

Abstract
Epicardial-derived cells (EPDCs) are involved in the regulation of myocardial growth and coronary vascularization and are critically important for proper development of the atrioventricular (AV) valves. SOX9 is a transcription factor expressed in a variety of epithelial and mesenchymal cells in the developing heart, including EPDCs. To determine the role of SOX9 in epicardial development, an epicardial-specific Sox9 knockout mouse model was generated. Deleting Sox9 from the epicardial cell lineage impairs the ability of EPDCs to invade both the ventricular myocardium and the developing AV valves. After birth, the mitral valves of these mice become myxomatous with associated abnormalities in extracellular matrix organization. This phenotype is reminiscent of that seen in humans with myxomatous mitral valve disease (MVD). An RNA-seq analysis was conducted in an effort to identify genes associated with this myxomatous degeneration. From this experiment, Cd109 was identified as a gene associated with myxomatous valve pathogenesis in this model. Cd109 has never been described in the context of heart development or valve disease. This study highlights the importance of SOX9 in the regulation of epicardial cell invasion—emphasizing the importance of EPDCs in regulating AV valve development and homeostasis—and reports a novel expression profile of Cd109, a gene with previously unknown relevance in heart development.
Keywords: Epicardium, Myxomatous Valve, Mitral Valve, Heart Development, SOX9, CD109
Graphical Abstract
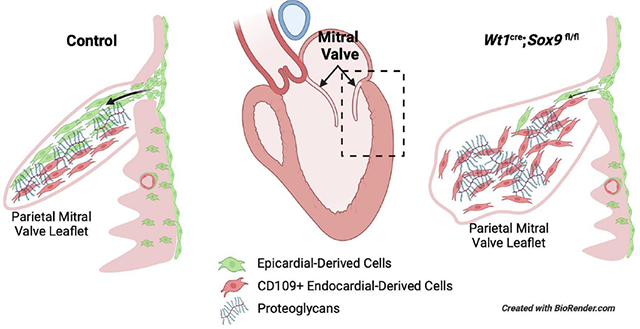
1. Introduction
The epicardium originates from the proepicardium, a heterogeneous cluster of cells situated at the venous pole of the developing heart [1]. Around murine embryonic day (E) 9.0, epicardial cells begin to migrate over the developing myocardium to form an epithelial monolayer surrounding the heart by E10.5 [1]. A subset of these cells undergo an epicardial-to-mesenchymal transformation (EpiMT) thereby giving rise to an invasive population of epicardial-derived cells (EPDCs). Beginning around E12.5, EPDCs begin to migrate into the underlying myocardium where they differentiate into cardiac fibroblasts, vascular smooth muscle cells, and coronary endothelial cells [2–5]. The epicardium and EPDCs provide paracrine signals, including fibroblast growth factors (FGFs) and insulin-like growth factors (IGFs), which are important for regulating myocardial growth, myocardial compaction, and coronary vessel patterning [6–8]. At the atrioventricular (AV) junction, a subpopulation of EPDCs accumulates forming the AV sulcus. These AV-EPDCs subsequently migrate through the AV junctional myocardium in response to localized bone morphogenic protein (BMP) signaling [9] and form the annulus fibrosus, separating atrial from ventricular myocardium [10, 11]. The AV-EPDCs then invade and populate the endocardial-derived lateral AV cushions where they eventually comprise over 50% of all the valve mesenchymal cells [11, 12]. In contrast, few AV-EPDCs are found in the AV valve leaflets that derive from the major AV cushions [11]. The critical importance of EPDCs for the proper development and maturation of the AV valves is demonstrated by the fact that perturbing the migration of AV-EPDCs into the leaflets leads to postnatal valve abnormalities [12, 13].
The transcription factor SOX9 (SRY-related high mobility group box 9) is an important regulator of epithelial-to-mesenchymal transformation (EMT), cell migration, chondrogenesis, and extracellular matrix (ECM) production [14–16]. Within the endocardial cell lineage, SOX9 is required for the proper development of the AV cushions by promoting the endocardial-to-mesenchymal transformation (EndMT) that gives rise to the mesenchymal cell population of the endocardial cushions—the precursors to the AV valves—as well as for the expansion of the endocardial-derived cushion mesenchyme after EndMT [17, 18]. SOX9 modulates the expression of other transcription factors such as TWIST1 and HAND2 known to be important for EndMT and valve development [19]. SOX9 is regulated through BMP signaling [20–22], a pathway critically important in the development of tissues at the AV junction. Localized expression of BMP2 by the AV junctional myocardium promotes EndMT and proliferation in the endocardial cushions as well as EpiMT at the AV sulcus [12, 23, 24]. Given the association between SOX9 and BMP signaling, the role of BMP signaling in AV-EPDC behavior, and the significance of SOX9 in the development of the AV valves, we decided to investigate the role of SOX9 in the development of the epicardial lineage with a particular focus on the contribution of EPDCs to the tissues of the AV junction and developing AV valves. To this end, we conditionally deleted Sox9 from the epicardial lineage in mice using the previously described epicardial-specific Wt1cre transgenic line [11].
Here, we report evidence that SOX9 promotes epicardial cell invasion during heart development. Deletion of Sox9 leads to a reduction in EPDC invasion into the ventricular myocardial wall as well as reduced EPDC contribution to the tissues of the AV junction. Postnatal specimens develop a mitral valve phenotype reminiscent of myxomatous valve disease in humans. Combined, our data highlight the requirement of SOX9 for proper epicardial invasion—emphasizing the importance of EPDCs in regulating valve development and homeostasis—and led to the identification of Cd109 as a novel candidate gene associated with AV valve and coronary system development. We propose that CD109 may be involved in the pathogenic process associated with myxomatous valve disease.
2. Materials and Methods
2.1. Animal Procedures
All experiments using mice were conducted in adherence with NIH guidelines (Guide for the Care and Use of Laboratory Animals), and according to protocols approved by the MUSC Institutional Animal Care and Use Committee (IACUC) under protocol ID number IACUC-2020-01140. The Tie2Cre and mWt1/IRES/GFP-Cre (Wt1Cre) mice were described previously [11, 25]. The B6.129S7-Sox9tm2Crm/J (Sox9fl/fl) mouse, the B6.129(Cg)-Gt(Rosa)26Sortm4(ACTB-tdTomato,EGFPLuo/J (ROSA26mTmG dual fluorescence lineage trace reporter expressing membrane bound tdTomato or eGFP, abbreviated R26mG) mouse, and the B6N.126-Gt(Rosa)26Sortm1(CAG-tdTomat*,-EGFP*)Ees/J (ROSA26nTnG dual fluorescence lineage trace reporter expressing nuclear tdTomato or eGFP, abbreviated R26nG) mouse were obtained from the Jackson Laboratory. Epicardial-specific SOX9 knockout animals (Wt1cre;Sox9fl/fl;R26mG, shorthanded SOX9EKO for “epicardial knockout” in graphs) and littermate controls (Wt1cre;Sox9fl/+;R26mG or Wt1cre;Sox9+/+;R26mG) animals were generated by crossing Wt1cre;Sox9fl/+ males with Sox9fl/+;R26mG or Sox9fl/fl;R26mG females. Mice heterozygous at the Sox9 locus (Wt1cre;Sox9fl/+;R26mG) do not present with any of the described Wt1cre;Sox9fl/fl;R26mG phenotypes and were thus deemed appropriate controls for histological and immunofluorescence analyses. Mice were euthanized by decapitation (embryonic and neonatal stages) or by isoflurane induction followed by cervical dislocation in accordance with NIH guidelines. Embryos were considered day 0.5 at midday on the day of vaginal plug detection, and staging was confirmed upon isolation using distinctive features described in The House Mouse: Atlas of Embryonic Development [26]. Both male and female specimens were generated, and combined data for both sexes are shown.
2.2. Immunofluorescence Staining and Imaging
Embryos and postnatal hearts were fixed in 4% paraformaldehyde (PFA) either for 4 hours at room temperature or overnight at 4°C. They were processed through a series of graded alcohols to dehydrate the tissue, cleared in toluene, and embedded in paraffin. Hematoxylin and Eosin (H&E) or immunohistochemical staining was performed on 5um tissue sections. Slides were deparaffinized in xylenes and rehydrated through graded alcohols. Antigen retrieval was performed by boiling slides in a pressure cooker in citric acid-based antigen unmasking solution (Vector, H3300) for 1 minute. Sections were incubated for 30 minutes with 1% bovine serum albumin (BSA) prior to immunostaining to minimize non-specific binding of primary antibodies. Sections were incubated in primary antibody overnight at 4°C, washed in PBS, and incubated with secondary antibody for 1 hour at room temperature. Serial dilutions were performed to determine optimal antibody concentrations, and secondary only controls were used to confirm signal specificity for the primary antibody. Primary and secondary antibodies and dilutions can be found in Supplemental Table 1. Slides were coverslipped with SlowFade Gold Antifade Reagent with DAPI (Invitrogen). Brightfield images were acquired using Olympus BX40. Fluorescence images were acquired with Zeiss AxioImager II, Leica TCS SP5, and Leica TCS SP8 microscopes.
2.3. Wholemount Immunofluorescent Staining
Embryonic hearts were fixed in 4% PFA overnight at 4°C. They were washed in PBS with 1% Triton X-100 3 times for 1 hour. Hearts were then incubated for 1 hour in PBS with 5% fetal bovine serum (FBS) and 1% Triton X-100. Primary antibody was diluted in blocking solution and incubation was done at 4°C overnight. 3 washes for 1 hour each were performed before incubation in secondary antibody at 4°C overnight. After 3 additional washes, hearts were stored at 4°C in glycerol until imaging.
2.4. Image Analysis and Quantification
For histological quantifications, the measure tool within the CellSens Standard imaging software was used to assess compact myocardium thickness and AV sulcus area. 8-10 sections were analyzed per specimen, and 6 fields of view were analyzed per section for thickness measurements. Researchers were blinded to genotype during measurements. For immunofluorescence images, technical adjustments to subtract background were performed in a consistent manner by applying linear adjustments of brightness and contrast equally to the entire image. For cell number and proliferation quantifications, CellProfiler 3.1.8 was used to identify and count DAPI or Ki67-positive nuclei in cropped images containing the compact myocardium, the AV sulcus, or the parietal mitral valve leaflet. 4-6 sections were analyzed per specimen. Fluorescence area of GFP and DAPI were calculated in CellProfiler 3.1.8 for quantification of EPDC invasion in cropped images containing compact myocardium or parietal mitral valve leaflet. For EPDC invasion into the myocardial wall, 4 sections were analyzed per specimen, and 6 fields of view were analyzed per section. Postnatal mitral valve morphometrics were analyzed on H&E stained sections through the entire mitral valve and volume and area per section statistics calculated in AMIRA 3D software. All data was analyzed and visualized in GraphPad Prism 9.
2.5. 3D Reconstruction of Microdissected Leaflets
Parietal mitral valve leaflets were microdissected from P5 hearts of Wt1cre;Sox9fl/fl;R26mG and littermate control mice and suspended in a droplet of immersion oil for immobilization. A Leica TMS SP5 microscope was used to take Z-stack images through the leaflet. 3D reconstructions of GFP and tdTomato expression and material statistics were generated using Amira 3D software. Researchers were blinded to genotype until after quantification.
