI] [PubMed] [Google Scholar]
- 44.Tang F, Chen Z, Ciszewski C, Setty M, Solus J, Tretiakova M, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. Journal of Experimental Medicine. 2009. Mar 16;206(3):707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sollid LM, Jabri B. Is celiac disease an autoimmune disorder? Current Opinion in Immunology. 2005. Dec 1;17(6):595–600. [DOI] [PubMed] [Google Scholar]
- 46.Lindeman I, Zhou C, Eggesbø LM, Miao Z, Polak J, Lundin KEA, et al. Longevity, clonal relationship, and transcriptional program of celiac disease–specific plasma cells. Journal of Experimental Medicine. 2021. Feb 1;218(2):e20200852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iversen R, Sollid LM. The Immunobiology and Pathogenesis of Celiac Disease. Annu Rev Pathol Mech Dis. 2023. Jan 24;18(1):47–70. [DOI] [PubMed] [Google Scholar]
- 48.Di Niro R, Mesin L, Zheng NY, Stamnaes J, Morrissey M, Lee JH, et al. High abundance of plasma cells secreting transglutaminase 2–specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012. Mar;18(3):441–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Høydahl LS, Richter L, Frick R, Snir O, Gunnarsen KS, Landsverk OJB, et al. Plasma Cells Are the Most Abundant Gluten Peptide MHC-expressing Cells in Inflamed Intestinal Tissues From Patients With Celiac Disease. Gastroenterology. 2019. Apr;156(5):1428–1439.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong SW, Krueger PD, Osum KC, Dileepan T, Herman A, Mueller DL, et al. Immune tolerance of food is mediated by layers of CD4+ T cell dysfunction. Nature. 2022. Jul;607(7920):762–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barro F, Iehisa JCM, Giménez MJ, García-Molina MD, Ozuna CV, Comino I, et al. Targeting of prolamins by RNAi in bread wheat: effectiveness of seven silencing-fragment combinations for obtaining lines devoid of coeliac disease epitopes from highly immunogenic gliadins. Plant Biotechnol J. 2016. Mar;14(3):986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guzmán-López MH, Sánchez-León S, Marín-Sanz M, Comino I, Segura V, Vaquero L, et al. Oral Consumption of Bread from an RNAi Wheat Line with Strongly Silenced Gliadins Elicits No Immunogenic Response in a Pilot Study with Celiac Disease Patients. Nutrients. 2021. Dec 18;13(12):4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinier M, Fuhrmann G, Galipeau HJ, Rivard N, Murray JA, David CS, et al. The Copolymer P(HEMA-co-SS) Binds Gluten and Reduces Immune Response in Gluten-Sensitized Mice and Human Tissues. Gastroenterology. 2012. Feb;142(2):316–325.e12. [DOI] [PubMed] [Google Scholar]
- 54.McCarville JL, Nisemblat Y, Galipeau HJ, Jury J, Tabakman R, Cohen A, et al. BL-7010 Demonstrates Specific Binding to Gliadin and Reduces Gluten-Associated Pathology in a Chronic Mouse Model of Gliadin Sensitivity. D’Auria S, editor. PLoS ONE 2014. Nov 3;9(11):e109972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gujral N, Löbenberg R, Suresh M, Sunwoo H. In-Vitro and In-Vivo Binding Activity of Chicken Egg Yolk Immunoglobulin Y (IgY) against Gliadin in Food Matrix. J Agric Food Chem. 2012. Mar 28;60(12):3166–72. [DOI] [PubMed] [Google Scholar]
- 56.Sample DA, Sunwoo HH, Huynh HQ, Rylance HL, Robert CL, Xu BW, et al. AGY, a Novel Egg Yolk-Derived Anti-gliadin Antibody, Is Safe for Patients with Celiac Disease. Dig Dis Sci. 2017. May;62(5):1277–85. [DOI] [PubMed] [Google Scholar]
- 57.Gass J, Bethune MT, Siegel M, Spencer A, Khosla C. Combination Enzyme Therapy for Gastric Digestion of Dietary Gluten in Patients With Celiac Sprue. Gastroenterology. 2007. Aug;133(2):472–80. [DOI] [PubMed] [Google Scholar]
