

SUMMARY
We report an in situ vaccination, adaptable to nearly any type of cancer, that combines radiotherapy targeting one tumor and intratumoral injection of this site with tumor-specific antibody and interleukin-2 (IL-2; 3xTx). In a phase I clinical trial, administration of 3xTx (with an immunocytokine fusion of tumor-specific antibody and IL-2, hu14.18-IL2) to subjects with metastatic melanoma increases peripheral CD8+ T cell effector polyfunctionality. This suggests the potential for 3xTx to promote antitumor immunity against metastatic tumors. In poorly immunogenic syngeneic murine melanoma or head and neck carcinoma models, 3xTx stimulates CD8+ T cell-mediated antitumor responses at targeted and non-targeted tumors. During 3xTx treatment, natural killer (NK) cells promote CTLA4+ regulatory T cell (Treg) apoptosis in non-targeted tumors. This is dependent on NK cell expression of CD86, which is upregulated downstream of KLRK1. NK cell depletion increases Treg infiltration, diminishing CD8+ T cell-dependent antitumor response. These findings demonstrate that NK cells sustain and propagate CD8+ T cell immunity following 3xTx.
Graphical abstract
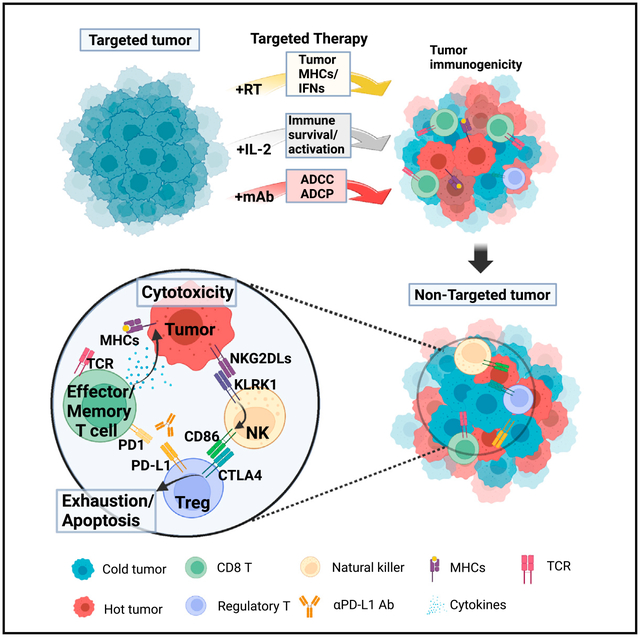
In brief
Jin et al. developed an in situ vaccination approach that combines radiation and intratumoral injection of a tumor-specific antibody and IL-2 (3xTx). This targeted 3xTx regimen elicits CD8+ T cell immunity supported by NK cell-mediated regulatory T cell antagonism in non-targeted tumors.
INTRODUCTION
Cytotoxic T lymphocytes (CTLs) play a central role in the adaptive antitumor immune response. CTLs enable specific destruction of tumor cells upon T cell receptor recognition of tumor antigens displayed by MHC class I.1,2 In patients with immunogenic tumors, in which there is an endogenous antitumor CTL response, immune-checkpoint inhibitors (e.g., anti-PD-1, anti-PD-L1, and anti-CTLA-4) may reverse mechanisms of tumor immune evasion, enabling complete, durable control of metastatic disease.3,4 However, poorly immunogenic tumors may not be effectively recognized by a patient’s adaptive immune system, rendering immune-checkpoint inhibition ineffective.5 While some immunologically “cold” tumors may have few mutation-associated neoantigens, rendering any adaptive immune recognition unlikely, others arise from potentially reversible phenotypic characteristics of the tumor cells or tumor microenvironment, which limit tumor immune infiltration, antigen presentation, MHC class I expression, or T cell activation.6,7 In situ vaccination is a therapeutic strategy that may overcome such phenotypic challenges by converting a poorly immunogenic tumor into a site for enhanced antigen cross-presentation, thereby enabling development of endogenous tumor-specific CTL immunity.8
Moderate-dose, hypofractionated radiation therapy can induce an in situ vaccine effect,9 activation of a type I interferon response, increased immune cell infiltration, MHC class I upregulation on radiated tumor cells, induction of immunogenic tumor cell death, and enhanced antigen cross-presentation by dendritic cells. Despite these effects, radiotherapy alone has not been demonstrated to boost clinical response rate to immune-checkpoint inhibition.10,11 One reason that radiation alone may exert a detectable in situ vaccine response but fail to mount an effective clinical antitumor response is that focal radiation may prime antitumor CTL immunity but not adequately sustain and propagate this to non-radiated tumor sites where CTL-suppression mechanisms remain intact.
Tumor-specific CTL immunity, once activated, is shaped by the propagating, sustaining, or inhibitory influences of tumor stroma, cytokines, and other immune lineages.12–14 Among those immune lineages, roles have been well described for helper and regulatory T cells (Tregs), B cells, and myeloid-derived suppressor cells,15 but the role(s) of natural killer (NK) cells in influencing tumor-specific CTL immunity has not been fully clarified. We hypothesized that a three-component therapy composed of radiotherapy followed by intratumoral injection of interleukin-2 (IL-2) and tumor-specific monoclonal antibody (mAb) into the radiated tumor site (3xTx) may augment the in situ vaccination by engaging NK cells in a manner that critically supports the development and propagation of tumor-specific CTL immunity while limiting the potential for resistance.
RESULTS
In situ vaccination with tumor-directed radiation and intratumoral hu14.18-IL2 induces a polyfunctional CD8+ T cell response in patients with melanoma
Tumor-specific mAb can enhance adaptive immunity by promoting antibody-dependent tumor antigen uptake and presentation and can stimulate antibody-dependent cell-mediated cytotoxicity (ADCC) by NK cells.16 Both NK cells and CTLs are potently activated by IL-2.17 We previously reported enhancement of the in situ vaccine effect of radiotherapy by combining radiotherapy that targeted a single tumor site with intratumoral injection of that site delivering hu14.18-IL2, an immunocytokine that results from genetic fusion of a tumor-specific mAb and IL-2.18 hu14.18-IL2 has an intact IgG linked to functional IL-2 and can induce ADCC via FcγR- and IL-2R-expressing immune cells.19,20 hu14.18 targets disialoganglioside D2 (GD2), a cell-surface antigen overexpressed on melanoma cells. Using GD2-expressing syngeneic murine melanoma models, the combination of radiotherapy and intratumoral hu14.18-IL2 led to regression of both targeted (GD2+) and non-targeted (GD2−) tumors distant from the treated site and improved survival.18 To begin evaluating this approach clinically, we initiated a phase I clinical trial in patients with metastatic melanoma (NCT03958383). Patients who had progressed following standard-of-care frontline treatment were treated with radiotherapy (8 Gy ×3) targeting a single tumor site followed by three daily intratumoral injections of this site with 1 mg/m2 hu14.18-IL2 starting 4 days after completion of radiotherapy (Figure 1A). A total of six patients were enrolled. However, due to a change in corporate ownership of hu14.18-IL2, accrual to this clinical trial was temporarily suspended after treatment of three patients with radiotherapy + intratumoral hu14.18-IL2. Clinical outcomes for these patients will be reported separately.
Figure 1. In situ vaccination with tumor-directed radiation and intratumoral hu14.18-IL2 induces a polyfunctional CD8+ T cell response in patients with melanoma.
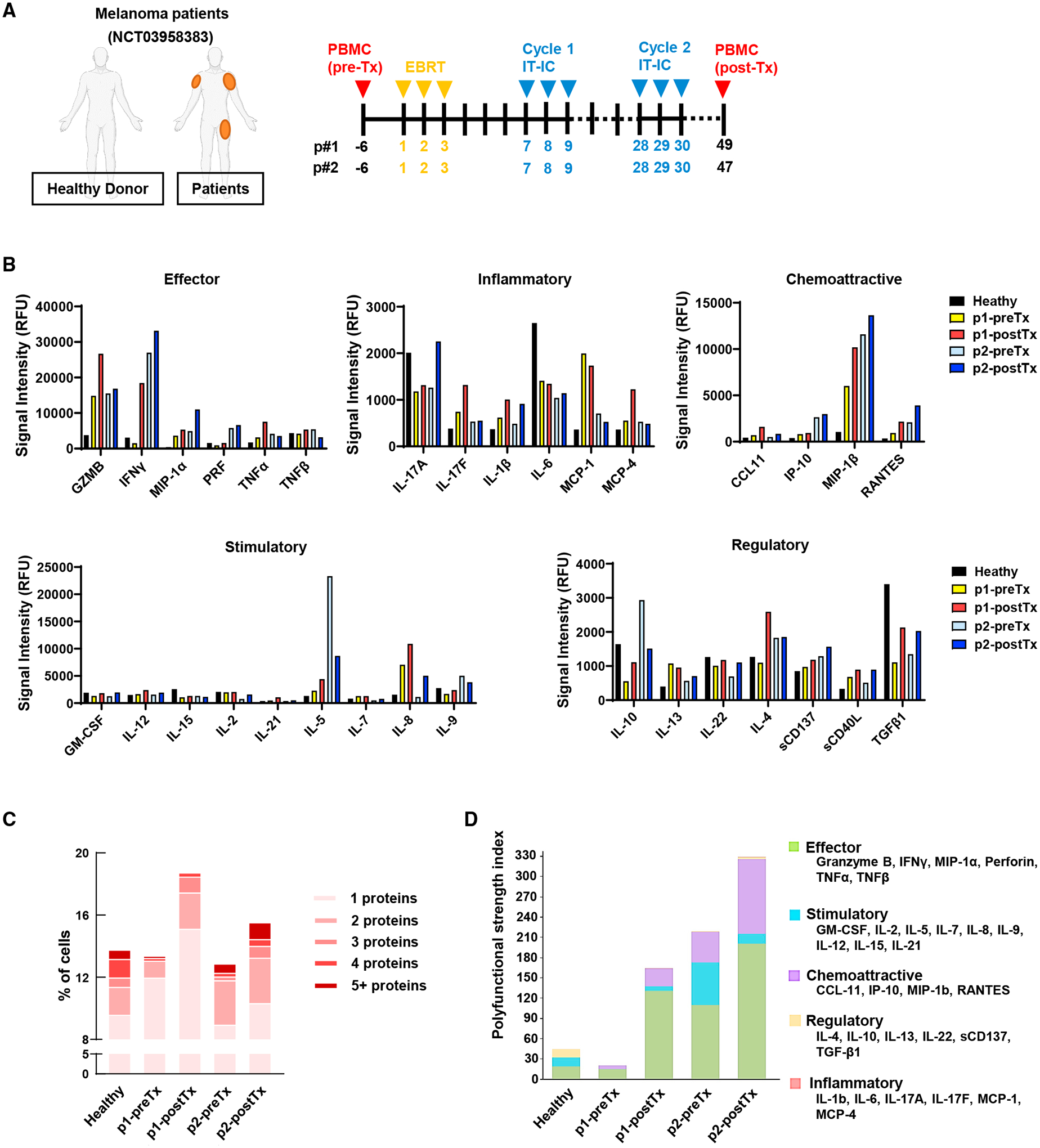
(A–D) Melanoma patients received local tumor radiation (EBRT) and intratumoral injection of that site with hu14.18-IL2 (IT-IC). Single-cell secretomes were obtained from CD8+ T cells isolated from PBMCs collected before (pre-Tx) or after (post-Tx) treatment or from a healthy donor. (A) Treatment regimen. (B) Relative fluorescence intensity (RFU) of individual proteins. (C) Multiple protein detection in single-cell secretomes. (D) Polyfunctional strength index (PSI) of CD8+ T cells.
To screen for treatment-induced phenotypic changes in CTLs, peripheral blood mononuclear cells (PBMCs) were collected from patients 6 days prior to radiotherapy (pre-Tx) and 18–20 days after the second hu14.18-IL2 injection cycle was completed (cycle 2, post-Tx) as indicated in Figure 1A. While these paired samples were available for just two patients because of the pause in the clinical trial, we sought to use these samples to screen for potential changes to CD8+ T cells that might suggest clinical effect in human patients to be further pursued in our murine models. A prior clinical trial evaluating an IL-2-based therapy in patients with metastatic melanoma (NCT02983045) demonstrated a strong and statistically significant correlation between treatment-induced expansion of polyfunctional CD8+ T cells and progression-free survival.21 Using the same approach, we evaluated proteomic changes in CD8+ T cells from pre-Tx and post-Tx PBMCs in patients treated with radiotherapy and intratumoral hu14.18-IL2 using a PhenomeX IsoCode single-cell cytokine secretome chip. The expression of effector proteins (GZMB and IFN-γ) was detected most strongly in CD8+ T cells from post-Tx PBMCs (Figures 1B and S1A). The expression of inflammatory (IL-17A, IL-17F, and IL-1β), chemoattractive (MIP-1β and RANTES), and stimulatory proteins (GM-CSF, IL-12, and IL-8) showed a similar increase. Expression of regulatory/inhibitory proteins (IL-22, IL-4, sCD137, sCD40L, and TGF-β) also increased following treatment, but their signal strength was much lower than the effector signal intensity.
Polyfunctional CD8+ T cells that express multiple cytokines in single-cell secretion analyses have been associated with favorable immunotherapy response in metastatic melanoma patients.21 We observed increased single- or multiple-cytokine co-production in CD8+ T cells from post-Tx PBMCs compared with pre-Tx specimens and compared with healthy donor specimens (Figures 1C and S1B). Polyfunctional activation topology principal-component analysis (PCA) plots revealed a comparable secretome grouping between the healthy donor and the pre-TX patients and that post-Tx CD8+ T cells had increased cytokine secretion frequency (circle size) compared with pre-Tx (Figure S1C). Polyfunctional strength index (PSI) analysis indicated that post-Tx CD8+ T cells had an increase in effector and chemoattractive PSI compared with pre-Tx (Figure 1D). Although the available patient numbers were small, these results suggest that delivering an in situ vaccination with radiotherapy and intratumoral hu14.18-IL2 to a single tumor site in patients with metastatic melanoma has the potential to activate phenotypic changes in the peripherally circulating population of CD8+ T cells. We further evaluated this finding mechanistically using murine models.
Durable tumor control with tumor-specific mAb and intratumoral injection of IL-2 in radiated murine tumors
We evaluated a more universal, “off-the-shelf,” and readily translatable approach to optimizing the in situ vaccine effect of radiotherapy than antibody-cytokine fusions such as hu14.18-IL2. Tumor-specific mAbs are widely available for most cancers, and the co-injection of these with a cytokine such as IL-2 could have broad clinical applications. Thus, we investigated a three-component regimen (3xTx) combining focal radiotherapy to a single tumor site and intratumoral injection of this site with IL-2 and a tumor-specific mAb at the same time. To begin, we evaluated the efficacy of this 3xTx regimen in mice bearing a poorly immunogenic, syngeneic B78 melanoma tumor engrafted 3 weeks prior to treatment. For this GD2+ melanoma model, we used hu14.18 as the tumor-specific mAb component of 3xTx (Figure 2A). The 3xTx regimen resulted in tumor regression and improved survival compared with control groups receiving single-agent treatment or dual treatment combinations (Figures 2B–2E). Forty-four percent of 3xTx-treated mice were rendered disease free (Figure 2F), and all of these demonstrated evidence of immune memory, as re-engrafted B78 tumor cells were rejected by all treated mice that had previously shown a complete response (tumor free), compared with 100% tumor development in tumor-naive mice (Figure 2G).
Figure 2. Durable tumor control with tumor-specific mAb and intratumoral injection of IL-2 in radiated murine tumors.
