


Abstract
A 24 triplet TGG·CCA repeat array shows length- and orientation-dependent propagation when present in the plasmid pUC18. When TGG24 is present as template for leading-strand synthesis, plasmid recovery is normal in all strains tested. However, when it acts as template for lagging-strand synthesis, plasmid propagation is seriously compromised. Plasmids carrying deletions in the 5′ side of this sequence can be isolated and products carrying 15 TGG triplets do not significantly interfere with plasmid propagation. Mutations in sbcCD, mutS and recA significantly improve the recovery of plasmids with TGG24 on the lagging-strand template. These findings suggest that TGG24 can fold into a structure that can interfere with DNA replication in vivo but that TGG15 cannot. Furthermore, since the presence of the MutS and SbcCD proteins are required for propagation interference, it is likely that stabilisation of mismatched base pairs and secondary structure cleavage are implicated. In contrast, there is no correlation of triplet repeat expansion and deletion instability with predicted DNA folding. These results argue for a dissociation of the factors affecting DNA fragility from those affecting trinucleotide repeat expansion–contraction instability.
INTRODUCTION
Non-random DNA sequences have the potential to adopt a number of unusual secondary structures that may interfere with DNA synthesis and/or cause instability. Palindromic sequences and some tandem repeats can form hairpins and pseudo-hairpins, oligopurine–oligopyrimidine sequences can form triplex h-DNA, alternating pyrimidine–purine sequences may form left-handed z-DNA, and G-rich sequences may form quadruplex structures. The processing of such folding anomalies in Escherichia coli has recently been reviewed (1).
The protein with a central role in processing folded DNA in E.coli is SbcCD. This is a nuclease that has been shown to cleave hairpin DNA in vitro (2) and to generate double-strand breaks at sites of palindromic DNA in vivo (3). These double-strand breaks can be repaired by homologous recombination with the sister chromosome (3,4) but can also lead to inhibition of replication if cleavage outstrips the ability of the cell to repair breaks (5,6). The eukaryotic homologues of SbcCD are known as Rad50/Mre11 (7). The eukaryotic complex, which includes the Xrs2/NbsI gene products, plays a central role in the response to double-strand breaks, and in double-strand break formation in yeast meiosis (8,9). In Saccharomyces cerevisiae the protein has been implicated in meiotic double-strand break formation and processing (10–12), mitotic homologous recombination (for discussion, see 13), non-homologous end-joining and telomere maintenance (14–19). Biochemical investigation of the human complex (hRad50/hMre11) has revealed a hairpin cleavage activity reminiscent of that observed for SbcCD (20,21).
The MutS protein is the key component of the methyl-directed mismatch repair system of E.coli which can recognise and bind to mismatched base pairs (22). Following this binding, MutL binds the MutS–DNA complex and allows MutH to nick a DNA strand at a hemi-methylated GATC site to initiate repair. In eukaryotes, two mismatch repair complexes exist that include MutS and MutL homologues, and it has been proposed that strand discrimination is accomplished by recognition of discontinuities that exist on the newly synthesised DNA strand (23).
Trinucleotide repeats are unstable and expansions of CAG·CTG, GAA·TTC and CGG·CCG repeats at particular chromosomal locations have been shown to cause at least 12 human genetic diseases (24). Furthermore, CGG·CCG repeats are responsible for folate-sensitive fragile sites (25). The mechanisms of instability are not yet understood but one of the arguments commonly raised is that non-B secondary structures lead to aberrant DNA replication or recombination that gives rise to instability (26). XGG repeats (CGG, AGG and TGG) have been shown to fold into various intrastrand and interstrand DNA secondary structures, including pseudo-hairpins, triplexes and quadruplexes (27 and references therein). CGG repeats can form both hairpins and quadruplexes under conditions where AGG and TGG repeats form only quadruplexes (27). Pause sites for DNA synthesis in vitro suggest that AGG repeats can also form intrastrand triplex structures (27) and these triplexes can be induced or stabilised by the binding of HMG protein (28–33). Although CGG is a disease-associated repeat, AGG and TGG repeats are not currently known to be associated with any trinucleotide expansion diseases. Nevertheless, XGG sequences are likely to share biological properties.
