

Abstract
INTRODUCTION
Biomarkers remain mostly unavailable for non‐Alzheimer's disease neuropathological changes (non‐ADNC) such as transactive response DNA‐binding protein 43 (TDP‐43) proteinopathy, Lewy body disease (LBD), and cerebral amyloid angiopathy (CAA).
METHODS
A multilabel non‐ADNC classifier using magnetic resonance imaging (MRI) signatures was developed for TDP‐43, LBD, and CAA in an autopsy‐confirmed cohort (N = 214).
RESULTS
A model using demographic, genetic, clinical, MRI, and ADNC variables (amyloid positive [Aβ+] and tau+) in autopsy‐confirmed participants showed accuracies of 84% for TDP‐43, 81% for LBD, and 81% to 93% for CAA, outperforming reference models without MRI and ADNC biomarkers. In an ADNI cohort (296 cognitively unimpaired, 401 mild cognitive impairment, 188 dementia), Aβ and tau explained 33% to 43% of variance in cognitive decline; imputed non‐ADNC explained an additional 16% to 26%. Accounting for non‐ADNC decreased the required sample size to detect a 30% effect on cognitive decline by up to 28%.
DISCUSSION
Our results lead to a better understanding of the factors that influence cognitive decline and may lead to improvements in AD clinical trial design.
Keywords: Alzheimer's disease, CAA, Lewy body, TDP‐43
1. BACKGROUND
Alzheimer's disease (AD) is defined neuropathologically by the presence of amyloid beta (Aβ) plaques and tau tangles. 1 The recent advent of positron emission tomography (PET) tracers and biofluid assays for Aβ and tau has enabled the use of these markers as inclusion criteria in clinical trials, ensuring that participants satisfy the biological definition of AD and are not clinical phenocopies. 2 However, neuropathological studies have also established that, in sporadic AD, the presence of Aβ and tau pathology very frequently is accompanied by other pathologies, including α‐synuclein‐containing Lewy body disease (LBD), transactive response DNA‐binding protein 43 (TDP‐43) inclusions, and Aβ in the form of cerebral amyloid angiopathy (CAA). 3 , 4 , 5 , 6 , 7 Indeed, in cases where AD is the primary pathologic diagnosis, only a minority of cases have “pure” AD neuropathological changes (ADNC) at autopsy. 3 , 8 , 9
With advancing age, the number of neuropathological changes found in combination with ADNC increases, with reports of co‐occurring pathologies ranging from 50% to 100%. 3 , 4 , 5 , 6 , 7 , 10 , 11 , 12 , 13 Clinicopathological research on ADNC has revealed that each major co‐occurring pathology can cause distinct and often accelerated progression of the disease compared to ADNC alone. 6 , 14 , 15 , 16 , 17 , 18 , 19 While ADNC has been shown to be responsible for about 50% of the observed cognitive decline on average, the contribution at the individual level varies widely from 22% to 100%, with comorbid LBD, TDP‐43, and vascular pathologies contributing up to 41%, 24%, and 16% to 20%, respectively, of decline 15 .
The presence of comorbid pathologies is thus an important factor in disease‐modifying AD clinical trials, in two respects. First, statistically, the varying pathological contributions contribute to an increased variance in longitudinal trajectories, in both placebo and treatment arms. Second, biologically, candidate therapeutics targeting Aβ or tau, even if effective, will have a ceiling of maximum response that is variable across participants. For example, an efficacious Aβ‐targeted therapy may only attenuate the component of longitudinal decline that is driven by Aβ. If this is only a minor contribution to the overall clinical decline in some participants, the overall effect is to reduce the mean difference from placebo and increase the variance in the treatment arm(s).
Currently, biomarker‐based enrichment for pathology has been integrated in many ongoing clinical trials 20 ; however, in vivo imaging and fluid biomarkers of neuropathologic changes have been well established only for Aβ and tau. As a result, the presence (or absence) of comorbid non‐ADNC (e.g., LBD, TDP‐43, and CAA) remains unknown while individuals are still alive and participating in clinical trials. By including individuals with hidden non‐ADNC, such trials are at risk of incorporating significant error into their modeling of clinical progression estimates, likely underestimating the biological effectiveness of any molecular treatment targeted to ADNC alone. Nonetheless, excluding all individuals with non‐ADNC may not be the best strategy. A trial that includes only participants with pure ADNC would not accurately reflect the general population and could limit the scope of the approved target population, unless broader eligibility criteria are evaluated in subsequent trials or eventual clinical practice. Therefore, tools for efficient phenotyping to identify both ADNC and non‐ADNC will be crucial to accurately measure the effectiveness of these treatments.
Our first goal was to develop a data analytic tool to impute the presence of comorbid non‐ADNC (LBD, TDP‐43, CAA) in living individuals. Antemortem in vivo MRI and autopsy‐confirmed neuropathology evaluations for ADNC and non‐ADNC were combined from three leading AD cohorts: ADNI, 21 Open Access Series of Imaging Studies‐3 (OASIS‐3), 22 and National Alzheimer's Coordinating Center (NACC). 23 We evaluated the degree to which the presence of comorbid non‐ADNC in individuals 50 years old and older is detectable using macrostructural brain changes measured by widely available brain MRIs together with demographic, genetic, and clinical assessments, and Aβ and tau biomarkers. Our second goal was to assess the value of the imputed presence of non‐ADNC by estimating the variance in cognitive decline in ADNI participants explained by non‐ADNC. Subsequently we incorporated these data to estimate the required sample size in early‐AD clinical trial scenarios designed to detect treatment effects on cognitive decline.
2. METHODS
2.1. Study participants
Four cohorts of participants were used: (1) main study cohort with autopsy confirmation of ADNC (Aβ plaques and neurofibrillary tau tangles) and non‐ADNC (ie, LBD, TDP‐43, and CAA), (2) target clinical study cohort, (3) early‐AD clinical cohort, and (4) extended cross‐sectional neuroimaging sample.
The main study cohort for the development and validation of models to detect the presence of non‐ADNC included participants from ADNI (data download on February 2023; N = 88), OASIS‐3 (July 2021 data release; N = 59), and NACC (December 2020 data release; N = 67). To meet inclusion criteria, these study participants had undergone (1) a neuropathologic evaluation of ADNC and non‐ADNC (LB, TDP‐43, and CAA) as captured in the NACC Neuropathology (NP) Form version 10 (2014); (2) antemortem structural MRI; and (3) clinical and cognitive assessments cross‐sectionally within 6 months of the last antemortem MRI, with the last clinical assessment yielding a clinical diagnosis of dementia due to AD, mild cognitive impairment (MCI), or cognitively unimpaired (CU). The sample size from NACC is limited as data collection on regional TDP‐43 pathology began with NP Form version 10, and not all NACC Alzheimer's Disease Research Centers (ADRCs) report TDP‐43 pathology data. Therefore, the NACC data were limited to two ADRC cohorts where balanced TDP‐43 positivity data were available.
The target clinical study cohort to assess the value of the imputed presence of non‐ADNC by estimating the variance in cognitive decline was composed of N = 872 ADNI participants who met the following criteria: (1) had PET biomarker assessment for the presence of AD Aβ pathology and cerebral spinal fluid (CSF) p‐tau181 biomarker assessment for the presence of AD tau pathology; (2) underwent antemortem structural MRI; (3) had clinical and cognitive assessments cross‐sectionally within 6 months of MRI; and (4) were not included in the main study cohort (ie, no autopsy confirmed neuropathology assessment).
An early‐AD clinical cohort was used to estimate required sample size for different clinical trial scenarios. This cohort included ADNI participants from the target clinical study cohort who were aged 50 to 85 years and had a clinical diagnosis of MCI or dementia due to AD, a Clinical Dementia Rating (CDR) of 0.5 or 1, a Mini‐Mental State Examination (MMSE) score of 24 to 30, biomarker evidence of AD Aβ pathology based on Aβ PET, and 2‐year longitudinal cognitive and clinical assessments.
Additionally, an extended cross‐sectional neuroimaging sample of CU participants from each cohort (N = 558 ADNI, N = 327 OASIS‐3, and N = 326 NACC) was used for harmonization of structural brain MRI volumetrics as described below.