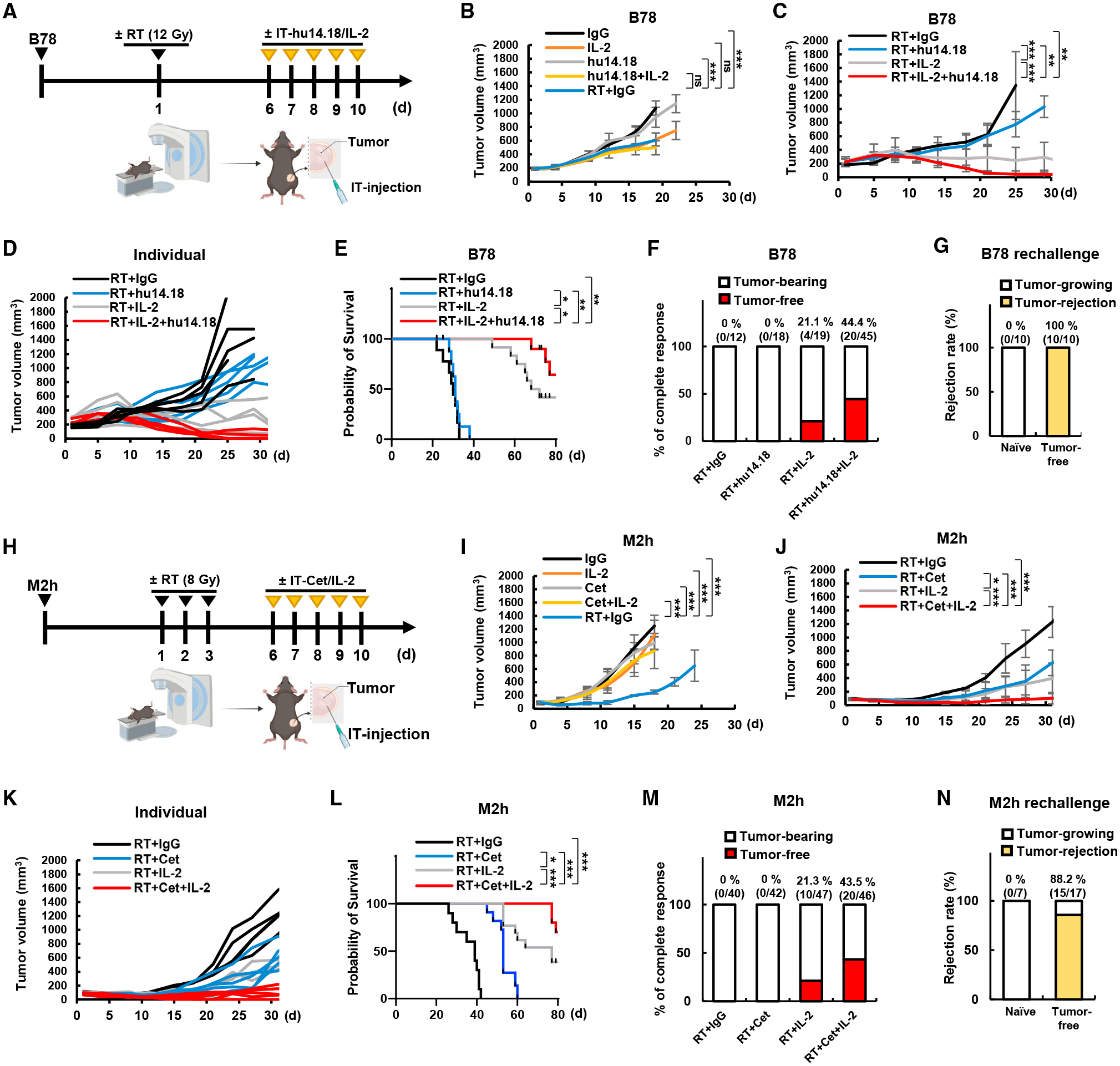
(A–F) Mice were engrafted with B78 cells in the right flank and then treated when mean tumor volume reached 170–230 mm3. (A) Treatment regimen. (B and C) Mean tumor volume and (D) individual tumor volume (n = 6). (E) Survival data (n = 8) and (F) accumulated complete response data (n = 12–45).
(G) Rejection rate of B78 tumor injection on naive mice or mice cured by 3xTx (n = 10).
(H–M) Mice were engrafted with M2h cells in the right flank and then treated when mean tumor volume reached 80–100 mm3. (H) Treatment regimen. (I and J) Mean tumor volume and (K) individual tumor volume (n = 5). (L) Survival data (n = 10) and (M) accumulated complete response data (n = 40–47).
(N) Rejection rate of M2h tumor injection on naive mice or mice cured by 3xTx (n = 7–17). Tumor growth curves represent the mean value and error bars indicate SD. ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
To test the generalizability of these observations, we used a poorly immunogenic murine model of head and neck squamous cell carcinoma (HNSCC), MOC2. We previously reported on the development of a huEGFR-expressing variant of this model (M2h), which can be targeted by the anti-human EGFR antibody cetuximab.22 Because cetuximab targets human EGFR but not murine EGFR, this model allows for evaluation of the immune-mediated effects of cetuximab bound to the M2h cell surface in the absence of confounding effects on the EGFR signaling blockade or radiosensitization. Using therapy dosing and timing optimized in our prior studies with M2h, we treated syngeneic C57BL/6 mice bearing a single flank M2h tumor with local radiotherapy (8 Gy ×3) and intratumoral IL-2 and cetuximab (Figure 2H). Mice receiving the 3xTx regimen exhibited durable tumor regression and improved survival compared with single or dual combination treatment regimens (Figures 2I–2L). As in the B78 model, 3xTx rendered 43% of M2h-bearing mice disease free (Figure 2M), and 88% of these mice exhibited evidence of immune memory (Figure 2N).
3xTx activates CD8+ T cell immunity to achieve an in situ vaccine effect
We next evaluated whether 3xTx activated an adaptive, systemic CTL response. In the spontaneously metastatic M2h model (Figures 3A and 3B), progression at the primary site in mice receiving IL-2 alone led to death between day 20 and day 25, before manifesting visible metastases (Figure 3B). Radiotherapy + IgG to the flank tumor provided sufficient survival time to develop grossly visible metastatic disease (Figure 3A). Among mice treated with radiotherapy + IgG, 86% developed lung metastases, whereas no lung metastases were observed in mice treated with 3xTx (Figure 3B). Consistent with a non-toxic induction of the antitumor response, 3xTx mice exhibited stable to increasing body weight, whereas mice receiving radiotherapy + IgG lost weight, likely due to disease progression (Figure 3C).
Figure 3. 3xTx activates CD8+ T cell immunity to achieve an in situ vaccine effect.
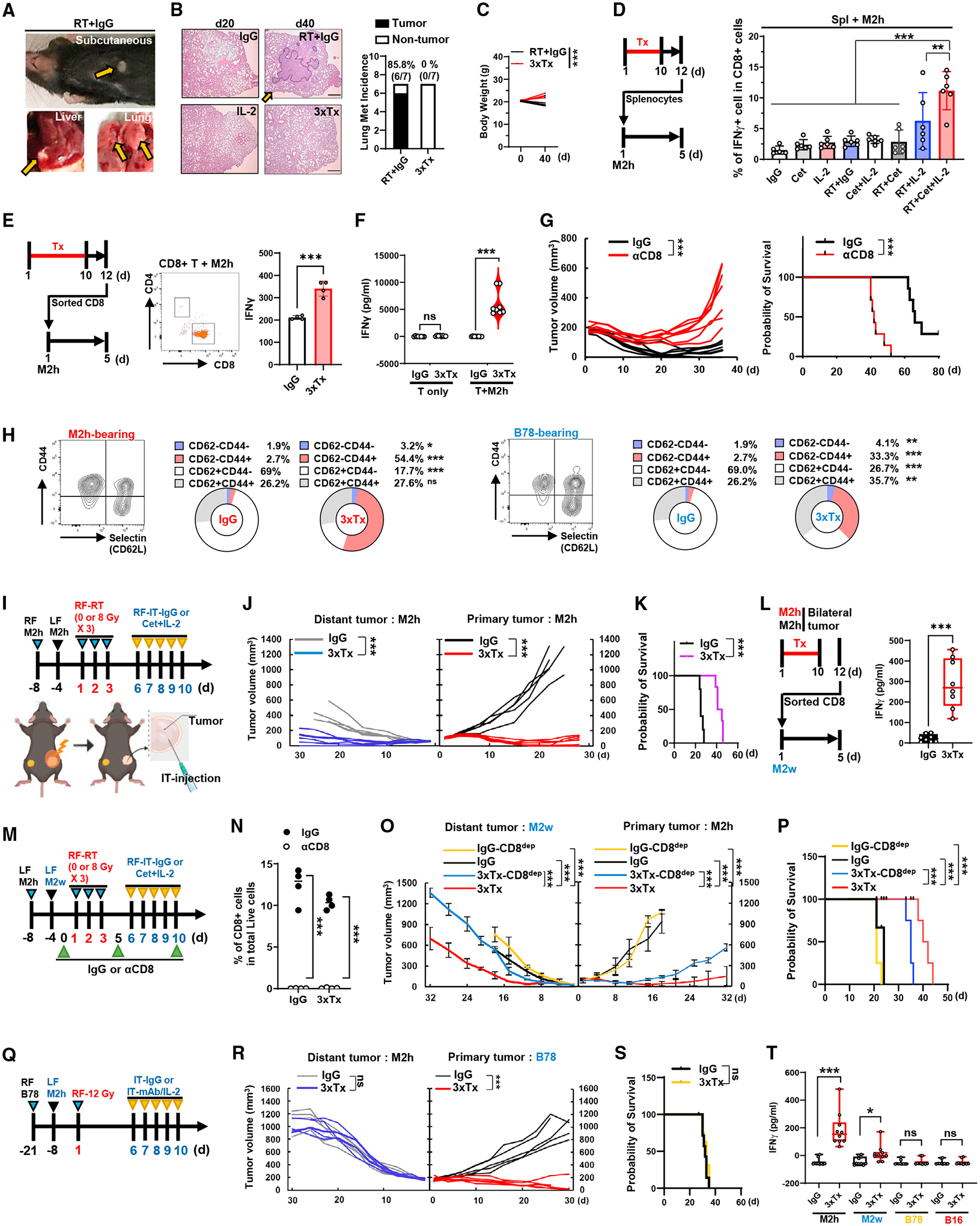
(A and B) Mice with an M2h flank tumor were treated as in Figure 2H. (A) Visible tumor metastases at day 40. (B) H&E staining of lung tissue (left) and quantification of lung metastases (right). Scale bar, 200 μm (n = 7). The orange arrows indicate tumors.
(C) Total body weight (n = 5).
(D–F) Spleens or spleen-sorted CD8+ T cells from M2h-bearing mice were collected and co-cultured with M2h for 5 days. (D) Quantification of IFN-γ+ cells in splenic CD8+ cells (n = 5–6). (E) Analysis of IFN-γ mean fluorescence intensity (MFI) in sorted CD8+ cells (n = 4). (F) Secreted IFN-γ from sorted CD8+ cells co-cultured with or without M2h (n = 4–8).
(G) Individual tumor volume (left) and survival data (right) from 3xTx-treated M2h-bearing mice that received IgG or anti-CD8a antibody (n = 7).
(H) CD8+ T cell subset in PBMCs collected from M2h- or B78-bearing mice after treatment (n = 3).
(I–L) Bilateral-tumor-bearing mice received 3xTx or IgG on the primary tumor. (I) Treatment regimen. (J) Individual tumor volume and (K) survival data (n = 5). (L) Secreted IFN-γ from co-cultured spleen-sorted CD8+ T cells and M2w (n = 8).
(M–P) Bilateral-tumor-bearing mice (M2h and M2w) received IgG or 3xTx on the primary tumor with IgG or anti-CD8a antibody (CD8dep). (M) Treatment regimen. (N) CD8+ T cell depletion efficacy in PBMCs (day 7). (O) Mean tumor volume and (P) survival data (n = 4).
(Q–T) Bilateral-tumor-bearing mice (B78 and M2h) received IgG or 3xTx on the primary tumor. (Q) Treatment regimen. (R) Individual tumor volume and (S) survival data (n = 5–7). (T) Secreted IFN-γ after co-culture of spleen-sorted CD8+ cells and tumor cells (n = 10). Data represent the mean value. Error bars indicate SD. ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
To assess systemic T cell immunity induced by 3xTx in M2h-tumor-bearing mice, splenocytes isolated on day 12 after initiation of treatment (and 2 days after the last treatment) were co-cultured for 5 days with pre-plated M2h cells (Figure 3D, left). CD8+ T cells from splenocytes of 3xTx-treated mice showed a greater frequency of IFN-γ+ cells upon co-culture with M2h cells compared with those from splenocytes of single- or dual-agent-treated mice (Figure 3D, right, and S2A). To clarify the IFN-γ induction by CD8+ T cells, sorted CD8+ T cells isolated from splenocytes were co-cultured with M2h cells (Figure 3E, left). CD8+ T cells from 3xTx-treated mice produced IFN-γ upon co-culture with M2h cells (Figure 3E, right). 3xTx did not induce CD8+ splenocyte IFN-γ production when not co-cultured with M2h cells; thus, this reflected not a baseline change, but a 3xTx-induced CD8+ T cell response to M2h cells (Figure 3F). In M2h-bearing mice, CD8+ T cell depletion resulted in reduced tumor regression and survival compared with 3xTx in mice that were not depleted of CD8+ T cells, confirming the role of CD8+ T cells in the therapeutic response (Figure 3G).
We isolated PBMCs from mice bearing either B78 or M2h tumors and treated with 3xTx (using the appropriate tumor-specific mAb for the respective tumor model). This treatment resulted in increased relative proportions of peripheral effector/memory T cells (L-selectin/CD62− CD44+) among CD8+ T cells (Figures 3H and S2B). We further investigated whether host IFN-γ expression was required for the efficacy of 3xTx. M2h tumor-bearing mice lacking the IFN-γ gene (IFNgtm1Ts) responded to 3xTx with reduced tumor regression and survival compared with wild-type (WT) mice (Figures S2C and S2D). These results demonstrate that a systemic tumor-reactive CTL response is generated in mice following 3xTx, consistent with an in situ vaccine effect.
Given that 3xTx targeting a single site elicited a systemic CTL response, we tested whether this therapy would also convey immunity against established distant tumor sites present at the time of 3xTx administration but not directly targeted by radiotherapy or intratumoral injection. To model established metastases present at a distant site, we generated a bilateral, two-tumor model, engrafting a “primary” M2h tumor on the right flank 8 days prior to treatment and then a second “distant” M2h tumor on the left flank 4 days prior to treatment (Figure 3I). Engrafting at different times was performed to confirm selection of mice that had not developed an endogenous antitumor immune response following primary tumor engraftment (13% of mice were not randomized to any treatment group due to failure to develop a distant tumor). In this bilateral tumor model, 3xTx targeting the primary tumor improved tumor response at both the primary and the distant sites (Figure 3J) and thus significantly improved overall survival (Figure 3K). Sorted splenic CD8+ T cells from 3xTx treatment of mice bearing an M2h tumor increased IFN-γ release upon co-culture with MOC2 WT (M2w) cells (Figure 3L), indicating that 3xTx-induced immunity was not solely dependent on huEGFR expression.
