

SUMMARY
Phenotypic heterogeneity in monogenic neurodevelopmental disorders can arise from differential severity of variants underlying disease, but how distinct alleles drive variable disease presentation is not well understood. Here, we investigate missense mutations in DNA methyltransferase 3A (DNMT3A), a DNA methyltransferase associated with overgrowth, intellectual disability, and autism, to uncover molecular correlates of phenotypic heterogeneity. We generate a Dnmt3aP900L/+ mouse mimicking a mutation with mild to moderate severity and compare phenotypic and epigenomic effects with a severe R878H mutation. P900L mutants exhibit core growth and behavioral phenotypes shared across models but show subtle epigenomic changes, while R878H mutants display extensive disruptions. We identify mutation-specific dysregulated genes that may contribute to variable disease severity. Shared transcriptomic disruption identified across mutations overlaps dysregulation observed in other developmental disorder models and likely drives common phenotypes. Together, our findings define central drivers of DNMT3A disorders and illustrate how variable epigenomic disruption contributes to phenotypic heterogeneity in neurodevelopmental disease.
Graphical Abstract
In brief
Beard et al. characterize a DNMT3A mutant mouse carrying a moderate severity mutation, comparing it with a severe mutation to uncover epigenomic and transcriptional drivers of phenotypic variability. They utilize these mice to identify DNA-methylation-dependent enhancer dysfunction, gene expression changes, and concordant gene dysregulation with other neurodevelopmental disorder models.
INTRODUCTION
As clinical sequencing becomes widely implemented, numerous causative variants have been identified in individual disease genes. A subset of genes associated with phenotypically heterogeneous intellectual disability and autism spectrum disorder (ASD) primarily present with missense variants rather than truncating mutations.1–3 For these genes, studies of multiple disease-causing alleles can identify heterogeneity and molecular differences between variants that drive diversity in clinical presentations while also establishing shared effects across alleles to define core pathways to be targeted for therapeutics.
DNA methyltransferase 3A (DNMT3A)-associated neurodevelopmental disorders display phenotypic heterogeneity that is likely driven by the diversity of missense mutations associated with disease. Heterozygous DNMT3A mutations cause Tatton-Brown Rahman syndrome (TBRS), an overgrowth and intellectual disability disorder typified by macrocephaly, a distinct facial gestalt, obesity, and comorbid ASD.4,5 DNMT3A mutations have also been identified in patient cohorts with a primary diagnosis of ASD.2,6,7 Disease mutations are frequently missense mutations, with truncations and deletions underrepresented compared to chance estimates.4,8 In vitro studies have demonstrated that mutations disrupt protein function through a variety of mechanisms8–10 and may lead to varying degrees of loss of function; however, it remains unclear to what extent these mutations drive differential phenotypes in vivo.
Disease-associated DNMT3A mutations impact multiple aspects of nervous system development and function. DNMT3A is expressed both embryonically and postnatally, contributing to genomic imprinting and maturation of the nervous system.11–13 DNMT3A peaks in expression in postnatal neurons (1–3 weeks old in mice)14,15 to establish uniquely high levels of non-CpG methylation in these cells relative to other somatic cell types.14,16–18 This methylation, occurring predominantly at CA dinucleotides (mCA), is sensitive to the expression levels of DNMT3A. For example, heterozygous loss of DNMT3A (knockout [KO]/+) causes 50% reduction in mCA genome-wide, while modest overexpression of DNMT3A leads to excess deposition of this mark.9,19 A major function of mCA is to recruit the methyl-binding protein MeCP2 to regulate the activity of enhancers20,21 and tune the expression of large numbers of genes, allowing neurons to dynamically respond to activity and maintain cell-type-specific gene expression.13,17,22
Mouse models of DNMT3A mutations have established phenotypes associated with human DNMT3A disorders, but missense alleles have not been systematically assessed in the brain. Analysis of DNMT3A heterozygous deletion mice (KO/+) detected growth and behavioral deficits that mirror aspects of human disease and identified underlying alterations in neuronal DNA methylation hypothesized to drive these effects.9,23 In addition, the mouse model of the human R882H mutation demonstrated more severe behavioral disruption than the KO/+.9,24 However, studies of R882H in acute myeloid leukemia suggest that this mutation results in dominant-negative effects not observed for other mutations,24–26 and no model representing the majority of “typical” missense mutations causing partial loss of function has been systematically analyzed in vivo. Therefore, core deficits shared by the majority of disease-associated DNMT3A missense mutations remain undefined, and the molecular underpinnings driving a spectrum of severity have not been assessed.
Here, we addressed these gaps in knowledge by generating and characterizing a mouse model of the human DNMT3A P904L mutation. Using these mice, which represent a class of missense mutations partially disrupting the methyltransferase activity of DNMT3A,4,9 we define core deficits observed across DNMT3A models, including overgrowth, obesity, altered communication, and reduced neuronal DNA methylation. Comparing this new model to a mouse model of the human R882H mutation, we identify distinct phenotypic, epigenomic, and transcriptional effects of these two DNMT3A missense mutations in vivo. Finally, we establish molecular pathways and etiology shared within DNMT3A models and use these core effects to explore convergent molecular mechanisms contributing to nervous system disruption across related neurodevelopmental disorders.
RESULTS
P900L heterozygous mutant mice exhibit obesity and bone length overgrowth
To explore effects caused by missense mutations in DNMT3A and to characterize a “typical” mutation, we generated a constitutive Dnmt3aP900L/+ (P900L) mutant mouse mimicking the recurrent human P904L mutation that shows robust loss-of-function effects in vitro9 (Figures S1A and S1B). P900L mutant mice did not display severe changes in general health (Figure S1C) and had similar expression of DNMT3A mRNA; however, a subtle reduction in protein expression was observed (Figure S1D), indicating that the mutant protein is expressed but that a subset of effects may be caused by reduced DNMT3A levels. With this model in hand, we assessed phenotypes caused by the P900L mutation.
Patients with DNMT3A mutations exhibit overgrowth (defined as being +2 standard deviations above mean height), macrocephaly, and a distinctive facial gestalt4; therefore, we probed for homologous morphological changes in the P900L model. Human height is correlated with leg bone length,27 so we measured femur length using X-ray imaging and found that P900L femurs were significantly longer than those of wild-type (WT) littermates (Figures 1A and 1B). Skull morphology analysis using microcomputed tomography (μCT) imaging detected no increases in skull size, with few subtle changes in distances between skull Euclidian landmarks (Figures 1C and 1D). Thus, the P900L model exhibits significant changes in long bone length without overall changes in skull size or shape.
Figure 1. P900L mutants have increases in bone length and progressive obesity.
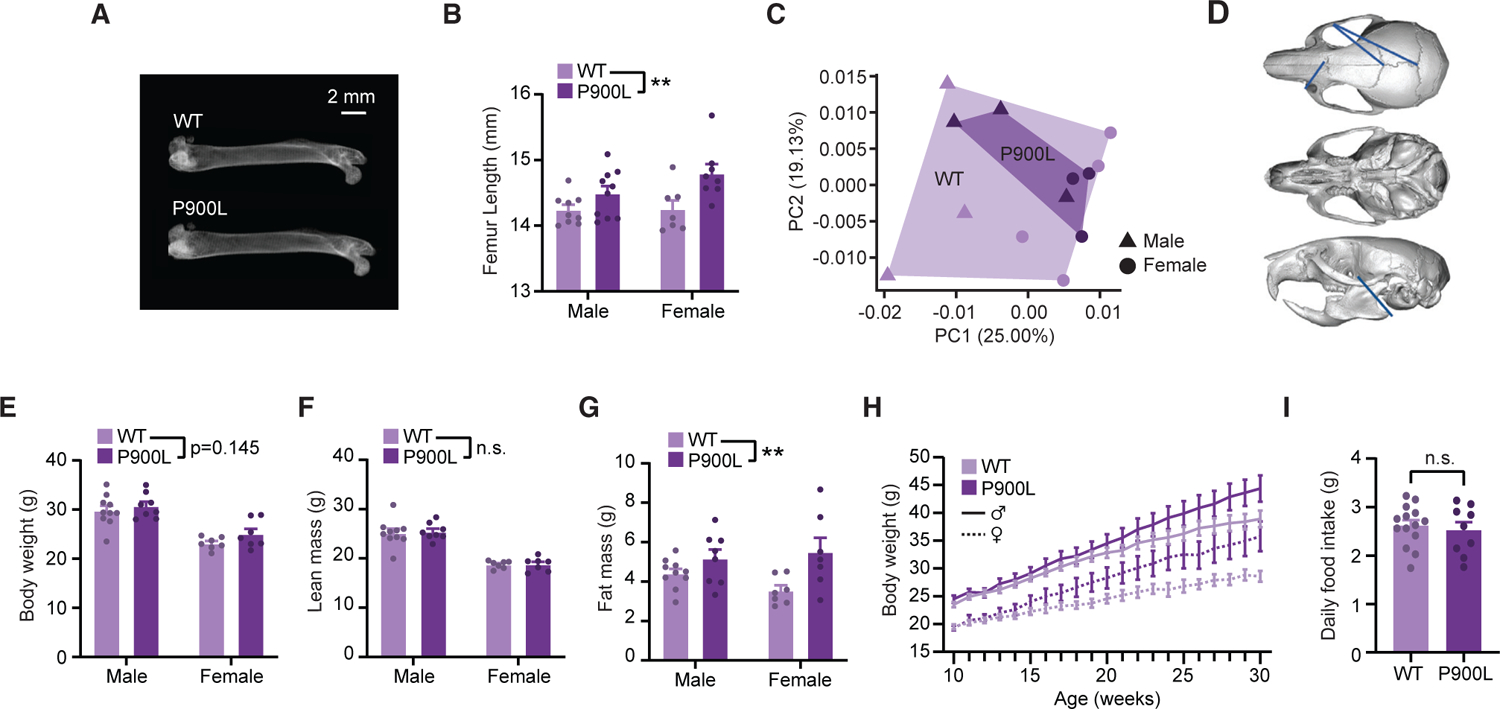
(A) Representative dual X-ray image of femurs isolated from 30-week-old WT and P900L littermates.
(B) Quantification of femur length.
(C) Principal-component analysis of skull landmark distances.
(D) Example image of reconstructed skull from μCT imaging with significant linear distances shown. Blue lines indicate distances that were significantly longer in the WT compared to the P900L. No distances were significantly longer in the P900L.
(E) Quantification of body weight.
(F and G) EchoMRI measures of lean mass and fat mass.
(H) Body weights of animals on a high-fat diet measured weekly for 20 weeks (three-way repeated measures ANOVA; genotype p = 0.022; genotype by time p < 0.0001).
(I) Daily food intake between 30-week-old WT and P900L animals fed a high-fat diet for 20 weeks (unpaired t test p = 0.624). Results are expressed as mean ± SEM. No significant sex-genotype interactions observed. Detailed statistics and sample sizes are in Table S1.
Obesity is an emerging phenotype in the TBRS clinical population with potential impacts on patient health. We measured body weight and used EchoMRI to measure body mass content in adult P900L animals. Mutants displayed increased body weight trends at 30–35 weeks and significantly increased fat mass with no change in lean mass, indicating an obesity phenotype (Figures 1E–1G). High-fat diets can exacerbate progressive weight gain24; therefore, we measured weight gain in animals on a high-fat diet and found a significant increase in weight in P900L animals compared to WT littermates (Figure 1H). Notably, no increase in food consumption was detected (Figure 1I). These results suggest that the P900L mutant model exhibits a progressive obesity phenotype possibly driven by metabolic or cellular changes rather than altered feeding behavior.
These findings indicate that the P900L mutant has increases in long bone length and body fat, suggesting that DNMT3A-associated overgrowth and obesity are consistent across multiple mutations and can be studied in mice.9,24,28 The P900L has no dramatic differences in skull size or shape, suggesting that DNMT3A mice are not ideal for investigating TBRS-associated cranial phenotypes. In contrast, the reproducible increase in long bone length and body fat across this and other mouse models indicates conserved DNMT3A-dependent processes affecting body fat and skeletal development.
DNMT3A mutant mice have decreased brain volume in late adulthood
Macrocephaly is a common phenotype in TBRS, and other structural brain changes such as ventriculomegaly and Chiari malformation have been observed4,5; however, brain size and structure in mice carrying DNMT3A mutations have yet to be investigated. We therefore interrogated brain size and structure in P900L adult mice using magnetic resonance imaging (MRI) (Figure 2A). Because brain anatomical phenotypes have not been previously investigated in DNMT3A mutant mice, we also assessed Dnmt3aR878H/+ (R878H) mice mimicking the severe R882H mutation.24 Given that DNMT3A has roles embryonically and in early postnatal development,11–13,29 we measured brain volume prior to the postnatal increase in DNMT3A expression (postnatal day 10 [P10]) and in adult mice (8 weeks). No gross structural changes were detected, and mutants had similar brain volumes as WT at both time points (Figures 2B–2E). To further evaluate potential alterations in cellular makeup in DNMT3A mutants, we performed histology, measuring cerebral volume and stereologically quantifying cell counts (Figure 2F). In concordance with our MRI results, we found no changes in cell counts (Figures 2G and 2H) or cerebral volume (Figures 2I and 2J) in either DNMT3A mutant at 8 weeks of age. These results suggest that developmental human brain overgrowth phenotypes are not recapitulated in mouse models.
Figure 2. DNMT3A mutations cause reductions in brain volume and cortical thickness in aged mice.
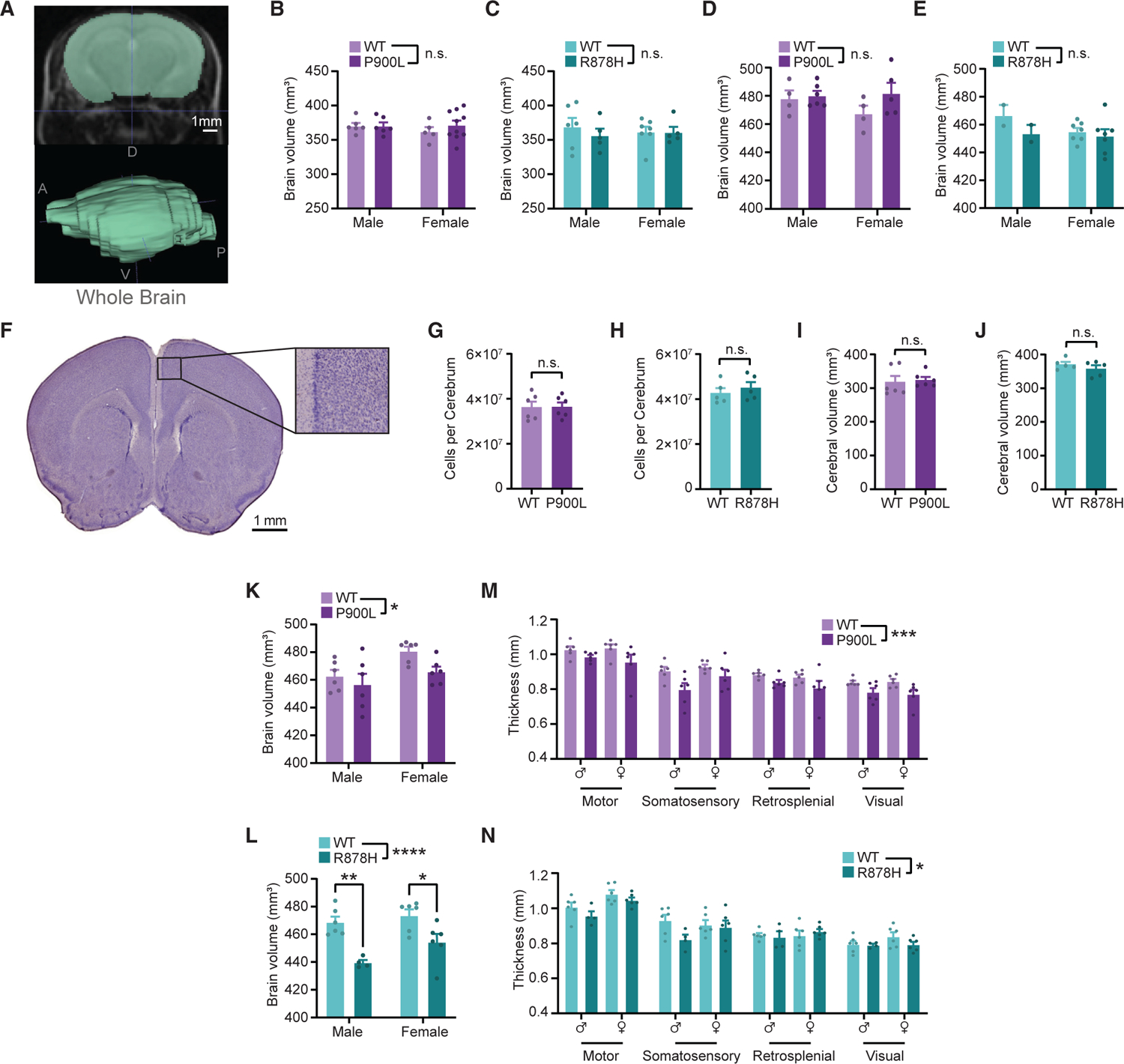
(A) Representative MRI image (top) and whole-brain segmentation (bottom). D, dorsal; V, ventral; A, anterior; P, posterior.