2.2. Neuropathological assessment
The selection criteria for the classification of AD and other pathologies in this study were based on the NACC Neuropathology (NP) version 10 guidelines, which were previously applied to cases from the ADNI, OASIS‐3, and NACC cohorts. 9 , 24 These criteria include the maximal density of neocortical diffuse plaques, the maximal density of neocortical neuritic plaques, Thal Aβ phase, and Braak stage for neurofibrillary degeneration. Participants were considered Aβ‐positive (Aβ+) if they had moderate or frequent neuritic plaques according to Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria and tau‐positive (tau+) if their Braak stage for neurofibrillary degeneration was equal to or greater than three. Furthermore, the assessment of ADNC involved the utilization of an “ABC” scoring system: Aβ plaques (A) by the method of Thal phases, neurofibrillary tangle stage (B) by the method of Braak, and neuritic plaque score (C) by the method of CERAD. The combination of A, B, and C scores was designated as “Not,” “Low,” “Intermediate,” or “High” ADNC, according to previously established criteria. 25 , 26 Specifically, when the A score was 0, it indicated the absence of ADNC, denoted as “Not.” ADNC was considered “Low” when the Braak stage I‐II, with A score ranging from 1 to 3, regardless of C score, or when the Braak stage III‐VI, with an A score of 1 and C score of 0 to 1. Within the Braak stage III‐IV range, ADNC was classified as “Intermediate” when there was an A score of 1 combined with a C score of 2 to 3, or an A score of 2 to 3 with any C score. Similarly, for Braak stage V‐VI, ADNC was categorized as “Intermediate” when there was an A score of 1 with a C score of 2 to 3, an A score of 2 with any C score, or an A score of 3 with a C score of 0 to 1. Lastly, ADNC was labeled as “High” when Braak stage V‐VI corresponded to an A score of 3 and a C score of 2 to 3.
RESEARCH IN CONTEXT
Systematic review: The authors searched PubMed, Google Scholar, and reference lists of relevant research articles. They found that co‐pathologies such as LBD, TDP‐43, and CAA were commonly observed in older individuals with AD, even in those with confirmed Aβ and tau pathology, and significantly contribute to cognitive and clinical decline. Unfortunately, there are currently few reliable methods to detect these co‐pathologies antemortem.
Interpretation: The authors used an MRI‐based classifier for co‐pathologies, which demonstrated that these co‐pathologies play a significant role in cognitive decline among older individuals. This improved understanding of the factors that influence cognitive decline could potentially lead to advancements in AD clinical trial design.
Future directions: Future work should further validate the proposed modeling approach for detecting the presence of co‐pathologies in larger and diverse autopsy‐confirmed cohorts. Meanwhile, the proposed model and similar approaches could provide the field with a better understanding of how co‐pathologies may impact clinical trials.
TDP‐43‐immunoreactive inclusions were evaluated in five brain regions: spinal cord, amygdala, hippocampus, entorhinal cortex/inferior temporal cortex, and frontal neocortex, with the response categories “no,” “yes,” “not assessed,” and “missing/unknown.” Due to the large number of “not assessed” or “missing/unknown” values in the spinal cord region, TDP‐43 positivity (ie, TDP‐43+) was determined if there was any form of TDP‐43 inclusions in the amygdala, hippocampus, entorhinal/inferior temporal cortex, or neocortex. 27
Assessment of LBD staging was done through α‐synuclein immunohistochemistry. Due to a high prevalence of cases with LBD primarily confined to the amygdala and/or limbic brain regions with minimal or no involvement of brainstem, LBD was considered present (i.e., LBD+) if the individual showed any evidence of amygdala‐predominant, limbic transitional, or neocortical diffuse LBD according to the Consortium on Dementia with Lewy Bodies criteria. 28
The CAA rating was determined using a topographic method. A score of 0 indicated no presence of Aβ in the leptomeningeal or superficial cortical blood vessels. A score of “mild” reflected trace to scattered positivity in either the leptomeningeal or cortical blood vessels. A score of “moderate” indicated the presence of circumferential, robust staining Aβ deposits in at least some vessels in the leptomeninges or neocortex. A score of “severe” corresponded to widespread circumferential Aβ positivity in many leptomeningeal and superficial cortical vessels. Participants were deemed CAA‐positive (i.e., CAA+) if they had moderate to severe Aβ positivity in parenchymal and/or leptomeningeal vessels. The analysis of CAA positivity was then repeated, comparing cases with mild, moderate, or severe CAA to those without any CAA.
2.3. Clinical assessments
The demographic information of each participant included age at structural brain MRI and sex. Apoliporpotein E (APOE) genotyping was performed using DNA from blood samples. Dose‐dependent effect of APOE ε2 and ε4 alleles were separately considered as potential predictors of non‐ADNC positivity in this study.
The candidate global cognitive assessments were limited to the CDR Sum of Boxes (CDR‐SB) and the MMSE based on a 30‐point questionnaire. In addition, the Alzheimer's Disease Assessment Scale‐Cognitive subscale 13‐item (ADAS‐Cog13) and a modified version of the preclinical Alzheimer's cognitive composite (mPACC) 29 were considered as the cognitive outcome measures in assessment of variance explained by ADNC and non‐ADNC and sample size calculation for early‐AD clinical trial scenarios. 30
2.4. Structural MRI acquisition and processing
The structural MRI data used in this study consisted of three‐dimensional MP‐RAGE or IR‐SPGR T1‐weighted images obtained from either 1.5T or 3T MRI scanners. A multi‐atlas segmentation (MUSE) framework was applied to parcellate the anatomical brain structures and calculate gray matter volumetrics from the structural MRI. 31 This framework uses a diverse ensemble of warped atlases, as opposed to a model‐based average with uniform regional labels, to estimate regional tissue volumes in each structural MRI. The approach allows for the optimal estimation of volumes, regardless of image acquisition parameters and quality, by maintaining the consistency of segmentations.
To address interscanner and imaging protocol differences, the ComBat‐GAM harmonization technique was used to remove study protocol‐ and scanner‐associated variability in volumetric measures of 113 regions of interest (ROIs) listed in Supplementary Material Table S1. 32 The study cohorts were divided into batches based on imaging protocol and scanner properties, such as 1.5T ADNI‐1, 3T ADNI‐GO/2, and 3T ADNI‐3 subcohorts within ADNI, 3T Siemens Biograph and 3T Siemens TrioTim subcohorts within OASIS‐3, and 1.5 GE Discovery MR 750 and 1.5 GE Signa subcohorts within NACC. Within each batch, individual ROI volume estimates were normalized using the residuals of a least‐squares‐derived linear regression between raw volumes and intracranial volume (ICV), based on the corresponding CU data. 33 The batch‐specific harmonization parameters were then estimated based on the variability observed within and across CU groups while considering normal variance due to age, sex, and APOE ε4 carrier status, using a penalized nonlinear term to describe the age effect. These batch‐specific parameters were then applied to the entire imaging dataset.
2.5. Data modeling and statistical analysis
The demographic, clinical, and biomarker characteristics were compared using two‐sample t tests for continuous variables and chi‐squared tests for categorical variables.
Primary predictor variables for constructing a multilabel random forest (RF) classifier to detect non‐ADNC (LBD, TDP‐43, and CAA) outcomes simultaneously were as follows: ICV‐adjusted and harmonized regional volumetric estimates from automated MUSE parcellation; demographics (age and sex); number of APOE alleles (ε2 and ε4 separately); MMSE; CDR; end‐of‐life ADNC status (Aβ positivity and tau positivity separately); and potential interaction with number of APOE alleles, sex, and AD pathology status. The RF classifier was chosen due to its classification ability, flexibility in handling mixed features, ability to prevent overfitting, handling of “small n large p” problems, complex interactions, and interpretability. 34 We adopted a multilabel approach as research indicates that the presence of different non‐AD pathological changes is not necessarily mutually exclusive. The model was regularized with a penalty term constraint using the structural connectivity‐derived Laplacian matrix 35 and controlled for antemortem time interval—the time between in vivo MRI and death. A fivefold cross‐validation approach was used for final model validation. Model performance was evaluated using area under the receiver operating characteristic (ROC) curve (AUC), classification accuracy (ACC), positive predictive value (PPV), and negative predictive value (NPV) for each non‐ADNC label. Reference multilabel RF classifiers were constructed to assess the added value of volumetric and ADNC variables (Aβ positivity and tau positivity) separately.
As the chance of additional pathology developing after in vivo MRI increases with longer antemortem intervals, we assessed the model performance for subgroups of participants with different ranges of antemortem intervals to assess the potential influence of antemortem interval on presence of non‐ADNC predictions.
