

Abstract
Much progress has been made in the development of methods to both create compounds that contain C–F bonds and to functionalize C–F bonds. As such, C–F bonds are becoming common and versatile synthetic functional handles. This review summarizes the advantages of defluorinative functionalization reactions for small molecule synthesis. The coverage is organized by the type of carbon framework the fluorine is attached to for mono- and polyfluorinated motifs. The main challenges, opportunities and advancements of defluorinative functionalization is discussed for each class of organofluorine. Most of the text focuses on case studies that illustrate how defluorofunctionalization can improve routes to synthetic targets or how the properties of C–F bonds enable unique mechanisms and reactions. The broader goal is to showcase the opportunities for incorporating and exploiting C–F bonds in the design of synthetic routes, improvement of specific reactions and advent of new methods.
Keywords: Fluorine, Defluorofunctionalization, C–F activation, Synthesis design, Synthetic methods
Graphical Abstract
1. Introduction
Organofluorine chemicals are abundantly available from commercial suppliers and through well-established synthetic protocols.[1] Reliable access to these compounds in modular and scalable fashions is the result of tremendous development in fluorination methods as well as the invention and use of fluorinated building blocks.[2] These efforts are generally motivated by the widespread use of fluorine incorporation as a means to modulate a molecule’s properties, especially within medicinal, agrochemical, materials and commercial product (e.g., refrigerants and polyfluoroalkyl substances) applications.[3] In these contexts, fluorine’s high electronegativity and the high strength of C–F bonds can improve a compound’s metabolic or oxidative stability, lipophilicity and solubility, and can modulate key properties such as pKa or conformational preferences.[4]
Meanwhile, chemists have made significant advances in C–F bond functionalization methodology, a task often inspired by the fundamental challenges associated with activating such strong bonds. This progress is the subject of numerous reviews that are often dedicated to a specific class of C–F bond (e.g., aryl fluorides) or mechanistic strategy (e.g., metal catalysis).[5–7] However, given the increasing availability of fluorinated chemicals and advances in C–F functionalization, an overview of the key synthetic advantages that motivate defluorinative methods and applications would be useful. This review aims to address this topic by answering the overarching question: “What is the advantage of purchasing or preparing a fluorinated chemical if the fluorine is to be removed in a subsequent reaction?”. The answers to this question can generally be classified as benefits to either an overall synthetic route or to the mechanism of a desired reaction.
Synthetic route advantages of defluorofunctionalization typically stem either from the availability of a fluorinated starting material or the chemoselectivity of a targeted reaction. For example, certain small molecule organofluorines are less expensive than alternately halogenated analogues. Strong C–F bonds are also inert under many standard reaction conditions and can often be carried through multistep syntheses for downstream functionalization, unlike other C–X bonds.[4] Alternatively, chemoselective activation of functionalized or multihalogenated organofluorines allows for regioselective multistep syntheses.[8] Defluorofunctionalization is also advantageous when the reaction generates a new, but more valuable, fluorinated product. This is exemplified by methods that activate C–F bonds for isomerization or insertion processes in order to transform simple organofluorines into complex fluorinated products. Similarly, the preparation or purchase of polyfluorinated starting materials for use in partial defluorofunctionalization can be a cost-effective and modular route to fluorine-containing compounds.[9]
There are several mechanistic features of defluorinative reactions that can enable desired transformations over alternative organohalide starting materials. Due to the small size of fluorine and the high bond polarization, certain C–F bonds (e.g. acyl and aryl fluorides) undergo uniquely facile substitution reactions with anionic and sterically-encumbered nucleophiles. The polarization also leads to low-lying C–F anti-bonding orbitals that stabilize β-carbanions and radicals, an enabling feature for addition/fluoride elimination sequences of allyl and vinyl fluorides.[10] Defluorination also frequently generates fluoride byproducts that play a pivotal role in reaction mechanisms, often as bases for pronucleophile activation or generation of unstable anionic species in situ.[11] Similarly, metal-catalyzed defluorinative reactions can proceed via metal fluoride complexes that react with a reagent for reaction propagation.[12] C–F bonds are also often reactive towards Lewis acids and can generate strong fluorine bonds with a reagent or catalyst (e.g., Si–F bond formation).[13] Lewis acidic activation can therefore enable non-traditional substitution selectivity and provide a driving force for thermodynamically challenging reactions or those that proceed through high-energy intermediates (e.g., aryl cation generation).
This review showcases the route and mechanistic advantages of C–F defluorofunctionalization for small molecule synthesis. The content is organized by the type of carbon framework the fluorine is attached to, as summarized in Scheme 1. The coverage does not include applications to material sciences[14], defluorination for environmental remediation[15], reactions with biomolecules[16] or commonplace defluorinative processes of commercial reagents (e.g., difluorocarbene generation from TMSCF3; TMS = trimethylsilyl).[17] The goal of this review is to highlight the main challenges, advances and applications of each type of C–F bond to guide synthetic chemists and to motivate researchers to devise new methods for C–F functionalization. The discussion is intended to demonstrate general reactivity patterns of C–F bonds as well as the key advantages of defluorination through specific examples. This non-comprehensive review often summarizes and represents the work of many researchers through a single example. We have attempted to select reactions that demonstrate as many benefits of defluorination as possible and refer readers to other reviews for more detailed coverage.[5–7]
Scheme 1.
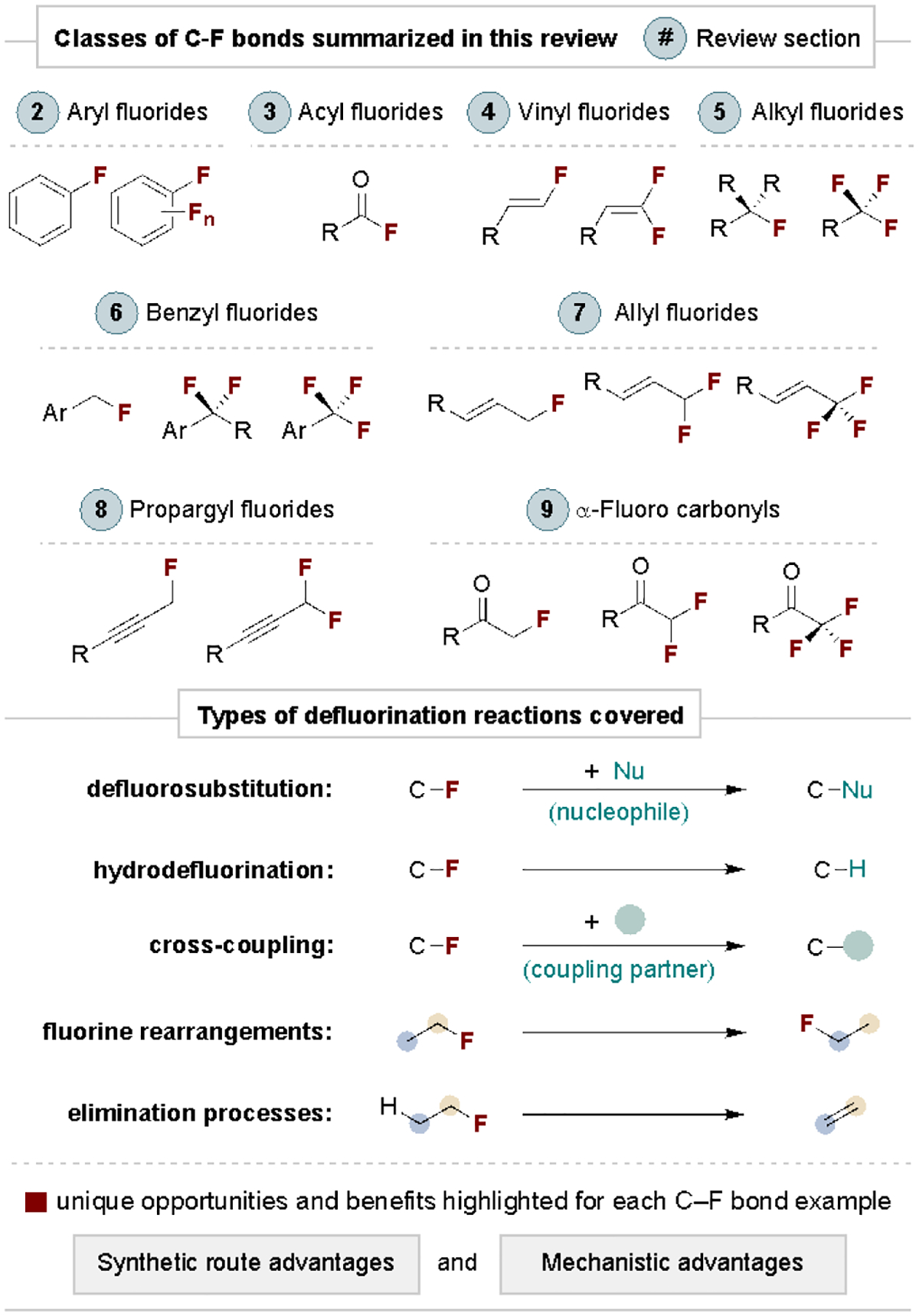
Schematic summary of the scope of this Review.
2. Aryl Fluoride Defluorofunctionalization
Simple aryl fluorides that contain one or two additional functional groups (e.g., halogens, nitro groups, esters and nitriles) are inexpensive commercial compounds. More complex fluoroarenes are generally less available and require independent synthesis, often via C–X fluorination, deoxyfluorination or Balz-Schiemann reactions.[18] Thus, most advantageous aryl defluorination methods employ more accessible, simple starting materials where the C–F bond offers unique reactivity compared to other halogens. Polyfluoroarenes represent a separate class of commercially available aryl fluoride that are used to access partially fluorinated arenes as an alternative to aryl fluorination methods.[9b,c] As mono- and polyfluoroarenes possess different challenges and opportunities, they are discussed separately in this section.
2.1. Defluorinative substitution reactions
C–F bonds are generally more reactive than other C–X bonds towards nucleophilic aromatic substitution reactions (SNAr) that can occur through stepwise or concerted pathways (Scheme 2a).[19] This trend often enables nucleophilic defluorosubstitution on electron-neutral arenes without the need for metal catalysis. This is an advantage for large-scale syntheses, including alkoxide SNAr reactions used to prepare the pharmaceuticals clomiphene and duloxetine (Scheme 2b).[20] Many nucleophiles and arenes are suitable for defluorinative SNAr reactions; weak nucleophiles require electron-deficient fluoroarenes, while strong nucleophiles can substitute electron-rich arenes. This trend is exemplified by a cyanation reaction reported by Jenkins and coworkers[21] and an α-arylation of alkyl nitriles reported by Caron and coworkers at Pfizer[22] in Scheme 2c. Aryl fluorides can be used to increase the yields or scope of many types of substitution reactions. For example, their use in N-heterocyclic carbene (NHC)-catalyzed aldehyde C–H arylation reactions outperform other aryl halides (Scheme 2d).[23] The ability of aryl fluorides to engage in concerted SNAr reactions with strong nucleophiles also enables regioselective substitution of electron-rich fluorides, as seen in Scheme 2c and in recently developed silylation reactions (Scheme 2e).[24]
Scheme 2.
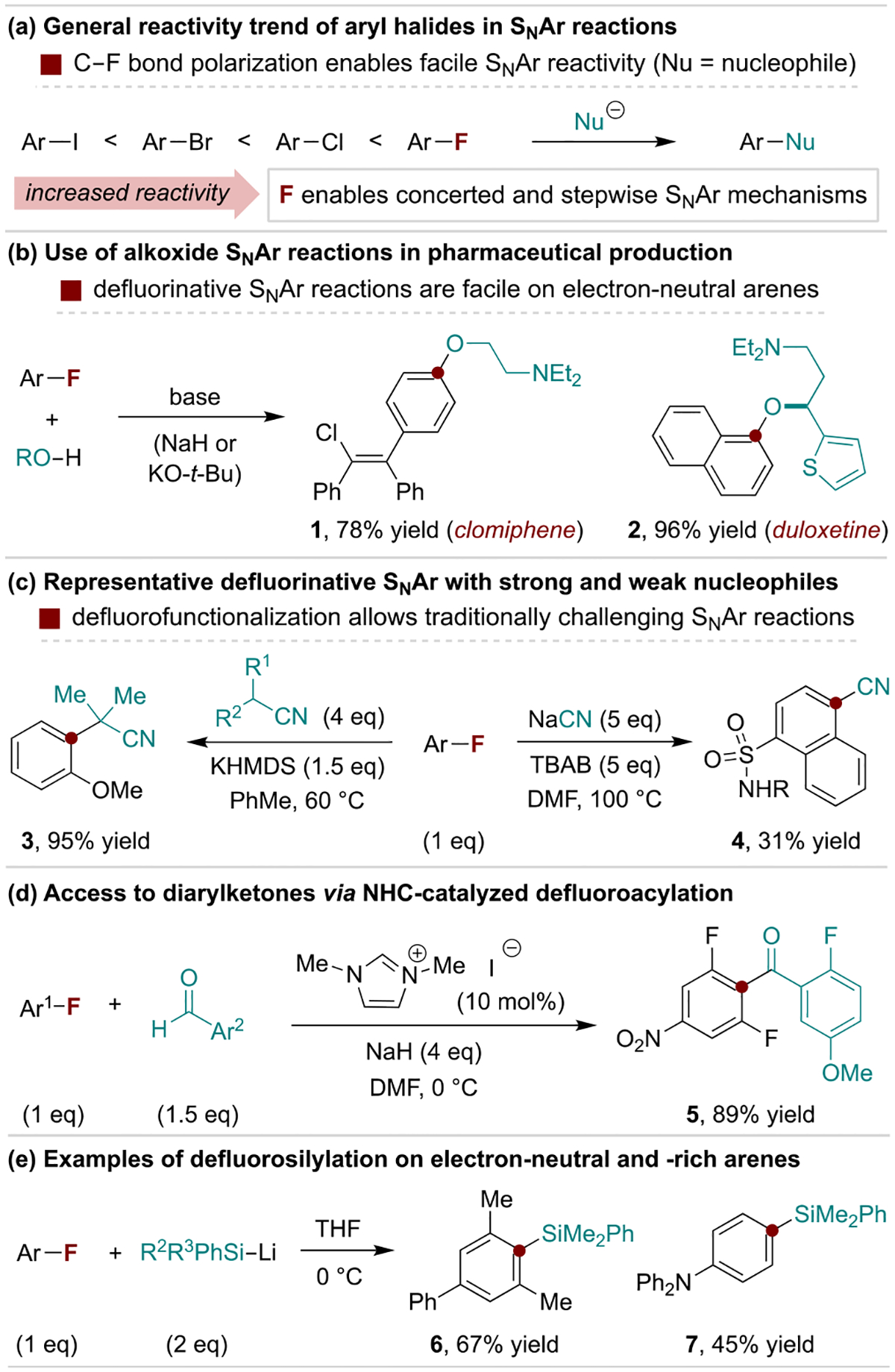
Selected uses of fluoroarenes in direct substitution reactions. KHMDS, potassium bis(trimethylsilyl)amide; TBAB, tetrabutylammonium bromide.
2.2. Fluorine-guided regioselective arene synthesis
Polyhalogenated arenes containing at least one fluorine are commercially available and enable assembly of substituted arenes via chemoselective C–X functionalization. While SNAr reactions preferentially occur with C–F bonds over other C–X bonds, the inverse trend is generally observed for metal-catalyzed coupling reactions.[8] This utility is illustrated in Scheme 3a for the regioselective synthesis of a complex biaryl system from 1-bromo-2-fluoro-4-iodobenzene by chemists at AstraZeneca.[25] C–F bonds can also control positional selectivity via alternative mechanisms, including acidifying effects that guide arene deprotonation.[26] This concept is illustrated in Scheme 3b, where scientists at GSK used 2-chloro-4-fluoropyridine for fluorine-directed 3-deprotonation/acylation, followed by sequential C–F amination and C–Cl methylation en route to HIV-1 integrase inhibitor candidates.[27] Fluoroarenes with ortho-withdrawing substituents (e.g., nitriles, esters and ketones) are readily available and useful for tandem SNAr/cyclization reactions to prepare benzo-fused heteroarenes. This sequence was used by chemists at Pfizer wherein a 2,4-difluoroaryl ketone forms an indazole via hydrazine condensation/SNAr, followed by facile defluoroalkylation en route to a potential treatment for inflammatory diseases (Scheme 3c).[28]
Scheme 3.
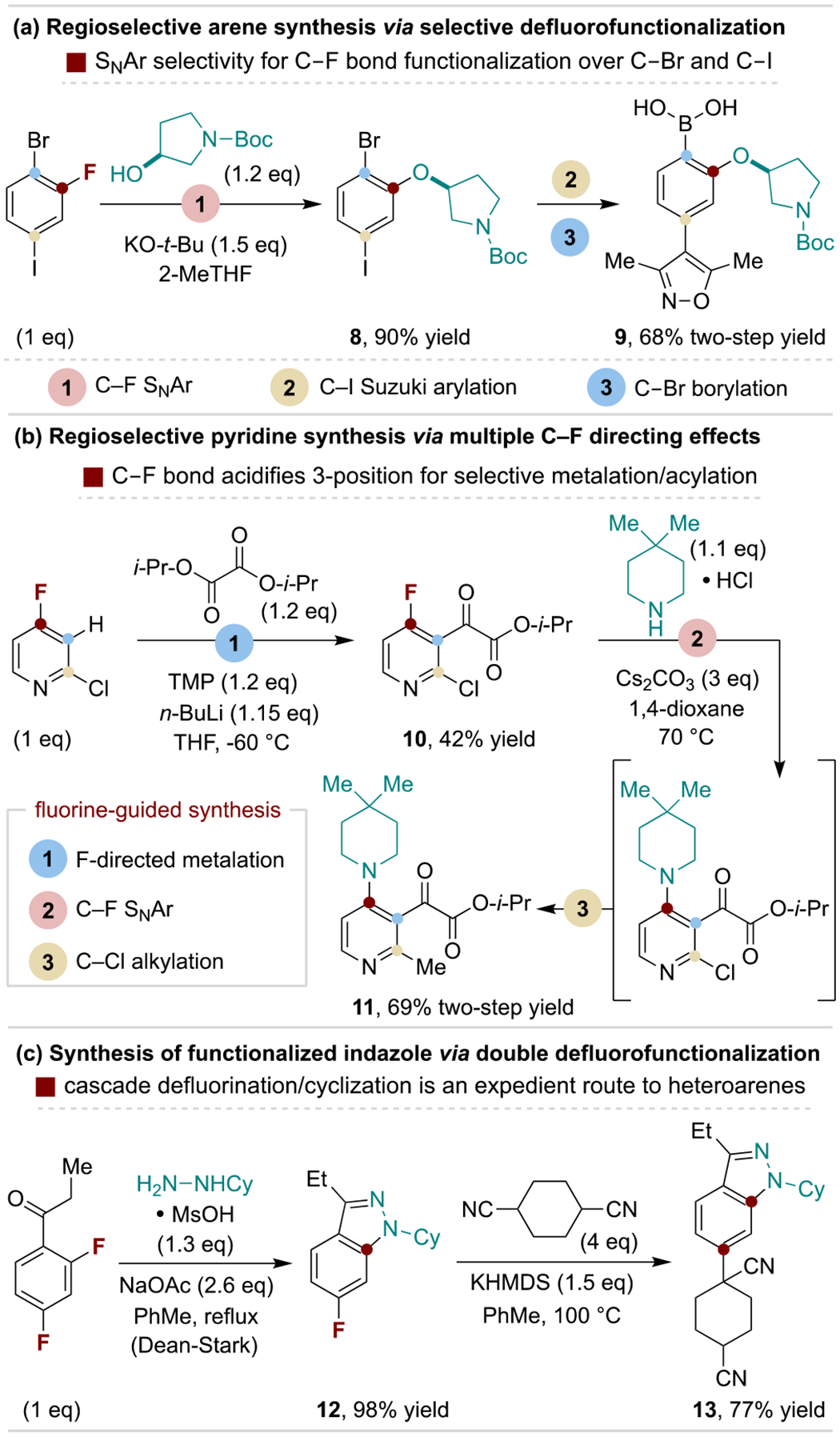
Selected uses of fluoroarenes in regioselective arene synthesis. TMP, 2,2,6,6-tetremethylpiperidine; Cy, cyclohexyl.
2.3. Defluorinative access to phosphine ligands
An additional valuable application of defluorinative substitution reactions is their use for preparing mono- and bisphosphines, including commercial ligands. Most commonly, this is achieved through base-promoted SNAr reactions of phosphines and simple fluoroarenes, with an array of representative ligands shown in Scheme 4a.[29] Alternatively, the acidifying effect of fluorine can promote facile metalation and fluoride elimination en route to versatile aryne intermediates. Scheme 4b demonstrates this strategy for production of the biaryl fragment of the BrettPhos ligand, a common route to privileged Buchwald-type biaryl phosphine ligands.[30] We note that the use of aryl fluorides as aryne precursors has found wide application in other contexts, including heteroarene, complex molecule and natural product syntheses.[31]
Scheme 4.
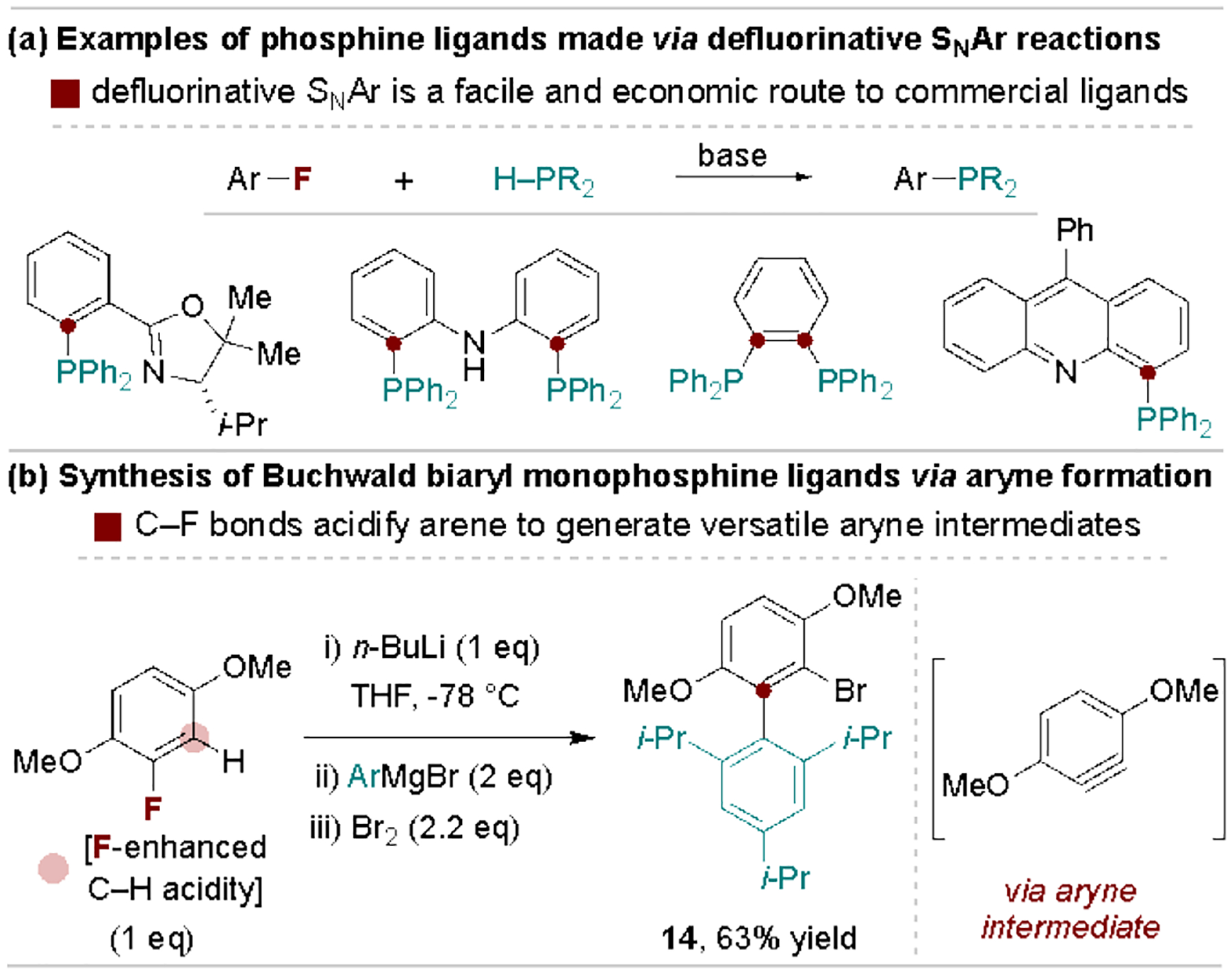
Examples of fluoroarenes used in phosphine ligand syntheses.
2.4. Alternative types of defluorinative SNAr reactions
Given the availability of fluoroarenes, there is strong motivation to develop new and complementary mechanistic strategies for defluorinative substitution reactions. To this end, Nicewicz and coworkers developed a photoredox-catalyzed SNAr method that operates via single electron oxidation of electron-rich to -neutral fluoroarenes to generate electrophilic aryl radical cation intermediates as outlined in Scheme 5a.[32] This protocol enables defluorinative SNAr in the presence of other halogens and can be used for radiofluorination of fluorinated drugs.[33] Lambert and coworkers developed a related method via an electrophotochemical strategy that tolerates base-sensitive functional groups, including aldehydes and traditionally SNAr-active heteroaryl halides (Scheme 5a, bottom).[34] An alternative photochemical process, disclosed by Larionov and coworkers, uses UV light to promote defluoroborylation of electron-rich aryl fluorides and is proposed to proceed through an aryl radical cation intermediate from C–F bond homolysis (Scheme 5b).[35] Another emerging strategy for activation of aryl fluorides is the catalytic use of metals that coordinate to the arene to form electrophilic π-metal complexes. The Shi Group has made significant advances in this regard using Ru and Rh catalysts for alcohol, water and amine SNAr reactions that selectively substitute electron-rich aryl fluorides as illustrated in Scheme 5c.[36]
Scheme 5.
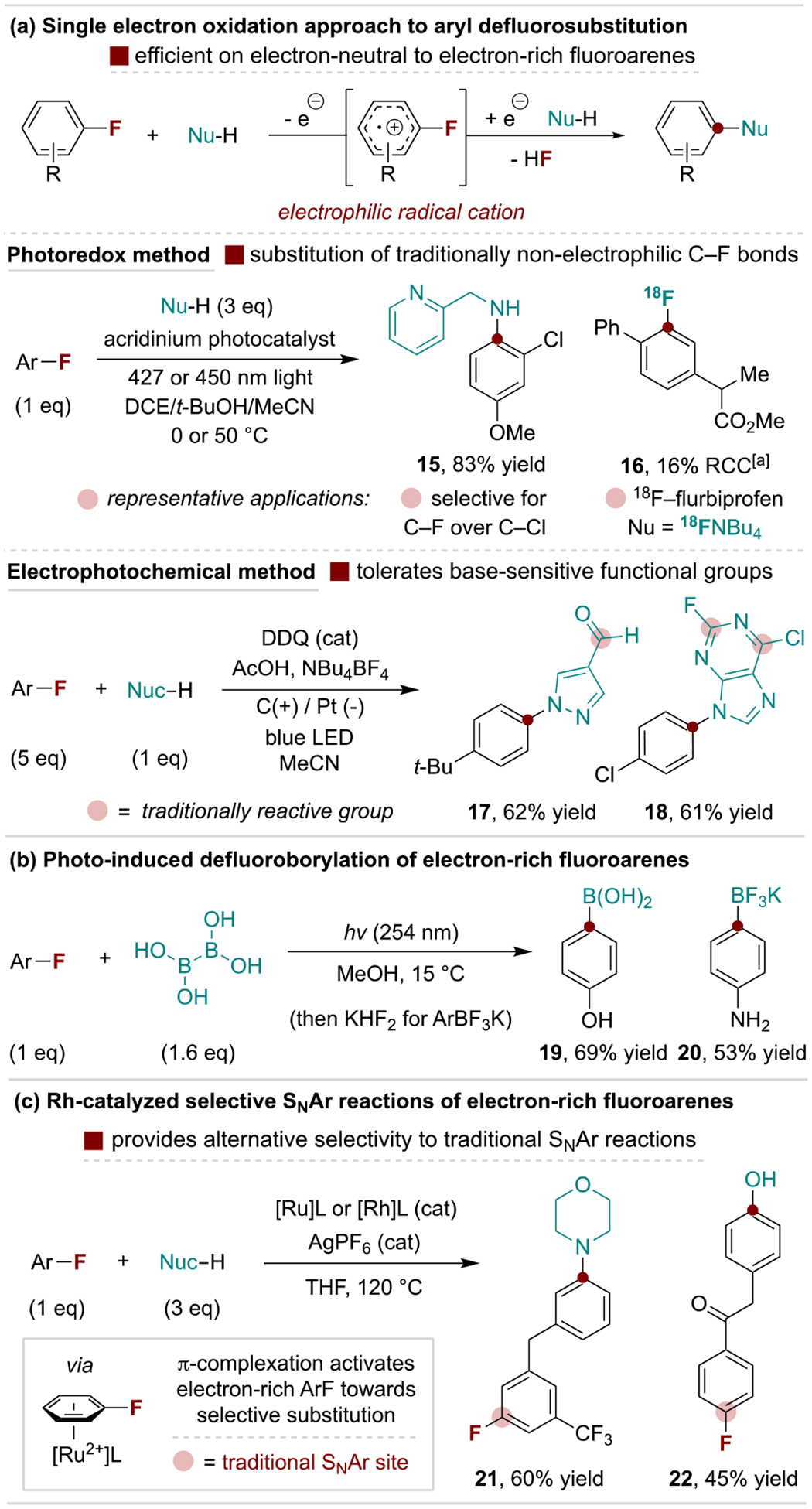
Selected new types of defluorinative SNAr reactions. DCE, 1,2-dichloroethane; DDQ, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. [a] Radiochemical conversion (RCC).
2.5. Metal and Lewis acid-catalyzed C–C bond-forming reactions
Fluoroarenes are active towards catalytic C–C bond-forming reactions via transition metal[6c,d] and Lewis acid[13] activation of C–F bonds. Although other C–X bonds are more prone towards oxidative addition processes, significant progress has been made in the use of C–F bonds for cross-coupling reactions for net defluoroarylation, -alkylation and -borylation.[37] These reactions are most advantageous when the fluoroarene substrate is inexpensive or if polyfunctionalized arenes are used for chemoselective syntheses. Representative conditions for such methods are shown in Scheme 6a through a distinctly selective defluorinative coupling reaction developed by Manabe and coworkers.[38] Here, Ni-catalyzed Kumada coupling selectively occurs at C–F bonds adjacent to alcohol and amine directing substituents over weaker C–X bonds. In addition to oxidative addition by transition metals, C–F bonds can be activated by strong Lewis acids to generate highly reactive aryl cation intermediates. This strategy enables a variety of useful arylation reactions, including intramolecular Friedel-Crafts-type arylation, as illustrated by Siegel and coworkers in Scheme 6b.[39] This protocol has also been applied to the synthesis of Buckybowl derivatives.[40] In a further advance, Nelson and coworkers discovered that C–F activation of ortho-silylated fluoroarenes generates discrete aryl cations that undergo intermolecular C–H insertion reactions with unactivated alkanes (Scheme 6c).[41]
Scheme 6.
