

Abstract
Here, we report the development of cobalt(I)-catalyzed regioselective allylic alkylation reactions of tertiary allyl carbonates with 1,3-dicarbonyl compounds. A family of well-defined tetrahedral cobalt(I) complexes bearing commercially available bidentate bis(phosphine) ligands [(P,P)Co(PPh3)Cl] are synthesized and explored as catalysts in allylic alkylation reactions. The catalyst [(dppp)Co(PPh3)Cl] (dppp = 1,3-Bis(diphenylphosphino)propane) enables the alkylation of a large variety of tertiary allyl carbonates with high yields and excellent regioselectivity for the branched product. Remarkably, this methodology is selective for the activation of tertiary allyl carbonates even in the presence of secondary allyl carbonates. This contrasts with the selectivity observed in cobalt-catalyzed allylic alkylations enabled by visible light photocatalysis. Mechanistic insights by means of experimental and computational investigations support a Co(I)/Co(III) catalytic cycle.
Keywords: Cobalt Catalysis, Allylic alkylation reaction, Reaction mechanism, Coordination cobalt(I) complexes, Theoretical calculations
Graphical Abstract

A cobalt(I)-catalyzed regioselective allylic alkylation of tertiary allylic carbonates with 1,3-dicarbonyl compounds is described. The alkylated products are obtained in high yields and with excellent selectivity towards the kinetic branched regioisomer. Mechanistic insights by combining experimental and computational investigations support a Co(I)/Co(III) catalytic cycle.
Introduction
Transition metal-catalyzed nucleophilic allylic substitutions are well-established methods for the construction of carbon–carbon and carbon–heteroatom bonds in organic synthesis.[1] The introduction of soft carbon-based nucleophiles, such as malonates, have been traditionally dominated by palladium (as in the Tsuji-Trost reaction),[1] rhodium[2] or iridium,[3] whereas reactions using hard carbon-based nucleophiles (RMgX, RZnX, R2Zn, RLi) are usually catalyzed by copper.[4] Recent advances in cobalt-catalyzed nucleophilic allylic substitutions illustrate that high levels of regio- and enantioselective control have also been achieved with this metal.[5] Selectivity towards the branched product is desirable when seeking for methods to synthesize quaternary stereogenic centers employing achiral nucleophiles.[6] In this regard, cobalt enables high regioselectivity towards branched addition products with stabilized enolates,[5a, 5c] similar to the heavier group 9 metals, rhodium and iridium.[7] This contrasts with cobalt-catalyzed reactions of hard carbon-based nucleophiles with allylic electrophiles since, in these cases, the linear product is usually the major regioisomer.[5d, 5e, 8] Although cobalt-catalyzed asymmetric allylic substitution reactions are underdeveloped, these precedents prompt the interest for exploring Co as a more abundant and environmentally benign alternative for such transformations, with specific aims at finding complementary reactivity to the well-established methods.
The mechanistic scenario depicted for cobalt-catalyzed allylic substitutions is largely extrapolated from the knowledge obtained with other metal catalysts. Thus, it has been proposed that cobalt-catalyzed allylic substitutions with soft carbon-nucleophiles proceed via Co(I)/Co(III) catalytic cycles (Figure 1). Most mechanistic scenarios converge on the suggestion that a cobalt(I) species is likely the active catalytic intermediate that enables the activation of the electrophile, which is commonly an allyl carbonate. The catalytic cycle is closed by the nucleophilic attack to the putative π-allylcobalt(III) intermediate, which regenerates the Co(I) and delivers the product that contains a new functional group at the allylic position. Thus, most methods for allylic substitution reactions require reducing conditions to form in situ low-valent cobalt species as the active catalytic intermediates from Co(II) precursors.[5b, 5c, 9] However, mechanistic studies in allylic alkylations catalyzed by well-defined Co(I) complexes are still limited.[5a, 5b, 10] It is worth noting that cobalt(II) complexes have been shown to catalyze the allylation of 1,3-dicarbonyl compounds with allyl acetates, though with limited efficiency in comparison with the reductive methods.[11]
Figure 1.

Previous work on cobalt-catalyzed allylic substitution reactions.
As part of our interest in the field of low-valent base-metal catalysis (Fe, Co, Ni),[12] we aimed at the development of well-defined cobalt(I) catalysts bearing commercially available bidentate bis(phosphine) complexes and to investigate their catalytic activity and the mechanism in allylic alkylations reactions with 1,3-dicarbonyl compounds. The results obtained in this work shed light on the reaction mechanism of cobalt(I)-catalyzed allylic substitution reactions and provide a blueprint for rational catalyst design.
