

Abstract
In the past two decades, significant progress has been made in uncovering the biological function of selenium. Selenium, an essential trace element is required for the biogenesis of selenocysteine, which is then incorporated into selenoproteins. These selenoproteins have emerged as central regulators of cellular antioxidant capacity and maintenance of redox homeostasis. This review provides a comprehensive examination of the multifaceted functions of selenoproteins, with a particular emphasis on their contributions to cellular antioxidant capacity. Additionally, we highlight the promising potential of targeting selenoproteins and the biogenesis of selenocysteine as avenues for therapeutic intervention in cancer. By understanding the intricate relationship between selenium, selenoproteins, and ROS, insights can be gained to develop therapies that exploit the inherent vulnerabilities of cancer cells.
Keywords: Selenium, selenoprotein, redox, oxidative stress, ROS, ferroptosis
Reactive Oxygen Species Emerge as Novel Therapeutic Targets in Cancer
Oncogenic transformation is accompanied by a host of changes to cellular genetics, epigenetics, metabolism, and to the cellular environment. An increase in reactive oxygen species (ROS) and dysregulation of redox homeostasis and signaling are hallmarks of cancer [1,2]. Mitochondria are major consumers of cellular oxygen, reducing divalent oxygen to produce carbon dioxide and water. However, reduced forms of oxygen including superoxide (O2−) and hydrogen peroxide (H2O2) can be generated as reactive byproducts of mitochondrial respiration, later producing highly reactive hydroxyl radicals (HO•) via the iron (Fe) dependent Fenton reaction. Thus the mitochondrial dysfunction often observed in cancer cells generates an environment primed for formation of high levels of ROS.
Observations of elevated levels of ROS in cancer cells initially led to hypotheses that treatments to reduce ROS in cancer could provide therapeutic benefit [3,4]. While elevated levels of ROS can increase protein, lipid, and DNA oxidation [5], ROS can also activate a variety of signaling pathways such as the DNA damage response (DDR), iron homeostasis, and the anti-oxidant and anti-inflammatory responses [6], supporting key aspects of oncogenesis such as cell proliferation, survival, and metastasis [7,8]. Thus, initial clinical trials were conceived to neutralize ROS in cancer utilizing antioxidant therapy [9] and uses of adjuvant antioxidant supplements persists into treatment strategies today with many reported studies of diets high in vitamins and antioxidants reducing chemotherapy toxicity and correlating with increases in survival [10]. However, antioxidant therapy rapidly emerged as a double-edged sword. Several cancers with extremely high native levels of ROS, such as melanoma, are able to become more aggressive and invasive following adjuvant antioxidant therapy [11]. In these cancers ROS are both necessary for growth through addiction to the ROS mediated signaling cascade, and limiting for growth due to the effects of oxidative stress [12].
Over the past several years the field has shifted from attempting to reduce ROS levels to instead using them as a targetable vulnerability [13,14]. Cancer cells require a robust antioxidant defense system to protect them from the high levels of ROS generated by altered metabolism and extrinsic stresses. Selenoproteins, enzymes that selectively include the amino acid selenocysteine, make up major classes of antioxidant proteins critical for the protection of cancer cells to elevated ROS.
There are 25 selenoproteins encoded within the human genome, with the majority of selenoproteins involved in cellular antioxidant capacity [15]. Nearly all glutathione peroxidase (GPx) and thioredoxin reductase (TrxR) enzymes fall into this category as a catalytic selenocysteine is essential for their activities. Studies replacing the catalytic selenocysteine of GPx4 for the sulfur containing cysteine result in highly reduced protein activity and increased susceptibility toward peroxide-induced cell death [16]. Therefore, selenoproteins and their biogenesis may serve as novel targets to induce synthetic vulnerabilities in ROS dependent cancers. In this review we will discuss the role of selenium in human biology and cancer.
Selenium - The Essential Poison and The Selenium-Cancer Hypothesis
Selenium, a trace metal, was discovered in 1817 [17]. Its essential role as a micronutrient for Escherichia coli enzyme activity was confirmed in 1954 [18], followed by the discovery of its importance for animals in 1957 [19,20]. Deficiency in selenium can lead to Keshan Disease (a cardiomyopathy) and Kashin-Beck Disease (a severe rheumatoid arthritis-like osteoarticular disorder) [21–24] (Box 1). However, intake of more than 400 μg/day of selenium can cause acute toxicity [25]. Currently, the US Department of Agriculture recommends a daily intake of 55 μg of selenium to prevent disease caused by selenium deficiency. In Finland, where selenium soil levels are naturally low, there is government mandated selenium supplementation in fertilizers to prevent dietary deficiency in the general population [26,27]. Interestingly, selenium supplementation has been recently implicated in viral defense, as Finland observed significantly lower death rates from the SARS-CoV-2 pandemic compared to neighboring countries with similar infection rates and similarly low native soil selenium levels [28].
Box 1. Historical perspectives on the role of selenium in cancer prevention.
Shamberger and Frost’s influential letter to the Canadian Medical Association Journal in 1969 established an inverse correlation between selenium concentration in forage crops and cancer death rates in the U.S., which aligned with a study in 1977 showing a similar inverse correlation of selenium soil concentration and cancer risk when measuring selenium blood levels across several U.S. cities and 27 countries [32]. The NPCT was published in 1996 by Clark and Combs as a large-scale, double-blind, randomized, placebo-controlled cancer prevention trial that was initially designed to measure recurrence of nonmelanoma skin cancer over a 10-year period through dietary intake of 200 μg/day of selenium in the form of selenized yeast tablets. However the trial ultimately demonstrated significant reductions (HR = 0.61) in colon, prostate, and lung cancer incidence through this dietary intervention [33]. Following the success of the blinded portion of the study, the trial was then unblinded and participants were followed for several more years. Subsequent analysis of the unblinded portion of the study refined the benefits of selenium supplementation to males with baseline levels of plasma selenium <121 μg/L, with the largest cancer prevention effects of selenium supplementation seen in prostate cancer [31].
Since the early 2000s, numerous studies have attempted to replicate the NPCT findings, yielding varying results. However, it is possible that these results stem from missteps in patient selection, specifically baseline plasma selenium levels [37]. The NPCT specifically recruited patients from regions with low selenium, resulting in a study cohort with a mean plasma selenium level of 114μg/l. In contrast, several follow-up studies, including SELECT, selected patients with median plasma selenium levels as high as 143μg/l, exceeding the beneficial range of selenium supplementation as defined by the NPCT (<121μg/l). While the negative results of SELECT and other selenium supplementation trials have temporarily halted most selenium-focused studies, these results underscore the dual nature of the benefits and risks associated with selenium supplementation.
Early research in the 1940s reported that high dietary selenium intake led to the development of liver tumors in rats [29], causing global concerns about selenium consumption. However, subsequent studies demonstrated protective effects against tumors in various cancer models, leading to further investigations [30]. The Nutritional Prevention of Cancer Trial (NPCT) in 1996 ultimately defined low plasma selenium levels (<121 μg/l)[31] as an increased risk factor for the development of prostate cancer (Box 1).
These findings prompted the National Institutes of Health (NIH) to conduct the Selenium and Vitamin E Cancer Prevention Trial (SELECT) [34]. This trial investigated selenium (in the form of L-selenomethionine, as this was determined to be the major selenium species in the NPCT) and vitamin E supplementation (as a general antioxidant) as a preventive measure for prostate cancer in a large-scale, placebo-controlled study. Despite being one of the largest human cancer prevention trials ever performed, the group receiving both vitamin E and selenium demonstrated a significantly increased risk of prostate cancer [35,36] (Box 1).
Selenium, as an essential poison, has a narrow window of benefit for human health (Figure 1). When measuring selenium blood plasma levels, too little selenium (<70μg/l) is associated with deficiency diseases, low plasma selenium (<120μg/l) is linked to an increased risk of cancer, selenium and vitamin E supplementation with levels >140μg/l can significantly increase cancer risk, elevated plasma selenium levels (>400μg/l) are associated with increased risks of selenosis, and highly elevated plasma levels (>1000μg/l) are seen in cases of acute toxicity [38]. Optimal baseline selenium levels have been established at 110–135μg/l correlating with a plateau in production of plasma selenoproteins at 130μg/l [39]. Interestingly, other further large scale studies have observed decreases in cancer mortality risk with increases in plasma selenium up to a level of 130μg/l, with increases in mortality risk observed at levels >150μg/l [40]. Overall, these studies suggest that the association between cancer risk and selenium follows the trends observed with ROS in cancer. In the early stages of tumorigenesis, elevated levels of ROS may increase the risk of cancer progression due to their ability to cause DNA damage and promote genomic instability. Thus antioxidant and/or selenium supplementation may provide significant benefits in the prevention of cancer. However, in established tumors, ROS are often elevated and the tumor must limit ROS to survive. Consequently, supplementation of an established tumor with antioxidants may lead to worsened outcomes. Given that many selenoproteins with antioxidant roles exhibit increased translation following selenium supplementation [41–44] it is a distinct possibility that observed effects of selenium supplementation are due to increased production of selenoproteins.