2.6. RNA-Sequencing
Total RNA was isolated from micro-dissected parietal leaflets from n=6 P5 control (Sox9fl/fl) and epicardial-knockout (Wt1cre;Sox9fl/fl) speciments using the RNeasy Micro Kit (QIAGEN) with RNase free DNAse digestion (QIAGEN) to prevent genomic DNA contamination according to manufacturer’s protocol. RNA integrity and concentration were determined by Bioanalyzer (Agilent 2100) utilizing samples with RINs of >8. Total RNA samples were submitted to a commercial provider (Genewiz-Azenta) for paired-end sequencing at 150b read length and 20-30M reads per sample. Sequence data (fastq files) was obtained and processed using Partek® Flow® software. Pre-alignment quality assessment showed that all samples had high quality scores, with most generated data scoring >30 Phred and positional quality consistent throughout the 150b read length. Alignment was done with an implementation of STAR [27] version 2.7.8a using mouse genome mm10. Quantification was done using Partek E/M annotation model for RefSeq transcripts 95 with the following settings: 1) strict paired-end compatibility, 2) require junctions to match introns, and 3) minimum reads of 10. Given that samples may contain small traces of contaminating myocardium due to imprecise microdissection, we assessed expression levels of myocardial genes, including 28 genes linked to “regulation of the force of heart contraction” (Gene Ontology GO:0002026). Hierarchical clustering of expression profiles for these genes indicated that three samples clustered apart from the others based, in part, on elevated expression levels of Myh6, Myh7, Pln, and Ryr2. These samples were judged to have appreciable amounts of contaminating myocardium and were therefore excluded from further analysis. Differential expression analysis was done with DESeq2 [28], yielding fold changes and False Dicovery Rate (FDR) step up adjusted p-values for pairwise comparison of control (Sox9fl/fl) and Wt1cre;Sox9fl/fl parietal leaflets. FDR threshold was set at either padj≤0.1 or padj≤0.3, with a fold change (FC) threshold of >1.3. These thresholds were chosen with the aim of capturing high-probability differentially expressed genes (DEGs) with the lower padj threshold of padj≤0.1, while avoiding the loss of false negatives with the more permissive threshold of padj≤0.3, thereby providing a larger list of genes for downstream functional analysis. Functional analysis was conducted using ToppFun [29]. Raw data (fastq files) from this study are archived in NCBI Gene Expression Omnibus (accession number GSE236187).
2.7. Echocardiography and Electrocardiogram Analysis
Echocardiography and isochronal capture of electrocardiogram was performed as previously described [30].
2.8. Statistics
Statistical analyses were conducted in GraphPad Prism 9 using Student’s unpaired t-test for hypothesis testing. P-values of less than 0.05 were considered significant, indicated by asterisks (*< 0.05; **<0.01; ***<0.001). P-values between 0.05 and 0.1 are displayed. Graphical points represent the mean values of each specimen plotted against the group mean, with error bars depicting ±s.e.m. or ±s.d. as indicated in figure descriptions. n=3-4 per genotype unless otherwise indicated.
3. Results
3.1. Epicardial SOX9 expression in the developing heart
SOX9 is expressed in various cell populations of the developing heart, including the endocardial-derived mesenchyme, cardiac neural crest-derived cells, and the mesenchyme derived from the Second Heart Field [18, 31, 32]. To specifically establish the spatiotemporal expression profile of SOX9 in the epicardial cell lineage, we conducted immunofluorescent labeling of serially sectioned hearts at different stages of development. In order to delineate/identify the epicardium and EPDCs, the Wt1cre mouse was used in combination with the ROSA26mT/mG (abbreviated R26mG) dual-fluorescence transgenic reporter mouse, essentially as described before [11]. In this Wt1cre;R26mG model, cells in the epicardial cell lineage can be identified by their GFP expression (detected immunofluorescently).
In the earliest stage of epicardial development, embryonic day (E) 9.5, SOX9 is expressed in the proepicardium, early epicardium, and mesenchymal cells within the AV cushions (Fig. 1A,A’). These same groups of cells maintain SOX9 expression as the epicardium begins to envelop the heart at E10.5 (Fig. 1B). As expected, these cells in both the proepicardium and the early epicardium also express the mesothelial transcription factor WT1 (Fig. 1C). Not all WT1-expressing cells in the proepicardium have undergone cre-mediated recombination of the R26mG reporter allele resulting in GFP expression. However, the majority of epicardial cells which have contacted the myocardium by E10.5 do express GFP (Fig. 1B,C). Importantly, at E12.5, SOX9 expression can be seen in epicardial cells which begin to undergo EpiMT to invade the ventricular myocardial wall (Fig. 1D). Additionally, AV-EPDCs in the sulcus upregulate SOX9 expression as they migrate towards the AV junctional myocardium (Fig. 1E). This expression profile through early epicardial development suggests a role for SOX9 in EpiMT and EPDC invasion behavior.
Figure 1:
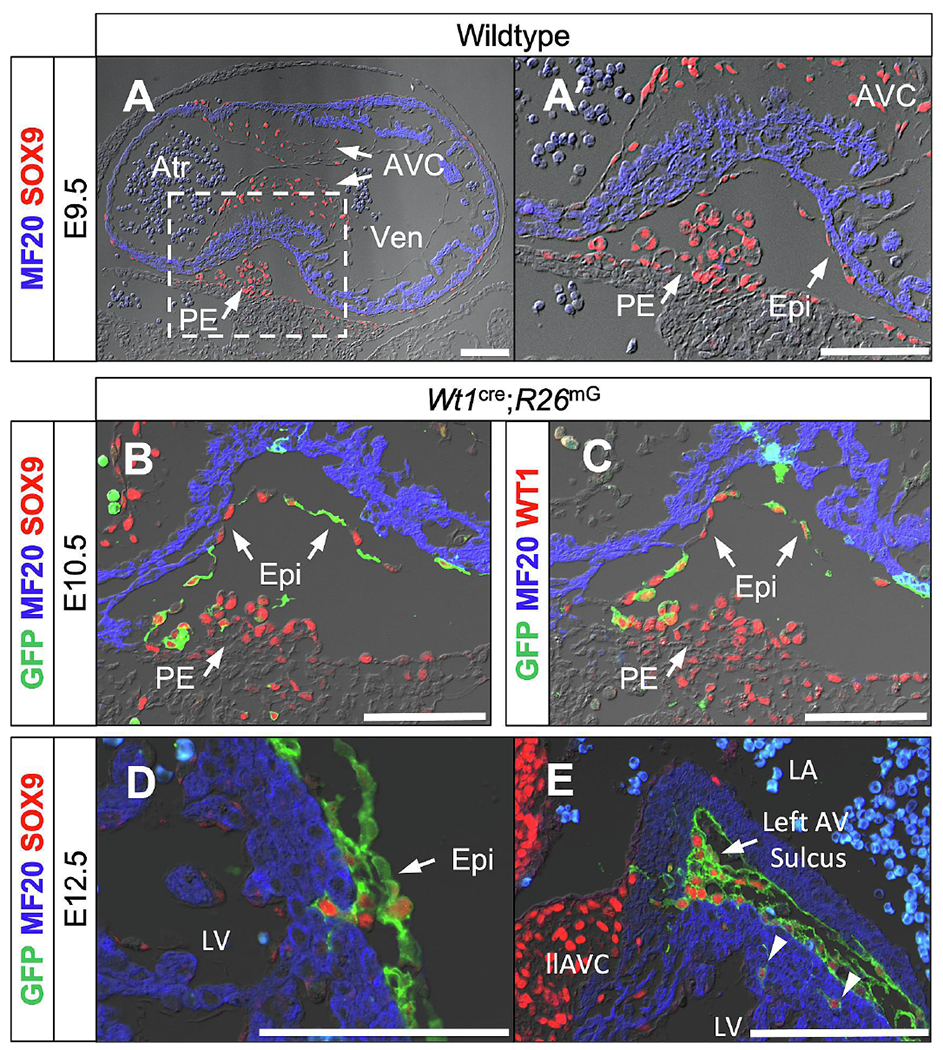
Epicardial SOX9 expression in the developing heart. (A,A’) SOX9 (red) expression in the proepicardium and the early epicardium as it contacts and envelops the myocardium (MF20, blue) at E9.5 in wildtype embryos. (B,C) SOX9 (red in B) expression and mesothelial transcription factor Wilms Tumor 1 (WT1, red in C) expression in the proepicardium and epicardial cells enveloping the myocardium at E10.5. Note that R26mG GFP reporter expression (green) is not in all proepicardial WT1-expressing cells at this timepoint, but that the majority of epicardial cells have undergone Wt1cre-mediated recombination. (D,E) SOX9 expression in epicardial cells undergoing EpiMT and migrating into the myocardium of the left ventricle (D, arrowheads in E) and AV junctional myocardium at the AV sulcus (E) at E12.5. Scale bars = 100um. (Atr, common atrium; AVC, atrioventricular cushion; Epi, epicardium; llAVC, left lateral atrioventricular cushion; LV, left ventricle; PE, proepicardium; RV, right ventricle; Ven, common ventricle)
3.2. Creating an epicardial-specific Sox9 knockout mouse model
To determine the importance of SOX9 in the various epicardial subpopulations, we generated an epicardial-specific Sox9 knockout mouse model using the Wt1cre mouse in combination with a floxed Sox9 (Sox9fl/fl) mouse [11, 33]. In order to determine the fate of the EPDCs after Sox9 deletion, the R26mG reporter construct was crossed in as well [12, 34]. This allowed us to determine how deleting Sox9 affected the spatial distribution of EPDCs in Wt1cre;Sox9fl/fl;R26mG mice. Mice heterozygous at the Sox9 locus (Wt1cre;Sox9fl/+;R26mG) did not present with any of the phenotypes described in their knockout littermates (see below) and were used as controls for our analyses.
3.3. Loss of SOX9 in the epicardial lineage results in impaired development of the compact layer of the ventricular wall
The epicardium and EPDCs provide paracrine signals to the myocardium such as insulin like growth factors (IGFs) and fibroblast growth factors (FGFs) that regulate myocardial growth and maturation [6, 35]. As SOX9 has been reported to modulate the actions of several of these growth factors [36], we sought to examine the effect of epicardial-specific loss of SOX9 on myocardial growth. At E12, we did not observe significant differences in the thickness of the compact myocardium of Wt1cre;Sox9fl/fl;R26mG specimens compared to littermate controls (Fig. 2A–C). There was no significant difference in the percentage of proliferating cardiomyocytes in the compact myocardium of Wt1cre;Sox9fl/fl;R26mG specimens compared to littermate controls at E12 as determined by KI-67 immunolabeling (Fig. 2D). These results suggest that SOX9 in the epicardium proper does not play an essential role in the initial development of the compact myocardium. In two out of four Wt1cre;Sox9fl/fl;R26mG specimens examined at this time point, regions of epicardial detachment were observed (Supplemental Fig. S1). At E14.5 this phenotype was not observed any longer. When we examined the thickness of the compact myocardial layers of the left and right ventricles in Wt1cre;Sox9fl/fl;R26mG mice at E14.5, they were found to be significantly thinner compared to controls (Fig. 2E–G). To determine if this thinner myocardium was due to decreased cardiomyocyte proliferation, we analyzed the percentage of proliferating cardiomyocytes by KI-67 and MF20 immunolabeling. Wt1cre;Sox9fl/fl;R26mG specimens were found to have a reduced percentage of proliferating cardiomyocytes compared to controls (Fig. 2H).