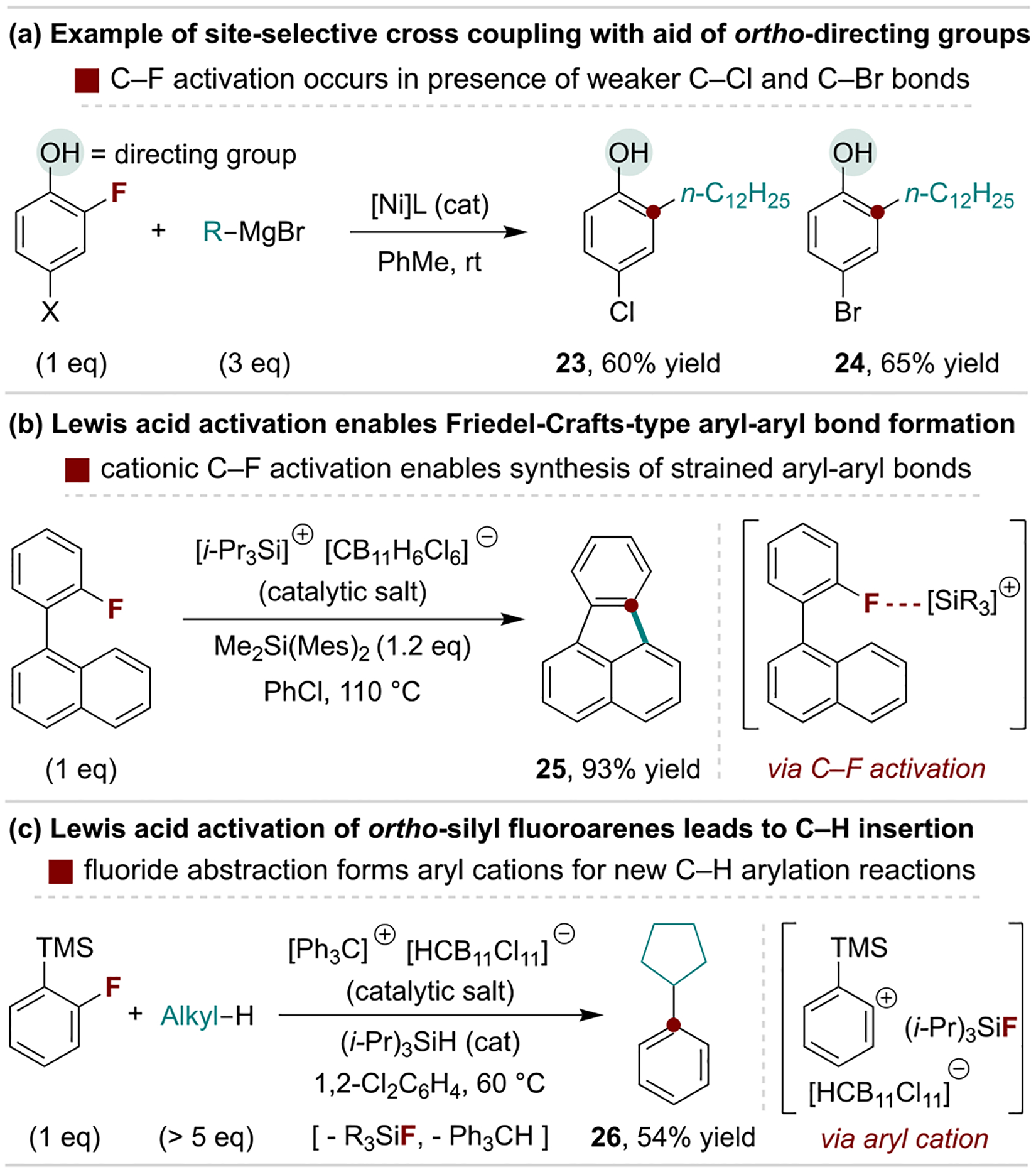
Catalytic defluorinative C–C coupling reactions of aryl fluorides. Mes, mesityl.
2.6. Defluorinative SNAr of polyfluoroarenes
Polyfluoroarenes participate in facile SNAr reactions under mild conditions with many types of common nucleophiles (e.g., C-, O-, N-, Si-based nucleophiles) as a modular route to fluorinated arenes (Scheme 7a). Their use in this regard has been studied extensively and the selectivity of consecutive defluorofunctionalization steps is the subject of various reports.[9b,c,g] Here, we discuss representative uses and provide extended examples of substrate scopes to illustrate the selectivity trends observed for each reaction. As an alternative to the use of organometallic nucleophiles[42], Wu, Yang and coworkers discovered that phosphonium ylides react with polyfluoroarenes to achieve net defluoroalkylation (Scheme 7b).[43] C–F substitution typically occurs para to an electron-withdrawing group, while a second substitution will occur at the ortho-position. Scheme 7c shows products accessible via base-promoted defluoroarylation of nitroalkanes, a process developed by Weaver and coworkers to access fluorinated benzylamine end products.[44] This includes examples that form regioisomeric mixtures (31 and 32) to illustrate how subtle effects between the nucleophile, base and substrate can impact site-selectivity, an important consideration and challenge for non-symmetrical fluorinated arenes. We note that the release of fluoride in SNAr reactions is also commonly used to activate silylated pronucleophiles, especially those that generate carbon-based nucleophiles (e.g., aryl and alkynyl carbanions and enolates).[45]
Scheme 7.
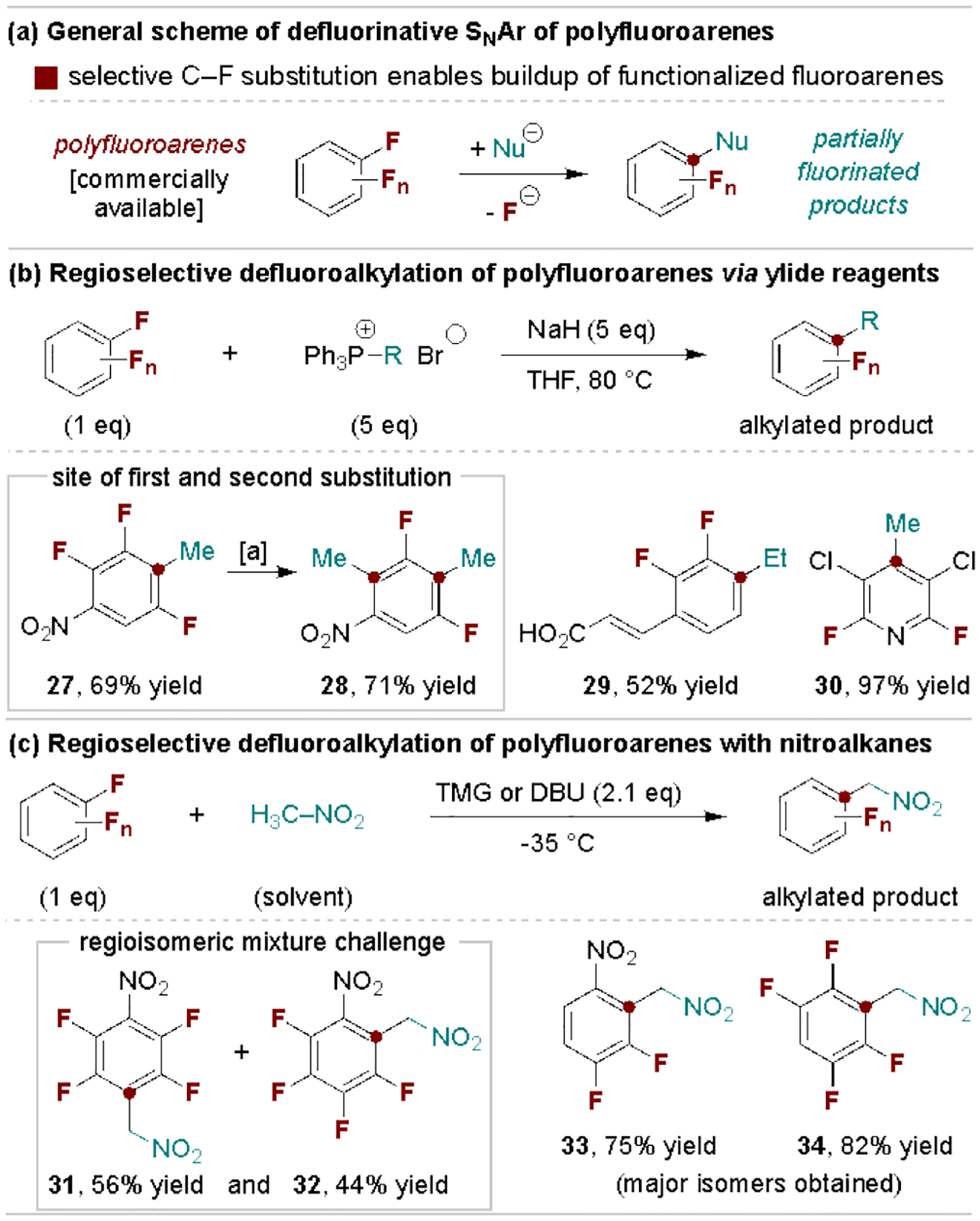
Selective SNAr defluorofunctionalization of polyfluoroarenes. TMG, 1,1,3,3-tetramethylguanidine; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene. [a] Use of excess ylide reagent for double defluoroalkylation.
2.7. Metal-catalyzed defluorination of polyfluoroarenes
A variety of metal-catalyzed cross-coupling reactions are operative on polyfluorinated arenes (Scheme 8a). Generally, these occur either at the traditionally most electrophilic C–F bond (e.g., para to an electron-withdrawing group) or at a C–F bond ortho to a directing group.[9b,g] These trends are represented in Schemes 8b and 8c for a defluorinative Suzuki coupling protocol and a directed C–H arylation method, respectively.[46] Beyond C–C coupling reactions, defluoroborylation is extensively developed as a means to install versatile C–B bonds onto fluorinated arenes. Defluoroborylation is possible using multiple mechanistic strategies that each display unique selectivity trends, including nucleophilic substitution[47], photopromoted processes[48] or via metal-catalyzed coupling as illustrated in Scheme 8d.[49]
Scheme 8.
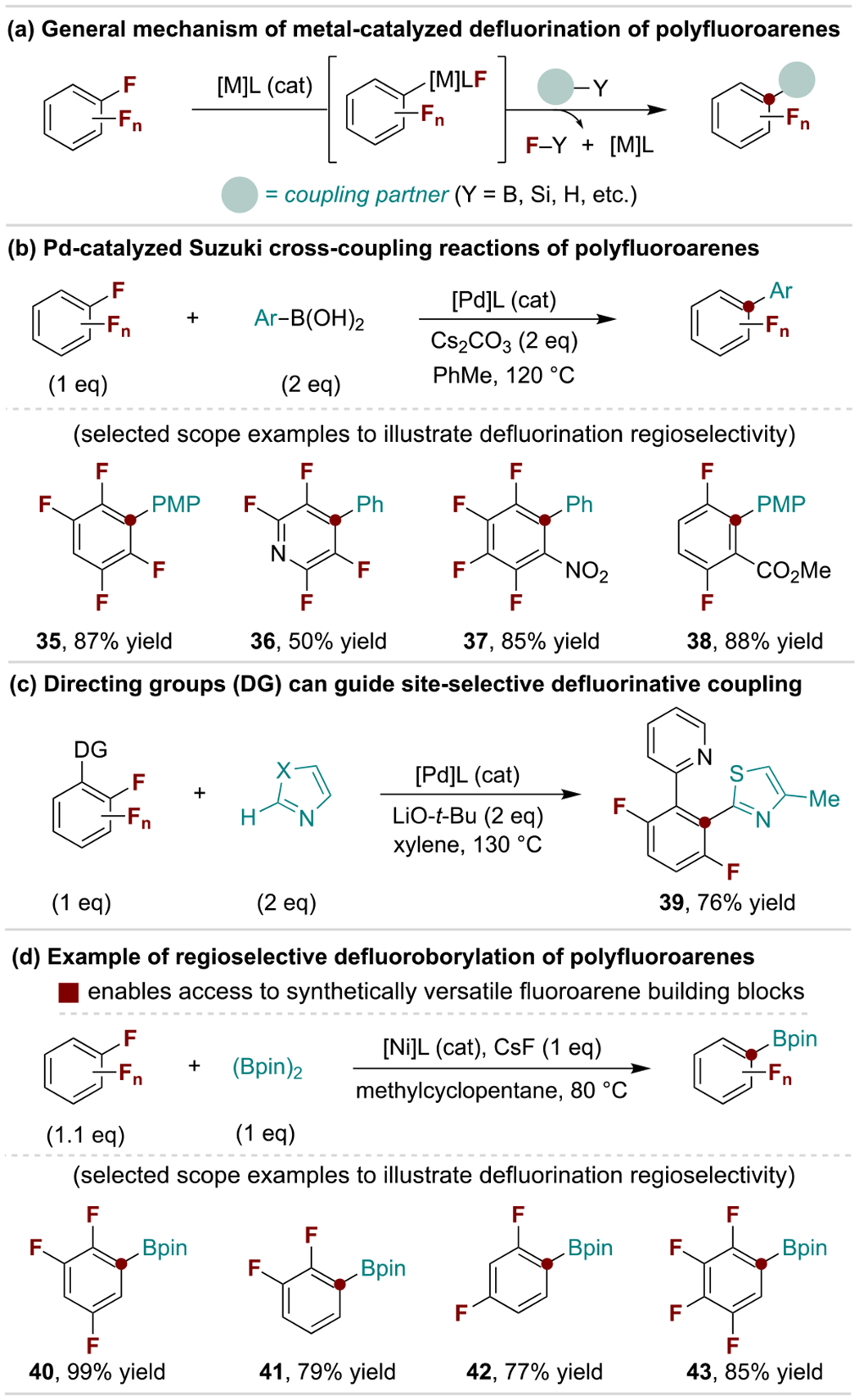
Selected metal-catalyzed coupling reactions of polyfluoroarenes. PMP, para-methoxyphenyl; pin, pinacolato.
2.8. Defluorinative radical coupling of polyfluoroarenes
Polyfluoroarenes are highly reactive substrates for radical-based coupling reactions as they can engage in diverse mechanistic processes, including direct addition of radical species, reduction to a radical anion intermediate for coupling or tandem reduction/C–F cleavage to form an aryl radical (Scheme 9a).[50] These pathways are typically promoted photochemically and enable a wide range of defluorinative C–C coupling reactions. Scheme 9b provides a selection of products and their origin to summarize the possible types and sites of radical defluorofunctionalization.[51] Analysis of this scope suggests that radical C–C coupling reactions to polyfluoroarenes is most suitable for highly electron-deficient systems (e.g., perfluoropyridine or tetra- and pentafluorobenzenes).
Scheme 9.
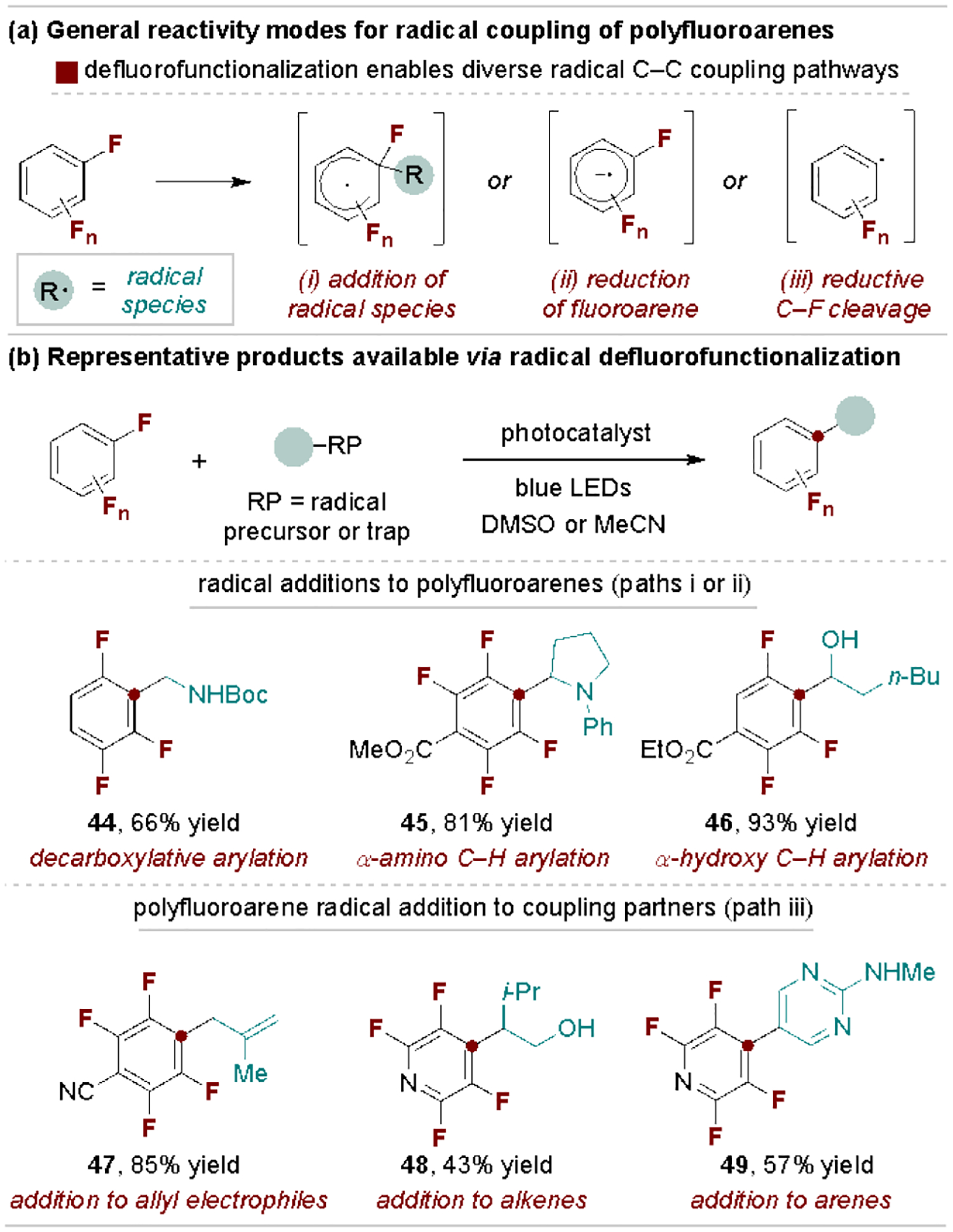
Selected products via polyfluoroarene radical coupling reactions.
2.9. Selective hydrodefluorination of polyfluoroarenes
Selective hydrodefluorination represents another well-developed tool to access partially fluorinated arenes.[52] In this regard, Weaver and coworkers reported a photocatalytic method that is useful for a wide scope of fluoroarenes, with representative examples shown for both single and double C–F reductions (Scheme 10a).[53] This reaction operates on less-activated fluoroarenes compared to the reductive defluorinative C–C coupling methods shown in Scheme 9. For more activated fluoroarenes, the Weaver group also developed a simple NaBH4-promoted hydrodefluorination protocol.[54] The high electrophilicity of polyfluoroarenes can also be exploited to enable facile defluorinative SNAr reactions prior to removal of excess fluorine atoms, a concept illustrated in Scheme 10b en route to a bispyridine diamine ligand.[55] Critically, direct amination of 4-halopyridines could not successfully prepare the targeted product 54; instead, selective SNAr on 2,4,6-trifluoropyridine followed by Ti-catalyzed hydrodefluorination was required.
Scheme 10.
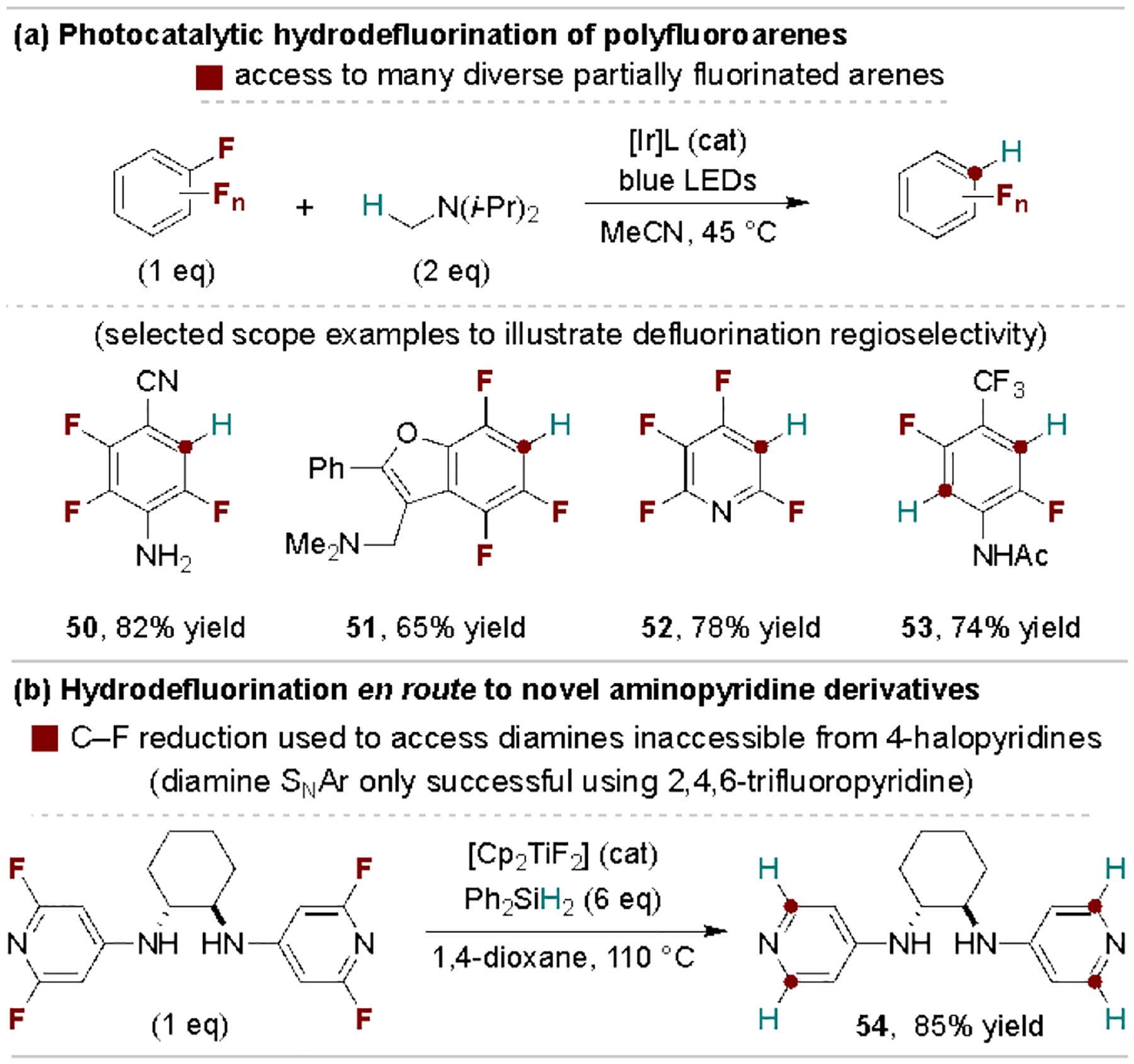
Examples of selective hydrodefluorination of polyfluoroarenes.
2.10. Application of polyfluoroarene defluorofunctionalization
The overarching motivation of polyfluoroarene defluorofunctionalization is to provide controlled access to multisubstituted or partially fluorinated arenes. Weaver and coworkers have made considerable advances on this subject, often described as “fluorine sculpting”.[9g,56] Scheme 11 provides three examples of sequential defluorofunctionalization of inexpensive polyfluoroarenes en route to more valuable products.[51c,54] This compares favorably to alternately halogenated starting materials or intermediates that could map onto the products but are more expensive or unavailable.
Scheme 11.
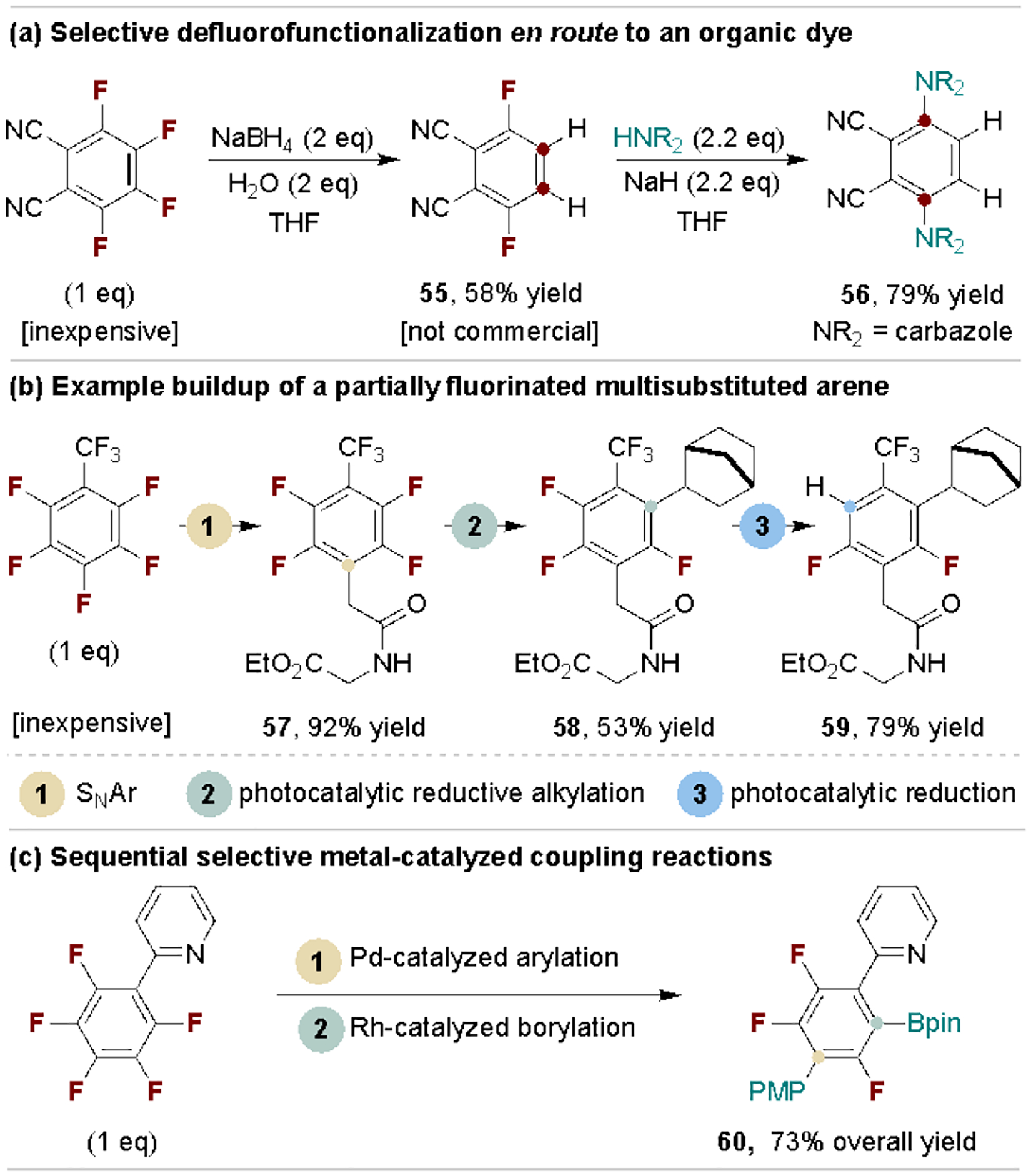
Applications of polyfluoroarene sequential C–F substitution.
3. Acyl Fluoride Defluorofunctionalization
Acyl fluorides, while generally not commercially available, are easily prepared from the corresponding carboxylic acid or acyl chloride using common fluorination reagents.[57] Acyl fluorides possess increased stability towards hydrolysis relative to acyl chlorides and can be purified on silica gel. The electrophilicity of acyl fluorides is comparable to an activated ester (e.g., an anhydride), but lacks the accompanying steric encumbrance, thus providing a balance between stability and reactivity that manifests in several unique applications summarized here. Acyl fluorides (“RCOF”) are versatile synthons for both defluorinative acylation (“RCO–“) and decarbonylative (“R–“) reactions that are discussed below. Upon reaction with a nucleophile in situ, acyl fluorides can serve as a useful source of anhydrous fluoride anions, a topic not covered here.[58]
3.1. General use as acyl electrophiles
There are several scenarios where preparation of acyl fluorides provides synthetic advantages over conventional electrophiles (e.g., acyl chlorides or in situ generated activated acids). The small size of fluorine enables acylation of hindered nucleophiles, especially anionic O- and N-species as illustrated in Scheme 12a. Bulky lithium amides react with acyl fluorides to generate sterically-hindered amides; in contrast, acyl chlorides exhibit lower yields and undergo aromatic metalation side reactions.[59] Similarly, acyl fluorides are often uniquely effective for challenging O-acylation reactions with complex alkoxides, a feature frequently exploited in natural product synthesis as exemplified by Fürstner and coworkers’ synthesis of aspercyclides (63).[60] It should also be noted that acyl fluorides typically undergo selective O-acylation of enolates over competing C-acylation.[61] The released fluoride anion in defluorinative acylation can be used to activate silylated derivatives of weakly nucleophilic coupling partners. This is illustrated for a reaction with heteroaryl amines in Scheme 12a (bottom), where attempts at coupling acyl chlorides with non-silylated amines led to low yields and mass balance.[62] A separate advantage of fluoride byproducts discovered by Rovis and coworkers is illustrated in Scheme 12b for the reductive acylation of chiral lactones.[63] Here, acyl fluorides significantly outperform chlorides as the authors discovered that DIBAL-Cl byproduct promotes product (or intermediate) decomposition, whereas less Lewis acidic DIBAL-F does not.
Scheme 12.
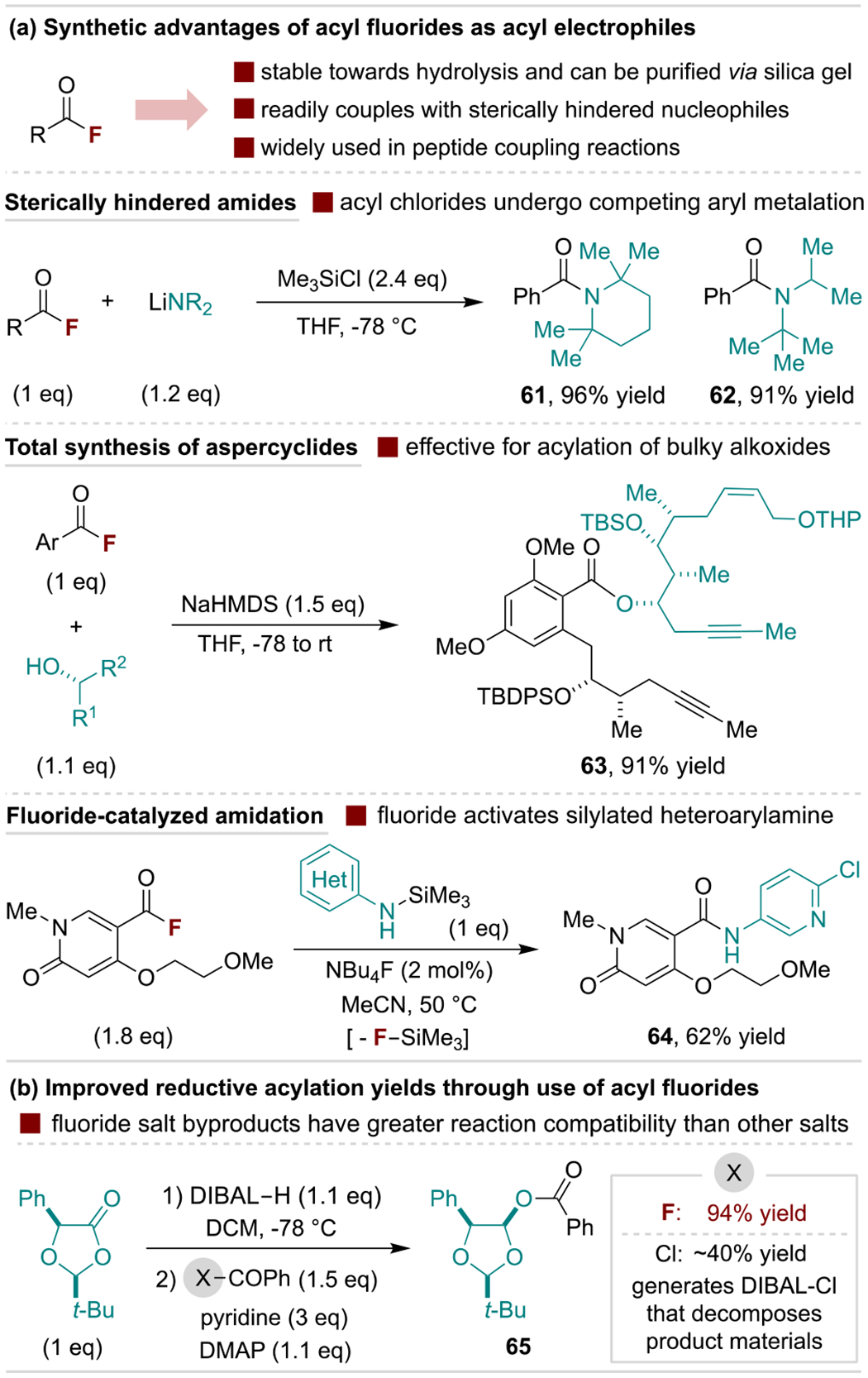
Representative utility of acyl fluorides as electrophiles. TBS, tert-butyldimethylsilyl; THP, tetrahydropyranyl; DIBAL, diisobutylaluminum; diDMAP, 4-(dimethylamino)pyridine.
3.2. Defluorinative peptide coupling
Carpino and coworkers pioneered the development of amino acid fluorides that are now extensively used to overcome a variety of challenges in peptide coupling reactions.[64] The advantages of acyl fluorides in this context have been the subject of several expert reviews, and here we summarize their utility through several specific applications.[65] Amino acid fluorides with diverse N-protecting groups are stable and thus attractive preactivated reagents ideally suited for solution- and solid-phase peptide synthesis.[66] Critically, amino acid fluorides are less susceptible towards oxazolone formation as compared to other activated acids, and are thus often used to enable challenging couplings without racemization of α-stereocenters, as demonstrated by an example from the Danishefsky Group in Scheme 13a.[67] The utility of acyl fluorides for hindered amide formation (discussed above) is widely exploited in peptide synthesis, as illustrated by Nicolaou and coworkers’ synthesis of tubulysins (Scheme 13b).[68] Another unique feature of defluoroamidation reactions is that they often do not require basic additives since the HF byproduct does not inhibit amine nucleophilicity (in contrast to HCl).[69] These features were exploited by Schafmeister and coworkers to prepare extremely hindered α,α,N-trialkyl dipeptides in hexafluoroisopropanol (HFIP), where the absence of base prevents formation of undesired ester side products (Scheme 13c).[70]
Scheme 13.