- 58.Siegel M, Bethune MT, Gass J, Ehren J, Xia J, Johannsen A, et al. Rational design of combination enzyme therapy for celiac sprue. Chem Biol. 2006. Jun;13(6):649–58. [DOI] [PubMed] [Google Scholar]
- 59.Tye-Din JA, Anderson RP, Ffrench RA, Brown GJ, Hodsman P, Siegel M, et al. The effects of ALV003 pre-digestion of gluten on immune response and symptoms in celiac disease in vivo. Clinical Immunology. 2010. Mar;134(3):289–95. [DOI] [PubMed] [Google Scholar]
- 60.Siegel M, Garber ME, Spencer AG, Botwick W, Kumar P, Williams RN, et al. Safety, Tolerability, and Activity of ALV003: Results from Two Phase 1 Single, Escalating-Dose Clinical Trials. Dig Dis Sci. 2012. Feb;57(2):440–50. [DOI] [PubMed] [Google Scholar]
- 61.Lähdeaho ML, Kaukinen K, Laurila K, Vuotikka P, Koivurova OP, Kärjä-Lahdensuu T, et al. Glutenase ALV003 Attenuates Gluten-Induced Mucosal Injury in Patients With Celiac Disease. Gastroenterology. 2014. Jun;146(7):1649–58. [DOI] [PubMed] [Google Scholar]
- 62.Murray JA, Syage JA, Wu TT, Dickason MA, Ramos AG, Van Dyke C, et al. Latiglutenase Protects the Mucosa and Attenuates Symptom Severity in Patients With Celiac Disease Exposed to a Gluten Challenge. Gastroenterology. 2022. Dec;163(6):1510–1521.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murray JA, Kelly CP, Green PHR, Marcantonio A, Wu TT, Mäki M, et al. No Difference Between Latiglutenase and Placebo in Reducing Villous Atrophy or Improving Symptoms in Patients With Symptomatic Celiac Disease. Gastroenterology. 2017. Mar;152(4):787–798.e2. [DOI] [PubMed] [Google Scholar]
- 64.Syage JA, Murray JA, Green PHR, Khosla C. Latiglutenase Improves Symptoms in Seropositive Celiac Disease Patients While on a Gluten-Free Diet. Dig Dis Sci. 2017. Sep;62(9):2428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitea C, Havenaar R, Drijfhout JW, Edens L, Dekking L, Koning F. Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: implications for coeliac disease. Gut. 2007. May 14;57(1):25–32. [DOI] [PubMed] [Google Scholar]
- 66.Pultz IS, Hill M, Vitanza JM, Wolf C, Saaby L, Liu T, et al. Gluten Degradation, Pharmacokinetics, Safety, and Tolerability of TAK-062, an Engineered Enzyme to Treat Celiac Disease. Gastroenterology. 2021. Jul;161(1):81–93.e3. [DOI] [PubMed] [Google Scholar]
- 67.Wolf C, Siegel JB, Tinberg C, Camarca A, Gianfrani C, Paski S, et al. Engineering of Kuma030: A Gliadin Peptidase That Rapidly Degrades Immunogenic Gliadin Peptides in Gastric Conditions. J Am Chem Soc. 2015. Oct 14;137(40):13106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tack GJ. Consumption of gluten with gluten-degrading enzyme by celiac patients: A pilotstudy. WJG. 2013;19(35):5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacology & Therapeutics. 2007. Aug 1;115(2):232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Büchold C, Hils M, Gerlach U, Weber J, Pelzer C, Heil A, et al. Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells. 2022. May 17;11(10):1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuppan D, Mäki M, Lundin KEA, Isola J, Friesing-Sosnik T, Taavela J, et al. A Randomized Trial of a Transglutaminase 2 Inhibitor for Celiac Disease. N Engl J Med. 2021. Jul 1;385(1):35–45. [DOI] [PubMed] [Google Scholar]
- 72.Kapoerchan VV, Wiesner M, Overhand M, Van Der Marel GA, Koning F, Overkleeft HS. Design of azidoproline containing gluten peptides to suppress CD4+ T-cell responses associated with Celiac disease. Bioorganic & Medicinal Chemistry. 2008. Feb 15;16(4):2053–62. [DOI] [PubMed] [Google Scholar]