Figure 2:
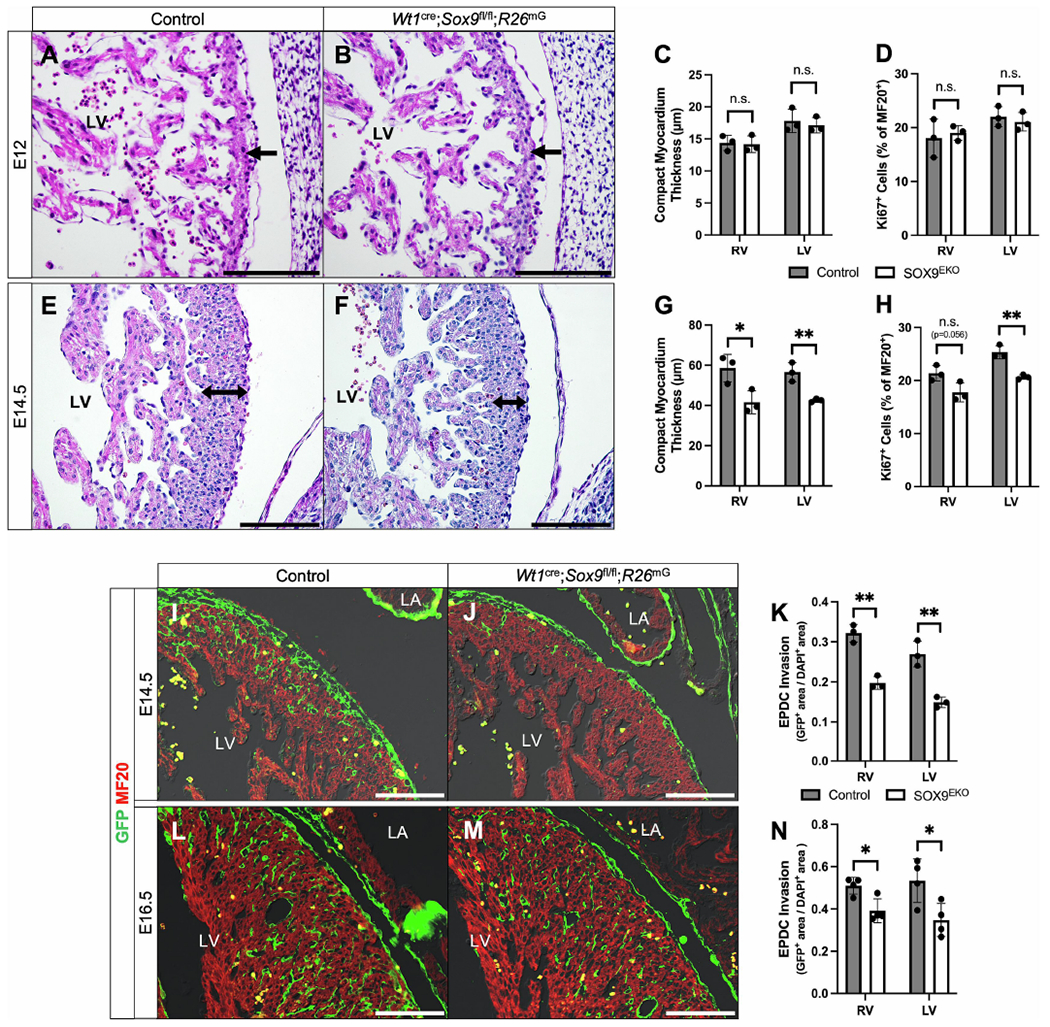
SOX9 in the epicardium is required for proper myocardial growth. (A-H) Compact myocardial thickness and proliferation measurements in control and Wt1cre;Sox9fl/fl;R26mG specimens at E12 (A-D) and E14.5 (E-H). At E12, there was no difference in the thickness of the compact myocardium (black arrows) between control hearts (A) and littermate Wt1cre;Sox9fl/fl;R26mG hearts (B), quantified in (C) (n=3). Proliferation of cardiomyocytes as calculated by KI67 index was no different at this stage (D). However, at E14.5, Wt1cre;Sox9fl/fl;R26mG mice (F) had reduced compact myocardial thickness (double black arrows) relative to controls (E). Quantified in (G), the compact myocardium of the right and left ventricle of Wt1cre;Sox9fl/fl;R26mG hearts were 29% and 25% thinner than controls, respectively (n=3). This phenotype was accompanied by a 16% and 20% reduction in cardiomyocyte proliferation in the right and left ventricle, respectively (H) (n=3). (I-N) Immunofluorescent stains show EPDCs (GFP, green) invading the ventricular myocardium (MF20, red) in control (I,L) and Wt1cre;Sox9fl/fl;R26mG (J,M) hearts at E14.5 (I,J) and E16.5 (L,M), quantified in (K, n=3) and (N, n=4), respectively. At E14.5, there was an approximately 40% reduction in the number of EPDCs that had invaded the myocardium in comparison to controls (I). At E16.5, this decrease was approximately 30% (L). Datapoints represent the average value for each specimen and are plotted against the group average for each genotype. Error bars indicate mean ±s.e.m., unpaired t-test, *p<0.05, **p<0.01. Scale bars = 100um. (LA, left atrium; LV, left ventricle).
Importantly, E12.5 is the timepoint during which EPDCs begin to invade the compact myocardial layer of the ventricular wall (Fig. 1D, arrowheads in 1E). Since SOX9 has been implicated in the regulation of cell invasion [15, 37, 38], we next examined the effect of loss of SOX9 on EPDC invasion into the underlying myocardium. At E14.5 there was a significant reduction in the number of EPDCs that had invaded the compact layer of the ventricular walls in Wt1cre;Sox9fl/fl;R26mG specimens when compared to controls (Fig. 2I–K). Significantly reduced numbers of EPDCs were also observed in the ventricular walls of E16.5 Wt1cre;Sox9fl/fl;R26mG specimens when compared to controls. (Fig. 2L-N). However, the thin myocardial phenotype was no longer observed at this stage (Supplemental Fig. S2). Overall, these data support that SOX9 is required for the normal invasion of EPDCs into the compact layer of the ventricular myocardium, which is in turn important for proper compact myocardial proliferation and maturation. Echocardiographic analysis of Wt1cre;Sox9fl/fl;R26mG and control specimens at 2 and 4 months of age did not reveal any significant differences in cardiac function resulting from the delayed myocardial maturation during embryonic development (Supplemental Table 2).
3.4. Loss of SOX9 may preferentially affect the differentiation of the epicardial-derived interstitial fibroblast lineage
EPDCs that migrate into the myocardium primarily differentiate into two lineages: interstitial fibroblasts and pericytes/vascular smooth muscle cells (VSMCs) [5]. Genes such as Tcf21, Pdgfra, and the Hippo kinases Lats1/2 have all been specifically tied to epicardial-derived fibroblast differentiation [37, 39, 40], while Tbx18 and Pdgfrb have been implicated in VSMC differentiation [37, 41, 42], highlighting distinct regulatory mechanisms at play in the differentiation of epicardial derivatives. Qualitative assessment of the mesenchymal marker PDGFRα required for epicardial-derived fibroblast differentiation revealed that at E14.5, there was a notable reduction in the number of PDGFRα-positive EPDCs in the myocardial wall of Wt1cre;Sox9fl/fl;R26mG specimens in comparison to controls (Supplemental Fig. S3 A,B). When we examined the expression patterns of the smooth muscle cell marker alpha-smooth muscle actin (αSMA) around the coronary arteries as they begin to mature at E16.5, there was no distinguishable difference between Wt1cre;Sox9fl/fl;R26mG mice and controls (Supplemental Fig. S3 C,D). The coronary vessels in both control and Wt1cre;Sox9fl/fl;R26mG specimens were surrounded by GFP-positive αSMA-expressing epicardial-derived VSMCs (Supplemental Fig. S3 C,D). These observations suggest that loss of SOX9 may preferentially affect the differentiation of the epicardial-derived interstitial fibroblasts.
3.5. Loss of SOX9 results in hypoplasia of the AV sulcus
SOX9 is important for EndMT generating the endocardial-derived mesenchyme of the AV cushions [18] and for the expansion of this population of cells [17]. Since similar events are also responsible for the formation of the epicardial-derived mesenchymal cells in the AV sulcus (i.e. EpiMT and the expansion of the population of AV-EPDCs), we turned our attention to this specific area at the AV junction. Immunolabeling for SOX9 in control hearts revealed upregulation of SOX9 expression by EPDCs as they undergo EpiMT and migrate towards the AV junctional myocardium (Fig. 1E, Fig. 3A,A’, Supplemental Fig. S4). The EPDCs in the AV sulcus of Wt1cre;Sox9fl/fl;R26mG specimens do not, as expected, express SOX9 (Fig. 3B,B’, Supplemental Fig. S4). At E14.5, the contribution of EPDCs to the lateral AV cushion is negligible in both control and Wt1cre;Sox9fl/fl;R26mG specimens (Fig. 3A,B arrowheads). Analyses of the right and left AV sulci in Wt1cre;Sox9fl/fl;R26mG mice revealed significant reductions in overall size and in the number of AV-EPDCs in comparison to controls at E12 (Supplemental Fig. S4) and E14.5 (Fig. 3C–H).