In this work, we have shown that when a TGG24 strand serves as the template for lagging-strand synthesis, plasmid propagation is prevented in the presence of mutS and sbcCD gene products. We argue that this is caused by MutS binding to mismatches formed in the folding of the TGG-repeat strand and by cleavage of the folded structure by the SbcCD nuclease. In mutS or sbcCD mutants a mutation in recA further facilitates plasmid replication and reduces the frequency of large deletions, suggesting that RecA protein may also play a role in preventing plasmid replication or in promoting deletions into flanking DNA. In contrast, small-scale expansions and deletions of triplet repeats are not significantly affected by the potential for secondary structure formation and we argue that these results suggest a dissociation between the requirements for repeated DNA to act as a fragile site and for expansion–contraction instability within the trinucleotide repeat array.
MATERIALS AND METHODS
Bacterial strains
The bacterial strains used are listed in Table 1.
Table 1. Escherichia coli strains used in this study.
| Strain | Relevant genotype | Source |
|---|---|---|
| JM83 |
mut+ rec+ sbcCD+ |
(34) |
| DL733 |
JM83 sbcCD::KmR |
(42) |
| DL887 |
JM83 recA::CmR |
P1 transduction of JM83 |
| |
|
Origin of recA::CmR (43) |
| DL888 |
JM83 recA::CmR |
P1 transduction of DL887 |
| |
sbcCD::KmR |
Origin of recA::CmR (43) |
| DL902 |
JM83 mutS::miniTn10 |
P1 transduction of JM83 |
| |
|
Origin of mutS:: miniTn10 (44) |
| DL905 |
JM83 mutS::miniTn10 |
P1 transduction of DL733 |
| |
sbcCD::KmR |
Origin of mutS:: miniTn10 (44) |
| DL916 |
JM83 mutL::Tn10 |
P1 transduction of JM83 |
| |
|
Origin of mutL::Tn10 (Frank Stahl) |
| DL1084 |
JM83 mutS::miniTn10 |
P1 transduction of DL902 |
| recA::CmR | Origin of recA::CmR (43) |
Plasmids
The plasmids used were derivatives of pUC18 (34). The plasmid pCCA24 with the sequence (CCA)24 (on the lagging strand) cloned in the EcoRI site of pUC18 plasmid was obtained from C. Abbott (University of Edinburgh). Inversion of the trinucleotide repeat array to generate plasmid pTGG24 was performed by cleavage with EcoRI followed by ligation.
Enzymes, antibiotics and biochemicals
EcoRI and PstI were purchased from Promega (Madison, WI), T4 DNA ligase from New England Biolabs (Beverly, MA), ampicillin from Boehringer Mannheim (East Sussex, UK).
Media and bacterial cultivation
Luria–Bertani (LB) broth was used and ampicillin was added at a concentration of 100 µg/ml. Cultivation of cells was at 37°C. Transformation was carried out by the CaCl2 method (35). Transduction was performed as described by Sambrook et al. (35).
Isolation of plasmid DNAs and agarose gel electrophoresis
Plasmid DNA was prepared using the method and kit provided by Qiagen (West Sussex, UK). DNA cycle-sequencing was performed using a kit from PE Applied Biosystems (Warrington, UK). Agarose gel electrophoresis was on 0.8% gels (Flowgen, Staffordshire, UK).
Examination of the repeat tract instability
Populations of plasmids were examined using the method of Schmidt et al. (36). Briefly, monomeric plasmid DNA was used to transform E.coli strains of interest and plasmid DNA prepared from a population of transformants. Cells from approximately 500 primary transformants were harvested in 5 ml LB broth and 50 µl of this suspension was diluted into 5 ml LB broth and grown for 24 h. This corresponds to approximately 30 generations of cell growth. Plasmid DNA was isolated and cleaved with EcoRI. The fragments were end-labelled with [35S]dATP using DNA polymerase I Klenow fragment and resolved on 5% native polyacrylamide gels. Bands were visualised using either X-ray film or a Molecular Dynamics PhosphorImager.
RESULTS
Orientation-dependent propagation of TGG·CCA repeats: implication of mutS and recA
To investigate the propagation and instability of TGG·CCA repeats in E.coli, we constructed the two orientations of this trinucleotide repeat array at the EcoRI site of the plasmid pUC18 (see Materials and Methods). When rec+ or recA mutant cells were transformed with each of the plasmids, a difference in behaviour was immediately obvious. Plasmids with the TGG repeat on the lagging-strand template gave rise to variable sized colonies ranging from normal to small. In contrast, plasmids with the CCA repeat on the lagging-strand template were all of an approximately uniform normal size. In order to investigate the basis of this difference, plasmid DNA was prepared from overnight cultures derived from small and normal sized colonies and analysed by agarose gel electrophoresis. As can be seen in Figure 1A, DNA of plasmids containing the CCA repeat on the lagging-strand template was easily prepared. However, DNA from the colonies with the TGG repeat on the lagging-strand template was barely visible even from cultures derived from normal sized colonies. This suggested that plasmid propagation was seriously compromised by the TGG repeat on the lagging-strand template. In order to determine whether the presence of MutS might influence this orientation-dependent propagation, transformations of mutS and mutS recA strains were performed and DNA was isolated as before. The mutS strain still revealed variable colony size when the TGG repeat was the template for the lagging strand, but the mutS recA strain gave rise to only normal sized colonies. DNA analysis revealed that some improved recovery of DNA from the normal sized colonies could be obtained in the mutS host and that normal recovery was possible from the mutS recA strain.