(B and C) Quantification of whole-brain volume from animals at postnatal day 10 (P10).
(D and E) Quantification of whole-brain volume from adult animals (8 weeks).
(F) Representative histology image from 8-week-old WT animal showing hematoxylin staining.
(G–J) Quantification of cell counts (G and H) and volume (I and J) of whole cerebrums of 8-week-old animals.
(K and L) Quantification of whole-brain volume from aged adult animals (30–35 weeks).
(M and N) Quantification of cortical thickness across various regions (three-way repeated measures ANOVA; M, P900L: genotype p < 0.0001, region p < 0.0001; N, R878H genotype p < 0.05, region p < 0.0001).
Results are expressed as mean ± SEM with individual animals shown. Genotype effect from two-way ANOVA and significant within-sex comparisons with Sidak’s multiple testing correction are shown for MRI data. Student’s t test results are shown from histology experiments. Detailed statistics and sample sizes are in Table S1. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Because we identified late-adulthood changes in body weight and fat in the P900L mutant model that could not be detected at 8 weeks, we assessed changes in brain volume late in adulthood. MRI in aged animals (30–35 weeks) revealed significantly reduced brain volume in late adulthood for both mutants (Figures 2K and 2L). To better understand brain regions contributing to these effects and to investigate any accompanying changes in connectivity, we manually segmented the corpus callosum, ventricles, and cortical thickness (Figure S2A). No significant differences in corpus callosum size were observed (Figures S2B and S2C); however, P900L mutants, but not R878H mutants, exhibited a subtle, significant decrease in the fractional anisotropy, indicating potential changes in white matter integrity or organization (Figures S2D and S2E). R878H mutants, but not P900L mutants, had subtle reductions in ventricular volume (Figures S2F and S2G). Both mutants demonstrated broad reductions in cortical thickness (Figures 2M and 2N). In total, these findings indicate that DNMT3A mutant models exhibit reduced brain size late in adulthood, suggesting a progressive phenotype that warrants future investigation.
P900L mutants exhibit early communication and tactile sensitivity deficits
Like numerous intellectual disability and ASD syndromes, DNMT3A disorders present variable behavioral deficits, and the molecular drivers of this diversity are poorly understood. To define shared and distinct cognitive phenotypes, we assessed behavioral domains disrupted in other DNMT3A models, including activity, exploration, and anxiety-like behaviors.9,18,24 In measures of activity and natural digging behaviors using the open field and marble burying assays, P900L mutants traveled similar distances compared to WT littermates (Figure 3A) and had no differences in digging behavior (Figure 3B). No motor, coordination, or sensorimotor phenotypes were observed (Figures S3A–S3H). Assessment of anxiety-like behaviors measured by time spent in the center of an open field and in open arms of the elevated plus maze showed no significant differences for P900L mutants (Figures 3C and 3D). These results indicate that exploration, motor, and anxiety phenotypes are not shared across all DNMT3A models.
Figure 3. P900L mutants show no activity or anxiety-like phenotypes but display changes in communication and tactile behaviors.

(A) Measurement of movement in an open field assay over 60 min.
(B) Marble burying behavior over a 30 min period.
(C) Time in the center of an open field assay.
(D) Time in the open arms of an elevated plus maze.
(E and F) Time spent freezing in a conditioned fear assay when in the environmental context alone (E) and in response to the tone (cue) stimuli (F).
(G and H) Morris water maze path lengths during cued trials (G) when escape platform was visible and during place trials (H) when the platform was no longer visible.
(I and J) Time in quadrants of the Morris water maze after the platform was removed (I) and number of crossings over the previous location of the escape platform (J).
(K) Preference index in a 3-chamber social approach assay for a mouse vs. an object and for a novel mouse vs. a familiar one. Preference indexes range from −1 to 1.
(L) Tube test percentage of bouts won, indicating that WT and P900L animals were equally likely to win bouts.
(M) Number of ultrasonic vocalizations of P900L and WT pups.
(N) Preference index for the novel object during a tactile novel object recognition assay for WT and P900L animals.
(O) Preference index for a visually distinct novel object for P900L and WT animals.
(P) Tube test percentage of bouts won, indicating that R878H animals won significantly more bouts than WT animals.
(Q) Number of ultrasonic vocalizations of WT and R878H pups.
(R) Preference index for the novel object during a tactile novel object recognition task for WT and R878H animals.
(S) Preference index for visually distinct novel object for R878H and WT animals.
Graphs indicate mean ± SEM. Detailed statistics and sample sizes are in Table S1. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Intellectual disability is a central phenotype in TBRS that may be variably present in individuals with DNMT3A mutations identified through studies of ASD. We therefore tested P900L mutants in conditioned fear and Morris water maze assays to assess aversive associative memory and spatial learning and memory. In conditioned fear, P900L mutants displayed normal responses to aversive stimuli and normal contextual and cued fear memory (Figures 3E and 3F; Figure S3I). P900L mutants also showed normal spatial learning in the Morris water maze assay following a slight difference upon initial task exposure (Figures 3G–3J). Notably, the absence of robust learning and memory deficits in P900L mutants appears to mirror some individuals with the homologous human P904L mutation that do not have intellectual disability diagnoses.30
Because DNMT3A mutations are associated with ASD,2,6 we next assessed phenotypes detected in mouse models of autism.31,32 In a three-chamber social approach assay,33 the P900L mutants and WT littermates showed similar preferences for exploring a conspecific over an object and for exploring a novel conspecific over a familiar mouse, with no change in overall distance traveled (Figure 3K; Figure S3J), suggesting no change in social preference or novelty. P900L and WT mice both won a similar number of bouts in the tube test, indicating no broad changes to social dominance or hierarchies (Figure 3L; Figure S3K). However, when we measured isolation-induced vocalizations in mouse pups,34 we found that mutant pups make significantly fewer calls when removed from the nest, indicating deficits in early communication behaviors (Figure 3M). Other major motor, developmental, and respiratory metrics remain otherwise unchanged (Figures S3L–S3N; Table S1). These results show that the P900L mutation causes significant deficits in neonatal communication behavior.
Disrupted somatosensory processing is implicated as a driver of autistic phenotypes, and mice carrying mutations in ASD-associated genes have been shown to have deficits in tactile discrimination.35,36 Therefore, we measured tactile discrimination using a textured novel object recognition task (NORT) in which mice explore objects that are visually indistinguishable but differ in texture. While WT mice showed a preference to explore a novel tactile object, this preference was lost in P900L mutants (Figure 3N; Figure S3O). To test the specificity of this phenotype, we repeated this task using visually and physically distinct objects and found that mutant and WT littermates displayed similar novel object preferences (Figure 3O; Figure S3P), indicating that P900L mutants have alterations specifically in tactile discrimination rather than broad deficits in associative memory or novelty-seeking behaviors.
The R878H mutation generally causes more severe behavioral deficits than those observed here in the P900L mutant24; however, the R878H mutant has not been tested for social or tactile phenotypes. We therefore tested R878H mutants for changes in social hierarchies, ultrasonic vocalizations, and tactile discrimination. Mutants displayed changes in social hierarchies, with mutants overwhelmingly winning bouts in the tube test (Figure 3P). Importantly, this behavior is not driven by increased body weight or lack of activity, as mutants did not have a significant difference in body weight at the time of testing, and the mutant mouse actively pushed the WT mouse out of the tube (Figures S3Q–S3R). Ultrasonic vocalizations were significantly reduced in R878H mutants compared to WT littermates, indicating decreases in pup communication (Figure 3Q). Mutants showed similar motor and respiratory measures but did weigh significantly less than WT littermates during this developmental window (Figures S3S–S3U; Table S1); thus, developmental delay may potentially be contributing to this phenotype. Tactile discrimination assays revealed no preference for novel tactile objects in R878H mutants and no preference for visually distinct novel objects, indicating a possible broad disruption of associative learning and memory (Figures 3R and 3S; Figures S3V and S3W).
Together, our findings indicate that the P900L mutation does not cause deficits in activity, exploration, or anxiety-like behaviors, contrasting previous findings for R878H mutants24 and Dnmt3aKO/+ mice9 and suggesting that these are not phenotypes universally associated with DNMT3A disruption. We also identify robust changes in social hierarchies in the R878H mutant, which, in combination with previous work,24 supports uniquely strong phenotypes in this mutant. These results indicate that R878H and KO/+ mutations are more severe than the P900L, but all mutants display disease-relevant behavioral deficits such as decreases in ultrasonic vocalizations and loss of preference for novel tactile objects. These shared phenotypes may be important measures for future work investigating cellular and molecular changes that contribute to disease.
Alterations in DNA methylation mirror differential phenotypic severity in DNMT3A mutants
Given the critical role DNA methylation plays in nervous system function, we hypothesized that altered methylation in the brain is a central driver of disease and that differential disruption of methylation between mutants underlies variable phenotypic severity. We therefore examined how P900L and R878H mutations affect DNA methylation in the brain by using whole-genome bisulfite sequencing (WGBS) across multiple brain regions. P900L mutants displayed a ~50% reduction of genome-wide mCA, while R878H mutants exhibited more severe (~75%) mCA reduction (Figure 4A). In contrast, global mCG levels showed trending reductions in P900L mutants and small but significant reductions in R878H mutants (Figure 4B). Thus, DNMT3A mutations cause widespread reductions of neuronal methylation, with mCA levels particularly sensitive to disruption.
Figure 4. DNMT3A mutants have significant changes to DNA methylation, with more extreme changes in the R878H mutant compared to the P900L.
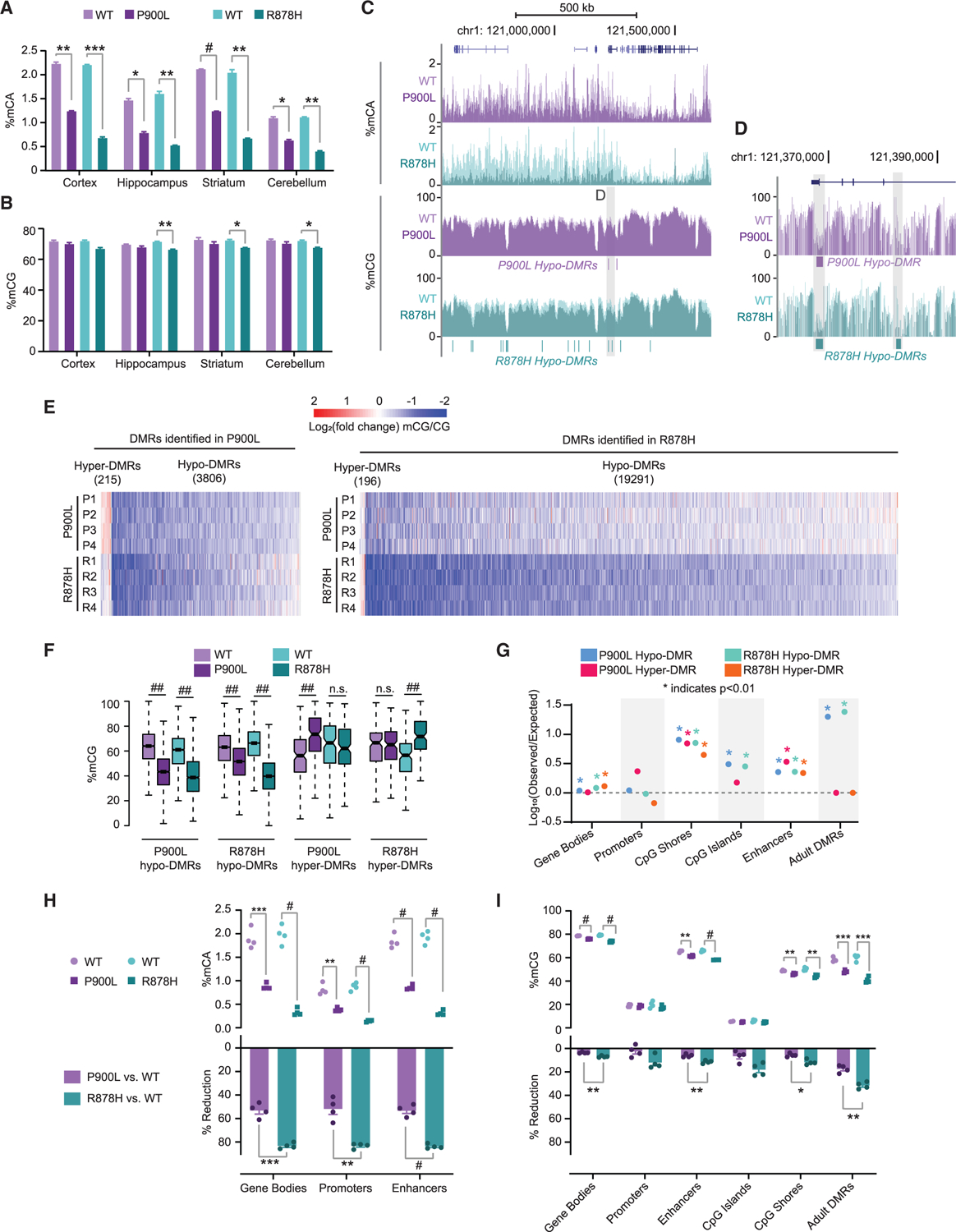
(A and B) Average genome-wide (A) percentage of mCA and (B) percentage of mCG from brain regions.
(C) Representative genome browser view showing percentages of mCA and mCG.
(D) Zoom browser view showing changes in mCG at hypo-DMRs.
(E) Heatmap of CG-DMRs identified in P900L and R878H mutants vs. their WT littermates. Log2(fold change) mCG/CG indicated between each littermate pair for each DMR.
(F) Average mCG in each genotype at DMRs called in both mutant strains.