Figure 1. Relevance of human plasma selenium levels to human health.
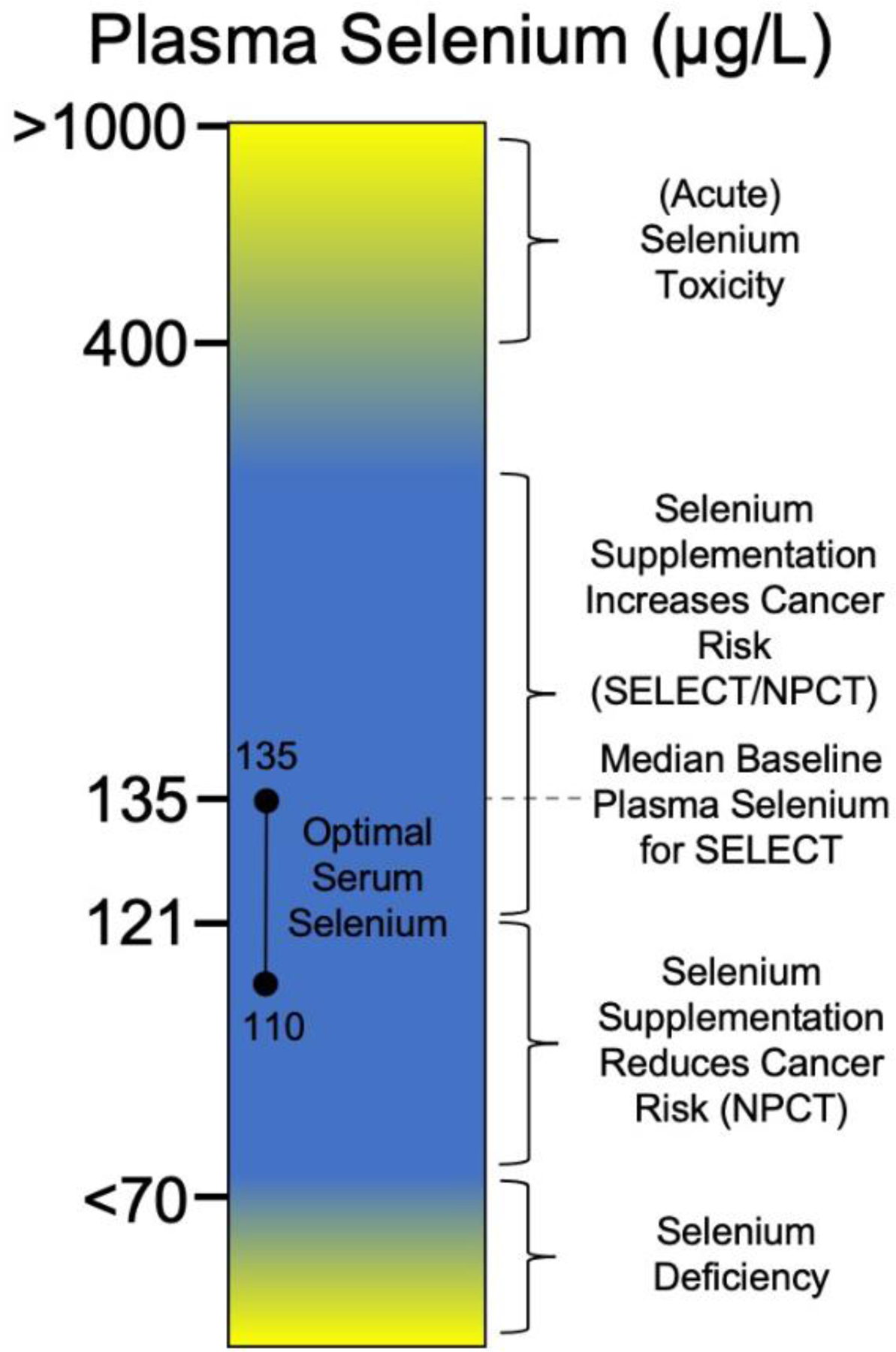
A particular focus on key findings from selenium supplementation trials for cancer prevention. See [23,25,31,33,35,36,38,39].
Cysteine versus Selenocysteine
A fundamental question that arises concerning selenoproteins is “why?”. Cysteine is an abundant and readily incorporated amino acid, yet this small class of enzymes specifically requires incorporation of a highly toxic trace mineral. From a chemical perspective, selenocysteine offers several unique advantages over cysteine. Selenium, positioned as the 34th element on the periodic table, resides one row below sulfur, the 16th element. Although sulfur and selenium share the same valence shell electron configuration, selenium’s larger atomic radius grants it superior nucleophilic properties compared to sulfur, resulting in heightened reactivity. This distinction becomes apparent when examining the pKa values of the amino acids: selenocysteine possesses a pKa of 5.24, whereas cysteine exhibits a pKa of 8.25. In a cellular environment with a pH of approximately 7.4, selenocysteine predominantly exists in a deprotonated state, while cysteine is primarily protonated[45,46]. Consequently, these different protonation states lead to significantly disparate rates of reactivity.
Furthermore, the divergent reactivity between selenocysteine and cysteine contributes to considerably reduced rates of terminal oxidation for selenocysteine enzymes [45]. In the ACS article “Why Nature Chose Selenium”, Reich and Hondal state that “almost all chemical reactions involving selenium are faster in comparison to the same reaction with sulfur” [45]. While it is tempting to conclude that nature selected selenium to replace sulfur due to its heightened chemical reactivity, thereby expediting enzymatic reactions, their viewpoint diverges from this notion, suggesting that nature specifically chose selenium for its unique capacity to engage with oxygen and ROS in a readily reversible manner. In a biological system, terminal oxidation of an enzymatic active site would require a complete resynthesis of the affected enzyme, leading us into discussion of nature’s redox machinery: selenoproteins.
Mammalian Selenoproteins
Glutathione Peroxidases (GPx1–4,6)
The functions of GPx in cancer have been extensively reviewed [47,48]. GPx enzymes play a crucial role in detoxifying RO) by utilizing glutathione (GSH) as a reducing agent (Figure 2). GPx1–4 and 6 are selenoproteins while GPx5, 7 and 8 are cysteine variants. All GPx enzymes operate through a conserved catalytic mechanism, relying on a tetrad of conserved amino acids: Sec/Cys, Gln, Trp, and Asn [49]. While structurally and functionally similar, GPx enzymes diversify their functions through differences in active site structure, substrate accommodation, tissue expression and subcellular localization [50]. GPx1 was the first characterized selenoprotein and is expressed ubiquitously across most cell types with cytoplasmic localization, making it one of the most active and critical cellular antioxidant defense enzymes [51]. GPx2 is found in the intestinal epithelium and is often referred to as the intestinal GPx. In colorectal adenocarcinoma loss of GPx2 has been linked to the development of microsatellite instability and immune infiltration [52]. GPx3 is secreted in plasma and has been implicated as a tumor suppressor gene in breast and lung cancers [53,54]. While the exact function of GPx3 is under debate as glutathione is rapidly catabolized in the plasma, it has been hypothesized that GPx3 may be active at hubs of ROS generation and may localize to basement membranes, providing antioxidant capability to the cellular microenvironment [55]. GPx4, originally called Phospholipid Hydroperoxide Glutathione Peroxidase (PHGPX) is essential for lipid peroxide detoxification and has emerged as a central regulator of ferroptosis [56], a non-apoptotic form of regulated cell death that has been extensively reviewed elsewhere. While the primary function of GPx4 is cytoplasmic lipid peroxide detoxification, GPx4 also has a nuclear isoform that can act as protein thiol peroxidase [57] and a mitochondrial isoform that protects against mitochondrial dependent ferroptosis [58]. Recently a R152H missense mutation in GPx4 was found in a patient with sedaghation-type spondylometaphyseal dysplasia (SSM) [59]. This mutation provided additional structural basis for GPx4 activity and further demonstrated the central role of GPx4 in human health. Little is known about GPx6 besides that it is expressed during embryonic development and in the olfactory epithelium [60].