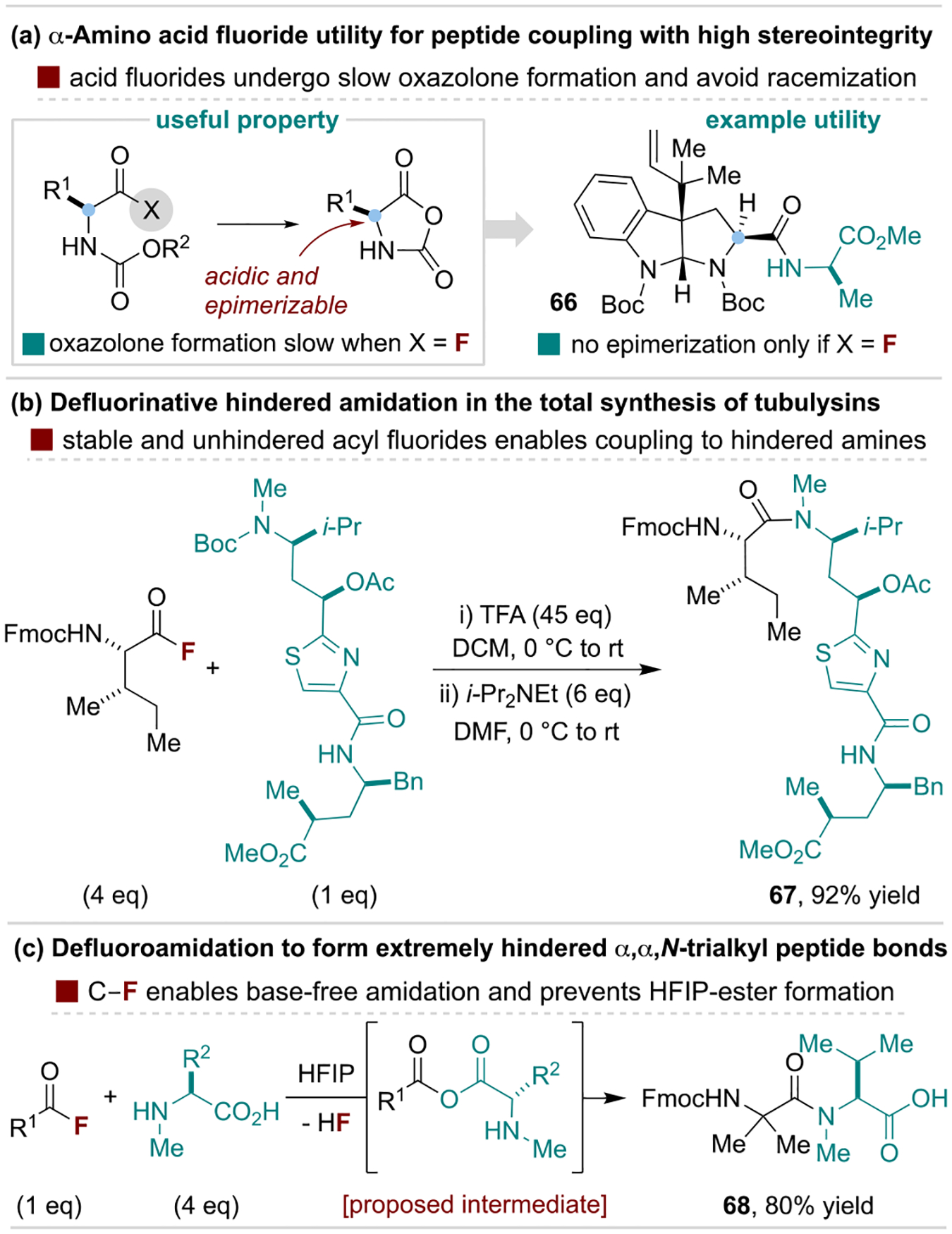
Representative utility of acyl fluorides in peptide coupling reactions. TFA, trifluoroacetic acid.
3.3. Defluorinative acyl cross-coupling
The availability of acyl fluorides makes them attractive, bench-stable electrophiles for acyl cross-coupling reactions (Scheme 14a, top).[71] In 2004, Rovis and coworkers disclosed the first use of acyl fluorides in this regard for Ni-catalyzed Negishi coupling reactions as a catalytic alternative to organolithium or -magnesium addition reactions to activated acids (e.g., Weinreb amides) (Scheme 14a, middle).[72] Importantly, base-sensitive functional groups (e.g., β-alkoxy ketones) are well-tolerated and epimerizable α-stereocenters retain high stereopurity. Weix and coworkers expanded such defluorinative coupling reactions in 2020 with a Ni-catalyzed reductive process using activated amine coupling partners (Scheme 14a, bottom).[73] This deaminative protocol affords dialkyl ketone products from carboxylic acid and amine precursors, as opposed to the more traditional amide products formed through C–N coupling.[74] The authors proposed acyl fluorides are effective for this method as fluoride salt byproducts do not interfere with catalytic intermediates, unlike thioesters which produce thiolate anions that can poison metal catalysts.
Scheme 14.
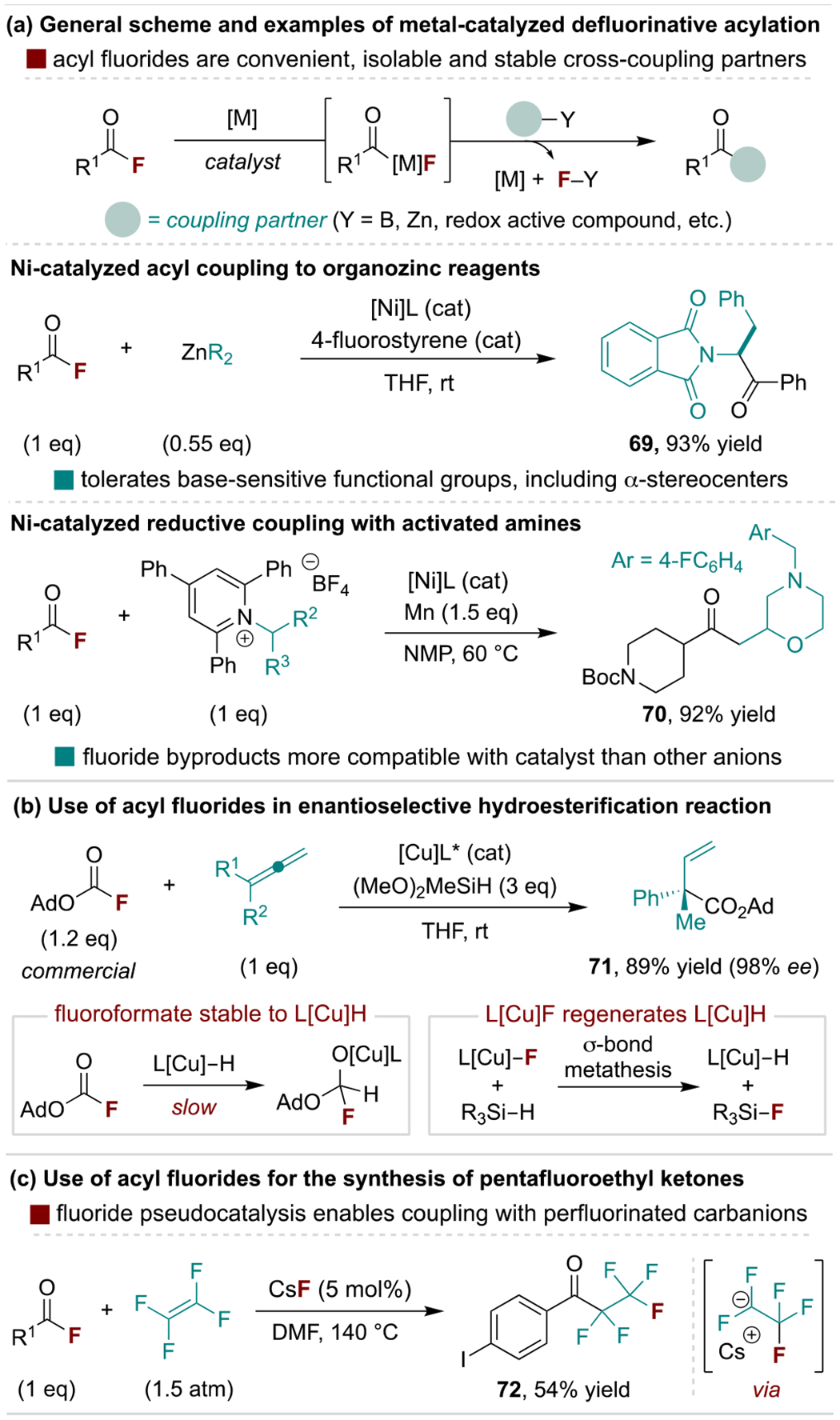
Representative use of acyl fluorides in C–C coupling reactions. Ad, adamantyl.
Acyl fluorides and their carbamyl and formyl derivatives have several features that enable metal-catalyzed alkene functionalization reactions. For example, Buchwald and coworkers developed a CuH-catalyzed enantioselective hydroesterification reaction of allenes using commercially available adamantyl fluoroformate (Scheme 14b).[75] Here, the use of the bulky fluoroformate allows for selective allene hydrocupration over carbonyl reduction to generate a nucleophilic allyl copper species. Following subsequent addition/fluoride elimination with the fluoroformate, a CuF intermediate can then directly react with the hydrosilane to regenerate the CuH catalyst.[76] Acyl fluorides are similarly useful for related Cu-catalyzed borylacylation reactions and in Ni-catalyzed carbocarbamoylation processes where the chloride analogues are prone to undesired decarbonylation or reduction.[77] A separate application of acyl fluorides in coupling reactions is the use of pseudo-catalytic fluoride anions to generate unstable nucleophilic coupling partners in situ, as demonstrated in a pentafluoroethylation process reported by Ogoshi and coworkers (Scheme 14c).[78]
3.4. Defluorinative synthesis of N-trifluoromethylamide derivatives
In 2019, Schoenebeck and coworkers developed the first general and modular synthetic approach to N-trifluoromethyl amides that relies on defluorinative acyl coupling.[79] This route proceeds via nucleophilic fluorination of isothiocyanates to form anionic N-trifluoromethyl intermediates, followed by reaction with triphosgene and an additional equivalent of fluoride to generate N-trifluoromethyl carbamoyl fluoride building blocks (Scheme 15, top). The authors demonstrated that nucleophilic defluorinative reactions of the carbamoyl fluorides can provide access to a diverse array of structurally complex N-trifluoromethyl amides, (thio)carbamates, ureas and formamides (Scheme 15, bottom).[80] These previously difficult-to-access structures are useful analogues of N-Me or N-H amides as the N-CF3 derivatives can have improved stability towards basic and oxidative conditions.
Scheme 15.
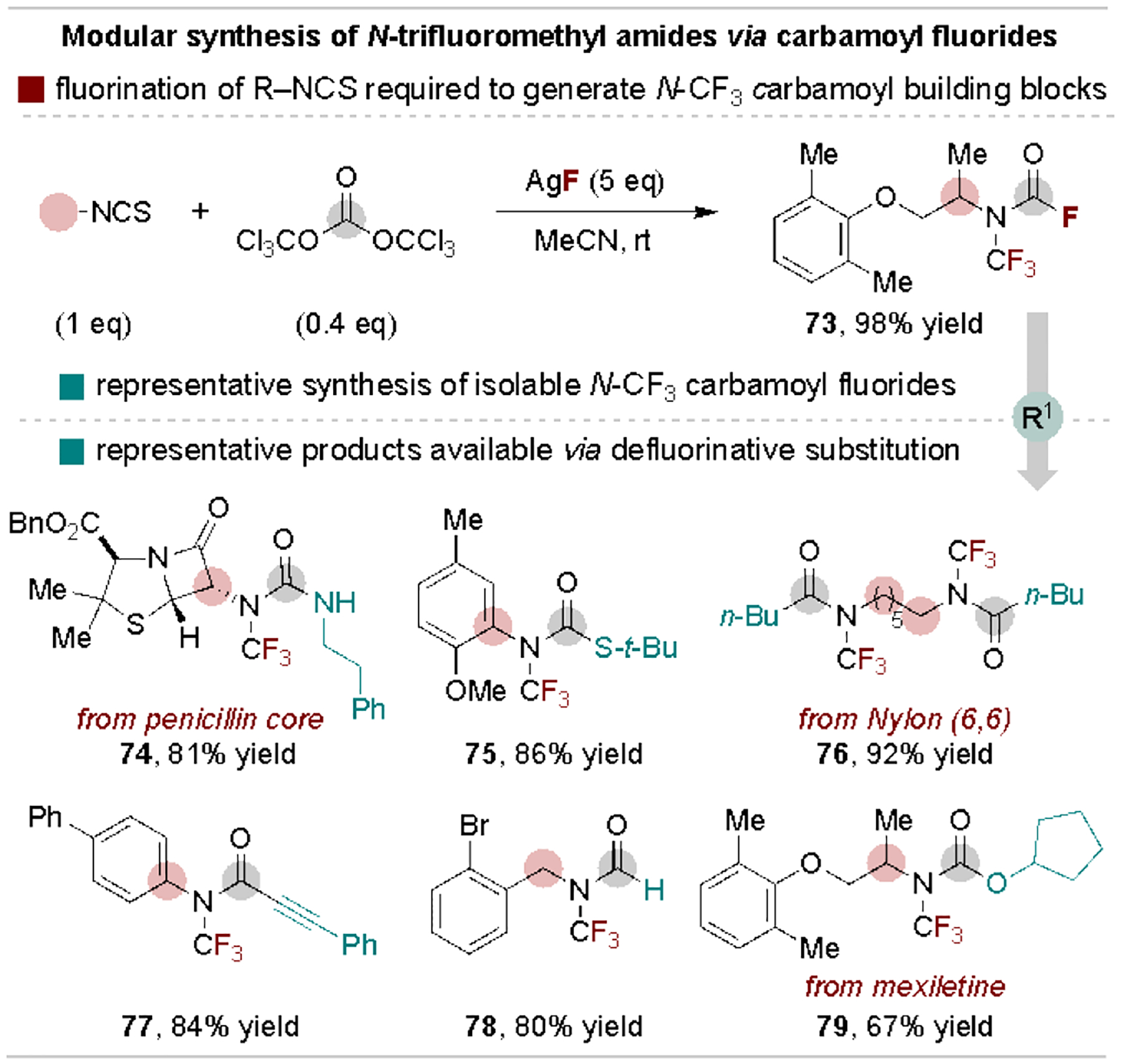
Defluorinative reactions to form N-trifluoromethylamide derivatives.
3.5. Acyl fluorides in organocatalytic reactions
Acyl fluorides have been strategically used in organocatalytic acylation methods where the released fluoride anion has influence over key intermediates. For example, Jacobsen and coworkers disclosed an enantioselective acylation of silyl ketene acetals using a dual catalytic system consisting of an achiral Lewis base and a chiral thiourea (Scheme 16a).[81] The high stereoinduction relies on strong H-bonding between the fluoride and chiral thiourea, which is ion paired with the electrophilic N-acyl ammonium electrophile during the key C–C bond-forming step. The fluoride anion also likely activates the silyl ketene acetal in this process, as both the yield and enantioselectivity are lower when acyl chloride and anhydride electrophiles are used. Acyl fluorides are also widely employed as isolable activated acid derivatives for NHC-catalyzed methods, both for asymmetric and non-asymmetric reactions.[82] Here, we illustrate a recent application where the fluorine from the acyl fluoride is incorporated into the product via a dual photoredox/NHC-catalyzed fluoroacylation reaction developed by Studer and coworkers (Scheme 16b).[83] A related transformation, developed by Tobisu and coworkers, uses phosphorous Lewis bases to catalyze alkynoate fluoroacylation that is proposed to operate via a P(III)/P(V) redox cycling pathway (Scheme 16c).[84]
Scheme 16.
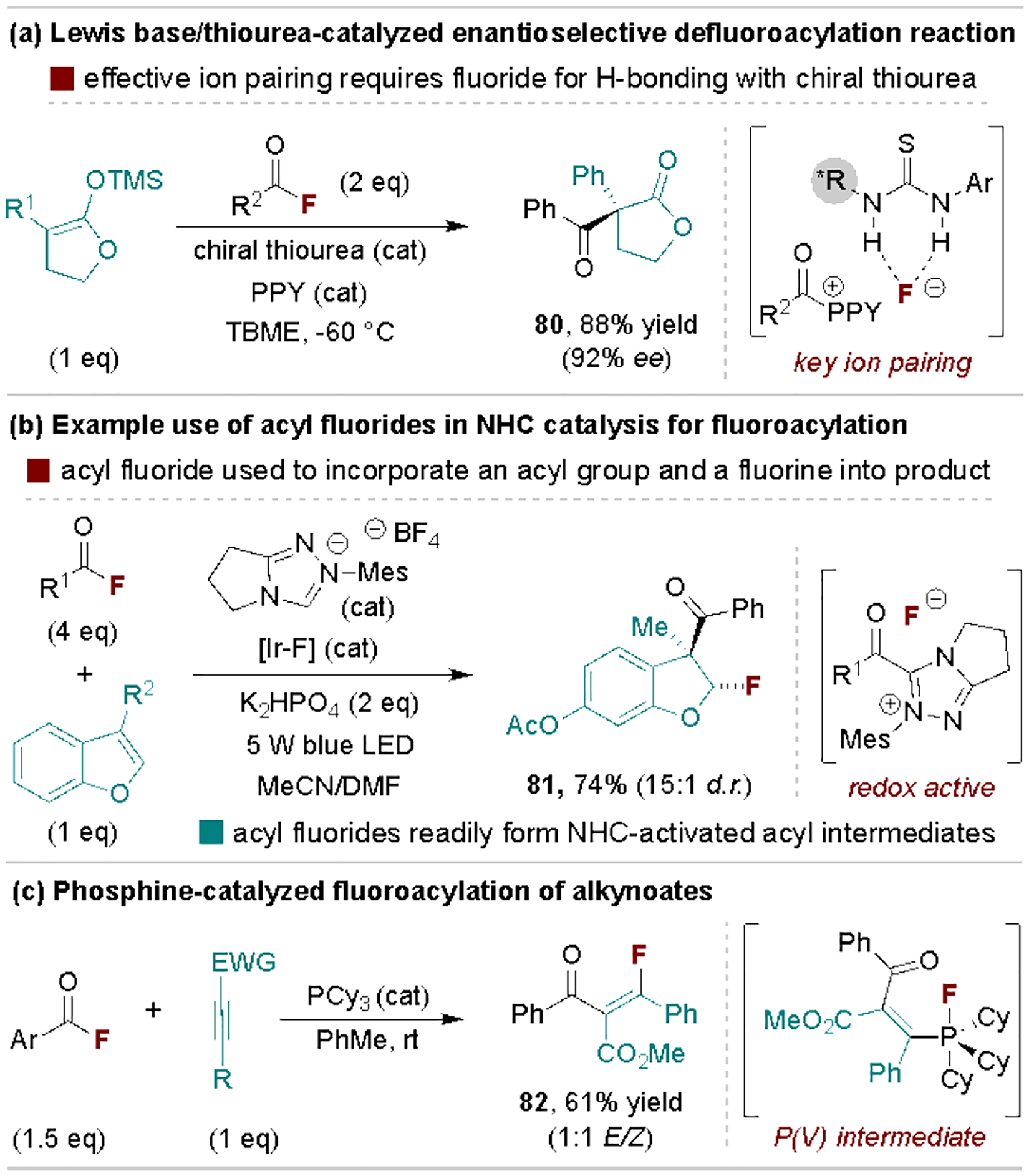
Representative defluorinative acylation reactions in organocatalysis. TBME, tert-butyl methyl ether; PPY, 4-pyrrolidinopyridine.
3.6. Defluorinative and decarbonylative coupling reactions
Carboxylic acid functional groups are attractive synthetic handles as they are abundant in both commercial reagents and complex molecules.[85] As such, their activation to acid fluorides is emerging as an advantageous approach in metal-catalyzed tandem defluorination/decarbonylation processes for formal decarboxylative coupling reactions.[86] Here, C–F activation via oxidative addition generates an acyl metal species that contains a fluoride that can directly engage in downstream mechanistic steps to enable unique coupling reactions (Scheme 17a). Ogiwara and Sakai recently published a comprehensive review on acyl fluorides in late transition metal-catalysis.[71] Here, we highlight applications that are critically reliant on the use of acyl fluorides over alternative acyl electrophiles.
Scheme 17.
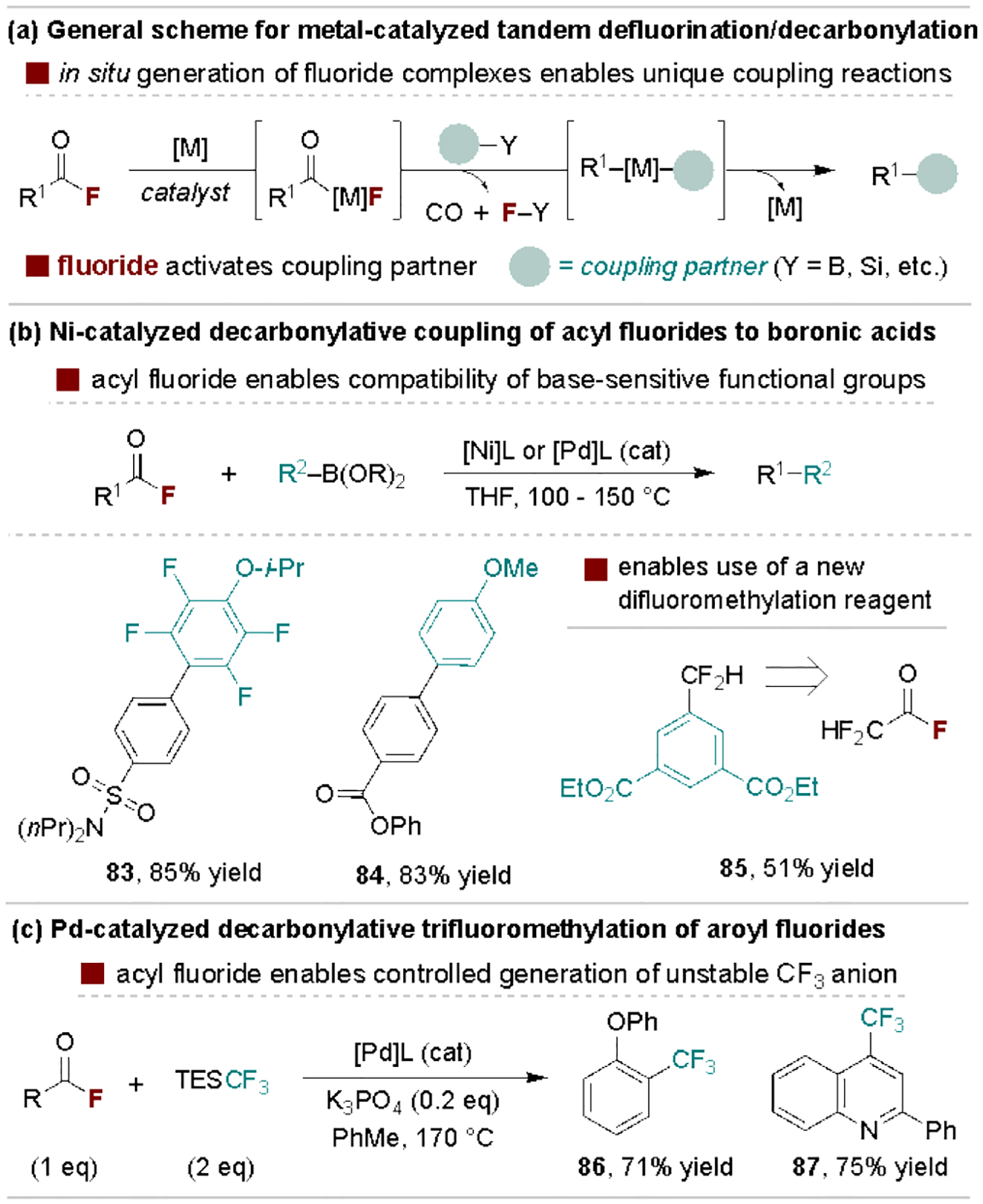
Representative applications of tandem defluorinative/decarbonylative coupling reactions.
Sanford and coworkers reported a key advance in 2018 wherein acyl fluorides allow for Ni-catalyzed decarbonylative boronic acid coupling reactions in the absence of added base (Scheme 17b).[87] Here, the boronic acid undergoes transmetalation with the ArNiF intermediate, enabling access to functionalized biaryl compounds that are challenging to prepare using traditional Suzuki coupling reactions (e.g., those containing ketones, aryl halides or polyfluoroaryl groups), which require stoichiometric strong base. The Sanford Group exploited a similar mechanism for the use of difluoroacetyl fluoride as a difluoromethylation reagent for aryl boronates (85), providing an attractive surrogate to Si- and Ag-based fluoroalkylation partners.[88] Alternatively, Schoenebeck and coworkers in 2018 disclosed a related method that uses Si-based fluoroalkylation reagents, wherein acyl defluorination enables a precisely tuned rate of fluoride activation of TES‒CF3 (TES = triethylsilyl) to transfer unstable “CF3” anions as needed (Scheme 17c).[89] Representative additional uses of the in situ generated fluoride include activation of diboron reagents and hydrosilanes, as well as aryl C–H activation for a variety of decarbonylative reactions.[90]
4. Vinyl Fluoride Defluorofunctionalization
This section highlights synthetic uses of monofluoroalkene and 1,1-difluoroalkene defluorofunctionalization. In general, internal monofluoroalkenes are more challenging to prepare than 1,1-difluoroalkenes and find relatively less use in advantageous defluorinative synthesis.[91] In fact, one of the most attractive routes to monofluoroalkenes is through defluorinative coupling reactions of 1,1-difluoroalkenes.[92] Monofluoroalkenes are also accessible through alkyne hydrofluorination[93] or metal-catalyzed vinyl halide fluorination protocols.[94] 1,1-Difluoroalkenes are readily prepared via difluoroolefination of carbonyls, as well as addition/fluoride elimination reactions of trifluoromethyl-substituted alkenes.[9a,92] Therefore, this section highlights uses of specific classes of abundantly available monofluoroalkenes and representative methods for monodefluorinative reactions of vinyl difluorides.
4.1. Monofluoroalkene defluorofunctionalization
Hydrofluoroolefins (HFOs) are heavily fluorinated alkenes used as environmentally-friendly refrigerants, making them abundantly available precursors to more complex or versatile fluorinated building blocks.[95] These and related vinyl fluorides undergo regioselective addition of nucleophilic species to generate anionic or organometallic intermediates prone to fluoride elimination (Scheme 18a). Crimmin and coworkers developed silyl-lithium and -magnesium reagents for such processes on HFOs to produce fluorinated vinylsilanes (Scheme 18b, top).[96] An alternative Cu-catalyzed variant was reported by Ogoshi and coworkers wherein the released fluoride aids in activation of a Bpin–SiMe2Ph coupling partner (Scheme 18b, bottom).[97] Gem-difluoroalkene units are also represented in HFOs and undergo similar transformations, including substitution with O- and N-based nucleophiles to access fluorinated vinyl ethers and amines.[98] Metal-catalyzed cross-coupling reactions of vinyl fluorides are well developed, although such protocols typically possess similar reactivity to other common vinyl (pseudo)halides.[99] A unique mechanistic pathway available to fluoroalkenes is their activation via strong Lewis acids to generate vinyl electrophile equivalents for reactions that do not map onto the aforementioned substitution processes. For example, Nishimoto and Yasuda employed α-fluorostyrene derivatives as electrophiles to couple with fluoride-activated silyl enol ether pronucleophiles (Scheme 18c).[100]
Scheme 18.
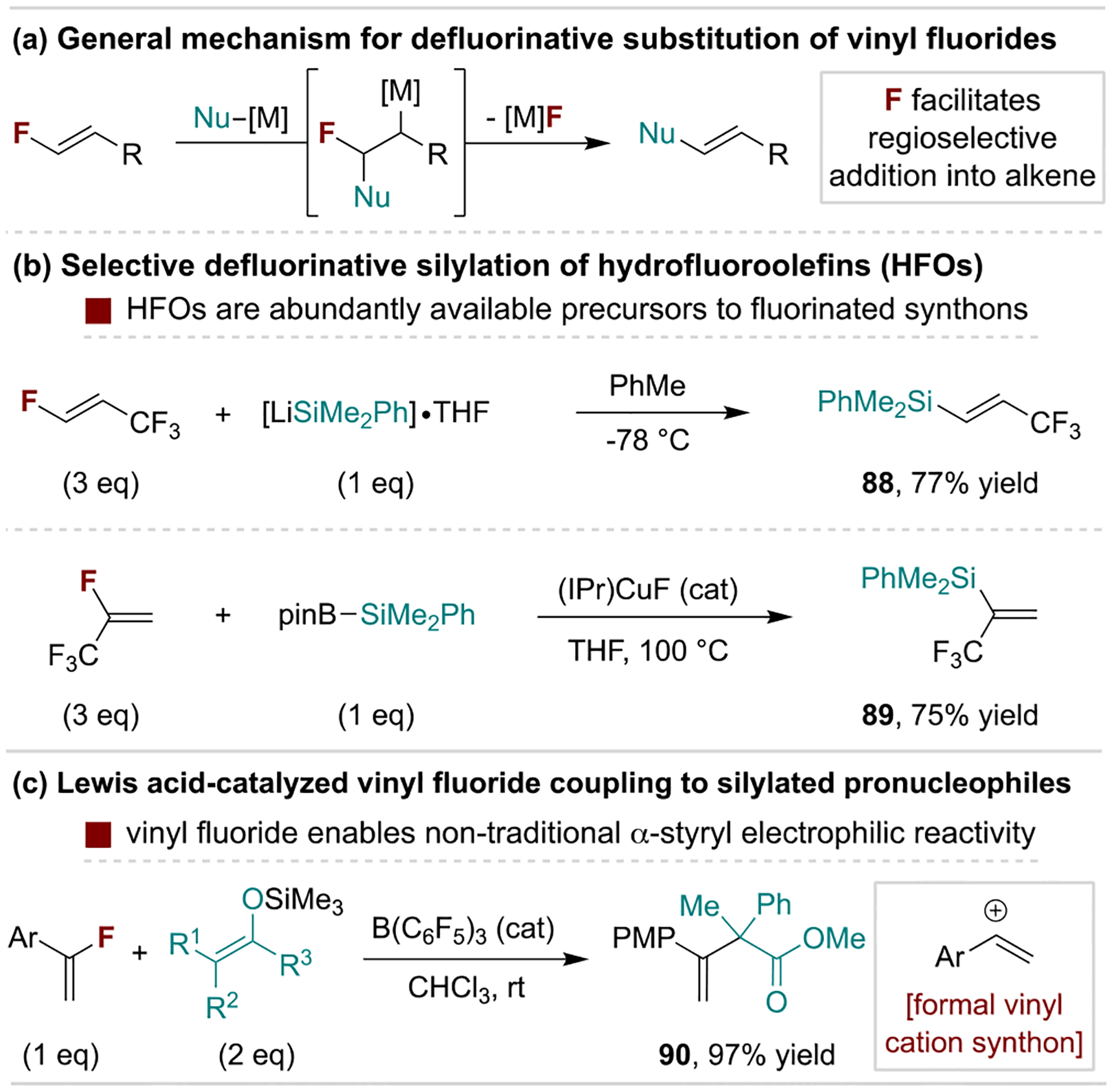
Example uses of monofluoroalkene defluorofunctionalization. IPr, 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene).
4.2. Gem-difluoroalkene general properties
Gem-difluoroalkenes are highly susceptible to addition reactions at the fluorinated carbon with nucleophilic and radical species, often resulting in fluoride elimination. The dramatic increase in reactivity compared to nonfluorinated alkenes is attributed to the following properties: (1) fluorine activates the π-bond via inductive effects; (2) fluorine’s lone pairs have repulsive interactions with the alkene’s π-electrons; (3) C–F anti-bonding orbitals stabilize β-carbanions through negative hyperconjugation; and (4) fluoride can serve as a leaving group.[9a,92] Therefore, addition/elimination sequences on gem-difluoroalkenes are versatile and well-developed routes to monofluoroalkenes. Such products are valuable as vinyl fluorides can act as peptide bioisosteres by mimicking an amide’s charge distribution and dipole moment while being less prone to hydrolysis.[101]
4.3. Nucleophilic gem-difluoroalkene defluorinative reactions
The substantial increase in electrophilicity of gem-difluoroalkenes has been used extensively in simple anionic fluoride substitution reactions (Scheme 19a). These include the addition of relatively weak nucleophiles into β,β-difluorostyrene derivatives[102] and stronger nucleophiles that can also add to alkyl-substituted 1,1-difluoroalkenes.[103] The utility of such reactivity is exemplified by straightforward routes to monofluoroalkenes via addition of Grignard and organolithium reagents to β,β-difluorostyrene derivatives and 1,1-difluoroalkenes, respectively (Scheme 19b).[104] The polarization of gem-difluoroalkenes also promotes nontraditional regioselectivity for cyclization reactions, as evidenced by 5-endo-trig processes that produce diverse fluorinated heterocycles (Scheme 19c).[105] Similar addition/elimination reactions occur with monofluoroalkenes, although additional activation by electron-deficient substituents is required.[106]
Scheme 19.