The validated model was applied to the target clinical ADNI cohort, and the frequency of positivity for each non‐ADNC was calculated. We determined the extent to which ADNC (Aβ‐positivity evidence from Aβ‐PET and tau‐positivity evidence from CSF p‐tau181 levels separately) and non‐ADNC explain the variance in change in outcome measures of mPACC, ADAS‐Cog13, and CDR‐SB. Specifically, a reference linear mixed‐effects model was created, and ADNC and non‐ADNC markers and their interactions were added in a series of models to estimate the between‐subject variation in cognitive decline explained by the corresponding pathologies after accounting for age, sex, years of education, APOE ε4 carrier status, and cerebrovascular white matter lesion burden. It is important to acknowledge that the target clinical ADNI cohort, which excluded participants with vascular pathology etiologies, had limited assessments of vascular risk factors and outcomes beyond the study entry visit. However, given the known association between vascular risk factors and white matter lesions, we chose to focus on white matter lesion burden as the primary cerebrovascular imaging marker, as it was available for all subjects in the cohort through fluid attenuated inversion recovery (FLAIR) MRI data. These lesions are recognized markers of cerebrovascular disease and can impact cognitive function and the risk of developing AD. 36 , 37
Sample size estimates were calculated for two early‐AD clinical trial enrichment scenarios to achieve 80% power in detecting treatment effect sizes of 20% to 50% in 18‐month placebo‐controlled trials with cognitive outcome assessment of ADAS‐Cog13, performed every 6 months. Both enrichment scenarios considered demographic and clinical inclusion criteria (ie, 50 to 85 years of age, a clinical diagnosis of MCI or dementia due to AD, CDR of 0.5 or 1, MMSE score of 24 to 30) and screened for Aβ biomarker positivity (global cortical florbetapir standardized uptake value ratio [SUVR] > 1.11, 38 corresponding to 20 centiloids). Participants were further screened for biomarker evidence of AD tau pathology (CSF p‐tau181 > 24 pg/mL 39 ) in the second enrichment scenario. CSF concentrations of p‐tau181 was measured using Elecsys CSF immunoassays on a cobas e 601 analyzer at the University of Pennsylvania by the ADNI Biomarkers Core.
The sample size estimates were calculated for assumed treatment effect sizes on overall clinical decline using linear mixed‐effects model analysis that applied standard methods described elsewhere. 40 The reference model for the fixed effects included the age at baseline. Fixed effects further included scores for the presence of non‐ADNC to account for variance due to comorbid non‐ADNC.
The use of the model as an additional screening tool was also evaluated. In these scenarios, hypothetical patients were screened out based on the presence of different non‐ADNC combinations, under the assumption that the treatment being tested would slow ADNC‐related changes only. Both the sample sizes and the associated screen fail rates—an important practical tradeoff in clinical trials—were calculated.
3. RESULTS
3.1. Main study participants
Participants (N = 214) who underwent autopsy and had available in vivo MRI and clinical assessments, as well as post mortem NACC Neuropathology version 10 reports with complete ADNC, TDP‐43, LBD, and CAA assessments, were included in the main study cohort for model development and validation (Table 1). Of these participants, 63% had a high level of ADNC (“High ADNC”), 12% had an intermediate level (“Intermediate ADNC”), 20% had a low level (“Low ADNC”), and 5% had no ADNC (“Not AD”). TDP‐43, LBD, and CAA were present in 43%, 40%, and 80% of the participants, respectively. Additionally, 98% of those with intermediate or high ADNC and 71% of those with either no amyloid plaques or low ADNC had one or more comorbid pathologies, as illustrated by the Venn diagrams in Supplementary Material Figure S1. More than one co‐pathology was present in 33% of individuals with no or low ADNC and 66% of individuals with intermediate or high ADNC.
TABLE 1.
Demographic, clinical, and neuropathologic characteristics of the participants included in the main study cohort for model development and validation.
| Characteristics | ADNI | OASIS‐3 | NACC | Overall |
|---|---|---|---|---|
| N | 88 | 59 | 67 | 214 |
| Age at death, years (SD) | 83.0 (7.1) | 83.6 (8.2) | 81.6 (8.4) | 82.7 (7.8) |
| Female, n (%) | 23 (26) | 23 (39) | 35 (52) | 81 (38) |
| Age at last MRI, years (SD) | 80.0 (7.0) | 78.4 (8.6) | 75.7 (8.4) | 78.2 (8.1) |
| Cognitively unimpaired at last MRI, n (%) | 11 (12) | 18 (31) | 10 (15) | 39 (18) |
| Mild cognitive impairment at last MRI, n (%) | 33 (38) | 0 (0) | 7 (10) | 40 (19) |
| Dementia at last MRI, n (%) | 44 (50) | 41 (69) | 50 (75) | 135 (63) |
| Mini‐Mental State Examination (MMSE) at last MRI, mean (SD) | 24.0 (4.9) | 25.5 (3.8) | 19.1 (6.6) | 22.9 (5.8) |
| Clinical dementia rating at last MRI, mean (SD) | 0.8 (0.6) | 0.6 (0.5) | 1.2 (0.8) | 0.9 (0.7) |
| APOE genotype, n (%) | ||||
| ε2/ε3 | 3 (3) | 7 (12) | 4 (6) | 14 (6) |
| ε2/ε4 | 2 (2) | 3 (5) | 2 (3) | 7 (3) |
| ε3/ε3 | 34 (39) | 22 (37) | 24 (36) | 80 (38) |
| ε3/ε4 | 35 (40) | 22 (37) | 27 (40) | 84 (39) |
| ε4/ε4 | 14 (16) | 5 (9) | 10 (15) | 29 (14) |
| AD neuropathological changes (ADNC) a , b | ||||
| Not, n (%) | 4 (5) | 2 (3) | 4 (6) | 10 (5) |
| Low, n (%) | 17 (19) | 16 (27) | 9 (13) | 42 (20) |
| Intermediate, n (%) | 8 (9) | 10 (17) | 8 (12) | 26 (12) |
| High, n (%) | 59 (67) | 31 (53) | 46 (69) | 136 (63) |
| TDP‐43 immunoreactive inclusions, simplified staging c | ||||
| None, n (%) | 46 (52) | 37 (63) | 38 (57) | 121 (57) |
| Amygdala only, n (%) | 4 (4) | 4 (7) | 6 (9) | 14 (6) |
| + Hippocampus, n (%) | 26 (30) | 12 (20) | 20 (30) | 58 (27) |
| + Middle frontal gyrus, n (%) | 12 (14) | 6 (10) | 3 (4) | 21 (10) |
| Lewy body pathology consensus criteria d | ||||
| None, n (%) | 43 (49) | 36 (61) | 42 (63) | 121 (56) |
| Olfactory only, n (%) | 3 (3) | 4 (7) | 1 (2) | 8 (4) |
| Amygdala predominant, n (%) | 12 (14) | 5 (9) | 9 (13) | 26 (12) |
| Limbic, n (%) | 8 (9) | 2 (3) | 11 (16) | 24 (11) |
| Neocortical, n (%) | 22 (25) | 12 (20) | 4 (6) | 35 (16) |
| Cerebral amyloid angiopathy | ||||
| None, n (%) | 14 (16) | 12 (20) | 16 (24) | 42 (20) |
| Mild, n (%) | 37 (42) | 26 (44) | 22 (33) | 85 (40) |
| Moderate, n (%) | 20 (23) | 14 (24) | 16 (24) | 50 (23) |
| Severe, n (%) | 17 (19) | 7 (12) | 13 (19) | 37 (17) |
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's & Dementia the Journal of the Alzheimer's Association: 2012;8:1‐13.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta neuropathologica. 2012;123:1‐11.
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503‐27.
Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G, et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post‐mortem brains: a multi‐centre study. Acta neuropathologica. 2021;141:159‐72.
3.2. In vivo classification of comorbid non‐ADNC
The full classifier model, which included demographic, clinical, number of APOE alleles (ε2 and ε4 separately), ADNC status (Aβ positivity and tau positivity separately), and MRI volumetric variables, as well as being assessed with cross‐validation within the main autopsy‐confirmed study cohort, had an accuracy of 81% [75%, 86%], with 78% PPV and 83% NPV, resulting in an AUC of 0.90 [0.86, 0.94] for TDP‐43 positivity (Figure 1). For LBD positivity, the model had an accuracy of 81% [75%, 86%], with 78% PPV, 83% NPV, and an AUC of 0.91 [0.87, 0.94]. The accuracy of CAA positivity in differentiating cases with moderate to severe CAA from those with no to mild CAA was 76% [70%, 82%], with 75% PPV, 77% NPV, and an AUC of 0.87 [0.82, 0.92]. CAA positivity in differentiating cases with mild to severe CAA to those without any CAA yielded a classification accuracy of 92% [88%, 95%], with 94% PPV, 84% NPV, and an AUC of 0.94 [0.90, 0.97]. In a separate sensitivity analysis, we evaluated the performance of the proposed multilabel imputation model in detecting the presence of a specific non‐ADNC pathology in the presence of other non‐ADNC pathologies. For instance, we focused on detecting the presence of TDP‐43 in individuals who were LBD‐positive or CAA‐positive. Comparing the model's performance in the presence of other co‐pathologies to its performance regardless of the presence of other co‐pathologies, we observed slight reductions in the AUC estimates by up to 0.03 units (0.87 vs 0.90 for TDP‐43; 0.90 vs 0.91 for LBD; 0.85 vs 0.87 for CAA in differentiating cases with moderate to severe CAA from those with no to mild CAA; 0.92 vs 0.94 for CAA in differentiating cases with mild to severe CAA to those without any CAA); however, these differences in performance were not statistically significant (DeLong p value of .57, .96, .60, and .80, respectively).