Figure 2. Conserved catalytic cycle of glutathione peroxidases.
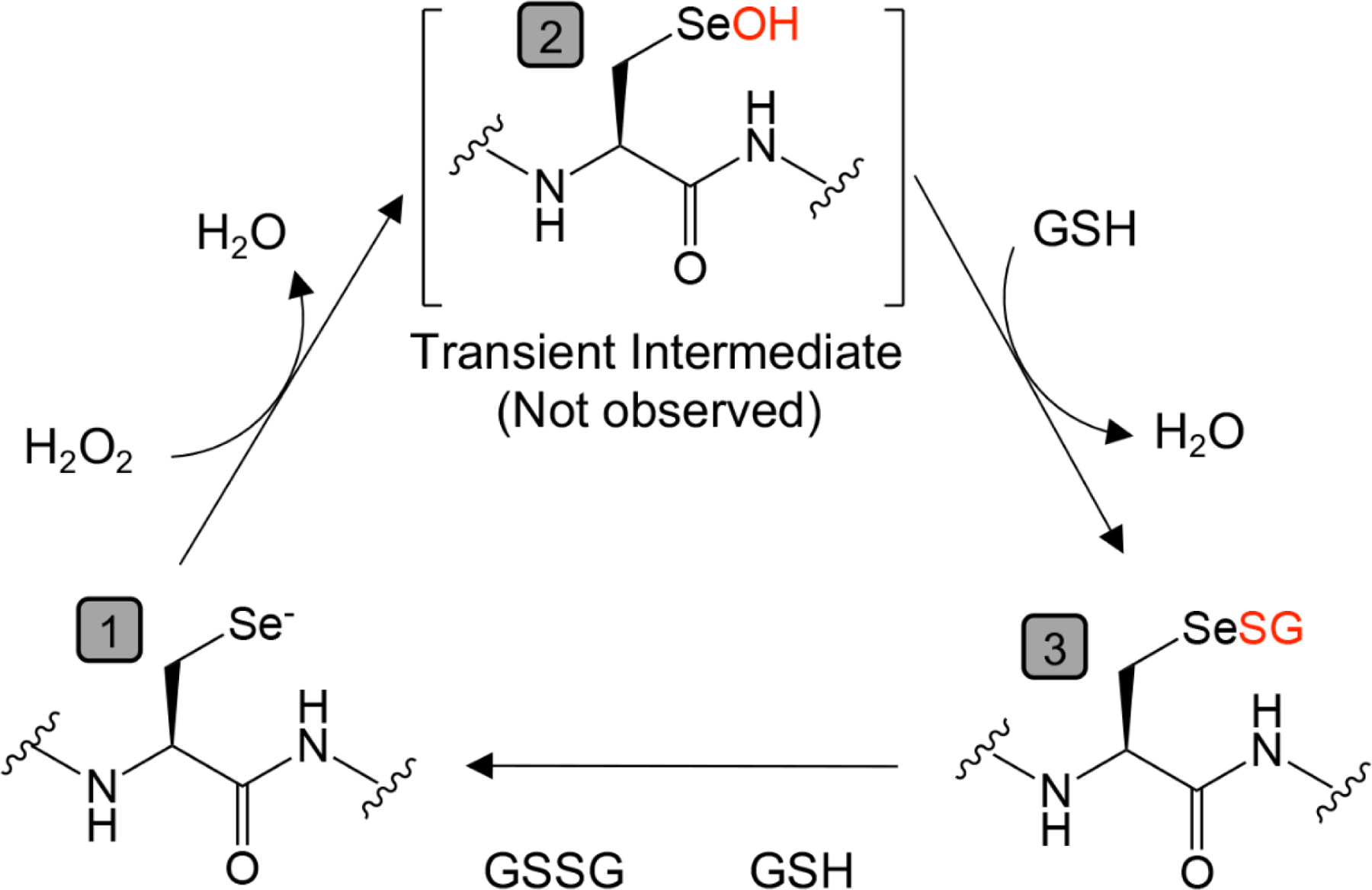
Starting from (1), the catalytic selenocysteine exists in a base state as a selenol (Se−) which quickly reacts with hydrogen peroxide to generate (2) selenenic acid (SeOH), a temporary intermediate that is rapidly replaced by reduced glutathione (GSH) to form (3) a selenenyl sulfide adduct (SeSG). The enzyme is subsequently regenerated to (1) through its reaction with a second GSH, resulting in the production of oxidized glutathione (GSSG).
Thioredoxin Reductases (TXNRD1–3)
Thiorexodins are small ~12kDa redox regulatory enzymes with two active site internal cysteine residues, capable of forming cis and trans disulfide bridges to assist in protein folding as well as a multitude of cellular processes. Thioredoxins are a key component of cellular redox homeostasis and are essential for the maintenance of a reductive environment through reduction of oxidized cysteine residues. While we will not discuss the function of the thioredoxin system in depth here, we recommend these excellent reviews to the reader[61–63]. Instead, we will focus on the FAD and NADP(H) dependent thioredoxin recycling systems, Thioredoxin Reductases (TrxRs) (Figure 3). There are three TrxRs in the human genome (TXNRD1–3), all of which are selenoproteins. Similarly to GPxs, TrxRs also display specific function due to subcellular localization. TrxR1 is cytosolic, TrxR2 is mitochondrial localized, and TrxR3 is testes-specific[60].
Figure 3. Conserved biological mechanism of thioredoxin.
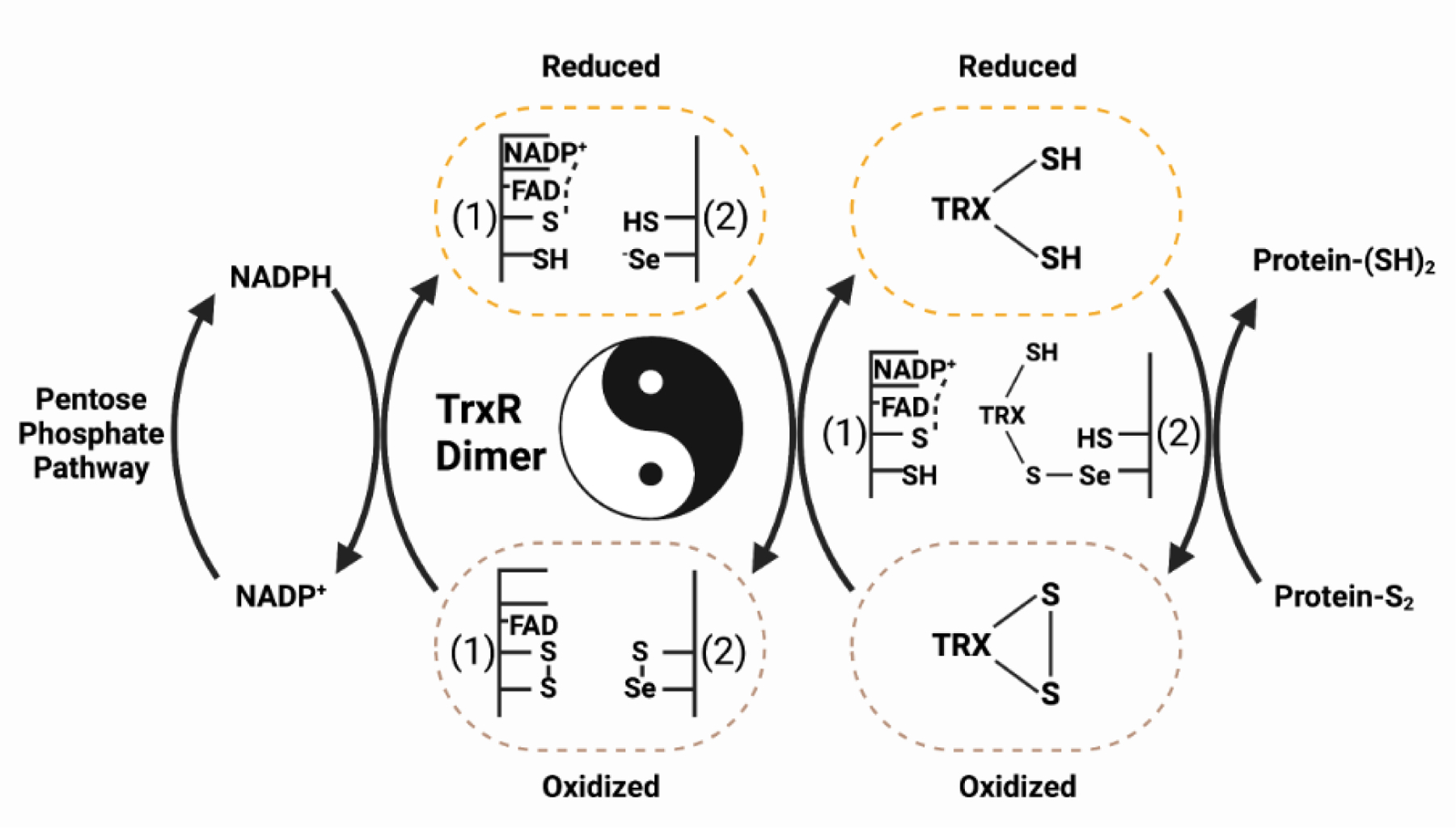
The thioredoxin pathway allows electrons from metabolism to cycle through the redox machinery, thereby maintaining a reduced cellular environment. From left to right, NADPH generated from the pentose phosphate metabolic pathway binds to a dimer of oxidized thioredoxin reductase (TrxR). Next, the TrxR dimer forms a yin-yang orientation where the “head” of protein 1 (1) binds into the “tail” of protein 2 (2) to reduce a Se-S bond mediated through an FAD cofactor. This process is performed in duplicate with the “tail” of (1) binding into the “head” of (2) (not shown). Third, the reduced TrxR dimer can then recycle oxidized thioredoxin by binding to the selenocysteine of the reduced TrxR. The resulting electron shuttle restores thioredoxin to its reduced form, thus regaining its cellular redox capabilities.