Figure 1.

(A) Recovery of DNA from mutS and recA strains. Lane 1, pCCA24 recovered from mut+ rec+ (JM83); lane 2, pTGG24 recovered from a large colony of mut+ rec+ (JM83); lane 3, pTGG24 recovered from a small colony of mut+ rec+ (JM83); lane 4, pCCA24 recovered from mutS (DL902); lane 5, pTGG24 recovered from a large colony of mutS (DL902); lane 6, pTGG24 recovered from a small colony of mutS (DL902); lane 7, pCCA24 recovered from recA (DL887); lane 8, pTGG24 recovered from a large colony of recA (DL887); lane 9, pTGG24 recovered from a small colony of recA (DL887); lane 10, pCCA24 recovered from mutS recA (DL1084); lane 11, pTGG24 recovered from a large colony of mutS recA (DL1084); lane 12, pCCA24 cut with PstI to mark the position of linear monomeric DNA. (B) Recovery of DNA in mutS and mutL strains from pooled colonies and after 24 h of cultivation in liquid. Lane 1, pTGG24 recovered from a pool of ~500–700 colonies of mutL (DL916); lane 2, pTGG24 recovered from 24 h cultivation in liquid of mutL pooled colonies; lane 3, pTGG24 recovered from a pool of ~500–700 colonies of mutS (DL902); lane 4, pTGG24 recovered from 24 h cultivation in liquid of mutS pooled colonies; lane 5, pTGG24 recovered from a pool of ~500–700 colonies of mut+ rec+ (JM83); lane 6, pTGG24 recovered from 24 h cultivation in liquid of mut+ rec+ pooled colonies.
To confirm that the inability to isolate DNA was caused by plasmid loss during propagation, DNA was prepared from pooled colonies taken directly from the initial transformation plates. Since selection for ampicillin resistance is good on plates it was expected that recovery of plasmid would be significantly improved compared to the analysis of overnight cultures shown in Figure 1A. This suspension of pooled colonies was also diluted 100-fold into broth and grown for 24 h in liquid and DNA prepared again. This period of growth in liquid was expected to allow plasmid loss after exhaustion of the ampicillin present initially. As shown in Figure 1B the mutS mutation reduces plasmid loss relative to mut+. Furthermore, a mutL mutation does not prevent plasmid loss suggesting that the effect of mutS is not caused by a deficiency in mismatch repair but is due to an absence of the MutS.
In order to further quantify the inhibition of propagation of the plasmids with the TGG sequence on the template for the lagging strand, colonies obtained after transformation were inoculated into broth and grown overnight. They were then plated in the presence and absence of ampicillin. This test is sensitive to differences in propagation stability between very unstable situations but does not measure instability at the more stable end of the spectrum. This is because a >100-fold lowering of plasmid copy number is needed before this form of segregational instability comes into play over a short period of cultivation, and long periods of cultivation could not be used because of selection for repeat array deletion. The mut+ rec+ strain JM83 showed extreme propagation instability with 79% plasmid-free cells arising. The recA derivative showed 35% loss, the mutL strain showed 40% loss whereas the mutS and the mutS recA strains showed no significant loss. All these values represent the average of five cultures. Assuming a Poisson distribution of plasmids per cell, the wild-type situation represents a copy number of 0.2, recA a copy number of 1.0, mutL a copy number of 0.9 and mutS (and mutS recA) a copy number between 3 and infinity (but not measurable by this method).