FIGURE 1.
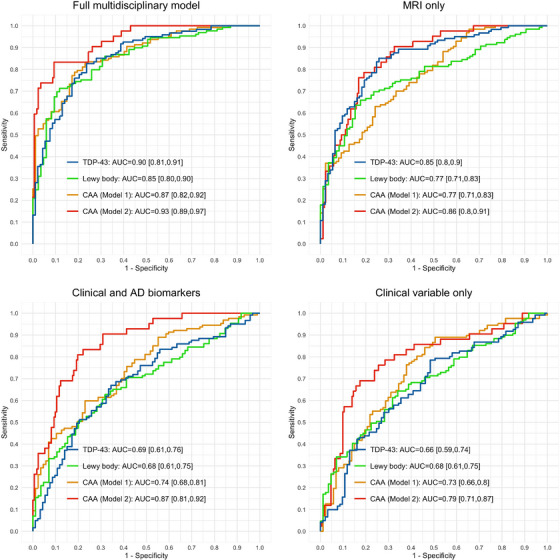
ROC analysis of comorbid non‐ADNC positivity prediction in autopsy‐confirmed main study cohort of CU elderly individuals, individuals with MCI, and individuals with dementia due to AD. The full multidisciplinary model used harmonized ICV‐adjusted regional volumetric estimates from automated MUSE parcellation, demographics, number of APOE alleles (ε2 and ε4 separately), MMSE, CDR, and end‐of‐life ADNC status (Aβ positivity and tau positivity separately) as primary predictor variables. Reference classifiers were constructed to assess the added value of ADNC biomarkers together with MRI volumetrics. TDP‐43 positivity: none versus inclusions in amygdala, hippocampus, entorhinal/inferior temporal cortex, or neocortex. LBD positivity: none versus amygdala predominant, limbic (transitional), or neocortical (diffuse) LBD pathology. CAA positivity: none or mild versus moderate or severe CAA in Model 1; none versus mild, moderate, or severe CAA in Model 2.
The reference model, which was limited to demographic, clinical, number of APOE alleles (ε2 and ε4 separately), and ADNC status without MRI volumetric variables, and trained and cross‐validated on the same main autopsy confirmed study cohort, performed significantly worse in differentiating positivity for all three comorbid non‐ADNC considered in this study. Specifically, the AUC for TDP‐43 was 0.69 compared to 0.90; that for LBD was 0.68 compared to 0.91; and that for CAA was 0.74 compared to 0.87 in differentiating cases with moderate to severe CAA from those with no to mild CAA, and 0.87 compared to 0.94 in differentiating cases with mild to severe CAA from those without any CAA (DeLong z = 5.8, 6.7, 4.8, and 3.8, respectively). When the reference model was further limited to demographic, clinical, and number of APOE alleles (ε2 and ε4 separately) variables only, the performance in identifying TDP‐43, LBD, moderate to severe CAA, and mild to severe CAA further decreased with AUC values of 0.66, 0.68, 0.73, and 0.79, respectively.
The AUC for in vivo detection of the comorbid non‐ADNC in the subsets of the study cohort with different antemortem interval limits is shown in Supplementary Material Figure S2. The performance for detecting the presence of TDP‐43, LBD, and mild to severe CAA was stable with varying antemortem intervals, but the performance for distinguishing moderate to severe CAA pathology from mild CAA or no CAA was slightly better with longer antemortem intervals, which might be due to significantly increased sample size rather than the antemortem time interval per se. Additionally, the estimated antemortem model scores for the prediction of the presence of comorbid non‐ADNC were associated with the stages of the TDP‐43, LB, and CAA defined at neuropathology assessment (Supplementary Material Figure S3).
According to the 95% confidence intervals of the coefficient estimates for the model variables, the presence of TDP‐43 at autopsy was associated with relatively greater atrophy in the limbic system and functionally connected regions of the salience and language networks, together with relatively lower levels of atrophy in key default mode network regions as illustrated in Figure 2A and detailed in Supplementary Material. The antemortem MRI volumetric signature for the presence of LBD at autopsy included relatively greater atrophy in parts of the limbic system, basal ganglia, and precuneus but relatively lower atrophy in the brain stem, primary visual cortex, and dorsolateral prefrontal cortex (Figure 2B; Supplementary Material). Similarly, relatively greater antemortem atrophy estimates predominantly in the limbic system, salience and sensorimotor network brain regions, together with relatively lower atrophy in the ventromedial prefrontal, primary visual, and parts of the sensorimotor and executive control network cortices (Figure 2C; Supplementary Material), were associated with the presence of CAA at autopsy.
FIGURE 2.
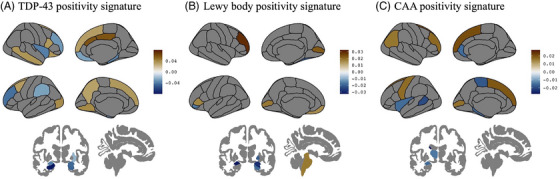
Antemortem MRI volumetric signatures associated with the presence of (A) TDP‐43, (B) LBD, and (C) CAA pathology at autopsy. A detailed list of anatomical regions is provided in the Supplementary Material. Color map illustrates the coefficient estimates of the statistically significant (based on 95% confidence intervals) regional volumetric variables in predicting the presence of the corresponding comorbid non‐AD pathology.
3.3. Predicted presence of comorbid non‐AD pathologies in clinically diagnosed ADNI participants
The target cross‐sectional ADNI study cohort of N = 872 included 298 individuals who were CU, 376 individuals with MCI, and 198 individuals with a dementia diagnosis at the time of MRI used for imputation of the presence of non‐ADNC (Table 2). According to biomarker evidence from Aβ PET (florbetapir) and CSF p‐tau181, 24% of the CU participants, 43% of the MCI participants, and 80% of the dementia participants had biomarker evidence for the presence of both Aβ and p‐tau pathologies.
TABLE 2.
Demographic, clinical, and neuropathologic characteristics of the ADNI participants without neuropathology confirmation.
| Characteristics | Cognitively unimpaired | MCI | Dementia |
|---|---|---|---|
| N | 298 | 376 | 198 |
| Age at MRI, years (SD) | 73.4 (6.7) | 72.2 (7.6) | 74.2 (7.9) |
| Female, n (%) | 166 (56) | 178 (44) | 74 (39) |
| MMSE at MRI, mean (SD) | 28.9 (1.4) | 27.2 (2.9) | 23.0 (3.8) |
| Clinical dementia rating at MRI, mean (SD) | 0.1 (0.2) | 0.5 (0.2) | 0.9 (0.6) |
| At least one copy of APOE ε4 allele, n (%) | 86 (29) | 175 (44) | 131 (70) |
| At least one copy of APOE ε2 allele, n (%) | 40 (14) | 33 (8) | 11 (6) |
| White matter hyperintensity lesion burden (% ICV), mean (SD) | 0.46 (0.76) | 0.62 (0.80) | 0.62 (0.70) |
| Modified Hachinski score*, n (%) | |||
| 0 | 158 (53) | 166 (44) | 97 (49) |
| 1 | 126 (42) | 186 (49) | 91 (46) |
| 2 | 10 (3) | 10 (3) | 6 (3) |
| 3 | 4 (1) | 11 (3) | 3 (2) |
| 4 | 0 (0) | 3 (1) | 1 (< 1) |
| Biomarker evidence for AD pathology | |||
| Aβ PET SUVR, mean (SD) | 1.1 (0.2) | 1.2 (0.2) | 1.4 (0.2) |
| PET Aβ+, n (%) | 100 (34) | 210 (52) | 166 (88) |
| CSF p‐tau181, mean (SD) | 21.5 (9.2) | 25.8 (14.1) | 36.4 (15.5) |
| CSF p‐tau181+, n (%) | 91 (31) | 169 (42) | 156 (83) |
The multilabel non‐AD pathology classifier identified 12% of the CU participants as having TDP‐43, 4% as having LBD, 25% as having moderate to severe CAA, and 77% as having mild to severe CAA (Figure 3). The estimated TDP‐43 positivity rates were 2.5 to 3.9 times higher in participants with a clinical diagnosis of MCI (30%) and dementia (47%), respectively. The estimated LBD positivity rates were 3.8 to 9.2 times higher in participants with a clinical diagnosis of MCI (15%) and dementia (37%), respectively. While the estimated moderate to severe and mild to severe CAA positivity rates within MCI (28% and 84%) were similar to those of CU participants, the estimated CAA positivity rates were 1.25 to 1.36 times greater within dementia (34% moderate to severe CAA and 95% mild to severe CAA) compared to those of CU participants.