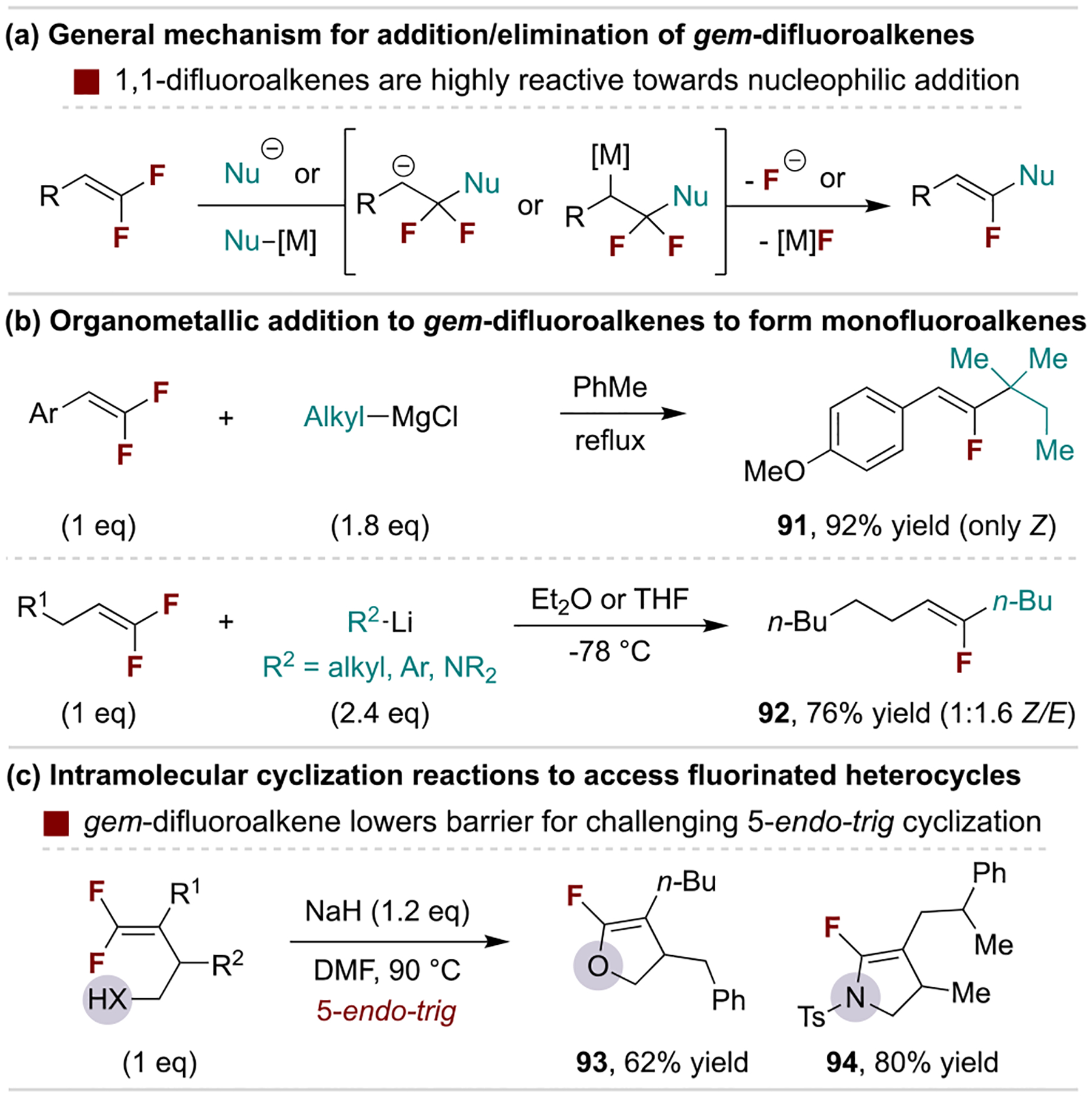
Example defluorinative substitution of gem-difluoroalkenes.
4.4. Gem-difluoroalkene defluoroborylation and -silylation
Metal-catalyzed defluoroborylation and -silylation methods provide access to synthetically useful B- and Si-substituted fluoroalkenes. These reactions proceed via boryl- and silylmetalation followed by β-fluoride elimination and are amenable to β,β-difluorostyrene derivatives and 1,1-difluoroalkenes (Scheme 20a).[107] This methodology was recently used by chemists at Janssen to make a (Z)-borylated fluoroalkene (97) as a key intermediate in the synthesis of fluoroalkene-containing anticancer drug candidates (Scheme 20b).[108] An advantage of this route is that the gem-difluoroalkene starting material is accessed via fluoride elimination of the readily available corresponding trifluoromethyl alkane. In an alternative application, Shi and coworkers developed a tandem defluoroborylation/protodeborylation protocol as an attractive route to terminal (Z)-fluoroalkenes (Scheme 20c).[109] A related defluorocarboxylation process with CO2 has also been developed.[110]
Scheme 20.
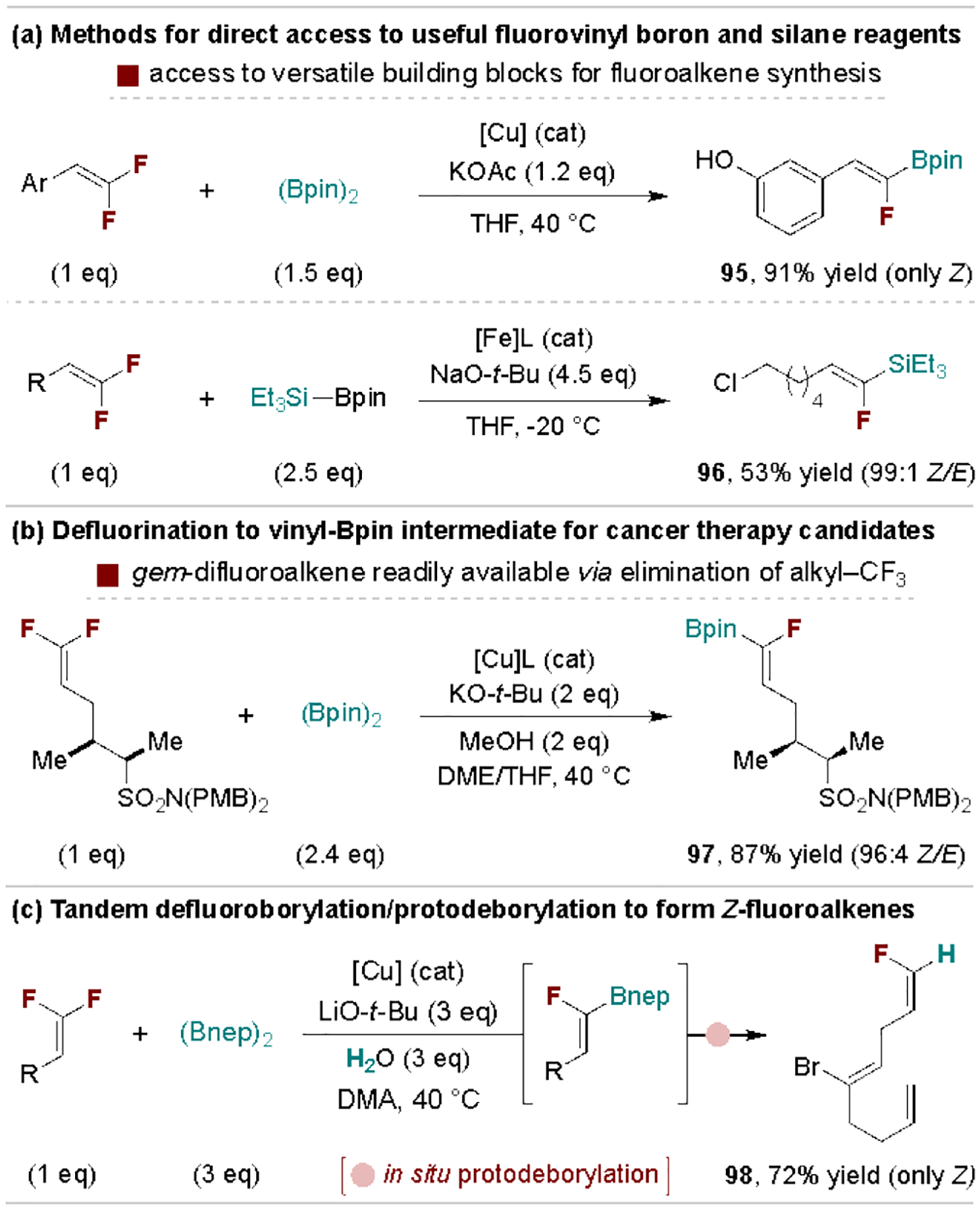
Representative access to and uses of boryl- and silylfluoroalkenes. PMB, para-methoxybenzyl nep, neopentylglycolato.
4.5. Gem-difluoroalkene catalytic defluorinative C–C coupling reactions
In addition to direct reactions with Grignard and organolithium reagents, gem-difluoroalkenes (especially β,β-difluorostyrene derivatives) undergo many types of catalytic defluorinative C–C coupling reactions.[111] Ogoshi and coworkers reported an early example of this reactivity for the Pd-catalyzed coupling of arylzinc reagents to tetrafluoroethylene.[112] Subsequent work by the authors[113], and the Toste Group[114], led to base-free defluorinative coupling reactions with boronic acids as represented in Scheme 21a. Here, the ligated PdF byproduct propagates the reaction through direct transmetalation with the aryl boronic acid. This feature improves the tolerance of base-sensitive functional groups such as aldehydes, as shown in the synthesis of a precursor to an imatinib fluoroalkene analogue (99). Gong, Fu and coworkers reported a Ni-catalyzed reductive coupling method that expands defluorinative coupling to alkyl halide partners (Scheme 21b).[115] β,β-Difluorostyrene derivatives are also extensively used to intercept carbon-centered radical intermediates prior to reductive fluoride elimination (Scheme 21c, top). Two examples are provided in Scheme 21c, including alkyl radical generation via Zn-promoted reductive decarboxylation of redox-active esters and photoredox-promoted oxidative decarboxylation of α-heteroatom carboxylic acids.[116]
Scheme 21.
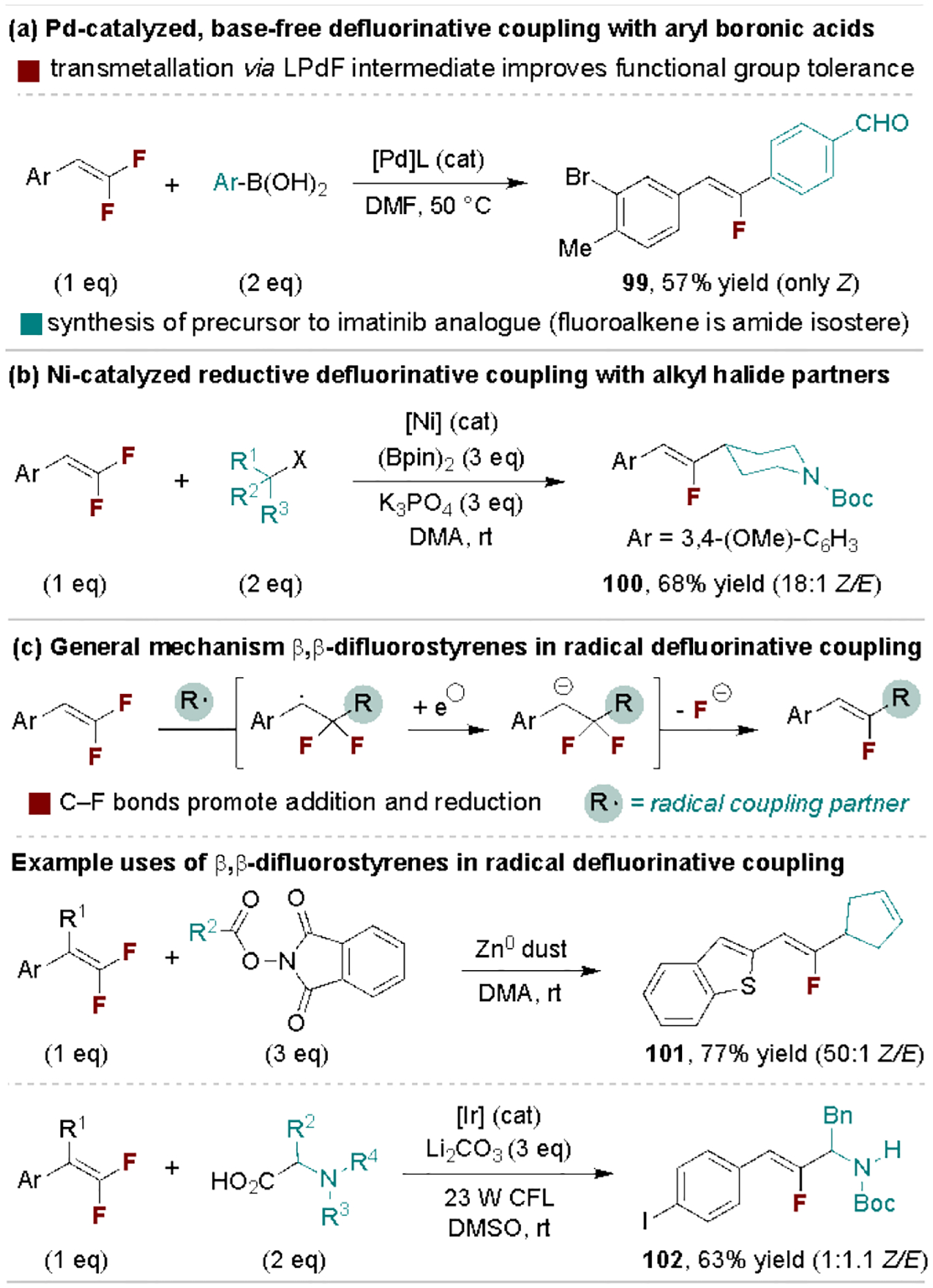
Catalytic defluorinative C–C coupling of β,β-difluorostyrenes. CFL, compact fluorescent lamp.
5. Alkyl Fluoride Defluorofunctionalization
Here, we highlight the advantages of defluorofunctionalization on unactivated aliphatic mono-, di- and trifluorides.[117] Given the high bond strength of aliphatic C–F bonds, synthetic applications via direct nucleophilic substitution, metal insertion and reductive activation approaches are relatively uncommon. Lewis acids, however, readily activate such C–F bonds, even in the presence of traditionally more electrophilic functional groups.[13] This can be useful as alkyl fluorides are easily synthesized via deoxyfluorination of the corresponding alcohol or carbonyl to access monofluorides and difluorides, respectively.[118] Monofluorides can also be made via fluoride substitution reactions and difluorides via nucleophilic addition into difluorocarbene equivalents.[2,119] In contrast, alkyl trifluorides require more specialized syntheses using trifluoromethyl reagents (e.g., TMSCF3), unless the -CF3 group is already incorporated into a simple commercial starting material.[120]
5.1. Alkyl monofluoride defluorination
Aliphatic fluorides undergo Lewis acid-promoted substitution reactions through either a direct process or carbocation intermediate, depending on the substitution pattern of the carbon backbone (Scheme 22a). However, in the absence of Lewis acids, alkyl fluorides possess relatively low electrophilicity, allowing C–F bonds to be carried through multistep syntheses prior to being converted into more reactive C‒X bonds at a later stage. This process critically relies on Lewis acidic activation of the C–F bond, which can be accomplished using aluminum halides or lithium iodide (Scheme 22b).[121] Hilmersson and coworkers further exploited a Lewis acidic strategy with a La-silylamide salt to achieve selective amination of alkyl fluorides in the presence of traditionally more reactive aldehyde and alkyl chloride groups (Scheme 22c).[122] Martin and coworkers developed a related LiHMDS-promoted defluorosilylation protocol that formally converts the electrophilic alkyl fluorides into nucleophilic alkyl silanes.[24a] The Lewis basicity of alkyl fluorides was also found to enable their direct reaction with phenylmagnesium chloride, where Mg is proposed to function as a Lewis acid to activate the C‒F bond without the need for a metal catalyst (Scheme 22d).[123] Alternatively, transition metal catalysis can be used to expand the range of C‒F bond coupling reactions to include alkyl Grignard and other organometallic reagents.[124] For example, Iwasakai, Kambe and coworkers demonstrated an alkyl fluoride can be carried through a multistep synthesis prior to Co-catalyzed alkylation (Scheme 22e).[125]
Scheme 22.
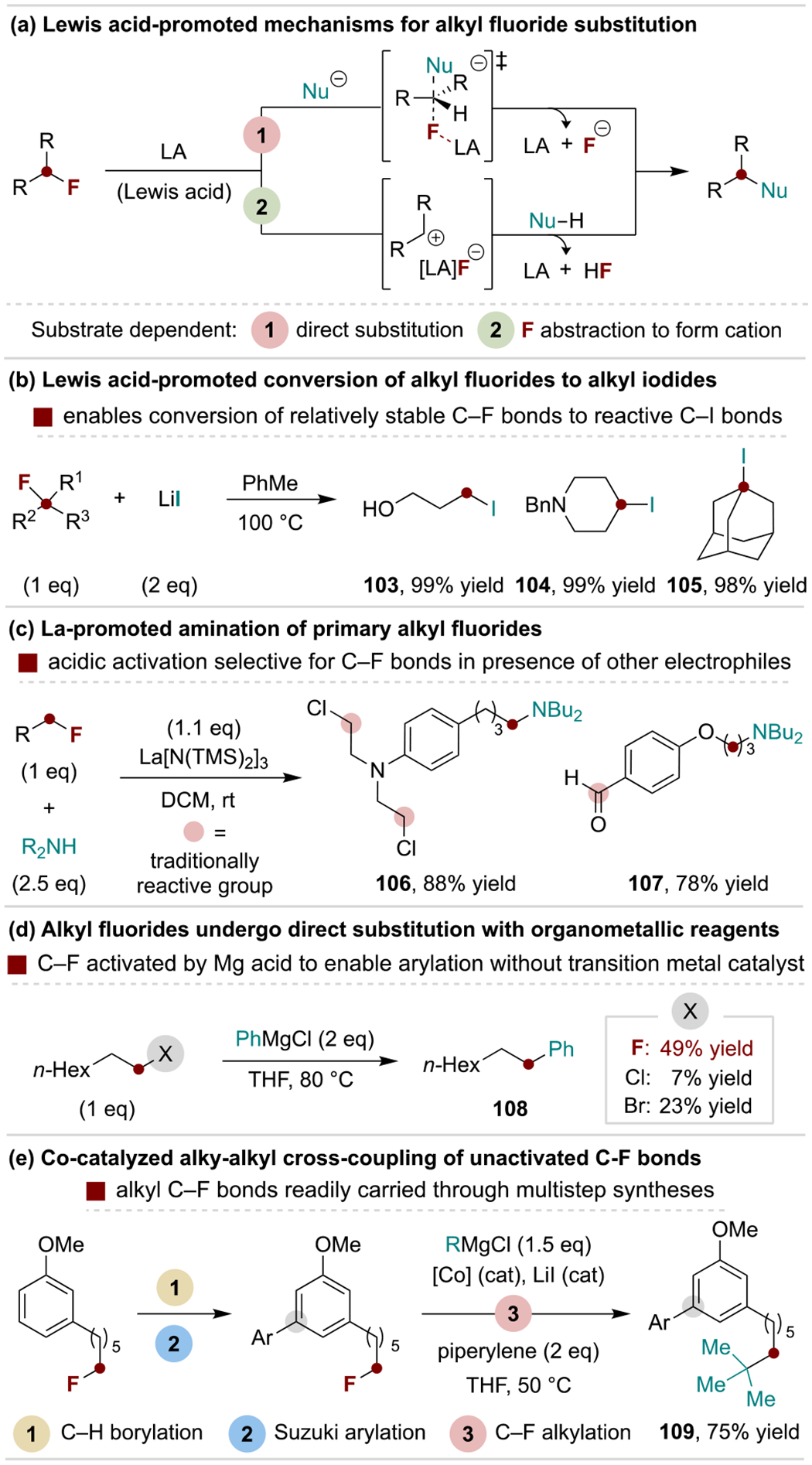
Representative examples for substitution of alkyl monofluorides.
The preceding methods rely on activation of the C–F bond for coupling to relatively strong nucleophiles, whereas alkyl fluorides that lead to stable carbocations (e.g., tertiary alkyl fluorides) can be activated via Lewis acids for substitution with weaker (pro)nucleophiles.[13] To complement protocols that rely on stoichiometric Lewis acid activators, Moran and coworkers developed B(C6F5)3•H2O-catalyzed coupling reactions of tertiary alkyl fluorides with a wide range of (pro)nucleophiles (Scheme 23a).[126] This reaction is selective for alkyl fluorides and is proposed to proceed through autocatalysis with in situ generated HF. An elegant application of this method was reported by Shibata and coworkers for the synthesis of spirocyclic motifs via double C‒F bond arylation (114).[127] In these processes, the released fluoride can also activate other pronucleophiles, such as silylated alkynes in a reaction reported by Young and coworkers (Scheme 23b).[128]
Scheme 23.
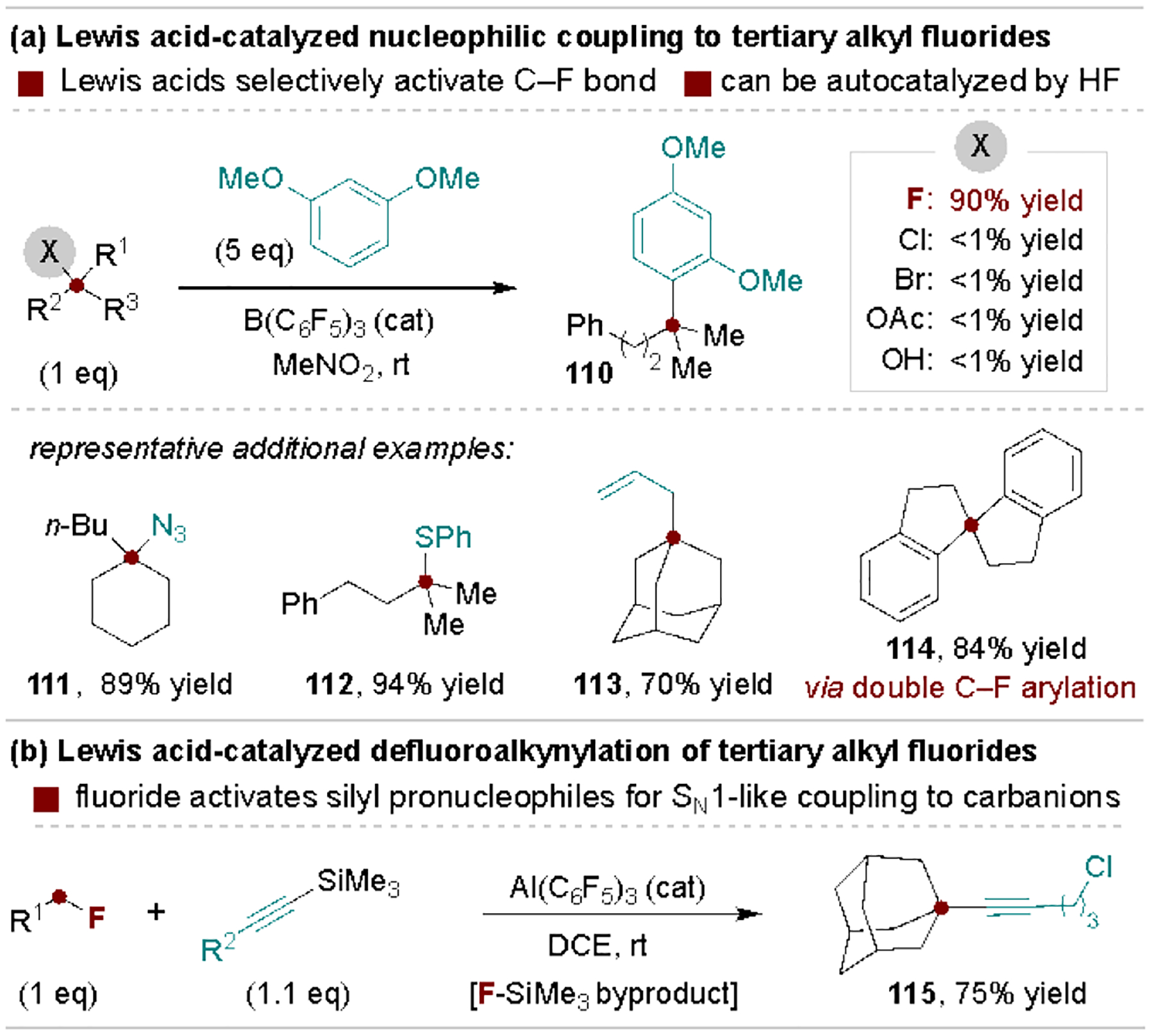
Examples of Lewis acid-promoted substitution of alkyl fluorides.
5.2. Monoselective activation of alkyl polyfluorides
Young and coworkers also pioneered a frustrated Lewis pair approach for installing diversifiable functional groups via defluorination of alkyl polyfluorides (Scheme 24).[129] This strategy is general to many classes of alkyl polyfluorides and installs versatile pyridinium or sulfonium groups for derivatization via nucleophilic, metal-catalyzed or radical-based coupling protocols. This method is powerful due to its ability to selectively activate a single C–F bond, as further fluoride removal from the cationic complex is prohibitively difficult.
Scheme 24.
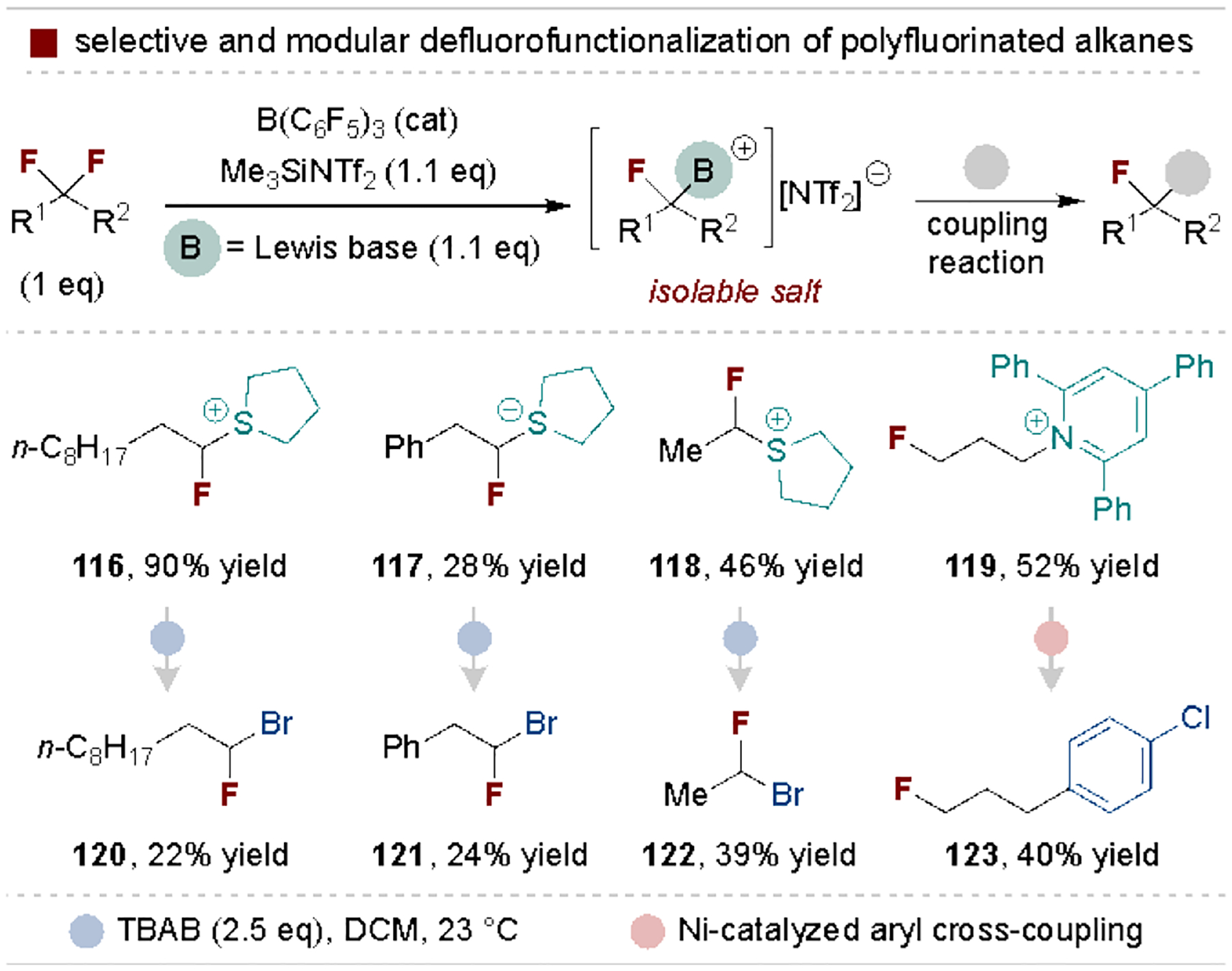
Selective defluorofunctionalization of alkyl polyfluorides via frustrated Lewis pair chemistry. Tf, triflyl.
5.3. Alkyl trifluoride defluorofunctionalization
Unactivated alkyl trifluorides with β-C–H bonds undergo elimination with strong bases (e.g., KHMDS)[108] to generate valuable gem-difluoroalkenes. Additionally, if the -CF3 group is adjacent to a relatively acidic proton, tandem elimination and addition processes allow for controlled substitutions of C–F bonds. This cascade process can be exploited in a variety of contexts such as net-defluoroalkylation of β-trifluoromethyl amides[130] and in the synthesis of fluorinated heteroarenes (Scheme 25a).[131] An additional noteworthy elimination protocol is the Zn-promoted reduction of α-bromotrifluoromethylalkanes, as reported by He and coworkers.[132] In this reaction, liquid 2-bromo-3,3,3-trifluoromethylpropene functions as a surrogate for gaseous 3,3,3-trifluoropropene in radical defluoroalkylation reactions (Scheme 25b).[133]
Scheme 25.
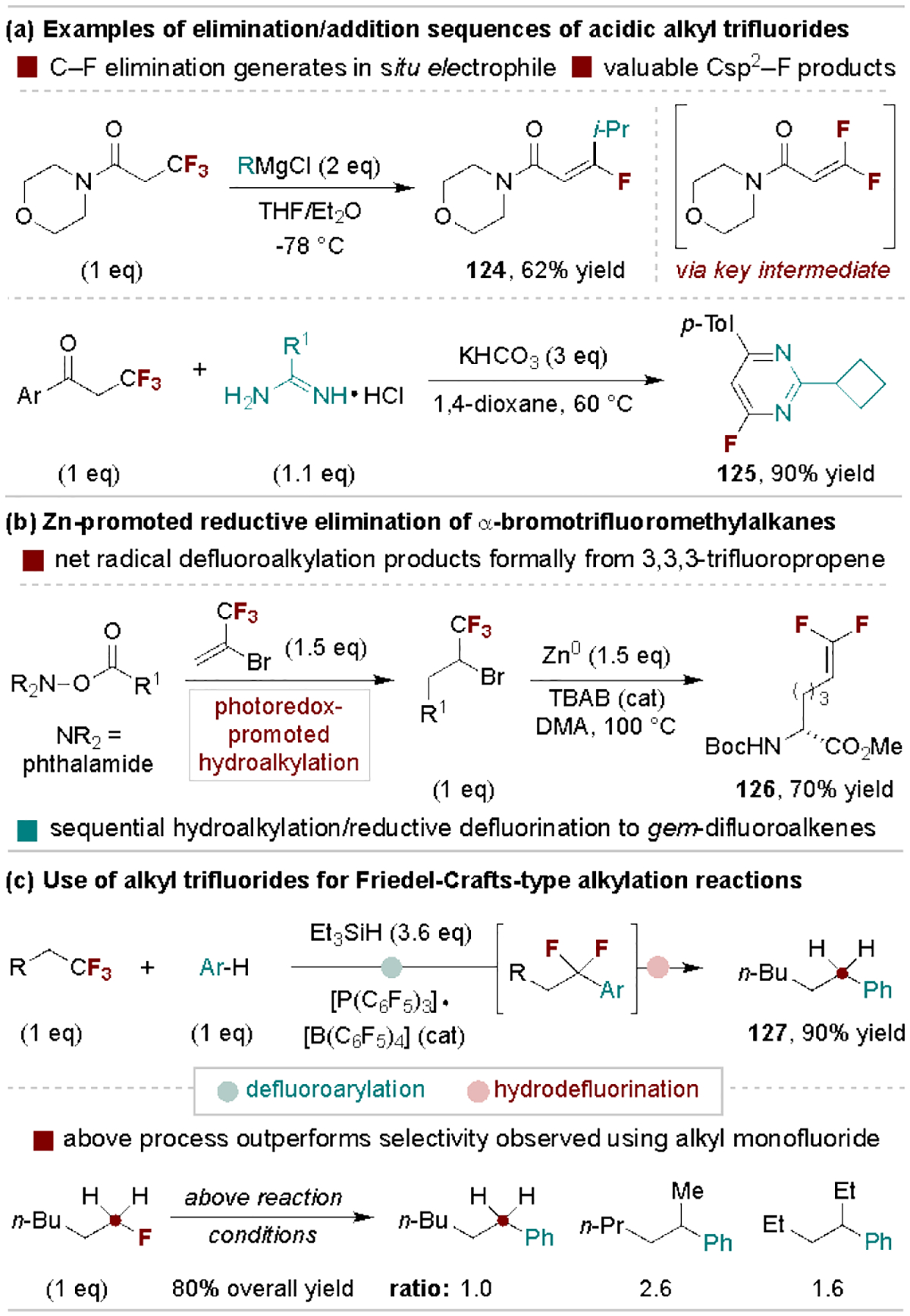
Representative defluorofunctionalization of aliphatic trifluorides.