Orientation-dependent propagation of TGG·CCA repeats: implication of sbcCD
In order to test whether the SbcCD nuclease was implicated in preventing the propagation of TGG repeats on the lagging-strand template, transformation of sbcCD mutant cells was carried out. As can be seen in Figure 2A, plasmid DNA was recovered from sbcCD and recA sbcCD mutants irrespective of the orientation of the TGG·CCA repeat tract. As observed previously, plasmids with the TGG repeats on the lagging-strand template were not well recovered from rec+ and recA strains. As expected from these results, and those presented in Figure 1, plasmid DNA was recovered in both orientations from an sbcCD mutS double mutant (Fig. 2B). This is consistent with the fact that the plasmid containing these repeats was initially obtained with the TGG repeats on the leading-strand template and that inversion of the repeat tract generated a clone producing very little DNA in wild-type cells (see Materials and Methods).
Figure 2.

(A) Recovery of DNA from recA and sbcCD strains. Lane 1, pCCA24 recovered from mut+ rec+ (JM83); lane 2, pTGG24 recovered from mut+ rec+ (JM83); lane 3, pCCA24 recovered from recA (DL887); lane 4, pTGG24 recovered from recA (DL887); lane 5, pCCA24 recovered from sbcCD (DL733); lane 6, pTGG24 recovered from a large colony of sbcCD (DL733); lane 7, pCCA24 recovered from recA sbcCD (DL888); lane 8, pTGG24 recovered from recA sbcCD (DL888). (B) Recovery of DNA from mutS and mutS sbcCD strains. Lane 1, pCCA24 recovered from mutS (DL902); lane 2, pTGG24 recovered from mutS (DL902); lane 3, pCCA24 recovered from mutS sbcCD (DL905); lane 4, pTGG24 recovered from mutS sbcCD (DL905); lane 5, pCCA24 cut with PstI as a marker. (C) Recovery of DNA in sbcCD, recA sbcCD and mutS sbcCD strains from pooled colonies and after 24 h of cultivation in liquid. Lane 1, pCCA24 cut with PstI as a marker; lane 2, pTGG24 recovered from a pool of ~500–700 colonies of sbcCD (DL733); lane 3, pTGG24 recovered from 24 h cultivation in liquid of sbcCD pooled colonies; lane 4, pTGG24 recovered from a pool of ~500–700 colonies of recA sbcCD (DL888); lane 5, pTGG24 recovered from 24 h cultivation in liquid of recA sbcCD pooled colonies; lane 6, pTGG24 recovered from a pool of ~500–700 colonies of mutS sbcCD (DL905); lane 7, pTGG24 recovered from 24 h cultivation in liquid of mutS sbcCD pooled colonies; lane 8, pTGG24 recovered from a pool of ~500–700 colonies of mut+ rec+ (JM83); lane 9, pTGG24 recovered from 24 h cultivation in liquid of mut+ rec+ pooled colonies.
To test for the loss of plasmids during propagation in liquid, DNA prepared from pooled colonies, taken directly from the initial transformation plates, was compared to DNA prepared after 24 h cultivation in liquid. It can be seen in Figure 2C that plasmid DNA was recovered from sbcCD, sbcCD recA and sbcCD mutS strains but not from rec+ mut+ cells.
Propagation stability was confirmed by plating overnight cultures on media with and without ampicillin. As with the mutS strain no propagation instability was detected for either the sbcCD or sbcCD recA strains suggesting (as for mutS) a copy number of between 3 and infinity but not measurable by this method.
Length-dependent propagation of TGG·CCA repeats
A plasmid containing 15 TGG·CCA repeats with the TGG sequence on the lagging-strand template was isolated as a spontaneous deletion product in the mut+ rec+ sbcCD+ strain JM83. This plasmid was used to re-transform JM83 to test whether this shorter length of repeat could be propagated in the presence of MutS, RecA and SbcCD. As shown in Figure 3, DNA of this plasmid could be recovered in this strain. This argues that a threshold for stable structure formation recognised by SbcCD in the presence of MutS protein occurs between 15 and 24 triplets. Sequencing of the repeat array revealed that the deletion that had generated the shorter repeat array had eliminated the EcoRI site on the 5′ side of the TGG sequence (on the template for lagging strand). This is consistent with a deletion mechanism where 15 TGG repeats had been copied on the lagging strand prior to dissociation of the newly synthesised strand from its template. Reannealing to a sequence downstream of the repeat array would have allowed deletion. This mechanism is very reminiscent of strand-slippage from inside a palindromic sequence to a target sequence downstream that has been observed to occur preferentially on the lagging strand (37–39).
Figure 3.