(G) Overlap analysis of DMRs with genomic regions of interest. No point indicated for R878H hyper-DMRs CpG islands due to 0 resampled DMRs overlapping.
(H) Average mCA level at regions of interest (top) and percentage of reduction of mCA between WT and mutants (bottom).
(I) Average mCG level at regions of interest (top) and percentage of reduction of mCG between WT and mutants (bottom).
Bar graphs indicate mean ± SEM. Notched box and whisker plots indicate median, interquartile, and confidence interval of median. All groups n = 4, 2 male, 2 female. Detailed statistics and sample sizes are in Table S1. *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; ##p < 2e–10.
To systematically assess altered DNA methylation and its potential impact on gene regulation, we next interrogated methylation changes at kilobase-scale regions, including enhancers and gene bodies. We performed high-depth WGBS in the cerebral cortex, as this region is involved in behavioral processes disrupted in TBRS. This analysis confirmed broad mCA reductions without profound global reductions in mCG (Figure 4C). To uncover site-specific DNA methylation changes, we identified CG differentially methylated regions (DMRs) between sex-matched littermate pairs for both mutants. We detected 19,487 DMRs (196 hyper- and 19,291 hypo-DMRs) in the R878H mutant and 4,021 DMRs (215 hyper- and 3,906 hypo-DMRs) in the P900L mutant (Figures 4D and 4E). While more dramatic methylation changes occur in the R878H mutant, hypo-DMRs called in one mutant were generally hypo-methylated in the other, and the hypo-DMRs from both mutants significantly overlapped, demonstrating a broad concordance of effects with differing magnitude of impacts (Figures 4E and 4F; Figure S4A). Hyper-DMRs were not consistent between mutations, suggesting that these effects are stochastic or secondary to DNMT3A disruption (Figure 4F). DMRs fall in gene regulatory regions more than chance estimates, especially at CpG shores, enhancers, and regions that gain methylation during postnatal neuronal maturation (adult DMRs14) (Figure 4G). Furthermore, genes containing hypo-DMRs in both mutants are enriched for Gene Ontology (GO) terms such as neurogenesis and nervous system development (Figure S4B). Thus, both mutants contain hypo-DMRs in critical neuronal gene regulatory regions, with R878H mutants displaying more severe effects than P900L mutants.
We next quantified overall levels of mCA and mCG across a number of regions of interest such as gene bodies, promoters, and enhancers. We found significant reductions in mCA and mCG at gene bodies and enhancers and significant changes in mCA at promoters (Figures 4H and 4I). R878H mutants displayed significantly larger reductions of mCA and mCG than P900L mutants across all regions (Figures 4H and 4I), further highlighting the increased severity of the R878H mutation. These results indicate that both mutants exhibit loss of DNA methylation at critical genome regulatory regions that have the potential to affect enhancer activity and gene expression.
Altered enhancer histone acetylation corresponds with DNA methylation loss in P900L and R878H mutants
Enhancers are cis-regulatory elements that regulate gene expression, and DNA methylation at both CA and CG sites modulates enhancer activity through effects on transcription factor binding and recruitment of methyl-DNA binding factors.37–39 We examined enhancer activity using chromatin immunoprecipitation sequencing (ChIP-seq) analysis of histone H3 lysine 27 acetylation (H3K27ac; a histone modification correlated with active enhancers40) in the cortex of 8-week-old P900L and R878H mutants to determine if enhancer disruption mirrors differential DNA methylation changes. Profiles of acetylation in mutants and associated controls showed enrichment at promoters and putative enhancers consistent with published cortical profiles (Figures 5A and 5B; Figure S4C).20 Quantification of differential acetylation by EdgeR did not detect significantly altered enhancers in the P900L mutant, while 29 up- and 29 downregulated enhancers were observed in the R878H mutant. However, enhancers containing hypo-CG DMRs showed significant increases in H3K27ac in the R878H mutant and a trend toward upregulation in the P900L mutant (Figure 5C), indicating that there may be effects on activity in enhancers containing DMRs.
Figure 5. Methylation changes in DNMT3A mutants disrupt enhancer H3K27 acetylation.
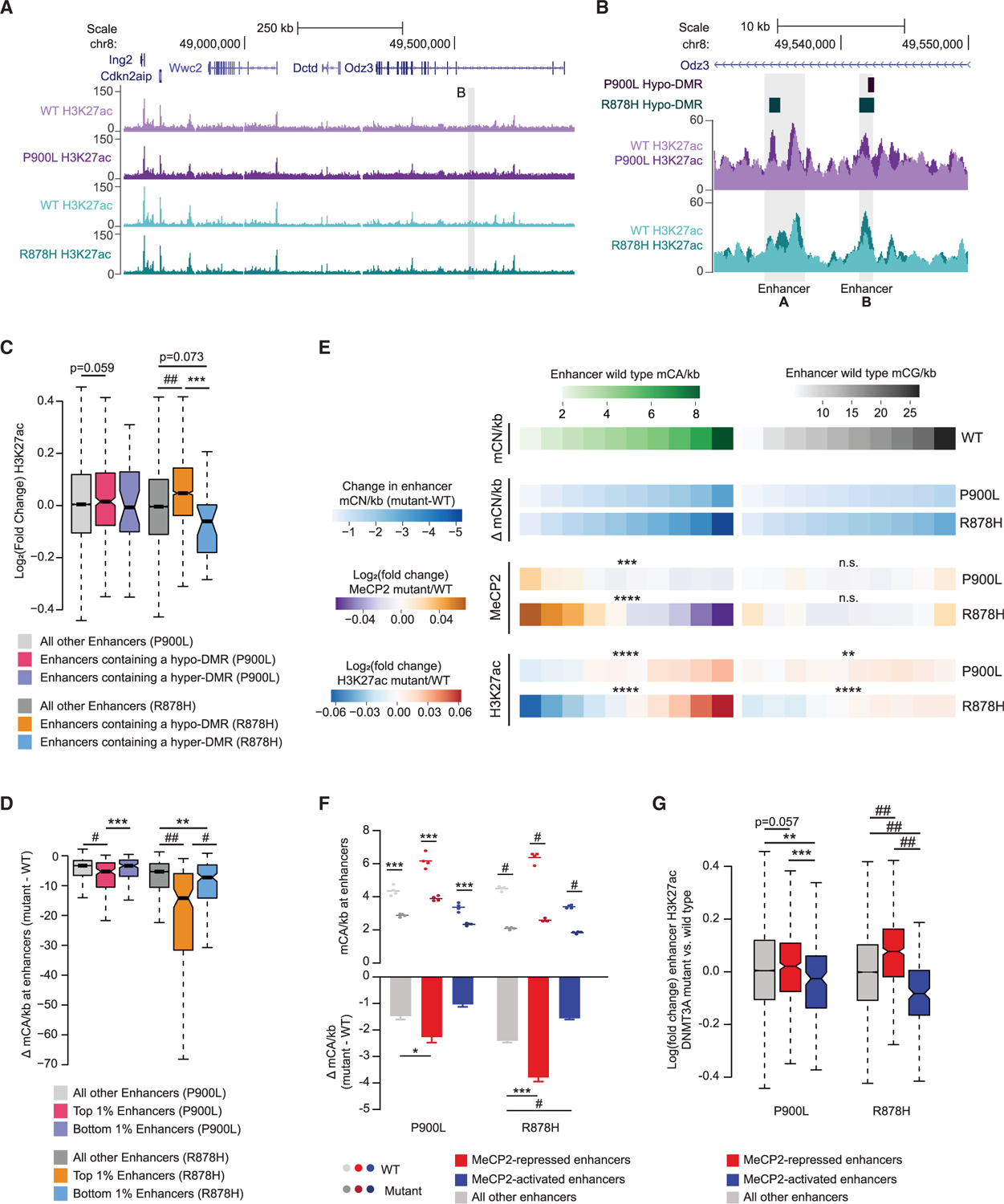
(A) Genome browser view of H3K27ac ChIP-seq data. Gray highlighted region indicates the zoomed region shown in (B).
(B) Zoomed genome browser view of H3K27ac ChIP-seq data with overlapping DMR regions and enhancer regions.
(C) Log2 fold changes of H3K27ac at enhancers containing DMRs called in each mutant.
(D) Change in mCA/kb (mutant – WT) for the top and bottom 1% of enhancers.
(E) Genome-wide deciles of WT mCA or mCG sites per kilobase at enhancers and the loss of methylation, change in MeCP2 binding, and fold change in H3K27ac at these sites in both P900L and R878H mutants. Significance of correlation between mCN/kb and measure of interest are shown.
(F) Mean mCA sites per kilobase in DNMT3A mutants and their WT littermates (top) and the change in mCA sites per kilobase between mutants and WT littermates (bottom) at enhancers significantly dysregulated in MeCP2 mutants.20
(G) Log2 fold changes in H3K27ac between mutants and WT littermates at enhancers significantly dysregulated in MeCP2 mutants.20
Notched box and whisker plots indicate median, interquartile, and confidence interval of median. Bar graphs indicate mean ± SEM. Detailed statistics and sample sizes are in Table S1. *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; ##p < 2e–10.
DNA methylation disruption can have widespread subtle effects that are not detected by significance calling in epigenomic analysis9,20; therefore, we focused our analysis on the most significant 1% of upregulated and downregulated enhancers, allowing for comparison of both mutants using a similarly sized group of enhancers. These enhancers demonstrate concordance, with enhancers up- or downregulated in one mutant also significantly dysregulated in the other (Figures S4D and S4E). To examine the relationship between mCA and enhancer H3K27ac, we measured the changes in mCA at these enhancers and observed a more dramatic loss of mCA in the most upregulated enhancers compared to other enhancers in both mutants (Figure 5D). These findings suggest that mCA changes contribute to altered enhancer activity, indicating a shared mechanism potentially affecting gene expression.
Our enhancer analysis suggests more dramatic changes in the R878H mutant; however, both mutants exhibited significant disruption of DNA methylation broadly across the genome. Therefore, we asked if enhancer changes correlate with genome-wide differences in DNA methylation and assessed if these changes differentially occur in P900L and R878H mutants. One mechanism of enhancer regulation is through MeCP2 binding to enhancer DNA methylation20,21; therefore, we used ChIP-seq to measure changes in MeCP2 binding at enhancers genome-wide. Enhancers with a high density of WT mCA sites (mCA/kb) exhibited the largest loss of mCA and MeCP2 binding in both mutants and the greatest increases in H3K27ac (Figure 5E). Enhancers with low WT levels of mCA showed the smallest reductions in mCA in mutants and a resulting decrease in relative H3K27ac. While the P900L mutants had significant effects, the R878H mutation caused larger disruptions of mCA and H3K27ac. Genome-wide mCG-driven changes in enhancer activity are more subtle, implying that perhaps only a subset of enhancers with robust mCG differences are affected (Figure 5E). Together, these data suggest that enhancers are sensitive to mCA changes in part due to MeCP2-mediated regulation.
To further investigate MeCP2-mediated enhancer regulation, we asked if enhancers most sensitive to MeCP2 disruption20 are disrupted in DNMT3A mutants and if the R878H mutants exhibit more severe effects at these enhancers than the P900L mutant. Enhancers repressed by MeCP2 displayed greater loss of mCA in both mutants compared to other enhancers (Figure 5F), accompanied by a corresponding increase in H3K27ac at these enhancers for both mutants (Figure 5G), with a more pronounced effect in the R878H mutant. Together, these findings demonstrate overlaps between enhancer disruption driven by mutation of MeCP2 and by loss of DNMT3A-dependent methylation, demonstrating shared effects between these two epigenetic regulators.
Core disruption of growth genes across DNMT3A mutants with mutation-specific effects on synaptic and protein processing genes
DNA methylation and enhancer activity are critical for tuning neuronal transcription necessary for development and function of the nervous system, and gene expression changes resulting from disrupted methylation likely contribute to disease phenotypes. Therefore, we next used RNA-seq of cerebral cortex from 8-week-old animals to define transcriptional alterations in DNMT3A mutants. P900L mutants displayed fewer significant genes (Figure 6A: 444 up, 182 down) compared to R878H mutants (Figure 6B: 797 up, 960 down), mirroring the more severe epigenomic and behavioral effects in the R878H mutant. Gene expression changes in both mutants are concordant with a model of conditional deletion of DNMT3A from postmitotic neurons,20 indicating that these gene expression changes are related to the postnatal neuronal function of DNMT3A (Figure S5A). We used PANTHER to identify the GO terms enriched in dysregulated genes to uncover the biological processes that may be most affected in these mutants. Neuronal functions related to cell adhesion and axon guidance were enriched in P900L-upregulated genes, and no terms were significantly enriched in P900L-downregulated genes (Figure 6C). Genes disrupted in R878H mutants were associated with protein folding, phospholipid translocation, and cell-cell recognition and assembly (Figure 6C). We also assessed the enrichment of mammalian phenotype ontology gene sets41 and further identified enrichments related to abnormal synaptic transmission and cognitive phenotypes in both mutants (Figure S5B). The distinct processes disrupted in these mutants may help explain the variable presentation of phenotypes; the P900L mutant has subtle changes in specific behavioral tasks, perhaps driven by changes in synaptic and axonal genes, whereas the R878H mutant shows widespread behavioral disruption corresponding with dramatic transcriptional changes involving fundamental biological processes such as protein folding and phospholipid translocation.
Figure 6. Mutation-specific changes in transcription indicate unique disruption in synaptic and protein processing gene networks.

(A) Volcano plot of DESeq2 log2 fold changes in P900L vs. WT cortex.
(B) Volcano plot of DESeq2 log2 fold changes in R878H vs. WT cortex.
(C) Top PANTHER Gene Ontology (biological process) terms enriched in each differentially expressed gene list. No significant terms were identified in the P900L-downregulated gene list.
(D) Scatterplot of DESeq2 log2 fold changes in each mutant highlighting P900L- and R878H-specific upregulated gene sets. Specific genes were defined as significantly upregulated in one mutant and either significantly unchanged (nominal p > 0.5) or downregulated in the other (fold change < 0).
(E) Top PANTHER Gene Ontology biological process terms enriched in P900L-specific and R878H-specific upregulated gene lists.
To more directly assess transcriptomic differences that could lead to distinct phenotypes between mutants, we defined mutant-specific genes as genes upregulated in one mutant and either unchanged or downregulated in the other mutant (Figure 6D) and again used PANTHER to identify enriched GO terms. We focused first on upregulated gene lists, as these may be the most direct targets from loss of DNA methylation. The P900L-specific upregulated genes showed enrichments of cell adhesion and axonal projection processes, whereas the R878H-specific upregulated genes showed enrichments related to protein folding (Figure 6E). The R878H-specific downregulated terms were primarily related to glutamatergic synaptic transmission, cell-cell adhesion, and phospholipid translocation, and no significantly enriched terms were associated with the P900L-specific downregulated genes (Figures S5C and S5D). This further suggests that transcriptional disruption in the P900L mutant affects sensitive neuronal processes, whereas dysregulated genes in the R878H mutant are potential indicators of more widespread cellular distress in addition to neuronal disruption.