Another distinct application of alkyl trifluorides is their use as effective surrogates for primary electrophiles in Friedel-Crafts reactions as shown in Scheme 25c.[134] In this process, developed by Stephan and coworkers, the trifluoromethyl group functions as a highly selective primary electrophile for arylation via a difluoro-substituted carbocation, followed by tandem reduction of the difluorobenzylic intermediate with triethylsilane. The authors found this process provides excellent linear selectivity as compared to the use of terminal alkyl monofluoride electrophiles.
6. Benzyl Fluoride Defluorofunctionalization
Benzyl monofluorides are easily accessible from the corresponding benzyl alcohol or halide via nucleophilic fluorination, or through C–H fluorination methods from the corresponding alkylarene.[135] In general, benzyl fluorides are less electrophilic than more common benzylic electrophiles, which makes defluorofunctionalization synthetically advantageous only in certain contexts. In contrast, benzyl trifluorides (also described as trifluoromethylarenes or ArCF3) are abundantly available from commercial sources and are privileged in bioactive compounds.[3a,b] Non-commercial perfluoroalkyl arenes can also be prepared via cross-coupling reactions, radical perfluoroalkylation, oxidative fluorination of the methylarene or Cu-mediated Sandmeyer trifluoromethylation.[135,136] Due to their prevalence and availability, monoselective defluorofunctionalization of trifluoromethyl- and perfluoroalkylarenes is an attractive synthetic route to fluorinated benzylic substructures.[137] Here, we highlight the main mechanistic strategies that have emerged to enable the use of benzylic C‒F bonds as synthetic handles.[138]
6.1. Benzyl monofluoride defluorofunctionalization
Benzyl C–F bonds can be activated via Lewis and Brønsted acid catalysts and provide complementary reactivity to other benzyl halides. Their fundamental reactivity in this regard for coupling with heteroatom and carbon-based nucleophiles has been studied in depth by the Paquin Group and others.[139] Scheme 26a highlights the utility of such reactivity within the context of Stahl and coworkers’ protocol for Cu-catalyzed, NFSI-mediated benzylic C–H fluorination.[140] Here, C–H fluorination serves as a gateway for benzylic coupling to a broad range of nucleophiles, even in the presence of electrophilic alkyl bromides and chlorides. Preparation of benzyl fluoride starting materials is also worthwhile when they are used to generate more valuable fluorinated products, as illustrated by a Lewis acid-catalyzed carbene insertion protocol reported by Nishimoto, Yasuda and coworkers (Scheme 26b).[141]
Scheme 26.
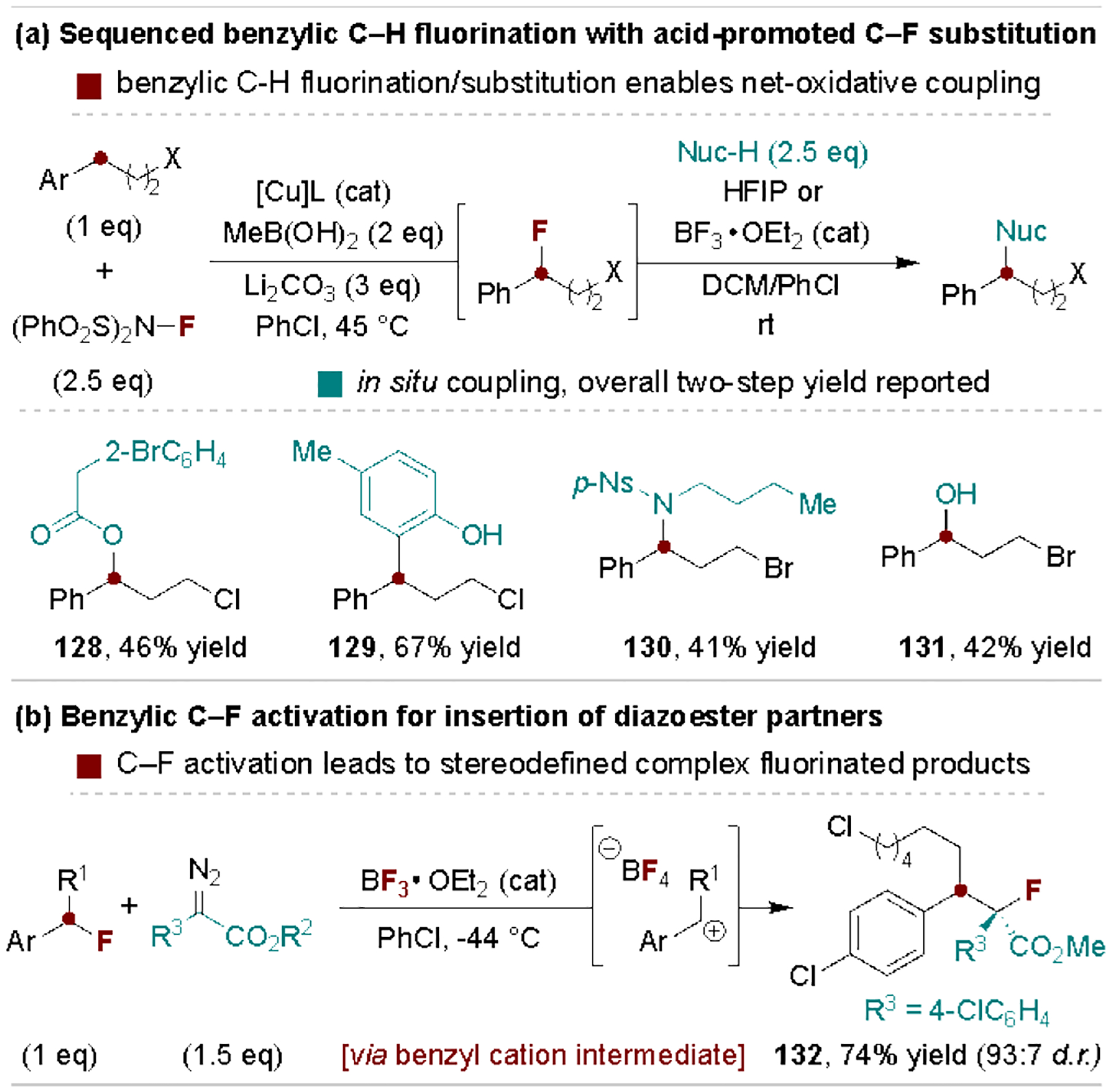
Example utility of benzylic monofluoride defluorofunctionalization.
6.2. Selective defluorofunctionalization of benzylic polyfluorides
Here, we discuss the utility and recent advances for the monoselective C–F bond substitution of benzylic polyfluorides, including difluoroalkyl, trifluoromethyl and perfluoroalkyl derivatives. The advantages of these starting materials stem from their abundance and availability relative to prefunctionalized motifs (e.g., ArCF2–Br). The resulting products, especially α,α-difluorobenzylic derivatives, are highly sought after in medicinal chemistry as benzylic fluorination can address metabolic stability concerns of analogous C–H bonds.[4b] This substructure also functions as a less oxidizable bioisostere of aryl ethers.[101b] Given the abundance of trifluoromethylarenes, their selective defluorofunctionalization could improve the economics of large-scale syntheses and enhance access to fluorinated libraries. It should also be noted that typically “inert” perfluoroalkyl groups can be carried through multistep syntheses and are frequently found in complex bioactive molecules, making their use in late-stage derivatization attractive. Despite these advantages, general protocols for monoselective defluorofunctionalization have only recently emerged due to the mechanistic difficulty associated with such processes.[137a] The central challenge is that C–F bond strengths of perfluoroalkyl arenes (e.g., PhCF3: 115 kcal/mol) decrease upon removal of each fluorine atom (e.g., PhCH2F: 95 kcal/mol), thus rendering it difficult to activate a C–F bond in the starting material while leaving a weaker C–F bond in the product intact.[142]
In 1989, monoselective trifluoromethylarene defluorofunctionalization was achieved electrochemically by Troupel and coworkers.[143] This process operates via reduction of the trifluoromethylarene to an aryl radical anion that undergoes mesolytic C–F cleavage to form a benzylic radical. This radical is further reduced to a difluorobenzylic carbanion that can be captured in situ with chlorosilane or carbonyl electrophiles (Scheme 27a, top).[144] This early work was highly influential as it illustrated that monoselective defluorofunctionalization could be achieved under single electron reducing conditions as the ArCF3 starting materials are easier to reduce than the ArCF2R products.[145] Recently, Lennox and coworkers developed an improved electrochemical method that expands the scope of hydrodefluorination to enable the direct conversion of bioactive and electron-rich trifluoromethylarenes to difluoromethyl variants (Scheme 27a, bottom).[146]
Scheme 27.
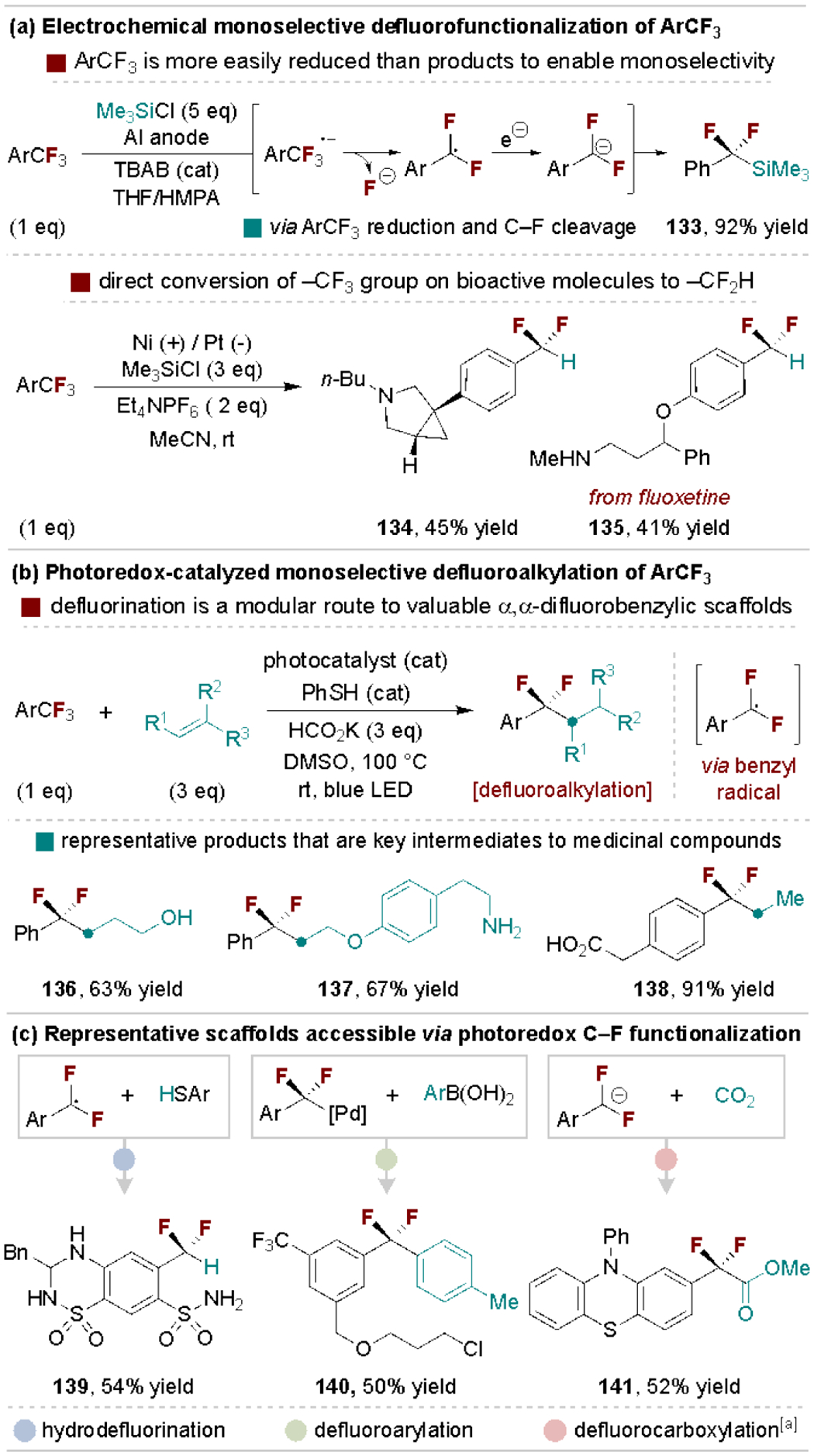
Representative electrochemical and photochemical ArCF3 defluorofunctionalization strategies and their products. HMPA, hexamethylphosphoramide. [a] Methyl ester isolated following treatment of reaction mixture with iodomethane.
In 2017 and 2018, the König[147] and Jui[148] Groups independently reported photoredox-catalyzed, monoselective defluoroalkylation of electron-deficient trifluoromethylarenes that also exploits a decrease in reduction potential upon C–F substitution. Here, ArCF3 reduction and C–F cleavage generate a difluorobenzylic radical that is intercepted by an alkene. These methods are represented in Scheme 27b by an improved protocol, developed by Jui and coworkers in 2019, that includes electron-neutral trifluoromethylarenes and facilitates rapid access to key fragments of medicinal compounds.[149] Following these pioneering reports, photopromoted processes have been expanded to include hydrodefluorination, defluoroarylation and defluorocarboxylation, as summarized in Scheme 27c.[150] It is likely that future work will continue to exploit benzylic C–F bonds as radical precursors for other coupling reactions.[151]
Lewis acid C–F bond activation strategies of polyfluoroalkylarenes typically lead to multiple C–F substitutions.[152] Yoshida, Hosoya and coworkers addressed this challenge through the generation of ortho-silylium ions on trifluoromethylarenes that abstract a single fluoride to generate versatile difluorobenzylic cation intermediates (Scheme 28a).[153] Young and coworkers disclosed a major breakthrough in 2018 through their use of frustrated Lewis pairs, previously discussed in Section 5.2[129], as a general defluorofunctionalization method of di- and trifluoromethylarenes to form pyridinium and phosphonium salts (Scheme 28b).[154] The obtained salts are versatile for diverse coupling methods as represented in Scheme 28b. This approach also enables 18F radiofluorination of di- and trifluoromethyl groups in bioactive polyfluoroalkylarenes for potential use in Positron Emission Tomography (PET) imaging (Scheme 28c).[155],[156]
Scheme 28.
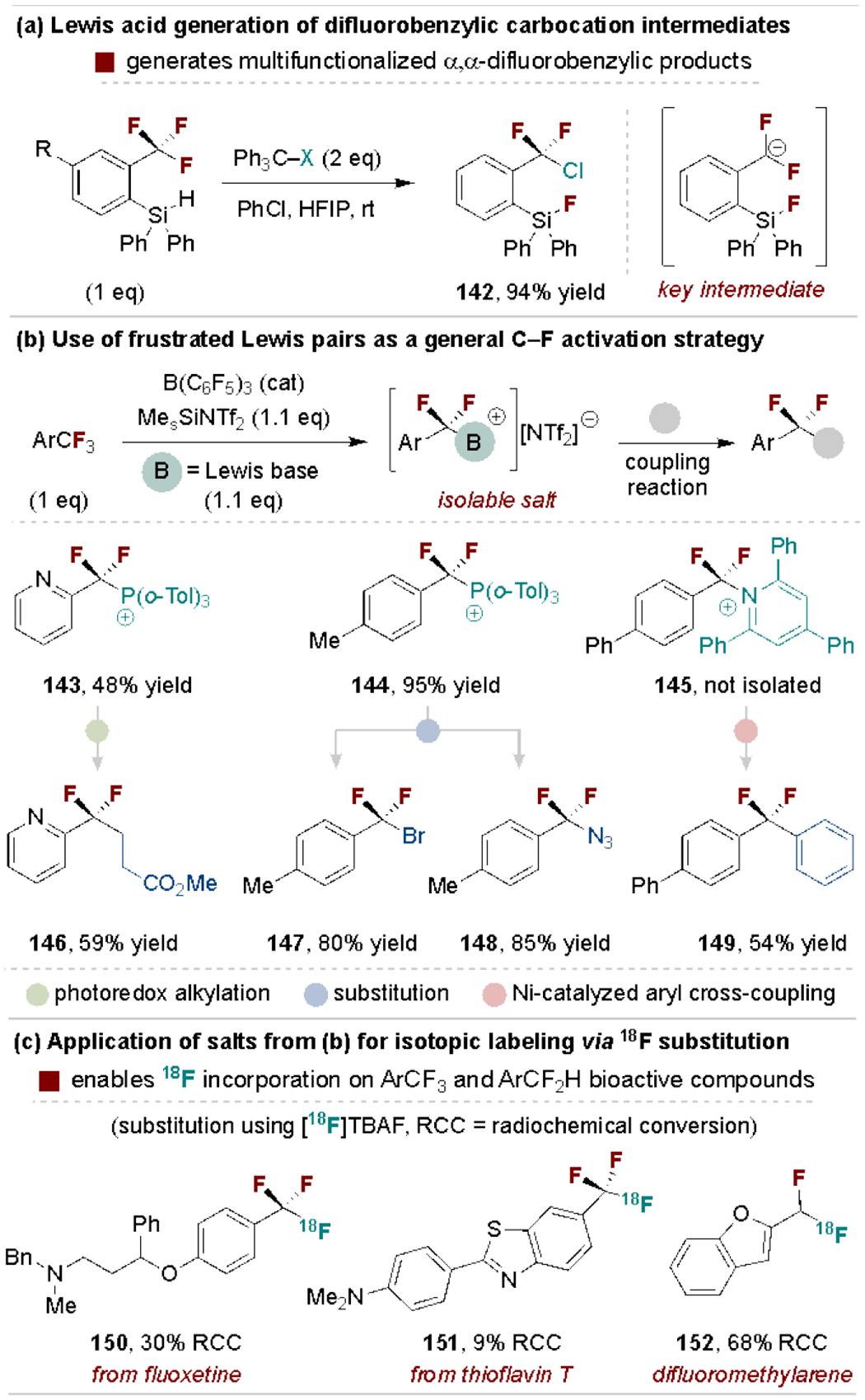
Lewis acid-mediated selective ArCF3 defluorofunctionalization. Tol, toluoyl, TBAF, tetrabutylammonium fluoride.
In 2019, our group discovered a fluoride-promoted defluoroallylation reaction of electron-deficient trifluoromethylarenes using allyltrimethylsilanes, which is proposed to proceed via single electron transfer (SET) from an anionic silicate intermediate to the ArCF3 (Scheme 29a).[157] The utility of this method was demonstrated through gram-scale reactions and derivatization of the allyl group into diverse difluoroalkyl substituents. Our group subsequently expanded the scope of accessible ArCF2R products via a Lewis base-promoted reductive coupling reaction with formamides (Scheme 29b).[158] This reaction is mediated by HSi(TMS)3 and proceeds via tandem defluorosilylation and nucleophilic addition to formamides to form synthetically versatile silylated hemiaminals. Jiao and coworkers developed an alternative protocol for defluoroallylation of CF3-substituted N-heteroarenes (Scheme 29c).[159] Here, the N-heteroarene and B2pin2 react to reduce a C–F bond, thus generating a difluorobenzylic anion that undergoes enantioselective allylation using a chiral Ir catalyst.
Scheme 29.
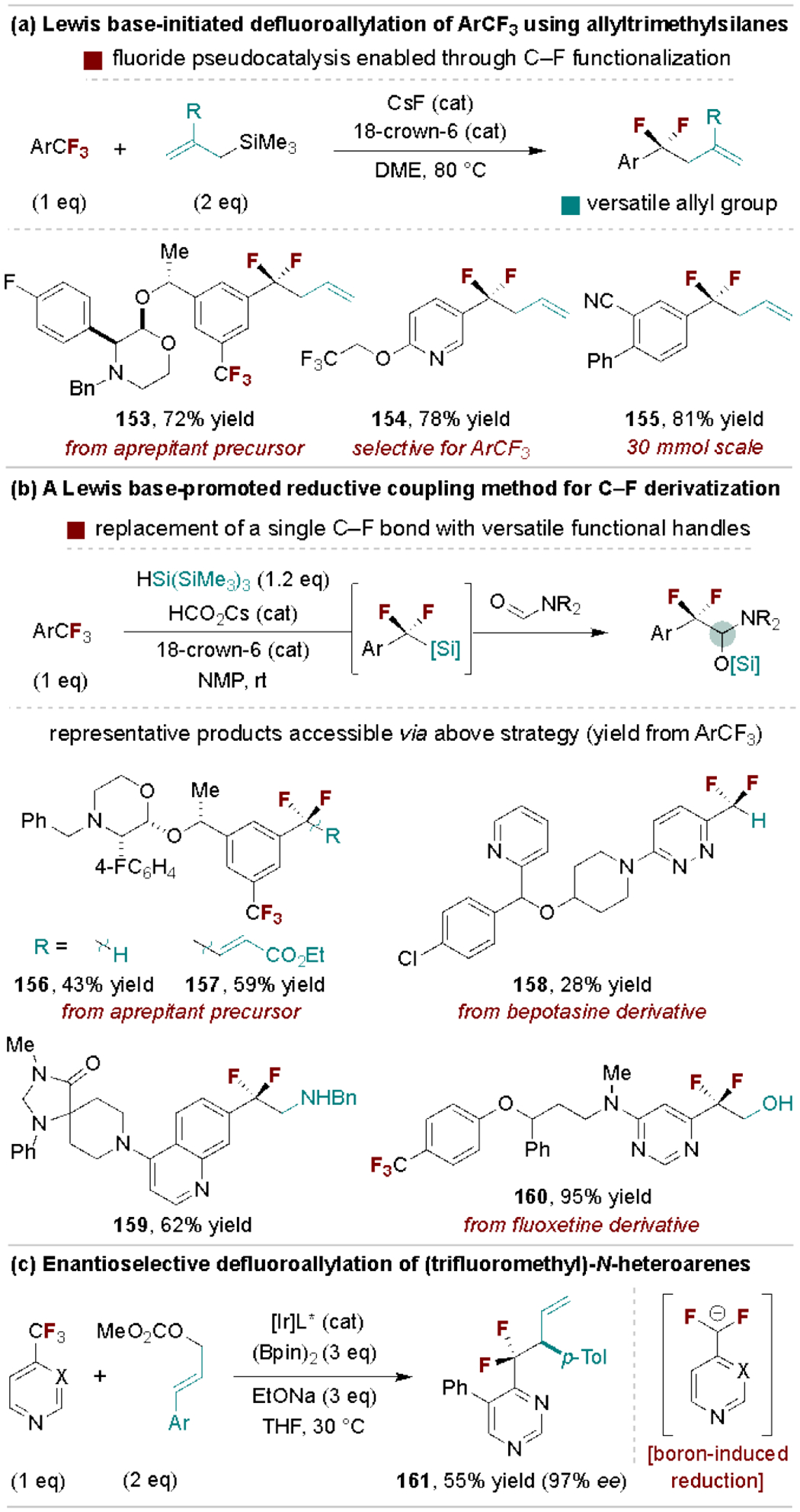
Base-promoted selective ArCF3 defluorofunctionalization. DME, 1,2-dimethoxyethane.
6.3. Exhaustive defluorofunctionalization of trifluoromethylarenes
The use of trifluoromethyl groups as oxidized carbon synthons can provide unique routes to commonly desired non-fluorinated aryl substituents. For example, an exhaustive defluoroalkylation protocol was developed by Ozerov and coworkers as a potentially valuable means to study peralkylated analogues of –CF3 groups (Scheme 30).[160] Related acid-catalyzed processes using hydride equivalents (e.g., hydrosilanes) are similarly amenable to transformation of aryl –CF3 substituents into –CH3 groups[13c, 160b]. Additionally, acidic activation of trifluoromethylarenes promotes electrophilic coupling with arenes, alcohols and amines en route to diaryl ketones, esters and amides, respectively.[161] These methods effectively allow the –CF3 group to serve as a protected carboxylic acid.
Scheme 30.
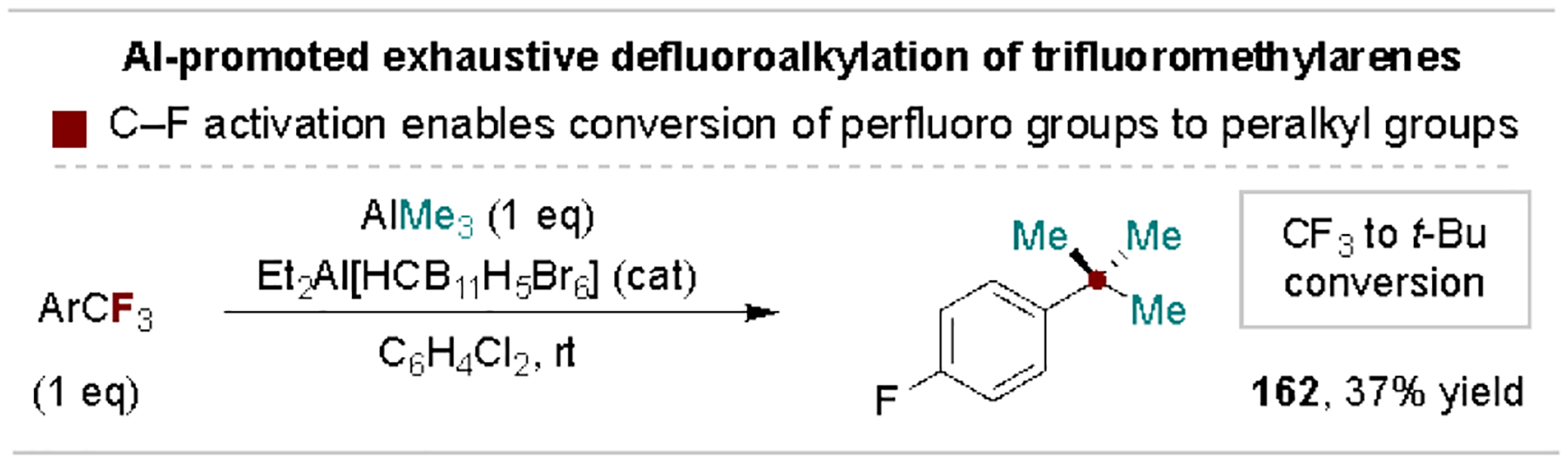
Exhaustive defluoroalkylation of trifluoromethylarenes.
7. Allyl Fluoride Defluorofunctionalization
Allylic monofluorides are readily available via nucleophilic fluorination of common allyl precursors, although their use in defluorofunctionalization reactions is advantageous only in certain contexts.[162] Meanwhile, α,α-difluoroalkyl- and α-perfluoroalkyl alkenes are available in modular fashion and engage in diverse defluorofunctionalization reactions.[163] Routes to these starting materials include olefination reactions of α-fluorinated ketones and metal-catalyzed coupling reactions of fluorinated vinyl reagents.[162] An overview of defluorinative coupling approaches to access valuable fluoroalkenes and α-fluorinated alkenes is discussed here.
7.1. Asymmetric substitution reactions of allylic monofluorides
Allyl fluorides are useful electrophiles in asymmetric substitution reactions where catalyst-controlled fluoride release leads to the activation of a pronucleophile. This strategy ensures the active nucleophile is generated only when the electrophilic chiral allyl intermediate is present. For example, the Shibata[164] and Vilotijevic[165] Groups developed chiral Lewis base-catalyzed substitution reactions of racemic allylic fluorides as shown in Scheme 31a. Here, the addition of the chiral base releases fluoride that activates silylated pronucleophiles for subsequent attack of the allyl ammonium intermediate. Similarly, Trost and coworkers reported Pd-catalyzed reactions of unactivated allylic fluorides to enable coupling to unstable perfluoroalkyl carbanions or sulfone pronucleophiles en route to enantioenriched cycloalkane products (Scheme 31b).[166]
Scheme 31.
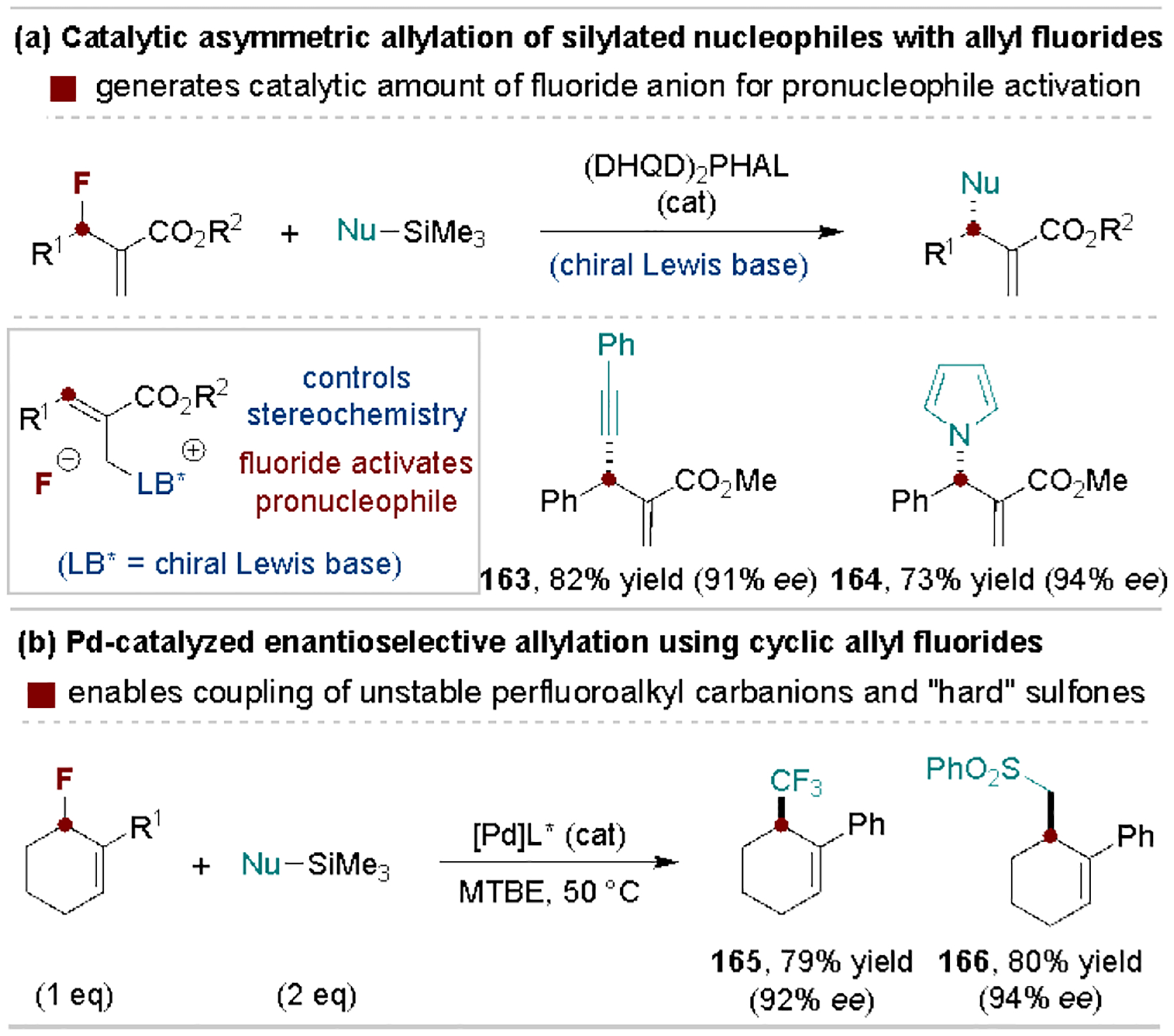
Representative asymmetric C–F allyl substitution reactions. (DHQD)2PHAL, hydroquinidine 1,4-phthalazinediyl diether.