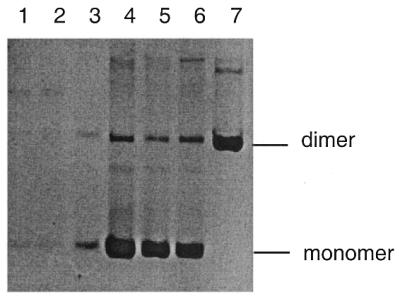
Recovery of pTGG24 and pTGG15 from mut+ sbcCD+ rec+ cells (JM83). Lanes 1–3, three pTGG24 transformants; lanes 4–6, three pTGG15 transformants; lane 7, dimer of pCCA24 as a marker.
Deletions extending into flanking sequence
The frequency of this type of deletion, extending from within the repeat array to a site within the flanking DNA, could be assessed by cleavage of plasmid DNA with EcoRI and PstI. Since the primary repeat array was flanked with EcoRI sites and a deletion removing the EcoRI site 5′ to the TGGs on the lagging-strand template would leave the 3′ EcoRI site intact, a small EcoRI fragment of 78 bp was diagnostic of the conservation of both sites, whereas the absence of such a fragment accompanied by the generation of a 75–85 bp EcoRI–PstI fragment was consistent with a 5′ deletion. This test was used to screen over 100 DNA preparations for the likelihood of 5′ deletions. In six cases, sequencing was used to confirm that the deletions were in the 5′ end of the TGG array and revealed that three to nine repeats had been deleted in different plasmids. As can be seen in Table 2, 5′ deletions are quite common in all the strains tested. This may reflect the difficulty in replicating this sequence even in permissive strains and a selective advantage of deletion products. The lowest level of deletion products is seen in the recA mutS and recA sbcCD mutants. We have not established whether the stabilising effect of recA is due to the promotion of deletions in a recA+ host or a reduction in selective disadvantage of the intact array in the absence of RecA. The latter possibility is suggested by the improved recovery of plasmid in the recA mutS strain relative to its mutS counterpart (Fig. 1A). In all cases of deletions extending from within the repeat array to a sequence outside the array, the EcoRI site deleted was that located 5′ of the TGG sequence on the lagging-stand template. This strongly suggests that the newly synthesised lagging strand enters the trinucleotide repeat sequence, stalls, dissociates and reanneals downstream in unique DNA, as demonstrated previously for palindromes (37–39).
Table 2. Analysis of 5′ deletion instability.
| Host | Relevant genotype | Clones with intact repeat tractsa | Clones with 5′ EcoRI sites deletedb | Total clones analysed | % Deletant |
|---|---|---|---|---|---|
| DL902 | mutS | 9 | 15 | 24 | 63 |
| DL733 | sbcCD | 13 | 17 | 30 | 57 |
| DL905 | mutS sbcCD | 12 | 13 | 15 | 52 |
| DL1084 | recA mutS | 16 | 5 | 21 | 24 |
| DL888 | recA sbcCD | 18 | 8 | 26 | 31 |
aIntact repeats as revealed by the presence of the 78 bp EcoRI fragment including the TGG repeats.
b5′ Deletions as revealed by the absence of the 78 bp EcoRI fragment including the TGG repeats but the presence of a 75–85 bp EcoRI–PstI fragment.
Expansion and contraction instability of the repeat array
In order to assess the contribution of secondary structure formation to instability within the repeat array, we measured array instability using the population method developed by Schmidt et al. (36). After cleavage with EcoRI and end-labelling, the expansion and deletion products were separated by native polyacrylamide gel electrophoresis. Because of the difficulties in propagating the TGG repeats on the lagging strand template, we chose to examine instability in mutS recA and sbcCD recA strains. In these strains recovery of plasmid is possible for both orientations of the repeat sequence but the predictions for the persistence of secondary structures are different. It is predicted that in the mutS recA strain, the absence of MutS will remove any stabilisation of folded structures and the presence of the SbcCD nuclease will destroy any secondary structures that do form. In contrast, in the sbcCD recA strain it is predicted that structures will be stabilised by MutS binding and the absence of the SbcCD nuclease will contribute to their persistence. As can be seen in Figure 4, there is no substantial difference in the overall instability in either strain. Furthermore, there is not a substantial orientation dependence in instability as would have been predicted if TGG repeat secondary structures played a major role in expansion and contraction within the repeat array. The mutS recA strain does show filling-in of the +1 and –1 triplet bands as has been seen for CAG·CTG repeats (36), confirming that mismatch repair deficiency allows single triplet mispairs to escape correction.
Figure 4.