Figure 3:
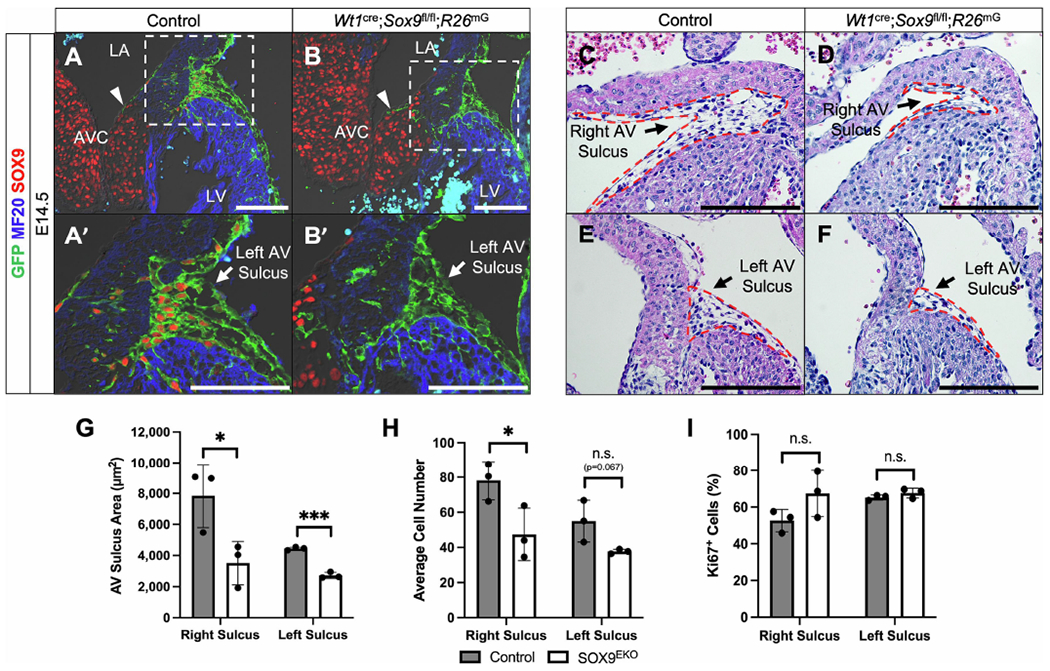
Deletion of Sox9 from the epicardium results in hypoplastic AV sulcus. (A-B’) Immunostaining reveals the transcription factor SOX9 (red) is upregulated in EPDCs (GFP, green) as AV epicardial cells undergo EpiMT and migrate towards the AV junctional myocardium (MF20, blue) in control (Wt1cre;Sox9fl/+;R26mG) (A,A’) but not Wt1cre;Sox9fl/fl;R26mG specimens (B,B’) at E14.5. Note that few EPDCs have invaded the parietal leaflet at this stage (arrowheads in A,B). (C-F) H&E stains show that, compared to control hearts (C,E), Wt1cre;Sox9fl/fl;R26mG specimens (D,F) have a reduced AV sulcus size (red dotted lines). (G,H) Quantification of the sulcus areas and cell numbers show a significant reduction in both the right and left AV sulci in Wt1cre;Sox9;R26mG specimens compared to controls (n=3). (I) Quantification of proliferation within the AV sulcus shows no difference in the percentage of proliferating cells between Wt1cre;Sox9fl/fl;R26mG specimens and controls (n=3). Datapoints represent the average value for each specimen and are plotted against the group average for each genotype. Error bars indicate mean ±s.e.m. (G,H) or mean ±s.d. (I), unpaired t-test, *p<0.05, ***p<0.001. Scale bars = 100um. (AVC, atrioventricular cushion; LA, left atrium; LV, left ventricle)
We next looked at markers of proliferation (KI-67) and apoptosis (cleaved caspase-3) to determine if the smaller AV sulci in Wt1cre;Sox9fl/fl;R26mG specimens were a result of a reduction in EPDC proliferation and/or an increase in EPDC apoptosis. No significant difference was observed in the percentage of proliferating cells within the sulcus of control and Wt1cre;Sox9fl/fl;R26mG hearts at either E12 (Supplemental Fig. S4) nor E14.5 (Fig. 3I). In addition, no cleaved caspase-3 expressing cells were observed within the sulcus at E14.5 (Supplemental Fig. S5), indicating that the smaller sulcus in Wt1cre;Sox9fl/fl;R26mG mice did not result from increased apoptosis. The fact that the AV sulcus was hypocellular with no significant difference in the percentage of proliferating or apoptotic cells suggested that the presence of fewer cells in the AV sulci of Wt1cre;Sox9fl/fl;R26mG mice was the result of impaired EpiMT. However, it is possible that the smaller sulcus size could be due to a reduction in the proliferation of AV-EPDCs at a timepoint earlier than E12.
3.6. Loss of SOX9 in the epicardial lineage results in reduced EPDC contribution to parietal AV valve leaflets and annulus fibrosis
We previously showed that EPDCs contribute to the parietal leaflets of the AV valves, but not to the leaflets that derive from the major AV cushions [11]. Additionally, we reported that perturbing EPDC contribution to parietal leaflets through epicardial-specifc deletion of the BMP receptor ALK3 led to valve abnormalities [12]. Given the similarities between the AV sulcus phenotype of our previously published Wt1cre;Alk3fl/fl;R26mG model and our Wt1cre;Sox9fl/fl;R26mG specimens, we hypothesized that the loss of SOX9 would also decrease the contribution of EPDCs to the parietal AV valve leaflets. To test this hypothesis, we examined the mitral valves of E16.5 hearts from control and Wt1cre;Sox9fl/fl;R26mG specimens. Immunofluorescence analyses revealed that the number of EPDCs in the parietal mitral valve leaflet of Wt1cre;Sox9fl/fl;R26mG specimens was reduced by more than 50% when compared to controls (Fig. 4A–C). There was, however, no significant difference in the average overall cell number in the parietal leaflet of control and knockout mitral valves (Fig. 4D), indicating a compensatory expansion of a different cell lineage, most likely from endocardial and/or hematopoietic origin.
Figure 4:
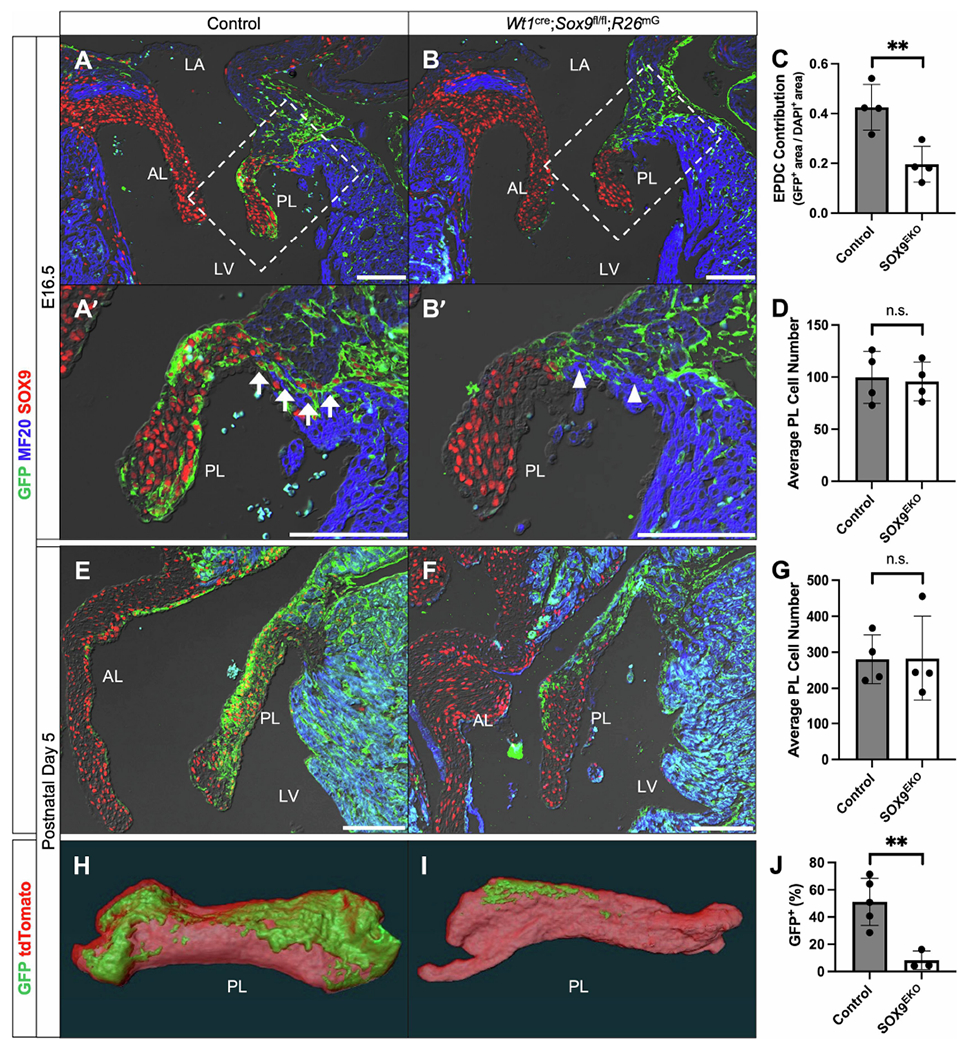
Deletion of Sox9 from the epicardium results in reduced EPDC contribution to parietal mitral valve leaflet. (A-B’) Immunostaining reveals a dramatic reduction in the number of EPDCs (GFP, green) that populate the parietal mitral valve leaflet (PL) in Wt1cre;Sox9fl/fl;R26mG specimens (B,B’) compared to controls (Wt1cre;Sox9fl/+;R26mG) (A,A’) at E16.5. A continuous line of EPDCs form the annulus fibrosis in control specimens (arrows), while Wt1cre;Sox9fl/fl;R26mG specimens display regions of continuity between atrial and ventricular myocardium (arrowheads). (C-D) Quantifications reveal that, while there is no significant difference in the average number of cells in the parietal leaflet (D), Wt1cre;Sox9fl/fl;R26mG specimens have a more than 50% reduction in the contribution of EPDCs compared to controls at E16.5 (C) (n=4). (E-F) Immunostaining at P5 shows that the reduction of EPDCs (GFP, green) in Wt1cre;Sox9fl/fl;R26mG specimens (F) compared to controls (E) persists into early postnatal stages. Notably, at both E16.5 and P5, non-EPDCs (GFP-negative) within the leaflets of Wt1cre;Sox9fl/fl;R26mG specimens still express SOX9. (G) Quantifications show no significant difference in the average number of cells in the parietal leaflet at P5 (n=4). (H-I) Three-dimensional reconstructions of GFP and tdTomato fluorescence in microdissected wholemount parietal leaflets reveal decreased contribution of EPDCs to the parietal leaflet of Wt1cre;Sox9fl/fl;R26mG specimens (I) compared to controls (H). (J) Quantification of the GFP-positive area shows a reduction of EPDC contribution from over 50% in controls to less than 10% in Wt1cre;Sox9fl/fl;R26mG parietal leaflets (n=5 control, n=3 Wt1cre;Sox9fl/fl;R26mG). Datapoints represent the average value for each specimen and are plotted against the group average for each genotype. Error bars indicate mean ±s.e.m. (C,D,G) or mean ±s.d. (J), unpaired t-test, *p<0.05, **p<0.01. Scale bars = 100um. (AL, anterior leaflet; LA, left atrium; LV, left ventricle; PL, parietal leaflet)
The annulus fibrosis is an epicardial-derived, fibrous tissue boundary between atrial and ventricular myocardium at the AV junction important for maintaining electrical discontinuity and preventing ventricular pre-excitation [1, 43]. EPDCs secrete collagens, periostin, and other arrhythmogenic matrix components important for maintaining this border [11, 43]. While control specimens had a continuous line of EPDCs separating the atria and ventricles by E16.5 (Fig. 4A’, arrows), Wt1cre;Sox9fl/fl;R26mG specimens did not all have this same distinct border (Fig. 4B’, arrowheads). To examine if this observation resulted in any functional defects postnatally, we performed ECG analyses on Wt1cre;Sox9fl/fl;R26mG and littermate-matched control specimens at 2 and 4 months of age. No conduction abnormalities (differences in PR interval, QRS duration, or RR interval) were observed in Wt1cre;Sox9fl/fl;R26mG compared to controls (Supplemental Table 2).
3.7. Wt1cre;Sox9fl/fl;R26mG specimens develop myxomatous mitral valves postnatally
We next examined the mitral valves of control and Wt1cre;Sox9fl/fl;R26mG specimens at an early postnatal timepoint. Consistent with embryonic observations, the parietal leaflets of Wt1cre;Sox9fl/fl;R26mG at postnatal day 5 (P5) showed a dramatic reduction in the number of EPDCs when compared to littermate controls (Fig. 4E,F). As was the case at E16.5, despite this reduction, the overall number of cells within the leaflets was comparable to that of the controls (Fig. 4G). Three-dimensional reconstructions of GFP expression in micro-dissected parietal leaflets from control and Wt1cre;Sox9fl/fl;R26mG mitral valves revealed a decrease in GFP expression from over 50% of the total leaflet volume in controls to less than 10% in knockouts (Fig. 4H–J). No indications of valve abnormalities at a structural level were observed in Wt1cre;Sox9fl/fl;R26mG specimens at P5.