Results and Discussion
The combination of cobalt salts and bidentate bis(phosphine) ligands has been shown to give access to highly efficient catalysts for a variety of hydrogenations, alkene isomerizations, hydroborations, cycloadditions, Alder-Ene and hydroacylation reactions, among others, under reducing conditions.[13] On the other hand, the isolation of Co(I) complexes bearing bidentate bis(phosphine) ligands showcases that cobalt can adopt different geometries depending on the ligand, the stoichiometry and reduction conditions.[5a, 13i, 14] In this regard, in order to control the geometry of the Co(I) catalysts bearing bidentate bis(phosphine) ligands we sought to prepare tetracoordinate Co(I) complexes by ligand substitution reaction at CoCl(PPh3)3.[15] CoCl(PPh3)3 is a convenient Co(I) precursor since it is commercially available or, alternatively, can be readily prepared in gram-scale in the laboratory. This strategy enables the synthesis of well-defined tetrahedral Co(I) complexes and a systematic evaluation of their catalytic activity.
Our study began with the synthesis of tetracoordinate Co(I) chloride complexes bearing bidentate phosphine ligands that present a variety of natural bite angles (from 83° for dppbz to 111° for Xantphos).[16] As shown in Figure 2a, the direct coordination of several bidentate phosphine ligands (and concomitant dissociation of two PPh3) to CoCl(PPh3)3 enabled the efficient synthesis of distorted tetrahedral Co(I) complexes [(P,P)Co(PPh3)Cl] in yields ranging from 75% to 96%. These cobalt(I) complexes are reasonably stable to air and moisture in solid state, although they get oxidized rapidly in solution, similarly to their Co(I) precursor. Gratifyingly, we obtained the solid state structure for six of these complexes (1Co-7Co, Figure 3).[17] A closer look at the structures of 6Co and 7Co that bear bis(phosphine) ligands with large bite angles (dppf and Xantphos respectively)[18] show slightly distorted tetrahedral geometries. On the other hand, bidentate ligands with smaller bite angles led to highly distorted tetrahedral geometries in complexes 1Co, 2Co and 3Co. Similarly, complex 4Co, which contains the more electron-donating bis(phosphine) ligand dcpe, also exhibits a distorted tetrahedral geometry, featuring a slightly larger bite angle and larger Co–P bond lengths in comparison with 3Co bearing dppe ligand.[19]
Figure 2.

a) Synthesis of well-defined tetrahedral Co(I) chloride catalysts by ligand substitution reaction. b) Synthesis of cationic Co(I) complexes by anion exchange with Na[BArF4] from Co(I) chloride precursors.
Figure 3.

Solid-state structures of complexes 1Co, 2Co, 3Co, 4Co, 6Co, 7Co, 11Co and 12Co and selected bond lengths and angles. For 2Co, 3Co and 11Co the bond lengths are an average of 2, 3 and 3 independent crystallographic molecules, respectively. Displacement ellipsoids are shown at 50% probability; hydrogen atoms are omitted for clarity.
The Co(I) chloride complexes 1Co-7Co are convenient precursors to synthesize cationic Co(I) complexes bearing bidentate bis(phosphine) ligands [(P,P)Co(η6-C7H8)][BArF4] (BArF4 = B[(3,5-(CF3)2)C6H3]4)). Thus, chloride exchange with Na[BArF4] in aromatic solvents at complexes 1Co, 2Co and 5Co afforded complexes 8Co-10Co in good yields (Figure 2b). Our two step synthesis from CoCl(PPh3)3 precursor is a synthetic alternative to previously reported procedures towards [(P,P)Co(η6-C7H8)]+ complexes.[13i, 13l, 14b, 20] Recrystallization in benzene:pentane afforded suitable single crystals for X-ray diffraction (XRD) analysis for complexes [(dppbz)Co(η6-C6H6)][BArF4] (11Co) and [(dppe)Co(η6-C6H6)][BArF4] 12Co, which are shown in Figure 3.
Next, we evaluated the catalytic activity of the isolated cobalt(I) complexes in allylic alkylation reactions with soft carbon-based nucleophiles. Particularly, we explored the reaction of allylic carbonate 1aOCO2Me and diethyl malonate 2 using 10 mol% of Co(I) precatalyst and 20 mol% of NaBF4 as additive in MeCN, at 60 °C for 24 h (Figure 4). We observed a strong influence of the bite angle of the bis(phosphine) ligand on the reaction yield.[21] While Co(I) precatalysts bearing dppf and Xantphos gave no conversion, the reaction proceeded with good yields (49–77%) when using Co(I) complexes featuring bis(phosphines) with smaller bite angles such as dppe, dppen and dppbz. A similar observation was reported for the enantioselective reverse prenylation of β-ketoesters.[5a] The coordination of the more electron-donating dcpe ligand to the Co(I) center in [(dcpe)Co(PPh3)Cl] (4Co) catalyst ceases the catalytic activity. A control experiment with catalyst (dppp)CoCl2, where the metal is in oxidation state +2, gave no conversion (see Table SI-1 in the Supporting Information (SI)).
Figure 4.