We investigated the efficacy of each treatment component of 3xTx in a bilateral-tumor-bearing mouse model (Figure S2E, left) and found that 3xTx was the most effective in reducing targeted tumor and non-targeted tumor growth (Figure S2E, right). 3xTx produced the highest increase in CD8+ T cell infiltration, and the ratio of effector memory T cells significantly increased in both radiation therapy (RT) + IL-2 and 3xTx treatment groups (Figures S2F and S2G). We hypothesized that the higher CD8+ T cell infiltration with increased effector/memory T cells in 3xTx-treated tumors compared with RT + IL-2 resulted from cetuximab treatment, as stimulation of other immune cells by a tumor-specific antibody may in turn further stimulate CD8+ T cells. Tumor infiltration by NK cells was unchanged after 3xTx, but compared with RT + IL-2, 3xTx significantly increased the mean fluorescence intensity (MFI) of IFN-γ and TNF-α among NK cells (Figure S2H). As IFN-γ plays a critical role in T cell proliferation, activation, and tumor antigen expression,23–26 tumor-specific antibody-mediated activation of Fc-receptor-positive cells has the potential to increase CD8+ T cell infiltration and memory impact.27,28 Interestingly, along with NK cells (CD3− NK1.1+), 3xTx-treated tumors harbor CD4+ T cells (CD90+CD3+CD4+), CD8+ T cells (CD90+CD3+CD8+), Tregs (CD4+CD25+FOXP3+), B cells(B220+MHCII+Siglec H−), neutrophils (CD11b+Ly6G+ and CD11b+Ly6G+CD64+), eosinophils (CD11b+Si-flec F+), monocytes (CD11b+CCR2+Ly6C+), classical type 1 dendritic cells (DCs; cDC1; CD11b+CD11c+MHCII+XCR1+), classical type 2 DCs (cDC2; CD11b+CD11c+MHCII+CD172a+), and plasma-cytoid DCs (pDC; CD11b+CD11c+B220+Siglec H+). However, macrophages (CD11b+F480+CD64+) were not detected (Figures S2I–S2L).29–34 Taken together, these results suggest that the in situ vaccine effect of 3xTx stimulates a complex antitumor immune response. We evaluated the immunologic mechanisms underlying the effectiveness of 3xTx in non-targeted distant tumors. To do this, we used our M2h/M2w two-tumor model to test CD8+ T cell response during local primary tumor treatment (Figures 3M–3P). 3xTx targeting the M2h primary tumor resulted in antitumor response at both tumor sites and improved survival (Figures 3O and 3P). Depletion of CD8+ T cells reduced tumor response at the radiated primary M2h tumor and eliminated response at the distant M2w tumor, resulting in reduced survival compared with 3xTx in mice that were not depleted of CD8+ T cells (Figures 3N–3P).
To further evaluate the specificity of the antitumor immunity generated by 3xTx, we engrafted mice with a right-flank B78 tumor and a left-flank M2h tumor (Figures 3Q–3T). Because B78 grows relatively slowly compared with M2h, B78 was engrafted 2 weeks prior to M2h to allow tumor growth evaluation. In this setting, 3xTx targeting the right-flank B78 tumor (12 Gy/intratumoral IL-2 + hu14.18) showed a lack of antitumor response at the distant M2h tumor site, resulting in no survival benefit despite a significant response at the targeted B78 tumor site (Figures 3R and 3S). Reciprocally, in this same model, 3xTx targeting M2h (8 Gy ×3/intratumoral IL-2 + cetuximab) had no effect on the growth of a non-targeted B78 tumor (Figures S2M and S2N), resulting in loss of survival benefit compared with M2h-M2w bilateral-tumor-bearing mice (Figure S2O). As in M2h-M2h bilateral-tumor-bearing mice treated with 3xTx, using splenocytes from M2h-B78 bilateral-tumor-bearing mice treated at the M2h tumor with 3xTx, we observed CD8+ T cell production of IFN-γ following co-culture with M2w or M2h cells but not upon co-culture with B78 cells or their parental B16 melanoma cells (Figure 3T). These observations indicate that 3xTx leads to tumor-specific T cell immunity in both models, shown to be CD8 mediated for M2h, resulting in a durable, systemic antitumor response.
Tregs and PD-L1/PD-1 are negative regulators of distant-tumor response after primary-tumor 3xTx
Given the clinical use of intravenous (i.v.) delivery for both IL-2 and tumor-specific antibodies such as cetuximab, we investigated the treatment efficacy of i.v.-3xTx in bilateral-tumor-bearing mice. We found that i.v.-3xTx reduced tumor growth in the primary but not the distant tumor (Figures S3A and S3B). Despite the lack of distant-tumor response, i.v.-3xTx still provided significantly improved survival benefit (Figures S3A and S3B). In addition, i.v.-3xTx showed increased CD4 T cells, CD8 T cells, NK cells, effector memory CD8 T cells, and Treg cells in the distant tumor compared with i.v.-IgG (Figure S3C). This suggests that i.v.-3xTx can elicit potentially clinically beneficial antitumor responses, although toxicity of systemic IL-2 may limit this approach.
To assess mechanisms of response and resistance to the systemic antitumor immune response following 3xTx, we performed bulk RNA sequencing (RNA-seq) of distant tumors in the bilateral M2h/M2w tumor model (Figure 4A). Compared with sham/IgG-treated mice, 3xTx mice showed increased expression of 1,432 genes and decreased expression of 1,679 genes and developed distinct PCA profiles (Figures 4B and 4C). Gene set enrichment analysis comparing 3xTx with sham/IgG gene expression from distant tumors showed significant positive enrichment of “natural killer cell-mediated cytotoxicity” and “T cell receptor signaling” KEGG pathways (Figure 4D). Consistent with KEGG pathway analysis, Gene Ontology (GO) analysis indicated a significantly upregulated immune response, including inflammatory response, IFN-γ signaling, and response to IFN-γ-related genes in distant tumors following 3xTx treatment (Figures 4E–4G).
Figure 4. Treg and PD-L1/PD-1 are negative regulators of distant-tumor response after primary-tumor 3xTx.
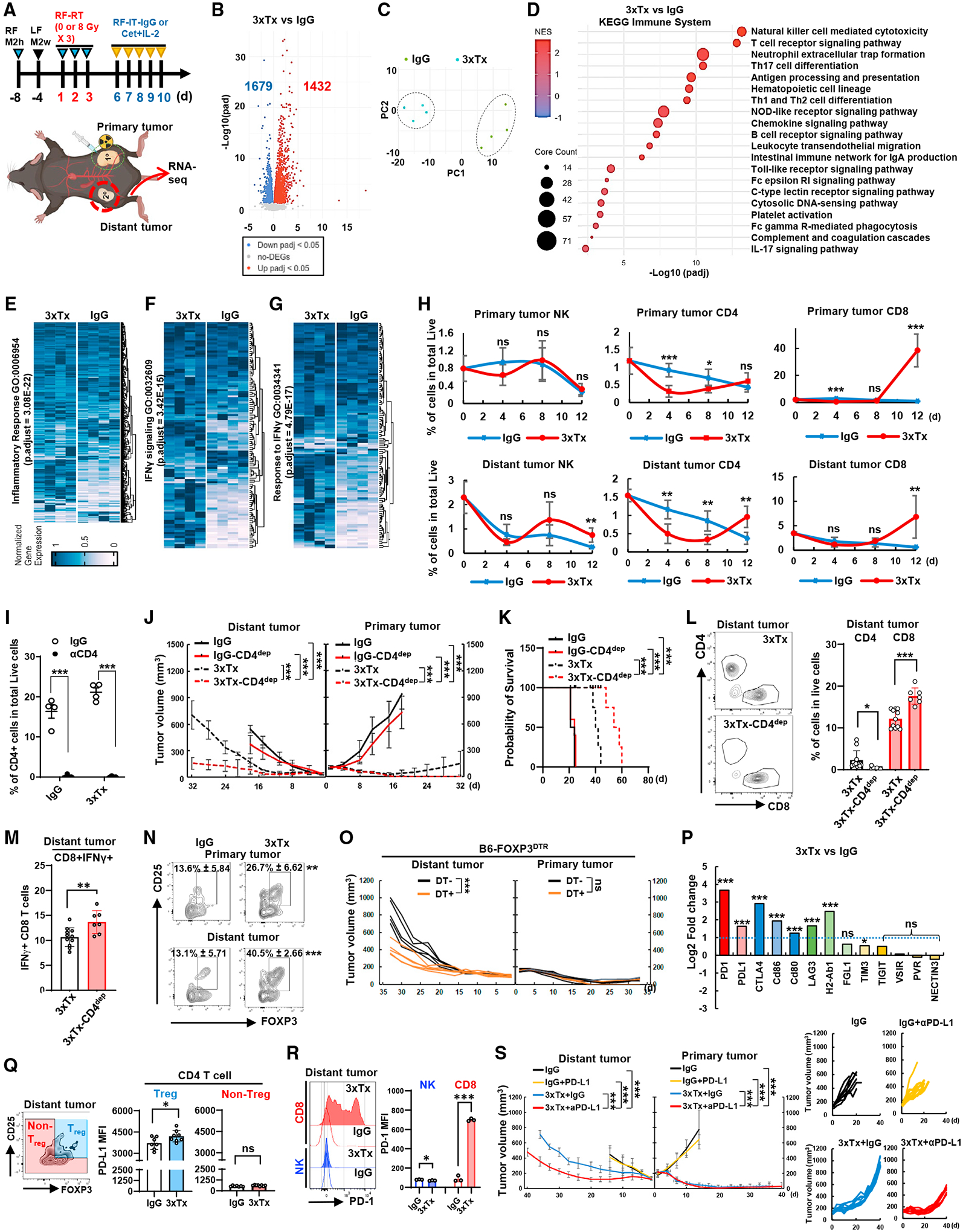
(A–G) Bilateral-tumor-bearing mice (M2h-RF and M2w-LF) received IgG or 3xTx on the primary tumor (n = 4). (A) M2w distant tumors were dissected on day 12 and subjected to RNA-seq. (B) Volcano plot of gene expression change. DEGs, differentially expressed genes; padj, adjusted p value. (C) PCA plot. (D) KEGG pathway analysis.
(E–G) GO analysis of inflammatory response, IFN-γ signaling, and response to IFN-γ.
(H) Time course of tumor infiltration of NK1.1+, CD4+, or CD8+ T cells (n = 5–6).
(I–N) Bilateral-tumor-bearing mice (M2h-RF and M2w-LF) received IgG or 3xTx on the primary tumor. During treatment, IgG (3xTx) or anti-CD4 antibody (3xTx-CD4dep) was injected (n = 4). (I) CD4+ T cell depletion efficacy in PBMCs (day 7). (J) Mean tumor volume and (K) survival data. (L and M) Analysis of CD4+ and CD8+ T cell infiltration on day 12 and IFN-γ+CD8+ T cells (n = 6–11). (N) Analysis of regulatory T cell (Treg; CD4+CD25+FOXP3+) infiltration on day 12 (n = 6).
(O) Individual tumor volumes from the bilateral-tumor (M2h-RF and M2w-LF)-bearing Foxp3DTR knockin mice treated with 3xTx on the primary tumor with or without diphtheria toxin (DT) injection (n = 5).
(P) Expression of immune-checkpoint receptor/ligand pooled from RNA-seq.
(Q) Analysis of PD-L1 expression in Treg or non-Treg (n = 7–8).
(R) Analysis of PD-1 expression in NK or CD8+ T cells (n = 3).
(S) Mean tumor volume (left) and individual tumor volumes (right) from 3xTx on the primary tumor with IgG or anti-PD-L1 antibody injection in bilateral-tumor (M2h-RF and M2w-LF)-bearing mice. Data represent the mean value. Error bars indicate SD. ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Given the bulk RNA-seq data showing evidence of NK cell and T cell immune responses in distant tumors following 3xTx, we evaluated lymphocyte infiltration (Figures 4H and S3D). On day 4, just after completion of radiation, we observed no significant change in primary tumor infiltration by NK1.1+ cells and a decrease in both CD4+ and CD8+ T cells. At this same time in the distant tumor, CD4+ T cells decreased, with no change in the levels of NK1.1+ or CD8+ T cells compared with specimens from mice treated with sham radiotherapy and non-specific control IgG injections. On day 8 of treatment, compared with control-treated mice, CD4+ T cells decreased in both the primary and the distant tumor in the 3xTx group, whereas no changes were observed in primary or distant tumor infiltration by NK1.1+ or CD8+ T cells. On day 12 after 3xTx, NK1.1+, CD4+, and CD8+ cell numbers were increased in the distant tumor compared with control-treated mice.
We evaluated the effect of CD4+ T cells on 3xTx using CD4+ T cell depletion (Figure 4I). Compared with non-CD4+-depleted mice, CD4+ T cell depletion improved tumor growth suppression and survival after 3xTx (Figures 4J and 4K). Notably, CD4+ T cell depletion significantly increased CD8+ T cell infiltration in the distant tumor and increased the proportion of these cells expressing IFN-γ (Figures 4L and 4M). These results support the hypothesis that tumor-infiltrating CD4+ T cells have a regulatory role antagonizing CD8+ T cells.35 In fact, following 3xTx, the proportion of Treg (CD25+/FOXP3+) increased among tumor-infiltrating CD4+ cells in both the primary and the distant tumors on day 12 compared with control-treated mice (Figure 4N).
To assess factors enhancing T cell migration to distant tumors, we evaluated chemokine genes at the distant tumor by RNA-seq. CXCL10, CXCL11, CCL5, CCL8, and CXCL9 were significantly upregulated (Figure S3E). In addition, we observed upregulation of CXCR3, a receptor of CXCL9, CXCL10, and CXCL11, in the distant tumor and on the surface of circulating Tregs and CD8+ T cells following 3xTx (Figure S3F).
To determine whether FOXP3-expressing cells suppressed the response to 3xTx, we used FOXP3DTR knockin mice bearing bilateral right-flank M2h and left-flank M2w. In this model, which allows selective depletion of FOXP3+ cells upon injection of diphtheria toxin (DT), distant-tumor response improved with 3xTx following Treg depletion (Figure 4O). Given that Treg express co-inhibitory molecules that can suppress CTL functions,36 we evaluated immune-checkpoint ligand/receptor genes from distant-tumor RNA-seq data from day 12 tumors. PD-1, PD-L1, CD86, CD80, LAG3, and H2-Ab1 were significantly upregulated (Figure 4P).37 We focused on the most increased, PD-1 and its ligand PD-L1, and confirmed increased PD-L1 expression on Treg compared with non-Treg in the distant tumor following 3xTx (Figure 4Q).
Interestingly, upon 3xTx, PD-1 expression increased markedly on CD8+ T cells in the distant tumor; however, PD-1 expression on NK1.1+ cells decreased significantly at the same time (Figure 4R). To determine whether PD-1/PD-L1 engagement was limiting antitumor response following 3xTx, we combined intraperitoneal injection of anti-PD-L1 blocking antibody or control IgG with 3xTx in the bilateral M2h-M2w tumor-bearing murine model. Combination of 3xTx with anti-PD-L1 significantly reduced tumor growth in the distant tumor (Figure 4S). Because Tregs constitutively express CTLA4, we next investigated anti-CTLA4 antibody efficacy with anti-PDL1 therapy following 3xTx (Figure S3G). While both 3xTx + anti-PDL1 and 3xTx + anti-CTLA4 provided strong antitumor response in the distant tumor, the addition of both anti-CTLA4 and anti-PD-L1 together to the 3xTx therapy regimen showed even greater antitumor response in the distant tumor compared with either 3xTx + anti-PDL1 or 3xTx + anti-CTLA4 (Figure S3G).