We next sought to determine the effect of reduced EPDC contribution on the morphology of adolescent and adult valves. The mitral valves of 20 Wt1cre;Sox9fl/fl;R26mG and 17 littermate-matched control specimens from 4-10 weeks of age were examined. While histological assessment of control specimens consistently revealed thin and compacted mitral valve leaflets (Fig. 5A,C), 60% of Wt1cre;Sox9fl/fl;R26mG specimens (12/20) displayed abnormalities with enlarged, misshapened leaflets, and disorganized ECM reminiscent of myxomatous valve degeneration (MVD) in the human (Fig. 5A–D). Quantitative analysis of total leaflet volume and area per section revealed an approximately 60% average increase in the size of the parietal leaflet in Wt1cre;Sox9fl/fl;R26mG specimens compared to controls (Fig. 5E,F). MVD is characterized by fibromyxomatous changes to the ECM that can lead to prolapse and mitral regurgitation [44, 45]. Altered metabolism and accumulation of collagens, proteoglycans, and glycosaminoglycans lead to leaflet enlargement and pathological changes to the ECM which can cause biomechanical incompetence [46, 47]. Immunofluorescent analyses of enlarged postnatal Wt1cre;Sox9fl/fl;R26mG valve leaflets revealed expanded expression of the chondroitin sulfate proteoglycan versican (VCAN) and a loss of the laminar organization of type 1 collagen (COL1) (Fig. 5G–J). All specimens with valvular abnormalities had myxomatous phenotypes in the parietal leaflet, while many had defects in both parietal and anterior leaflets, indicative of a pathogenic remodeling of the anterior leaflet secondary to the loss of EPDCs in the parietal leaflet. These observations highlight the importance of the epicardial lineage of VICs in maintaining proper leaflet integrity throughout valve maturation.
Figure 5:
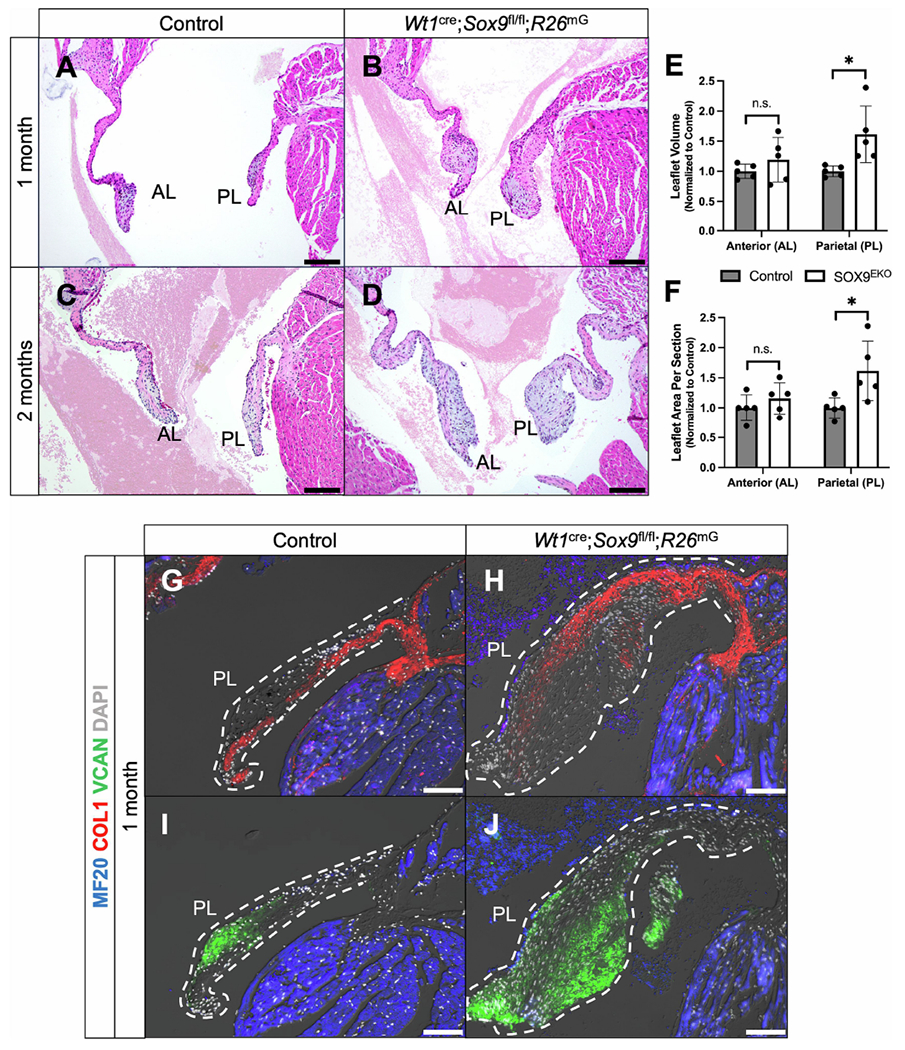
Wt1cre;Sox9fl/fl;R26mG specimens develop myxomatous mitral valve degeneration postnatally. (A-D) H&E stains show enlarged mitral valve leaflets in Wt1cre;Sox9fl/fl;R26mG specimens (B,D) compared to controls (A,C) at 1 month (A,B) and 2 months (C,D) postnatally. Blood in the ventricular and atrial lumen is obscured with a semi-transparent filter for clearer visualization of valve leaflets. For original images, see Supplemental Fig. S3. (E,F) Quantifications of the leaflet volume and area per section in postnatal specimens 4-10 weeks of age reveal an approximately 60% increase in the size of the parietal leaflet volume (E) and area per section (F) of Wt1cre;Sox9fl/fl;R26mG specimens compared to controls (n=5). (G-J) Immunostaining for VCAN (green) and COL1 (red) show expanded expression of VCAN and a loss of the laminar organization of COL1 in 1 month postnatal Wt1cre;Sox9fl/fl;R26mG specimens. Scale bars = 100um. (AL, anterior leaflet; PL, parietal leaflet)
3.8. RNA-sequencing reveals dysregulation of genes involved in valve development and homeostasis
A number of studies have highlighted the heterogeneity among populations of valve interstitial cells (VICs), a topic of great interest in valve pathobiology [48–51]. However, these studies examining the cellular make-up of the valves in mice and humans did not specifically address or take into account that EPDCs contribute the majority of VICs to the parietal leaflet of the mitral valve. The valve abnormalities observed in our earlier study of the Wt1cre;Alk3fl/fl;R26mG [12], as well as our data in the current study (Fig. 5), demonstrate the necessity of elucidating the specific role of this lineage in valve development and in maintaining valve homeostasis in health and disease. To gain further insight into the transcriptomic landscape of Wt1cre;Sox9fl/fl;R26mG parietal leaflets that lack a proper contribution of EPDCs, we performed bulk RNA-sequencing followed by differential expression analysis on RNA isolated from parietal leaflets of 5 control (Cre-negative Sox9fl/fl) and 4 littermate Wt1cre;Sox9fl/fl specimens at postnatal day 5 (Fig. 6A). This time point was strategically chosen to capture the changes that the parietal mitral valve leaflets of Wt1cre;Sox9fl/fl mice are undergoing at the early stages of valve pathogenesis prior to observations of myxomatous degeneration.
Figure 6:
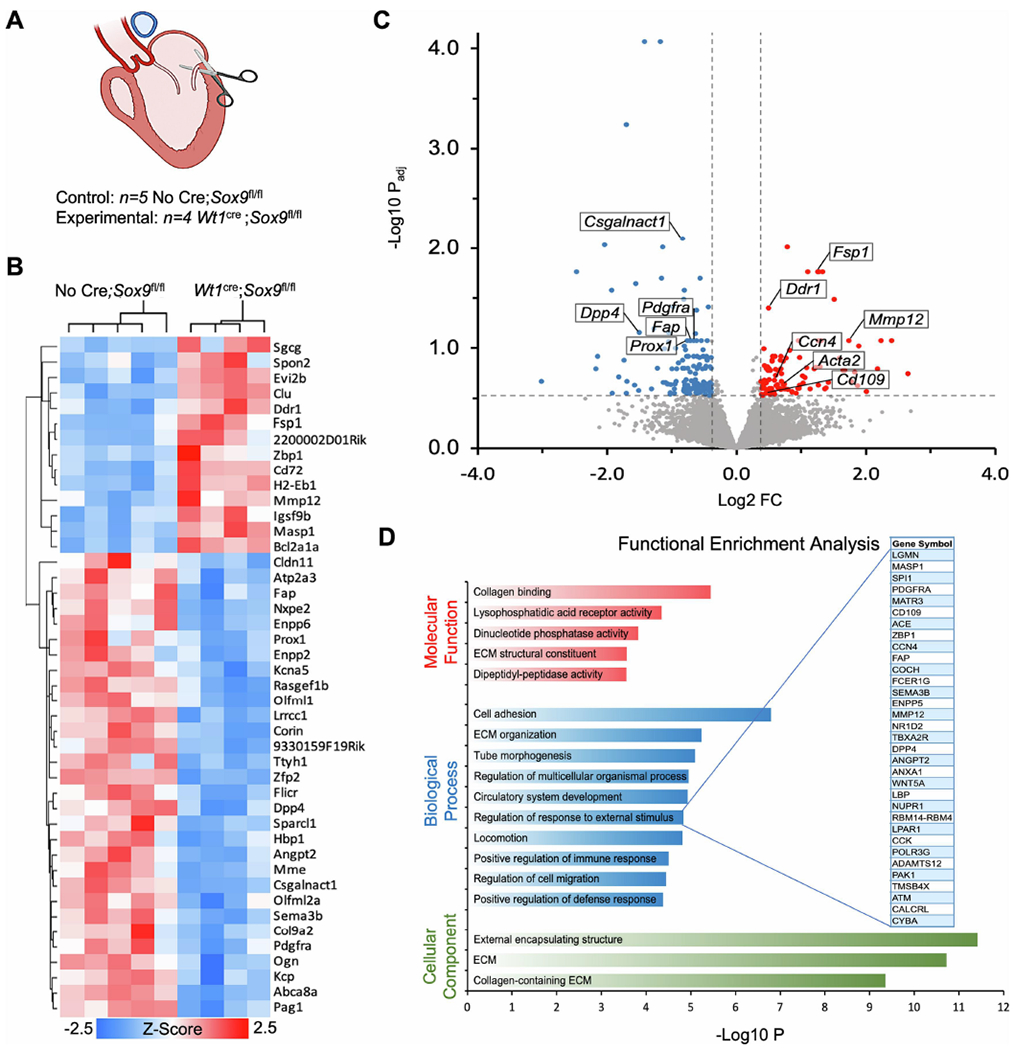
RNA-sequencing of Wt1cre;Sox9fl/fl;R26mG parietal leaflet show dysregulation in ECM organization. (A) Schematic depicting the parietal leaflet tissue that was microdissected for bulk RNA-sequencing at postnatal day 5. (B) A heat map showing hierarchical clustering of the 44 genes that were differentially expressed (DESeq2, padj<0.1, FC>1.3) between the parietal leaflets of Wt1cre;Sox9fl/fl mice compared to controls (No Cre;Sox9fl/fl). (C) Volcano plot showing 246 genes that were differentially expressed at a more permissive threshold (DESeq2, padj<0.3, FC>1.3). Labeled genes have been previously reported in the literature to have important roles in valve development, epicardial biology, and ECM organization. (D) Functional enrichment analysis in ToppFun using the differentially expressed genes from (C) as input reveals significant enrichment in a variety of molecular functions (red), biological processes (blue), and cellular components (green) related to ECM organization and cell migration. Genes from our data included in the term “Regulation of response to external stimulus” are expanded.