Screening of the catalytic activity of isolated (P,P)Co(PPh3)Cl complexes. Conditions: 1.5 equiv of diethyl malonate, 1 equiv of allyl carbonate (0.2 M), (P,P)Co(PPh3)Cl (10 mol%), NaBF4 (20 mol%), MeCN, 60 °C, 24 h. The conversion (in parenthesis) and the yield were determined by GC using undecane as internal standard. n.d. = not detected.
Among the catalysts tested, complex 5Co bearing dppp is the most efficient one under our standard reaction conditions, giving the product with 77% yield. In the absence of the additive NaBF4 the reaction yield drops to 21%. Other salts such NaBArF4, LiOTf or La(OTf)3 were tested as additives but lower yields were obtained (Figure 4). Our synthetic method enables the addition of 1,3-dicarbonyl compounds without the use of external base, in contrast with the common reaction conditions reported for nucleophilic allylic substitutions catalyzed by Pd, Rh, Ir.[1c, 1d, 2–3, 22] The performance of the cationic Co(I) complexes 8Co-10Co in catalysis was also tested. Similarly to the experiments with the Co(I) chloride complex 5Co, the addition of NaBF4 also had a positive impact in the yield (Figure 4). However, overall the highest yield was obtained when using complex 5Co, which was selected for further exploring the scope of the reaction.
As shown in Table 1, our method is applicable for a wide range of tertiary allyl carbonates 1 containing two aliphatic substituents to afford the branched product 3 with high regioselectivity (the linear product is not detected by 1H-NMR spectroscopy of the reaction crude). Allyl carbonates containing alkene (3h), ether (3c), silyl ether (3i), unprotected alcohol (3j), aryl and alkyl chlorides (3g and 3k), aryl fluorides (3f), amides (3i) and ester (3m, 3n) are suitable substrates. The method tolerates several heteroaromatics in the substrate such as pyrrole (3o), indole (3p), furan (3q), benzofuran (3r), thiophene (3s) and benzothiophene (3t). However, only secondary allyl carbonates containing an electron-deficient aryl group (3u, 3v, 3w) afford the branched alkylated product, and in low yields (27–30%). Other 1,3-diesters as dibenzyl malonate and diisopropyl malonate were suitable nucleophiles and they furnished the alkylated products 3x and 3y in 62% and 59% yields, respectively. The reaction using β-ketoesters such as ethyl 3-oxobutanoate and phenyl 3-oxobutanoate gave the corresponding branched product 3z and 3aa in 82% and 77% respectively. The reverse prenylation of β-ketoester 1ab containing the tetralone scaffold gave product 3ab in 69% yield. However, the ester group was required in the nucleophile since no conversion was observed when using β-diketones, as pentane-2,4-dione and 1,3-diphenylpropane-1,3-dione, and malononitrile derivatives (see SI for further details).
Table 1.
Screening of substrate scope.[a]
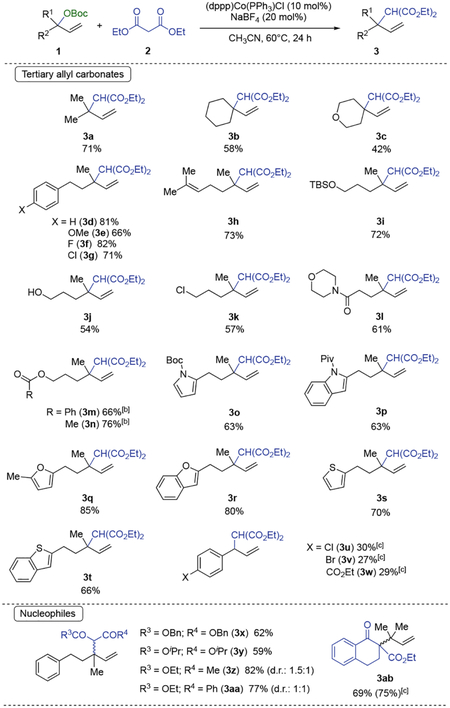
Conditions: 81.5 equiv of diethyl malonate, 1 equiv of allyl carbonate (0.2 M), (dppp)Co(PPh3)Cl (10 mol%), NaBF4 (20 mol%), MeCN, 60°C, 24 h. [b] with 20 mol% of (dppp)Co(PPh3)Cl. [c] Using the substrate containing the methyl carbonate instead of tert-butyl carbonate (Boc).
The cobalt(I)-catalyzed allylic alkylation protocol can be further simplified by preparing in-situ the (dppp)Co(PPh3)Cl (5Co) catalyst. Thus, a mixture of CoCl(PPh3)3 and dppp ligand in MeCN added directly to the reaction vessel that contains the allylic carbonate 1h, diethyl malonate and NaBF4 afforded the branched alkylated product 3h in 56% yield (70% conversion). This yield is lower in comparison with the yield obtained when using isolated 5Co as precatalyst (73% yield). We considered that PPh3 could be an inhibitor for the reaction. Indeed, addition of increasing amounts of PPh3 (from 0.5 to 5 equiv) to the standard reaction conditions using catalyst 5Co caused a decrease in the yield of 3h to 30% already after addition of 1 equiv (see Table SI-5), which corroborates this hypothesis. Similarly, the addition of PPh3 has also an inhibitory effect in the reactions catalyzed by the cationic Co(I) complexes [(P,P)Co(η6-C7H8)][BArF4] (see Table SI-6).