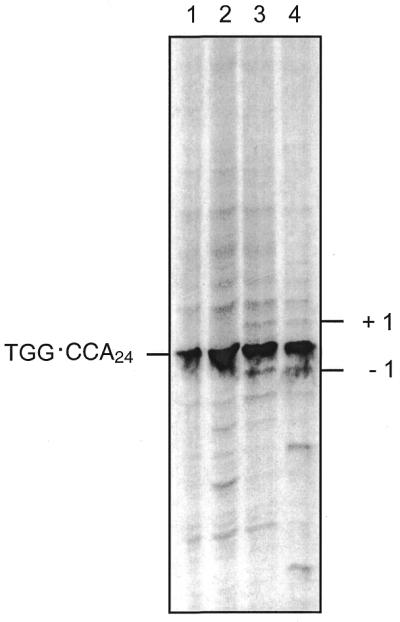
Analysis of expansion–contraction repeat instability. Expansion and contraction of the repeat array bounded by two EcoRI sites was analysed by EcoRI fragment size analysis as described in Materials and Methods. Because the visualisation of instability by this method requires the retention of both EcoRI sites, this method only measures changes that lie entirely within the repeat array. Lane 1, pCCA24 after 30 generations of propagation in recA sbcCD (DL888); lane 2, pTGG24 after 30 generations of propagation in recA sbcCD (DL888); lane 3, pCCA24 after 30 generations of propagation in recA mutS (DL1084); lane 4, pTGG24 after 30 generations of propagation in recA mutS (DL1084).
DISCUSSION
In this work we have shown that the propagation of a TGG·CCA repeat tract consisting of 24 triplets is strongly orientation dependent in rec+ and recA mutant strains. The orientation that causes inhibition of plasmid propagation is that where the TGG repeat strand forms the template for lagging-strand synthesis. This is consistent with observations in vitro that the G-rich strands of XGG repeats can form quadruplex structures under physiological conditions (27). We have observed that the propagation of this orientation of the repeated sequence is substantially relieved in a mutS but not a mutL strain. This argues that the presence of the MutS protein (and not active mismatch repair) contributes to propagation-inhibition and raises the possibility that MutS binding may promote or stabilise an unusual DNA secondary structure containing mismatches. This structure may be an intermediate in the folding pathway or the final structure itself. A small effect of mutL was detected on segregational instability and may be caused by stronger binding of the MutS–MutL complex than MutS alone.
Propagation is fully restored in a mutS recA mutant, arguing that the RecA protein directly or indirectly contributes to poor replication of this sequence in a mutS mutant. The role of RecA is unknown but it has been shown that TG-rich sequences are preferentially bound by RecA protein in SELEX experiments (40). TG repeats bind RecA strongly and can initiate pairing with homologous duplex but strand-exchange is substantially inhibited (41). It may be that RecA-promoted recombination is initiated between sister-strands but cannot be completed and this interferes with DNA replication. The effect of recA can also be seen in the segregational instability detected in a mut+ background. Here, both wild-type and recA strains show evidence of very low copy numbers but plasmid loss from the recA strain is lower than wild-type suggesting a slightly higher copy number.
Propagation is also restored in sbcCD and sbcCD recA mutants. These observations are significant since the SbcCD nuclease is known to attack and degrade folded DNA structures in vitro (2) and inhibits the propagation of long palindromic DNA sequences in vivo. The implication of SbcCD in the propagation of TGG repeats is good indirect evidence that they are forming an unusual DNA secondary structure in vivo. The propagation of these TGG repeats in sbcCD mutants that are mutS+ and in mutS mutants that are sbcCD+ argue that MutS binding favours the persistence of a secondary structure that can be cleaved by SbcCD. It must be the processing of the structure by SbcCD not its formation alone that results in propagation inhibition.
The nature of the secondary structure formed is not known but a substantial improvement in propagation is obtained by a deletion of nine repeats to generate a repeat array of 15 triplets. This argues that more than 15 of the 24 triplets in the original array are included in the structure that inhibits DNA replication. Given the strong inhibitory effect of quadruplex structures on DNA synthesis and the observation of substantial effects of length on stability in vitro (27) it is attractive to suggest that this type of structure may be responsible for the replication inhibition observed. Deletions of three to nine triplets at the 5′ end of the TGG strand are common and are presumably caused by the newly synthesised lagging strand stalling, dissociating and reannealing with a sequence downstream of the repeat array as has been shown to occur for palindromic sequences (37–39).