NK cells are necessary for distant antitumor response mediated by CD8+ T cells following in situ vaccination
As NK cell infiltration is enhanced in distant tumors following 3xTx, we depleted NK cells in mice bearing bilateral tumors and treated with 3xTx targeting the primary M2h tumor to assess NK cell necessity for the distant-tumor response (Figure 5A). Based on our prior studies showing that radiotherapy enhances NK cell-mediated ADCC,22 we initially expected that NK cell depletion might limit antitumor response at the primary M2h site and possibly at a distant M2h tumor site. NK cell depletion (3xTxNKdep) antagonized the response to 3xTx at the distant tumor sites compared with 3xTx in non-depleted mice (Figures 5B–5E). Interestingly, this effect was observed in distant M2h and M2w tumors, independent of distant tumor expression of the antigen (huEGFR) targeted by the tumor-specific antibody in 3xTx. Consistently, 3xTxNKdep reduced survival in 3xTx-treated mice with either an M2h or an M2w distant tumor site (Figures 5D and 5E). In mice bearing an M2h primary tumor and treated with 3xTx, engraftment of an M2w distant tumor at day 12 after treatment initiation was rejected 80% of the time; however, 3xTxNKdep reduced this efficacy to just 10% (Figure 5F).
Figure 5. NK cells are necessary for distant antitumor response mediated by CD8+ T cells following in situ vaccination.
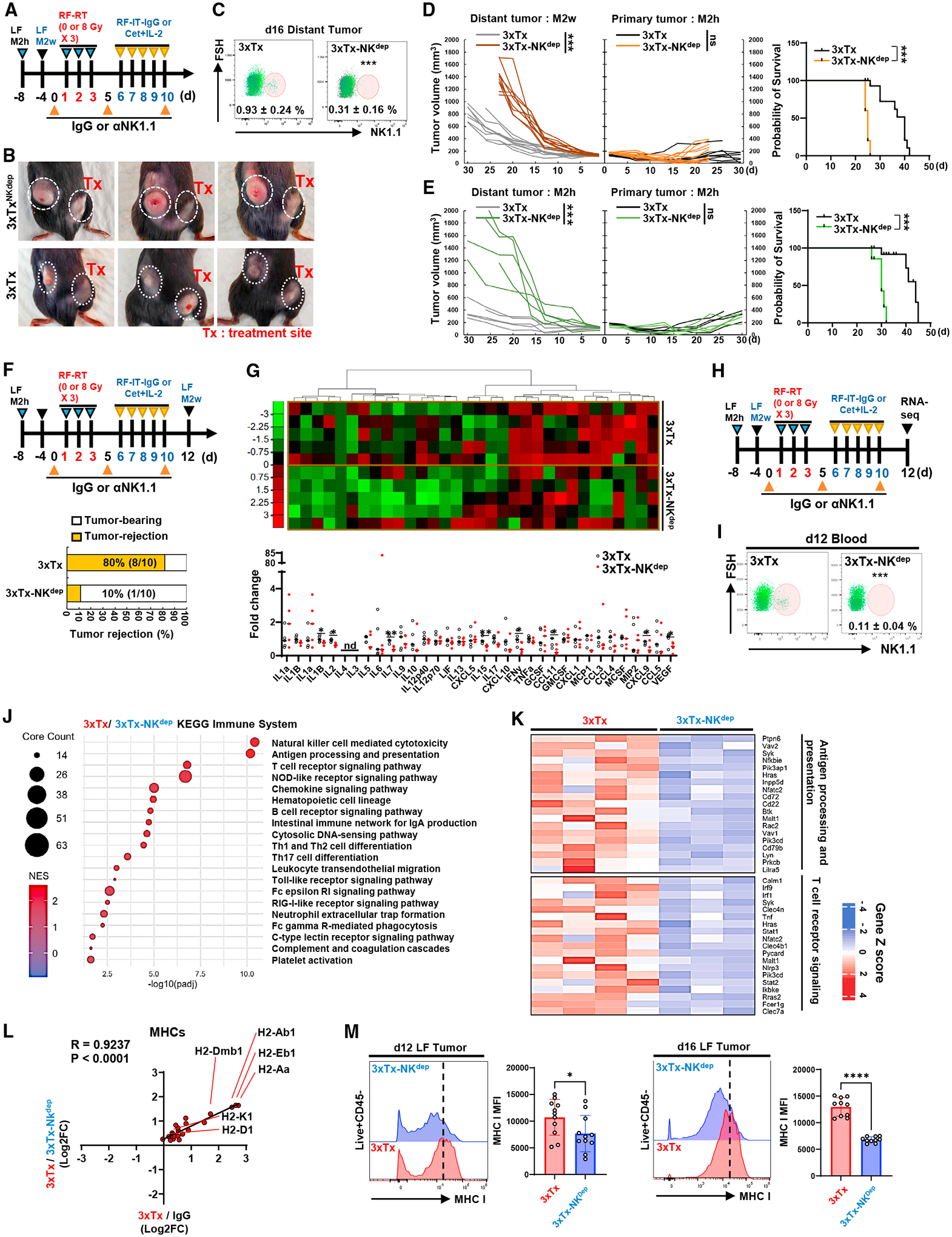
(A–E) Bilateral-tumor-bearing mice received 3xTx on the primary tumor. During treatment, IgG (3xTx) or anti-NK1.1 antibody (3xTx-NKdep) was injected intraperitoneally. (A) Treatment regimen. (B) Tumor-bearing mice. (C) NK cell depletion efficacy (n = 6–7). (D and E) Individual tumor volumes and survival data from bilateral-tumor-bearing mice (up, M2h-RF and M2w-LF; low, M2h-RF and M2h-LF) (n = 5 or 10).
(F) Tumor rejection rate 2 weeks after M2w injection into 3xTx-receiving M2h-bearing mice (n = 10).
(G) Analysis of cytokine/chemokine expression in the distant tumor (n = 5).
(H–K) RNA-seq was performed on distant tumors collected at day 12 after NK cell depletion during 3xTx on the primary tumor. (H) Treatment regimen. (I) NK cell depletion efficacy (n = 4). (J) KEGG pathway analysis. (K) GO analysis related to KEGG pathway (n = 3–4).
(L) Analysis of the correlation of MHC-related genes.
(M) Analysis of MHC class I MFI in live+CD45− cells from distant tumor (n = 10–11). Data represent the mean value. Error bars indicate SD. ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001; nd, not detected.
Multiplex cytokine analysis within the tumors demonstrated that NK depletion resulted in decreased production of T cell-activating or -recruiting cytokines IL-1β, IL-2, IL-7, IL-15, IFN-γ, CCL11, CXCL9, and VEGF (Figure 5G). To further evaluate NK cell-dependent effects in distant M2w tumors following 3xTx targeting a primary M2h tumor, we performed bulk RNA-seq on these distant tumors following 3xTxNKdep (Figure 5H) and confirmation of NK cell depletion at day 12 post-treatment initiation (Figure 5I). Gene set enrichment analysis of the KEGG pathway revealed that “T cell receptor signaling” and “antigen processing and presentation” pathways were the most positively enriched in the distant tumor following 3xTx compared with 3xTxNKdep (Figures 5J and 5K). Expression of genes related to MHC class I (H2-D1, H2-K1) and MHC class II (H2-Dmb1, -Aa, Eb1) was upregulated following 3xTx compared with IgG-control-treated and 3xTxNKdep-treated mice (Figure 5L). Consistently, 3xTxNKdep treatment in mice bearing bilateral M2h-M2w tumors showed reduced MHC class I expression on CD45− tumor and non-immune stroma cells at days 12 and 16 compared with distant tumors after 3xTx (Figures 5M and S4).
Depletion of NK cells reduces CD8+ T cell polyfunctionality
To clarify the roles of NK and T cell lineages, we sorted CD8+ T cells and NK cells from the spleens of mice bearing bilateral M2h-M2w tumors after 3xTx. These cells were intratumorally injected into M2w-bearing immunodeficient mice (NOD-scid IL2Rgnull). After 5 days, CD8+ T cells, but not NK cells, showed MHC class I induction in the tumor (Figure S5A). Next, we investigated whether NK cells were involved in CD8+ T cell immunity. We sorted CD8+ T cells from the spleens of 3xTx or 3xTxNKdep mice (Figures 6A and 6B) for co-culture with M2h or M2w cells. 3xTxNKdep slightly reduced the proportion of T effector/memory cells (Tems) upon co-culture with M2h or M2w (Figures S5B and S5C). IFN-γ and TNF-α induction in Tems compared with Tnaives from 3xTx mice increased upon co-culture, but 3xTxNKdep reduced this induction in both Tem and Tnaive cells (Figures 6C and 6D). Tem infiltration of distant tumors showed reduction on day 12, but not on day 16 (Figure S5D). Tumor-infiltrating Tem cells showed reduced IFN-γ - and TNF-α-positive cells without a change in LAG3-positive cells, an exhaustion marker (Figure 6E). NK cell depletion reduced the percentage of IFN-γ and TNF-α double-positive tumor-infiltrating T cells following 3xTx (Figure 6F). This suggests that NK cells may promote or sustain T cell effector functionality.
Figure 6. Depletion of NK cells reduces CD8+ T cell polyfunctionality.
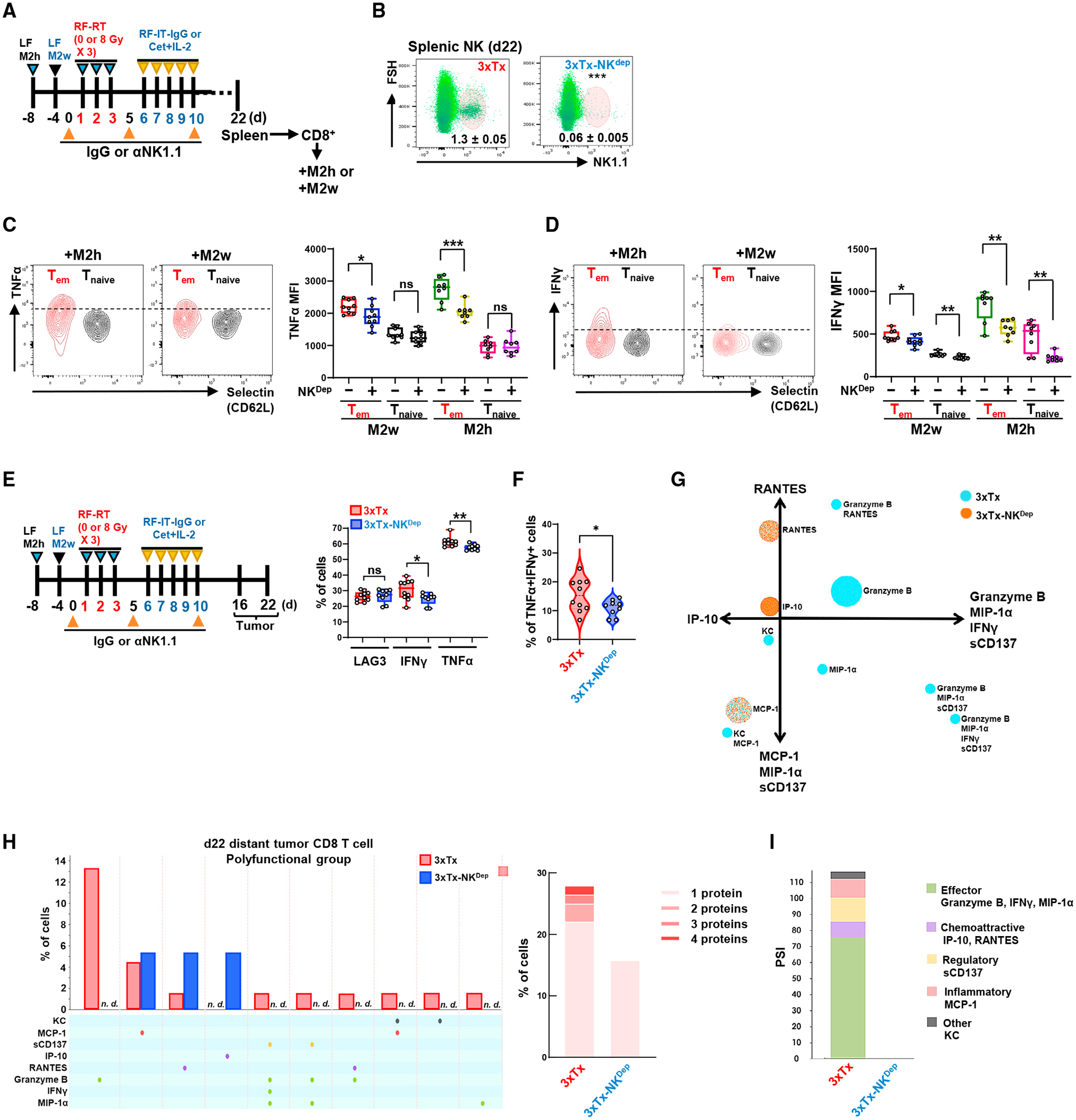
(A–D) Bilateral-tumor-bearing mice received 3xTx on the primary tumor. During treatment, IgG (3xTx) or anti-NK1.1 antibody (3xTx-NKdep) was injected intraperitoneally. On day 22, spleen-sorted CD8+ T cells were co-cultured with pre-plated M2w or M2h for 5 days. (A) Treatment regimen. (B) NK cell depletion efficacy (n = 6). (C) Analysis of TNF-α and (D) IFN-γ MFI in effector/memory CD8+ T cells (Tem; Selectin−CD44+) and naive CD8+ T cells (Tnaive; Selectin+CD44−) (n = 8–9). (E–I) Bilateral-tumor-bearing mice received 3xTx with IgG (3xTx) or anti-NK1.1 antibody (3xTx-NKdep). On day 16 or day 22, the distant tumor was collected and analyzed. (E) Analysis of LAG-, IFN-γ-, and TNF-α-positive or (F) IFN-γ and TNF-α double-positive cells of tumor-infiltrating Tem on day 16 (n = 9–13). (G) Polyfunctional activation topology PCA plot. (H) Single-cell polyfunctional group analysis (left) and multiple protein detection (right). (I) Polyfunctional strength index (PSI) of CD8+ T cells. Data represent the mean value. Error bars indicate SD. ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Using the same single-cell proteomics approach from our phase I clinical trial (Figure 1), we tested whether 3xTx activated a polyfunctional T cell response in the M2h-M2w bilateral-tumor-bearing mice and whether NK cells were necessary for this. Sorted CD8+ T cells from disaggregated distant M2w tumors (day 22) demonstrated the presence of multicytokine-secreting polyfunctional subsets of CD8+ T cells, and these were markedly reduced by NK cell depletion (Figure 6G). 3xTx consistently induced populations of CD8+ T cells that co-secreted cytokines that are predominantly associated with effector, stimulatory, or chemoattractive phenotypes, including Granzyme B, RANTES (CCL5), MIP-1α, and IFN-γ. Yet, inflammatory and regulatory cytokines, including keratinocyte-derived chemokine (KC), monocyte chemoattractant protein-1 (MCP-1), and sCD137, were also found on these polyfunctional cells, suggesting diverse functional roles during clonal expansion, including effector and memory response.38 In contrast, 3xTxNKdep-derived CD8+ T cell polyfunctionality was not detected (Figures 6H and 6I).