7.2. Allyl difluoride defluorofunctionalization
Difluoroallylic compounds engage in substitution reactions through the processes depicted in Scheme 32a, including via organometallic addition/elimination, direct SN2’, and metal π-allyl mechanisms. SN2’ reactions with strong nucleophiles are a simple route to vinyl fluorides with examples from Paquin and coworkers in Scheme 32b.[167] These products can serve as amide bioisosteres, as previously discussed in the Vinyl C‒F section.[101a] The scope of nucleophilic coupling partners can be expanded through metal-catalyzed addition/β-fluoride elimination reactions.[168] This reactivity and its utility are illustrated in Scheme 32c in an example reported by Taguchi, Yanai and coworkers for the assembly of peptide isosteres via Cu-catalyzed defluoroarylation with PhMgBr.[169] Hoveyda, Ito and coworkers developed an enantioselective Cu-catalyzed defluoroborylation protocol that generates chiral allylic boronates (Scheme 32d).[170] These products are useful reagents en route to chiral fluorinated products, including allylation reactions to enable net C‒F reductive coupling transformations. Hartwig and coworkers reported an alternative method to accomplish gem-difluoride desymmetrization via Ir-catalyzed defluoroallylation of enolate pronucleophiles to generate tertiary chiral fluorides (Scheme 32e).[171]
Scheme 32.
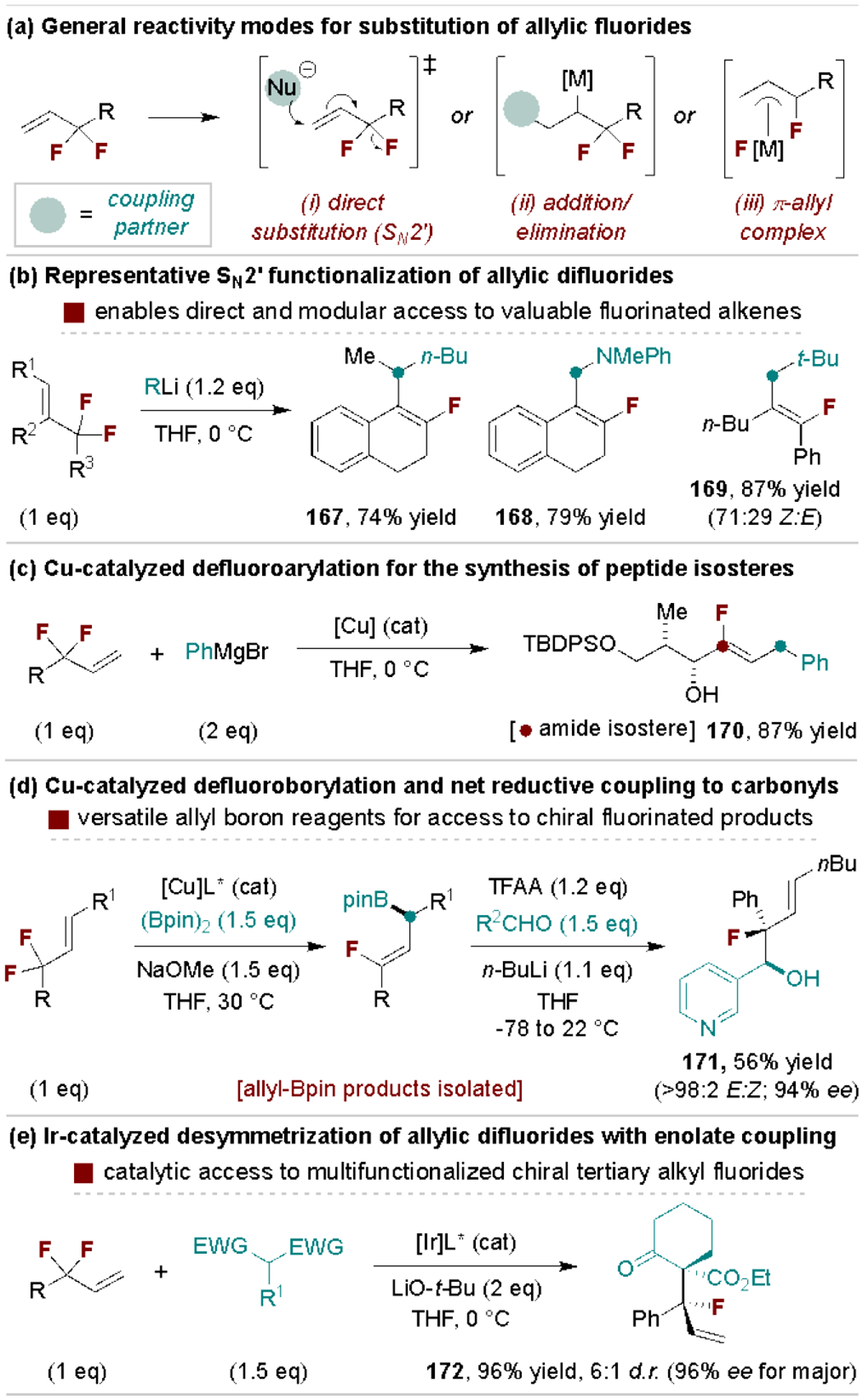
Representative allyl difluoride defluorofunctionalization methods. TBDPS, tert-butyldiphenylsilyl; TFAA, trifluoroacetic anhydride.
7.3. Net C‒F substitution of α-perfluoroalkyl alkenes
Direct C‒F bond substitution of α-perfluoroalkyl alkenes is relatively uncommon as these compounds are typically poor electrophiles for SN2 reactions.[172] However, Li and coworkers recently reported base-promoted reactions of α-trifluoromethylstyrene derivatives with phenols that are proposed to proceed via direct C–F substitution (Scheme 33a).[173] A separate case-specific approach for C‒F substitution occurs through a fluoride-catalyzed SN2’ defluoroallylation/Cope rearrangement cascade of α-trifluoromethylstyrene derivatives, reported by Feng and coworkers (Scheme 33b).[174] More general defluorination strategies are available through preparation of gem-difluorovinyl synthons that are either electrophilic or nucleophilic at the original –CF3 site. For example, the use of amines or thiols for the SN2’ reaction, followed by activation of the intermediate, generates potent electrophiles for net C–F coupling to nucleophiles (Scheme 33c).[175] Alternatively, defluorosilylation or -borylation of α-perfluoroalkylalkenes forms nucleophilic intermediates that react with electrophiles (in situ or in a subsequent step) at the fluorinated center, as illustrated by a direct C–F carboxylation reaction reported by Yu and coworkers (Scheme 33d).[110]
Scheme 33.
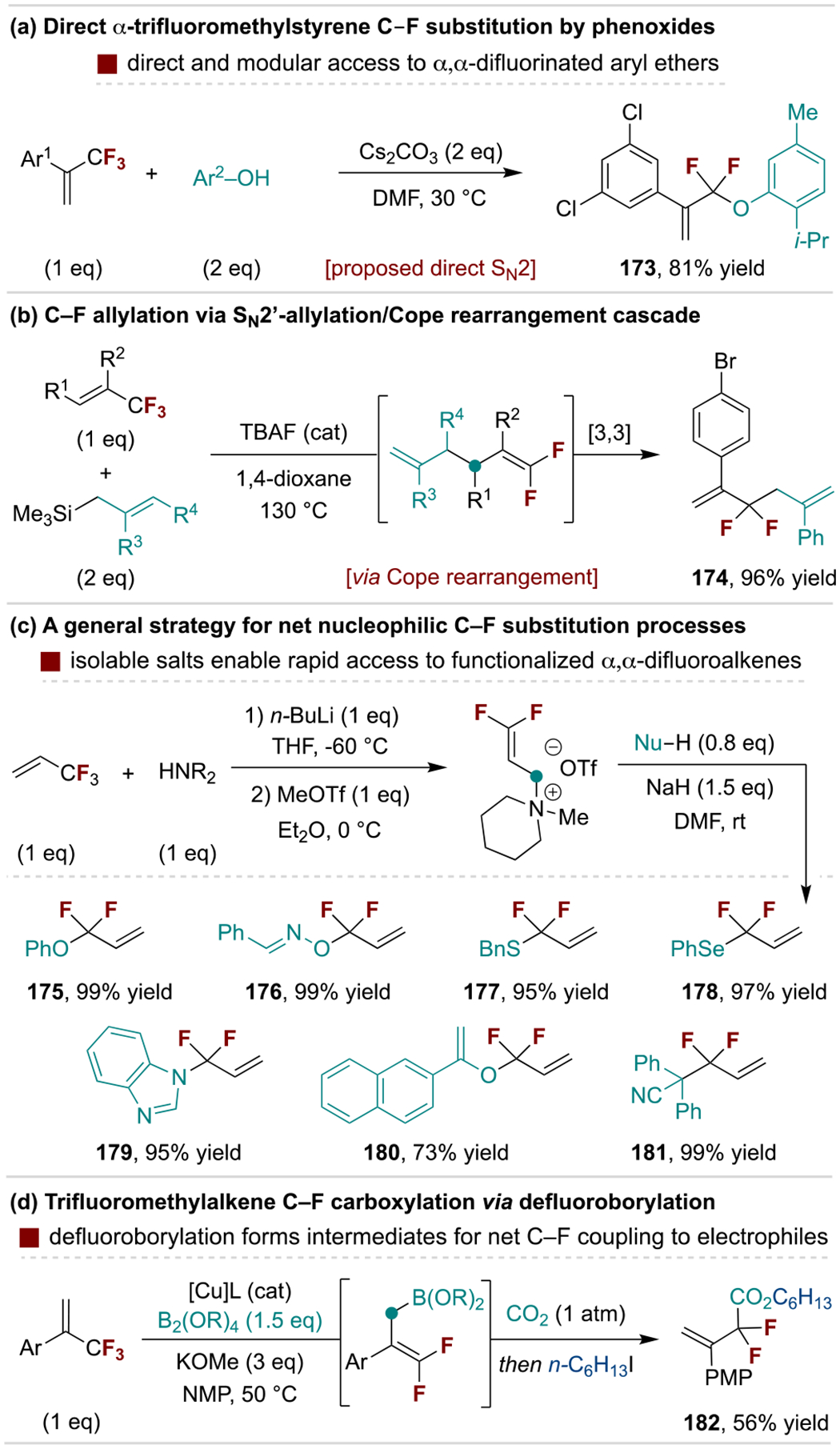
Examples of net C‒F substitution of α-perfluoroalkylalkenes.
7.4. Defluorofunctionalization to access fluorinated alkenes
α-Perfluoroalkyl alkenes readily react with nucleophiles, organometallic reagents and radical species through addition/fluoride elimination sequences to generate gem-difluoroalkenes.[176] These methods are valuable as 1,1-difluoroalkenes are useful building blocks and are also often studied as less-electrophilic isosteres for carbonyl groups, as fluorine’s small size and high electronegativity can mimic oxygen atom lone pairs.[101] Here, we highlight three modular approaches for accomplishing such defluorinative processes.
Common nucleophiles such as amines, organolithiums and silyl anions undergo SN2’ reactions with trifluoromethylalkenes to afford gem-difluoroalkenes (Scheme 34a).[177] This process can be incorporated into multistep sequences, as highlighted by base-promoted SN2’/cyclization/defluorination reactions between α-trifluoromethylstyrene derivatives and hydrazines to access 3-fluoropyrazoles developed by Ichikawa and coworkers (Scheme 34b).[178] Alternatively, Lewis or Brønsted acid catalysis can be used to remove fluorine or to protonate the alkene, respectively, and form cationic intermediates for reactions with mild nucleophiles; this is exemplified by the defluoroarylation process in Scheme 34c, also developed by the Ichikawa Group.[179]
Scheme 34.
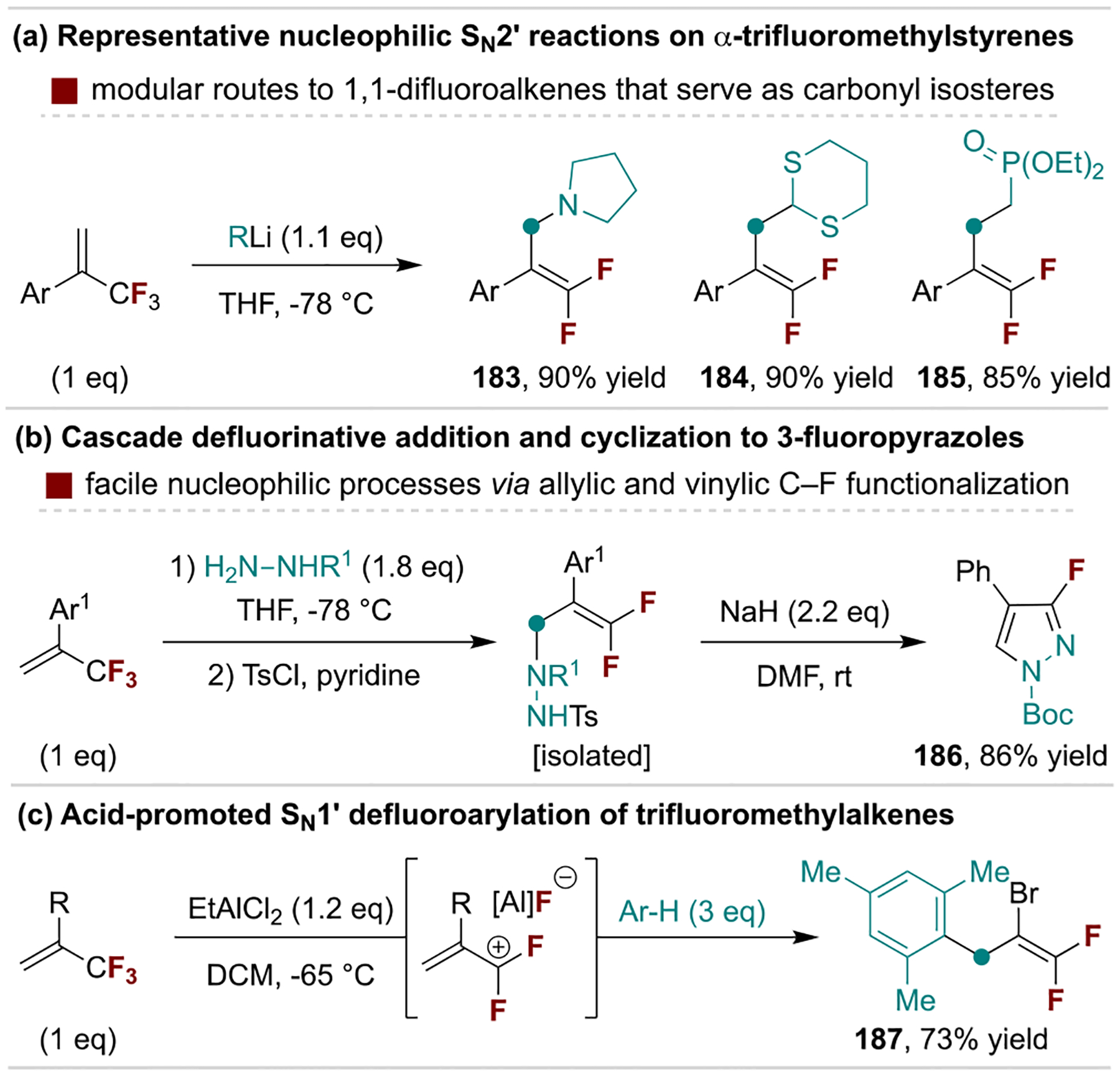
Representative anionic and cationic defluorinative reactions.
Metal-catalyzed addition/β-fluoride elimination sequences are widely developed for 1,1-difluoroalkene synthesis, with a survey of accessible products shown in Scheme 35.[168,180] Importantly, catalytic protocols for the enantioselective addition of aryl and alkyl groups provide access to difluoroalkene isosteres that map onto α-chiral carbonyl fragments. Furthermore, defluoroborylation and -silylation provide synthetically versatile chiral products.[181]
Scheme 35.
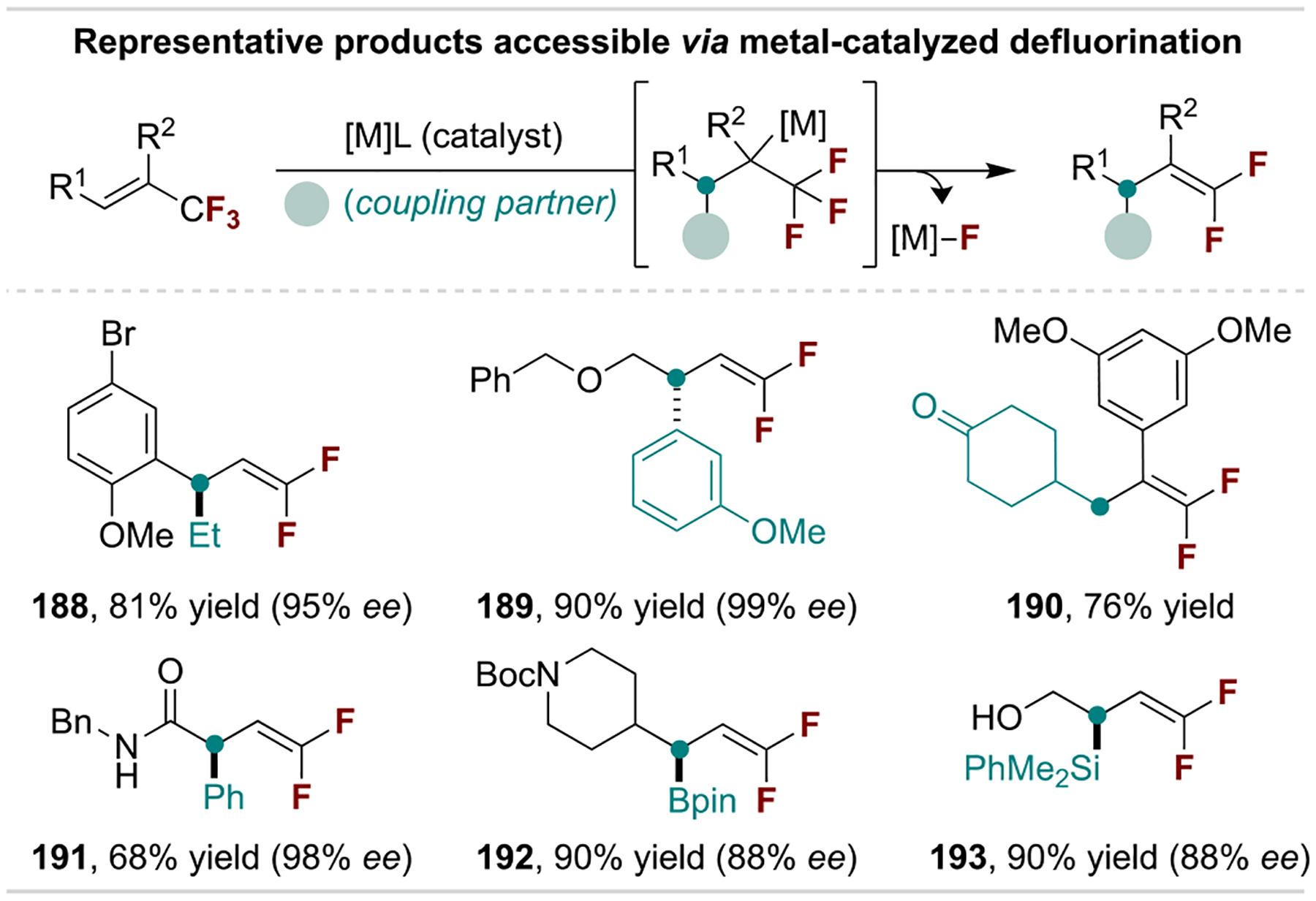
Representative products accessible via metal-catalyzed defluorination reactions.
Nucleophilic radical species easily add to α-perfluoroalkyl alkenes to generate radical intermediates that are reduced to carbanions prior to fluoride elimination (Scheme 36a).[6b] This process expands the range of defluorinative coupling partners beyond common nucleophiles and organometallic reagents as radical intermediates can be generated from many precursors, with several representative examples highlighted here. In 2017, Molander and coworkers disclosed a Ru-catalyzed photoredox strategy to generate alkyl radicals for defluoroalkylation of a wide range of trifluoromethyl-substituted alkenes (Scheme 36b).[182] Due to the mild conditions and the rapid rate of radical addition, this chemistry is compatible with complex biomolecules and can be used within DNA-encoded library synthesis.[183] A separate unique application is highlighted in Scheme 36c by Shen and coworkers, wherein the difluoroalkene intermediates undergo a photopromoted [2+2] cycloaddition to form gem-difluorooxetanes.[184] Electrochemical strategies can also be used to promote radical defluorocoupling reactions, as illustrated by Ni, Guo, Wang and coworkers through the use of Katritzky salts for deaminative alkylation (Scheme 36d).[185]
Scheme 36.
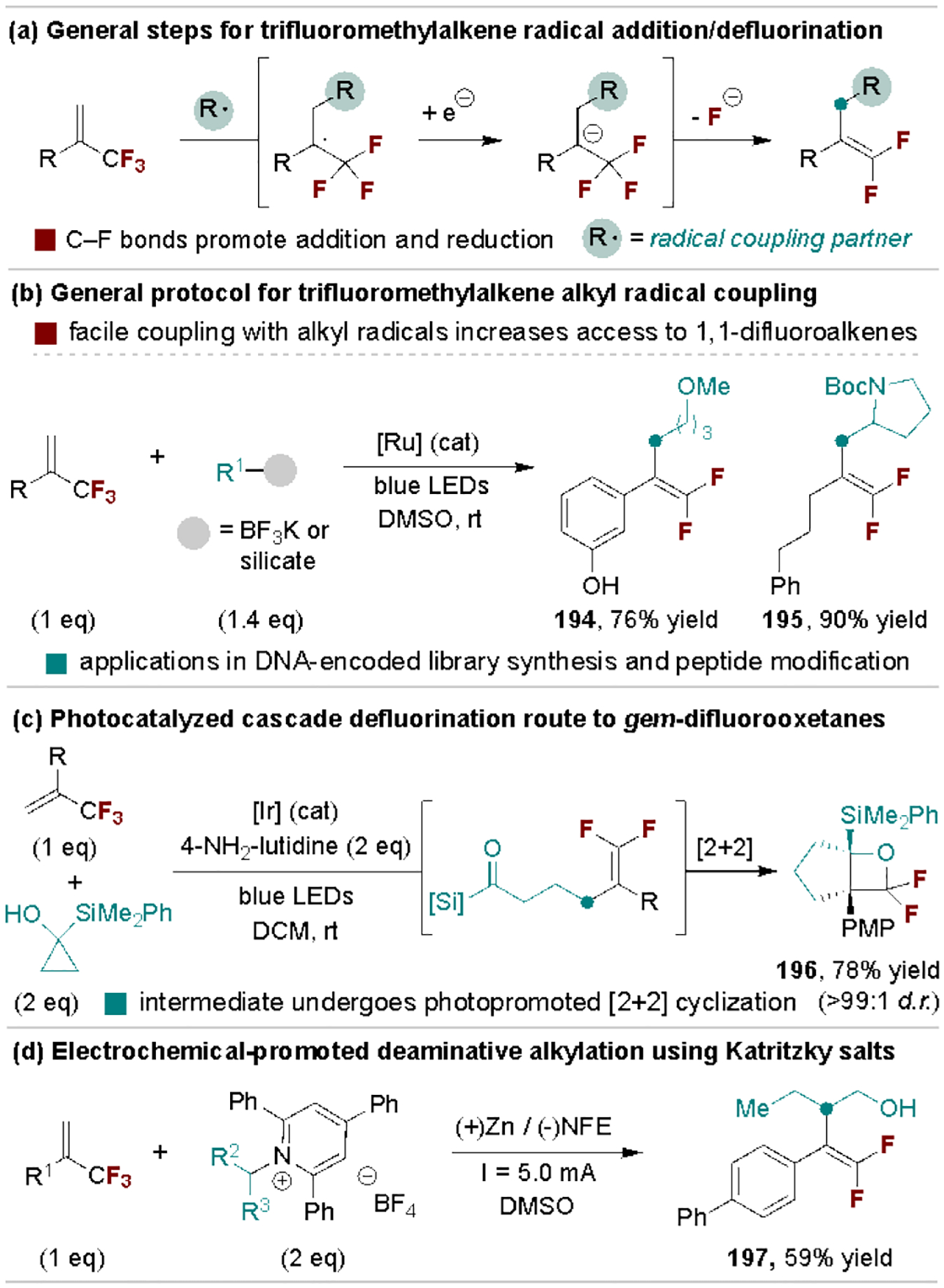
Representative radical coupling defluorinative reactions. NFE, nickel foamed electrode.
8. Propargyl Fluoride Defluorofunctionalization
Defluorinative reactions of propargyl fluorides are relatively less studied and utilized.[162] While propargylic mono- and difluoride defluorofunctionalizations have several synthetic advantages, trifluoride derivatives have thus far found less utility and are not discussed here. An overarching benefit of these methods is that they generate fluorinated products that can be cumbersome to prepare via alternative fluorination protocols. Propargylic fluorides are accessed in modular fashion via deoxyfluorination of the corresponding alcohol or ketone, available from alkynyl addition reactions to carbonyl electrophiles.[118] Sonogashira-type coupling for fluoroalkylation of terminal alkynes can also access these compounds.[186]
8.1. Propargyl monofluoride rearrangement
Zhao and coworkers developed a defluorinative, Meyer-Schuster-like rearrangement of propargylic monofluorides to furnish allylic gem-difluoride products (Scheme 37a).[187] Use of propargyl fluoride starting materials provides substantially higher yields over related electrophiles, such as propargyl bromides or alcohols. These products are valuable as allylic difluorination is often studied in medicinal chemistry as a means to inhibit metabolic oxidation of allylic C–H bonds; additionally, the α-iodoalkene handle allows further diversification.
Scheme 37.
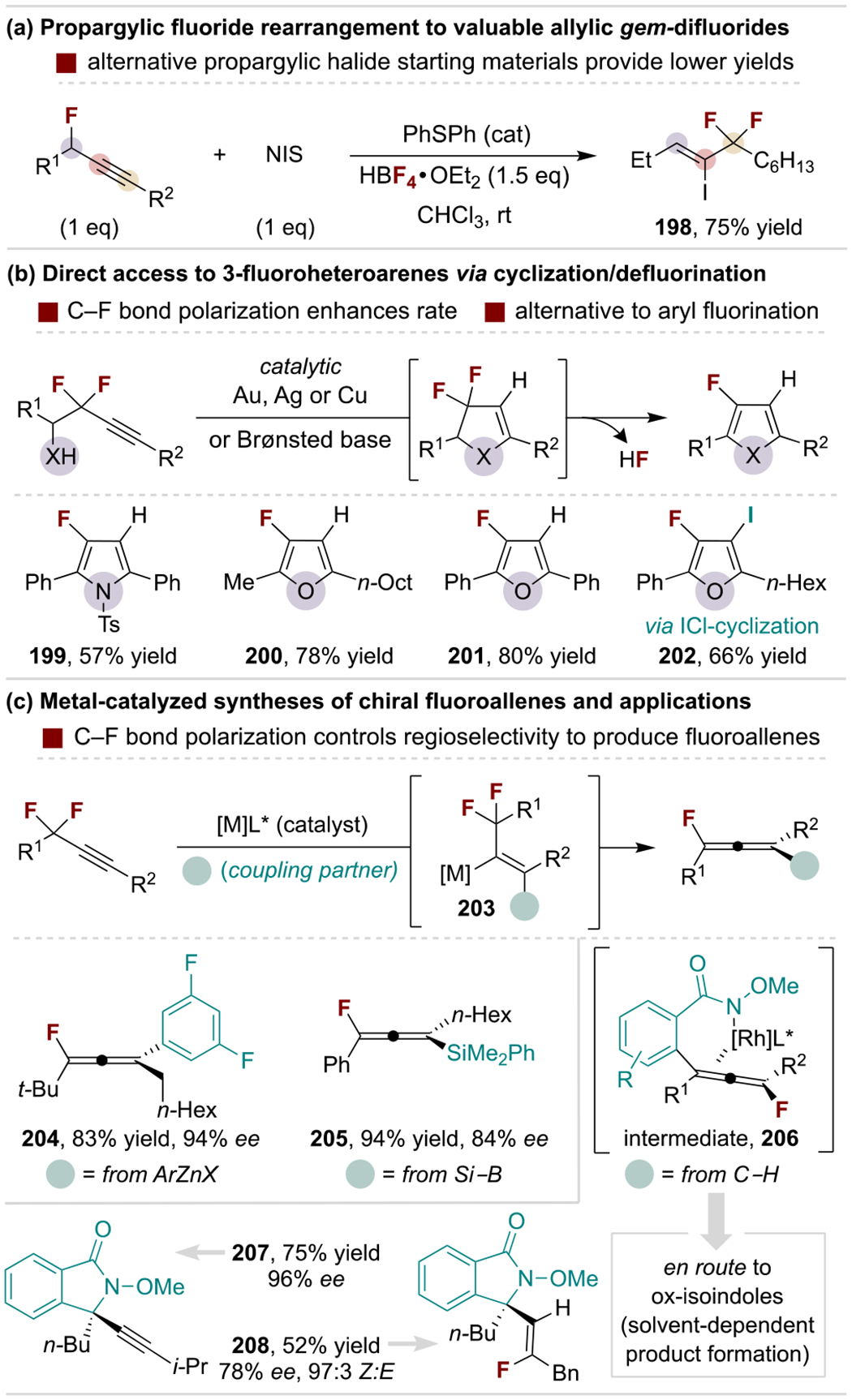
Selected uses of propargylic mono- and difluoride compounds. NIS, N-iodosuccinimide.
8.2. Defluorinative access to fluoroheteroarenes
The electronic polarization of propargylic fluorides activates the distal carbon of the alkyne for nucleophilic attack. This effect enhances the rates of 5-endo-dig cyclization reactions that are commonly paired with fluoride elimination to produce fluorinated heteroarenes (Scheme 37b).[188] Such processes can be catalyzed by Brønsted base or metal catalysts to provide an attractive alternative to routes that rely on aromatic fluorination.
8.3. Defluorinative access to chiral fluoroallenes
The polarization of propargylic gem-difluorides also facilitates regioselective intermolecular addition of organometallic species. For example, Hayashi and coworkers developed a Rh-catalyzed protocol for organozinc addition sequences that proceed through enantioselective β-fluoride elimination of the vinyl metal intermediate (203) to produce chiral fluoroallenes (Scheme 37c).[189] The reaction tolerates alkyl- and arylzinc reagents, while the gem-difluoro group requires a tertiary carbon substituent for high enantioselectivity. Toste, Liu and coworkers disclosed a related Cu-catalyzed silylation process to access Si-substituted chiral fluoroallenes.[190] Although underinvestigated, such chiral fluoroallenes have potential as versatile intermediates to more complex fluorinated products. However, related methods exploit fluoroallenes as intermediates within cascade reactions, with a representative example by Wang and coworkers shown in Scheme 37c.[191] Here, C–H activation and arylation/fluoride elimination generates fluoroallene 206 that undergoes N-addition to produce chiral ox-isoindoles.
9. α-Fluorocarbonyl Defluorofunctionalization
This section summarizes advantages of defluorination reactions of α-fluorocarbonyl compounds, including α-mono- and α-perfluorinated derivatives.[9d,e,f] Generally, α-monofluorocarbonyls are both less available and less reactive than alternate α-halo derivatives, and thus are only briefly discussed. In contrast, methods for selective defluorofunctionalization of α-polyfluoroalkylated carbonyl compounds are more prevalent and are covered in greater detail. Simple α-monofluorocarbonyls are typically prepared via fluoride substitution of α-halocarbonyls or through electrophilic α-fluorination of enolate equivalents.[192] α-Perfluorinated carbonyl derivatives can be accessed via nucleophilic fluoroalkylation of acyl electrophiles. Alternatively, acyl substitution reactions using common fluorinated building blocks, such as fluoroacetic acid or trifluoroacetic anhydride, provide α-fluorinated esters, amides and ketones.[2d]
9.1. Defluorination of α-monofluorocarbonyl compounds
Due to their relatively low electrophilicity, defluorofunctionalization of α-fluorocarbonyls is most advantageous when the stability of the reagent is of critical concern.[193] For example, Zhang, Yi, and coworkers exploited this property to enable base-promoted cyclization and fluoride elimination sequences en route to phenol and pyridine derivatives (Scheme 38).[194] Here, α-fluoro-β-keto esters productively react with α,β-unsaturated carbonyls, whereas use of α-chloro and α-bromo derivatives results in undesired elimination and alkylation side reactions.
Scheme 38.