MSRB1,SELENOH,SELENOO
Other key redox involved selenoproteins are the methionine sulfoxide reductase MSRB1 and the enzymes SELENOH and SELENOO. MSRB1 mediates reduction of oxidized methionine to protect the proteome from effects of ROS. MSRB1 activity has been implicated in overall redox homeostasis[64] as well as pro-inflammatory cytokine production in macrophages, implicating this enzyme in the inflammatory response[65]. SELENOH is a nuclear oxidoreductase with DNA binding activity capable of regulating p53 in response to oxidative stress[66]. SELENOO is the largest human selenoprotein and is selectively localized to the mitochondria. While the exact function of SELENOO is still unknown it has been implicated in redox homeostasis and is suspected to have an active kinase domain[67].
Other Mammalian Selenoproteins
While in this review we focus primarily on the 11 redox active selenoproteins, we will also briefly touch upon the other 14 selenoproteins encoded within the human genome. Selenoproteins F, K, M, N, S, and T have been implicated in ER homeostasis and utilize their oxidative capabilities in protein folding [68,69]. The enzymes DIO1–3 are essential for thyroid hormone metabolism and are likely responsible for some of the pathological presentation of selenium deficiency[70]. Selenoprotein P is a selenium chaperone as it contains 11 selenocysteine residues, is synthesized in the liver, and secreted to selectively carry selenium throughout the body[71,72]. SEPHS2 is a selenium donor utilized for the biosynthesis of selenoproteins and has been implicated in the cellular detoxification of selenide, a byproduct of selenocysteine biosynthesis [73,74]. Still the function of several selenoproteins, namely V and W, remain unknown. Broad classification, substrate specificity, and kinetic details of many selenoproteins remain difficult to study due to extreme difficulty in the production of these enzymes in recombinant or overexpression systems[75].
Inhibition of Selenoproteins for Cancer Therapy
To further explore the potential of selenoproteins as novel targets for cancer treatment, publicly available survival data was analyzed. Survival graphs and hazard ratios for all 25 selenoproteins across 27 different types of cancer were assessed. Using a strict significance cutoff of p < 0.05, the hazard ratios were generated as a heatmap and plotted using unguided hierarchical clustering (Figure 5, Key figure). While many significant survival correlations are found, minimal significant clustering of selenoprotein hazard ratios across different cancers was observed. This finding supports previous research suggesting that the selenoproteome is highly complex, and its involvement in cancer is highly context dependent. For example, cervical squamous cell carcinoma has multiple selenoprotein hazard ratios <1, indicating that higher expression correlates with increased survival. These data would suggest that cervical squamous cell carcinoma patients may benefit from selenium supplementation, in support of already underway clinical trials testing this hypothesis and already demonstrating reduced chemotherapy toxicity [76]. Furthermore, liver hepatocellular carcinoma has multiple selenoprotein hazard ratios >1, indicating negative correlations between selenoprotein expression and survival. Liver hepatocellular carcinoma is a known ROS-driven cancer with recent data pointing to a complex regulatory role of TrxR1 in development [77]. These observations may help explain the conflicting results found in the existing literature on selenoproteins, as significant and contrasting survival correlations of the same gene across various cancer types are observed in this dataset.
Figure 5. Key figure. Hazard ratios of selenoproteins across cancer types.
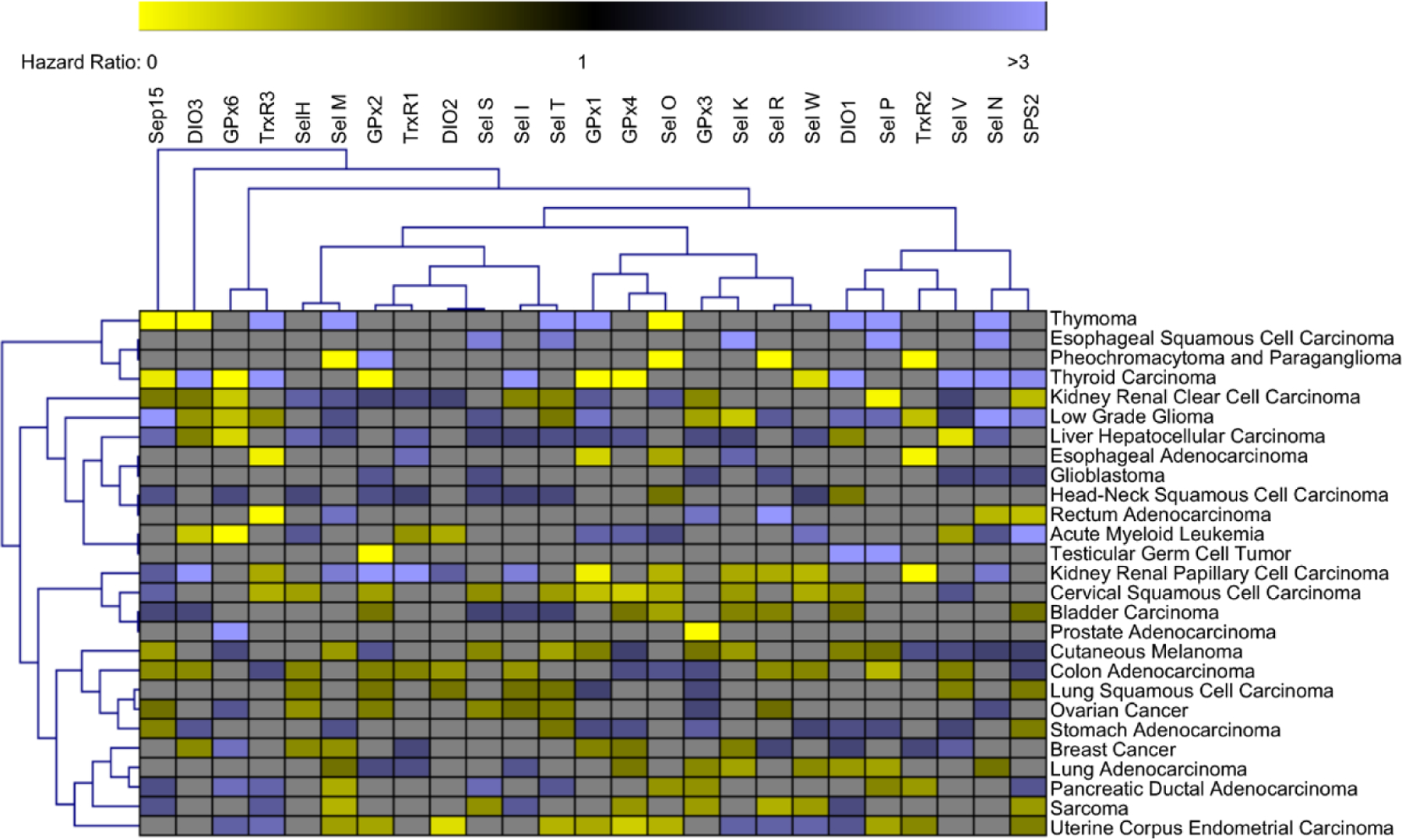
Unbiased hierarchal clustering was used for visualization of statistically significant (p<0.05) selenoprotein hazard ratios across various cancers with branches representing statistically similar groupings of genes or cancers. A hazard ratio of 1 indicates no difference between groups (high vs low expression of selected gene). Hazard ratios >1 (Blue) indicates correlation between higher expression and lower survival of the indicated gene. Hazard ratios <1 (Yellow) indicates correlation between lower expression and higher survival of the indicated gene. Hazard ratios with nonsignificant correlations (p>0.05) were not included in the analysis and are represented as gray boxes. Several cancers such as liver hepatocellular carcinoma, glioblastoma, and head-neck squamous cell carcinoma have multiple selenoprotein hazard ratios > 1 indicating that efforts to reduce selenoprotein expression may provide therapeutic benefit. Other cancers such as cervical squamous cell carcinoma and uterine corpus endometrial carcinoma have multiple selenoprotein hazard ratios < 1 indicating that efforts to boost selenoprotein expression may provide therapeutic benefit. However, throughout the analysis of 25 selenoproteins across 27 cancers the only cancer with a net positive or negative survival correlation with selenoprotein expression is glioblastoma. Furthermore, many selenoproteins have significant and opposite correlations with patient survival across different cancer types. This data highlights the complexity and context dependent role of selenoproteins across different cancer types.
