

Abstract
Aberrant neurodevelopment in Down syndrome (DS)—caused by triplication of human chromosome 21—is commonly attributed to gene dosage imbalance, linking overexpression of trisomic genes with disrupted developmental processes, with DYRK1A particularly implicated. We hypothesized that regional brain DYRK1A protein overexpression in trisomic mice varies over development in sex-specific patterns that may be distinct from Dyrk1a transcription, and reduction of Dyrk1a copy number from 3 to 2 in otherwise trisomic mice reduces DYRK1A, independent of other trisomic genes. DYRK1A overexpression varied with age, sex, and brain region, with peak overexpression on postnatal day (P) 6 in both sexes. Sex-dependent differences were also evident from P15-P24. Reducing Dyrk1a copy number confirmed that these differences depended on Dyrk1a gene dosage and not other trisomic genes. Trisomic Dyrk1a mRNA and protein expression were not highly correlated. Sex-specific patterns of DYRK1A overexpression during trisomic neurodevelopment may provide mechanistic targets for therapeutic intervention in DS.
Keywords: Down syndrome, Neonatal, Development, Cerebral cortex, Cerebellum, Hippocampus
1. Introduction
Trisomy of genes on human chromosome 21 (Hsa21)—the smallest human chromosome with 46.7 Mbp of DNA that contains 233 protein coding, 423 noncoding, and 188 pseudogenes (ensembl.org)—leads to the ~80 clinical phenotypes associated with DS (Antonarakis et al., 2020). How dosage imbalance of Hsa21 genes causes DS is not known. The relationship between trisomic genes and DS phenotypes is complex and likely involves multiple genes, genetic regions, or genetic mechanisms associated with particular DS phenotypes (Korbel et al., 2009; Lyle et al., 2009) that emerge over time as a consequence of altered developmental expression of trisomic genes (Roper and Reeves, 2006). There are multiple hypotheses of how trisomy 21 (Ts21) causes DS phenotypes including: the pervasively accepted effect of an approximate 1.5-fold increase in expression of trisomic genes in cells and tissues across development (gene dosage overexpression); that certain overexpressed trisomic genes are most important to cause phenotypes and expression of others are modulated (dosage compensation); or that trisomy may directly or indirectly affect homeostasis in cells, tissues, organs and systems (Ait Yahya-Graison et al., 2007; Antonarakis et al., 2020; Duchon et al., 2021; Hwang et al., 2021; Kojima and Cimini, 2019; Moyer et al., 2021). These different hypotheses may not be mutually exclusive in the causation of DS phenotypes and may be evaluated differently if considering all the genes at dosage imbalance or a select number of genes.
Genetic mouse models of DS provide an essential means to understand genotype-phenotype relationships in DS, yet the models may vary according to phenotype and triplicated genomic content (Antonarakis et al., 2020; Duchon and Herault, 2016; Herault et al., 2017). The Ts65Dn DS mouse model has been widely used and offers a third copy of approximately half of the Hsa21 orthologs on a freely segregating extra marker chromosome (Reeves et al., 1995). Duplications of homologous mouse chromosomal regions have generated Dp1Yey and many other models with unique genetic contributions to understand DS-related phenotypes (Herault et al., 2017). TcMAC21 is a new DS mouse model with a near complete human chromosome 21 and DS-like abnormalities (Kazuki et al., 2022). The Ts65Dn DS mouse model demonstrates embryonic and early postnatal gene expression, neuroanatomical, and developmental behavioral deficits like those observed in DS, whereas other DS models, including Dp1Yey and TcMAC21, do not have or have not yet been tested for these phenotypes (Aziz et al., 2018; Goodliffe et al., 2016).