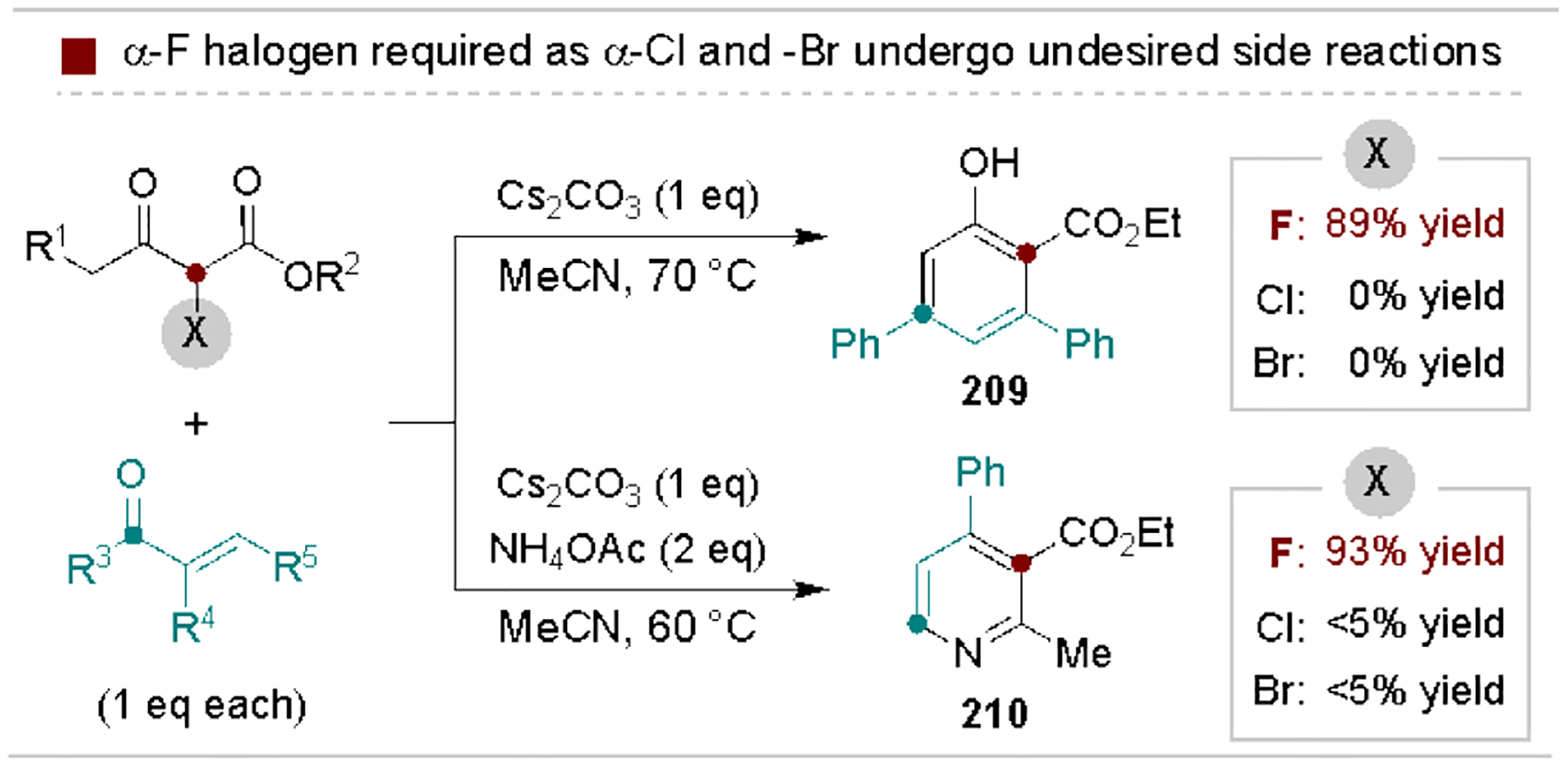
Use of α-monofluorocarbonyls in (hetero)arene synthesis.
9.2. α-Perfluorocarbonyl defluorination
α-Perfluoroalkyl carbonyl compounds are frequently used in monodefluorinative coupling reactions to assemble α-fluorinated ketone and acid derivatives.[9d,e,f] Such products are valuable as they can provide increased metabolic and thermal stability relative to the des-fluoro variants.[101] However, a mechanistic challenge for monodefluorofunctionalization is that C–F bond strengths of α-fluorinated carbonyls decrease upon removal of each fluorine (e.g., C–F BDE for an α,α,α-trifluoroamide is ~119 kcal/mol, while an α-monofluoroamide is ~106 kcal/mol).[195] To achieve monoselectivity, reductive activation strategies are commonly employed to create products that are more difficult to reduce than the starting materials. It should be noted that many of the products discussed below can also be formed from a brominated precursor (e.g., α-bromo-α,α-difluoro acyl compounds); however, these materials are often more expensive than α-perfluorinated variants.
Reduction of α-polyfluorinated carbonyls forms ketyl radical species that undergo further reduction and β-fluoride elimination to generate enolate intermediates. This sequence can be promoted by reducing metals (e.g., Mg or SmI2)[196] or through electrochemical and photochemical processes, while the enolate intermediates are readily captured by in situ electrophiles, most commonly chlorosilanes (Scheme 39a).[197] The fluorinated silyl enol ether products are useful for further α-functionalization, including halogenation. A notable application of this process is the use of 18F-substitution of α-iodo-polyfluorocarbonyl intermediates to achieve net isotopic labeling of trifluoromethyl ketones in bioactive molecules for PET imaging (Scheme 39b).[198] Recently, Lennox and coworkers developed an improved hydrodefluorination protocol via an electrochemical strategy; as illustrated in Scheme 39c, this method offers improved yields and substrate tolerance over direct use of reducing metals (e.g., Mg).[199] Reductively-generated α,α-difluoroenolates can also be intercepted by proton sources, carbonyl electrophiles and CO2 (Scheme 39d) to access a wider range of products.[150c, 200] An alternative reductive coupling strategy was reported by Ogashi and coworkers wherein carbonyl borylcupration leads to a Cu enolate that couples with an expanded scope of carbonyl partners (Scheme 39e).[201]
Scheme 39.
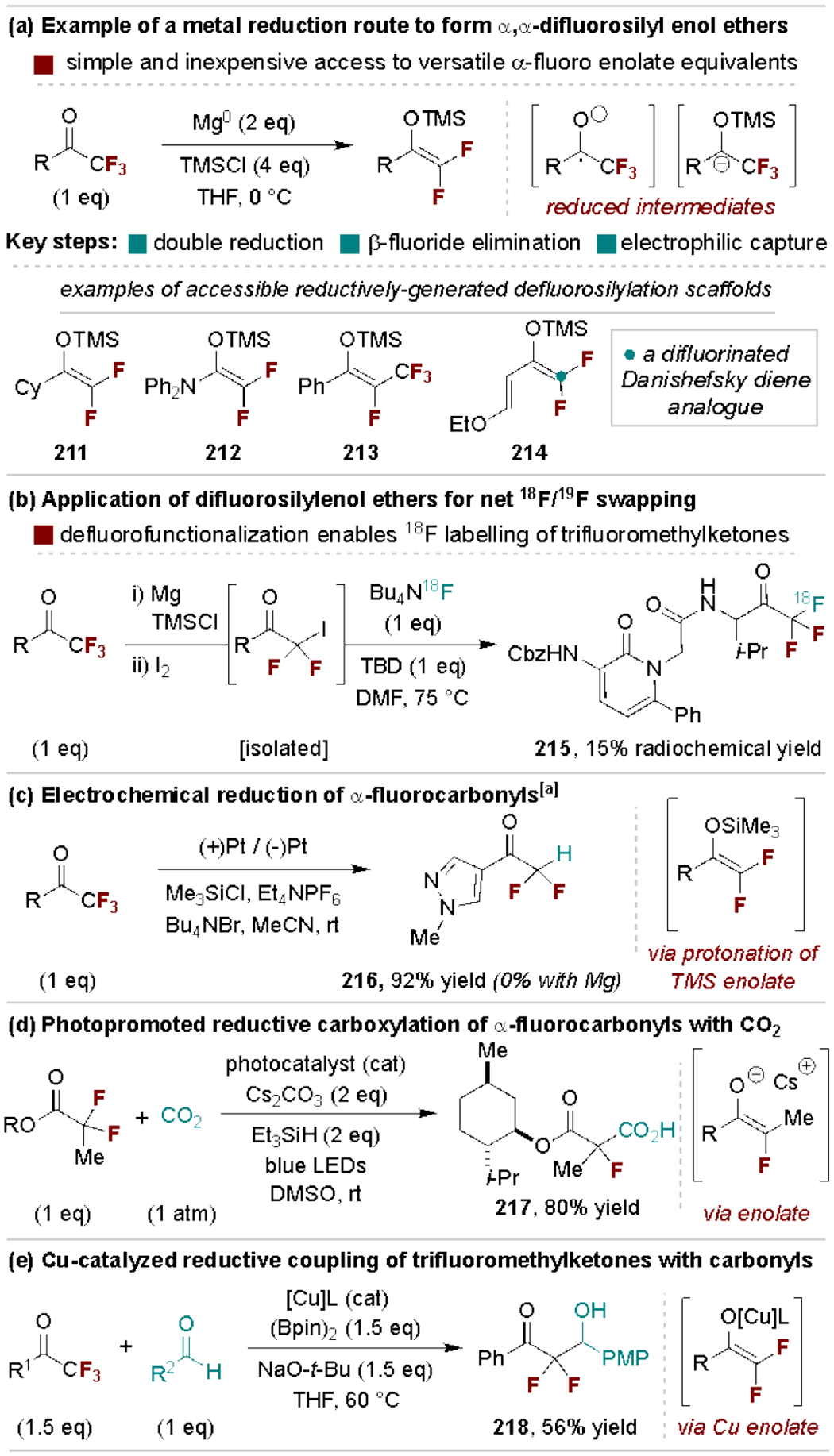
Representative applications of reductively-generated difluorosilylenol ethers and difluoroenolates. TBD, 1,5,7-triazabicyclo[4.4.0]dec-5-ene. [a] Abbreviated conditions are shown, see ref [199] for full details.
Several recent reports have exploited spin-center shift processes of the initially reduced carbonyl radical ion intermediate wherein mesolytic cleavage generates an α-radical species for coupling reactions. For example, Zhang, Houk and coworkers reported that 4-(N,N-dimethylamino)pyridine/BH3 acts as a reductant, with a thiol hydrogen atom transfer catalyst, to promote defluoroalkylation of trifluoroacetates and acetamides with alkene partners (Scheme 40a, top).[195] Glorius and coworkers also developed a related photopromoted protocol (Scheme 40a, bottom).[202] These approaches are further applicable to α,α-difluorinated starting materials, and they can be used to achieve selective hydrodefluorination via hydrogen atom transfer to the initial α-radical intermediate. Additional coupling partners have since been shown to engage in α-radical capture, as represented by Molander and coworkers’ report for defluorinative coupling to indoles (Scheme 40b).[203]
Scheme 40.
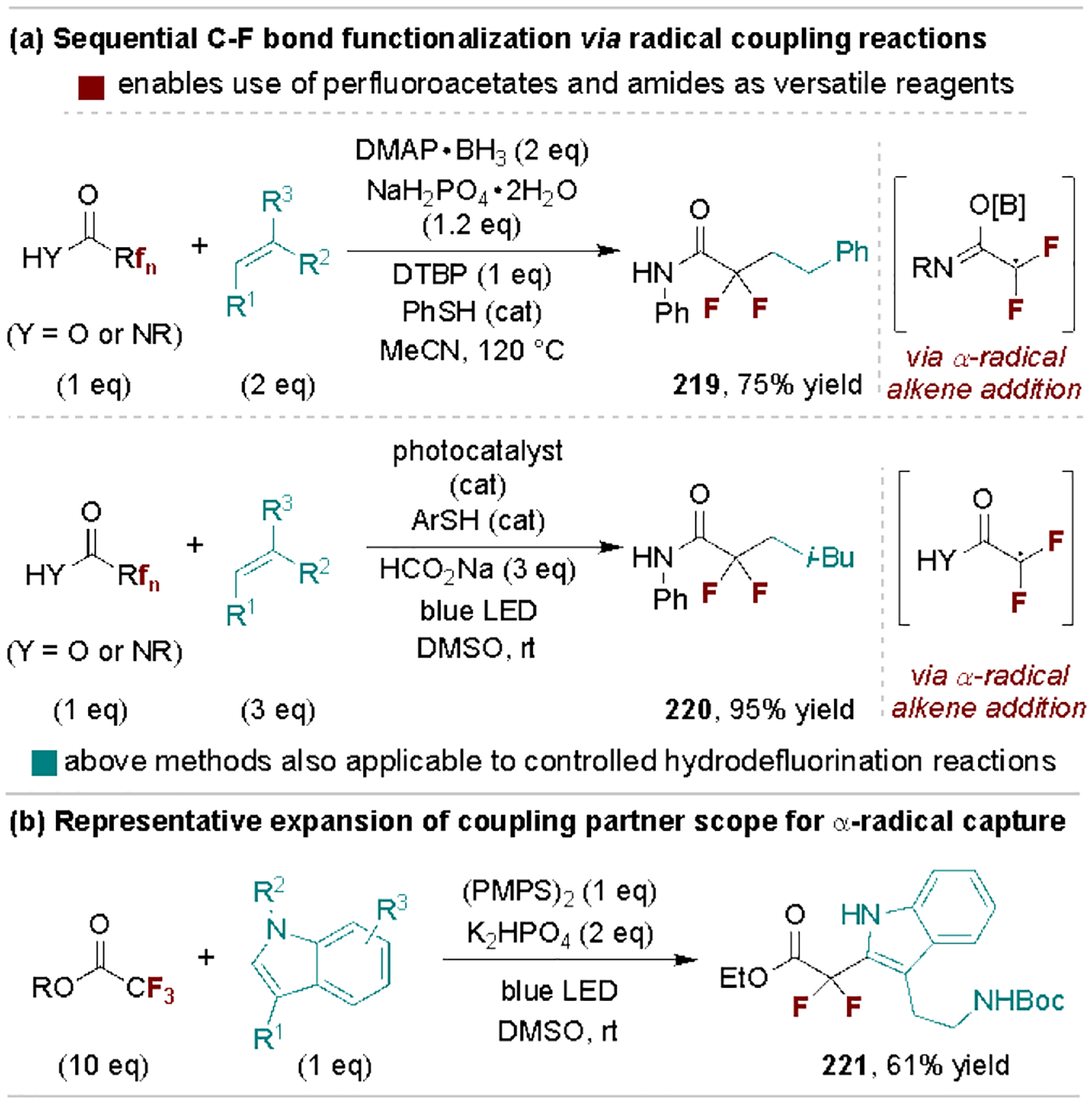
Defluorinative coupling reactions via α-carbonyl radical species. DTBP, di-tert-butyl peroxide.
10. Summary and Outlook
Chemists have made significant advances within defluorinative functionalization chemistry that enable C–F bonds to be treated as versatile functional handles. This progress has introduced multiple mechanistic strategies that can activate strong C–F bonds and control selectivity (e.g., site- and mono-) for a diverse array of organofluorine motifs. Critically, these methods do not simply serve as an alternative to the use of other halogenated compounds that are frequently more available and reactive; rather, defluorofunctionalization offers distinct advantages that enable reaction mechanisms, synthetic routes and applications that are not available via other approaches. In this regard, this review demonstrates how the strategic use of C–F bonds can provide valuable opportunities to streamline syntheses, improve reaction efficiencies and enable new reactivity.
Moving forward, we hope this overview motivates chemists to discover additional innovative strategies for C–F functionalization and to apply them towards methods that improve chemical synthesis. As illustrated herein, there are diverse mechanistic approaches available to activate C–F bonds, including through anionic, cationic, reductive, metal-catalyzed and radical-based processes. Given this rich reactivity, we anticipate the synthetic value of organofluorine compounds will continue to grow. To this end, we expect that improving selectivity for C–F bond activation in the presence of multiple functional groups, continuing to address selectivity challenges for polyfluorinated compounds, and harnessing the unique mechanistic possibilities of C–F bonds and fluoride byproducts, will be critical. When paired with the increasing availability of fluorinated chemicals and their commercial applications, these advances will not only impact chemical synthesis but could inspire future uses of organofluorines.
Acknowledgements
Support was provided by the National Institute of General Medical Sciences of the U. S. National Institutes of Health under award number R35GM138350. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. J. S. B. thanks the Research Corporation for Science Advancement (Cottrell Scholar Award) and L. V. H. thanks the U. S. National Science Foundation for a Graduate Research Fellowship (#1840343).
Biographies

Leidy V. Hooker received her B.S. in Chemistry from Montana State University in Bozeman, Montana in 2019 where she conducted undergraduate research with Sharon R. Neufeldt. She then began her Ph.D. studies with Jeffrey S. Bandar at Colorado State University in Fort Collins, Colorado where she is currently a fifth-year graduate student and a National Science Foundation fellowship recipient.

Jeff Bandar received his B.A. in Chemistry from Saint John’s University in Collegeville, Minnesota in 2009. He then conducted Ph.D. studies with Tristan H. Lambert at Columbia University from 2009 to 2014, followed by postdoctoral work with Stephen L. Buchwald at Massachusetts Institute of Technology. In 2017, Jeff started his independent research career at Colorado State University where his group focuses on the discovery and development of new base-promoted synthetic methods.
References
- [1].(a) Hiyama T in Organofluorine Compounds, Springer; ,1st Ed., Berlin, Heidelberg, 2000; [Google Scholar]; (b) Szabó K, Selander N in Organofluorine Chemistry: Synthesis, Modeling, and Applications, Wiley-VCH, 2021. [Google Scholar]
- [2].(a) For selected reviews on the synthesis of organofluorines: Liang T, Neumann CN, Ritter T, Angew. Chem. Int. Ed 2013, 52, 8214–8264; [DOI] [PubMed] [Google Scholar]; (b) Champagne PA, Desroches J, Hamel J-D, Vandamme M, Paquin J-F, Chem. Rev 2015, 115, 9073–9174; [DOI] [PubMed] [Google Scholar]; (c) Szpera R, Moseley DFJ, Smith LB, Sterling AJ, Gouverneur V, Angew. Chem. Int. Ed 2019, 58, 14824–14848; [DOI] [PubMed] [Google Scholar]; (d) Caron S, Org. Process Res. Dev 2020, 24, 470–480; [Google Scholar]; (e) Neumann CN, Ritter T, Angew. Chem. Int. Ed 2015, 54, 3216–3221; [DOI] [PubMed] [Google Scholar]; (f) Bui TT, Hong WP, Kim H-K, J. Fluor. Chem 2021, 247, 109794; [Google Scholar]; (g) Britton R, Gouverneur V, Lin J-H, Meanwell M, Ni C, Pupo G, Xiao J-C, Hu J, Nat. Rev. Methods Primers 2021, 1, 47; [Google Scholar]; (h) Zou Z, Zhang W, Wang Y, Pan Y, Org. Chem. Front 2021, 8, 2786–2798. [Google Scholar]
- [3].(a) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H, Chem. Rev 2014, 114, 2432–2506; [DOI] [PubMed] [Google Scholar]; (b) Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA, J. Med. Chem 2015, 58, 8315–8359; [DOI] [PubMed] [Google Scholar]; (c) Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Aceña JL, Soloshonok VA, Izawa K, Liu H, Chem. Rev 2016, 116, 422–518; [DOI] [PubMed] [Google Scholar]; (d) Berger R, Resnati G, Metrangolo P, Weber E, Hulliger J, Chem. Soc. Rev 2011, 3496–3508; [DOI] [PubMed] [Google Scholar]; (e) Jeschke P, ChemBioChem 2004, 5, 570–589; [DOI] [PubMed] [Google Scholar]; (f) Kotthoff M, Müller J, Jürling H, Schlummer M, Fiedler D, Environ. Sci. Pollut. Res 2015, 22, 14546–14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) O’Hagan D, Chem. Soc. Rev 2008, 37, 308–319; [DOI] [PubMed] [Google Scholar]; (b) Johnson BM, Shu Y-Z, Zhuo X, Meanwell NA, J. Med. Chem 2020, 63, 6315–6386. [DOI] [PubMed] [Google Scholar]
- [5].(a) For comprehensive reviews on C–F functionalization: Amii H, Uneyama K, Chem. Rev 2009, 109, 2119–2183; [DOI] [PubMed] [Google Scholar]; (b) Fuchibe K, Fujita T, Ichikawa J in C‒F Bond Activation Reactions: Comprehensive Organometallic Chemistry IV, Elsevier, 2022. [Google Scholar]
- [6].(a) For comprehensive reviews on C–F functionalization specific to a mechanistic strategy: Ai H-J, Ma X, Song Q, Wu X-F, Sci. China Chem 2021, 64, 1630–1659; [Google Scholar]; (b) Wang Z, Sun Y, Shen L-Y, Yang W-C, Meng F, Li P, Org. Chem. Front 2022, 9, 853–873; [Google Scholar]; (c) Kiplinger JL, Richmond TG, Osterberg CE, Chem. Rev 1994, 94, 373–431; [Google Scholar]; (d) Muthuvel K, Gandhi T, ChemCatChem 2022, 14, e202101579; [Google Scholar]; (e) Röckl JL, Robertson EL, Lundberg H, Org. Biomol. Chem 2022, 20, 6707–6720; [DOI] [PubMed] [Google Scholar]; (f) Chen W, Bakewell C, Crimmin MR, Synthesis 2017, 49, 810–821. [Google Scholar]
- [7].Reviews on C–F functionalization specific to a class of substrate are referenced within their corresponding section of this review.
- [8].(a) Palani V, Perea MA, Sarpong R, Chem. Rev 2022, 122, 10126–10169; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schröter S, Stock C, Bach T, Tetrahedron 2005, 61, 2245–2267; [Google Scholar]; (c) Norman JP, Neufeldt SR, ACS Catal 2022, 12, 12014–12026; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kapdi AR, Prajapati D, RCS Adv 2014, 4, 41245–41259. [Google Scholar]
- [9].(a) For reviews on C–F functionalization of polyfluorinated compounds: Xu W, Zhang Q, Shao Q, Xia C, Wu M, Asian J. Org. Chem 2021, 10, 2454–2472; [Google Scholar]; (b) Ahrens T, Kohlmann J, Ahrens M, Braun T, Chem. Rev 2015, 115, 931–972; [DOI] [PubMed] [Google Scholar]; (c) Sun AD, Love JA, Dalton, Trans 2010, 39, 10362–10374; [DOI] [PubMed] [Google Scholar]; (d) Ma X, Song Q, Chem. Soc. Rev 2020, 49, 9197–9219; [DOI] [PubMed] [Google Scholar]; (e) Yan G, Qiu K, Guo M, Org. Chem. Front 2021, 8, 3915–3942; [Google Scholar]; (f) Li S, Shu W, Chem. Commun 2022, 58, 1066–1077; [DOI] [PubMed] [Google Scholar]; g) Weaver JD, Senaweera S, Tetrahedron 2014, 70, 7413–7428. [Google Scholar]
- [10].(a) Hunter L, Beilstein J. Org. Chem 2010, 6, 38; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alabugin IV, Kuhn L, Krivoshchapov NV, Mehaffy P, Medvedev MG, Chem. Soc. Rev 2021, 50, 10212–10252. [DOI] [PubMed] [Google Scholar]
- [11].Lewis Base Catalysis in Organic Synthesis, Vol. 1, 2, 3 (Eds.: Vedejs E, Denmark SE), Wiley-VCH, 2016. [DOI] [PubMed] [Google Scholar]
- [12].(a) Grimaud L, Jutand A, Synthesis, 2017, 49, 1182–1189; [Google Scholar]; (b) Hartwig JF in Organotransition Metal Chemistry: From Bonding to Catalysis, Springer Link, University Science Books Mill Valley, 2010, Ed. 1. [Google Scholar]
- [13].(a) Stahl T, Klare HFT, Oestreich M, ACS Catal 2013, 3, 1578–1587; [Google Scholar]; (b) Klare HFT, Albers L, Süsse L, Keess S, Müller T, Oestreich M, Chem. Rev 2021, 121, 5889–5985; [DOI] [PubMed] [Google Scholar]; (c) Douvris C, Ozerov OV, Science 2008, 321, 1188–1190. [DOI] [PubMed] [Google Scholar]
- [14].(a) Gautam R, Geniza I, Iacono ST, Friesen CM, Jennings AR, Molecules 2022, 27, 1616; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gardiner J, Australian J Chem 2014, 68, 13–22. [Google Scholar]
- [15].Meegoda JN, Kewalramani JA, Li B, Marsh RW, Int. J. Environ. Res. Public Health 2020, 17, 8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].(a) Brittain WDG, Coxon CR, Chem. Eur. J 2022, 28, 10–23; [Google Scholar]; (b) Dax K, Albert M, Ortner J, Paul BJ, Carbohydrate Research 2000, 327, 47–86. [DOI] [PubMed] [Google Scholar]
- [17].(a) Wang F, Luo T, Hu J, Wang Y, Krishnan HS, Jog PV, Ganesh SK, Prakash GKS, Olah GA, Angew. Chem. Int. Ed 2011, 50, 7153–7157; [DOI] [PubMed] [Google Scholar]; (b) Liu X, Wang M, Liu Q, Chem. Rev 2015, 115, 683–730. [DOI] [PubMed] [Google Scholar]
- [18].(a) LaBerge NA, Love JA in Activation and Formation of Aromatic C–F Bonds. In: Braun T, Hughes R (eds) Organometallic Fluorine Chemistry. Topics in Organometallic Chemistry, 2015, Vol. 52, Springer, Cham.; [Google Scholar]; (b) Campbell MG, Ritter T, Chem. Rev 2015, 115, 612–633. [DOI] [PubMed] [Google Scholar]
- [19].(a) Bunnett JF, Zahler RE, Chem. Rev 1951, 49, 273–412; [Google Scholar]; (b) Chae SY, Choi JY, Kim Y, Nguyen DLT, Joo O-S, Angew. Chem. Int. Ed 2019, 58, 16331–16388. [Google Scholar]
- [20].(a) Karadeolian A, Emmett M, Souza F, Chung A, Blazecka P, Le Sueur R, Patel D, Zhao Y, Rey A, Green S, Org. Process Res. Dev 2022, 26, 1124–1129. [Google Scholar]; (b) Berglund RA, Org. Process Res. Dev 1997, 1, 328–330. [Google Scholar]
- [21].Jenkins TJ, Guan B, Dai M, Li G, Lightburn TE, Huang S, Freeze BS, Burdi DF, Jacutin-Porte S, Bennett R, Chen W, Minor C, Ghosh S, Blackburn C, Gigstad KM, Jones M, Kolbeck R, Yin W, Smith S, Cardillo D, Ocain TD, Harriman GC, J. Med. Chem 2007, 50, 566–584. [DOI] [PubMed] [Google Scholar]
- [22].Vasquez E, Wojcik JM, Caron S, J. Am. Chem. Soc 2000, 122, 712–713. [Google Scholar]
- [23].Suzuki Y, Ota S, Fukuta Y, Ueda Y, Sato M, J. Org. Chem 2008, 73, 2420–2423. [DOI] [PubMed] [Google Scholar]
- [24].(a) Liu X-W, Zarate C, Martin R, Angew. Chem. Int. Ed 2019, 58, 2064–2068; [DOI] [PubMed] [Google Scholar]; (b) Mallick S, Xu P, Würthwen E-U, Studer A, Angew. Chem. Int. Ed 2019, 58, 283–287 [DOI] [PubMed] [Google Scholar]
- [25].Murugan A, Bachu S, Manjunatha SG, Ramakrishnan R, Krishna Kadambar V, Reddy C, Rao V, Torlikonda, George S, Ramasubramanian S, Nambiar S, Org. Process Res. Dev 2014, 18, 646–651. [Google Scholar]
- [26].Schlosser M, Angew. Chem. Int. Ed 2006, 45, 5432–5446. [DOI] [PubMed] [Google Scholar]
- [27].Bobko MA, Elward JM, Naidu BN, Nieves-Quinones YE, Reiher CA, Su Q, Sun L, Woodward J, Xe S, Yang W, Yin Y, Org. Process Res. Dev 2023, 27, 217–226. [Google Scholar]
- [28].Vasquez E, Caron S, Org. Process Res. Dev 2001, 5, 587–592. [Google Scholar]
- [29].(a) Liang L-C, Chien P-S, Lin J-M, Huang M-H, Huang Y-L, Liao J-H, Organometallics 2006, 25, 1399–1411; [Google Scholar]; (b) Matveeva AG, Artyushin OI, Pasechnik MP, Stash AI, Vologzhanina AV, Matveev SV, Godovikov IA, Aysin RR, Moiseeva AA, Turanov AN, Karandashev VK, Brel VK, Polyhedron 2021, 198, 115085; [Google Scholar]; (c) Bélanger E, Pouliot M-F, Courtemancha M-A, Paquin J-F, J. Org. Chem 2012, 77, 317–331. [DOI] [PubMed] [Google Scholar]
- [30].Hoshiya N, Buchwald SL, Adv. Synth. Catal 2012, 354, 2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].(a) Larrosa I, Silva ID, Gómez PM, Hannen P, Ko E, Lenger SR, Linke SR, White AJP, Wilton D, Barrett AGM, J. Am. Chem. Soc 2006, 128, 14042–14043; [DOI] [PubMed] [Google Scholar]; (b) Sanz R, Fernández Y, Castroviejo MP, Pérez A, Fañanás FJ, J. Org. Chem 2006, 71, 6291–6294 [DOI] [PubMed] [Google Scholar]
- [32].Pistritto VA, Schutsbach-Horton ME, Nicewicz DA, J. Am. Chem. Soc 2020, 142, 17187–17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen W, Wang H, Tay NES, Pristritto VA, Li K-P, Wu Z, Nicewicz DA, Li Z, Nat. Chem 2022, 14, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang H, Lambert TH, Angew. Chem. Int. Ed 2020, 59, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mfuh AM, Doyle JD, Chhetri B, Arman HD, Larionov OV, J. Am. Chem. Soc 2016, 138, 2985–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].(a) Kang Q-K, Lin Y, Li Y. Xu L, Li K, Shi H, Angew. Chem. Int. Ed 2021, 60, 20391–20399; [DOI] [PubMed] [Google Scholar]; b) Kang Q-K, Lin Y, Li Y, Shi H, J. Am. Chem. Soc 2020, 142, 3706. [DOI] [PubMed] [Google Scholar]
- [37].Ma R, Hu H, Li X, Mao G, Song Y, Xin S, Catalysts 2022, 12, 1665. [Google Scholar]
- [38].Wang J-R, Manabe K, Org. Lett 2009, 11, 741–744. [DOI] [PubMed] [Google Scholar]
- [39].Allemann O, Duttwyler S, Romanato P, Baldridge KK, Siegel JS, Science 2011, 332, 574. [DOI] [PubMed] [Google Scholar]
- [40].Amsharov KY, Kabdulov MA, Jensen M, Angew. Chem. Int. Ed 2012, 51, 4594–4597. [DOI] [PubMed] [Google Scholar]
- [41].Shaq B, Bagdasarian AL, Popov S, Nelson HM, Science 2017, 355, 1403–1407. [DOI] [PubMed] [Google Scholar]
- [42].Sun Y, Sun H, Jia J, Du A, Li X, Organometallics, 2014, 33, 4, 1079–1081. [Google Scholar]
- [43].Lu W, Gao J, Yang J-K, Liu L, Zhao Y, Wu H-C, Chem. Sci 2014, 5, 1934. [Google Scholar]
- [44].Day JI, Weaver JD, J. Org. Chem 2017, 82, 6801–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].(a) Sanji T, Iyoda T, J. Am. Chem. Soc 2014, 136, 10238–10241; [DOI] [PubMed] [Google Scholar]; (b) Finck L, Oestreich M, Chem. Eur. J 2021, 27, 11061–11064; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ueno M, Yonemoto M, Hashimoto M, Wheatley AEH, Naka H, Kondo Y, Chem. Commun 2007, 2264–2266. [DOI] [PubMed] [Google Scholar]
- [46].(a) Luo Z-J, Zhao H-Y, Zhang X, Org. Lett 2018, 20, 2543–2546. [DOI] [PubMed] [Google Scholar]; (b) Yu D, Lu L, Shen Q, Org. Lett 2013, 15, 940–943. [DOI] [PubMed] [Google Scholar]
- [47].Landmann J, Hennig PT, Ignat’ev NV, Finze M, Chem. Sci 2017, 8, 5962–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xu W, Jiang H, Leng J, Ong H-W, Wu J, Angew. Chem. Int. Ed 2020, 59, 4009–4016. [DOI] [PubMed] [Google Scholar]
- [49].Zhou J, Kuntze-Fechner M, Bertermann R, Paul USD, Berthel JHJ, Friedrich A, Du Z, Marder TB, Radius U, J. Am. Chem. Soc 2016, 138, 5250–5253. [DOI] [PubMed] [Google Scholar]
- [50].Senaweera S, Weaver JD, Aldrichimica, Acta 2016, 49, 3, 45–54. [PMC free article] [PubMed] [Google Scholar]
- [51].(a) Sun X, Ritter T, Angew. Chem. Int. Ed 2021, 60, 10557–10562; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xie J, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 2017, 56, 7266–7270; [DOI] [PubMed] [Google Scholar]; (c) Singh A, Kubik JJ, Weaver JD, Chem. Sci 2015, 6, 7206–7212; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Priya S, Weaver JD, J. Am. Chem. Soc 2018, 140, 16020–16025; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Senaweera S, Weaver JD, J. Am. Chem. Soc 2016, 138, 2520–2523; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xu W, Shao Q, Xia C, Zhang Q, Xu Y, Liu Y, Wu M, Chem. Sci 2023, 14, 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].(a) Kuehnel MF, Lentz D, Braun T, Angew. Chem. Int. Ed 2013, 52, 3328–3348; [DOI] [PubMed] [Google Scholar]; (b) Whittlesey MK, Peris E, ACS Catal 2014, 4, 3152–3159; [Google Scholar]; (c) Yan G, Chem. Eur. J 2022, 28, e202200231. [DOI] [PubMed] [Google Scholar]
- [53].Senaweera SM, Singh A, Weaver JD, J. Am. Chem. Soc 2014, 136, 3002–3005. [DOI] [PubMed] [Google Scholar]
- [54].Schoch TD, Mondal M, Weaver JD, Org. Lett 2021, 23, 1588–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Podolan G, Lentz D, Reissig H-U, Angew. Chem. Int. Ed 2013, 52, 9491–9494. [DOI] [PubMed] [Google Scholar]
- [56].Kharbanda S, Weaver JD, J. Org. Chem 2023, 88, 6434–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].(a) Karbakhshzadeh A, Heravi MRP, Rahmani Z, Ebadi AG, Vessally E, J. Fluor, Chem 2021, 248, 109806; [Google Scholar]; (b) Gonay M, Batisse C, Paquin J-F, Synthesis 2021, 53, 653–665. [Google Scholar]
- [58].Cismesia MA, Ryan SJ, Bland DC, Sanford MS, J. Org. Chem 2017, 82, 5020–5056. [DOI] [PubMed] [Google Scholar]
- [59].Tryniszewski M, Barbasiewicz M, Synthesis 2022, 54, 1446–1460. [Google Scholar]
- [60].Pospίšil J, Müller C, Fürstner A, Chem. Eur. J 2009, 15, 5956–5968. [DOI] [PubMed] [Google Scholar]
- [61].Limat D, Schlosser M, Tetrahedron 1995, 51, 5799–5806. [Google Scholar]
- [62].Zamiri M, Grierson DS, Synthesis 2017, 49, 571–578. [Google Scholar]
- [63].Zhang Y, Rovis T, Org. Lett 2004, 6, 1877–1879. [DOI] [PubMed] [Google Scholar]
- [64].Wenschuh H, Bienert M, Beyermann M, Carpino LA, Acc. Chem. Res 1996, 29, 268–274. [Google Scholar]
- [65].Prabhu G, Narendra N, Basavaprabhu, Panduranga V, Sureshbabu VV, RSC Adv 2015, 5, 48331–48362. [Google Scholar]
- [66].Hjørringgaard CU, Pedersen JM, Vosegaard T, Nielsen NC, Skrydstrup T, J. Org. Chem 2009, 74, 1329–1332. [DOI] [PubMed] [Google Scholar]
- [67].Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ, J. Am. Chem. Soc 1999, 121, 11953–11963. [Google Scholar]
- [68].Nicolaou KC, Yin J, Mandal D, Erande RD, Klahn P, Jin M, Aujay M, Sandoval J, Gavrilyuk J, Vourloumis D, J. Am. Chem. Soc 2016, 138, 1698–1708. [DOI] [PubMed] [Google Scholar]