FIGURE 3.
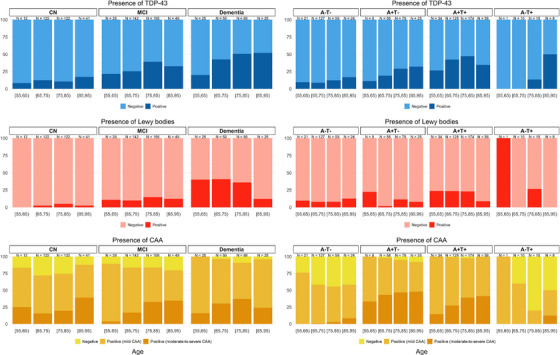
Estimated rates of TDP‐43, LB, and CAA positivity within clinical diagnostic groups and within PET Aβ (A) and CSF p‐tau181 (T) biomarker‐positive groups in target ADNI cohort without gold standard neuropathology assessments.
Among participants with biomarker evidence of PET Aβ and CSF p‐tau181 (A+T+), that is, suggesting intermediate to high ADNC, regardless of clinical diagnosis, TDP‐43 was imputed to be positive in 49% of participants, LBD pathology in 24%, moderate or severe CAA in 32%, and mild, moderate, or severe CAA in 98% (Figure 3). These rates of non‐AD pathologies imputed within A+T+ ADNI participants are in line with the previous clinicopathology literature. Specifically, 17% to 45% of ADNC cases at autopsy are accompanied by neocortical LBD. 5 , 6 , 10 , 11 , 12 , 41 , 42 The comorbidity rate for TDP‐43 (including limbic‐predominant age‐related TDP‐43 encephalopathy neuropathologic change, or LATE‐NC) is 35% to 58% based on multiple studies. 43 , 44 Moreover, in carefully characterized samples, CAA has been found to affect between 43% and 86% of individuals with ADNC, 5 , 6 with estimates of 85% to 95% of AD cases having at least some degree of CAA. 45 In this cohort, only N = 35 participants were A−T+, of which 17% were classified as LBD+, 17% TDP‐43+, 3% moderate or severe CAA+, and 40% mild, moderate, or severe CAA+. However, due to the limited sample size, the estimated rates of non‐ADNC positivity within different age groups are inconclusive (Figure 3).
3.4. Contribution of ADNC and non‐ADNC to cognitive and clinical decline
Within the target clinical cohort of ADNI participants, demographic factors (age, sex, years of education), AD risk factors (APOE ε4 carrier status and cerebrovascular white matter lesion burden), CSF biomarker of AD‐related p‐tau181, and PET Aβ burden, together with imputed scores for the presence of non‐ADNC (i.e., LBD, TDP‐43, and CAA), explained 50% to 61% of the variation in cognitive and clinical decline measured by mPACC, ADAS‐Cog13, and CDR‐SB (Table 3). While CSF biomarker of AD‐related p‐tau181 and PET Aβ burden explained 26% to 36% of the variation in cognitive and clinical decline after accounting for demographic factors, non‐ADNC explained an additional 24% to 25%, largely driven by LBD pathology. In isolation, demographic factors explained only 8% to 12% of the variation in decline in these cognitive and clinical outcome measures.
TABLE 3.
Between‐subject variances in decline in clinical and cognitive outcome measures explained by demographics (age, sex, years of education, APOE ε4 carrier status, and cerebrovascular white matter lesion burden), AD pathologies (PET Aβ and CSF p‐tau181 levels), and non‐AD pathologies (model estimated scores for presence of TDP‐43, LBD, and CAA).
| Total variance | Reduction | Percentage total variance explained | Percentage additional variance explained relative to demographics + AD biomarkers model | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Predictor | mPACC | ADAS‐Cog 13 | CDR‐SB | mPACC | ADAS‐Cog 13 | CDR‐SB | mPACC | ADAS‐Cog 13 | CDR‐SB | mPACC | ADAS‐Cog 13 | CDR‐SB |
| Reference model | 2.52 | 5.87 | 0.48 | − | − | − | − | − | − | − | − | − |
| Demographics | 2.31 | 5.21 | 0.42 | 0.21 | 0.66 | 0.05 | 8% | 11% | 12% | − | − | − |
| Demographics + ADNC | 1.71 | 3.75 | 0.35 | 0.81 | 2.11 | 0.12 | 32% | 36% | 26% | − | − | − |
| Demographics + ADNC + LBD + TDP43 + CAA | 1.09 | 2.30 | 0.24 | 1.43 | 3.57 | 0.24 | 57% | 61% | 50% | 25% | 25% | 24% |
| Demographics + ADNC + LBD | 1.21 | 2.49 | 0.25 | 1.31 | 3.37 | 0.22 | 52% | 57% | 47% | 20% | 21% | 21% |
| Demographics + ADNC + TDP‐43 | 1.73 | 3.82 | 0.35 | 0.79 | 2.05 | 0.12 | 31% | 35% | 26% | − | − | − |
| Demographics + ADNC + CAA | 1.67 | 3.72 | 0.35 | 0.84 | 2.15 | 0.13 | 34% | 37% | 26% | 2% | 1% | − |
3.5. Early‐AD clinical trial scenarios: Required sample size and screen fails
Figure 4 show the effect sizes needed to achieve 80% power with varying sample sizes for 18‐month early‐AD clinical trials. Overall, taking into account the presence of co‐pathologies at the individual level enhances statistical power and reduces the necessary sample sizes required to detect a slowing in the increase in ADAS‐Cog13 at any required effect size. Specifically, in an 18‐month placebo‐controlled trial, a sample size of 293 Aβ+ participants per group is required to detect a 30% slowing in overall increase in ADAS‐Cog13 with 80% power. However, when accounting for contributions of non‐ADNC, the sample size decreases to 212 Aβ+ participants per group to detect a 30% slowing in increase in ADAS‐Cog13. This equates to a 28% reduction in the required sample size. Similarly, to detect a 30% slowing in overall increase in ADAS‐Cog13 in a study of participants positive for both Aβ and p‐tau ADNC, the required sample size per group decreases by 24%, from 228 per group to 174, after accounting for contributions of non‐ADNC co‐pathologies at the subject level.
FIGURE 4.
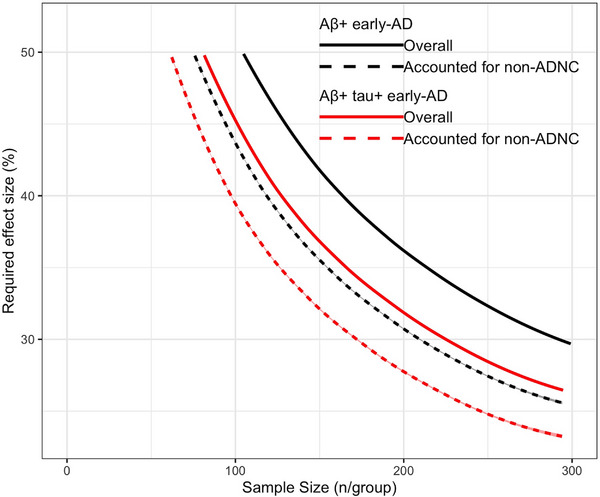
Required treatment effect sizes are plotted against sample sizes in two 18‐month early‐AD clinical trial schemes, screened for evidence of Aβ pathology only in black and for evidence of both Aβ and p‐tau pathology in red. The plotted curves show the combinations of the required treatment effect and sample size to achieve 80% power for ADAS‐Cog13 outcome measures based on overall estimates of decline in outcome measures after accounting for age at baseline in solid lines and after accounting for the contributions of the presence of non‐ADNC co‐pathologies at the subject level in dashed lines.