The selectivity of the cobalt(I)-catalyzed allylic alkylation reaction reported here is showcased by testing substrate 1ac, that contains both secondary and tertiary allyl carbonates (Figure 5). Under the standard reaction conditions, product 3ac is isolated in 76% yield, which indicates that the malonate is only incorporated at the tertiary allyl carbonate, whereas the secondary allyl carbonate remains unreacted. The observed selectivity pattern is complementary to the selectivity commonly reported for palladium-catalyzed Tsuji-Trost reaction.[1c, 1d, 22–23] In this regard, the reaction of diethyl malonate and substrate 1ac catalyzed by Pd(PPh3)4 afforded product 4 in 30%. Palladium(0) reacts with the secondary allyl carbonate to give the linear alkylated product but undergoes β-hydride elimination to form the 1,3-diene upon cleavage of the tertiary allyl carbonate.[24]
Figure 5.

Divergent reactivity between Pd(PPh3)4 and (dppp)Co(PPh3)Cl with allyl carbonates. Conditions: a) 1.5 equiv of diethyl malonate, 1 equiv of allyl carbonate (0.2 M), (dppp)Co(PPh3)Cl (10 mol%), NaBF4 (20 mol%), MeCN, 60°C, 24 h. b) 1.5 equiv of diethyl malonate, 1 equiv of allyl carbonate (0.1 M), Pd(PPh3)4 (5 mol%), THF, 30°C, 20 min.
The selectivity observed with isolated cobalt(I) precatalyst (dppp)Co(PPh3)Cl (5Co) in allylic alkylation reactions is also complementary to the selectivity achieved by the reported cobalt-catalyzed allylic alkylation under visible-light photoredox conditions (Figure 6).[5c] Reaction of diethyl malonate 2 with tertiary allylic carbonates 1d and 1ad catalyzed by CoBr2/dppp (5 mol%) and 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4Cz-IPN, 0.5 mol%) in the presence of iPr2NEt2 (1 equiv) in THF, 4Å MS, rt, 16 h under visible-light irradiation did not give conversion. In contrast, as previously reported, the visible light photocatalytic reaction of nucleophile 2 with secondary allyl carbonates 1ae and 1af afforded the corresponding alkylated products 3ae and 3af in 78% and 81% yield, respectively. Similar results were obtained when CoCl2 was used as Co(II) salt instead of CoBr2. Our protocol enables the activation of more sterically hindered tertiary allylic carbonates, differing from photocatalytic methods that are limited to secondary allylic carbonates. This likely indicates that the thermal activation with well-defined Co(I) precatalysts and the photocatalytic activation with the CoX2/dppp and 4Cz-IPN dual catalytic system proceed via different reaction mechanisms.
Figure 6.

Comparison of thermal conditions (method A) vs. visible-light photocatalysis (method B) catalyzed by cobalt. 4Cz-IPN = 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene.
Next, we focused on shedding light on the mechanism of the developed cobalt(I)-catalyzed allylic alkylation reaction by means of mechanistic probes in combination with theoretical calculations. We carried out a catalysis using substrate 1ag, which contains a cyclopropyl ring in α-position to the tertiary allyl carbonate. The corresponding branched alkylated product 3ag was isolated in 40% yield (Figure 7a). Importantly, the ring opening product was not detected, which rules out the intermediacy of long-lived allyl radicals. In addition, no conversion was observed in the reaction of Co(I) precatalyst 5Co with the allyl carbonate 1d without the addition of diethyl malonate in MeCN at 60°C (Figure 7a). These results contrast those recently reported by Meng and co-workers on the reductive cobalt-catalyzed enantioselective addition of allyl carbonates to aldehydes.[25] In that precedent, the use of phosphorous-oxazoline ligand in combination with CoI2 and Mn as a reductant was key for the success of the reaction. The formation of radical intermediates upon activation of the allyl carbonate was proposed based on the observation of the ring opening product of an allyl carbonate containing a cyclopropyl ring as radical probe. In addition, it was reported the isolation of the homodimerization product of the allyl carbonate in the absence of aldehyde.
Figure 7.

a) Mechanistic probes. b) Proposed Co-catalysed mechanism for regioselective allylic alkylation reaction. The energies shown were computed at the uB3LYP-D3/def2tzvpp-CPCM(acetonitrile)//uB3LYP-D3/def2SVP-CPCM(acetonitrile) level of theory. c) Relevant transition state structures. All energies in the scheme are in kcal/mol.