Differential expression analysis identified 44 DEGs with padj≤0.1 and FC>±1.3 (Fig. 6B). These 44 transcripts included genes known to be involved in ECM organization such as Dpp4, Fap, Ddr1, Csgalnact1, and Mmp12, as well as genes previously reported to be important for valve development and remodeling, such as Fsp1, Pdgfra, and Prox1 (Fig. 6B,C) [52–54]. In an effort to gain biologically informative insight into functional enrichments in our dataset, we input 246 DEGs with padj≤0.3 and FC>±1.3 into the ToppGene suite’s candidate gene identification tool ToppFun [29]. This analysis revealed enrichment in molecular functions including collagen binding and ECM structural constituent, and biological processes such as cell adhesion, ECM organization, regulation of response to external stimulus, and regulation of cell migration (Fig. 6D). Interestingly, within the enriched Biological Process term “regulation of response to external stimulus,” Wt1cre;Sox9fl/fl;R26mG leaflets showed upregulation of a number of genes reported to be positively associated with disease severity of myxomatous degeneration in canines, including Ccn4 (also called Wisp1), Mmp12, and Acta2 [55] (Fig. 6C,D). As a whole, our RNA-sequencing data suggest that the EPDC-depleted parietal leaflets of neonatal Wt1cre;Sox9fl/fl;R26mG mice have dysregulated processes of ECM remodeling conducive to the disorganization and accumulation of ECM observed in postnatal myxomatous valve leaflets. (For a complete list of DEGs with padj≤0.3 and FC>±1.3, see Supplemental Table 3).
3.9. Identification of Cd109 as a gene with novel implications for valve development and disease
Among the genes that were upregulated in Wt1cre;Sox9fl/fl;R26mG parietal leaflets compared to controls was Cd109, identified from the enriched Biological Process “regulation of response to external stimulus” (Fig. 6C,D, Supplemental Table 3). While CD109 has previously been reported to act as a Transforming Growth Factor Beta (TGF-β) co-receptor and signaling modulator in other settings [56–59], it has, unlike many other genes identified in our dataset, never been described in the context of cardiovascular development. Due to the involvement of TGF-β signaling in valve development and myxomatous valve disease pathogenesis [60–63], we decided to further investigate CD109 expression during heart development.
Immunofluorescent stainings revealed CD109 expression throughout both leaflets of the developing mitral valve at E15.5 (Fig. 7A). While EPDCs have begun to populate the parietal leaflet in control specimens (Fig. 7A), there are few EPDCs in the parietal leaflet of the Wt1cre;Sox9fl/fl;R26mG specimens (Fig. 7B). As EPDCs begin to populate the majority of the parietal leaflet in control specimens, CD109 expression appears to be restricted primarily to the non-epicardial, GFP-negative cells towards the distal tip of the leaflet (Fig. 7C). In contrast, the leaflets of Wt1cre;Sox9fl/fl;R26mG mice which contain significantly reduced numbers of AV-EPDCs displayed an expanded population of CD109-positive cells (Fig. 7D). This same pattern is observed in the early postnatal stages at P5 (Fig. 7E,F), confirming our RNA-seq observations. Additionally, immunofluorescent analyses revealed an expanded population of CD109-expressing cells in neonatal Wt1cre;Alk3fl/fl specimens compared to controls (Supplemental Fig. S7). This previously described model also has reduced EPDC contribution to the mitral valve, resulting in myxomatous valve abnormalities postnatally [12]. As anticipated, we did not observe any differences in CD109 expression in the anterior leaflet between control and Wt1cre;Sox9fl/fl;R26mG or Wt1cre;Alk3fl/fl specimens, as the cellular composition of this leaflet is not altered by genetic perturbations specific to the epicardial lineage (Fig. 7A,B, Supplemental Fig. S7A,B).
Figure 7:
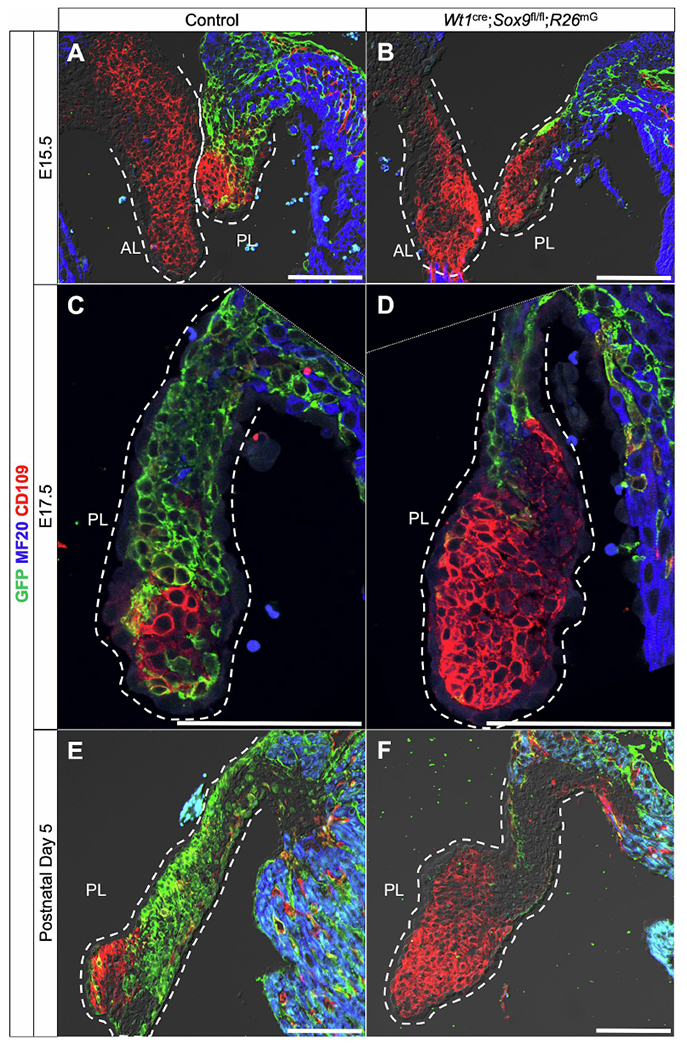
CD109 is upregulated in Wt1cre;Sox9fl/fl;R26mG parietal leaflets. Immunolabeling shows CD109 (red) is expressed in both leaflets of the mitral valve at E15.5 in both control and Wt1cre;Sox9fl/fl;R26mG specimens (A,B). Later in development at E17.5, CD109 expression becomes spatially restricted to a small population, primarily of non-EPDCs, in the distal tip of the parietal leaflet of control specimens (C), while Wt1cre;Sox9fl/fl;R26mG leaflets nearly devoid of EPDCs (green) show an expanded population of CD109-expressing cells (D). At P5, CD109 is similarly expressed by a population of non-EPDCs in the parietal leaflet which is expanded in Wt1cre;Sox9fl/fl;R26mG specimens (F) when compared to controls (E), confirming observations from RNA-seq data.
To determine the origin of these CD109-expressing cells, we next compared tissue sections from Wt1cre;R26mG specimens (Fig. 8A–B’) to sections from the endothelial-specific Tie2cre mouse crossed with R26nG mouse (in which GFP reporter expression is localized to the nucleus) (Fig. 8C–D’). In the mitral valve leaflets of E14.5 and E16.5 Tie2cre;R26nG embryos, nearly all CD109-expressing cells were co-labeled with GFP (Fig. 8C–D’), in contrast to Wt1cre;R26mG embryos in which most CD109-expressing cells are GFP-negative (Fig. 8A–B’). This complementary expression pattern in these two models confirms that CD109 in the mitral valve is preferentially expressed by the endocardial cell lineage (Fig. 8B’,D’). To our knowledge, this is the first protein that preferentially marks non-EPDCs within the developing mitral valve, and as such, could prove extremely useful as a marker in mouse models without lineage trace reporters. We also observed CD109 expression in the mesenchyme of diverse origins in the developing semilunar valves, similarly restricted spatially to the distal ends of the developing leaflets (Supplemental Fig. S8). This lineage-associated expression of CD109 in the AV valves combined with its reported role in modulating TGF-β-dependent ECM production [57, 58, 64] warrants further in vitro and in vivo investigations into the specific roles of CD109 in the context of valve development and disease pathogenesis.
Figure 8.