The evidence discussed above implies that DNA secondary structures form in TGG repeats when they are located on the template for lagging-strand synthesis and this leads to propagation inhibition that can be alleviated by large deletions. In contrast, small-scale expansion–contraction instability within the repeat tract does not appear to be substantially affected by secondary structure. Expansion–contraction instability is not particularly orientation dependent and is not particularly different in an sbcCD recA mutant relative to a mutS recA mutant. An sbcCD recA mutant is predicted to have substantially more folded DNA than a mutS recA mutant since the former lacks the enzyme SbcCD that destroys structures but contains the protein MutS that binds mismatches and potentially stabilises structures. The reverse is true of the latter.
These results argue for a dissociation between the phenomena of fragility and instability in trinucleotide repeat arrays. Our argument is that the inhibition of propagation that we observe in E.coli is akin to DNA fragility in human cells. The fragile nature of the DNA in E.coli is clearly illustrated by the effects of sbcCD, genes known to encode a nuclease that can attack and degrade folded DNA. The molecular nature of DNA fragility in human cells is not known but a role for nucleolytic degradation of folded DNA has not been ruled out. In contrast, expansion–deletion instability within the repeat array does not seem to be associated with the formation of secondary structures and is not influenced by SbcCD. A model outlining our understanding of these interactions is shown in Figure 5. It remains to be seen whether a similar dissociation between fragility and instability can be demonstrated in human cells.
Figure 5.

Illustration of how secondary structures may differentially affect genetic instability and chromosome fragility. Small-scale slippages may not be affected by the formation of secondary structures and arise simply through strand-slippage on a repetitive template. In contrast, DNA fragility may arise when DNA folds into unusual secondary structures such as hairpins, pseudo-hairpins, triplexes or quadruplexes. The ultimate cause of fragility may be site and organism dependent. We have shown that in E.coli propagation inhibition of TGG repeat arrays can simply be caused by the action of the nuclease SbcCD in conjunction with the mismatch binding protein MutS and the recombination protein RecA. The implication of SbcCD nuclease in propagation inhibition suggests a form of DNA fragility that may not be unique. In human chromosomes DNA fragility may also be initiated by DNA folding. This may initiate a series of events such as the repositioning of nucleosomes and may include the action of proteins such as hMsh1, hMsh2, hRad51 and hRad50/hMre11/Nbs1, the human homologues of MutS, RecA and SbcCD.
The severe problem in propagating TGG·CCA repeats in E.coli observed here and the tendency of this sequence to form quadruplexes that can inhibit DNA synthesis in vitro (27) suggest that this sequence may be intrinsically difficult to replicate. In this context, it is interesting to note that in the human genomic sequence currently available in databases, all the very long TGG·CCA microsatellites that can be found (up to 1 kb in length) are multiply interrupted. In fact, TGG15 is the longest uninterrupted human TGG repeat sequence deposited in the databases at the time of submission of this paper. It is possible that DNA fragility at TGG repeats has not been detected in human chromosomes because the potential fragility caused by this sequence is so severe that long uninterrupted arrays are strongly selected against.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr C. M. Abbott, University of Edinburgh, for providing the (TGG·CCA)24 trinucleotide repeat plasmid, and John Connelly and Gareth Cromie for invaluable suggestions and comments. This work is supported by a grant from the Medical Research Council (UK).
REFERENCES
- 1.Leach D.R.F. (1999) In Smith,P.J. and Jones,C.J. (eds), DNA Recombination and Repair. Oxford University Press, Oxford, UK, pp. 1–15.