The reaction of the enantiopure allyl carbonate (R)-1h with dibenzyl benzoate catalyzed by (dppp)Co(PPh3)Cl afforded the alkylated product 3ah in 75% yield as a racemate (Figure 7a). The loss of stereochemical information is also observed when using the chiral Co(I) catalyst formed in situ by combining (PPh3)3CoCl and (R,R)-Ph-BPE ligand. This loss of stereochemical information during catalysis could indicate a faster oxidative addition of Co(I) into the allyl carbonate and isomerization of the π-allylcobalt(III) intermediate in comparison with the next steps that lead to the product formation, which is in agreement with the theoretical calculations (vide infra). Finally, analysis of the headspace of the reaction vessel after catalysis confirmed the quantitative formation of CO2 (Figure SI–4).
In order to further understand the mechanism and the origin of the observed selectivity, we carried out dispersion-corrected density functional theory (DFT) calculations (see the SI for the details on the computational methods). For simplicity, we only show and discuss in detail the lowest energy pathway (see the SI for alternative mechanisms considered). As shown in Figure 7b, firstly an oxidative addition reaction of the cationic (dppp)Co(I)+ in the presence of the alkene 1aOCO2Me proceeds rapidly and irreversibly via a 3TS-A-B (barrier, ~7.4 kcal/mol) to form the corresponding Co(III) intermediate 3B. Alternative (dppp)Co(I) species (i.e., (dppp)Co(I)-Cl, (dppp)Co(I)-MeCN and (dppp)Co(I)-PPh3; Figure SI–41) were considered for the oxidative addition step, but the transition states were found to be much higher >12.0 kcal/mol). Then, the release of CO2 promoted by cobalt was considered from intermediate 3B, as CO2 was experimentally detected. Specifically, our calculations show that species 3B could undergo facile decarboxylation through the 3TS-B-B´ (barrier, ~16.4 kcal/mol) to form the corresponding Co(III) intermediate 3B’ and release of CO2. From 3B’, the calculations show that the coordination (and deprotonation) of dimethyl malonate to afford the π-allylcobalt(III) intermediate 3C and MeOH is thermodynamically favourable. In turn, intermediate 3C is prone to undergo a branched-selective reductive elimination (via 3TS-C-Dbranched) to furnish the desired product and restart the catalytic cycle. Notably, in agreement with experiments, the linear-selective reductive elimination (via 3TS-C-Dlinear) is significantly higher than branched-selective reductive elimination (ΔΔG‡ = 15.6 kcal mol−1 versus 9.2 kcal mol−1, Figure 7c). Noncovalent interaction (NCI) analysis revealed greater steric repulsions between the alkene substituents and the phenyl groups in the dppp ligand in 3TS-C-Dlinear in comparison to 3TS-C-Dbranched leading to exclusive regioselectivity (Figure SI–42). In addition, Natural Bond Orbital (NBO) analysis revealed that 3TS-C-Dbranched benefits from favorable donor–acceptor interaction energy from the lone pair (LP) on the cobalt atom to the neighboring antibond (BD*) of the C1–C2 bond (Figure SI–43). Finally, higher barriers were observed for secondary allylic carbonate systems, consistent with the experimental data (See SI for further details and discussion).
Conclusion
In conclusion, we have developed regioselective allylic alkylation reactions with soft carbon-nucleophiles catalyzed by well-defined tetrahedral bis(phosphine) cobalt(I) complexes. The reaction of 1,3-diesters and β-ketoesters compounds with tertiary allyl carbonates catalyzed by cobalt(I) proceeds under mild conditions and with excellent regioselectivity towards the branched alkylated product. Remarkably, this method enables the alkylation of tertiary allyl carbonates in the presence of secondary allyl carbonates. This unusual selectivity is complementary to the previously observed selectivity achieved in cobalt-catalyzed allylic alkylation reactions under visible light photoredox catalysis. Radical probe experiments in combination with DFT computational studies were employed to elucidate the Co(I)-catalyzed mechanistic pathway for this transformation. All the collected data support a Co(I)/Co(III) catalytic cycle. Theoretical studies provide a better understanding of the non-covalent interactions that induce regioselectivity in this reaction.