In 2014, the compound (1S, 3R)-RSL3 was characterized as a GPx4 inactivator capable of driving ferroptosis [56,78]. While the poor pharmacokinetics of RSL3 have prevented its transition to clinical settings, a new RSL3 derivative with greatly improved bioavailability and plasma half-life has recently been developed [79]. However, in addition to GPx4 there is evidence that RSL3 can bind to other targets including TrxR1 and its effects may not be strictly GPx related [80]. As RSL3 utilizes a chloroacetamide functional group to covalently bind to an active site selenocysteine, it is possible there are still undiscovered RSL3 targets to be found. Furthermore, it was demonstrated that the mechanism of action of ferroptosis inducing compounds RSL3, ML162, and FIN56 is chemical induction of GPx4 proteasomal degradation [81]. While the exact biochemical mechanisms of small molecule induced GPx4 degradation is unclear, recent advances in the understanding of targeted protein degradation may eventually lead to the development of the next generation of ferroptosis inducing compounds in a specific targeted degrader of GPx4.
TrxRs have been implicated in redox homeostasis across many cell types due to their inherent role in thioredoxin function and have emerged as promising cancer targets [62,63,82–84] with several developed inhibitors [85–88]. The most prominent TrxR inhibitor is the FDA approved gold containing compound Auranofin (Ridaura). While Ridaura is FDA certified for rheumatoid arthritis it has demonstrated potent anti-cancer activity and trials are underway to assess its effectiveness[89].
Additional mechanisms to target ferroptosis in cancer have discovered that the lipoprotein receptor LPR8 is an essential ferroptosis resistance gene [90,91]. Interestingly, LPR8 is a key member of the complex responsible for endocytosis of Selenoprotein P [92,93]. Once Selenoprotein P undergoes endocytosis it is rapidly degraded and selenocysteine lyase (SCLY) breaks down free selenocysteine to alanine[94], releasing selenium for use in synthesis of new selenoproteins, a topic that will be covered in more detail in the next section of this review.
Selenoprotein Biogenesis
While inhibitors are being developed to target specific selenoproteins, it is important not to overlook the biosynthetic cascade required for selenoprotein biogenesis, which could represent a novel and exciting area of research with the potential to broadly regulate the selenoproteome. Mammalian incorporation of selenocysteine is characterized by several unique features. First, selenocysteine does not have its own codon; instead, it reprograms a UGA-STOP codon through a complex translational process [95,96]. In all mammalian mRNA encoding selenoproteins, there is a conserved hairpin structure in the 3’ UTR known as the selenocysteine incorporation sequence (SECIS). This structure is recognized and bound by a multiprotein complex consisting of SECIS binding protein 2 (SECISBP2), Eukaryotic Elongation Factor Selenocysteine (EEFSEC), and tRNA selenocysteine 1 associated protein 1 (TRNAU1AP) [97–99]. The complex facilitates the positioning of tRNASec into the elongating ribosome [100]. Ribosomal protein L30 (RPL30), eukaryotic translation initiation factor 4A3 (eIF4A3), and nucleolin (NCL) also play roles in regulating selenocysteine insertion. Mutations in SECISBP2 have been associated with thyroid disorder and multisystem selenoprotein deficiency disorder[101–103].
Second, tRNASec stands out from other tRNA species as it lacks its own aminoacyl-tRNA synthetase (aaRS) [104]. Instead, tRNASec is initially aminoacylated as serine by the enzyme Seryl-tRNA synthetase 1 (SerRS) [105]. The resulting seryl-tRNASec is then phosphorylated by Phosphoseryl tRNA Kinase (PSTK) to create a phosphate leaving group for subsequent reactions [106]. The enzyme SEPSECS, in complex with the selenium donor SEPHS2, catalyzes the substitution of the oxygen moiety for selenium via a PLP intermediate [107]. However, even after synthesis, tRNASec must undergo additional post-transcriptional modifications before it can be used for translation.
While human tRNAs carry on average 13 post-transcriptional modifications per molecule [108], tRNASec is unique in its hypomodification status as it contains only four: mcm5U(m)34, i6A37, PseudoU55, and m1A58 [104]. PseudoUridine 55 and m1A58, which are critical for proper tRNA folding, are placed by PUS4 and TRM6/61, respectively. The remaining modifications occur in the anticodon region and are essential for proper codon recognition [109]. Isopentylation of A37 (i6A) by TRIT1 is commonly observed in conjunction with modification at position 34 and has moderate effects on selenoprotein translation [110], while modification of U34 is vital for selenoprotein translation [111,112]. The post-transcriptional modification of U34 involves a multifaceted enzymatic process. The Elongator Complex, through the radical SAM and acetyl-CoA dependent enzyme Elp3, introduces the chemical modification 5-carboxymethyl-Uridine (cm5U) [113]. Subsequently, cm5U is methylated by the tRNA methyltransferase AlkBH8, resulting in 5-methoxycarbonylmethyl-Uridine (mcm5U) [114]. Further modification at U34 can occur via 2’-O-methylation by the enzyme FTSJ1, although the function and regulation of this modification in mammalian systems are still unknown [115] (Figure 4).
Figure 4. Selenocysteine biosynthesis and post transcriptional modifications of tRNA-selenocysteine (tRNA[Ser]Sec).