While leveraging transcriptional and phenotypic differences between mutants offers insight into which gene sets contribute to distinct phenotypes, identifying shared effects across multiple DNMT3A mutant models can define central biology driving common disease phenotypes. Therefore, we used an existing disease-relevant cortical dataset from the DNMT3A heterozygous deletion mouse model together with our new transcriptomic data to identify shared disruption across TBRS models (Dnmt3aKO/+, Dnmt3aP900L/+, and Dnmt3aR878H/+). This analysis identified 228 upregulated and 160 downregulated genes that show concordant dysregulation across mutant strains (Figures 7A and 7B; Figures S5E and S5F). These genes are functionally linked to enhancers that are concordantly disrupted in the R878H mutants and trend toward similarly changed in the P900L mutants (Figure S5G). TBRS-upregulated genes are enriched for processes such as cellular and developmental growth, axon extension, and neural crest cell migration, while no terms were significantly enriched in the downregulated genes (Figure 7C). The dysregulated genes in these pathways may be critical in driving the overgrowth and behavioral phenotypes identified in individuals with DNMT3A disorders and represent strong candidates for future cellular and therapeutic studies.
Figure 7. Shared transcriptional changes across DNMT3A mutants indicate disruption of growth and synaptic processes.
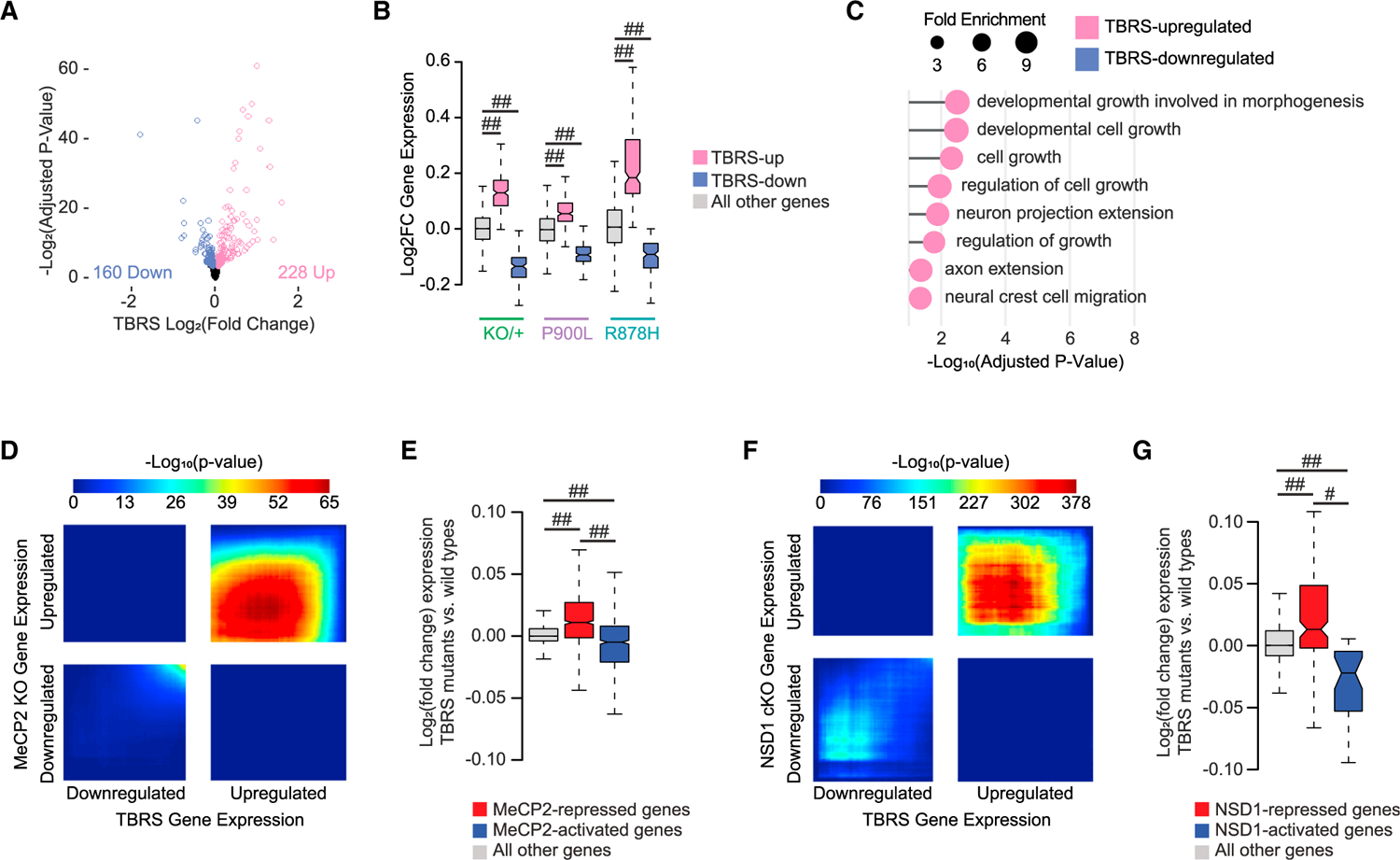
(A) Volcano plot of DESeq2 log2 fold changes from DNMT3A mutant vs. WT analysis between paired littermate data from Dnmt3aKO/+,9 Dnmt3aR878H/+, and Dnmt3aP900L/+ datasets (design = ~pair + group; contrast by group). Genes reaching a significance of false discovery rate (FDR) <0.1 are indicated.
(B) Log2 fold changes of gene expression within each mutant (KO/+, P900L, and R878H) of genes defined as differentially expressed in the combined TBRS mutant analysis.
(C) Most significant PANTHER Gene Ontology biological process terms enriched in the TBRS differentially expressed gene lists. No significant terms were identified in the TBRS-downregulated gene list.
(D) RRHO of transcriptome-wide gene expression changes in the cerebral cortex of TBRS mutants vs. MeCP2 KO mice.20
(E) Log2 fold changes in the TBRS mutants at genes significantly disrupted in MeCP2 mutants.20
(F) RRHO of transcriptome-wide gene expression changes in the cerebral cortex of TBRS mutants vs. NSD1 conditional KO mice.42
(G) Log2 fold changes in the TBRS mutants at genes significantly disrupted in NSD1 conditional KO (cKO) cortices.42
Notched box and whisker plots indicate median, interquartile, and confidence interval of median with significance from Wilcoxon rank-sum test shown. Detailed statistics and sample sizes are in Table S1. #p < 0.0001; ##p < 2e–10.
To measure concordant and distinct gene expression effects transcriptome-wide, we performed a rank-rank hypergeometric overlap (RRHO) analysis43 between the TBRS mutant models. This analysis revealed that the P900L and R878H datasets each demonstrated broad concordance with both the TBRS and Dnmt3aKO/+ datasets (Figures S5H–S5K). Interestingly, the genome-wide gene expression changes had poor concordance between the R878H and P900L mutants (Figure S5L), indicating broad-scale differences in transcriptomic disruption between these mutants. These mutations result in phenotypic severity that is either more or less severe than the KO/+, and the unique gene expression observed between P900L and R878H may correspond to the unique phenotypes observed.9,24 However, these mutants also each show strong concordance with the phenotypically intermediate KO/+ model, demonstrating shared disease-relevant effects. Thus, distinct gene expression effects may arise due to differences in the relative impact of each mutation on DNMT3A function; however, all mutants share disruption of core DNMT3A gene targets (Figure S5M). These findings highlight the necessity for using multiple mutant models together when defining the core disease-relevant transcriptional changes and differentiating these effects from potential allele-specific impacts.
Genes disrupted in TBRS models are shared across disorders that impact the neuronal methylome
Multiple neurodevelopmental diseases are caused by mutations in genes associated with the neuronal methylome (e.g., DNMT3A,4 MeCP2,44 NSD142), so we next asked if transcriptomic disruption is shared between multiple neurodevelopmental disorder models. Previous work has shown that deletions of MeCP2 and DNMT3A have overlapping gene expression patterns,9,20,29 and we have established that MeCP2-regulated enhancers are similarly disrupted in the P900L and R878H mutants. We performed RRHO analysis43 to measure transcriptome-wide correspondence between the TBRS mutant models and the MeCP2 KO and found significant overlaps in the concordant quadrants (Figure 7D). Additionally, genes significantly dysregulated in MeCP2 mutants are correspondingly disrupted in the TBRS mutants (Figures 7E; Figure S5N). Transcriptional overlap between these mutants further supports a shared molecular etiology between mutation of DNMT3A, which methylates the neuronal genome, and MeCP2, which binds that methylation to repress transcription.
Overlapping clinical phenotypes or shared biological pathways can be used to suggest other important candidate regulators of DNMT3A. One such candidate is NSD1, a histone methyltransferase associated with Sotos syndrome.45,46 A significant number of patients with NSD1 mutations display overgrowth and intellectual disability phenotypically similar to patients with TBRS,47 and recent work indicates that NSD1-deposited H3K36me2 directs DNMT3A to key genomic regions in neurons and other cell types.42,48,49 This led us to ask if there are shared effects between NSD1 mutants and TBRS models. RRHO comparison of cortical genes dysregulated in an NSD1 conditional KO model and aggregate TBRS effects indicates transcriptome-wide concordance (Figure 7F), and genes identified as dysregulated in the NSD1 mutant are similarly dysregulated in TBRS mutants (Figures 7G; Figure S5O). Strictly examining significantly dysregulated genes in the TBRS, MeCP2, and NSD1 datasets yields more modest overlaps compared to the genome-wide comparisons because of the small number of genes that reach genome-wide significance (Figures S5P and S5Q). Thus, these models share subtle effects across thousands of genes rather than a subset of highly significant effects. Together, these results indicate that the core gene expression disruption in DNMT3A disorders is shared with models of Rett syndrome and Sotos syndrome, demonstrating biological convergence and suggesting that the neuronal mCA pathway may be a useful target for candidate therapeutics for multiple disorders.
DISCUSSION
Neurodevelopmental disorders (NDDs) often present with varied phenotypes and numerous comorbidities, and the molecular mechanisms driving this spectrum of phenotypic heterogeneity have not been clearly identified. Additionally, a substantial number of mutations identified in some NDD-associated genes are missense rather than stop gains (e.g., KIF1A, MEFC2, CHD3), and the effects of these diverse mutations are not fully understood.1 Here, we studied missense mutations in DNMT3A to investigate the origins of clinically diverse phenotypes within one causative locus. Through this work, we not only identified a core set of phenotypes and shared genes that are central to DNMT3A disorders but also uncovered allele-specific variability of epigenomic disruptions driving distinct gene networks that may contribute to unique behavioral phenotypes. Furthermore, we detected transcriptional overlap between core DNMT3A gene expression effects and disruption of MeCP2 and NSD1, highlighting a potential point of convergence in disease etiology and therapeutic intervention.
In this study, we identified skeletal development and obesity phenotypes that are consistent across multiple DNMT3A mutations. The increase in long bone length shared between the P900L mutation and other mutants underscores the importance of DNMT3A in skeletal development and growth. P900L mutants also exhibit similar increases in body fat compared to other DNMT3A mutants,9,24,28 and we expand these observations by demonstrating that progressive increase in fat mass can occur without changes in feeding behavior or substantial decreases in exploratory behaviors, supporting the suggestion that other metabolic or cellular processes such as an expansion of adipocyte progenitors28 may be responsible for obesity in DNMT3A mutants.28 These findings reinforce the importance of DNMT3A in skeletal development and provide important context supporting the role of DNMT3A in obesity.
Our analysis of skull size and shape demonstrates that the P900L mutation does not exhibit changes in skull morphology, which is similar to other mouse models of TBRS but does not phenocopy the human disorder.4,9,24 We also did not identify increases in brain volumes early in development; however, we uncovered reductions in brain volume in aged mutant mice. This progressive decrease in brain volume could potentially be similar to other models of epigenetic disruption with aberrant microglial activation.50 As the clinical population diagnosed with TBRS has a mean age under 20, it remains unknown if this progressive decrease in brain volume is observed in patients.51 It will be important in future studies to investigate mechanisms driving this phenotype and to determine if similar processes are affected in the clinical population.
Humans with DNMT3A mutations range in clinical diagnoses from ASD to severe intellectual disability, and our characterization of the P900L model allowed us to identify behavioral domains with similar phenotypic heterogeneity. Previous work demonstrated that Dnmt3aKO/+ mice have reduced exploration and increased anxiety-like behaviors9 and that R878H mutant mice have more dramatic reductions in exploratory behavior and disruption of motor coordination.24 In contrast, the P900L mutant has no motor, exploratory, or anxiety-like changes, indicating that these phenotypes are not ubiquitous across all mouse models of DNMT3A disruption and instead may be a heterogeneous phenotype in these mutants. These findings clearly demonstrate differences in phenotype severity that we can compare to altered epigenomic and transcriptional effects in these models.
We expanded the established DNMT3A phenotypes in mice by assessing behaviors associated with ASD and identified disruption of communication and tactile discrimination shared across multiple mutations. Altered tactile discrimination is an emerging phenotype across multiple ASD models,35,36 indicating a potential mechanism contributing to behavioral disruption and highlighting the importance of DNMT3A in sensory processing. Our study also confirms that neonatal ultrasonic vocalizations are reproducibly sensitive to DNMT3A disruption, as demonstrated by the reduction in calls in P900L and R878H mutant pups and shown previously in KO/+ models.9 This work establishes these measures for future work testing therapeutics or identifying cellular mechanisms contributing to disruption.
We found that the P900L mutation causes a 50% reduction of cortical mCA, supporting the hypothesis that mCA levels are a sensitive readout of DNMT3A function. In contrast, the R878H mutation causes greater loss of mCA, demonstrating that it drives more dramatic effects than other mutations. While our results do not shed light on the exact mechanism leading to this effect, this in vivo result supports studies in the blood lineage indicating that R878H mutation is dominant negative.25 Differences in allele severity are further reflected by the increased number of DMRs in the R878H mutant compared to the P900L mutant, and we demonstrate that these methylation differences overlap key genomic regulatory elements such as gene bodies and enhancers. Notably, we found increased enhancer disruption in the R878H mutant that corresponds to larger changes in mCA at enhancers. This enhancer effect is similar to observations in MeCP2 mutants,20 and we demonstrate that DNMT3A mutants have concordant disruption of enhancers regulated by MeCP2. Together, these results demonstrate how disease-associated missense mutations in DNMT3A differentially disrupt numerous neuronal epigenomic processes and suggest a molecular mechanism driving the spectrum of phenotypic severity (summarized in Table S4).
Our work defined mutation-specific gene expression changes to gain insights into the cellular disruptions and biological pathways that may be driving the spectrum of disease phenotypes. The P900L mutation causes disruption of fine-tuned neuronal genes related to synaptic function and axonal guidance, suggesting that synapses, axon projections, and circuit connectivity may be disrupted in mutants. The R878H mutation causes more extensive transcriptomic disruption, altering gene networks involved in key cellular processes such as protein folding and molecular transport. These allele-specific transcriptomic effects suggest cellular mechanisms that may underlie the unique behavioral phenotypes and provide compelling candidates for future work on distinct cellular- and circuit-level effects in DNMT3A disorders.
Our characterization of transcriptional disruption in multiple models also allowed us to define a core set of neuronal genes most sensitive to DNMT3A mutation that may contribute to TBRS pathology. Upregulation of NDD-associated genes such as the Semaphorin family and Tbr1 suggest potential changes in axon guidance and migration in DNMT3A mutants, and these effects could be involved in disruption of ultrasonic vocalizations in mice.52–55 Downregulation of Sox21 and Gabrg1 suggests potential differences in GABAergic interneurons and precursors that may be playing a role in developmental delay.56–58 These genes defined in the combined DNMT3A mutant dataset provide experimental targets for future studies of underlying cortical mechanisms driving behavioral phenotypes.
Finally, our study detected transcriptional convergence between core gene dysregulation in TBRS models and mutations in other proteins in the neuronal-methylome pathway, supporting potential functional links between Sotos syndrome, TBRS, and Rett syndrome. The transcriptional similarities between disruption of DNMT3A and other epigenetic regulators highlight the importance of this pathway for neuronal gene regulation and indicate a therapeutic point of convergence across an entire class of NDDs.