Supplementary Material
Acknowledgements
Generous financial support to A.C. by the Philipps-Universität Marburg is gratefully acknowledged. We thank Paul H. Moths, Hannah Goerlach and Belén Pardo for preliminary work on the development of the catalytic methodology. We thank Prof. Eric L. Meggers for providing access to analytical instruments. We thank Niels N. Oehlmann and Dr. Johannes G. Rebelein for assisting us with the quantification of CO2 in the catalytic experiments. O.G is grateful for financial support provided by the National Institutes of Health National Science (R35GM137797), Camille and Henry Dreyfus Foundation and Welch Foundation (A-2102-20220331). We also acknowledge Texas A&M University HPRC (https://hprc.tamu.edu) for computational resources.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- [1].a) Hartwig JF, Organotransition metal chemistry. From bonding to catalysis, University Science Books, Sausalito, 2010; [Google Scholar]; b) Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, Springer, Berlin, Heidelberg, 2012; [Google Scholar]; c) Trost BM, Van Vranken DL, Chem. Rev 1996, 96, 395–422; [DOI] [PubMed] [Google Scholar]; d) Trost BM, Crawley ML, Chem. Rev 2003, 103, 2921–2944. [DOI] [PubMed] [Google Scholar]
- [2].a) Trost BM, Fullerton TJ, J. Am. Chem. Soc 1973, 95, 292–294; [Google Scholar]; b) Trost BM, Dietsch TJ, J. Am. Chem. Soc 1973, 95, 8200–8201; [Google Scholar]; c) Tsuji J, Minami I, Shimizu I, Tetrahedron Lett. 1983, 24, 1793–1796; [Google Scholar]; d) Trost BM, Tetrahedron 2015, 71, 5708–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Turnbull BWH, Evans PA, J. Org. Chem 2018, 83, 11463–11479. [DOI] [PubMed] [Google Scholar]
- [4].a) Cheng Q, Tu H-F, Zheng C, Qu J-P, Helmchen G, You S-L, Chem. Rev 2019, 119, 1855–1969; [DOI] [PubMed] [Google Scholar]; b) Qu J, Helmchen G, Acc. Chem. Res 2017, 50, 2539–2555; [DOI] [PubMed] [Google Scholar]; c) Hartwig JF, Stanley LM, Acc. Chem. Res 2010, 43, 1461–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL, Chem. Rev 2008, 108, 2824–2852; [DOI] [PubMed] [Google Scholar]; b) Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M, Chem. Rev 2008, 108, 2796–2823; [DOI] [PubMed] [Google Scholar]; c) Falciola CA, Alexakis A, Eur. J. Org. Chem 2008, 2008, 3765–3780; [Google Scholar]; d) Luchaco-Cullis CA, Mizutani H, Murphy KE, Hoveyda AH, Angew. Chem. Int. Ed 2001, 40, 1456–1460; [DOI] [PubMed] [Google Scholar]; e) Pérez M, Fañanás-Mastral M, Bos PH, Rudolph A, Harutyunyan SR, Feringa BL, Nat. Chem 2011, 3, 377–381; [DOI] [PubMed] [Google Scholar]; f) Tissot-Croset K, Polet D, Alexakis A, Angew. Chem. Int. Ed 2004, 43, 2426–2428. [DOI] [PubMed] [Google Scholar]
- [6].a) Sun M, Chen J-F, Chen S, Li C, Org. Lett 2019, 21, 1278–1282; [DOI] [PubMed] [Google Scholar]; b) Ghorai S, Chirke SS, Xu W-B, Chen J-F, Li C J. Am. Chem. Soc 2019, 141, 11430–11434; [DOI] [PubMed] [Google Scholar]; c) Takizawa K, Sekino T, Sato S, Yoshino T, Kojima M, Matsunaga S, Angew. Chem. Int. Ed 2019, 58, 9199–9203; [DOI] [PubMed] [Google Scholar]; d) Reddy CK, Knochel P, Angew. Chem. Int. Ed 1996, 35, 1700–1701; [Google Scholar]; e) Dunet G, Knochel P, Synlett 2007, 9, 1383–1386; [Google Scholar]; f) Ghorai S, Ur Rehman S, Xu W-B, Huang W-Y, Li C, Org. Lett 2020, 22, 3519–3523; [DOI] [PubMed] [Google Scholar]; g) Han J-F, Guo P, Zhang X-G, Liao J-B, Ye K-Y, Org. Biomol. Chem 2020, 18, 7740–7750. [DOI] [PubMed] [Google Scholar]