- 73.Huan J, Meza-Romero R, Mooney JL, Vandenbark AA, Offner H, Burrows GG. Single-chain recombinant HLA-DQ2.5/peptide molecules block α2-gliadin-specific pathogenic CD4+ T-cell proliferation and attenuate production of inflammatory cytokines: a potential therapy for celiac disease. Mucosal Immunology. 2011. Jan;4(1):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xia J, Bergseng E, Fleckenstein B, Siegel M, Kim CY, Khosla C, et al. Cyclic and dimeric gluten peptide analogues inhibiting DQ2-mediated antigen presentation in celiac disease. Bioorganic & Medicinal Chemistry. 2007. Oct;15(20):6565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frick R, Høydahl LS, Petersen J, du Pré MF, Kumari S, Berntsen G, et al. A high-affinity human TCR-like antibody detects celiac disease gluten peptide–MHC complexes and inhibits T cell activation. Sci Immunol. 2021. Aug 20;6(62):eabg4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lähdeaho ML, Scheinin M, Vuotikka P, Taavela J, Popp A, Laukkarinen J, et al. Safety and efficacy of AMG 714 in adults with coeliac disease exposed to gluten challenge: a phase 2a, randomised, double-blind, placebo-controlled study. The Lancet Gastroenterology & Hepatology. 2019. Dec;4(12):948–59. [DOI] [PubMed] [Google Scholar]
- 77.Cellier C, Bouma G, Van Gils T, Khater S, Malamut G, Crespo L, et al. Safety and efficacy of AMG 714 in patients with type 2 refractory coeliac disease: a phase 2a, randomised, double-blind, placebo-controlled, parallel-group study. The Lancet Gastroenterology & Hepatology. 2019. Dec;4(12):960–70. [DOI] [PubMed] [Google Scholar]
- 78.Wagner N, Löhler J, Kunkel EJ, Ley K, Leung E, Krissansen G, et al. Critical role for β7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996. Jul;382(6589):366–70. [DOI] [PubMed] [Google Scholar]
- 79.Immunic Reports Positive Results From Phase 1b Clinical Trial of IMU-856 in Celiac Disease, Providing Clinical Proof-of-Concept for New Therapeutic Approach to Gastrointestinal Disorders [Internet]. Immunic, Inc.; 2023. May. Available from: https://imux.com/immunic-reports-positive-results-from-phase-1b-clinical-trial-of-imu-856-in-celiac-disease-providing-clinical-proof-of-concept-for-new-therapeutic-approach-to-gastrointestinal-disorders/ [Google Scholar]
- 80.Freitag TL, Podojil JR, Pearson RM, Fokta FJ, Sahl C, Messing M, et al. Gliadin Nanoparticles Induce Immune Tolerance to Gliadin in Mouse Models of Celiac Disease. Gastroenterology. 2020. May;158(6):1667–1681.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCarthy DP, Hunter ZN, Chackerian B, Shea LD, Miller SD. Targeted immunomodulation using antigen-conjugated nanoparticles: Targeted immunomodulation. WIREs Nanomed Nanobiotechnol. 2014. May;6(3):298–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, et al. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. Journal of Autoimmunity. 2018. May;89:112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kelly CP, Murray JA, Leffler DA, Getts DR, Bledsoe AC, Smithson G, et al. TAK-101 Nanoparticles Induce Gluten-Specific Tolerance in Celiac Disease: A Randomized, Double- Blind, Placebo-Controlled Study. Gastroenterology. 2021. Jul;161(1):66–80.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Daveson AJM, Ee HC, Andrews JM, King T, Goldstein KE, Dzuris JL, et al. Epitope-Specific Immunotherapy Targeting CD4-Positive T Cells in Celiac Disease: Safety, Pharmacokinetics, and Effects on Intestinal Histology and Plasma Cytokines with Escalating Dose Regimens of Nexvax2 in a Randomized, Double-Blind, Placebo-Controlled Phase 1 Study. EBioMedicine. 2017. Dec;26:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chlubnová M, Christophersen AO, Sandve GKF, Lundin KEA, Jahnsen J, Dahal-Koirala S, et al. Identification of gluten T cell epitopes driving celiac disease. Sci Adv. 2023. Jan 27;9(4):eade5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goel G, King T, Daveson AJ, Andrews JM, Krishnarajah J, Krause R, et al. Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease: two randomised, double-blind, placebo-controlled phase 1 studies. The Lancet Gastroenterology & Hepatology. 2017. Jul;2(7):479–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tye-Din JA, Daveson AJM, Ee HC, Goel G, MacDougall J, Acaster S, et al. Elevated serum interleukin-2 after gluten correlates with symptoms and is a potential diagnostic biomarker for coeliac disease. Aliment Pharmacol Ther. 2019. Oct;50(8):901–10. [DOI] [PubMed] [Google Scholar]