Limitations of the study
Our study demonstrates epigenomic and transcriptional disruption in the cortex of P900L and R878H mutants with disease-relevant effects. However, our RNA-seq analysis of bulk tissue fails to resolve cell-type-specific effects or determine if DNMT3A disruption alters cell composition in the brain. As DNMT3A has been implicated in cellular differentiation,59,60 some transcriptional signals in our analysis may be driven by cellular composition changes.
While our work provides evidence linking the severity of altered DNA methylation and enhancer histone acetylation to the severity of transcriptional disruption and behavioral phenotypes, the impacts on enhancer acetylation are subtle, and other epigenetic mechanisms could also be affected by loss of DNA methylation. This includes alterations in additional histone modifications, transcription initiation, and chromatin looping that may contribute to transcriptional changes we observe in the DNMT3A mutants. Future work will be required to further assess if additional epigenetic mechanisms contribute to the transcriptomic dysregulation in TBRS mutants that we have introduced in this study.
While we have identified epigenomic and transcriptomic changes that are likely to underlie behavioral phenotypes, our study does not directly connect these effects to altered behaviors. Future studies interrogating the gene expression changes we observe here and assessing their impact in behavior-relevant circuits will be needed to begin to understand how transcriptomic alterations lead to specific changes in behavior.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the lead contact, Harrison Gabel (gabelh@wustl.edu).
Materials availability
The P900L mutant mouse line generated in this study will be available by request and deposited to be made available through Jackson Labs.
Data and code availability
Next generation sequencing data have been deposited to GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-alpha-Tubulin (EP1332Y) | Abcam | Cat# ab52866; RRID: AB_869989 |
| Mouse Anti-Dnmt3a Monoclonal Antibody, Clone 64B1446 | Abcam | Cat# ab13888; RRID: AB_300714 |
| IRDye 800CW Goat anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32211; RRID: AB_621843 |
| IRDye 800CW Goat anti-Mouse IgG antibody | LI-COR Biosciences | Cat# 926–32210; RRID: AB_621842 |
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Rabbit anti-MeCP2 | Chen et al.61 | N/A |
| Critical commercial assays | ||
| Mspa1I | NEB | Cat# R0577 |
| AllPrep DNA/RNA Kit | QIAGEN | Cat# 80284 |
| Ovation Ultralow Methyl-Seq Kit | Tecan | Cat# 0335–32 |
| Epitect Bisulfite Kit | Qiagen | Cat# 59824 |
| EZ DNA Methylation-Direct Kit | Zymo Research Corporation | Cat# D5020 |
| Accel-NGS Methyl-Seq DNA Library Kit | Swift Biosciences | Cat# 30024 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat# E7420 |
| NEBNext rRNA Depletion Kit (Human/Mouse/Rat) | NEB | Cat# E6310 |
| Accel-NGS 2S Plus DNA Library Kit (24 rxns) | Swift Biosciences | Ca#21024 |
| Deposited data | ||
| RNA-sequencing data | This paper | GEO: GSE225372 |
| ChIP-sequencing data (H3K27ac and MeCP2) | This paper | GEO: GSE225372 |
| Bisulfite-sequencing data | This paper | GEO: GSE225372 |
| Bisulfite-sequencing data | Lister et al.14 | GEO: GSE47966 |
| RNA- and ChIP-sequencing data | Clemens et al.20 | GEO: GSE123373 |
| RNA-sequencing data | Christian et al.9 | GEO: GSE147899 |
| RNA-sequencing data | Hamagami et al.42 | GEO: GSE212847 |
| Mus musculus mm9 genome assembly | UCSC | http://hgdownload.soe.ucsc.edu/goldenPath/mm9/ |
| Ensembl gene models | UCSC | https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Dnmt3a P900L/+ | This paper | N/A |
| Dnmt3a R878H/+ | Smith et al.24 | Provided by T. Ley |
| Oligonucleotides | ||
| Actb Forward | IDT | AAGGCCAACCGTGAAAAGAT |
| Actb Reverse | IDT | GTGGTACGACCAGAGGCATAC |
| Dnmt3a Forward | IDT | GGCCTTCTCGACTCCAGATG |
| Dnmt3a Reverse | IDT | TTCCTCTTCTCAGCTGGCAC |
| Dnmt3a P900L Region Forward | IDT | AGAGGGGCATTTATGGATGA |
| Dnmt3a P900L Region Reverse | IDT | GAGGGGCCTATTTTGCTTTT |
| Software and algorithms | ||
| DESeq2 (v1.14.1) | Love et al.62 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR (v3.16.5) | Robinson et al.63 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| BEDtools2 (v2.25.0) | Quinlan and Hall64 | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.5) | Langmead and Salzberg65 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | Dobin et al.66 | https://github.com/alexdobin/STAR |
| fastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim galore | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| BS-seeker2 | Guo et al.67 | https://github.com/BSSeeker/BSseeker2 |
| BSmooth | Hansen et al.68 | https://www.bioconductor.org/packages/release/bioc/html/bsseq.html |
| GraphPad Prism v9.4.1 | GraphPad by Dotmatics | https://www.graphpad.com/ |
| Avizo | ThermoFisher | http://www.vsg3d.com/ |
| ITK-SNAP | Yushkevich et al.69 | http://itksnap.org/ |
| PANTHER Gene Ontology (v17.0) | Mi and Thomas70; Thomas et al.71 | http://www.pantherdb.org/tools/compareToRefList.jsp |
| WebGestalt Gene Ontology | Liao et al.72 | https://www.webgestalt.org/ |
| RRHO2 | Cahill et al.43 | https://github.com/RRHO2/RRHO2 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animal husbandry
All animal protocols were approved by the Institutional Animal Care and Use Committee and the Animal Studies Committee of Washington University in St. Louis, and in accordance with guidelines from the National Institutes of Health (NIH). Mice were housed in a room on a 12:12 h light/dark cycle, with controlled room temperature (20°C–22°C) and relative humidity (50%). Home cages (36.2 × 17.1 × 13 cm) were individually ventilated and supplied with corncob bedding and standard laboratory chow (PicoLab Irradiated Rodent Diet 5053) and water unless otherwise specified. For experiments of progressive weight gain, male and female animals (P900L n = 18, 8 male, 10 female; WT n = 24, 12 male, 12 female) were given free access to the Tekkad High Fat Diet (Envigo; TD.88137; 42% Calories from Fat) instead of standard laboratory chow from 10 to 30 weeks of age. During this time, mice were weighed weekly. At 30 weeks of age, mice were single housed, and food was weighed every two days for a total of six days (3 timepoints) to measure food consumption. Unless otherwise specified, all mice were group-housed and adequate measures were taken to minimize animal pain or discomfort.
Transgenic animals
The DNMT3A P900L mouse model was generated using single guide RNAs (sgRNAs) to create a C→T substitution at chr12:3,907,719 (GRCm38/mm10 assembly). This mutation changed the proline CCG codon into a leucine CTG codon (Figure S1A). sgRNAs were cloned into the pX330 Cas9 plasmid (Addgene), and then transfected into N2A cells. Validation was done using the T7 enzyme assay by the Washington University School of Medicine Transgenic Vectors Core. sgRNAs were transcribed in vitro using MEGAShortScript (Ambion), and Cas9 mRNA was in vitro transcribed, G-capped and poly-A tailed using the mMessageMachine kit (Ambion). mRNA of the sgRNA and Cas9 were then injected into hybrid C57Bl/6J × CBA fertilized eggs at the mouse genetics core at Washington University School of Medicine. Founders were deep sequenced at expected cut sites to identify which alleles were present, and deep sequencing analyses of four kilobases surrounding the targeted region was used to ensure no off-target recombination events occurred. Founders were then crossed to C57BL6/J females (JAX Stock No. 000664) for 5–10 generations before experimental analysis.
To generate experimental animals, Dnmt3aR878H/+ (R878H) or Dnmt3aP900L/+ (P900L) male mice were crossed with C57BL6/J females (JAX Stock No. 000664). R878H and P900L females were not used for breeding to avoid social differences in mothering from mutant dams. Mice were genotyped with ear-, tail-, or toe- DNA by PCR for either R878H or P900L mutations. Mice were weighed at a variety of timepoints to assess growth. Experimental studies whenever applicable utilized both male and female animals, and always included littermate-paired control animals. Ages of mice for each analysis are detailed in Table S1.
METHOD DETAILS
P900L genotyping
To genotype for the P900L mutation, ear-, tail-, or toe- DNA was amplified using primers designed around the P900L mutation (F:AG AGGGGCATTTATGGATGA, R: GAGGGGCCTATTTTGCTTTT). The 706bp PCR product could then be Sanger Sequenced (Figure S1A) or digested using Mspa1I for an extended 3-h digestion time followed by the standard heat-shock inactivation. The wild-type sequence is susceptible to restriction enzyme digestion, leaving a 285bp and 421bp fragments, whereas the P900L mutation is not digested and will remain at 706bp (Figure S1B).
Bone length measurements
We chose to quantify long bones that may directly relate to the height phenotype seen in patients. Femurs were dissected from 30 to 35-week-old mice (P900L n = 18, 10 male, 8 female; WT n = 16, 9 male, 7 female) and scanned using a Faxitron Model UltraFocus100 Digital Radiography system at the Washington University Musculoskeletal Research Center. Image analysis was done using Faxitron Vision Software (Version 2.3.1). When analyzed with a two-way ANOVA, there was no significant sex effect.
Craniofacial morphological analyses
A total of 16 sex-matched littermate paired mice (P900L n = 8, 4 male, 4 female; WT n = 8, 4 male, 4 female) at 30–35 weeks of age were fixed with intracardiac perfusions of 4% paraformaldehyde. Whole mouse heads were scanned using a Scanco μCT40 machine at the Musculoskeletal Research Center at Washington University in St. Louis. Image processing was performed as previously described.9,73 Briefly, CT images were converted to 8-bit and surface reconstructions were acquired in Avizo (http://www.vsg3d.com/). 35 landmarks were collected from surface reconstructions of the cranium and mandible using Avizo. Principal components were identified from generalized Procrustes analysis in Geomorph package in R and Morphologika software as previously described.73 To identify specifically altered linear distances, landmark coordinates were natural log-transformed and analyzed with linear regression using Euclidean Distance Matrix Analysis (EDMA).
EchoMRI to measure body composition
Fat and lean mass measures of live WT and P900L mice were measured with whole-body quantitative magnetic resonance using an EchoMRI Body Composition Analyzer at the Washington University Diabetes Research Center. Experiments were performed as previously described.74 Briefly, animals of 30–35 weeks of age (P900L n = 15, 8 male, 7 female; WT n = 17, 10 male, 7 female) were placed in a plastic cylinder tube with a solid insert to limit movement. Signal in response to a low-intensity electromagnetic field was used to measure the relaxation of spin curves, allowing for the quantification of fat and lean tissue volume. Canola oil was used to standardize measurements between different recording days.
Magnetic resonance imaging (MRI) acquisition and Diffusion Tensor Imaging (DTI) analysis
Three cohorts of different ages were used for MRI experiments. At P10, twenty-seven animals were used for P900L experiments (P900L n = 16, 6 male, 10 female; WT n = 11, 6 male, 5 female) and twenty-three were used for R878H experiments (R878H n = 10, 5 male, 5 female; WT n = 13, 6 male, 7 female). At 8 weeks, nineteen animals were used for P900L experiments (P900L n = 11, 6 male, 5 female; WT n = 8, 4 male, 4 female), and eighteen animals were used for R878H experiments (R878H n = 9, 2 male, 7 female; WT n = 9, 2 male, 7 female). At 30–35 weeks, total of twenty-four animals were used P900L experiments (WT n = 12, 6 males, 6 females; P900L n = 12, 6 males, 6 females), and twenty-four animals for R878H experiments (WT n = 12, 6 males, 6 females; R878H n = 12, 6 males, 6 females). Imaging and analysis were performed as described previously.31 In brief, isoflurane-anesthetized animals were scanned with a small-animal MR scanner built around an Oxford Instruments 4.7T horizontal-bore superconducting magnet equipped with an Agilent/Varian DirectDrive console. Data were collected using a laboratory-built actively decoupled 7.5-cm ID volume coil (transmit)/1.5-cm OD surface coil (receive) RF coil pair. Mouse respiratory rate and body temperature (rectal probe) were measured with a Small Animal Instruments (SAI, Stony Brook, NY) monitoring and gating unit.
T2-weighted trans-axial images (T2W), collected with a 2D fast spin-echo multi-slice (FSEMS) sequence, were used for structural and volumetric analyses. Diffusion Tensor Imaging (DTI), which measures the directional movement of water along and perpendicular to axons (fractional anisotropy: FA), provided a measure of white-matter track integrity. DTI data were collected using a multi-echo, spin-echo diffusion-weighted sequence with 25-direction diffusion encoding, max b-value = 2200 s/mm2, as described previously.31 Two echoes were collected per scan, with an echo spacing of 23.4 ms, and combined offline to increase signal-to-noise ratio (SNR), resulting in an SNR improvement of ~1.4x compared with a single echo.
DTI data were analyzed as described previously31 according to the standard MR diffusion equation75 using purpose-written MATLAB software. Eigenvalues (λ1, λ2, λ3) corresponding to the diffusion coefficients in three orthogonal directions, and parametric maps of apparent diffusion coefficient (ADC), axial diffusion (Daxial), radial diffusion (Dradial), and fractional anisotropy (FA) were calculated according to standard methods.76,77 Parametric maps were converted into NIfTI (.nii) files for inspection and segmentation using ITK-SNAP (www.itksnap.org).69 Segmentation was performed blinded to strain, sex, or genotype, and consistency was assessed by re-segmenting blinded data files.
Stereological quantification of hematoxylin-stained nuclei
A total of twelve animals were used for P900L experiments (WT n = 6, 3 males, 3 females; P900L n = 6, 3 males, 3 females) and ten animals were used for R878H experiments (WT n = 5, 2 males, 3 females; R878H n = 5, 2 males, 3 females). Perfused brains from P56 mice were vibratome sectioned in the coronal plane at 75μm, using a DSK Microslicer DTK-1000N vibratome. Every 16th section between the olfactory bulb and cerebellum was slide mounted overnight and submerged in two changes of xylene for 10 min each. The tissue was then re-hydrated in two 5-min changes of absolute alcohol followed by changes of 95% alcohol and 70% alcohol for 2 min each. The slides were washed with distilled water, placed in the nuclear stain hematoxylin (Gill’s No. 1, GHS132-1L, Millipore Sigma) for 8 min, and washed with two changes of distilled water. Lastly, the slides were dipped in 0.02% ammonia water for 10 s, washed with running distilled water for 10 min, placed in a 60°C oven to dry for 8 min, and cover-slipped with DPX mountant. The optical fractionator method was then used to stereologically count cell nuclei using Stereoinvestigator Software (v 2019.1.3, MBF Bioscience, Williston, Vermont, USA) connected to a QImaging 2000R camera and a Labophot-2 Nikon microscope with motorized stage. A rater, blind to treatment, quantified cell nuclei on the hemisection with volume and counts multiplied by two to get total estimates per cerebrum.
Behavioral analyses
Mice for behavioral testing were housed in mixed genotype home cages with 2–5 animals per cage, and all tests were performed during the light cycle. All experimenters were female and were blinded to genotype during testing. For increased experimental rigor and reproducibility, we used separate cohorts of mice to ensure quality and consistency in any observed phenotypes. Adult testing was performed when mice were 2–4 months of age.