- 2.Connelly J.C., Kirkham,L.A. and Leach,D.R.F. (1998) Proc. Natl Acad. Sci. USA, 95, 7969–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leach D.R.F., Okely,E.A. and Pinder,D.J. (1997) Mol. Microbiol., 26, 597–606. [DOI] [PubMed] [Google Scholar]
- 4.Cromie G.A., Millar,C.B., Schmidt,K.H. and Leach,D.R.F. (2000) Genetics, 154, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindsey J.C. and Leach,D.R.F. (1989) J. Mol. Biol., 206, 7024–7027. [Google Scholar]
- 6.Shurvinton C.E., Stahl,M.M. and Stahl,F.W. (1987) Proc. Natl Acad. Sci. USA, 84, 1624–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharples G.J. and Leach,D.R.F. (1995) Mol. Microbiol., 17, 1215–1217. [DOI] [PubMed] [Google Scholar]
- 8.Haber J.E. (1998) Cell, 95, 583–586. [DOI] [PubMed] [Google Scholar]
- 9.Ohta K., Nicolas,A., Furuse,M., Nabetani,A., Ogawa,H. and Shibata,T. (1998) Proc. Natl Acad. Sci. USA, 95, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alani E., Subbiah,S. and Kleckner,N. (1989). Genetics, 122, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov E.L., Korolev,V.G. and Fabre,F. (1992) Genetics, 132, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ajimura M.M., Leem,S.H. and Ogawa,H. (1993) Genetics, 133, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paques F. and Haber,J.E. (1999) Microbiol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsukamoto Y. and Ikeda,H. (1998) Genes Cells, 3, 135–144. [DOI] [PubMed] [Google Scholar]
- 15.Kironmai K.M. and Muniyappa,K. (1997) Genes Cells, 2, 443–455. [DOI] [PubMed] [Google Scholar]
- 16.Nugent C.I., Bosco,G., Ross,L.O., Evans,S.K., Salinger,A.P., Moore,J.K., Haber,J.E. and Lundblad,V. (1998) Curr. Biol., 8, 657–660. [DOI] [PubMed] [Google Scholar]
- 17.Chamankhah M. and Xiao,W. (1999) Nucleic Acids Res., 27, 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le S., Moore,J.K., Haber,J.E. and Greider,C.W. (1999) Genetics, 152, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreau S., Ferguson,J.R. and Symington,L.S. (1999) Mol. Cell. Biol., 19, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paull T.T. and Gellert,M. (1998) Mol. Cell., 1, 969–979. [DOI] [PubMed] [Google Scholar]
- 21.Paull T.T. and Gellert,M. (1999) Genes Dev., 13, 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Modrich P. (1991) Annu. Rev. Genet., 25, 229–253. [DOI] [PubMed] [Google Scholar]
- 23.Jiricny J. (1998) EMBO J., 17, 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards R.I. and Sutherland,G.R. (1997) Trends Biochem. Sci., 22, 432–436. [DOI] [PubMed] [Google Scholar]
- 25.Sutherland G.R., Baker,E. and Richards,R.I. (1998) Trends Genet., 14, 501–506. [DOI] [PubMed] [Google Scholar]
- 26.McMurray C.T. (1999) Proc. Natl Acad. Sci. USA, 96, 1823–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Usdin K. (1998) Nucleic Acids Res., 26, 4078–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruss A., Moretto,V., Ehrlich,S.D., Duwat,P. and Dabert,P. (1991) J. Biol. Chem., 266, 6667–6669. [PubMed] [Google Scholar]
- 29.Suda T., Mishima,Y., Asakura,H. and Kominami,R. (1995) Nucleic Acids Res., 23, 3771–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suda T., Mishima,Y., Takayanagi,K., Asakura,H., Odani,S. and Kominami,R. (1996) Nucleic Acids Res., 24, 4733–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takayanagi K., Mishima,Y. and Kominami,R. (1997) DNA Res., 4, 241–247. [DOI] [PubMed] [Google Scholar]
- 32.Mishima Y., Kaizu,H. and Kominami,R. (1997) J. Biol. Chem., 272, 26578–26584. [DOI] [PubMed] [Google Scholar]
- 33.Mishima Y., Suda,T. and Kominami,R. (1996) J. Biochem., 119, 805–810. [DOI] [PubMed] [Google Scholar]
- 34.Yanisch-Perron C., Viera,J., and Messing,J. (1985) Gene, 33, 103–119. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Schmidt K.H., Abbott,C. and Leach,D.R.F. (2000) Mol. Microbiol., 35, 463–471. [DOI] [PubMed] [Google Scholar]
- 37.Trinh T.Q. and Sinden,R.R. (1991) Nature, 352, 544–547. [DOI] [PubMed] [Google Scholar]
- 38.Rosche W.A., Trinh,T.Q. and Sinden,R.R. (1995) J. Bacteriol., 177, 4385–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinder D.J., Blake,C.E., Lindsey,J.C. and Leach,D.R.F. (1998) Mol. Microbiol., 28, 719–727. [DOI] [PubMed] [Google Scholar]
- 40.Tracy R.B. and Kowalczykowski,S.C. (1996) Genes Dev., 10, 1890–1903. [DOI] [PubMed] [Google Scholar]
- 41.Dutreix M. (1997) J. Mol. Biol., 273, 105–113. [DOI] [PubMed] [Google Scholar]
- 42.Connelly J.C., de Leau,E.S., Okely,E.A. and Leach,D.R.F. (1997) J. Biol. Chem., 272, 19819–19826. [DOI] [PubMed] [Google Scholar]
- 43.Wertman K.F., Wyman,A.R. and Botstein,D. (1986) Gene, 49, 253–262. [DOI] [PubMed] [Google Scholar]
- 44.Miller J.H. (1991) A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.