Table 4 summarizes the results of modeling various scenarios for the biomarker‐based enrichment of non‐ADNC pathology for an 18‐month placebo‐controlled trial involving an early‐AD population with biomarker‐confirmed AD Aβ+ and p‐tau+ pathologies. The reference scenario (scenario 0) involved participants with varying comorbid non‐ADNC but with no selection for it. The ADNC‐driven contribution to the slowing rate, estimated based on an overall 30% slowing in the increase in ADAS‐Cog13 in scenario 0, was 51%, assuming the imputed presence of non‐ADNC within this reference population. This estimated rate was then used to determine the required overall slowing rate for scenarios 1 to 3, where participants were selectively screened out based on the presence of comorbid non‐ADNC and their combinations, also based on the imputed presence of non‐ADNC within the corresponding population. Accordingly, the required overall rate of slowing for scenario 1, wherein participants with any comorbid non‐ADNC were screened out, was 51%. The required overall rate of slowing for scenario 2, wherein participants with more than one comorbid non‐ADNC were screened out, was 33%. The required overall rate of slowing for scenario 3, wherein participants with comorbid LBD pathology were screened out regardless of the presence or absence of comorbid TDP‐43 or CAA, was 46%. While screening out participants with any comorbid non‐ADNC led to a high additional screen fail rate of 78%, the required sample size in this scenario was only 39% of that required in the reference scenario. In contrast, a scenario of screening out only the participants with more than one comorbid non‐ADNC and the scenario of screening out only the participants with comorbid LBD pathology both involved a smaller additional screen fail rate of 18% to 19%, but with gains in the relative sample size of 15% and 38%, respectively.
TABLE 4.
Scenarios of biomarker‐based enrichment for comorbid pathology in placebo‐controlled 18‐month trial on early‐AD population with biomarker‐confirmed AD Aβ and p‐tau pathologies.
| Scenarios | Overall rate of slowing | ADNC‐attributed rate of slowing | Additional screen failure rate (%) | Annual rate of decline | Sample size (n/group) | Relative sample size |
|---|---|---|---|---|---|---|
| No selection for comorbid non‐ADNC | 30% | 51% | 0% | 2.39 ± 2.64 | 226 | 100% |
| Screen out patients with any non‐ADNC | 51% | 51% | 78% | 2.17 ± 2.61 | 88 | 39% |
| Screen out patients with > 1 non‐ADNC | 33% | 51% | 19% | 2.24 ± 2.82 | 191 | 85% |
| Screen out patients positive for LBD | 46% | 51% | 18% | 1.89 ± 2.69 | 139 | 62% |
Note. The reference cohort (scenario 0) participants had variable comorbid non‐ADNC (no selection for comorbid non‐ADNC). Within the reference cohort, each participant's imputed presence of non‐ADNC was accounted for to estimate rate of slowing in ADAS‐Cog13 progression that would be attributed to ADNC, given a 30% slowing in the overall ADAS‐Cog13 progression. The estimated ADNC‐attributed slowing rate was then used to estimate the required overall slowing rate for scenarios 1 to 3, wherein some participants were selectively excluded based on the presence of one or more comorbid non‐ADNC pathologies as specified. Sample size to detect the required rate of slowing in overall rate of ADAS‐Cog13 progression with 80% power along with the estimated relative sample size compared to reference scenario, additional screen failure rate, and annual overall rate of decline are reported.
4. DISCUSSION
Co‐pathologies such as LBD, TDP‐43, and CAA are frequently observed in older individuals with AD, even in those with confirmed Aβ and tau pathology, and significantly contribute to cognitive and clinical decline. Treatments targeting just one pathology will thus have variable and reduced overall measurable efficacy when applied to such populations compared with an assumed population with ADNC and no co‐pathologies. We identified imaging signatures of these co‐pathologies, which cannot currently be measured with biomarkers, and used these signatures to determine the extent to which these co‐pathologies account for the variance in cognitive decline. We demonstrated that when these signatures are used as covariates in a clinical trial design, power is increased, resulting in a reduction of the required sample size. Tools to identify individuals with comorbid non‐ADNC would be enabling technologies for a precision medicine approach to clinical trials in sporadic AD and may reduce sample sizes through use as covariates in statistical analysis or as additional screening criteria.
This study used cross‐sectional structural MRI, demographics, clinical measures, number of APOE alleles (ε2 and ε4 separately), and AD Aβ and p‐tau pathology to accurately identify individuals with comorbid pathological changes (accuracy: 76% to 92%; PPV: 75% to 94%; NPV: 77% to 84%). The model outperformed the reference models, which only used demographics, clinical measures, number of APOE alleles, and (separately or jointly) AD pathology information, without MRI volumetric variables, with a 0.08‐ to 0.22‐unit improvement in AUC. Without in vivo biomarkers to measure the presence and severity of these co‐pathologies, at present, TDP‐43 pathology can only be accurately predicted in individuals with C9orf72 and GRN gene mutations or with neuropathological verification for individuals without a family history. Fluid biomarker studies have reported the association of elevated CSF TDP‐43 levels with amyotrophic lateral sclerosis and rapid progression of frontotemporal dementia. 46 However, since TDP‐43 has a low concentration in CSF and mainly originates from blood, its correlation with pathological accumulation and neurodegeneration in the brain remains controversial. 47 Similarly, α‐synuclein markers in CSF, detected using assays based on seeded aggregation of recombinant α‐synuclein, have shown promising results as indicative biomarkers for LBD 48 and the presence of α‐synuclein pathology in patients with Parkinson's disease. 49 Plans are under way for ADNI CSF samples to be analyzed by this assay, and the results will be used to further validate our approach to detect the presence of LBD. Other indicative biomarkers, such as reduced basal ganglia dopamine transporter uptake by PET or single‐photon emission CT, and supportive imaging biomarkers, such as relative preservation of medial temporal lobe structures, reduced occipital activity, and posterior cingulate island glucose metabolism sign, 50 support the diagnosis of LBD. Although these biomarkers may have prognostic implications, they do not inform the presence of underlying pathology. Similarly, the Boston criteria are a diagnostic tool for probable‐CAA based on clinical and MRI information. 51 The Boston criteria have high diagnostic accuracy (74.5% sensitivity; 95.0% specificity; AUC 0.85) but have not been validated for use in asymptomatic individuals or individuals with a full range of neurodegenerative pathologies. Our proposed computational approach for detecting CAA showed superior performance to the Boston criteria (AUC 0.95 vs 0.85) with greater sensitivity (97.7% vs 74.5%) but lower specificity (76.2% vs 95.0%). The poor specificity may be due to the fact that the majority of the study cohort was identified with mild to severe CAA at autopsy. When the class sizes are very different, classification algorithms may favor the majority class, resulting in poor accuracy in minority class prediction. Despite the trade‐off between sensitivity and specificity compared to the Boston criteria, which rely on ratings of multiple MRI modalities to identify cerebrovascular imaging changes visually, the proposed computational model for CAA may be useful as an automated and scalable screening tool for comorbid CAA in dementia studies.
We found that incorporating MRI volumetric information improved classification performance compared to the models using only non‐imaging variables separately or jointly with AD biomarker variables. The implications of ADNC in the context of predicting non‐ADNC are still incompletely understood, so our results suggesting ADNC biomarkers do not improve the predictive value of non‐imaging models require further investigation. For example, Aβ‐PET scans reveal Aβ brain deposition in over 50% of LBD cases, reducing its ability to distinguish between ADNC or LBD as the etiologic process underlying an individual's dementia. 52 However, some reports suggest that TDP‐43 may be driven by neuritic Aβ plaques, meaning more neuritic plaques may lead to greater likelihood of TDP‐43 proteinopathy. 27 Since PET and CSF biomarkers were not widely available in our autopsy‐confirmed study cohort, we used binary Aβ and p‐tau positivity status as predictive variables in our models. Future studies are needed to determine whether severity and burden of Aβ and p‐tau pathologies beyond positivity status can further predict the presence of non‐ADNC.