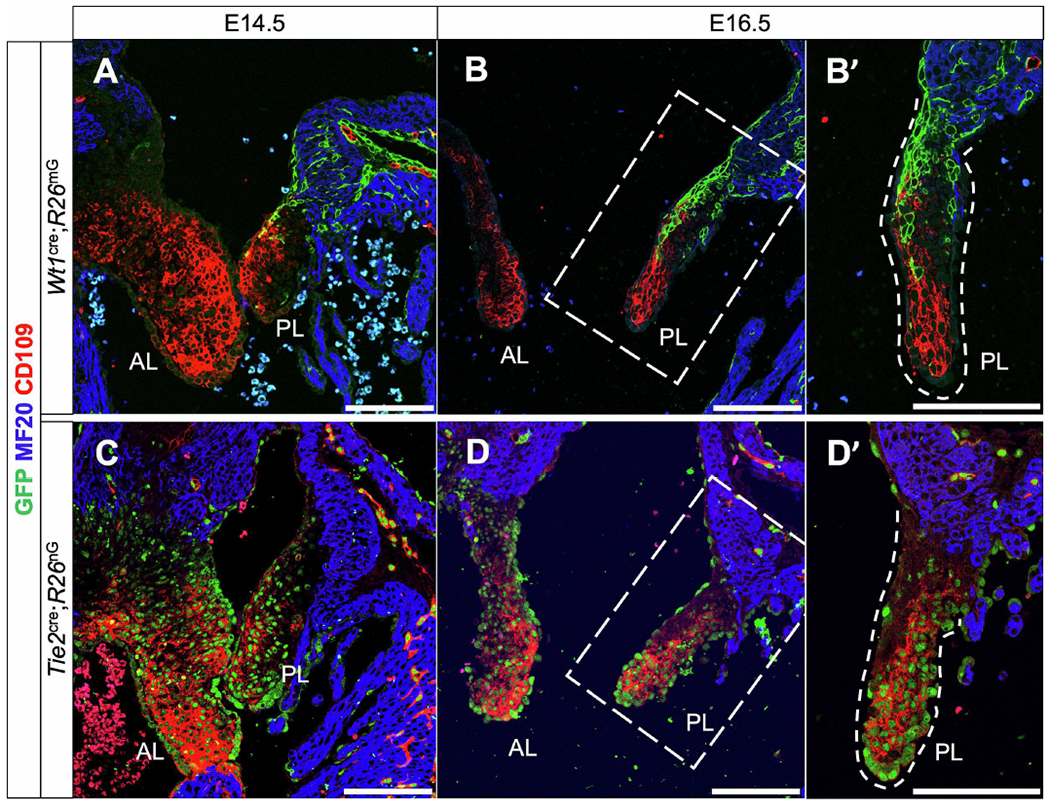
CD109 is expressed preferentially by cells derived from the endocardium. (A-B’) Immunolabeling of Wt1cre;R26mG specimens early in valve development show that CD109 is expressed by valve mesenchymal cells throughout the developing leaflets at E14.5 (A), and progressively restricted to the distal regions of the developing parietal leaflet at E16.5 (B,B’). (C-D’) To more conclusively determine the developmental origin of these CD109-expressing cells, we immunolabeled an endothelial-specific Tie2cre;R26nG specimen for CD109 (red) and GFP (green) in which nuclear-localized GFP expression indicates endothelial origin. CD109 appears to be expressed preferentially by a subpopulation of mesenchymal cells originating from EndMT during valve development. Note in (D’) the CD109-negative area on the atrial-facing side of the parietal leaflet that is also GFP-negative. These are likely EPDCs which are not expressing CD109. Scale bars = 100um (AL, anterior leaflet; PL, parietal leaflet)
3.10. CD109 is expressed by endothelial cells of the coronary vasculature
We next directed our attention to CD109 expression in other regions of the developing heart. Examining immunolabeled sections from control embryos, we observed that EPDCs and CD109-expressing cells were often found in close proximity, with a heterogeneity of GFP and CD109 single- and double-positive cells scattered throughout the ventricular myocardial wall (Supplemental Fig. S9A). At E17.5, CD109 clearly marked the endothelial lining of the developing coronary arteries, with epicardial-derived VSMCs surrounding a ring of CD109-positive endothelial cells (Supplemental Fig. S9A,A’). In order to determine the nature of the CD109-positive cells within the ventricular myocardial wall, we performed wholemount immunofluorescent labeling for CD109 on control E17.5 hearts. CD109 marked a continuous network of vasculature (Supplemental Fig. S9B). Additionally, immuolabeling for endothelial marker PECAM1 and CD109 in Tie2cre;R26nG embryos confirms the vascular endothelial identity of CD109-positive cells within the ventricular wall (Supplemental Fig. S9C). Overall, these results suggest a role for CD109 in the development and/or function of the coronary endothelium. Further investigations beyond the scope of the present study will investigate mechanisms behind the role of CD109 in cardiovascular development and disease.
4. Discussion
Previous studies using various transgenic cell lineage-specific cre-driver mouse models have identified essential roles for SOX9 in cardiovascular development. [17, 18, 31]. Here, we report the first epicardial-specific SOX9 knockout using the Wt1cre mouse in combination with the Sox9 floxed mouse, showing a critical role for SOX9 in the invasion of EPDCs into the developing heart.
Loss of SOX9 did not severely affect the initial establishment of the epicardium proper, nor was the compact myocardial layer affected in Wt1cre;Sox9fl/fl;R26mG embryos at E12. However, the reduction in the number of EPDCs invading the ventricular compact myocardium as a result of the loss of SOX9 led to a decrease in compact myocardial thickness and cardiomyocyte proliferation at E14.5. This provides evidence that the invasion of EPDCs into the ventricular myocardial wall, under the regulation of SOX9, is necessary for normal myocardial growth and maturation. The epicardium has been previously described to regulate myocardial growth both through growth factor signaling [6, 35], as well as through signals which coordinate myocardial growth with the vascularization of the developing heart [65]. While it is possible that the reduction in myocardial thickness observed in the present report is due to aberrations in epicardial contribution to the development of the coronary system, we did not observe any differences between Wt1cre;Sox9fl/fl;R26mG and control embryos in the epicardial-derived pericyte or VSMC lineages surrounding the developing vasculature at E16.5. Additionally, we observed a decrease of PDGFRα-positive cells invading the myocardium at E14.5 in Wt1cre;Sox9fl/fl;R26mG specimens. Loss of PDGFRα in the epicardium has been shown to specifically lead to a decrease in epicardial-derived interstitial fibroblasts [37]. Taken together, these observations suggest that the loss of SOX9 in EPDCs may be primarily affecting the epicardial-derived interstitial fibroblasts. We cannot rule out that SOX9 may promote the expansion of EPDCs after EpiMT in a manner similar to how it controls the development of the endocardial-derived mesenchyme of the cushions [17]. Further investigation is required to elucidate the specific mechanisms by which loss of SOX9 results in decreased EPDCs in the myocardial wall.
In addition to its role in regulating EPDC invasion into the ventricular myocardium, we also demonstrate that SOX9 regulates EPDC contribution to the tissues of the AV junction. In Wt1cre;Sox9fl/fl;R26mG mice, the AV sulcus is hypoplastic and contains fewer cells than in control specimens. However, no differences in proliferation nor apoptosis were observed within the sulcus of Wt1cre;Sox9fl/fl;R26mG in comparison to controls. Hypoplastic sulci without differences in proliferation or apoptosis were also reported in the Myh6cre;Tbx2fl/fl mouse and the Wt1cre;Alk3fl/fl mouse [12, 66] in which reduced EpiMT was implicated. These data together suggest that SOX9 is required for proper EpiMT that gives rise to the AV sulcus. However, we cannot rule out the possibility that the smaller sulci could be due to a reduction in the proliferation of AV-EPDCs at a timepoint earlier than E12.
This report also builds on previous evidence [12] to confirm an essential role for EPDCs in the regulation of normal AV valve development. The invasion of EPDCs into the parietal leaflets of the developing AV valves takes place beginning around E14.5 [11] at an important stage of AV cushion remodeling and valve compaction [67–69]. Late fetal valve remodeling is critical for establishment of the laminar organization of ECM components. Perturbations of fetal valve development have been associated with myxomatous valve degeneration, a disease characterized by the disorganization and aberrant accumulation of ECM components [61, 70]. Myxomatous valve degeneration is a leading cause of mitral valve prolapse (MVP) and/or mitral regurgitation, a common valve disease that can result in severe complications and even sudden cardiac death [71]. In Wt1cre;Sox9fl/fl;R26mG mice, there is a dramatic reduction in the contribution of EPDCs to the parietal mitral valve leaflet beginning as early as E16.5, and persisting into early postnatal stages. While fetal development of the valves appears normal at a structural level, we observed postnatal valve abnormalities reminiscent of myxomatous valve degeneration in more than half of specimens examined between 4-10 weeks of postnatal life. Wt1cre;Sox9fl/fl;R26mG postnatal valves exhibited aberrant accumulation of VCAN and loss of ECM boundary interfaces which can be attributed to the lack of proper EPDC contribution during development, suggesting a critical role for EPDCs in maintaining mitral valve homeostasis.
Gene expression analysis of parietal leaflets from P5 Wt1cre;Sox9fl/fl and control specimens revealed dysregulation of genes associated with ECM organization including upregulation of the fibroblast markers Acta2 and Fsp1 and collagen receptor Ddr1, as well as downregulation of proteases such as Dpp4 and Fap. These dipeptidyl-metallopeptidases have been reported to form heterodimers which retain both collagenase and gelatinase activity [72, 73], and Dpp4 ECM proteolysis activity has been associated with EPDC migration [40]. Interestingly, our data showed upregulation of genes positively associated with myxomatous degeneration severity in canines, including the WNT1-inducible signaling protein Ccn4, the matrix elastase Mmp12, and the activated fibroblast marker Acta2 [55]. We also observed downregulation of genes associated with valve endothelial integrity that have been implicated in myxomatous valve degeneration including Prox1 and Pdgfra [52, 74]. This suggests a crosstalk between valve interstitial EPDCs and valve endothelial cells important for maintaining ECM homeostasis. Overall, the data reported here indicate that the proper contribution of EPDCs to the parietal leaflet during key windows of valve development is important for ECM organization.
Perhaps the most significant new finding of this report is the identification of CD109 as a novel player in cardiovascular development, and its potential relevance to the pathogenesis of myxomatous valve degeneration in our model. RNA-sequencing followed up with immunofluorescence demonstrated that there is an increased presence of CD109-expressing cells in response to the reduction of EPDCs in the Wt1cre;Sox9fl/fl;R26mG mouse, presumably marking the expansion of the endocardial-derived valve interstitial lineage. Supporting this hypothesis, we also saw an increased population of CD109 positive cells in the parietal leaflet of Wt1cre;Alk3fl/fl specimens, another previously described mouse model where reduced EPDC invasion leads to myxomatous valve abnormalities [12]. Immunostaining for CD109 in Tie2cre;R26nG hearts revealed that the vast majority of CD109-expressing cells within the valve are of endocardial origin. However, not all endocardial-derived VICs express CD109, which highlights the heterogeneity of cell populations during valve development [48, 49]. A previous study reported that the mesenchyme in the distal portion of the developing AV leaflets underlying the endocardial lining is a region of active processing of proteoglycans such as VCAN during valve compaction and remodeling [75]. The CD109-expressing cells we observed in the distal regions of the developing valve leaflets may represent a population of cells with a role in ECM remodeling.
Moreover, we report in this study that CD109 is also expressed by coronary endothelial cells during coronary vascular development. While a specific role for CD109 in coronary development has not yet been investigated, recent genome-wide analyses discovered a variant in an intronic region of Cd109 (rs56171536) that is associated with coronary artery disease and myocardial infarction [76, 77], highlighting the potential relevance of CD109 in coronary homeostasis. This is the first study to describe CD109 expression in the developing mouse heart, and future investigations are needed to examine its potential roles in valve and coronary system development.
In conclusion, using transgenic mouse models, we have identified a role for SOX9 in the control of epicardial-derived cell invasion during heart development that is essential for maintaining AV valve homeostasis. We further identified Cd109 as a gene associated with the endocardial/endothelial lineage that may play important roles in valve and coronary system development.
Supplementary Material
Highlights.
SOX9 controls epicardial-derived cell invasion into the developing heart
Epicardial-derived cells are needed to maintain atrioventricular valve homeostasis
Cd109 is identified as novel gene associated with valve development and disease
5. Acknowledgements
This research was supported by grants from NIH: R01-HL122906 (A.W., R.A.N., J.L.B., R.W., R.D., J.D.), R01-HL162913 (A.W., R.A.N., R.M., R.W.), R01-HL149696 (R.A.N.), R01-HL13546 (R.A.N.), R01-HL148728 (G.T.), R01-MD015395 (P.R.), T32-GM132055 (H.T., A.T.), T32-HL007260 (A.B.H.), as well as American Heart Association Predoctoral Fellowship PRE-1014420 (A.B.H.). Bioinformatics and technical service was provided by the MUSC Molecular Analytics Core that is supported by P20 GM103499 (J.L.B.) and MUSC’s Office of the Vice President for Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors did not use generative AI or AI-assisted technologies in the development of this manuscript.