KLRK1-dependent induction of CD86 expression in NK cells triggers CD86+ NK cell-dependent apoptotic response in CTLA+ Tregs in the tumor microenvironment
Intriguingly, 3xTxNKdep increased the infiltration of CD4+ T cells in the non-targeted tumor (Figure 7A), as well as the proportion of Tregs among CD4+ T cells (Figure 7B), and these Tregs exhibited higher expression of CTLA4 compared with other CD4+ T cells (Figure 7C), suggesting that NK cells decreased Treg cells in the tumor.39 From bulk RNA-seq analysis (Figures 4A and 5H), we interrogated NK-related 3xTx-induced genes abrogated by 3xTxNKdep (3xTx-NKdep/3xTx) (Figure 7D). Among those 437 genes, we identified and focused on KLRK1 and CD86. Intriguingly, KLRK1 and CD86 mRNA expression was correlated in both melanoma and HNSCC patients (Figure 7E). Murine NK cells consistently increased CD86 expression in the presence of tumor cells, but KLRK1-knockout (KO) NK cells did not (Figure 7F). This was independent of NK cell expression of FcγR.
Figure 7. KLRK1-dependent induction of CD86 expression in NK cells triggers CD86+ NK cell-dependent apoptotic response in CTLA+ Tregs in the tumor microenvironment.
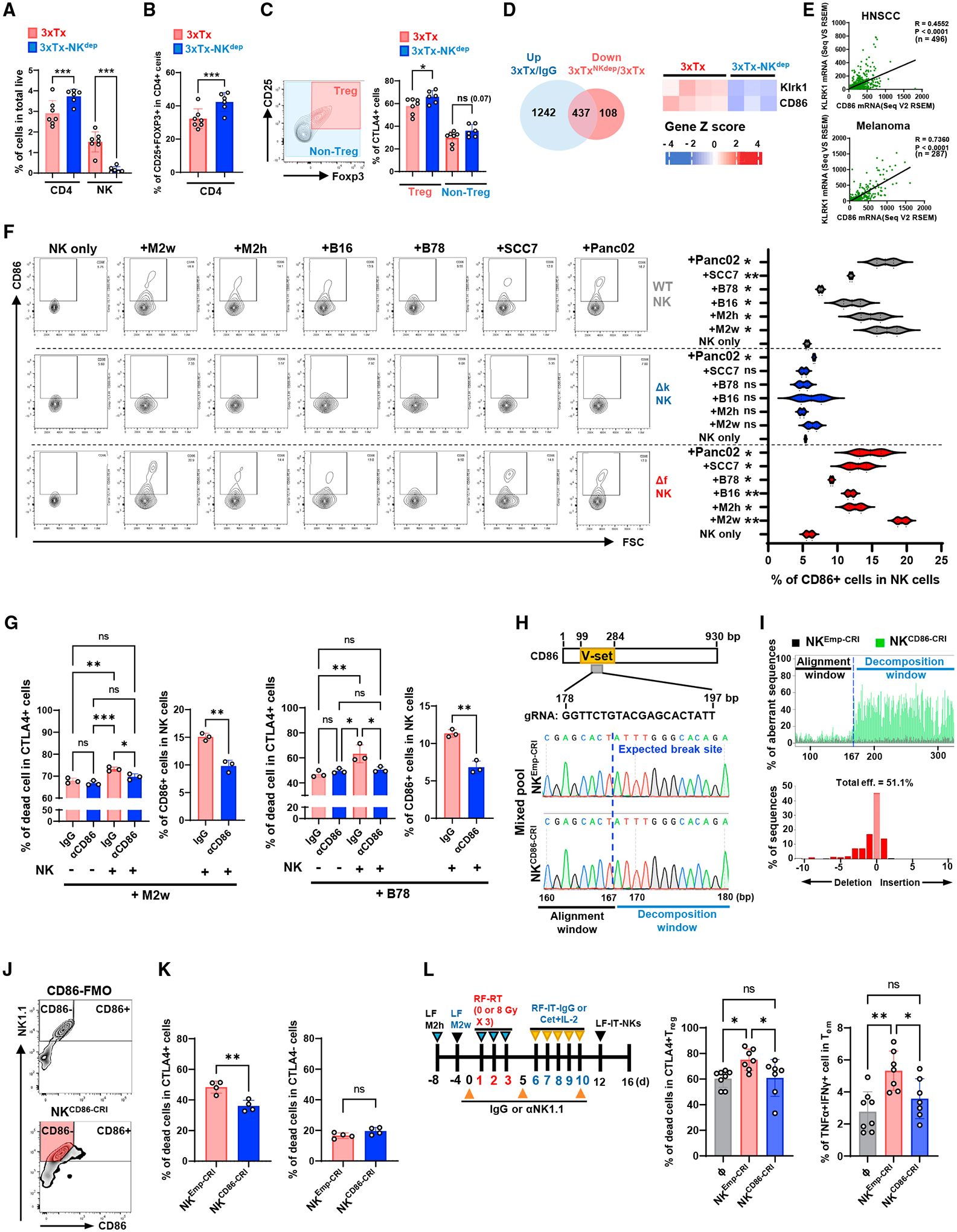
(A–C) Bilateral-tumor-bearing mice received 3xTx on the primary tumor. During treatment, IgG (3xTx) or anti-NK1.1 antibody (3xTx-NKdep) was injected intraperitoneally. Distant tumors were collected on day 12 and analyzed. Analysis of (A) CD4+, NK, (B) Treg (CD25+FOXP3+), and (C) CTLA4+ cells in Tregs (n = 6–7).
(D) Venn diagram (left) pooled from distant-tumor RNA-seq and a heatmap of Klrk1 and CD86 expression (right).
(E) Analysis of correlations between KLRK1 and CD86 mRNA expression in melanoma patients or HNSCC patients.
(F) CD86+ cell analysis in NK cells after tumor cell co-culture (n = 2).
(G) Analysis of dead cells in CD4+CTLA4+ cells after NK cell and tumor cell co-culture (n = 3).
(H–L) CRISPR-Cas9 genomic CD86-deleted NK cells (NKCD86-CRI) or control NK cells (NKEmpty-CRI) were generated. (H) Guide RNA targeting site (up; gRNA) and genomic DNA sequencing results (low). (I) CD86 depletion aberrant efficiency (eff). (J) Sorting strategy for CD86-deficient NK cells (red-colored area). (K) Analysis of dead cells in CD4+CTLA4+ cells after NK cell and tumor cell co-culture (n = 4). (L) Treatment regimen for NK cell administration after 3xTx on bilateral-tumor-bearing mice (left) and analysis of dead+CTLA4+ Tregs and TNF-α+IFN-γ+ double-positive Tems (n = 7–8). ⧞, PBS. Data represent the mean value. Error bars indicate SD. ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
To evaluate the potential for a direct cytotoxic effect of NK cells against Tregs, we generated Treg-enriched cells by IL-2 stimulation on sorted CD4+ T cells. IL-2 stimulation increased CTLA4 expression in Tregs only (Figure S6A). IL-2-stimulated, CFSE-labeled CD4+ T cells together with MOC2 tumor cells were cultured with either NK cells sorted from naive mouse spleen or PBS. In areas where tumor cells were present, CD4+ T cells increased expression of the apoptosis marker Annexin V in response to co-culture with NK cells (Figure S6B). Blocking the CD86-CTLA4 interaction using a CD86-neutralization antibody during co-culture reduced NK-mediated cytotoxicity of CTLA4-expressing CD4+ T cells in the presence of M2h or B78 (Figures 7G, S6C, and S6D). Interestingly, CTLA4-negative cells also showed a small but significant increase in cell death in the presence of NK cells and either M2w or B78 cells that was independent of CD86 (Figure S6D). Irradiation of tumor cells prior to co-culture increased the percentage of dead CTLA4-expressing cells, and this effect was abrogated by a CD86-blocking antibody (Figure S6E). To confirm the efficacy of the CD86-blocking antibody (fluorophore unconjugated), we performed flow cytometry with a fluorophore-conjugated CD86 antibody (phycoerythrin [PE] labeled). The unconjugated anti-CD86 antibody effectively blocked fluorophore-conjugated antibody-bound NK cells in the presence of either M2w or B78 cells (Figure S6H) without causing NK cell death (Figure S6I).
Because NF-κB is reported to stimulate transcription of CD86, and the NKG2D pathway can stimulate NF-κB signaling,40–42 we investigated whether this mechanism might contribute to CD86 expression. We analyzed the tumor cell-surface expression of ULBP1, one of the NKG2D ligands (Figure 6SJ). Next, we stimulated NK cells sorted from naive mouse spleen with recombinant ULBP1, which increased CD86 mRNA in NK cells and could be blocked with an NF-κB transcription inhibitor (Figure S6K).
To investigate whether CD86-expressing NK cells could regulate Treg viability,43 freshly sorted NK cells from the naive mouse spleen were engineered using CRISPR-Cas9 to generate a genomic CD86 deletion (Figure 7H). The guide RNA (gRNA) sequences targeted the functional domain V-set, which is critical to the interaction of CD86 with CTLA4.44 Sanger sequencing identified CD86 genomic DNA alteration in the surrounding target sequence and showed 51.1% of aberrant sequences in the CRISPR-CD86-engineered cells (CD86-CRI) compared with CRISPR-empty-engineered cells (Emp-CRI) (Figure 7I). Because of the difficulty of single-clone expansion of gene-modulated NK cells, we sorted NK cells that did not express surface CD86 using flow cytometry (Figure 7J, red-colored area). Co-culture with a combination of MOC2 tumor cells and CD86-CRI NK cells decreased the dead+CTLA4+CD4+ T cells compared with a co-culture of MOC2 tumor cells and Emp-CRI NK cells (Figure 7K).
To evaluate this mechanism in vivo, we delivered 3xTx to the right-flank tumor in NK-depleted mice bearing bilateral tumors, RF-M2h and LF-M2w (Figure 7L). On day 12 after initiation of treatment, Emp-CRI NK cells or CD86-CRI NK cells were injected into the untreated, distant left-flank tumor. After 5 days, flow cytometry on the distant tumor revealed decreased dead+ CTLA4+ Tregs and Treg number in tumors injected with Emp-CRI NK cells compared with those not injected with NK cells or injected with CD86-CRI NK cells (Figures 7L and S6L). Consistently, Emp-CRI NK cells showed no change on Tem infiltration, but increased the levels of polyfunctional TNF-α+IFN-γ+CD8+ Tem cells in these tumors compared with tumors that were not injected with NK cells or those injected with CRI-CD86 NK cells (Figures 7L and S6M). These findings shed light on an important role of NK cells in propagating CD8+ T cell antitumor immunity.
DISCUSSION
While radiotherapy alone can elicit an in situ vaccine response,45 it has not been demonstrated to enhance clinical response to immune-checkpoint blockade.11 Likewise, IL-2 has been extensively explored as an important factor in antitumor therapies, and while high-dose IL-2 has been effective in some cancers, such as melanoma, it can lead to severe side effects such as vascular leakage.46,47 Low doses, however, have been less effective and can lead to Treg increases.48 Therefore, to augment the in situ vaccine effect of radiotherapy, we combined it with tumor-specific antibody and intratumoral injection of IL-2 in the radiated tumor microenvironment. This 3xTx immunotherapy regimen combines off-the-shelf treatments readily available for most cancers to elicit an in situ vaccination that converts a targeted tumor into a site for priming systemic, adaptive, antitumor, CD8+ T cell immunity.
NK cells were critical to the 3xTx in situ vaccine response. That role extended to the distant tumors that were not directly treated by 3xTx, even in tumors that lacked the antigen targeted by the tumor-specific antibody. The mechanism of the NK cell roles differed between the primary and the distant tumors, with depletion studies demonstrating a necessary role for NK cells as a positive regulator of adaptive antitumor CD8+ T cell immunity at the distant tumor but not at the directly targeted tumor. Because the NK cell response at the distant tumor was independent of 3xTx tumor-specific antibody, ADCC may not be required for distant-tumor 3xTx response. These are important features for an effective vaccine response, as metastatic sites might vary in the expression of a tumor-specific antibody-targeted antigen.
Notably, NK cell infiltration did not significantly increase at the distant tumor until the relatively late day 12 time point after 3xTx completion, which coincided with CD8+ T cell infiltration. This was consistent with a role for NK cells in supporting the adaptive CD8+ T cell response in a chaperone role. CD8 depletion completely abrogated the distant tumor response to 3xTx while only partially impairing the local tumor response, suggesting that the adaptive response to 3xTx required antigen presentation to stimulate Tem distant-tumor response. This is supported by a lack of response of an M2h distant tumor site to 3xTx on a B78 tumor. In contrast to NK cells, adaptive T cell response required tumor-specific antibody. NK depletion during 3xTx had no effect on primary tumor response but impaired distant tumor response and thus survival. NK depletion also reduced MHC class I expression and release of IFN-γ from CD8+ T cells. Our data are consistent with a model in which FcγR-expressing cell populations other than NK cells are involved in the priming of in situ vaccination following 3xTx, whereas NK cells are critical in propagating this response.
Effector/memory and polyfunctional T cells have emerged as critical mediators of antitumor immunity and antitumor responses.21,49–52 Antigen recognition by these cells initiates production of IFN-γ,53 a positive regulator of CD8+ T cell proliferation and trafficking and of tumor cell MHC class I expression.23,54 Our data indicated that 3xTx-promoted distant-tumor infiltration by Tems required NK cells to sustain effector/memory and polyfunctional T cell differentiation. While the mechanism by which NK cells might support T cell polyfunctionality during 3xTx is not yet known, the increase in IFN-γ released by NK cells during 3xTx and the requirement of IFN-γ for effective tumor response and increased survival following 3xTx in mice suggest that IFN-γ is a critical intermediary signal between NK and T cells. These observations may bear relevance not only for in situ vaccine approaches but also for optimizing adoptive and chimeric antigen receptor T cell therapies. Because CD8+ T cell response to 3xTx is antagonized by recruitment of Tregs and upregulation of PD-1/PD-L1 in the distant tumor, we amplified 3xTx effectiveness by anti-PD-L1 or anti-CTLA-4 immune-checkpoint blockade. Our results demonstrate potential for 3xTx to enhance antitumor response in poorly immunogenic tumors that do not respond to immune-checkpoint blockade alone, possibly through NK cell support of effector T cell activity.
The role of NK cells in supporting CD8+ T cell immunity has been noted in other studies55–59; however, the mechanism has not been fully elucidated. Some reports identified effects of NK cells antagonizing or depleting activated CD4+ T cells or macrophages.16,60 Here, we observe that NK cells can directly antagonize and destroy tumor-infiltrating Tregs that are expanded by IL-2 stimulation61,62 via a CD86-mediated pathway, which leads to apoptosis in CTLA4+ Tregs. We postulate the following mechanistic model: 3xTx primes a tumor-specific CD8+ T cell response that can be propagated to distant tumor sites, but this requires overcoming inhibitory effects of Tregs at these locations. This can be achieved by NK cells, activated by 3xTx and perhaps drawn to distant tumor sites by paracrine signaling from CD8+ T cells, resulting in KLRK1-dependent upregulation of CD86 on NK cells, possibly depending on tumor cell expression of NKG2D ligands. Notably, we and others have observed that radiation upregulates the expression of NKG2D ligands on the surface of tumor cells,22,63 and this may suggest a role for radiation in priming the T cell chaperone effect of NK cells. CD86+ NK cells elicit a direct cytotoxic effect on CTLA4+ Tregs, which enables the propagation of antitumor immune response by tumor-specific polyfunctional CD8+ T cells at tumor sites throughout the body following 3xTx in situ vaccination.
Among the limitations of our approach is the use of intratumoral injection, which is more challenging than i.v. delivery in clinical applications but produced improved response in both the targeted and the non-targeted tumor. While tumor-specific antibody in 3xTx can be delivered systemically, IL-2 is delivered by intratumoral injection to enhance efficacy and limit toxicity.64 Intratumoral approaches have received US Food and Drug Administration (FDA) approval65 and can be applied in patients with metastatic disease. Valuable future studies include optimization of an effective 3xTx regimen with the smallest number of intratumoral injections.