Maternal isolation-induced ultrasonic vocalizations
Pup ultrasonic vocalization (USV) measurements were performed to assess early social communicative behavior as previously described.31 Ninety-seven animals were used for P900L experiments (n = 47 WT, 19 males and 28 females; n = 50 P900L, 30 males and 20 females), and ninety-two animals were used for R878H experiments (n = 51 WT, 24 males, 27 females; n = 41 R878H, 19 males and 22 females). Recordings were done at postnatal days 5, 7, and 9. In brief, adults were removed from the nest and home-cages were placed in a warming box (~33°C) 10 min before recording began. Body temperature was recorded immediately before placing pups in a dark, enclosed chamber for 3-min recordings. Following the USV recording, pups were weighed and returned to their nest. Frequency sonograms were prepared and analyzed in MATLAB as previously described.9,31 Within-subjects repeated measures ANOVA were used to assess significance, and no significant differences occurred between sexes for any vocalization measures, therefore data were combined between sexes.
Marble burying
WT (n = 13; 8 male, 5 female) and litter matched P900L (n = 13; 8 male, 5 female) mice were used for marble burying as previously described.9 In brief, 8-week-old mice were placed in a transparent enclosure (28.5 cm × 17.5 cm × 12 cm) with clean aspen bedding and 20 dark blue marbles evenly spaced in a 4 × 5 grid on top of the bedding. Animals were allowed to explore freely for 30 min, and the number of buried marbles were counted every 5 min by two independent blinded observers. Marbles were considered “buried” if they were at least two-thirds covered by bedding. Enclosure and marbles were cleaned thoroughly between animals. Data was analyzed with a within-subjects repeated measured ANOVA, and no sex effect was observed so data was combined between sexes.
Three-chamber social approach
Eighteen litter-matched animals that were 10–12 weeks old were used in the 3-chamber social approach assay (P900L n = 9, 5 male, 4 female; WT n = 9, 4 male, 5 female) as previously described.78 Briefly, mice were acclimated to a clear acrylic rectangular apparatus (60 cm × 40.5 cm), which was separated into three chambers by walls with sliding doors (6 cm × 6 cm). The apparatus was placed in an isolated, quiet room with low light (270 lux) to minimize stress. Both side chambers contained an inverted cup. Testing consisted of three 10-min phases: during the first phase, the mouse freely explored all chambers, in the second phase a conspecific mouse was added to one of the cups (mouse vs. object), and in the third phase a novel conspecific was added to the remaining empty cup (novel vs. familiar). During all phases, the test mouse was allowed to freely explore, and all stimulus mice were sex-matched conspecifics. A digital video camera was used to record sessions, location of mice in the apparatus was analyzed. Between experimental animals, 70% ethanol was used to clean the apparatus. As mice rapidly habituate to this task,78 only the first 5 min of each phase was used for analysis.
Social dominance tube test
Tube test was conducted to assess social hierarchy behavior as previously described.31 For P900L experiments, ninety-four animals were used (n = 47 WT, 24 males and 23 females; n = 47 P900L, 24 males and 23 females) across three experimental cohorts, and one cohort of thirty-four mice was used for R878H experiments (n = 17 WT, 9 males, 8 females; n = 17 WT, 9 males, 8 females). In brief, mice were allowed to learn to traverse the clear acrylic tube apparatus on days 1 and 2 of the task. On days 3–5, sex-matched pairs of WT and mutant mice were tested on dominance bouts, avoiding cage mate pairings. A new WT-mutant pairing was used each day, allowing for three distinct matchups for each animal. During bouts, animals were allowed to enter the tubes while separated from each other with an acrylic divider. A bout begins when the divider was removed and concluded when one mouse fully backed out of the tube or when 2 min passed. The animal remaining in the tube was considered the winner of the bout (dominant) and the animal that exited the tube was the loser (submissive). Active wins were defined as the winner pushing the other animal from the tube, whereas passive wins were defined as the winner refusing to move and the loser backing out of the tube. The tube was cleaned with a 0.02% chlorhexidine solution between bouts. Bout recordings were scored by a blinded observer. A two-tailed binomial test was performed on numbers of bouts won, with a null hypothesis that 50% of bouts would be won by each genotype.
Novel object recognition – tactile
Novel Object Recognition-Tactile (NORT) was used to measure general and tactile associative memory adapted from previous work.35,36 Briefly, the task consisted of five consecutive days including two initial habituation trials, NORT testing, a third habitation trial, and NOR testing. During habituation trials, mice were allowed to freely explore the empty acrylic apparatus (26 × 26 cm or 40 × 40 cm) for 10 min under white light (75–100 lux). During NORT testing, the mice received a learning trial to freely explore two matching acrylic 4cm cubes that were either both smooth or both textured. Following a 5-min inter-trial interval (ITI) in which the animals were removed to holding cages, the mice received a 3-min test trial during which one of the cubes was replaced with a novel cube identical in appearance to the original object but with different tactile properties (smooth vs. textured). NOR was conducted the same as NORT except the objects differed visually, tactilely, and in size and materials, and the ITI was 50 min. The objects consisted of a ½ inch diameter white PVC standing pipe measuring 14 cm tall surrounded by a metal spiral and a 3D-printed blue block measuring 14.4 cm × 5 cm × 2.5 cm. For both NORT and NOR, object type and side on which the novel object was presented was counterbalanced across groups. The movement of the mice was tracked with ANY-maze Software (Stoelting, Co.). The outcomes analyzed included total distance traveled and time spent investigating the objects, defined as the nose within 10 mm zone surrounding the object and pointing toward the object, excluding any time the mouse was climbing on the object. All objects and the apparatus were cleaned with 0.02% chlorhexidine between trials.
One-hour locomotor activity
P900L (n = 21, 11 male and 10 female) and litter-matched WT (n = 21, 10 male and 11 female) mice were used for the remainder of behavioral tests, which were performed by the Intellectual and Developmental Disabilities Research Center Animal Behavior Subunit at Washington University in St. Louis. Locomotor activity was measured using the SmartFrame Home Cage System (Kinder Scientific) in a transparent polystyrene enclosure (47.6 cm × 25.4 cm × 20.6 cm) by measuring photobeam breaks, as previously described.79 Total ambulatory movement, vertical rearing behavior, and time spent in a 33 cm × 11 cm central zone were measured. Total ambulations reflects larger animal movements, such as when the subject changes its entire body position on the grid and is calculated when a new beam break occurs and the anchor beam in that dimension is released. Due to the nature of the algorithm of how these beam breaks are calculated, this measure does not have units.
Sensorimotor battery
Walking initiation, balance (ledge and platform tests), volitional movement (pole and inclined screens), and strength (inverted screen) were measured as described previously.31 For the walking initiation test, mice were placed on the surface in the center of a 21 cm × 21 cm square marked with tape and the time for the mouse to leave the square was recorded. During the balance tests, the time the mouse remained on an elevated plexiglass ledge (0.75 cm wide) or small circular wooden platform (3.0 cm in diameter) was recorded. During the Pole test, mice were placed at the top of a vertical pole pointing upwards, and the time for the mouse to turn and descend the pole was recorded. During the inclined screen tests, the mouse was placed head-down on an elevated mesh grid, and the time to climb up the grid was recorded. During the inverted screen test, a mouse was placed on an elevated mesh grid, which was then inverted 180°, and the time to fall was measured. Tests lasted for 1 min, except for the pole test which lasted 2 min. Data used for analysis are an average of two trials done on subsequent days.
Continuous and accelerating rotarod
Balance and coordination were assessed using the rotarod test (Rotamex-5, Columbus Instruments, Columbus, OH) as previously described,79 using both constant rotation (5 rpm, 60 s maximum) and acceleration rotation (5–20 rpm, 180 s maximum) trials. Three sessions of testing consisting of two trials each were conducted, and trials were averaged. To focus the task on coordination rather than learning, testing sessions were separated by 4 days.
Morris water maze
To assess spatial learning, we performed the Morris Water Maze, consisting of cued trials, place trials, and probe trials as previously described.79 Animals were placed in a large water-filled pool, and time and distance to reach an escape platform were measured (ANY-maze, Stoelting). Maximum trial duration was 1 min. During cued trials, there was a visible escape platform that was moved to new locations for each trial, and the mice experienced 4 trials per day (separated by 30-min inter-trial-intervals) across 2 days. Performance was analyzed in 2-trial blocks, with trials averaged. Three days later, animals were tested in place trials in which the escape platform was submerged in a consistent location, and there were numerous distal visual cues available. Place trials occurred daily for 5 days, consisting of 2 blocks of 2 consecutive trials. Trials within blocks were separated by a 30-s interval, and blocks were separated by 2 h. Mice were released in different areas of the maze and required to use visual cues to find the hidden platform. Trial data were averaged across the trials within each day. One hour after the final place trial occurred, the probe trial took place, in which the platform was removed entirely. The mouse was released from the quadrant opposite to the learned platform location and allowed to swim in the task for 1 min. Time spent in each quadrant, and the number of crossings over the zone the platform was previously in were recorded. All genotypes spent significantly more time in the target quadrant than could be expected by chance (25%), and these statistics are included in Table S1.
Elevated plus maze
Elevated plus maze tests were done as previously described.80 In brief, the elevated apparatus contains a central platform (5.5 cm × 5.5 cm) with four arms extending from the central platform (each 36 cm × 6.1 cm). Two opposing arms were open and two have 15 cm tall opaque Plexiglas walls. Test sessions were conducted in a dimly lit environment with in which the mouse was able to freely explore the apparatus for 5 min. Position was measured with beam-breaks and time, distance, and entries into each zone were recorded and analyzed (MotoMonitor, Kinder Scientific).
Conditioned fear
Fear conditioning was performed as previously described.80 Briefly, mice were habituated to an acrylic chamber (26 cm × 18 cm × 18 cm) that contained a metal grid floor, a LED light which remained on during trials, and a chamber odorant. During the training day, baseline measurements of freezing behavior were collected for 2 min. Then, once per minute, three training rounds occurred in which a 20-s 80 dB tone sounded for 20 s. During the last 2 s of the tone (conditioned stimulus) a 1.0 mA foot-shock (unconditioned stimulus) occurred. The next day, contextual fear was tested, in which the animals were placed in the same chamber with the same odorant with the testing light illuminated but no tones or shocks delivered. The following day, cued fear was tested, in which the animals were placed in a new opaque box with a new odorant. After a 2-min baseline period with no tone, the same 80 dB tone was played for the remainder of the 8-min trial. During all trials, freezing behavior were recorded and analyzed.
DNMT3A protein and RNA expression
Cortex tissue from P900L and WT animals (2 weeks old) were dissected in ice-cold PBS, flash frozen with liquid nitrogen, and stored at −80°C. Half of the cortex was used for protein expression measurement with western blotting, and the other half was used for RNA expression via RT-qPCR. Expression was assessed at 2 weeks of age because this is a timepoint with high postnatal expression.
Western blotting
Western blotting was performed as previously described.9 WT and P900L (n = 8/genotype, 4 males, 4 females) half-cortexes were homogenized with protease inhibitors (Buffer: 10mM HEPES pH 7.9, 10mM KCl, 1.5mM MgCl2, 1mM DTT, 10mM EDTA), and 1% SDS was added prior to boiling the samples for 10 min at 95°C. Subsequently, samples were spun at 15,000g for 10 min, and supernatant was run through a Wizard Column (Fisher, Wizard Minipreps Mini Columns, PRA7211), and protein concentration was measured using a Bradford assay. Samples were diluted in LDS sample buffer with 5% β-mercaptoethanol and boiled for 5 min before being run on a gel. An 8% acrylamide gel was used, and samples were run for 60 min at 125V before being transferred to a nitrocellulose membrane. Blots were blocked for 1 h at room temperature in TBS-T with 3% bovine serum albumin, then immunostained with anti-DNMT3A (Abcam, 1:1000, ab13888) or anti-α-Tubulin (Abcam, 1:1000, ab52866) for 12–16 h at 4°C. After washing membranes, they were incubated with secondary antibodies for 1 h at room temperature in light-protected boxes (IRDye 800CW Goat anti-Rabbit, or IRDye 800CW Goat anti-Mouse, LI-COR Biosciences, 1:15,000, product numbers: 926–32211 and 926–32210 respectively). Primary and secondary antibodies were diluted in 3% Bovine Serum Albumin in TBS-T. Blots were imaged using the LiCOR Odyssey XCL system and quantified using Image Studio Lite software (LI-COR Biosciences). DNMT3A and α-Tubulin levels were normalized to a standard curve, and protein levels are expressed as normalized DNMT3A values divided by normalized α-Tubulin values to enable comparison of DNMT3A levels between blots. Significance was assessed using an unpaired Student’s T Test.
qRT-PCR
RNA from WT and P900L (n = 5/genotype, 3 males, 2 females) half-cortexes were isolated using the AllPrep DNA/RNA kits (QIAGEN, 80284), and RNAs were reverse transcribed using the using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). DNMT3A and ACTB were measured by qPCR using the Power SYBR Green PCR Master Mix and primers for ACTB (F:AAGGCC AACCGTGAAAAGAT, R:GTGGTACGACCAGAGGCATAC) or DNMT3A (F:GGCCTTCTCGACTCCAGATG, R:TTCCTCTTCTCAGCTG GCAC). The Ct of each primer set in each sample was calculated, and relative quantity was determined by comparing to a standard curve and then normalizing the DNMT3A signal to the ACTB signal.
Whole genome bisulfite sequencing
Global methylation across brain regions
300ng of DNA was isolated from brain tissue from 8-week animals (n = 2/sex/genotype/mutation/region) using the AllPrep DNA/RNA kit (QIAGEN, 80284). DNA was then fragmented for 45 s with the Covaris S220 sonicator (10% Duty Factory, 175 Peak Incidence Power, 200 cycles per burst, milliTUBE 200μL AFA Fiber). To select for long DNA inserts, DNA was purified using 0.7 volumes of Agencourt Beads. A small amount of Lambda DNA was spiked in to allow for estimation of non-conversion rates. To prepare bisulfite DNA libraries, we used the Tecan Ovation Ultralow Methyl-Seq Kit (Tecan, 0335-32) and the Epitect Bisulfite Kit (Qiagen, 59824). Alternate bisulfite conversion cycling conditions were used to ensure lowest possible non-conversion rate ([95°C, 5 min; 60°C, 20 min] × 4 cycles, 20°C hold). Libraries were PCR amplified 11–13 cycles and pooled for low-depth sequencing at the Washington University in St. Louis Center for Genomic Science. Libraries were sequenced using a MiSeq 2×150 and sequenced at an average depth of 0.018x genomic coverage (average 0.2M reads per sample). Sequencing data were processed as described below, and genome-wide averages of mCA and mCG were analyzed using a paired Student’s T Test with Bonferroni correction.
Deep sequencing of cortical DNA methylation
50ng of DNA isolated from a total of sixteen 8-week cortex samples (n = 2/sex/genotype/mutation) and fragmented for 45 s using the Covaris E220 sonicator (10% Duty Factory, 175 Peak Incidence Power, 200 cycles per burst, milliTUBE 200μL AFA Fiber) and purified using 0.7 volumes of SPRISelect Beads (Beckman Coulter Life Sciences). A small amount of Lambda DNA was spiked in to allow for estimation of non-conversion rates. DNA was then bisulfite converted using the EZ DNA Methylation-Direct Kit (Zymo Research Corporation, D5020) using extended bisulfite conversion incubation to ensure lowest possible non-conversion rates (98°C, 8 min; 64°C, 4 h 15 min). Samples were either stored overnight at −20°C, or libraries were immediately prepared using the Accel-NGS Methyl-Seq DNA Library Kit (Swift, 30024) with combinatorial dual indexes (Swift, 38096) as instructed, using 10 cycles of final amplification. Libraries were pooled and sequenced at the Genome Technology Access Center at the Washington University McDonnell Genome Institute using the NovaSeq 6000 2×150. An average sequencing depth of 10x genomic coverage (average 144M reads per sample) were obtained per sample.
Chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation library generation
Chromatin immunoprecipitation was performed as previously described.20 Cerebral cortex was dissected on ice in PBS from DNMT3A mutants and their WT littermates at 8-weeks old and flash-frozen in liquid nitrogen prior to storage at −80°C. Chromatin were fragmented with the Covaris E220 sonicator (5% Duty Factory, 140 Peak Incidence Power, 200 cycles per burst, milliTUBE 1mL AFA Fiber). ChIP was performed with H3K27ac antibody (0.1μg; Abcam, ab4729; n = 2/sex/genotype/mutation; a total of 4 WT and 4 mutants in P900L litters, and a total of 4 WT and 4 mutants in R878H litters) or a MeCP2 antibody (5μL serum per IP; described in Chen et al.,61; n = 2 per genotype), and libraries were generated using Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences). Pooled libraries were sequenced using a NovaSeq 6000 with the Genome Technology Access Center at Washington University in St. Louis, typically yielding 20–50 million (average: 34 million) single-end reads per sample.
RNA sequencing
RNA sequencing library generation
Total RNA isolation was carried out as previously described.20 In brief, cerebral cortex was dissected in ice-cold PBS from P900L or R878H mutants and their respective WT littermates at 8 weeks of age (n = 7 pairs, 3 male, 4 female). Cortex was lysed in RLT buffer and RNA was isolated using the AllPrep DNA/RNA kit (QIAGEN, 80284). RNA libraries were generated from 250ng of RNA with NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) using a modified amplification protocol (37°C, 15 min; 98°C, 30 s; [98°C, 10 s; 65°C, 30 s; 72°C, 30 s]×13; 72°C, 5 min; 4°C hold). RNA libraries were pooled at a final concentration of 10nM and sequenced using Illumina NextSeq-High 1×75bp with the Center for Genome Sciences at Washington University in St. Louis, typically yielding 15–30 million single-end reads per sample.
Experimental design
Sample sizes were chosen based upon previously published studies using similar techniques. Statistical tests and exclusion criteria (values beyond 2 standard deviations of the group mean) were similar to that of previously published studies and indicated in the appropriate methods. For all animal experiments, experimenters were blinded to genotype during data collection. No treatment conditions were used, so no samples or animals were allocated to experimental groups and no randomization was needed. Tests that assume equal variance were only run if group variances were similar, otherwise alternative tests were used.
QUANTIFICATION AND STATISTICAL ANALYSIS
Detailed information regarding statistical tests, sample sizes, sample information, and statistical outputs for all figures in this study are included in Table S1.
Statistical analysis for behavioral tests
Behavioral data were analyzed and plotted using GraphPad Prism 9.4.1. No consistent genotype by sex interaction effects were observed for any behavioral tests, therefore data were collapsed across sex. Statistical testing was performed using planned assay-specific methods, such as using unpaired Student’s T-Tests for single parameter comparisons between genotypes, and within-subjects two-way or three-way repeated-measures ANOVA for comparisons across timepoints. Individual timepoints within repeated measures tests were evaluated using Sidak’s multiple comparisons test. Unless otherwise noted, bar graphs and line graphs indicate mean ± SEM.
Whole genome bisulfite analysis
Analysis of bisulfite sequencing was performed as described previously.9,20 Reads were adapter-trimmed, mapped to mm9, deduplicated, and called for methylation using BS-seeker2.67 Bedtools map -o sum was used to assess methylation across regions, summing the number of reads mapped to Cs (interpreted as mC after bisulfite conversion) and then dividing by the sum of Cs and Ts (indicating C) at that region. %mC values from biological replicates were averaged together. Though our methods should maximize the amount of efficient bisulfite conversion, a small percentage of unmethylated cytosines can are called as methylated due to nonconversion (0.2–0.3%). To adjust for nonconversion rate, regions were adjusted by the % methylation measured in Lambda spike-ins per sample, similar to previous analysis.14 If corrected region values were below 0, the %mC value was set to 0. Due to background nonconversion, lowly methylated regions (e.g., mCA at CpG islands or promoters) are not expected to show the same percentage reduction in methylation as higher mCA regions.
Differentially methylated region detection
We used BSmooth68 on four biological replicates of P900L or R878H and their sex-matched WT littermates to call differential CpG methylated regions. CG sites were then filtered, requiring >2x genomic coverage in all replicates. Differentially methylated regions (DMRs) were called using a statistical threshold of t-stat >2.0, requiring length >100 bp, and biological replicate consistency (i.e., for hypomethylated regions, all WT mCG/CG values must be higher than mutant mCG/mCG values). Data fit the assumptions and requirements for BSmooth and fisher’s exact testing. Resampling for overlap analysis was done using bedtools shuffle. Significance of DMR overlap with genomic regions of interest was assessed with a Chi-Squared test with expected proportions of overlapping and nonoverlapping measured by resampling DMRs. Hypo- and Hyper-DMR overlap analysis from both mutants was performed using a Fisher’s Exact Test compared to resampled DMRs. Only hypo-methylated DMRs from both mutations overlapped, with no other DMR groups having significant overlaps.
Chromatin immunoprecipitation analysis
ChIP sequencing analysis was performed as previously described.20 In brief, reads were mapped to mm9 with bowtie2,65 and deduplicated with picardtools MarkDuplicates. Bedtools coverage -counts64 was used to assess H3K27ac and MeCP2 signal at the various genomic regions examined.20 edgeR was then used to determine differential H3K27ac signal between WT and mutant animals.63 Significant differentially acetylated regions were called using FDR<0.1. No enhancers in the P900L comparison reached statistical significance, and in the R878H comparison, 29 enhancers were upregulated and 29 were downregulated. These results are similar to previous work in the DNMT3A KO/+9 suggesting that methylation has subtle effects over a large population of enhancers. To generate a robust population of enhancers for analysis, the top and bottom most significant (lowest p value) 1% of enhancers were used in the up- and down-regulated directions within each mutant strain. This analysis included 167 upregulated and 160 downregulated P900L enhancers, and 163 upregulated and 164 downregulated R878H enhancers. Data were normalized to read count and visualized using the UCSC genome browser.81 To link enhancer activity with gene expression, we measured H3K27ac signal at candidate cis-regulatory elements (cCREs) linked to neuronal gene expression changes.82
RNA sequencing processing
RNA sequencing analysis was performed as previously described.20 Briefly, raw FASTQ files were trimmed with Trim Galore and rRNA sequences were filtered and removed with Bowtie. Remaining reads were aligned to mm9 using STAR,66 and uniquely mapping reads were converted to BED files and separated into intronic and exonic reads. These exonic BED files were used to assess gene counts using bedtools coverage -counts.64
Differential gene expression
DESeq2 was used to identify differentially expressed genes between mutants and their WT littermates.62 To control for batch, sex, and litter, paired analysis was done using a design = ~ pair + genotype, and contrasted by genotype for all analysis. Though all libraries were processed in groups that contained P900L and R878H pairs, P900L and R878H datasets were analyzed separately. Significantly dysregulated genes were called when FDR<0.05, and Log2 Fold changes were corrected using ashr.83 Mutant-specific genes were defined as significantly regulated in one direction in one mutant, and either being unchanged (nominal p value >0.5) or regulated in the opposite direction in the other mutant. Differentially expressed gene sets are included in Table S2.
Defining shared TBRS genes
RNA-seq data in the DNMT3A KO/+ (n = 7 pairs; 4 male, 3 female) from Christian et al., 2020 were analyzed in addition to with P900L (n = 7 pairs; 4 male, 3 female) and R878H (n = 7 pairs; 4 male, 3 female) datasets. All datasets were generated from 8-week cortex and processed using similar methods. Datasets were combined, and littermate pairwise genotype comparisons were made using DESeq2 across all WT and mutant animals (design = ~ pair + group and contrasted by group; group defined as WT or mutant with no indication of origin dataset). As this analysis combines multiple studies, we utilized an FDR<0.1 to call significant differentially expressed genes.
PANTHER gene ontology analysis
Gene set enrichment analysis was done using the PANTHER Overrepresentation Test (Version 17.0, Released 2022-02-22).70,71 Analyzed lists (e.g., significantly upregulated genes in the P900L mutant) were compared to a reference list of all expressed genes in our study (defined as genes with more than an average of 10 counts in both WT littermate datasets). Analysis identified PANTHER GO-slim Biological Process terms and used a Fisher test with FDR correction. A subset of the most significant PANTHER terms is shown in figures with full PANTHER results (for gene set FDR<0.1) in Table S3.
WebGestalt phenotype ontology analysis
Further functional gene set enrichment analysis was done using the WebGestalt Over-representation analysis (Released 2019-01-14).72 Analyzed lists (e.g., significantly upregulated genes in the P900L mutant) were compared to the Mammalian Phenotype Ontology list (accessed on 2018-11-14) measured against a genomic reference gene list. Up to 10 significant (FDR<0.05) phenotype terms per gene list are shown in figures.
Rank-rank hypergeometric overlap (RRHO) analysis
For each mutant-WT pair, a ranked gene list was created using a gene score calculated as −log10(p value) * sign (log2Fold Change) using the DESeq2 results for that gene. RRHO2_initialize() was used to generate RRHO object, and RRHO2_heatmap() was used to generate a heatmap of overlapping genes between different mutants.
Overlap analysis
To calculate significance overlaps for differentially expressed gene lists displayed in Venn diagrams, a Fisher’s Exact test was performed between the observed overlaps and the expected overlaps, and p value is shown. Expected overlap was generated by averaging the overlaps observed in randomly shuffled gene sets of identical size (50,000 re-samplings). Expected overlap between three lists was generated through similar means using 10,000 re-samplings.
Supplementary Material
Highlights.
DNMT3A mutant phenotypes correlate with epigenomic and transcriptomic disruption
Analyzing multiple DNMT3A mutations reveals core disease-relevant genomic effects
DNMT3A mutants show concordant gene dysregulation with Rett and Sotos syndrome mice
ACKNOWLEDGMENTS
We thank T. Ley, A. Smith, and the TBRS community for reagents and discussion. We also thank the IDDRC@WUSTL Animal Behavior Subunit; C. Semenkovich and S. Adak at the Washington University Diabetes Research Center; M. Brodt and the Washington University Musculoskeletal Research Center; and M. Haywood for assistance landmarking skulls. We thank our funding sources: NIH-NICHD F31HD100098 (to D.C.B.), NIH-NICHD P50HD103525 (to the IDDRC@WUSTL), and The Simons Foundation Autism Research Initiative, NIH-NIMH R01MH117405, and NIH-NINDS R01NS04102 (to H.W.G.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113411.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Wang T, Hoekzema K, Vecchio D, Wu H, Sulovari A, Coe BP, Gillentine MA, Wilfert AB, Perez-Jurado LA, Kvarnung M, et al. (2020). Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun 11, 4932. 10.1038/s41467-020-18723-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, de Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23. 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake AM, Dougherty JD, Lein ES, Hormozdiari F, Bernier RA, and Eichler EE (2019). Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat. Genet 51, 106–116. 10.1038/s41588-018-0288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, Baralle D, Barge-Schaapveld D, Blyth M, Bouma M, et al. (2018). The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 3, 46. 10.12688/wellcomeopenres.14430.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, del Vecchio Duarte S, Zachariou A, Hanks S, O’Brien E, Aksglaede L, et al. (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet 46, 385–388. 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, di Lullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plummer JT, Gordon AJ, and Levitt P (2016). The genetic intersection of neurodevelopmental disorders and shared medical comorbidities - relations that translate from bench to bedside. Front. Psychiatry 7, 142. 10.3389/fpsyt.2016.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang YH, Chen CW, Sundaramurthy V, S1abicki M, Hao D, Watson CJ, Tovy A, Reyes JM, Dakhova O, Crovetti BR, et al. (2022). Systematic Profiling of DNMT3A Variants Reveals Protein Instability Mediated by the DCAF8 E3 Ubiquitin Ligase Adaptor. Cancer Discov. 12, 220–235. 10.1158/2159-8290.CD-21-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christian DL, Wu DY, Martin JR, Moore JR, Liu YR, Clemens AW, Nettles SA, Kirkland NM, Papouin T, Hill CA, et al. (2020). DNMT3A Haploinsufficiency Results in Behavioral Deficits and Global Epigenomic Dysregulation Shared across Neurodevelopmental Disorders. Cell Rep. 33, 108416. 10.1016/j.celrep.2020.108416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lue NZ, Garcia EM, Ngan KC, Lee C, Doench JG, and Liau BB (2023). Base editor scanning charts the DNMT3A activity landscape. Nat. Chem. Biol 19, 176–186. 10.1038/s41589-022-01167-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okano M, Bell DW, Haber DA, and Li E (1999). DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 99, 247–257. 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 12.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, and Sasaki H (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Letters to Nature 429, 900–903. [DOI] [PubMed] [Google Scholar]
- 13.Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, and Greenberg ME (2017). Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164.e16. 10.1016/j.cell.2017.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng J, Chang H, Li E, and Fan G (2005). Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res 79, 734–746. 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- 16.Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci 17, 215–222. 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen S, Meletis K, Fu D, Jhaveri S, and Jaenisch R (2007). Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn 236, 1663–1676. 10.1002/dvdy.21176. [DOI] [PubMed] [Google Scholar]