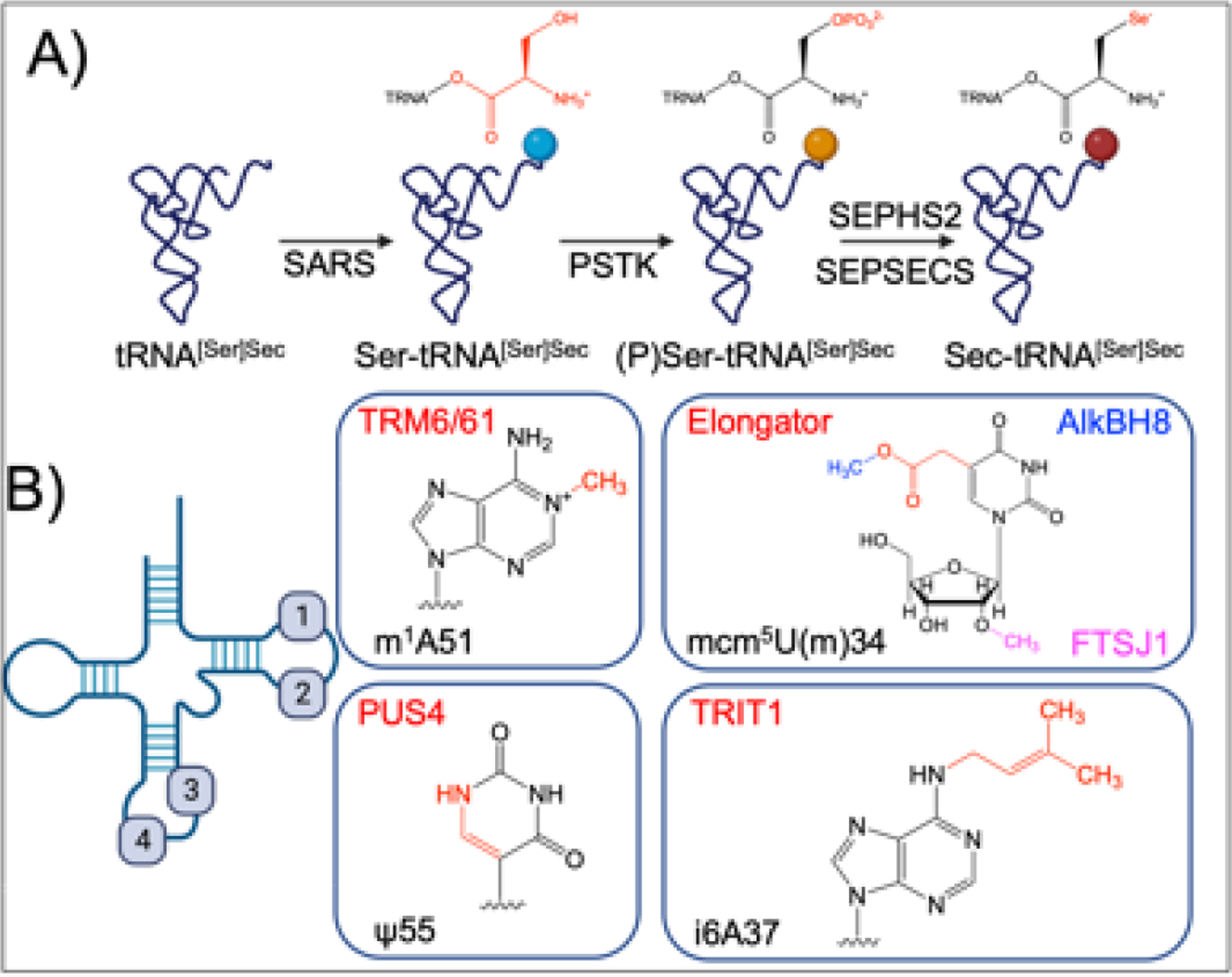
(A) Biogenesis of selenocysteine. tRNA-sec is initially aminoacylated with serine by seryl-tRNA synthetase (SARS). Phosphoseryl tRNA Kinase (PSTK) phosphorylates Ser-tRNA[Ser]Sec , allowing for substitution of the oxygen for a selenium by selenophosphate synthetase 2 (SEPHS2) and (Sep (O-Phosphoserine) TRNA:Sec (Selenocysteine) TRNA Synthase) SEPSECS, forming selenocysteine on the tRNA. (B) Post transcriptional modifications of tRNA-sec. tRNA-sec contains four post transcriptional modifications, 1-methyladenosine (m1A) 51 placed by the tRNA (adenine(58)-N(1))-methyltransferase non-catalytic subunit (TRM6) and TRNA (Adenine-N(1)-)-Methyltransferase Catalytic Subunit (TRM61), Pseudouridine (ψ) 55 placed by PseudoUridine Synthase 4 (PUS4), N6-isopentlyadenosine (i6A) placed by tRNA isopentyltransferase 1 (TRIT1), and 5-methoxycarbonylmethyl-(2’-O-methyl)-uridine (mcm5U(m)) placed in conjunction by the Elongator Complex (cm5), AlkB Homolog 8, tRNA methyltransferase (AlkBH8) (mcm5), and FtsJ RNA 2’-O-Methyltransferase 1 (FTSJ1) (Um). While mcm5 is essential for selenoprotein translation, the necessity for 2’-O-methylation is variable through poorly understood mechanisms.
The biosynthesis of tRNASec represents a complex and coordinated system for incorporating selenocysteine into a limited number of enzymes. This system presents a unique opportunity: by disrupting the mechanisms of tRNASec biogenesis, post-transcriptional modification, or translational incorporation, it may be possible to dysregulate selenoproteins on a global scale as seen in genetic mutations of tRNASec[116]. While caution must be exercised when modulating the selenoproteome, our data and historical perspectives strongly suggest that this enzymatic cascade offers multiple novel therapeutic targets capable of modulating cellular redox homeostasis.
Concluding Remarks
Understanding the role of ROS and redox signaling in cancer has evolved significantly. Initially, the focus was on reducing ROS levels in cancer cells through antioxidant therapy. However, it became evident that ROS also play important roles in cancer progression and survival, activating signaling pathways essential for oncogenesis. This realization led to a shift in perspective, with ROS being recognized as targetable vulnerabilities in cancer cells. Selenoproteins, which incorporate the amino acid selenocysteine, have emerged as crucial players in cellular antioxidant defense mechanisms. GPx and TrxR are two major classes of selenoproteins involved in ROS detoxification. GPx enzymes protect cells from oxidative damage by utilizing glutathione as a reducing agent, while TrxRs maintain redox homeostasis by recycling oxidized thioredoxin. Recent advances have shed light on the role of selenoproteins in cancer biology. GPx4 has emerged as a central regulator of ferroptosis, a form of regulated cell death that shows promise as a therapeutic target in various cancers. Inhibitors targeting GPx4 and TrxRs have demonstrated anticancer activity in preclinical studies, with some compounds showing potential for clinical development. While links between inhibition of selenoprotein function and inducing cancer cell death have been uncovered, there are still gaps in our knowledge of how to design therapies that target selenoprotein function or selenocysteine biogenesis (see Outstanding questions). Moreover, the field of selenium and selenoprotein research in cancer is complex and evolving. The dual nature of selenium’s effects, as both a beneficial micronutrient and a potential toxin, highlights the importance of understanding its role within a narrow range. The specific functions of selenoproteins in cancer cells and their potential as therapeutic targets provide exciting avenues for future research and the development of novel anticancer strategies. Further investigations into the redox biology of selenium and selenoproteins will undoubtedly contribute to our understanding of cancer biology and may lead to the development of innovative approaches to cancer treatment.
Outstanding Questions.
Do the benefits of selenium supplementation result from increases in plasma levels of selenoprotein P?
Given the narrow window between benefit and toxicity, can we create a comprehensive guideline for ideal plasma selenium levels?
Why do different tumor cell types and normal cells display unique dependencies on selenoproteins and selenocysteine biogenesis enzymes?
What are the redundant and distinct functions for redox active selenoproteins in cancer progression?
Will specific selenoprotein inhibitors become clinically viable?
Will targeting the selenocysteine biogenesis pathway to alter the selenoproteome show promise as a viable therapeutic strategy?
Highlights.
Reactive oxygen species (ROS) are tightly regulated to promote tumor growth.
Disruption of key ROS detoxification enzymes or pathways induce ferroptosis, a non-apoptotic cell death pathway.
Many key ROS detoxification enzymes are selenoproteins.
The incorporation of selenocysteine into proteins involves a complex multistep mechanism.
The selenocysteine biogenesis pathway is emerging as a novel target for cancer treatment as its disruption is capable of inducing ferroptosis
Acknowledgements
This work was funded by NIH grants: R01CA148828, R01CA245546, and R01DK095201 (Y.M.S) and R01GM117141 (M.K.)