- 88.Christophersen A, Lund EG, Snir O, Solà E, Kanduri C, Dahal-Koirala S, et al. Distinct phenotype of CD4+ T cells driving celiac disease identified in multiple autoimmune conditions. Nat Med. 2019. May;25(5):734–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klareskog L, Rönnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to Citrullinated Proteins in Rheumatoid Arthritis. Annu Rev Immunol. 2008. Apr 1;26(1):651–75. [DOI] [PubMed] [Google Scholar]
- 90.Freire R, Ingano L, Serena G, Cetinbas M, Anselmo A, Sapone A, et al. Human gut derivedorganoids provide model to study gluten response and effects of microbiota-derived molecules in celiac disease. Sci Rep. 2019. May 7;9(1):7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guz-Mark A, Perets TT, Biran N, Jack Y, Zevit N, Silbermintz A, et al. Gluten Immunogenic Peptides Are Not Correlated With Reported Adherence to Gluten-Free Diet in Children With Celiac Disease. Journal of Pediatric Gastroenterology & Nutrition. 2023. Aug;77(2):244–8. [DOI] [PubMed] [Google Scholar]
- 92.Duggan JM. Coeliac disease: the great imitator. Medical Journal of Australia. 2004. May;180(10):524–6. [DOI] [PubMed] [Google Scholar]
- 93.Vivas S, Ruiz De Morales JM, Fernandez M, Hernando M, Herrero B, Casqueiro J, et al. Age-Related Clinical, Serological, and Histopathological Features of Celiac Disease. The American Journal of Gastroenterology. 2008. Sep;103(9):2360–5. [DOI] [PubMed] [Google Scholar]
- 94.Lebwohl B, Rubio-Tapia A. Epidemiology, Presentation, and Diagnosis of Celiac Disease. Gastroenterology. 2021. Jan;160(1):63–75. [DOI] [PubMed] [Google Scholar]
- 95.Hopper AD, Cross SS, Hurlstone DP, McAlindon ME, Lobo AJ, Hadjivassiliou M, et al. Preendoscopy serological testing for coeliac disease: evaluation of a clinical decision tool. BMJ. 2007. Apr 7;334(7596):729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rubio-Tapia A, Hill ID, Semrad C, Kelly CP, Greer KB, Limketkai BN, et al. American College of Gastroenterology Guidelines Update: Diagnosis and Management of Celiac Disease. Am J Gastroenterol. 2023. Jan;118(1):59–76. [DOI] [PubMed] [Google Scholar]
- 97.Husby S, Koletzko S, Korponay-Szabó IR, Mearin ML, Phillips A, Shamir R, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition Guidelines for the Diagnosis of Coeliac Disease. Journal of Pediatric Gastroenterology & Nutrition. 2012. Jan;54(1):136–60. [DOI] [PubMed] [Google Scholar]
- 98.Rubio-Tapia A, Rahim MW, See JA, Lahr BD, Wu TT, Murray JA. Mucosal Recovery and Mortality in Adults With Celiac Disease After Treatment With a Gluten-Free Diet. American Journal of Gastroenterology. 2010. Jun;105(6):1412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zanini B, Lanzarotto F, Mora A, Bertolazzi S, Turini D, Cesana B, et al. Five year time course of celiac disease serology during gluten free diet: results of a community based “CDWatch” program. Digestive and Liver Disease. 2010. Dec;42(12):865–70. [DOI] [PubMed] [Google Scholar]
- 100.Catassi C, Fabiani E, Iacono G, D’Agate C, Francavilla R, Biagi F, et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. The American Journal of Clinical Nutrition. 2007. Jan 1;85(1):160–6. [DOI] [PubMed] [Google Scholar]