REFERENCES
- 1.Viragh S and Challice CE, The origin of the epicardium and the embryonic myocardial circulation in the mouse. The Anatomical Record, 1981.201(1): p. 157–168. [DOI] [PubMed] [Google Scholar]
- 2.Wessels A and Pérez-Pomares JM, The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat Rec A Discov Mol Cell Evol Biol, 2004. 276(1): p. 43–57. [DOI] [PubMed] [Google Scholar]
- 3.Cao Y, Duca S, and Cao J, Epicardium in Heart Development. Cold Spring Harb Perspect Biol, 2020. 12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gittenberger-de Groot AC, Winter EM, and Poelmann RE, Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J Cell Mol Med, 2010.14(5): p. 1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quijada P, Trembley MA, and Small EM, The Role of the Epicardium During Heart Development and Repair. Circ Res, 2020. 126(3): p. 377–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li P, et al. , IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development, 2011.138(9): p. 1795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Den Akker NM, et al. , Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Dev Dyn, 2005. 233(4): p. 1579–88. [DOI] [PubMed] [Google Scholar]
- 8.Trembley MA, et al. , Myocardin-related transcription factors control the motility of epicardium-derived cells and the maturation of coronary vessels. Development, 2015. 142(1): p. 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaussin V, et al. , Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ Res, 2005. 97(3): p. 219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolditz DP, et al. , Epicardium-Derived Cells in Development of Annulus Fibrosis and Persistence of Accessory Pathways. Circulation, 2008. 117(12): p. 1508–1517. [DOI] [PubMed] [Google Scholar]
- 11.Wessels A, et al. , Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol, 2012. 366(2): p. 111–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lockhart MM, et al. , Alk3 mediated Bmp signaling controls the contribution of epicardially derived cells to the tissues of the atrioventricular junction. Dev Biol, 2014. 396(1): p. 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolters R, et al. , Role of the Epicardium in the Development of the Atrioventricular Valves and Its Relevance to the Pathogenesis of Myxomatous Valve Disease. J Cardiovasc Dev Dis, 2021. 8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacraz GPA, et al. , Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis During Ischemic Injury. Circulation, 2017. 136(15): p. 1396–1409. [DOI] [PubMed] [Google Scholar]
- 15.Huang JQ, et al. , SOX9 drives the epithelial-mesenchymal transition in non-small-cell lung cancer through the Wnt/β-catenin pathway. J Transl Med, 2019. 17(1): p. 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefebvre V, Angelozzi M, and Haseeb A, SOX9 in cartilage development and disease. Curr Opin Cell Biol, 2019. 61: p. 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lincoln J, et al. , Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Developmental Biology, 2007. 305(1): p. 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiyama H, et al. , Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci U S A, 2004. 101(17): p. 6502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garside VC, et al. , SOX9 modulates the expression of key transcription factors required for heart valve development. Development, 2015. 142(24): p. 4340–50. [DOI] [PubMed] [Google Scholar]
- 20.Lincoln J, Alfieri CM, and Yutzey KE, BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev Biol, 2006. 292(2): p. 292–302. [DOI] [PubMed] [Google Scholar]
- 21.Zehentner BK, Dony C, and Burtscher H, The transcription factor Sox9 is involved in BMP-2 signaling. J Bone Miner Res, 1999. 14(10): p. 1734–41. [DOI] [PubMed] [Google Scholar]
- 22.Pan Q, et al. , Sox9, a key transcription factor of bone morphogenetic protein-2-induced chondrogenesis, is activated through BMP pathway and a CCAAT box in the proximal promoter. J Cell Physiol, 2008. 217(1): p. 228–41. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, et al. , Myocardial β-Catenin-BMP2 signaling promotes mesenchymal cell proliferation during endocardial cushion formation. J Mol Cell Cardiol, 2018. 123: p. 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma L, et al. , Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development, 2005. 132(24): p. 5601–11. [DOI] [PubMed] [Google Scholar]
- 25.Kisanuki YY, et al. , Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol, 2001.230(2): p. 230–42. [DOI] [PubMed] [Google Scholar]
- 26.Theiler K, The House Mouse: Atlas of Embryonic Development. 1989, New York: Springer-Verlag. [Google Scholar]
- 27.Dobin A, et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 2013. 29(1): p. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014. 15(12): p. 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, et al. , Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics, 2007. 8: p. 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan P, et al. , The prevalent I686T human variant and loss-of-function mutations in the cardiomyocyte-specific kinase gene TNNI3K cause adverse contractility and concentric remodeling in mice. Hum Mol Genet, 2021. 29(21): p. 3504–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deepe RN, et al. , Sox9 Expression in the Second Heart Field; A Morphological Assessment of the Importance to Cardiac Development with Emphasis on Atrioventricular Septation. J Cardiovasc Dev Dis, 2022. 9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, et al. , Fibroblasts in an endocardial fibroelastosis disease model mainly originate from mesenchymal derivatives of epicardium. Cell Res, 2017. 27(9): p. 1157–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akiyama H, et al. , The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev, 2002. 16(21): p. 2813–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muzumdar MD, et al. , A global double-fluorescent Cre reporter mouse. Genesis, 2007. 45(9): p. 593–605. [DOI] [PubMed] [Google Scholar]
- 35.Lavine KJ, et al. , Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell, 2005. 8(1): p. 85–95. [DOI] [PubMed] [Google Scholar]
- 36.Shi S, et al. , Role of sox9 in growth factor regulation of articular chondrocytes. J Cell Biochem, 2015. 116(7): p. 1391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith CL, et al. , Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res, 2011.108(12): p. e15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao B, et al. SOX9 promotes nasopharyngeal carcinoma cell proliferation, migration and invasion through BMP2 and mTOR signaling. Gene, 2019. 715: p. 144017. [DOI] [PubMed] [Google Scholar]
- 39.Acharya A, et al. , The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development, 2012.139(12): p. 2139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao Y, et al. , Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev Cell, 2018. 45(2): p. 153–169.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu SP, et al. , Tbx18 regulates development of the epicardium and coronary vessels. Dev Biol, 2013. 383(2): p. 307–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mellgren AM, et al. , Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res, 2008. 103(12): p. 1393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou B, et al. , Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol, 2010. 338(2): p. 251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabkin E, et al. , Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation, 2001. 104(21): p. 2525–32. [DOI] [PubMed] [Google Scholar]
- 45.Hayek E, Gring CN, and Griffin BP, Mitral valve prolapse. Lancet, 2005. 365(9458): p. 507–18. [DOI] [PubMed] [Google Scholar]
- 46.Gupta V, et al. , Abundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc Pathol, 2009. 18(4): p. 191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kodigepalli KM, et al. , Biology and Biomechanics of the Heart Valve Extracellular Matrix. J Cardiovasc Dev Dis, 2020. 7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu M and Song J, Single-cell RNA sequencing reveals the diversity and biology of valve cells in cardiac valve disease. J Cardiol, 2023. 81(1): p. 49–56. [DOI] [PubMed] [Google Scholar]
- 49.Horne TE, et al. , Dynamic Heterogeneity of the Heart Valve Interstitial Cell Population in Mitral Valve Health and Disease. J Cardiovasc Dev Dis, 2015. 2(3): p. 214–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hulin A, et al. , Maturation of heart valve cell populations during postnatal remodeling. Development, 2019. 146(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu AC, Joag VR, and Gotlieb AI, The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol, 2007.171(5): p. 1407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moore K, et al. , PDGFRα: Expression and Function during Mitral Valve Morphogenesis. J Cardiovasc Dev Dis, 2021.8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Donnell A and Yutzey K, Prox1+ Endothelial Cells in Heart Valve Development and Homeostasis. The FASEB Journal, 2021. 35(S1). [Google Scholar]
- 54.Cano-Ballesteros S, et al. , Fsp1 cardiac embryonic expression delineates atrioventricular endocardial cushion, coronary venous and lymphatic valve development. J Anat, 2021.238(2): p. 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Markby GR, et al. , Disease Severity-Associated Gene Expression in Canine Myxomatous Mitral Valve Disease Is Dominated by TGFβ Signaling. Front Genet, 2020. 11: p. 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finnson KW, et al. , Identification of CD109 as part of the TGF-beta receptor system in human keratinocytes. Faseb j, 2006. 20(9): p. 1525–7. [DOI] [PubMed] [Google Scholar]
- 57.Vorstenbosch J, et al. , Overexpression of CD109 in the Epidermis Differentially Regulates ALK1 Versus ALK5 Signaling and Modulates Extracellular Matrix Synthesis in the Skin. J Invest Dermatol, 2017. 137(3): p. 641–649. [DOI] [PubMed] [Google Scholar]
- 58.Man XY, et al. , CD109, a TGF-β co-receptor, attenuates extracellular matrix production in scleroderma skin fibroblasts. Arthritis Res Ther, 2012. 14(3): p. R144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang JM, et al. , CD109 attenuates TGF-β1 signaling and enhances EGF signaling in SK-MG-1 human glioblastoma cells. Biochem Biophys Res Commun, 2015. 459(2): p. 252–258. [DOI] [PubMed] [Google Scholar]
- 60.Tang Q, et al. , The Role of Transforming Growth Factor-β Signaling in Myxomatous Mitral Valve Degeneration. Front Cardiovasc Med, 2022. 9: p. 872288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ng CM, et al. , TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest, 2004. 114(11): p. 1586–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oyama MA, et al. , Comparative pathology of human and canine myxomatous mitral valve degeneration: 5HT and TGF-β mechanisms. Cardiovasc Pathol, 2020. 46: p. 107196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Azhar M, et al. , Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev, 2003. 14(5): p. 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winocour S, et al. , CD109, a novel TGF-β antagonist, decreases fibrotic responses in a hypoxic wound model. Exp Dermatol, 2014. 23(7): p. 475–9. [DOI] [PubMed] [Google Scholar]
- 65.Olivey HE and Svensson EC, Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res, 2010. 106(5): p. 818–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aanhaanen WT, et al. , Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J Clin Invest, 2011. 121 (2): p. 534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kruithof BP, Krawitz SA, and Gaussin V, Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev Biol, 2007. 302(1): p. 208–17. [DOI] [PubMed] [Google Scholar]
- 68.de Vlaming A, et al. , Atrioventricular valve development: new perspectives on an old theme. Differentiation, 2012. 84(1): p. 103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hinton RB and Yutzey KE, Heart valve structure and function in development and disease. Annu Rev Physiol, 2011. 73: p. 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sauls K, et al. , Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc Res, 2012. 96(1): p. 109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nkomo VT, et al. , Burden of valvular heart diseases: a population-based study. The Lancet, 2006. 368(9540): p. 1005–1011. [DOI] [PubMed] [Google Scholar]
- 72.Zhang HE, et al. , Identification of Novel Natural Substrates of Fibroblast Activation Protein-alpha by Differential Degradomics and Proteomics. Mol Cell Proteomics, 2019. 18(1): p. 65–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wagner L, et al. , Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homologue (DASH) proteins. Clin Exp Immunol, 2016. 184(3): p. 265–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho Y-C, et al. , PROX1 inhibits PDGF-B expression to prevent myxomatous degeneration of heart valves. bioRxiv, 2023: p. 2023.05.10.540284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kern CB, et al. , Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn, 2006. 235(8): p. 2238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koyama S, et al. , Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet, 2020. 52(11): p. 1169–1177. [DOI] [PubMed] [Google Scholar]
- 77.Sakaue S, et al. , A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet, 2021. 53(10): p. 1415–1424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.