- 19.Swahari V, Nakamura A, Hollville E, Stroud H, Simon JM, Ptacek TS, Beck M.v., Flowers C, Guo J, Plestant C, et al. (2021). MicroRNA-29 is an essential regulator of brain maturation through regulation of CH methylation. Cell Rep. 35, 108946. 10.1016/j.celrep.2021.108946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clemens AW, Wu DY, Moore JR, Christian DL, Zhao G, and Gabel HW (2020). MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol. Cell 77, 279–293.e8. 10.1016/j.molcel.2019.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stroud H, Yang MG, Tsitohay YN, Davis CP, Sherman MA, Hrvatin S, Ling E, and Greenberg ME (2020). An Activity-Mediated Transition in Transcription in Early Postnatal Neurons. J. Clean. Prod 107, 874–890.e8. 10.1016/j.neuron.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tovy A, Rosas C, Gaikwad AS, Medrano G, Zhang L, Reyes JM, Huang YH, Arakawa T, Kurtz K, Conneely SE, et al. (2022). Perturbed hematopoiesis in individuals with germline DNMT3A overgrowth Tatton-Brown-Rahman syndrome. Haematologica 107, 887–898. 10.3324/haematol.2021.278990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith AM, LaValle TA, Shinawi M, Ramakrishnan SM, Abel HJ, Hill CA, Kirkland NM, Rettig MP, Helton NM, Heath SE, et al. (2021). Functional and epigenetic phenotypes of humans and mice with DNMT3A Overgrowth Syndrome. Nat. Commun 12, 4549. 10.1038/s41467-021-24800-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK, and Ley TJ (2014). The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 25, 442–454. 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emperle M, Dukatz M, Kunert S, Holzer K, Rajavelu A, Jurkowska RZ, and Jeltsch A (2018). The DNMT3A R882H mutation does not cause dominant negative effects in purified mixed DNMT3A/R882H complexes. Sci. Rep 8, 13242. 10.1038/s41598-018-31635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duyar I, and Pelin C (2003). Body height estimation based on tibia length in different stature groups. Am. J. Phys. Anthropol 122, 23–27. 10.1002/ajpa.10257. [DOI] [PubMed] [Google Scholar]
- 28.Tovy A, Reyes JM, Zhang L, Huang Y-H, Rosas C, Daquinag AC, Guzman A, Ramabadran R, Chen C-W, Gu T, et al. (2022). Constitutive loss of DNMT3A causes morbid obesity through misregulation of adipogenesis. Elife 11, e72359. 10.7554/eLife. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavery LA, Ure K, Wan Y-W, Luo C, Trostle AJ, Wang W, Jin H, Lopez J, Lucero J, Durham MA, et al. (2020). Losing Dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting Rett syndrome. Elife 9, e52981. 10.7554/eLife.52981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Lambo ME, Ge X, Dearborn JT, Liu Y, McCullough KB, Swift RG, Tabachnick DR, Tian L, Noguchi K, et al. (2021). A MYT1L syndrome mouse model recapitulates patient phenotypes and reveals altered brain development due to disrupted neuronal maturation. Neuron 109, 3775–3792.e14. 10.1016/j.neuron.2021.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han KA, Yoon TH, Shin J, Um JW, and Ko J (2020). Differentially altered social dominance- and cooperative-like behaviors in Shank2- and Shank3-mutant mice. Mol. Autism 11, 87. 10.1186/s13229-020-00392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, and Crawley JN (2004). Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 3, 287–302. 10.1111/j.1601-183X.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 34.Barnes TD, Rieger MA, Dougherty JD, and Holy TE (2017). Group and individual variability in mouse pup isolation calls recorded on the same day show stability. Front. Behav. Neurosci 11, 243. 10.3389/fnbeh.2017.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, and Ginty DD (2016). Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 166, 299–313. 10.1016/j.cell.2016.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orefice LL, Mosko JR, Morency DT, Wells MF, Tasnim A, Mozeika SM, Ye M, Chirila AM, Emanuel AJ, Rankin G, et al. (2019). Targeting Peripheral Somatosensory Neurons to Improve Tactile-Related Phenotypes in ASD Models. Cell 178, 867–886.e24. 10.1016/j.cell.2019.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clemens AW, and Gabel HW (2020). Emerging Insights into the Distinctive Neuronal Methylome (Preprint at Elsevier Ltd; ), 10.1016/j.tig.2020.07.009. 10.1016/j.tig.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozlenkov A, Roussos P, Timashpolsky A, Barbu M, Rudchenko S, Bibikova M, Klotzle B, Byne W, Lyddon R, di Narzo AF, et al. (2014). Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42, 109–127. 10.1093/nar/gkt838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giacoman-Lozano M, Meléndez-Ramírez C, Martinez-Ledesma E, Cuevas-Diaz Duran R, and Velasco I (2022). Epigenetics of Neural Differentiation: Spotlight on Enhancers (Preprint at Frontiers Media S.A.), 10.3389/fcell.2022.1001701. 10.3389/fcell.2022.1001701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Vasaikar S, Shi Z, Greer M, and Zhang B (2017). WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137. 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamagami N, Wu DY, Clemens AW, Nettles SA, Li A, and Gabel HW (2023). NSD1 deposits histone H3 lysine 36 dimethylation to pattern non-CG DNA methylation in neurons. Mol. Cell 83, 1412–1428.e7. 10.1016/j.molcel.2023.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cahill KM, Huo Z, Tseng GC, Logan RW, and Seney ML (2018). Improved identification of concordant and discordant gene expression signatures using an updated rank-rank hypergeometric overlap approach. Sci. Rep 8, 9588. 10.1038/s41598-018-27903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tillotson R, Cholewa-Waclaw J, Chhatbar K, Connelly JC, Kirschner SA, Webb S, Koerner M.v., Selfridge J, Kelly DA, de Sousa D, et al. (2021). Neuronal non-CG methylation is an essential target for MeCP2 function. Mol. Cell 81, 1260–1275.e12. 10.1016/j.molcel.2021.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tatton-Brown K, Douglas J, Coleman K, Ve Baujat G, Cole TRP, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, et al. (2005). Genotype-Phenotype Associations in Sotos Syndrome: An Analysis of 266 Individuals with NSD1 Aberrations. Am. J. Hum. Genet 77, 193–204. 10.1086/432082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saugier-Veber P, Bonnet C, Afenjar A, Drouin-Garraud V, Coubes C, Fehrenbach S, Holder-Espinasse M, Roume J, Malan V, Portnoi M-F, et al. (2007). Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome. Hum. Mutat 28, 1098–1107. 10.1002/humu.20568. [DOI] [PubMed] [Google Scholar]
- 47.Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, Elliott A, Wylie H, Ardissone A, Rittinger O, et al. (2017). Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. Am. J. Hum. Genet 100, 725–736. 10.1016/j.ajhg.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, and Jeltsch A (2019). H3K36me2/3 Binding and DNA Binding of the DNA Methyltransferase DNMT3A PWWP Domain Both Contribute to its Chromatin Interaction. J. Mol. Biol 431, 5063–5074. 10.1016/j.jmb.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 49.Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khazaei S, Chen CCL, Andrade AF, Kabir N, Azarafshar P, Morcos SM, França JA, Lopes M, Lund PJ, Danieau G, et al. (2023). Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration. Cell 186, 1162–1178.e20. 10.1016/j.cell.2023.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferris MA, Smith AM, Heath SE, Duncavage EJ, Oberley M, Freyer D, Wynn R, Douzgou S, Maris JM, Reilly AF, et al. (2022). DNMT3A overgrowth syndrome is associated with the development of hematopoietic malignancies in children and young adults. Blood 139, 461–464. 10.1182/blood.2021014052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao XF, Kohen R, Parent R, Duan Y, Fisher GL, Korn MJ, Ji L, Wan G, Jin J, Püschel AW, et al. (2018). PlexinA2 Forward Signaling through Rap1 GTPases Regulates Dentate Gyrus Development and Schizophrenia-like Behaviors. Cell Rep. 22, 456–470. 10.1016/j.celrep.2017.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Co M, Barnard RA, Jahncke JN, Grindstaff S, Fedorov LM, Adey AC, Wright KM, and O’Roak BJ (2022). Shared and Distinct Functional Effects of Patient-Specific Tbr1 Mutations on Cortical Development. J. Neurosci 42, 7166–7181. 10.1523/JNEUROSCI.0409-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sollis E, den Hoed J, Quevedo M, Estruch SB, Vino A, Dekkers DHW, Demmers JAA, Poot R, Deriziotis P, and Fisher SE (2023). Characterization of the TBR1 interactome: variants associated with neurodevelopmental disorders disrupt novel protein interactions. Hum. Mol. Genet 32, 1497–1510. 10.1093/hmg/ddac311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fazel Darbandi S, Robinson Schwartz SE, Pai ELL, Everitt A, Turner ML, Cheyette BNR, Willsey AJ, State MW, Sohal VS, and Rubenstein JLR (2020). Enhancing WNT Signaling Restores Cortical Neuronal Spine Maturation and Synaptogenesis in Tbr1 Mutants. Cell Rep. 31, 107495. 10.1016/j.celrep.2020.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makrides N, Panayiotou E, Fanis P, Karaiskos C, Lapathitis G, and Malas S (2018). Sequential role of SOXB2 factors in GABAergic neuron specification of the dorsal midbrain. Front. Mol. Neurosci 11, 152. 10.3389/fnmol.2018.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polan MB, Pastore MT, Steingass K, Hashimoto S, Thrush DL, Pyatt R, Reshmi S, Gastier-Foster JM, Astbury C, and McBride KL (2014). Neurodevelopmental disorders among individuals with duplication of 4p13 to 4p12 containing a GABA A receptor subunit gene cluster. Eur. J. Hum. Genet 22, 105–109. 10.1038/ejhg.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams A, Cooney E, Segal G, Narayanan S, Morand M, and Agadi S (2022). GABRG1 variant as a potential novel cause of epileptic encephalopathy, hypotonia, and global developmental delay. Am. J. Med. Genet 188, 3546–3549. 10.1002/ajmg.a.62969. [DOI] [PubMed] [Google Scholar]
- 59.Wu Z, Huang K, Yu J, Le T, Namihira M, Liu Y, Zhang J, Xue Z, Cheng L, and Fan G (2012). Dnmt3a regulates both proliferation and differentiation of mouse neural stem cells. J. Neurosci. Res 90, 1883–1891. 10.1002/jnr.23077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet 44, 23–31. 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, and Greenberg ME (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen P-Y, and Pellegrini M (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 14, 774. 10.1186/1471-2164-14-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hansen KD, Langmead B, and Irizarry RA (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13, R83. 10.1186/gb-2012-13-10-R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, and Gerig G (2006). User-guided 3D active contour segmentation of anatomical structures: Significantly improved efficiency and reliability. Neuroimage 31, 1116–1128. 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 70.Mi H, and Thomas P (2009). PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol 563, 123–140. 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou LP, and Mi H (2022). PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 31, 8–22. 10.1002/pro.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205. 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hill CA, Martínez-Abadías N, Motch SM, Austin JR, Wang Y, Jabs EW, Richtsmeier JT, and Aldridge K (2013). Postnatal brain and skull growth in an Apert syndrome mouse model. Am. J. Med. Genet 161A, 745–757. 10.1002/ajmg.a.35805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nixon JP, Zhang M, Wang C, Kuskowski MA, Novak CM, Levine JA, Billington CJ, and Kotz CM (2010). Evaluation of a quantitative magnetic resonance imaging system for whole body composition analysis in rodents. Obesity 18, 1652–1659. 10.1038/oby.2009.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stejskal EO, and Tanner JE (1965). Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys 42, 288–292. 10.1063/1.1695690. [DOI] [Google Scholar]
- 76.Basser PJ, and Pierpaoli C (2011). Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J. Magn. Reson 213, 560–570. 10.1016/j.jmr.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 77.Mori S, and Tournier J-D (2014). Introduction to Diffusion Tensor Imaging and Higher Order Models, 2nd ed. (Elsevier Science; ). [Google Scholar]
- 78.Manno R, Witte J, and Papouin T (2020). A Modular Setup to Run a Large Line of Behavioral Testing in Mice in a Single Space. Curr. Protoc. Neurosci 93, e102. 10.1002/cpns.102. [DOI] [PubMed] [Google Scholar]
- 79.Maloney SE, Yuede CM, Creeley CE, Williams SL, Huffman JN, Taylor GT, Noguchi KN, and Wozniak DF (2019). Repeated neonatal isoflurane exposures in the mouse induce apoptotic degenerative changes in the brain and relatively mild long-term behavioral deficits. Sci. Rep 9, 2779. 10.1038/s41598-019-39174-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maloney SE, Rieger MA, Al-Hasani R, Bruchas MR, Wozniak DF, and Dougherty JD (2019). Loss of CELF6 RNA binding protein impairs cocaine conditioned place preference and contextual fear conditioning. Genes Brain Behav. 18, e12593. 10.1111/gbb.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, et al. (2019). The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47, D853–D858. 10.1093/nar/gky1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li YE, Preissl S, Hou X, Zhang Z, Zhang K, Qiu Y, Poirion OB, Li B, Chiou J, Liu H, et al. (2021). An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136. 10.1038/s41586-021-03604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stephens M (2017). False discovery rates: a new deal. Biostatistics 18, 275–294. 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next generation sequencing data have been deposited to GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-alpha-Tubulin (EP1332Y) | Abcam | Cat# ab52866; RRID: AB_869989 |
| Mouse Anti-Dnmt3a Monoclonal Antibody, Clone 64B1446 | Abcam | Cat# ab13888; RRID: AB_300714 |
| IRDye 800CW Goat anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32211; RRID: AB_621843 |
| IRDye 800CW Goat anti-Mouse IgG antibody | LI-COR Biosciences | Cat# 926–32210; RRID: AB_621842 |
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Rabbit anti-MeCP2 | Chen et al.61 | N/A |
| Critical commercial assays | ||
| Mspa1I | NEB | Cat# R0577 |
| AllPrep DNA/RNA Kit | QIAGEN | Cat# 80284 |
| Ovation Ultralow Methyl-Seq Kit | Tecan | Cat# 0335–32 |
| Epitect Bisulfite Kit | Qiagen | Cat# 59824 |
| EZ DNA Methylation-Direct Kit | Zymo Research Corporation | Cat# D5020 |
| Accel-NGS Methyl-Seq DNA Library Kit | Swift Biosciences | Cat# 30024 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat# E7420 |
| NEBNext rRNA Depletion Kit (Human/Mouse/Rat) | NEB | Cat# E6310 |
| Accel-NGS 2S Plus DNA Library Kit (24 rxns) | Swift Biosciences | Ca#21024 |
| Deposited data | ||
| RNA-sequencing data | This paper | GEO: GSE225372 |
| ChIP-sequencing data (H3K27ac and MeCP2) | This paper | GEO: GSE225372 |
| Bisulfite-sequencing data | This paper | GEO: GSE225372 |
| Bisulfite-sequencing data | Lister et al.14 | GEO: GSE47966 |
| RNA- and ChIP-sequencing data | Clemens et al.20 | GEO: GSE123373 |
| RNA-sequencing data | Christian et al.9 | GEO: GSE147899 |
| RNA-sequencing data | Hamagami et al.42 | GEO: GSE212847 |
| Mus musculus mm9 genome assembly | UCSC | http://hgdownload.soe.ucsc.edu/goldenPath/mm9/ |
| Ensembl gene models | UCSC | https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Dnmt3a P900L/+ | This paper | N/A |
| Dnmt3a R878H/+ | Smith et al.24 | Provided by T. Ley |
| Oligonucleotides | ||
| Actb Forward | IDT | AAGGCCAACCGTGAAAAGAT |
| Actb Reverse | IDT | GTGGTACGACCAGAGGCATAC |
| Dnmt3a Forward | IDT | GGCCTTCTCGACTCCAGATG |
| Dnmt3a Reverse | IDT | TTCCTCTTCTCAGCTGGCAC |
| Dnmt3a P900L Region Forward | IDT | AGAGGGGCATTTATGGATGA |
| Dnmt3a P900L Region Reverse | IDT | GAGGGGCCTATTTTGCTTTT |
| Software and algorithms | ||
| DESeq2 (v1.14.1) | Love et al.62 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR (v3.16.5) | Robinson et al.63 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| BEDtools2 (v2.25.0) | Quinlan and Hall64 | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.5) | Langmead and Salzberg65 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | Dobin et al.66 | https://github.com/alexdobin/STAR |
| fastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim galore | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| BS-seeker2 | Guo et al.67 | https://github.com/BSSeeker/BSseeker2 |
| BSmooth | Hansen et al.68 | https://www.bioconductor.org/packages/release/bioc/html/bsseq.html |
| GraphPad Prism v9.4.1 | GraphPad by Dotmatics | https://www.graphpad.com/ |
| Avizo | ThermoFisher | http://www.vsg3d.com/ |
| ITK-SNAP | Yushkevich et al.69 | http://itksnap.org/ |
| PANTHER Gene Ontology (v17.0) | Mi and Thomas70; Thomas et al.71 | http://www.pantherdb.org/tools/compareToRefList.jsp |
| WebGestalt Gene Ontology | Liao et al.72 | https://www.webgestalt.org/ |
| RRHO2 | Cahill et al.43 | https://github.com/RRHO2/RRHO2 |