Other important future directions include investigating additional therapeutic combinations that may further build upon 3xTx efficacy. The critical role for NK cells in sustaining Tem responses against distant tumors following in situ tumor vaccination suggests additional therapeutic benefit through the development of treatments that specifically target the interaction between NK cells and Tems and/or Tregs.
Limitations of the study
We studied the propagation of adaptive T cell priming to non-targeted tumors in humans and mice, but the bulk of our mechanistic data arises from syngeneic murine models. Future studies must assess these mechanisms in additional clinical settings, as our human study data include very few patients. Although we identified a major mechanism of NK cells in propagating antitumor T cell response in the presence of Tregs, additional cellular crosstalk between effector and suppressor immune lineages remains to be explored.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zachary Morris (zmorris@humonc.wisc.edu).
Materials availability
This study did not newly generate new materials.
Data and code availability
All code are publicly available. All data including supplementary information are available from the lead contact upon request.
Bulk RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database under the accession code GSE209945; access for reviewers: mbqjymwwzfwpnet
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Melanoma patient specimens
The clinical trial UW16134 was approved by the Health Science Institutional Review Board (IRB) that serves the University of Wisconsin Hospital and Clinics (NCT 03958383). Written informed consents were obtained from all participants. Eligibility requirements included patients with melanoma that was advanced (stage IV) or that could not be removed by surgery and was considered surgically incurable. Patients were treated with radiotherapy (8 Gy × 3 fractions) targeting a single tumor site followed by three daily intratumoral injections of this site with 1mg/m2 hu14.18-IL2 starting 4 days after completion of radiotherapy. PBMCs were collected from patients 6 days prior to radiotherapy (preTx) and 18–20 days after the second hu14.18-IL2 injection cycle (Cycle 2, postTx). PBMC collection was conducted under this IRB approved clinical trial protocol. Whole blood from patients was collected in EDTA-coated vacutainer tubes. After centrifugation at 300 G for 20 min at room temperature, the PBMC layer was isolated and aliquoted at 10 million cell/ml in cryopreservation media containing 90 % of heat-inactivated fetal bovine serum (FBS, Life Technology) and 10 % dimethyl sulfoxide (DMSO, Sigma-Aldrich) and stored in liquid nitrogen until use. Healthy PBMCs (Donor-222183430) were obtained from Stem Cell (200–0093).
METHOD DETAILS
Single-cell multiplex secretome study
Patient specimens single cell preparation: Cryopreserved PBMCs were thawed and cultured with RPMI 1640 (Corning) containing IL2 (10 ng/ml, eBioscience), 10 % FBS (Life Technologies) and 100 U/ml penicillin/streptomycin (Life Technologies) overnight at 5 % CO2, 37°C. CD8+ T cells were isolated using a CD8+ T cell microbead sorting kit (Miltenyi Biotec). The sorted CD8+ T cells were further cultured overnight on an anti-CD3 antibody-coated 96-well plate (10 μg/ml, eBioscience) with soluble anti-CD28 antibody (5 μg/ml, eBioscience) stimulation.
Mouse tumor single-cell preparation: Distant tumors were collected and dissociated using a Miltenyi gentle-MACS Octo Dissociator in RPMI 1640 (Corning) media (5 ml) containing DNase (500 μg, Sigma-Aldrich) and collagenase (5 mg, Sigma-Aldrich) for 30 min, 37°C. Dissociated single-cell suspensions were strained (70 μm pore size, Corning) and centrifuged (300 G, 10 min) to remove enzymes. Single-cells were resuspended in 4 ml of RPMI 1640 (Corning) and carefully transferred to a Ficoll-containing tube (3 ml, Sigma-Aldrich). The cells were centrifuged (400 G, 30 min, room temperature) without brake. Mononuclear cells were collected and sorted using a CD8+ T cell microbead sorting kit (Miltenyi Biotec). The cells were further cultured as described in the patient specimens single cell preparation section.
After 24 hours, the cells were collected and stained with Alexa Fluor 647 anti-CD8 antibody (PhenomeX) before loading onto the Adaptive Immune IsoCode chip (PhenomeX). The live single-cell secretome was measured during 16 hours of incubation, and secreted cytokines were detected by fluorescence labeled antibodies. Cytokine-secreting cells, signal intensity, PCA plots, t-SNE plots, and PSI were calculated by IsoPeak software.21
Cell lines
The MOC2 murine oral cancer cell line was obtained from Dr. Ravindra Uppaluri (Brigham and Women’s Hospital and Dana-Farber Cancer Institute). The huEGFR-expressing MOC2 cells (M2h) were generated by human EGFR (NM_005228.3) via lentiviral transduction and selected for by puromycin resistance co-expressed via the pLV vector, designed in VectorBuilder. Cetuximab binding ability was previously evaluated.22 The B16 melanoma and GD2-expressing B78-D14 murine melanoma cell line (B78) were obtained from Dr. Ralph Reisfield (Scripps Research Institute), and hu14.18 binding ability was previously evaluated.66 MOC2-WT (M2w) or M2h was cultured in Dulbecco’s Modified Eagle Medium (DMEM; Corning) / Ham’s F12 (Corning) at a 2:1 mixture and supplemented with 5 % fetal bovine serum (FBS, Life Technologies), 100 U/ml of penicillin/streptomycin (Life Technologies), epidermal growth factor (5 ng/ml, Gibco), hydrocortisone (400 ng/ml, Sigma-Aldrich) and insulin (5 μg/ml, Sigma-Aldrich). Murine HNSCC SCC7 cell line was kindly provided by Dr. Stephen Schoenberger (La Jolla Institute) and Panc02 pancreatic cancer cell line was obtained from the National Cancer Institute. SCC7 cells were grown in RPMI containing 10 % FBS (Life Technologies), 100 U/ml penicillin/streptomycin (Life Technologies), and 1 μg/ml hydrocortisone (Sigma-Aldrich). Panc02, B78 and B16 cells were grown in RPMI 1640 (Panc02 and B78, Corning) or DMEM (B16, Corning) with 10 % FBS, penicillin/streptomycin (100 U/ml). Cells were incubated at 37 C in a 5% CO2 incubator. ATCC guidelines were followed for cell authentication using morphology monitoring, growth curve analysis, and testing for mycoplasma within 6 months of use.
Tumor-bearing models, tumor measurement, and rechallenge
All animals were housed following the Guide for Care and Use of Laboratory Mice, and studies were conducted under a protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee (IACUC). Six to eight-week-old female mice were purchased from Taconic (C57BL/6). C57BL/6-background transgenic mice (Six to eight-week-old female) including IFNγtm1Ts (IFNg KO), Foxp3tm3(DTR/GFP)Ayr (Foxp3DTR), Klrk1tm1Dhr (NKG2D KO), and Fcer1gtm1Rav (FcγR KO) were purchased from Jackson Laboratory. NSG mice (NOD-scid IL2Rgnull) were kindly gifted by Dr. Paul Lambert.
Single tumor-bearing mice model; M2h or B78 tumors were engrafted by subcutaneous flank injection (2 × 106 cells in 100 ml of PBS) into the right flank. When the mean tumor volume reached 80–100 mm3 (M2h) or 170–230 mm3 (B78), mice were randomized and selected for the in vivo experiments. Two tumor-bearing models; Tumor cells (M2h/M2w or B7) were subcutaneously injected into the left flank (1 × 106 cells) 3 days or 13 days after primary tumor injection (M2h or B78, right flank, 2 × 106 cells). Mice were randomized and selected prior to treatment once the mean tumor size reached the indicated tumor size (primary tumor; 100 – 150 mm3, distant tumor; 60 – 100 mm3) for each in vivo experiment.
Tumor sizes were measured using digital calipers twice weekly, and volume was calculated as (width2 × length)/2. After treatment initiation (day 1), tumor growth was measured until all mice in each group reached a terminal endpoint indicated by a tumor exceeding a maximum diameter of 20 mm, hunched posture, or veterinary recommendation, at which point mice were euthanized.
To evaluate immune memory acquirement, tumors were re-engrafted on the opposite flank on day 90 (left flank, 5 × 105 cells in 100 μl of PBS) from the initial primary treatment with the same tumor cell line they had been cured of. Age-matched naïve control mice were engrafted at the same time. Two weeks after engraftment, tumor-grown- or tumor-not-grown mice were counted and calculated (% of tumor rejection).
Local radiotherapy and immunotherapy
Radiation was delivered at 8 Gy or 12 Gy using a cabinet orthovoltage X-ray biological irradiator X-RAD 320 (Precision X-Ray Inc.) with the tumor exposed and the rest of the animal shielded using a custom lead block. For intratumoral injections, 50 μg of the anti-human EGFR antibody cetuximab (Eli Lilly), anti-human/mouse GD2 antibody hu14.18K322A (Children’s GMP, LLC facility, Memphis, TN), or non-specific human IgG (Sigma-Aldrich) was mixed with 75,000 U of human IL2 (NCI Repository) or PBS. Intratumoral injections were given to the radiated tumor daily from day 6 to 10, a timing we have previously optimized (10). Anti-murine PDL1 (10F.9G2™, BioXcell, RRID:AB_10949073, 100 μg) or anti-murine CTLA4 (9D9, BioXcell, RRID:AB_10949609, 100 μg) was given via intraperitoneal injection on days 0, 4, 8, 12, and 16.
Multiplex protein profiling
On day 12, after indicated treatment initiation, distant tumors (M2w) were collected and recorded. Collected tumors were placed in the tube containing ceramic bead (Fisher Brand) and added 5 μl/mg of Cell Lysis Buffer (Cell Signaling Technology), PMSF (Cell Signaling Technology), and HaltTM Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The mixture was homogenized using a Bead Ruptor Elite (OMNI) for 30 sec. The lysate was subjected to the multiplex immunoassay (MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel, MilliporeSigma) to determine the concentration of 32 cytokines/chemokines following the manufacturer’s instruction. After the multiplex was measured on the MAGPIX system (MilliporeSigma), proteins concentrations were inter-polated from curves constructed by protein standards and individual protein median fluorescence intensity (MFI) reads (MILLIPREX Analyst, MilliporeSigma). Protein concentrations were normalized by 3xTx and represented.
Immune cell sorting, transfer, and depletion study
Spleens collected from treated- or non-treated mice were mechanically digested using a syringe plunger and strained through a 70 μm strainer. Red Blood Cell (RBC) Lysing Buffer (Sigma-Aldrich) was added and incubated for 10 min in room temperature. After RBC lysis, selected immune cells (CD4/8 and NK cells) were sorted using MACS-negative selection (Miltenyi Biotec) according to the manufacture’s instruction. Sorted NK cells (1 × 106) and CD8+ T cells (1 × 106) were injected intratumorally into the M2w-bearing NSG mice.
Lymphocyte-specific depletion antibodies were obtained from BioXcell and delivered intraperitoneally using IgG isotype control, anti-NK1.1 (clone# PK136, BioXcell, RRID: AB_1107737, 50 μg), anti-CD4 (clone# GK1.5, BioXcell, RRID: AB_1107636, 300 μg), and anti-CD8a (clone# 2.43, BioXcell, RRID: AB_1125541, 300 μg) antibodies on days 0, 5, and 10. For the Foxp3+ cell depletion, Foxp3DTR mice received DT (1 μg, Sigma-Aldrich) intraperitoneally daily on d12 to d16.
Lung metastasis histology
Lung tissue was collected on d20 or d40, and fixed with 10% formalin in PBS at 4°C. After 24h, the samples were further incubated in 70% EtOH for 24h at 4°C. Next, the samples were embedded in paraffin, and the blocks were sectioned at 5 μm thickness using a Rotary Microtome (Leica). Then, the samples were H&E stained using Mayer’s Hematoxylin (Sigma) and Eosin (Leica) and mounted (Richard-Allan Scientific). Visible lung metastasis images were acquired using a microscope (Olympus BX41 and IX51) and analyzed using ImageJ.
Immune cell analysis using flow cytometry
Collected tumors were dissociated using a Miltenyi gentle-MACS Octo Dissociator in the presence of RPMI 1640 (Corning) containing Dnase (500μg, Sigma-Aldrich) and collagenase (5 mg, Sigma-Aldrich) for 30 min. Dissociated single cells suspensions were incubated with red blood cell lysing buffer (Sigma-Aldrich) and treated with CD16/32 antibody (BioLegend) to avoid tumor cell non-specific binding. The cells were then stained with live/dead cell staining using Ghost Red Dye 780 (Tonbo Biosciences). Cell surface antigens were labeled using flow cytometry detectible antibodies. Peripheral blood was collected from the submandibular vein, and RBCs were lysed using lysis buffer (BioLegend). Internal staining was conducted using BD Cytofix/Cytoperm™ according to the manufacturer’s instructions. Flow cytometry analysis was determined after bead compensation (UltraComp eBeadsTM, Invitrogen), calibration (Ultra Rainbow bead, Spherotech), and fluorescence minus one (FMO) gating methodology. Flow cytometry was performed using an Attune NxT Flow Cytometer (ThermoFisher), and the data were analyzed using FlowJo Software (BD). The antibodies used for Attune flow cytometry are listed in the STAR Methods (antibody name_Attune).
To investigate infiltrated CD45+ cell populations, tumors were collected, dissociated, and RBC-lysed as described above. Dissociated single cells were stained, and flow cytometry was performed using Cytek Aurora (Cytek Bioscience). Analysis was determined after dissociated single cell- or bead (UltraComp eBeadsTM, Invitrogen) compensation. Live+CD45+ cells were manually gated, and the cells were visualized by tSNE using FlowJo (v10.8.1).67,68 After visualizing the high-dimensional flow dataset, Flowsom analysis was performed to generate clusters and Median Marker Expression (MME) heatmap.34 Based on the MME heatmap, each cluster was analyzed. The antibodies used for the CyteK flow cytometer are listed in the STAR Methods (antibody name_Cytek).
T cell memory evaluation
The immune cells (2 × 105 cells) from whole splenocytes or MACS-sorted CD4+ or CD8+ cells (Miltenyi Biotec, negative selection) were co-cultured with pre-plated tumor cells (2 × 104 cells) in the 48-well plates. The cells were then allowed to incubate for 5 days in the presence of RPMI 1640 containing 10% heat-inactivated FBS and penicillin/streptomycin (100 U/ml). GolgiStopTM (4 μl/sample, BD) was added 5 hours before collecting cells for flow cytometry staining. Total cells were then prepped and treated with CD16/32 antibody (1 μg/ml, BioLegend), and cell staining for flow cytometry was conducted as described above. To detect secreted IFNγ, sorted CD8+ T cells (2 × 105 cells) were co-cultured with indicated tumor cells (B78, MOC2-WT, or MOC2-huEGFR, 2 × 104 cells) for 5 days in the 48-well plate. The supernatants were then collected and pelleted by centrifugation (2000 RPM, 4°C, 10 min) to separate debris. Separated supernatants were analyzed to quantify levels of IFNγ using ELISA (BioLegend) according to the manufacturer’s instructions.
CD86 CRISPR KO
crRNA and sNLS-spCas9-sNLS were obtained from IDT™. To generate genomic CD86 (NM_019388.3) knockout, the RNP complex containing crRNA and tracrRNA for murine CD86 was electroporated into spleen-sorted NK cells (1 × 106 cells, six to eight-week-old mouse, C57BL/6) using 4D Nucleofector™ with P3 primary Cell 4D-Nucleofector® X kit. After electroporation, the cells were maintained using RPMI 1640 (Corning) media containing IL-15 (500a ng/ml, PeproTech), 10 % FBS (Life Technologies), penicillin/streptomycin (100 U/ml, Life Technologies) in 96-well plates. After 3 days incubation at 37 C in a 5% CO2 incubator, genomic DNA was extracted using PureLink Genomic DNA mini kit (Invitrogen), subjected to Sanger sequencing, and the results were analyzed using SnapGene (http://www.snapgene.com) software.