The authors would like to acknowledge members of the Shah and Koutmos labs for editing and revision of the manuscript and associated figures
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
None are declared by the authors.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discov 2022; 12:31–46. [DOI] [PubMed] [Google Scholar]
- 3.Srinivas US, et al. ROS and the DNA damage response in cancer. Redox Biol 2019; 25:101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 1991; 51:794–798. [PubMed] [Google Scholar]
- 5.Juan CA, et al. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int J Mol Sci 2021; 22:4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooke MS, et al. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003; 17:1195–1214. [DOI] [PubMed] [Google Scholar]
- 7.Kumari S, et al. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 2018; 13:1177271918755391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayes JD, et al. Oxidative Stress in Cancer. Cancer Cell 2020; 38:167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh K, et al. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity - Exploring the armoury of obscurity. Saudi Pharm J SPJ Off Publ Saudi Pharm Soc 2018; 26:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simone CB, et al. Antioxidants and other nutrients do not interfere with chemotherapy or radiation therapy and can increase kill and increase survival, Part 2. Altern Ther Health Med 2007; 13:40–47. [PubMed] [Google Scholar]
- 11.Le Gal K, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 2015; 7:308re8. [DOI] [PubMed] [Google Scholar]
- 12.Piskounova E, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015; 527:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trachootham D, et al. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8:579–591. [DOI] [PubMed] [Google Scholar]
- 14.Zou Z, et al. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis Int J Program Cell Death 2017; 22:1321–1335. [DOI] [PubMed] [Google Scholar]
- 15.Mangiapane E, et al. Selenium and selenoproteins: an overview on different biological systems. Curr Protein Pept Sci 2014; 15:598–607. [DOI] [PubMed] [Google Scholar]
- 16.Ingold I, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018; 172:409–422.e21. [DOI] [PubMed] [Google Scholar]
- 17.Trofast J Berzelius’ Discovery of Selenium. Chem Int 2011; 33:16–19. [Google Scholar]
- 18.Pinsent J The need for selenite and molybdate in the formation of formate dehydrogenases by members of the Coliaerogenes group of bacteria. Biochem J 1954. 57:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patterson EL, et al. Effect of selenium in preventing exudative diathesis in chicks. Exp Biol Med 1957; 95:617–620. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz K, et al. Prevention of exudative diathesis in chicks by factor 3 and selenium. Exp Biol Med 1957; 95:617–620. [DOI] [PubMed] [Google Scholar]
- 21.Keshan Disease Research Group of the Chinese Academy of, Medical Sciences. Observations on effect of sodium selenite in prevention of Keshan disease. Chin Med J 1979; 92. [PubMed] [Google Scholar]
- 22.Tan JA, et al. The Keshan disease in China: a study of the geographical epidemiology. Acta Geogr Sin 1979; 34:85–104. [Google Scholar]
- 23.Hou J, et al. Suboptimal selenium supply--a continuing problem in Keshan disease areas in Heilongjiang province. Biol Trace Elem Res 2011; 143:1255–1263. [DOI] [PubMed] [Google Scholar]
- 24.Sokoloff L Kashin-Beck Disease: Current status. Nutr Rev 1988; 46:113–119. [DOI] [PubMed] [Google Scholar]
- 25.Nuttall KL. Evaluating Selenium Poisoning. Ann Clin Lab Sci 2006; 36:409–420. [PubMed] [Google Scholar]
- 26.Varo P, et al. Nationwide Selenium Supplementation in Finland—Effects on Diet, Blood and Tissue Levels, and Health. In: Burk RF, ed. Selenium in Biology and Human Health New York, NY: Springer; 1994:197–218. [Google Scholar]
- 27.Alfthan G, et al. Effects of nationwide addition of selenium to fertilizers on foods, and animal and human health in Finland: From deficiency to optimal selenium status of the population. J Trace Elem Med Biol 2015; 31:142–147. [DOI] [PubMed] [Google Scholar]
- 28.Ulfberg J, Stehlik R. (2021) Finland’s handling of selenium is a model in these times of coronavirus infections. Br J Nutr:1–2. [DOI] [PMC free article] [PubMed]
- 29.Nelson AA, et al. Liver tumors following cirrhosis caused by selenium in rat. Cancer Res 1943; 3:230–236. [Google Scholar]
- 30.Shamberger RJ, and Rudolph G Protection against cocarcinogenesis by antioxidants. Experientia 1966; 22:116. [DOI] [PubMed] [Google Scholar]
- 31.Duffield-Lillico AJ, et al. Baseline Characteristics and the Effect of Selenium Supplementation on Cancer Incidence in a Randomized Clinical Trial: A Summary Report of the Nutritional Prevention of Cancer Trial1. Cancer Epidemiol Biomarkers Prev 2002; 11:630–639. [PubMed] [Google Scholar]
- 32.Schrauzer GN, et al. Cancer mortality correlation studies III: Statistical association with dietary selenium intakes. Bioinorg Chem 1977; 7:23–31. [DOI] [PubMed] [Google Scholar]
- 33.Clark LC, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA 1996; 276:1957–1963. [PubMed] [Google Scholar]
- 34.Lippman SM, et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J Natl Cancer Inst 2005; 97:94–102. [DOI] [PubMed] [Google Scholar]
- 35.Lippman SM, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klein EA, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011; 306:1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kristal AR, et al. Baseline selenium status and effects of selenium and vitamin e supplementation on prostate cancer risk. J Natl Cancer Inst 2014; 106:djt456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacFarquhar JK, et al. Acute Selenium Toxicity Associated With a Dietary Supplement. Arch Intern Med 2010; 170:256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurst R, et al. Establishing optimal selenium status: results of a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr 2010; 91:923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bleys J, et al. Serum selenium levels and all-cause, cancer, and cardiovascular mortality among US adults. Arch Intern Med 2008; 168:404–410. [DOI] [PubMed] [Google Scholar]
- 41.Aderao GN, et al. Dietary selenium levels modulates antioxidant, cytokine and immune response and selenoproteins mRNA expression in rats under heat stress condition. J Trace Elem Med Biol 2023; 75:127105. [DOI] [PubMed] [Google Scholar]
- 42.Hofstee P, et al. Analysis of Selenoprotein Expression in Response to Dietary Selenium Deficiency During Pregnancy Indicates Tissue Specific Differential Expression in Mothers and Sex Specific Changes in the Fetus and Offspring. Int J Mol Sci 2020; 21:2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weeks BS, et al. Dietary selenium and selenoprotein function. Med Sci Monit Int Med J Exp Clin Res 2012; 18:RA127–RA132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsuji PA, et al. Dietary Selenium Levels Affect Selenoprotein Expression and Support the Interferon-γ and IL-6 Immune Response Pathways in Mice. Nutrients 2015; 7:6529–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reich HJ, Hondal RJ. Why Nature Chose Selenium. ACS Chem Biol 2016; 11:821–841. [DOI] [PubMed] [Google Scholar]
- 46.Serrão VHB, Scortecci JF. Why Selenocysteine Is Unique? Front Mol Biosci 2020; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta BBA - Gen Subj 2013; 1830:3289–3303. [DOI] [PubMed] [Google Scholar]
- 48.Lubos E, et al. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid Redox Signal 2011; 15:1957–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masuda R, et al. Modeling the Catalytic Cycle of Glutathione Peroxidase by Nuclear Magnetic Resonance Spectroscopic Analysis of Selenocysteine Selenenic Acids. J Am Chem Soc 2021; 143:6345–6350. [DOI] [PubMed] [Google Scholar]
- 50.Brigelius-Flohé R Tissue-specific functions of individual glutathione peroxidases. Free Radic Biol Med 1999; 27:951–965. [DOI] [PubMed] [Google Scholar]
- 51.Handy DE, Loscalzo J. The role of glutathione peroxidase-1 in health and disease. Free Radic Biol Med 2022; 188:146–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui Z, et al. Glutathione peroxidase 2: A key factor in the development of microsatellite instability in colon cancer. Pathol Res Pract 2023; 243:154372. [DOI] [PubMed] [Google Scholar]
- 53.An BC, et al. GPx3-mediated redox signaling arrests the cell cycle and acts as a tumor suppressor in lung cancer cell lines. PLOS ONE 2018; 13:e0204170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lou W, et al. Overexpression of GPX3, a potential biomarker for diagnosis and prognosis of breast cancer, inhibits progression of breast cancer cells in vitro. Cancer Cell Int 2020; 20:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang C, et al. Extracellular Glutathione Peroxidase GPx3 and Its Role in Cancer. Cancers 2020; 12:2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang WS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014; 156:317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conrad M, et al. The Nuclear Form of Phospholipid Hydroperoxide Glutathione Peroxidase Is a Protein Thiol Peroxidase Contributing to Sperm Chromatin Stability. Mol Cell Biol 2005; 25:7637–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tadokoro T, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2023; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith AC, et al. Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J Med Genet 2014; 51:470–474. [DOI] [PubMed] [Google Scholar]