- 101.Vici G, Belli L, Biondi M, Polzonetti V. Gluten free diet and nutrient deficiencies: A review. Clinical Nutrition. 2016. Dec;35(6):1236–41. [DOI] [PubMed] [Google Scholar]
- 102.Niland B, Cash BD. Health Benefits and Adverse Effects of a Gluten-Free Diet in Non-Celiac Disease Patients. Gastroenterol Hepatol (N Y). 2018. Feb;14(2):82–91. [PMC free article] [PubMed] [Google Scholar]
- 103.Roshan B, Leffler DA, Jamma S, Dennis M, Sheth S, Falchuk K, et al. The Incidence and Clinical Spectrum of Refractory Celiac Disease in a North American Referral Center. American Journal of Gastroenterology. 2011. May;106(5):923–8. [DOI] [PubMed] [Google Scholar]
- 104.Malamut G, Cellier C. Refractory Celiac Disease. Gastroenterology Clinics of North America. 2019. Mar;48(1):137–44. [DOI] [PubMed] [Google Scholar]
- 105.Freeman HJ. Adult Celiac Disease and Its Malignant Complications. Gut Liver. 2009. Dec 30;3(4):237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haas SV. THE VALUE OF THE BANANA IN THE TREATMENT OF CELIAC DISEASE. Arch Pediatr Adolesc Med. 1924. Oct 1;28(4):421. [Google Scholar]
- 107.Dicke WK, Weijers HA, Kamer JHVD. Coeliac Disease The Presence in Wheat of a Factor Having a Deleterious Effect in Cases of Coeliac Disease. Acta Paediatrica. 1953. Jan;42(1):34–42. [DOI] [PubMed] [Google Scholar]
- 108.Shiner M JEJUNAL-BIOPSY TUBE. The Lancet. 1956. Jan;267(6907):85. [DOI] [PubMed] [Google Scholar]
- 109.Crosby WH, Army US, Kugler HW. Intraluminal biopsy of the small intestine: The intestinal biopsy capsule. Digest Dis Sci. 1957. May;2(5):236–41. [DOI] [PubMed] [Google Scholar]
- 110.Frazer AC, Fletcher RF, Shaw B, Ross ConstanceAC, Sammons HG, Schneider R. GLUTEN-INDUCED ENTEROPATHY THE EFFECT OF PARTIALLY DIGESTED GLUTEN. The Lancet. 1959. Sep;274(7097):252–5. [DOI] [PubMed] [Google Scholar]
- 111.Berger E, Bürgin-Wolff A, Freudenberg E. Diagnostische Bewertung des Nachweises von Gliadin-Antikörpern bei Cöliakie. Klin Wochenschr. 1964. Aug;42(16):788–90. [DOI] [PubMed] [Google Scholar]
- 112.Marsh MN. Gluten, major histocompatibility complex, and the small intestine. Gastroenterology. 1992. Jan;102(1):330–54. [PubMed] [Google Scholar]
- 113.Marsh MN. Immunocytes, enterocytes and the lamina propria: an immunopathological framework of coeliac disease. J R Coll Physicians Lond. 1983. Oct;17(4):205–12. [PMC free article] [PubMed] [Google Scholar]
- 114.Chorzelski TP, Beutner EH, Sulej J, Tchorzewska H, Jablonska S, Kumar V, et al. IgA antiendomysium antibody. A new immunological marker of dermatitis herpetiformis and coeliac disease. Br J Dermatol. 1984. Oct;111(4):395–402. [DOI] [PubMed] [Google Scholar]
- 115.Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. The Journal of experimental medicine. 1989. Jan 1;169(1):345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lundin KE, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, et al. Gliadin-specific, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. The Journal of experimental medicine. 1993. Jul 1;178(1):187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997. Jul;3(7):797–801. [DOI] [PubMed] [Google Scholar]
- 118.Molberg Ø, Mcadam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998. Jun;4(6):713–7. [DOI] [PubMed] [Google Scholar]
- 119.Anderson RP. T cells in peripheral blood after gluten challenge in coeliac disease. Gut. 2005. Sep 1;54(9):1217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abadie V, Kim SM, Lejeune T, Palanski BA, Ernest JD, Tastet O, et al. IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease. Nature. 2020. Feb 27;578(7796):600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]