In the proposed computational models, the brain tissue volumetric variations influencing the predicted score for the presence of non‐ADNC were dominated by limbic brain regions and cortical networks. There is emerging literature investigating neuropathology‐related brain changes using antemortem structural MRI, describing associations between end‐of‐life presence of TDP‐43 pathological changes in AD with smaller hippocampus, 53 , 54 , 55 smaller amygdala, 53 , 55 , 56 and faster rates of hippocampal atrophy; 57 LBD with normal hippocampal volume, 58 , 59 , 60 smaller dorsal mesopontine gray matter, 58 , 59 , 60 smaller amygdala 58 , 59 , 61 and occipital hypoperfusion; 54 and CAA with tissue loss pronounced in the occipital, temporal, posterior parietal, and medial frontal regions independent of AD pathology. 62 , 63 It is important to note that our study does not explain the mechanisms behind the detected non‐ADNC‐related structural variations. One possible interpretation is that the MRI volumetric signatures are due in part to the direct toxicity of the non‐ADNC. There is considerable evidence from animal and in vitro studies demonstrating that TDP‐43, LBD, and CAA have adverse effects on neuronal function. The second possible interpretation, not mutually exclusive, comes from pathology studies showing that MRI volumetric changes represent cumulative neurodegeneration from all etiologies, that is, they are not specific for any single pathological process but rather reflect the complex interactions of different etiologies.
Clinical trials aimed at treating Aβ accumulation in individuals with MCI and mild to moderate AD clinical diagnoses have shown that removing Aβ only partially slows downstream effects on tau phosphorylation, neurodegeneration, and cognitive decline. 64 , 65 , 66 Consistent with this observation, our study and previous clinicopathologic reports 15 , 18 suggest that ADNC is the primary contributor to observed cognitive and clinical decline. However, our results further suggest that only a portion of the cognitive decline in these individuals can be attributed to Aβ and p‐tau, with non‐ADNC accounting for up to 25% of the variance in decline. Of the non‐ADNC studied, LBD was found to have the greatest impact on clinical decline compared to TDP‐43 and CAA.
In a hypothetical clinical trial scenario involving Aβ+ early‐AD participants, accounting for cognitive decline caused by non‐ADNC decreased the sample size required to detect an effect on clinical decline by up to 25%. This highlights how the heterogeneity of factors contributing to clinical outcome measures at the cohort level can impact the design of AD clinical trials. It is worth noting that interventions targeting ADNC may also result in a reduction of non‐ADNC burden, as some evidence suggests that ADNC may facilitate non‐ADNC pathologies, perhaps through mechanisms like cross‐seeding. 67 Based on our findings, biomarker‐based screening for comorbid non‐ADNC in an early‐AD population could reduce sample size up to 61% but comes with a higher additional screen fail rate of 78%. However, selectively screening out participants with comorbid LBD regardless of the presence or absence of comorbid TDP‐43 or CAA could lead to a smaller additional screen fail rate of 18% and a greater relative sample size gain of 38%. Alternative enrichment strategies, wherein AD biomarkers are personalized based on presence or absence of co‐pathologies, may have great potential for direct application in AD trials and will require further investigation through future modeling studies that incorporate multipathology and clinical symptom analysis.
This study has some limitations. Even though its data originated from three different observational studies, these observational studies involved convenience cohorts, and the predictive performance of our final classifier model was assessed using cross‐validation. Thus, the degree to which our findings apply to the general population is unknown. We acknowledge the potential selection bias in autopsy‐confirmed cohorts and their differences from populations typically targeted in early‐AD clinical trials. While our study does not exclusively focus on trial enrichment, it provides valuable insights into the presence and characteristics of non‐ADNC pathologies in older individuals, regardless of their eligibility for clinical trials. Additionally, the set of candidate independent non‐ADNC predictor variables considered in this study is not exhaustive. Most notably, the prediction model did not incorporate cerebrovascular white matter lesion burden as a predictor variable, because a substantial proportion of the study cohort lacked the necessary FLAIR MRI. Another limitation of the current study is the relatively small sample size due to the limited availability of imaging and pathology data on the same elderly individuals, especially with autopsy assessment of TDP‐43. This constrained our modeling to global binary classification, rather than regionally specific staging. This limitation is especially relevant for LBD and TDP‐43, as previous research identified region‐specific associations between these pathologies, AD pathology, and cognitive decline 67 .
CONFLICT OF INTEREST STATEMENT
D.T., R.J.P., and W.A.K. have nothing to disclose. O.Y., A.S, and A.J.S. are full‐time employees of Takeda Pharmaceutical Company Ltd. M.W.W. serves on editorial boards for Alzheimer's & Dementia and the Journal of Prevention of Alzheimer's Disease. He has served on advisory boards for Acumen Pharmaceuticals, Alzheon, Inc., Cerecin, Merck Sharp & Dohme Corp., and NC Registry for Brain Health. He also serves on the USC ACTC grant, which receives funding from Eisai for the AHEAD study. He has provided consulting to BioClinica, Boxer Capital, LLC, Cerecin, Inc., Clario, Dementia Society of Japan, Eisai, Guidepoint, Health and Wellness Partners, Indiana University, LCN Consulting, Merck Sharp & Dohme Corp., NC Registry for Brain Health, Prova Education, T3D Therapeutics, University of Southern California (USC), and WebMD. He has acted as a speaker/lecturer for China Association for Alzheimer's Disease (CAAD) and Taipei Medical University, as well as a speaker/lecturer with academic travel funding provided by AD/PD Congress, Cleveland Clinic, CTAD Congress, Foundation of Learning, Health Society (Japan), INSPIRE Project, University of Toulouse, Japan Society for Dementia Research, Korean Dementia Society, Merck Sharp & Dohme Corp., National Center for Geriatrics and Gerontology (NCGG; Japan), and USC. He holds stock options with Alzeca, Alzheon, Inc., ALZPath, Inc., and Anven. Dr Weiner received support for his research from the following funding sources: National Institutes of Health (NIH)/NINDS/National Institute on Aging (NIA), Department of Defense (DOD), California Department of Public Health (CDPH), University of Michigan, Siemens, Biogen, Hillblom Foundation, Alzheimer's Association, Johnson & Johnson, Kevin and Connie Shanahan, GE, VUmc, Australian Catholic University (HBI‐BHR), The Stroke Foundation, and the Veterans Administration. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All three datasets were approved by each cohort's respective institutional review board, and informed written consent was obtained from all participants.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health [NIH] grant U19AG024904, U01AG068057, R01AG058676, U24AG074855) and Department of Defense (DOD) ADNI (award W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging (NIA) and the National Institute of Biomedical Imaging and Bioengineering and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.;Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private‐sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California (USC). ADNI data are disseminated by the Laboratory for Neuro Imaging at USC.
Data used in preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
OASIS‐3: Longitudinal Multimodal Neuroimaging: Principal Investigators: T. Benzinger, D. Marcus, J. Morris; NIH P30 AG066444, P50 AG00561, P30 NS09857781, P01 AG026276, P01 AG003991, R01 AG043434, UL1 TR000448, and R01 EB009352. AV‐45 doses were provided by Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly.
The NACC database is funded by NIA/NIH grant U01 AG016976. The study was supported by NIH grants P01 AG003991, P30 AG028383, P30 AG013854, AG047366, P50 AG005133, and P50 AG005681. Data analysis for this study was partially funded by NIH/NIA U01AG024904, NIH/NIA U01AG068057‐01, NIH/NIA R01AG058676, and Takeda Pharmaceutical Company Ltd.