- [7].a) Liu Y, Han S-J, Liu W-B, Stoltz BM, Acc. Chem. Res 2015, 48, 740–751; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Süsse L, Stoltz BM, Chem. Rev 2021, 121, 4084–4099; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Butcher TW, Hartwig JF, Angew. Chem. Int. Ed 2018, 57, 13125–13129; [DOI] [PubMed] [Google Scholar]; d) Chen W, Chen M, Hartwig JF, J. Am. Chem. Soc 2014, 136, 15825–15828; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jiang X, Chen W, Hartwig JF, Angew. Chem. Int. Ed 2016, 55, 5819–5823; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chen W, Hartwig JF, J. Am. Chem. Soc 2014, 136, 377–382; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Fañanás-Mastral M, Pérez M, Bos PH, Rudolph A, Harutyunyan SR, Feringa BL, Angew. Chem. Int. Ed 2012, 51, 1922–1925; [DOI] [PubMed] [Google Scholar]; h) Hethcox JC, Shockley SE, Stoltz BM, Angew. Chem. Int. Ed 2018, 57, 8664–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Takeuchi R, Kashio M, J. Am. Chem. Soc 1998, 120, 8647–8655; [Google Scholar]; b) Moghadam FA, Hicks EF, Sercel ZP, Cusumano AQ, Bartberger MD, Stoltz BM, J. Am. Chem. Soc 2022, 144, 7983–7987; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Takeuchi R, Kashio M, Angew. Chem. Int. Ed. Engl 1997, 36, 263–265; [Google Scholar]; d) Takeuchi R, Kitamura N, New J. Chem 1998, 22, 659–660. [Google Scholar]
- [9].Keiya M, Hideki Y, Koichiro O, Chem. Lett 2004, 33, 832–833. [Google Scholar]
- [10].a) Gomes P, Gosmini C, Périchon J, J. Org. Chem 2003, 68, 1142–1145; [DOI] [PubMed] [Google Scholar]; b) Michigami K, Mita T, Sato Y, J. Am. Chem. Soc 2017, 139, 6094–6097; [DOI] [PubMed] [Google Scholar]; c) Gomes P, Gosmini C, Périchon J, Org. Lett 2003, 5, 1043–1045; [DOI] [PubMed] [Google Scholar]; d) Ang NWJ, Oliveira JCA, Ackermann L, Angew. Chem. Int. Ed 2020, 59, 12842–12847; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Qian X, Auffrant A, Felouat A, Gosmini C, Angew. Chem. Int. Ed 2011, 50, 10402–10405. [DOI] [PubMed] [Google Scholar]
- [11].a) Roustan JL, Mérour JY, Houlihan F, Tetrahedron Lett. 1979, 20, 3721–3724; [Google Scholar]; b) Tang T, Jones E, Wild T, Hazra A, Minteer SD, Sigman MS, J. Am. Chem. Soc 2022, 144, 20056–20066. [DOI] [PubMed] [Google Scholar]
- [12].Bhatia B, Reddy MM, Iqbal J, Tetrahedron Lett. 1993, 34, 6301–6304. [Google Scholar]
- [13].a) Claros M, Ungeheuer F, Franco F, Martin-Diaconescu V, Casitas A, Lloret-Fillol J, Angew. Chem. Int. Ed 2019, 58, 4869–4874; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yuan M, Song Z, Badir SO, Molander GA, Gutierrez O, J. Am. Chem. Soc 2020, 142, 7225–7234; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu L, Aguilera MC, Lee W, Youshaw CR, Neidig ML, Gutierrez O, Science 2021, 374, 432–439; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rentería-Gómez A, Lee W, Yin S, Davis M, Gogoi AR, Gutierrez O, ACS Catal. 2022, 12, 11547–11556. [Google Scholar]
- [14].a) Treutwein J, Hilt G, Angew. Chem. Int. Ed 2008, 47, 6811–6813; [DOI] [PubMed] [Google Scholar]; b) Hilt G, Paul A, Treutwein J, Org. Lett 2010, 12, 1536–1539; [DOI] [PubMed] [Google Scholar]; c) Schmidt A, Hilt G, Org. Lett 2013, 15, 2708–2711; [DOI] [PubMed] [Google Scholar]; d) Friedfeld MR, Shevlin M, Hoyt JM, Krska SW, Tudge MT, Chirik PJ, Science 2013, 342, 1076–1080; [DOI] [PubMed] [Google Scholar]; e) Yang J, Yoshikai N, J. Am. Chem. Soc 2014, 136, 16748–16751; [DOI] [PubMed] [Google Scholar]; f) Chen Q-A, Kim DK, Dong VM, J. Am. Chem. Soc 2014, 136, 3772–3775; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Santhoshkumar R, Mannathan S, Cheng C-H, J. Am. Chem. Soc 2015, 137, 16116–16120; [DOI] [PubMed] [Google Scholar]; h) Jing SM, Balasanthiran V, Pagar V, Gallucci JC, RajanBabu TV, J. Am. Chem. Soc 2017, 139, 18034–18043; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Friedfeld MR, Zhong H, Ruck RT, Shevlin M, Chirik PJ, Science 2018, 360, 888–893; [DOI] [PubMed] [Google Scholar]; j) Weber SM, Hilt G, Org. Lett 2019, 21, 4106–4110; [DOI] [PubMed] [Google Scholar]; k) Ding W, Yoshikai N, Angew. Chem. Int. Ed 2019, 58, 2500–2504; [DOI] [PubMed] [Google Scholar]; l) Zhong H, Friedfeld MR, Chirik PJ, Angew. Chem. Int. Ed 2019, 58, 9194–9198; [DOI] [PubMed] [Google Scholar]; m) Gray M, Hines MT, Parsutkar MM, Wahlstrom AJ, Brunelli NA, RajanBabu TV, ACS Catal. 