- 60.Shema R, et al. Synthetic lethal screening in the mammalian central nervous system identifies Gpx6 as a modulator of Huntington’s disease. Proc Natl Acad Sci U S A 2015; 112:268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 2000; 267:6102–6109. [DOI] [PubMed] [Google Scholar]
- 62.Bian M, et al. Targeting the Thioredoxin System as a Strategy for Cancer Therapy. J Med Chem 2019; 62:7309–7321. [DOI] [PubMed] [Google Scholar]
- 63.Bjørklund G, et al. The Role of the Thioredoxin System in Brain Diseases. Antioxid Basel Switz 2022; 11:2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tarrago L, et al. The selenoprotein methionine sulfoxide reductase B1 (MSRB1). Free Radic Biol Med 2022; 191:228–240. [DOI] [PubMed] [Google Scholar]
- 65.Lee BC, et al. Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci Rep 2017; 7:5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cox AG, et al. Selenoprotein H is an essential regulator of redox homeostasis that cooperates with p53 in development and tumorigenesis. Proc Natl Acad Sci 2016; 113:E5562–E5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han S-J, et al. Characterization of Mammalian Selenoprotein O: A Redox-Active Mitochondrial Protein. PLOS ONE 2014; 9:e95518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gromer S, et al. Human selenoproteins at a glance. Cell Mol Life Sci CMLS 2005; 62:2414–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Labunskyy VM, et al. Selenoproteins: molecular pathways and physiological roles. Physiol Rev 2014; 94:739–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luongo C, et al. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat Rev Endocrinol 2019; 15:479–488. [DOI] [PubMed] [Google Scholar]
- 71.Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr 2005; 25:215–235. [DOI] [PubMed] [Google Scholar]
- 72.Burk RF, Hill KE. Selenoprotein P – Expression, Functions, and Roles in Mammals. Biochim Biophys Acta 2009; 1790:1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nunziata C, et al. Structural analysis of human SEPHS2 protein, a selenocysteine machinery component, over-expressed in triple negative breast cancer. Sci Rep 2019; 9:16131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carlisle AE, et al. Selenium detoxification is required for cancer-cell survival. Nat Metab 2020; 2:603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thyer R, et al. Custom selenoprotein production enabled by laboratory evolution of recoded bacterial strains. Nat Biotechnol 2018; 36:624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang M, et al. Effects of selenium supplementation on concurrent chemoradiotherapy in patients with cervical cancer: A randomized, double-blind, placebo-parallel controlled phase II clinical trial. Front Nutr 2023; 10:1094081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McLoughlin MR, et al. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc Natl Acad Sci 2019; 116:11408–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sui X, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol 2018; 9:1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Randolph JT, et al. Discovery of a Potent Chloroacetamide GPX4 Inhibitor with Bioavailability to Enable Target Engagement in Mice, a Potential Tool Compound for Inducing Ferroptosis In Vivo. J Med Chem 2023; 66:3852–3865. [DOI] [PubMed] [Google Scholar]
- 80.Cheff DM, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol 2023; 62:102703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu H, et al. Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol 2022; 18:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arnér ESJ. Focus on mammalian thioredoxin reductases — Important selenoproteins with versatile functions. Biochim Biophys Acta BBA - Gen Subj 2009; 1790:495–526. [DOI] [PubMed] [Google Scholar]
- 83.Gencheva R, Arnér ESJ. Thioredoxin Reductase Inhibition for Cancer Therapy. Annu Rev Pharmacol Toxicol 2022; 62:177–196. [DOI] [PubMed] [Google Scholar]
- 84.Ghareeb H, Metanis N. The Thioredoxin System: A Promising Target for Cancer Drug Development. Chem – Eur J 2020; 26:10175–10184. [DOI] [PubMed] [Google Scholar]
- 85.Xi J, Tian L-L, Xi J, et al. Alterperylenol as a Novel Thioredoxin Reductase Inhibitor Induces Liver Cancer Cell Apoptosis and Ferroptosis. J Agric Food Chem 2022; 70:15763–15775. [DOI] [PubMed] [Google Scholar]
- 86.Tuladhar A, Rein KS. Manumycin A Is a Potent Inhibitor of Mammalian Thioredoxin Reductase-1 (TrxR-1). ACS Med Chem Lett 2018; 9:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang J, et al. Inhibition of Thioredoxin Reductase by Santamarine Conferring Anticancer Effect in HeLa Cells. Front Mol Biosci 2021; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chupakhin E, Krasavin M. Thioredoxin reductase inhibitors: updated patent review (2017-present). Expert Opin Ther Pat 2021; 31:745–758. [DOI] [PubMed] [Google Scholar]
- 89.Abdalbari FH, Telleria CM. The gold complex auranofin: new perspectives for cancer therapy. Discov Oncol 2021; 12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greenough MA, et al. Selective ferroptosis vulnerability due to familial Alzheimer’s disease presenilin mutations. Cell Death Differ 2022; 29:2123–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Z, et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol 2022; 18:751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olson GE, et al. Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J Biol Chem 2007; 282:12290–12297. [DOI] [PubMed] [Google Scholar]
- 93.Burk RF, et al. Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J Neurosci Off J Soc Neurosci 2007; 27:6207–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seale LA. Selenocysteine β-Lyase: Biochemistry, Regulation and Physiological Role of the Selenocysteine Decomposition Enzyme. Antioxidants 2019; 8:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Diamond A, et al. Structure and properties of a bovine liver UGA suppressor serine tRNA with a tryptophan anticodon. Cell 1981; 25:497–506. [DOI] [PubMed] [Google Scholar]
- 96.Leinfelder W, et al. Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature 1988; 331:723–725. [DOI] [PubMed] [Google Scholar]
- 97.Su D, et al. Selenocysteine insertion directed by the 3’-UTR SECIS element in Escherichia coli. Nucleic Acids Res 2005; 33:2486–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Low SC, et al. SECIS–SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J 2000; 19:6882–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berry M j., et al. Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J 1993; 12:3315–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fischer N, et al. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature 2016; 540:80–85. [DOI] [PubMed] [Google Scholar]
- 101.Dumitrescu AM, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet 2005; 37:1247–1252. [DOI] [PubMed] [Google Scholar]
- 102.Çatli G, et al. A Novel Homozygous Selenocysteine Insertion Sequence Binding Protein 2 (SECISBP2, SBP2) Gene Mutation in a Turkish Boy. Thyroid Off J Am Thyroid Assoc 2018; 28:1221–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fu J, et al. Clinical and Molecular Analysis in 2 Families With Novel Compound Heterozygous SBP2 (SECISBP2) Mutations. J Clin Endocrinol Metab 2020; 105:e6–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Serrão VHB, et al. The unique tRNASec and its role in selenocysteine biosynthesis. Amino Acids 2018; 50:1145–1167. [DOI] [PubMed] [Google Scholar]
- 105.Wang C, et al. SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis. Nucleic Acids Res 2015; 43:10534–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carlson BA, et al. Identification and characterization of phosphoseryl-tRNA[Ser]Sec kinase. Proc Natl Acad Sci U S A 2004; 101:12848–12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Palioura S, et al. The Human SepSecS-tRNASec Complex Reveals the Mechanism of Selenocysteine Formation. Science 2009; 325:321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pan T Modifications and functional genomics of human transfer RNA. Cell Res 2018; 28:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou J-B, et al. Modifications of the human tRNA anticodon loop and their associations with genetic diseases. Cell Mol Life Sci CMLS 2021; 78:7087–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fradejas-Villar N, et al. The Effect of tRNA[Ser]Sec Isopentenylation on Selenoprotein Expression. Int J Mol Sci 2021; 22:11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee MY, et al. Loss of epitranscriptomic control of selenocysteine utilization engages senescence and mitochondrial reprogramming. Redox Biol 2020; 28:101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Endres L, et al. Alkbh8 Regulates Selenocysteine-Protein Expression to Protect against Reactive Oxygen Species Damage. PLoS ONE 2015; 10:e0131335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karlsborn T, et al. Elongator, a conserved complex required for wobble uridine modifications in Eukaryotes. RNA Biol 2015; 11:1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fu D, et al. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol 2010; 30:2449–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li J, et al. Intellectual disability-associated gene ftsj1 is responsible for 2’-O-methylation of specific tRNAs. EMBO Rep 2020; 21:e50095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schoenmakers E, et al. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J Clin Invest 2016; 126:992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]