Tosun D, Yardibi O, Benzinger TLS, et al. Identifying individuals with non‐Alzheimer's disease co‐pathologies: A precision medicine approach to clinical trials in sporadic Alzheimer's disease. Alzheimer's Dement. 2024;20:421–436. 10.1002/alz.13447
REFERENCES
- 1. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 2. Cummings J. The role of biomarkers in Alzheimer's disease drug development. Adv Exp Med Biol. 2019;1118:29‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rabinovici GD, Carrillo MC, Forman M, et al. Multiple comorbid neuropathologies in the setting of Alzheimer's disease neuropathology and implications for drug development. Alzheimers Dement (New York, N Y). 2017;3:83‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer's dementia attributed to age‐related neuropathologies. Ann Neurol. 2019;85:114‐124. PMID: 30421454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas DX, Bajaj S, McRae‐McKee K, Hadjichrysanthou C, Anderson RM, Collinge J. Association of TDP‐43 proteinopathy, cerebral amyloid angiopathy, and Lewy bodies with cognitive impairment in individuals with or without Alzheimer's disease neuropathology. Sci Rep. 2020;10:14579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late‐onset Alzheimer's disease. Brain. 2021;144:2186‐2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suemoto CK, Ferretti‐Rebustini RE, Rodriguez RD, et al. Neuropathological diagnoses and clinical correlates in older adults in Brazil: a cross‐sectional study. PLoS Med. 2017;14:e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134:171‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi‐center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathol: JJSN. 2015;35:390‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain. 2018;141:2181‐2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kovacs GG, Milenkovic I, Wöhrer A, et al. Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol. 2013;126:365‐384. PMID: 23900711. [DOI] [PubMed] [Google Scholar]
- 13. Suemoto CK, Leite REP, Ferretti‐Rebustini REL, et al. Neuropathological lesions in the very old: results from a large Brazilian autopsy study. Brain Pathol. 2019;29:771‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson RS, Yang J, Yu L, et al. Postmortem neurodegenerative markers and trajectories of decline in cognitive systems. Neurology. 2019;92:e831‐e840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person‐specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83:74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu L, Wang T, Wilson RS, et al. Common age‐related neuropathologies and yearly variability in cognition. Ann Clin Transl Neurol. 2019;6:2140‐2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wennberg AM, Whitwell JL, Tosakulwong N, et al. The influence of tau, amyloid, alpha‐synuclein, TDP‐43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging. 2019;77:26‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boyle PA, Wang T, Yu L, et al. To what degree is late life cognitive decline driven by age‐related neuropathologies? Brain. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boyle PA, Yang J, Yu L, et al. Varied effects of age‐related neuropathologies on the trajectory of late life cognitive decline. Brain. 2017;140:804‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stocchetti N, Paterno R, Citerio G, Beretta L, Colombo A. Traumatic brain injury in an aging population. J Neurotrauma. 2012;29:1119‐1125. PMID: 22220762. [DOI] [PubMed] [Google Scholar]
- 21. Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer's Disease Neuroimaging Initiative 3: continued innovation for clinical trial improvement. Alzheimers Dementia: J Alzheimer's Assoc. 2017;13:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. LaMontagne PJ, Benzinger TL, Morris JC, et al. OASIS‐3: longitudinal neuroimaging, clinical, and cognitive dataset for normal aging and Alzheimer Disease. medRxiv. 2019. 2019.12.13.19014902. [Google Scholar]
- 23. Beekly DL, Ramos EM, Van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270‐277. PMID: 15592144. [PubMed] [Google Scholar]
- 24. Besser LM, Kukull WA, Teylan MA, et al. The revised national Alzheimer's coordinating center's neuropathology form‐available data and new analyses. J Neuropathol Exp Neurol. 2018;77:717‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dementia: J Alzheimer's Assoc. 2012;8:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017. [DOI] [PubMed] [Google Scholar]
- 29. Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite: measuring amyloid‐related decline. JAMA Neurol. 2014;71:961‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2020. Alzheimer Dementia: Transl Research Clin Intervent. 2020;6:e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doshi J, Erus G, Ou Y, et al. MUSE: mUlti‐atlas region Segmentation utilizing Ensembles of registration algorithms and parameters, and locally optimal atlas selection. Neuroimage. 2016;127:186‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pomponio R, Erus G, Habes M, et al. Harmonization of large MRI datasets for the analysis of brain imaging patterns throughout the lifespan. Neuroimage. 2020;208:116450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Voevodskaya O, Simmons A, Nordenskjöld R, et al. The effects of intracranial volume adjustment approaches on multiple regional MRI volumes in healthy aging and Alzheimer's disease. Front Aging Neurosci. 2014;6:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. MFn D, E C, Barro S, Amorim DG. Do we need hundreds of classifiers to solve real world classification problems? J Mach Learn Res. 2014;15:3133‐3181. [Google Scholar]
- 35. Karas M, Brzyski D, Dzemidzic M, et al. Brain connectivity‐informed regularization methods for regression. Stat Biosci. 2019;11:47‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chutinet A, Rost NS. White matter disease as a biomarker for long‐term cerebrovascular disease and dementia. Curr Treat Options Cardiovasc Med. 2014;16:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alosco ML, Sugarman MA, Besser LM, et al. A clinicopathological investigation of white matter hyperintensities and Alzheimer's disease neuropathology. J Alzheimer dis: JAD. 2018;63:1347‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landau S, Thomas B, Thurfjell L, et al. Amyloid PET imaging in Alzheimer's disease: a comparison of three radiotracers. Eur J Nucl Med Mol Imaging. 2014;41:1398‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blennow K, Shaw LM, Stomrud E, et al. Predicting clinical decline and conversion to Alzheimer's disease or dementia using novel Elecsys Aβ(1‐42), pTau and tTau CSF immunoassays. Sci Rep. 2019;9:19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. SaD I, Michael C. Power and sample size for longitudinal models in R – The longpower Package and Shiny App. The R Journal. 2022;14:264‐282. [Google Scholar]
- 41. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology. 2007;69:2197‐2204. [DOI] [PubMed] [Google Scholar]
- 42. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimer Research Ther. 2014;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kapasi A, Yu L, Boyle PA, Barnes LL, Bennett DA, Schneider JA. Limbic‐predominant age‐related TDP‐43 encephalopathy, ADNC pathology, and cognitive decline in aging. Neurology. 2020;95:e1951‐e1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP‐43 stage, mixed pathologies, and clinical Alzheimer's‐type dementia. Brain. 2016;139:2983‐2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jäkel L, De Kort AM, Klijn CJM, Schreuder F, Verbeek MM. Prevalence of cerebral amyloid angiopathy: a systematic review and meta‐analysis. Alzheimers Dementia: J Alzheimer's Assoc. 2022;18:10‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scialò C, Tran TH, Salzano G, et al. TDP‐43 real‐time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020;2:fcaa142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feneberg E, Steinacker P, Lehnert S, et al. Limited role of free TDP‐43 as a diagnostic tool in neurodegenerative diseases. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:351‐356. [DOI] [PubMed] [Google Scholar]
- 48. Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of parkinson disease by detection of α‐synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74:163‐172. [DOI] [PubMed] [Google Scholar]
- 49. Concha‐Marambio L, Pritzkow S, Shahnawaz M, Farris CM, Soto C. Seed amplification assay for the detection of pathologic alpha‐synuclein aggregates in cerebrospinal fluid. Nat Protoc. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Charidimou A, Boulouis G, Frosch MP, et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI‐neuropathology diagnostic accuracy study. Lancet Neurol. 2022;21:714‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petrou M, Dwamena BA, Foerster BR, et al. Amyloid deposition in Parkinson's disease and cognitive impairment: a systematic review. Mov Disord: Off J Mov Disord Soc. 2015;30:928‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Josephs KA, Murray ME, Whitwell JL, et al. Staging TDP‐43 pathology in Alzheimer's disease. Acta Neuropathol. 2014;127:441‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Toledo JB, Cairns NJ, Da X, et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun. 2013;1:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bejanin A, Murray ME, Martin P, et al. Antemortem volume loss mirrors TDP‐43 staging in older adults with non‐frontotemporal lobar degeneration. Brain. 2019;142:3621‐3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Makkinejad N, Schneider JA, Yu J, et al. Associations of amygdala volume and shape with transactive response DNA‐binding protein 43 (TDP‐43) pathology in a community cohort of older adults. Neurobiol Aging. 2019;77:104‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Josephs KA, Dickson DW, Tosakulwong N, et al. Rates of hippocampal atrophy and presence of post‐mortem TDP‐43 in patients with Alzheimer's disease: a longitudinal retrospective study. Lancet Neurol. 2017;16:917‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kantarci K, Ferman TJ, Boeve BF, et al. Focal atrophy on MRI and neuropathologic classification of dementia with Lewy bodies. Neurology. 2012;79:553‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging. 2012;33:2091‐2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Whitwell JL, Weigand SD, Shiung MM, et al. Focal atrophy in dementia with Lewy bodies on MRI: a distinct pattern from Alzheimer's disease. Brain. 2007;130:708‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Burton EJ, Mukaetova‐Ladinska EB, Perry RH, Jaros E, Barber R, O'Brien JT. Neuropathological correlates of volumetric MRI in autopsy‐confirmed Lewy body dementia. Neurobiol Aging. 2012;33:1228‐1236. PMID: 21353336. [DOI] [PubMed] [Google Scholar]
- 62. Fotiadis P, Van Rooden S, Van Der Grond J, et al. Cortical atrophy in patients with cerebral amyloid angiopathy: a case‐control study. Lancet Neurol. 2016;15:811‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Subotic A, McCreary CR, Saad F, et al. Cortical thickness and its association with clinical cognitive and neuroimaging markers in cerebral amyloid angiopathy. J Alzheimer Dis: JAD. 2021;81:1663‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9:197‐210. [DOI] [PubMed] [Google Scholar]
- 65. Van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2022. [DOI] [PubMed] [Google Scholar]
- 66. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384:1691‐1704. [DOI] [PubMed] [Google Scholar]
- 67. Robinson JL, Richardson H, Xie SX, et al. The development and convergence of co‐pathologies in Alzheimer's disease. Brain. 2021;144:953‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information