- [69].Wenshuch H, Beyermann M, El-Faham A, Ghassemi S, Carpino LA, Biernert M, J. Chem. Soc. Chem. Commun 1995, 669–670. [Google Scholar]
- [70].Brown ZZ, Schafmeister CE, J. Am. Chem. Soc 2008, 130, 14382–14383. [DOI] [PubMed] [Google Scholar]
- [71].Ogiwara Y, Sakai N, Angew. Chem. Int. Ed 2020, 59, 574–594. [DOI] [PubMed] [Google Scholar]
- [72].Zhang Y, Rovis T, J. Am. Chem. Soc 2004, 126, 15964–15965. [DOI] [PubMed] [Google Scholar]
- [73].Wang J, Hoerrner ME, Watson MP, Weix DJ, Angew. Chem. Int. Ed 2020, 59, 13484–13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li Y-N, Xiao F, Guo Y, Zeng Y-F, Eur. J. Org. Chem 2021, 1215–1228. [Google Scholar]
- [75].Feng S, Buchwald SL, J. Am. Chem. Soc 2021, 143, 4935–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liu RY, Buchwald SL, Acc. Chem. Res 2020, 53, 1229–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Boreux A, Indukuri K, Gagosz F, Riant O, ACS Catal 2017, 7, 8200–8204. [Google Scholar]
- [78].Ishida N, Iwamoto H, Sunagawa DE, Ohashi M, Ogoshi S, Synthesis 2021, 53, 3137–3143. [Google Scholar]
- [79].Scattolin T, Bouayad-Gervais S, Schoenebeck F, Nature 2019, 573, 102–107. [DOI] [PubMed] [Google Scholar]
- [80].(a) Zivkovic FG, Nielsen CD-T, Schoenebeck F, Angew. Chem. Int. Ed 2022, 61, e202213829; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nielsen CD-T, Zivkovic FG, Schoenebeck F, J. Am. Chem. Soc 2021, 143, 13029–13033. [DOI] [PubMed] [Google Scholar]
- [81].Birrell JA, Desrosiers J-N, Jacobsen EN, J. Am. Chem. Soc 2011, 133, 13872–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].(a) Flanigan DM, Romanov-Michailidis F, White NA, Rovis T, Chem. Rev 2015, 115, 9307–9387; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishii T, Nagao K, Ohmiya H, Chem. Sci 2020, 11, 5630–5636; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao C, Blaszczyk SA, Wang J, Green Synthesis and Catalysis 2021, 2, 198–215. [Google Scholar]
- [83].Yu X, Meng Q-Y, Daniliuc CG, Studer A, J. Am. Chem. Soc 2022, 144, 7072–7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fujimoto H, Kodama T, Tobisu M, J. Am. Chem. Soc 2020, 142, 17323–17328. [DOI] [PubMed] [Google Scholar]
- [85].Beil SB, Chen TQ, Intermaggio NE, MacMillan DWC, Acc. Chem. Res 2022, 55, 3481–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].(a) Rodrίgues N, Goossen LJ, Chem. Soc. Rev 2011, 40, 5030–5048; [DOI] [PubMed] [Google Scholar]; (b) Arshadi S, Ebrahimiasl S, Hosseinian A, Monfared A, Vessally E, RSC Adv 2019, 9, 8964–8976; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Laudadio G, Palkowitz MD, El-Hayek Ewing T, Baran PS, ACS Med. Chem. Lett 2022, 13, 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Malapit CA, Bour JR, Brigham CE, Sanford MS, Nature 2018, 563, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lalloo N, Malapit CA, Taimorry SM, Brigham CE, Sanford MS, J. Am. Chem. Soc 2021, 143, 18617–18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Keaveney ST, Schoenebeck F, Angew. Chem. Int. Ed 2018, 57, 4073–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].(a) Malapit CA, Bour JR, Laursen SR, Sanford MS, J. Am. Chem. Soc 2019, 141, 17322–17330; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ogiwara Y, Sakurai Y, Hattori H, Sakai N, Org. Lett 2018, 20, 4204–4208. [DOI] [PubMed] [Google Scholar]
- [91].(a) Drouin M, Hamel J-D, Paquin J-F, Synthesis 2018, 50, 881–955; [Google Scholar]; (b) Yanai H, Taguchi T, Eur. J. Org. Chem 2011, 5939–5954. [Google Scholar]
- [92].Chelucci G, Chem. Rev 2012, 112, 1344–1462. [DOI] [PubMed] [Google Scholar]
- [93].(a) Guo R, Qi X, Hengye X, Geaneotes P, Wang R, Liu P, Wang Y-M, Angew. Chem. Int. Ed 2020, 59, 16651–16660; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mulryan D, Rodwell J, Phillips NA, Crimmin MR, ACS Catal 2022, 12, 3411–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ye Y, Takada T, Buchwald SL, Angew. Chem. Int. Ed 2016, 55, 15559–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nair V, Int. J. Refrigeration 2021, 122, 156–170. [Google Scholar]
- [96].Coates G, Tan HY, Kalff C, White AJP, Crimmin MR, Angew. Chem. Int. Ed 2019, 58, 12514–12518. [DOI] [PubMed] [Google Scholar]
- [97].Sakaguchi H, Ohashi M, Ogoshi S, Angew. Chem. Int. Ed 2018, 57, 328–332. [DOI] [PubMed] [Google Scholar]
- [98].Hung M-H, Rozen S, Smart BE, J. Org. Chem 1994, 59, 4332–4335. [Google Scholar]
- [99].Shi H, Dai W, Wang B, Cao S, Organometallics 2018, 37, 459–463. [Google Scholar]
- [100].Yata T, Nishimoto Y, Yasuda M, Chem. Eur. J 2022, 28, e202103852. [DOI] [PubMed] [Google Scholar]
- [101].(a) Kumari S, Carmona AV, Tiwari AK, Trippier PC, J. Med. Chem 2020, 63, 12290–12358; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meanwell NA, J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- [102].Xiong Y, Zhang X, Huang T, Cao S, J. Org. Chem 2014, 79, 6395–6402. [DOI] [PubMed] [Google Scholar]
- [103].(a) Gao P, Wang G, Xi L, Wang M, Li S, Shi Z, Chin. J. Chem 2019, 37, 1009–1014; [Google Scholar]; (b) Ma Y, Reddy BRP, Bi X, Org. Lett 2019, 21, 9860–9863. [DOI] [PubMed] [Google Scholar]
- [104].(a) Dai W, Shi H, Zhao X, Cao S, Org. Lett 2016, 18, 4284–4287; [DOI] [PubMed] [Google Scholar]; (b) Sei-ichi H, Takeshi N, Nobuo I, Chem. Lett 1980, 9, 935–938. [Google Scholar]
- [105].Sakoda K, Wada Y, Ichikawa J, Synthesis 2002, 13, 1917–1936. [Google Scholar]
- [106].Zong Y, Ma Q, Tsui GC, Org. Lett 2021, 23, 6169–6173. [DOI] [PubMed] [Google Scholar]
- [107].(a) Sakaguchi H, Uetake Y, Ohashi M, Niwa T, Ogoshi S, J. Am. Chem. Soc 2017, 139, 12855–12862; [DOI] [PubMed] [Google Scholar]; (b) Zhang H, Wang E, Geng S, Liu Z, He Y, Peng Q, Feng Z, Angew. Chem. Int. Ed 2021, 60, 10211–10218. [DOI] [PubMed] [Google Scholar]
- [108].Jouffroy M, Pye P, Jerhaoui S, Chen W, Surkyn M, J. Org. Chem 2022, 87, 2136–2141. [DOI] [PubMed] [Google Scholar]
- [109].Hu J, Han X, Yuan Y, Shi Z, Angew. Chem. Int. Ed 2017, 56, 13342–13346. [DOI] [PubMed] [Google Scholar]
- [110].Yan S-S, Wu D-S, Ye J-H, Gong L, Zeng X, Ran C-K, Gui Y-Y, Li J, Yu D-G, ACS Catal 2019, 9, 6987–6992. [Google Scholar]
- [111].Koley S, Altman RA, Isr. J. Chem 2020, 60, 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ohashi M, Kambara T, Hatanaka T, Saijo H, Doi R, Ogoshi S, J. Am. Chem. Soc 2011, 133, 3256–3259. [DOI] [PubMed] [Google Scholar]
- [113].Ohasi M, Saijo H, Shibata M, Ogoshi S, Eur. J. Org. Chem 2013, 443–447. [Google Scholar]
- [114].Thornbury RT, Toste FD, Angew. Chem. Int. Ed 2016, 55, 11629–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lu X, Wang Y, Zhang B, Pi J-J, Wang X-X, Gong T-J, Xiao B, Fu Y, J. Am. Chem. Soc 2017, 139, 12632–12637. [DOI] [PubMed] [Google Scholar]
- [116].(a) Li J, Lefebvre Q, Yang H, Zhao Y, Fu H, Chem. Commun 2017, 53, 10299–10302; [DOI] [PubMed] [Google Scholar]; (b) Yu L, Tang M-L, Si C-H, Meng Z, Liang Y, Han J, Sun X, Org. Lett 2018, 20, 4579–4583. [DOI] [PubMed] [Google Scholar]
- [117].Shen Q, Huang Y-G, Liu C, Xiao J-C, Chen Q-Y, Guo Y, J. Fluor. Chem 2015, 179, 14–22. [Google Scholar]
- [118].(a) Aggarwal T, Sushmita , Verma AK, Org. Chem. Front 2021, 8, 6452–6468; [Google Scholar]; (b) Ni C, Hu M, Hu J, Chem. Rev 2015, 115, 765–825. [DOI] [PubMed] [Google Scholar]
- [119].Song H-X, Han Q-Y, Zhao C-L, Zhang C-P, Green, Chem, 2018, 20, 1662–1731. [Google Scholar]
- [120].Mandal D, Maji S, Pal T, Sinha SK, Maiti D, Chem. Commun 2022, 58, 10442–10468. [DOI] [PubMed] [Google Scholar]
- [121].(a) Balaraman K, Kyriazakos S, Palmer R, Thanzeel FY, Wolf C, Synthesis 2022, 54, 4320–4328; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Terao J, Begum SA, Shinohara Y, Tomita M, Kambe N, Chem. Commun 2007, 855–857. [DOI] [PubMed] [Google Scholar]
- [122].Janjetovic M, Träff AM, Hilmersson G, Chem. Eur. J 2015, 21, 3772–3777. [DOI] [PubMed] [Google Scholar]
- [123].Ishibashi T, Koga Y, Matsubara K, Org. Lett 2009, 11, 1765–1768. [DOI] [PubMed] [Google Scholar]
- [124].(a) Terao J, Ikumi A, Kuniyasu H, Kambe N, J. Am. Chem. Soc 2003, 125, 5646–5647; [DOI] [PubMed] [Google Scholar]; (b) Terao J, Todo H, Begum SA, Kuniyasu H, Kambe N, Angew. Chem. Int. Ed 2007, 46, 2086–2089. [DOI] [PubMed] [Google Scholar]
- [125].Iwasaki T, Yamashita K, Kuniyasu H, Kambe N, Org. Lett 2017, 19, 3691–3694. [DOI] [PubMed] [Google Scholar]
- [126].(a) Dryzhakov M, Moran J, ACS Catal 2016, 6, 3670–3673; [Google Scholar]; (b) Dryzhakov M, Richmond E, Li G, Moran J, J. Fluor. Chem 2017, 193, 45–51. [Google Scholar]
- [127].Wang J, Ogawa Y, Shibata N, iScience 2019, 17, 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Jaiswal AK, Goh KKK, Sung S, Young RD, Org. Lett 2017, 19, 1934–1937. [DOI] [PubMed] [Google Scholar]
- [129].Gupta R, Mandal D, Jaiswal AK, Young RD, Org. Lett 2021, 23, 1915–1920. [DOI] [PubMed] [Google Scholar]
- [130].Adam AT, Fronczek FR, Colby DA, Org. Lett 2020, 22, 2630–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Liu F, Zhang X, Qian Q, Yang C, Synthesis 2020, 52, 273–280. [Google Scholar]
- [132].Guo P, Tao M, Xu W-W, Wang A-J, Li W, Yao Q, Tong J, He C-Y, Org. Lett 2022, 24, 2143–2148. [DOI] [PubMed] [Google Scholar]
- [133].Zhou Q, Yan G, Adv. Synth. Catal 2022, 364, 1371–1387. [Google Scholar]
- [134].Zhu J, Perez M, Caputo CB, Stephan DW, Angew. Chem. Int. Ed 2016, 55, 1417–1421. [DOI] [PubMed] [Google Scholar]
- [135].Koperniku A, Liu H, Hurley PB, Eur. J. Org. Chem 2016, 871–886. [Google Scholar]
- [136].(a) Feng Z, Xiao Y-L, Zhang X, Acc. Chem. Res 2018, 51, 9, 2264–2278; [DOI] [PubMed] [Google Scholar]; (b) Dai J-J, Fang C, Xiao B, Yi J, Xu J, Liu Z-J, Lu X, Liu L, Fu Y, J. Am. Chem. Soc 2013, 135, 8436–8439. [DOI] [PubMed] [Google Scholar]
- [137].(a) Jaroschik F, Chem. Eur. J 2018, 24, 14572–14582; [DOI] [PubMed] [Google Scholar]; (b) Zhao F, Zhou W, Zuo Z, Adv. Synth. Catal 2021, 364, 234–267. [Google Scholar]
- [138].Hamel J-D, Paquin J-F, Chem. Commun 2018, 54, 10224–10239. [DOI] [PubMed] [Google Scholar]
- [139].(a) Champagne PA, Pomarole J, Thérien M-È, Benhassine Y, Beaulieu S, Legault CY, Paquin J-F, Org. Lett 2013, 15, 9, 2210–2213; [DOI] [PubMed] [Google Scholar]; (b) Champagne PA, Benhassine Y, Desroches J, Paquin J-F, Angew. Chem. Int. Ed 2014, 53, 13835–13839; [DOI] [PubMed] [Google Scholar]; (c) Willcox DR, Nichol GS, Thomas SP, ACS Catal 2021, 11, 3190–3197. [Google Scholar]
- [140].Vasilopoulos A, Golden DL, Buss JA, Stahl SS, Org. Lett 2020, 22, 5753–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wang F, Nishimoto Y, Yasuda M, J. Am. Chem. Soc 2021, 143, 20616–20621. [DOI] [PubMed] [Google Scholar]
- [142].(a) Luo Y-R in Comprehensive Handbook of Chemical Bond Energies, 2007, 1st Ed, CRC Press; [Google Scholar]; (b) Wilberg KB, Rablen PR, J. Am. Chem. Soc 1993, 115, 614–625. [Google Scholar]
- [143].Saboureau C, Troupel M, Sibille S, Périchon J, J. Chem. Soc., Chem. Commun 1989, 1138–1139. [Google Scholar]
- [144].(a) Clavel P, Lessene G, Biran C, Bordeau M, Roques N, Trévin S, de Montauzon D, J. Fluor. Chem 2001, 107, 301–310; [Google Scholar]; (b) Léger-Lambert M-P, Biran C, Serein-Spirau F, Bordeau M, Roques N, Marzouk H, Clavel P, Synthesis 1999, 829–834. [Google Scholar]
- [145].(a) Utsumi S, Katagiri T, Uneyama K, Tetrahedron 2012, 68, 1085–1091; [Google Scholar]; (b) Munoz SB, Ni C, Zhang Z, Wang F, Shao N, Mathew T, Olah GA, Prakash GKS, Eur. J. Org. Chem 2017, 16, 2322–2326. [Google Scholar]
- [146].Box JR, Avanthay ME, Poole DL, Lennox AJJ, Angew. Chem. Int. Ed 2023, 62, e202218195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chen K, Berg N, Gschwind R, König B, J. Am. Chem. Soc 2017, 139, 18444–18447. [DOI] [PubMed] [Google Scholar]
- [148].Wang H, Jui NT, J. Am. Chem. Soc 2018, 140, 163–166. [DOI] [PubMed] [Google Scholar]
- [149].Vogt DB, Seath CP, Wang H, Jui NT, J. Am. Chem. Soc 2017, 141, 13203–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].(a) Sap JBI, Straathof NJW, Knauber T, Meyer CF, Médebielle M, Buglioni L, Genicot C, Trabanco AA, Noël T, am Ende CW, Gouverneur V, J. Am. Chem. Soc 2020, 142, 9181–9187; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Luo Y-C, Tong F-F, Zhang Y, He C-Y, Zhang X, J. Am. Chem. Soc 2021, 143, 13971–13979; [DOI] [PubMed] [Google Scholar]; (c) Yan S-S, Liu S-H, Chen L, Bo Z-Y, Jing K, Gao T-Y, Yu B, Lan Y, Luo S-P, Yu D-G, Chem 2021, 7, 3099–3113. [Google Scholar]; (d) Sugihara N, Suzuki K, Nishimoto Y, Yasuda M, J. Am. Chem. Soc 2021, 143, 9308–9313. [DOI] [PubMed] [Google Scholar]
- [151].Simur TT, Ye T, Yu Y-J, Zhang F-L, Wang Y-F, Chin. Chem. Lett 2022, 33, 1193–1198. [Google Scholar]
- [152].Saito K, Umi T, Yamada T, Suga T, Akiyama T, Org. Biomol. Chem 2017, 15, 1767–1770. [DOI] [PubMed] [Google Scholar]
- [153].(a) Yoshida S, Shimomori K, Kim Y, Hosoya T, Angew. Chem. Int. Ed 2016, 55, 10406–10409; [DOI] [PubMed] [Google Scholar]; (b) Idogawa R, Kim Y, Shimomori K, Hosoya T, Yoshida S, Org. Lett 2020, 22, 9292–9297. [DOI] [PubMed] [Google Scholar]
- [154].Mandal D, Gupta R, Jaiswal AK, Young RD, J. Am. Chem. Soc 2020, 142, 2572–2578. [DOI] [PubMed] [Google Scholar]
- [155].Khanapur S, Lye K, Mandal D, Wee XJ, Robins EG, Young RD, Angew. Chem. Int. Ed 2022, 61, e202210917. [DOI] [PubMed] [Google Scholar]
- [156].Halder R, Ritter T, J. Org. Chem 2021, 86, 13873–13884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Luo C, Bandar JS, J. Am. Chem. Soc 2019, 141, 14120–14125. [DOI] [PubMed] [Google Scholar]
- [158].Wright SE, Bandar JS, J. Am. Chem. Soc 2022, 144, 13032–13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Zhou F-Y, Jiao L, Angew. Chem. Int. Ed 2022, 134, e202201102. [DOI] [PubMed] [Google Scholar]
- [160].(a) Gu W, Haneline MR, Douvris C, Ozerov OV, J. Am. Chem. Soc 2009, 131, 11203–11212; [DOI] [PubMed] [Google Scholar]; (b) Scott VJ, Ҫelenligil-Ҫetin R, Ozerov OV, J. Am. Chem. Soc 2005, 127, 2852–2853. [DOI] [PubMed] [Google Scholar]
- [161].Ikeda M, Matsuzawa T, Morita T, Hosoya T, Yoshida S, Chem. Eur. J 2020, 26, 12333–12337. [DOI] [PubMed] [Google Scholar]
- [162].(a) Pachecho MC, Purser S, Gouverneur V, Chem. Rev 2008, 108, 1943–1981; [DOI] [PubMed] [Google Scholar]; (b) Arimitsu S, Chem. Rec 2023, e202300021. [DOI] [PubMed] [Google Scholar]
- [163].Song S, Xu S, Chinese J Org. Chem 2023, 43, 411–425. [Google Scholar]
- [164].(a) Nishimine T, Taira H, Tokunaga E, Shiro M, Shibata N, Angew. Chem. Int. Ed 2016, 55, 359–363; [DOI] [PubMed] [Google Scholar]; (b) Okusu S, Okazaki H, Tokunaga E, Soloshonok VA, Shibata N, Angew. Chem. Int. Ed 2016, 55, 6744–6748; [DOI] [PubMed] [Google Scholar]; (c) Nishimine T, Taira H, Mori S, Matsubara O, Tokunaga E, Akiyama H, Soloshonok VA, Shibata N, Chem. Commun 2017, 53, 1128. [DOI] [PubMed] [Google Scholar]
- [165].(a) Zi Y, Lange M, Schultz C, Vilotijevic I, Angew. Chem. Int. Ed 2019, 58, 10727–10731; [DOI] [PubMed] [Google Scholar]; (b) Zi Y, Lange M, Vilotijevic I, Chem. Commun 2020, 56, 5689–5692. [DOI] [PubMed] [Google Scholar]
- [166].(a) Gholami H, Zell D, Trost BM, J. Am. Chem. Soc 2019, 141, 11446–11451; [DOI] [PubMed] [Google Scholar]; (b) Gholami H, Trost BM, Jiao Z, Chem. Sci 2021, 12, 10532–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].(a) Bergeron M, Johnson T, Paquin J-F, Angew. Chem. Int. Ed 2011, 50, 11112–11116; [DOI] [PubMed] [Google Scholar]; (b) Bergeron M, Guyader D, Paquin J-F, Org. Lett 2012, 14, 5888–5891. [DOI] [PubMed] [Google Scholar]
- [168].Paioti PHS, Gonsales SA, Xu S, Nikbakht A, Fager DC, Liu Q, Hoveyda AH, Angew. Chem. Int. Ed 2022, 61, e202208742. [DOI] [PubMed] [Google Scholar]
- [169].Watanabe D, Koura M, Saito A, Yanai H, Nakamura Y, Okada M, Sato A, Taguchi T, J. Fluor. Chem 2011, 132, 327–338. [Google Scholar]
- [170].(a) Akiyama S, Kubota K, Mikus MS, Paioti PHS, Romiti F, Liu Q, Zhou Y, Hoveyda AH, Ito H, Angew. Chem. Int. Ed 2019, 58, 11998–12003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Butcher TW, Yang JL, Hartwig JF, Org. Lett 2020, 22, 6805–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Butcher TW, Yang JL, Amberg WM, Watkins NB, Wilkinson ND, Hartwig JF, Nature 2020, 583, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Zeng H, Zhu C, Jiang H, Org. Lett 2019, 21, 1123–1133. [DOI] [PubMed] [Google Scholar]
- [173].Tian Q, Pei B, Wang C, Zhang C, Li Y, J. Org. Chem 2022, 87, 10908–10916. [DOI] [PubMed] [Google Scholar]
- [174].Zhu C, Sun M-M, Chen K, Liu H, Feng C, Angew. Chem. Int. Ed 2021, 60, 20237–20242. [DOI] [PubMed] [Google Scholar]
- [175].(a) Ye F, Spannenberg A, Neumann H, Wu L-W, Beller M, Nat. Commun 2021, 12, 3257; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Feng X-T, Ren J-X, Gao X, Min Q-Q, Zhang X, Angew. Chem. Int. Ed 2022, 61, e202210103; [DOI] [PubMed] [Google Scholar]; (c) Tang L, Liu Z-Y, She W, Feng C, Chem. Sci 2019, 10, 8701–8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Tan F, Yan G, Yu J, Chem. Commun 2019, 55, 13486–13505. [DOI] [PubMed] [Google Scholar]
- [177].Bégué J-P, Rock MH, Bonnet-Delpon D, J. Chem. Soc., Perkin Trans 1996, 1, 1409–1413. [Google Scholar]
- [178].Fuchibe K, Takahashi M, Ichikawa J, Angew. Chem. Int. Ed 2012, 51, 12059–12062. [DOI] [PubMed] [Google Scholar]
- [179].Fuchibe K, Hatta H, Oh K, Oki R, Ichikawa J, Angew. Chem. Int. Ed 2017, 56, 5890–5893. [DOI] [PubMed] [Google Scholar]
- [180].(a) Wang M, Pu X, Zhao Y, Wang P, Li Z, Zhu C, Shi S, J. Am. Chem. Soc 2018, 140, 9061–9065; [DOI] [PubMed] [Google Scholar]; (b) Huang Y, Hayashi T, J. Am. Chem. Soc 2016, 138, 12340–12343; [DOI] [PubMed] [Google Scholar]; (c) Jin Jang Y, Rose D, Mirabi B, Lautens M, Angew. Chem. Int. Ed 2018, 57, 16147–16151; [DOI] [PubMed] [Google Scholar]; (d) Yue W-J, Day CS, Martin R, J. Am. Chem. Soc 2021, 143, 6395–6400; [DOI] [PubMed] [Google Scholar]; (e) Lan Y, Yang F, Wang C, ACS Catal 2018, 8, 9245–9251; [Google Scholar]; (f) Zhang C, Lin Z, Zhu Y, Wang C, J. Am. Chem. Soc 2021, 143, 11602–11610. [DOI] [PubMed] [Google Scholar]
- [181].Paioti PHS, del Pozo J, Mikus MS, Lee J, Joo Koh M, Romiti F, Torker S, Hoveyda AH, J. Am. Chem. Soc 2019, 141, 19917–19934. [DOI] [PubMed] [Google Scholar]
- [182].Lang SB, Wiles RJ, Kelly CB, Molander GA, Angew. Chem. Int. Ed 2017, 56, 15073–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Yen-Pon E, Li L, Levitre G, Majhi J, McClain EJ, Voight EA, Crane EA, Molander GA, J. Am. Chem. Soc 2022, 144, 12184–12191; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Zhang Y, Niu Y, Guo Y, Wang J, Zhang Y, Liu S, Shen X, Angew. Chem. Int. Ed 2022, 61, e202212201. [DOI] [PubMed] [Google Scholar]
- [185].Liu Y, Tao X, Mao Y, Yuan X, Qiu J, Kong L, Ni S, Guo K, Wang Y, Pan Y, Nat Commun 2021, 12, 6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].An. L, Xu C, Zhang X, Nat. Commun 2017, 8, 1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Liao L, An R, Li H, Wu J-J, Zhao X, Angew. Chem. Int. Ed 2020, 59, 11010–11019. [DOI] [PubMed] [Google Scholar]
- [188].(a) Surmont R, Verniest G, De Kimpe N, Org. Lett 2009, 11, 2920–2923; [DOI] [PubMed] [Google Scholar]; (b) Hamel J-D, Paquin J-F, J. Fluor. Chem 2018, 216, 11–23; [Google Scholar]; (c) Sham HL, Betebenner DA, J. Chem. Soc., Chem. Commun 1991, 16, 1134–1135; [Google Scholar]; (d) Arimitsu S, Hammond GB, J. Org. Chem 2007, 72, 8559–8561; [DOI] [PubMed] [Google Scholar]; (e) Li P, Chai Z, Zhao G, Zhu S-Z, Synlett 2008, 16, 2547–2551. [Google Scholar]
- [189].Sheng Ng J, Hayashi T, Angew. Chem. Int. Ed 2021, 60, 20771–20775. [DOI] [PubMed] [Google Scholar]
- [190].O’Connor TJ, Khanh Mai B, Nafie J, Liu P, Toste FD, J. Am. Chem. Soc 2021, 143, 13759–13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Li T, Zhou C, Yan X, Wang J, Angew. Chem. Int. Ed 2018, 57, 4048–4052. [DOI] [PubMed] [Google Scholar]
- [192].(a) Rozatian N, Hodgson DRW, Chem. Commun 2021, 57, 683–712; [DOI] [PubMed] [Google Scholar]; (b) Lal GS, Pez GP, Syvret RG, Chem. Rev 1996, 96, 1737–1756. [DOI] [PubMed] [Google Scholar]
- [193].Lyu Z, Zhao Y, Buuh ZY, Gorman N, Goldman AR, Islam MS, Tang H-Y, Wang RE, J. Am. Chem. Soc 2021, 143, 1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].(a) Qian J, Yi W, Miao Y, Zhang J, Cai C, Zhang W, Org. Lett 2015, 17, 1090–1093; [DOI] [PubMed] [Google Scholar]; (b) Song Z, Huang X, Yi W, Zhang W, Org. Lett 2016, 18, 5640–5643. [DOI] [PubMed] [Google Scholar]
- [195].Yu Y-J, Zhang F-L, Peng T-Y, Wang C-L, Cheng J, Chen C, Houk KN, Wang Y-F, Science 2021, 371, 1232–1240. [DOI] [PubMed] [Google Scholar]
- [196].Hu J, Olah GA, Prakash GKS, J. Fluor. Chem 2001, 112, 355–360. [Google Scholar]
- [197].(a) Amii H, Kobayashi T, Hatamoto Y, Uneyama K, Chem. Commun 1999, 14, 1323–1324; [Google Scholar]; (b) Uneyama K, Mizutani G, Maeda K, Kato T, J. Org. Chem 1999, 64, 6717–6723; [DOI] [PubMed] [Google Scholar]; (c) Amii H, Kobayashi T, Terasawa H, Uneyama K, Org. Lett 2001, 3, 3103–3105. [DOI] [PubMed] [Google Scholar]
- [198].Meyer DN, Cortés González MA, Jiang X, Johansson-Holm L, Pourghasemi Lati M, Elgand M, Nordeman P, Antoni G, Szabó KJ, Chem. Commun 2021, 57, 8476–8479. [DOI] [PubMed] [Google Scholar]
- [199].Box JR, Atkins AP, Lennox AJJ, Chem. Sci 2021, 12, 10252–10258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Shen Z-J, Zhu C, Zhang X, Yang C, Rueping M, Guo L, Xia W, Angew. Chem. Int. Ed 2023, 62, e202217244. [DOI] [PubMed] [Google Scholar]
- [201].Doi R, Ohashi M, Ogoshi S, Angew. Chem. Int. Ed 2016, 55, 341–344. [DOI] [PubMed] [Google Scholar]
- [202].Ya J-H, Bellotti P, Heusel C, Glorius F, Angew. Chem. Int. Ed 2022, 61, e202115456. [DOI] [PubMed] [Google Scholar]
- [203].Schreiber ST, Granados A, Majhi J, Campbell MW, Patel S, Molander GA, Org. Lett 2022, 24, 46, 8542–8546. [DOI] [PMC free article] [PubMed] [Google Scholar]