CTLA4+Treg-enriched CD4+ T cell preparation, NK cell cytotoxicity study, and antibody competition assay
Spleen-sorted CD4+ T cells (1 × 106 cells, six to eight-week-old mouse, C57BL/6) were cultured in the RPMI 1650 (Corning) media containing 10% FBS (Life Technologies) and penicillin/streptomycin (100 U/ml, Life Technologies) with PBS or IL2 (300, 1000, or 5000 U/ml, NCI Repository) for 24 hours in a 96-well plate. The cells were collected and evaluated for Treg (CD25+FOXP3+) and CTLA4+ cell development after gating CD4+ T cells using flow cytometry (Figure S3A).
IL2 (5000 U/ml, 24 h)-stimulated CD4+ T cells (1 × 106 cells) were labeled with CFSE (5 μM, Invitrogen) according to the manufacturer’s instruction. Then, the cells (2 × 105 cells) were cultured with spleen-sorted NK cells (2 × 105 cells, six to eight-week-old mouse, C57BL/6) in the M2w- or B78-precultured plate (2 × 104 cells, 24 hours before) with CF-594-conjugated anti-Annexin V antibody (150 ng/ml, Biotium)-containing media (RPMI 1640 with 10% of FBS and 100 U/ml of penicillin/streptomycin). Fluorescence images were taken after 35h co-culture using the Incucyte Zoom live-cell monitoring system (Essen Bioscience) according to the manufacturer’s instructions.
To test CD86-dependent NK cell cytotoxicity, anti-mouse CD86 antibody (clone# A17199H, RRID: AB_2888740, 5 μg/ml, BioLegend) or Rat IgG2a isotype control (clone# RTK2758, RRID: AB_326523, 5 μg/ml, BioLegend) was added in the media (RPMI 1640 with 10% of FBS and 100 U/ml of penicillin/streptomycin) during co-culture of NK cell and CD4+ T cells (1:1 ratio, 1 × 105 cells). After 6 hours, the cells were collected and conducted a flow cytometry analysis to determine dead cells (GhostRed780-positive cells, Tonbo Biosciences) in total CD4+CTLA4+ cells. CD86-blocking efficacy cells were evaluated using PE-conjugated anti-mouse CD86 antibody (clone#A17199A, RRID: AB_2832567, BioLegend).
To test irradiated tumor impacts on CD86-dependent cytotoxicity, pre-plated M2w or B78 (2 × 104 cells, 24 hours before) was radiated (M2w; 0 or 8 Gy, B78 0 or 12 Gy) and cultured for 3 days. Then NK cells sorted from spleens (2 × 105 cells, six to eight-week-old mouse, C57BL/6) were co-cultured with the pre-plated tumor cells with anti-mouse CD86 antibody (clone# A17199H, RRID: AB_2888740, 5 μg/ml, BioLegend) or Rat IgG2a isotype control (clone# RTK2758, RRID: AB_326523, 5 μg/ml, BioLegend). IL-2-stimulated CD4+ T cells (sorted from spleen, 5000 U/ml, 24 h, 2 × 105 cells) were added in the culture plate. After 12 hours incubation, the cells were collected and flow cytometry was performed to quantify dead cells in CD4+CTLA4+ cells.
To test antibody blocking efficacy, spleen-sorted NK cells (2 × 105 cells, six to eight-week-old mouse, C57BL/6) were cultured in with preplated M2w- or B78-precultured plate (2 × 104 cells, 24 hours before) containing RPMI 1650 (Corning) media with 10% FBS (Life Technologies) and penicillin/streptomycin (100 U/ml, Life Technologies). Then, indicated dose of anti-mouse CD86 blocking antibody (clone# A17199H, RRID: AB_2888740, BioLegend) or Rat IgG2a isotype control (clone# RTK2758, RRID: AB_326523, 5 μg/ml, BioLegend) was added to the culture media for 6 hours. Then the cells were collected and flow cytometry was performed to quantify CD86 expression in NK cells using PE-conjugated anti-mouse CD86 antibody (clone#A17199A, RRID: AB_2832567, BioLegend).
ULBP1 and CD86 expression
M2w or B78 cells (2 × 105 cells) were cultured in a 6-well plate. After 24h incubation, the cells were washed with PBS and collected by scraping (FALCON). Collected cells were incubated at 4 °C with flow buffer (PBS-containing 2 mM EDTA (Sigma-Aldrich) and 2% FBS) with or without AF405 anti-ULBP1 antibody (clone #237104). After 30 min, flow cytometry was performed using an Attune NxT Flow Cytometer (ThermoFisher), and the data were analyzed using FlowJo Software (BD).
Tumor cells (M2w, M2h, B16, B78, SCC7, or Panc02; 1 × 105) were cultured in 12 well-plates. After 24h incubation, spleen-sorted NK cells (1 × 105 cells) were added. After 15 h incubation, total cells were collected and analyzed CD86 expression in NK1.1+ cells by flow cytometry.
NK cells sorted from spleen (1 × 106 cells, six to eight-week-old mouse, C57BL/6) were cultured in a 48-well plates (Corning) containing RPMI 1650 (Corning) media with 10% FBS (Life Technologies) and penicillin/streptomycin (100 U/ml, Life Technologies). Then the indicated dose of recombinant mouse ULBP-1 (R&D System™) or NF-κB transcriptional activity inhibitor (JSH 23, Abcam) was added to the culture media for 6 hours. The cells were then washed with PBS, and 1 ml of Trizol reagent (Invitrogen) was added. Total RNA was extracted by RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. Then, isolated RNA was subjected to complementary DNA (cDNA) synthesis using the QuantiTect Reverse Transcription Kit (QIAGEN) according to the manufacturer’s instructions. Quantitative PCR (qPCR) on the resulting cDNA was performed using PowerUp SYBR Green master mix (Applied Bio-systems) on a CFX96 Real-Time System (Bio-Rad). The following primers were used: mus musculus Cd86 (GenBank: NM_019388.3) forward: CTTACGGAAGCACCCACGAT, reverse-CGTCTCCACGGAAACAGCAT; mus musculus Hprt (GenBank: NM_013556.2) forward: AGCCTAAGATGAGCGCAAGT, reverse- GGCCACAGGACTAGAACACC. Relative CD86 mRNA expression of target genes was determined according to the 2–ΔΔCT method using Hprt as a reference gene.69
Transduced NK cells adoptive transfer
Bilateral tumor-bearing mice (RF-M2h, LF-M2w) were depleted of NK cells during RF-3xTx, as described above. NK cells (Empty-CRI or CD86-CRI cells) were prepared (2 × 105 cells in 100 μl of PBS) and intratumorally injected into the left flank tumor (M2w) on day 12. After 5 days, left flank tumors were collected, digested, washed, and stained for flow cytometry analysis to determine Treg number and CD8+ T cell-immunity.
Library and RNA-sequencing
Distant tumors were collected on d12. The tumor tissues were dissected and transferred to tubes containing 2.8 mm ceramic beads (Fisher Brand) with 1 ml of Trizol reagent (ThermoFisher). The samples were homogenized using a Bead Ruptor Elite (OMNI) for 45 s. RNA was isolated using Qiagen’s Rneasy Mini Kit according to the manufacturer’s instructions. 1 μg of RNA was used to make a library (Illumina) with UD indexing (Illumina). The prepared libraries were sequenced on an Illumina NOVAseq 6000 at the University of Wisconsin Biotechnology Center DNA Sequencing Facility. Skewer70 was used to trim adapter sequences from reads and to remove low quality bases from read 3′ ends before aligning to GRCm38.p6 using STAR.71
RNA-seq differential expression and pathway enrichment analysis
RSEM was used to calculate expected gene counts from aligned reads.72 Principal component analysis and Pearson correlation between vectors of gene counts that belong to biological replicates were employed to detect outlier libraries. Any samples with inter-replicate Pearson correlation less than 0.9 or which did not cluster with replicates by PCA were dropped from downstream analyses. Features representing rRNA, tRNA, and genes with an average gene count of less than 20 across all remaining samples were removed from differential expression analysis. Using R 4.0.5, gene count normalization and differential expression testing were performed by DESeq2 v1.30.1.73 ClusterProfiler v3.18.1 was used to perform gene set enrichment analysis of KEGG immune system pathways.74 ComplexHeatmap was used to generate heat maps of z-score normalized gene expression calculated from DESeq2 normalized gene counts.75
Previously published RNAseq data (mRNA expression z-scores relative to diploid samples-RNA Seq V2 RSEM) for HNSCC (TCGA, Firehose Legacy) and Skin Cutaneous Melanoma (TCGA, Firehose Legacy) were obtained from the cBioPortal (https://www.cbioportal.org). Correlation graph (linear regression) visualized by GraphPad Prism 9.3.1 with two-tailed test. RNA-seq data are available at GEO (GSE209945).
QUANTIFICATION AND STATISTICAL ANALYSIS
A p-value of < 0.05 was considered statistically significant. Statistical analysis was conducted using R (v. 4.1.1), the packages ‘lme4’ (v. 1.1.27.1) for tumor volume, and ‘survival’ (v. 3.2–13) for time to death subject to censoring. To analyze the longitudinal tumor growth data (volumes) for each experiment, log-transformed tumor growth over time was modeled and compared between treatment groups using linear mixed-effects models, in which individual samples were modeled as a random effect, while treatment groups, time, and their interaction were modeled as fixed effects. Specifically, the fixed-effect model matrices were parameterized such that all main and interactive effects between respective treatment groups and time were estimated. The intra-mouse correlation was accommodated via random intercepts. Tumor volumes with a value of 0 were inputted with half the value of the smallest recorded tumor volume. Relative ratios (RR) were reported for each treatment pairwise combination. Survival probability was estimated and plotted using the Kaplan-Meier method and compared using the log-rank test. Mice that were sacrificed/died beyond the 90-day experiment were censored. Observed differences among groups from flow cytometry and ELISA were compared using ANOVA and Tukey’s method for multiple comparisons. Two-tailed Student’s t-test was used for a two-sample comparison.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Anti bodies | ||
| Anti CD45 PE-Cy7_Attune | BioLegend | RRID: AB_312978 |
| Anti CD3 FITC_Attune | BioLegend | RRID: AB_312660 |
| Anti CD4 BV510_Attune | BioLegend | RRID: AB_2561388 |
| Anti CD8 PerCP-Cy5.5_Attune | BioLegend | RRID: AB_2075239 |
| Anti NK1.1 BV605_Attune | BioLegend | RRID: AB_2562273 |
| Anti CD25 BV711_Attune | BioLegend | RRID: AB_2564130 |
| Anti FOXP3 BV421_Attune | BioLegend | RRID: AB_2565933 |
| Anti CXCR3 APC_Attune | BioLegend | RRID: AB_2814077 |
| Anti CD44 BV605_Attune | BioLegend | RRID: AB_2562451 |
| Anti CD44 BV711_Attune | BioLegend | RRID: AB_2564214 |
| Anti CD62L AF700_Attune | BioLegend | RRID: AB_493719 |
| Anti H-2 FITC_Attune | BioLegend | RRID: AB_1236475 |
| Anti TNFα BV711_Attune | BioLegend | RRID: AB_2629800 |
| Anti TNFα PE_Attune | BioLegend | RRID: AB_315426 |
| Anti IFNγ APC_Attune | BioLegend | RRID: AB_315403 |
| Anti PD-L1 PerCP-Cy5.5_Attune | BioLegend | RRID: AB_2629831 |
| Anti PD-1 BV421_Attune | BioLegend | RRID: AB_10900085 |
| Anti CD86 blocking Ab_Attune | BioLegend | RRID: AB_2888740 |
| Anti CD86 PE_Attune | BioLegend | RRID: AB_2832567 |
| Anti CTLA4 BV421_Attune | BioLegend | RRID: AB_2616790 |
| Anti CTLA4 PE_Attune | BioLegend | RRID: AB_313254 |
| Anti CD45 BUV395_Cytek | BD | RRID: AB_2651134 |
| Anti CD4 BUV496_Cytek | BD | RRID: AB_2970666 |
| Anti Ly6G BUV563_Cytek | BD | Cat# 612921 |
| Anti CD11b BUV661_Cytek | BD | Cat# 612977 |
| Anti CD8a_Cytek | Invitrogen | Cat# 368-0081-82 |
| Anti B220_Cytek | BioLegend | RRID: AB_492877 |
| Anti CD25_Cytek | Invitrogen | Cat# 69-0251-82; RRID: AB_2762802 |
| Anti XCR1_Cytek | BioLegend | RRID: AB_566410 |
| Anti NK1.1_Cytek | BioLegend | RRID: AB_2563286 |
| Anti Siglec H_Cytek | BD | RRID: AB_2744231 |
| Anti Siglec F_Cytek | BD | RRID: AB_2738833 |
| Anti CD64 AF532_Cytek | Novus Biologicals | Cat# NB039151 |
| Anti Ly6c PerCP_Cytek | BioLegend | RRID: AB_10900235 |
| Anti MHC II BB700_Cytek | BD | RRID: AB_2743544 |
| Anti CCR2 PE_Cytek | BioLegend | RRID: AB_2616981 |
| Anti CD11c PE/Dazzle 594_Cytek | BioLegend | RRID: AB_2563654 |
| Anti F4/80 PE/Cy5_Cytek | BioLegend | RRID: AB_893494 |
| Anti CD172a PE/Cy7_Cytek | BioLegend | RRID: AB_2563545 |
| Anti FOXP3 FITC_Cytek | Invitrogen | Cat# 11-5773-82; RRID: AB_465243 |
| Anti CD3 APC_Cytek | BioLegend | RRID: AB_2561455 |
| Anti CD90 AF700_Cytek | R&D System | Cat# FAB7335N-100UG |
| Hu14.18K322A | St. Jude Children’s Research Hospital | N/A |
| Cetuximab (Erbitux, anti huEGFR antibody) | Eli Lilly | N/A |
| Anti PD-L1 (in vivo) | BioXcell | RRID: AB10949073 |
| Anti CTLA4 (in vivo) | BioXcell | RRID: AB10949609 |
| CD8 depletion antibody (in vivo) | BioXcell | RRID: AB1125541 |
| CD4 depletion antibody (in vivo) | BioXcell | RRID: AB1107636 |
| NK depletion antibody (in vivo) | BioXcell | RRID: AB1107737 |
| Biological samples | ||
| Healthy donor PBMCs | Stem Cell | Cat# 200–0093 |
| Patients PBMCs | This paper | UW16134 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant mouse ULBP1 | R&D System | Cat# 2588-MU |
| NF-κB transcription inhibitor (JSH 23) | Abcam | Cat# ab144824 |
| IL2 | National Cancer Institute | Ro 23–6019 |
| sNLS-spCas9-sNLS | IDT | Cat# 10017688 |
| Critical commercial assays | ||
| Multiplex immunoassay | MilliporeSigma | Cat# MCYTMAG-70K-PX32 |
| IFNγ ELISA assay | BioLegend | Cat# 430807 |
| Single cell secretomes-human | PhenomeX | Cat# ISOCODE-1001–4 |
| Single cell secretomes-mouse | PhenomeX | Cat# ISOCODE-1004–4 |
| TruSeq RNA UD Indexes | Illumina | Cat# 20020591 |
| TruSeq Stranded mRNA LP | Illumina | Cat# 20020595 |
| CD4+ cell isolation | Miltenyi Biotec | Cat# 130-104-454 |
| CD8+ cell isolation | Miltenyi Biotec | Cat# 130-104-075 |
| NK cell isolation | Miltenyi Biotec | Cat# 130-115-818 |
| Deposited data | ||
| RNA seq | This study | GEO : GSE209945 |
| Experimental models: Cell lines | ||
| MOC2 (M2w) | Brigham and Women’s Hospital and Dana-Farber Cancer Institute | Dr. Ravindra Uppaluri |
| MOC2-huEGFR (M2h) | University of Wisconsin | This study |
| B16 | Scripps Research Institute | Dr. Ralph Reisfield |
| B78 | Scripps Research Institute | Dr. Ralph Reisfield |
| SCC7 | La Jolla Institute | Dr. Stephen Schoenberger |
| Panc02 | National Cancer institute | This study |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Taconic | B6-F |
| IFNgtm1Ts | Jackson Laboratory | IMSR_JAX:002287 |
| FOXP3tm3(DTR/GFP)Ayr | Jackson Laboratory | IMSR_JAX:016958 |
| Klrk1tm1Dhr | Jackson Laboratory | IMSR_JAX:022733 |
| Fcer1gtm1Rav | Jackson Laboratory | IMSR_JAX:002847 |
| NOD-scid IL2Rgnull | University of Wisconsin | Dr. Paul Lambert |
| Oligonucleotides | ||
| Murine CD86 forward/reverse primers | IDT | N/A |
| Murine HPRT forward/reverse primers | IDT | N/A |
| Recombinant DNA | ||
| CRISPR CD86 gRNA | IDT | Mm.Cas9.CD86.1.AA |
| Software and algorithms | ||
| FlowJo | BD | N/A |
| cBioPortal | https://www.cbioportal.org | N/A |
| IsoPeak | https://isoplexis.com/videos/isospeak-software/ | N/A |
| R (Version 4.0.5 or 4.1.1) | https://www.r-project.org | N/A |
| DESeq2 Version 1.30.1 | https://bioconductor.org/ | N/A |
| ClusterPROFILER Version 3.18.1 | https://bioconductor.org/ | N/A |
| ComplexHeatmap | https://bioconductor.org/ | N/A |
| R packages ‘lme4’ | https://cran.r-project.org/web/packages/lme4/ | N/A |
| R packages ‘survival’ (Version 3.2–13) | https://cran.r-project.org/web/packages/survival | N/A |
| PRISM | GraphPad Software | N/A |
Highlights.