2020, 10, 4337–4348; [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Whyte A, Torelli A, Mirabi B, Prieto L, Rodríguez JF, Lautens M, J. Am. Chem. Soc 2020, 142, 9510–9517; [DOI] [PubMed] [Google Scholar]; o) Whyte A, Bajohr J, Torelli A, Lautens M, Angew. Chem. Int. Ed 2020, 59, 16409–16413; [DOI] [PubMed] [Google Scholar]; p) Hu M, Ge S, Nat. Comm 2020, 11, 765; [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Da Concepción E, Fernández I, Mascareñas JL, López F, Angew. Chem. Int. Ed 2021, 60, 8182–8188; [DOI] [PubMed] [Google Scholar]; r) Herbort JH, Lalisse RF, Hadad CM, RajanBabu TV, ACS Catal. 2021, 11, 9605–9617; [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Cristòfol À, Limburg B, Kleij AW, Angew. Chem. Int. Ed 2021, 60, 15266–15270; [DOI] [PubMed] [Google Scholar]; t) Zhang Y-D, Li X-Y, Mo Q-K, Shi W-B, Zhao J-B, Zhu S-F, Angew. Chem. Int. Ed 2022, 61, e202208473; For references on cobalt catalysis under reducing conditions with other P,P,N ligands see: [DOI] [PubMed] [Google Scholar]; u) Liu H, Xu M, Cai C, Chen J, Gu Y, Xia Y, Org. Lett 2020, 22, 1193–1198; [DOI] [PubMed] [Google Scholar]; v) Liu X, Rong X, Liu S, Lan Y, Liu Q, J. Am. Chem. Soc 2021, 143, 20633–20639; [DOI] [PubMed] [Google Scholar]; w) Rong X, Yang J, Liu S, Lan Y, Liu Q, CCS Chem. 2023, 5, 1293–1300. [Google Scholar]
- [15].a) Duvvuri K, Dewese KR, Parsutkar MM, Jing SM, Mehta MM, Gallucci JC, RajanBabu, J. Am. Chem. Soc 2019, 141, 7365–7375; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Parsutkar MM, Moore CE, RajanBabu TV, Dalton Trans. 2022, 51, 10148–10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Brennan MR, Kim D, Fout AR, Chem. Sci 2014, 5, 4831–4839; [Google Scholar]; b) Cassidy JM, Whitmire KH, Acta Cryst. 1991, C47, 2094–2098. [Google Scholar]
- [17].van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P, Chem. Rev 2000, 100, 2741–2770. [DOI] [PubMed] [Google Scholar]
- [18].CCDC 2278006, 2278007, 2278008, 2278009, 2278010, 2287011, 2278012 and 2278013 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. [Google Scholar]
- [19].Dierkes P, van Leeuwen PWNM, J. Chem. Soc., Dalton Trans 1999, 1519–1530. [Google Scholar]
- [20].a) Allman T, Goel RG, Can. J. Chem 1982, 60, 716–722; [Google Scholar]; b) Tolman CA, J. Am. Chem. Soc 1970, 92, 2953–2956. [Google Scholar]
- [21].Farmer ME, Ehehalt LE, Pabst TP, Tudge MT, Chirik PJ, Organometallics 2021, 40, 3599–3607. [Google Scholar]
- [22].For an example of ligand-controlled cobalt-catalyzed regiodivergent hydroboration of allenes, see: Wu C, Ge S, Chem. Sci 2020, 11, 2783–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pàmies O, Margalef J, Cañellas S, James J, Judge E, Guiry PJ, Moberg C, Bäckvall J-E, Pfaltz A, Pericàs MA, Diéguez M, Chem. Rev 2021, 121, 4373–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Trost BM, Schultz JE, Synthesis 2019, 51, 1–30; [Google Scholar]; b) Wang Y-N, Lu L-Q, Xiao W-J, Chem. Asian J 2018, 13, 2174–2183; [DOI] [PubMed] [Google Scholar]; c) Prétôt R, Pfaltz A, Angew. Chem. Int. Ed 1998, 37, 323–325; [DOI] [PubMed] [Google Scholar]; d) Cai A, Guo W, Martínez-Rodríguez L, Kleij AW, J. Am. Chem. Soc 2016, 138, 14194–14197; [DOI] [PubMed] [Google Scholar]; e) Dubovyk I, Watson IDG, Yudin AK, J. Org. Chem 2013, 78, 1559–1575; [DOI] [PubMed] [Google Scholar]; f) Nong Z-S, Zhu L, Wang T-C, Fan L-F, Wang P-S, Gong L-Z, Nat. Synth 2022, 1, 487–496. [Google Scholar]
- [25].a) Shimizu I, Sugiura T, Tsuji J, J. Org. Chem 1985, 50, 537–539; [Google Scholar]; b) Al-Masum M, Yamamoto Y, J. Am. Chem. Soc 1998, 120, 3809–3810; [Google Scholar]; c) Crouch IT, Dreier T, Frantz DE, Angew. Chem. Int. Ed 2011, 50, 6128–6132. [DOI] [PubMed] [Google Scholar]
- [26].Wang L, Wang L, Li M, Chong Q, Meng F, J. Am. Chem. Soc 2021, 143, 12755–12765. [DOI] [PubMed] [Google Scholar]
- [27].For a review on distortion/interaction analyses see: Bickelhaupt FM, Houk KN, Angew. Chem. Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.