In situ vaccine effect of radiation, antibody, and IL-2 injection (3xTx)
3xTx elicits enhanced infiltration of T cells and NK cells in non-targeted tumors
NK cell depletion increases regulatory T cell infiltration in non-targeted tumors
NK cells antagonize CTLA4+ Treg and elicit CD8+ T cell immunity
ACKNOWLEDGMENTS
We would like to thank the University of Wisconsin Carbone Cancer Center (UWCCC) flow cytometry and histology/pathology core personnel; the UWCCC Cancer Informatics Shared Resource, supported by P30 CA014520; the University of Wisconsin Small Animal Radiotherapy Facility; the University of Wisconsin Biotechnology Center DNA Sequencing Facility and Bioinformatics Resource Center for providing Illumina sequencing and data processing, and Tracy Berg for editing.
Funding for this work came from National Institutes of Health grants P30 CA014520, P50 DE026787, P50 CA278595, 1DP5DO024576, U01CA233102, P01CA250972, T32GM140935, F30CA250263, and TL1TR002375.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found onlin6e at https://doi.org/10.1016/j.celrep.2023.113556.
DECLARATION OF INTERESTS
Z.S.M. is a member of the scientific advisory boards for Archeus Technologies, NorthStar Medical Isotopes, and Seneca Therapeutics.
REFERENCES
- 1.O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MWL, et al. (2012). Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med 209, 1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galon J, and Bruni D (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Dis-cov 18, 197–218. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, Lao CD, Schadendorf D, Wagstaff J, Dummer R, et al. (2018). Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 19, 1480–1492. [DOI] [PubMed] [Google Scholar]
- 5.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJM, Krainer M, Houede N, Santos R, Mahammedi H, et al. (2014). Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yee C, and Greenberg P (2002). Modulating T-cell immunity to tumours: new strategies for monitoring T-cell responses. Nat. Rev. Cancer 2, 409–419. [DOI] [PubMed] [Google Scholar]
- 7.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marabelle A, Kohrt H, Caux C, and Levy R (2014). Intratumoral immunization: a new paradigm for cancer therapy. Clin. Cancer Res 20, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, and Demaria S (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McBride S, Sherman E, Tsai CJ, Baxi S, Aghalar J, Eng J, Zhi WI, McFarland D, Michel LS, Young R, et al. (2021). Randomized Phase II Trial of Nivolumab With Stereotactic Body Radiotherapy Versus Nivolumab Alone in Metastatic Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol 39, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, Dumoulin DW, Bahce I, Niemeijer ALN, de Langen AJ, et al. (2019). Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol 5, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suresh M, Whitmire JK, Harrington LE, Larsen CP, Pearson TC, Altman JD, and Ahmed R (2001). Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J. Immunol 167, 5565–5573. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Q, and Elson CO (2018). Adaptive immune education by gut microbiota antigens. Immunology 154, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milstein O, Hagin D, Lask A, Reich-Zeliger S, Shezen E, Ophir E, Eidelstein Y, Afik R, Antebi YE, Dustin ML, and Reisner Y (2011). CTLs respond with activation and granule secretion when serving as targets for T-cell recognition. Blood 117, 1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grivennikov SI, Greten FR, and Karin M (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivier E, Tomasello E, Baratin M, Walzer T, and Ugolini S (2008). Functions of natural killer cells. Nat. Immunol 9, 503–510. [DOI] [PubMed] [Google Scholar]
- 17.Liao W, Lin JX, and Leonard WJ (2013). Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 38, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris ZS, Guy EI, Francis DM, Gressett MM, Werner LR, Carmichael LL, Yang RK, Armstrong EA, Huang S, Navid F, et al. (2016). In Situ Tumor Vaccination by Combining Local Radiation and Tumor-Specific Antibody or Immunocytokine Treatments. Cancer Res 76, 3929–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hank JA, Robinson RR, Surfus J, Mueller BM, Reisfeld RA, Cheung NK, and Sondel PM (1990). Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant interleukin 2. Cancer Res 50, 5234–5239. [PubMed] [Google Scholar]
- 20.Gillies SD, Reilly EB, Lo KM, and Reisfeld RA (1992). Antibody-targeted interleukin 2 stimulates T-cell killing of autologous tumor cells. Proc. Natl. Acad. Sci. USA 89, 1428–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diab A, Tykodi SS, Daniels GA, Maio M, Curti BD, Lewis KD, Jang S, Kalinka E, Puzanov I, Spira AI, et al. (2021). Bempegaldesleukin Plus Nivolumab in First-Line Metastatic Melanoma. J. Clin. Oncol 39, 2914–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin WJ, Erbe AK, Schwarz CN, Jaquish AA, Anderson BR, Sriramaneni RN, Jagodinsky JC, Bates AM, Clark PA, Le T, et al. (2020). Tumor-Specific Antibody, Cetuximab, Enhances the In Situ Vaccine Effect of Radiation in Immunologically Cold Head and Neck Squamous Cell Carcinoma. Front. Immunol 11, 591139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitmire JK, Tan JT, and Whitton JL (2005). Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J. Exp. Med 201, 1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge MQ, Ho AWS, Tang Y, Wong KHS, Chua BYL, Gasser S, and Kemeny DM (2012). NK cells regulate CD8+ T cell priming and dendritic cell migration during influenza A infection by IFN-γ and perforin-dependent mechanisms. J. Immunol 189, 2099–2109. [DOI] [PubMed] [Google Scholar]
- 25.Tewari K, Nakayama Y, and Suresh M (2007). Role of direct effects of IFN-gamma on T cells in the regulation of CD8 T cell homeostasis. J. Immunol 179, 2115–2125. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Kohli K, Black RG, Yao L, Spadinger SM, He Q, Pillarisetty VG, Cranmer LD, Van Tine BA, Yee C, et al. (2019). Systemic Interferon-γ Increases MHC Class I Expression and T-cell Infiltration in Cold Tumors: Results of a Phase 0 Clinical Trial. Cancer Immunol. Res 7, 1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, Rispens T, and Vidarsson G (2017). Affinity of human IgG subclasses to mouse Fc gamma receptors. mAbs 9, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casey E, Bournazos S, Mo G, Mondello P, Tan KS, Ravetch JV, and Scheinberg DA (2018). A new mouse expressing human Fcγ receptors to better predict therapeutic efficacy of human anti-cancer antibodies. Leukemia 32, 547–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junker F, Gordon J, and Qureshi O (2020). Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol 11, 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheng J, Chen Q, Soncin I, Ng SL, Karjalainen K, and Ruedl C (2017). A Discrete Subset of Monocyte-Derived Cells among Typical Conventional Type 2 Dendritic Cells Can Efficiently Cross-Present. Cell Rep 21, 1203–1214. [DOI] [PubMed] [Google Scholar]
- 31.Balan S, Saxena M, and Bhardwaj N (2019). Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol 348, 1–68. [DOI] [PubMed] [Google Scholar]
- 32.Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, Tay A, Lum J, Kairi MFBM, Hwang YY, et al. (2019). Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat. Immunol 20, 852–864. [DOI] [PubMed] [Google Scholar]
- 33.Allen E, Bakke AC, Purtzer MZ, and Deodhar A (2002). Neutrophil CD64 expression: distinguishing acute inflammatory autoimmune disease from systemic infections. Ann. Rheum. Dis 61, 522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- 35.Hori S, Nomura T, and Sakaguchi S (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, and Sakaguchi S (2000). Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med 192, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He X, and Xu C (2020). Immune checkpoint signaling and cancer immunotherapy. Cell Res 30, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arens R, and Schoenberger SP (2010). Plasticity in programming of effector and memory CD8 T-cell formation. Immunol. Rev 235, 190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Read S, Malmström V, and Powrie F (2000). Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+) CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med 192, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, Wang Y, Xiong F, Guo C, Li Y, et al. (2019). Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 18, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanada S, Nishiyama C, Nakano N, Suzuki R, Maeda K, Hara M, Kitamura N, Ogawa H, and Okumura K (2011). Critical role of transcription factor PU.1 in the expression of CD80 and CD86 on dendritic cells. Blood 117, 2211–2222. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Liu Z, Jiang S, Cortesini R, Lederman S, and Suciu-Foca N (1999). T suppressor lymphocytes inhibit NF-kappa B-mediated transcription of CD86 gene in APC. J. Immunol 163, 6386–6392. [PubMed] [Google Scholar]
- 43.Krummel MF, and Allison JP (1996). CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med 183, 2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellis JH, Burden MN, Vinogradov DV, Linge C, and Crowe JS (1996). Interactions of CD80 and CD86 with CD28 and CTLA4. J. Immunol 156, 2700–2709. [PubMed] [Google Scholar]
- 45.Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. (2018). Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med 24, 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, et al. (1999). High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol 17, 2105–2116. [DOI] [PubMed] [Google Scholar]
- 47.Baluna R, and Vitetta ES (1997). Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology 37, 117–132. [DOI] [PubMed] [Google Scholar]
- 48.Klatzmann D, and Abbas AK (2015). The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol 15, 283–294. [DOI] [PubMed] [Google Scholar]
- 49.Nolz JC, Starbeck-Miller GR, and Harty JT (2011). Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions. Immunotherapy 3, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou S, Shao T, Mao T, Shi J, Sun J, Mei M, Tan X, and Qi H (2021). Virtual memory T cells orchestrate extralymphoid responses conducive to resident memory. Sci. Immunol 6, eabg9433. [DOI] [PubMed] [Google Scholar]
- 51.Youngblood B, Hale JS, and Ahmed R (2013). T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology 139, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakajima Y, Chamoto K, Oura T, and Honjo T (2021). Critical role of the CD44(low)CD62L(low) CD8(+) T cell subset in restoring antitumor immunity in aged mice. Proc. Natl. Acad. Sci. USA 118, e2103730118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samji T, and Khanna KM (2017). Understanding memory CD8(+) T cells. Immunol. Lett 185, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, Turley H, Dobbs N, Gatter K, Talbot DC, et al. (2003). Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin. Cancer Res 9, 84–92. [PubMed] [Google Scholar]
- 55.Schuster IS, Coudert JD, Andoniou CE, and Degli-Esposti MA (2016). Natural Regulators”: NK Cells as Modulators of T Cell Immunity. Front. Immunol 7, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mocikat R, Braumüller H, Gumy A, Egeter O, Ziegler H, Reusch U, Bubeck A, Louis J, Mailhammer R, Riethmüller G, et al. (2003). Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 19, 561–569. [DOI] [PubMed] [Google Scholar]
- 57.Nakayama M, Takeda K, Kawano M, Takai T, Ishii N, and Ogasawara K (2011). Natural killer (NK)-dendritic cell interactions generate MHC class II-dressed NK cells that regulate CD4+ T cells. Proc. Natl. Acad. Sci. USA 108, 18360–18365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, and Biron CA (2009). Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J. Exp. Med 206, 2235–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krebs P, Barnes MJ, Lampe K, Whitley K, Bahjat KS, Beutler B, Janssen E, and Hoebe K (2009). NK-cell-mediated killing of target cells triggers robust antigen-specific T-cell-mediated and humoral responses. Blood 113, 6593–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nielsen N, Ødum N, Ursø B, Lanier LL, and Spee P (2012). Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS One 7, e31959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, and Lafaille JJ (2002). Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J. Exp. Med 196, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY (2005). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol 6, 1142–1151. [DOI] [PubMed] [Google Scholar]
- 63.Kim JY, Son YO, Park SW, Bae JH, Chung JS, Kim HH, Chung BS, Kim SH, and Kang CD (2006). Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp. Mol. Med 38, 474–484. [DOI] [PubMed] [Google Scholar]
- 64.Yang RK, Kalogriopoulos NA, Rakhmilevich AL, Ranheim EA, Seo S, Kim K, Alderson KL, Gan J, Reisfeld RA, Gillies SD, et al. (2012). Intratumoral hu14.18-IL-2 (IC) induces local and systemic antitumor effects that involve both activated T and NK cells as well as enhanced IC retention. J. Immunol 189, 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pol J, Kroemer G, and Galluzzi L (2016). First oncolytic virus approved for melanoma immunotherapy. OncoImmunology 5, e1115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alderson KL, Luangrath M, Elsenheimer MM, Gillies SD, Navid F, Rakhmilevich AL, and Sondel PM (2013). Enhancement of the anti-melanoma response of Hu14.18K322A by alphaCD40 + CpG. Cancer Immunol. Immunother 62, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ujas TA, Obregon-Perko V, and Stowe AM (2023). A Guide on Analyzing Flow Cytometry Data Using Clustering Methods and Nonlinear Dimensionality Reduction (tSNE or UMAP). Methods Mol. Biol 2616, 231–249. [DOI] [PubMed] [Google Scholar]
- 68.Krishnarajah S, Ingelfinger F, Friebel E, Cansever D, Amorim A, Andreadou M, Bamert D, Litscher G, Lutz M, Mayoux M, et al. (2022). Single-cell profiling of immune system alterations in lymphoid, barrier and solid tissues in aged mice. Nat. Aging 2, 74–89. [DOI] [PubMed] [Google Scholar]
- 69.Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 70.Jiang H, Lei R, Ding SW, and Zhu S (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinf 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code are publicly available. All data including supplementary information are available from the lead contact upon request.
Bulk RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database under the accession code GSE209945; access for reviewers: mbqjymwwzfwpnet
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.