Cognitive impairment is a hallmark DS phenotype affecting all individuals with Ts21 (Antonarakis et al., 2020) with regions of disproportionately small brain volume including the cerebrum, cerebellum, and hippocampus (Aylward et al., 1999; Kesslak et al., 1994; Pinter et al., 2001; Raz et al., 1995; Weis et al., 1991). Dysregulated neurogenesis, atypical cell cycle regulation, and abnormal cellular differentiation are hypothesized to contribute to the developmental deficits in brain structures in individuals with DS (Contestabile et al., 2007; Contestabile et al., 2017; Guidi et al., 2008; Pinter et al., 2001; Stagni et al., 2018). Translational studies in DS mouse models have identified relevant cognitive phenotypes associated with candidate neural correlates involving developmental alterations in regional brain morphology, neuronal structure and connectivity, and synaptic plasticity (Belichenko et al., 2015; Belichenko et al., 2007; Contestabile et al., 2017; Edgin et al., 2012; Roper et al., 2020; Stringer et al., 2017b). These structural abnormalities and deficits in synaptic function and plasticity are implicated as primary causes of impaired learning, memory, and motor disabilities.
Sex differences in developmental phenotypes have been generally under-studied but some information has been reported in individuals with DS and in DS mouse models. For individuals with DS, girls are more typically in the above-average group on intelligence and adaptive function compared to boys (Marchal et al., 2016); eight-year-old girls had significantly higher developmental age than boys (van Gameren-Oosterom et al., 2011); in adolescents, males exhibited behavioral problems more often than females (van Gameren-Oosterom et al., 2013); in adults, women show higher cognitive abilities compared to men, and the frequency of profound intellectual disability was twice as high in men as women (Maatta et al., 2006). Men with DS had higher postmortem synaptic density of synaptophysin than females in areas of brain measured—except for the cerebellum in which women had a marginally higher density (Downes et al., 2008). Sex had a significant modulatory role in behavioral changes in environmental enrichment of Ts65Dn mice (Martinez-Cue et al., 2002). Female but not male Ts65Dn mice showed higher anxiety measures and altered defensive behaviors than control mice (Martinez-Cue et al., 2006).
Among the genes on Hsa21 that have been identified as possible regulators of neurogenesis and long-term cognitive function, a leading candidate is Dual-specificity tyrosine-phosphorylated regulated kinase 1A (DYRK1A). DYRK1A is a serine-threonine kinase that utilizes autophosphorylation to enhance activity and stabilize itself, regulates many downstream proteins and transcription factors, and has a crucial role during brain development (Ahn et al., 2006; Arron et al., 2006; Atas-Ozcan et al., 2021; Branchi et al., 2004; Canzonetta et al., 2008; Hammerle et al., 2011; Martinez de Lagran et al., 2004; Park and Chung, 2013; Yabut et al., 2010). Trisomic DYRK1A has been linked to aberrant brain development, brain pathology, and impaired cognitive phenotypes in individuals with DS and DS mouse models (Arron et al., 2006; Becker et al., 2014; Dowjat et al., 2007; Duchon and Herault, 2016; Liu et al., 2008). Trisomic Dyrk1a has been implicated in the development of cerebellar granule and Purkinje cell neurons by its influence on the Sonic Hedgehog pathway (Roper et al., 2006a) and in neuronal proliferation and cell cycle control in individuals with DS and DS mouse models (Stagni et al., 2018). DYRK1A has been identified as a target for therapeutic drug development in DS (Becker et al., 2014; De la Torre and Dierssen, 2012; Duchon and Herault, 2016). Subtracting one copy of Dyrk1a at conception from otherwise trisomic mice improved cognitive, neurological and Alzheimer disease-like phenotypes (Garcia-Cerro et al., 2014; Garcia-Cerro et al., 2017) and improved T-maze and contextual fear conditioning tests (Jiang et al., 2015). Reducing DYRK1A protein levels in the hippocampus of 2-month-old Ts65Dn mice by stereotaxic injection of AAV-shDyrk1a improved synaptic plasticity but only corrected thigmotaxic abnormalities in the Morris water maze (Altafaj et al., 2013). Pharmacological treatments to improve DS phenotypes in mouse models by reducing DYRK1A activity have been ultimately inconclusive due to the lack of information regarding developmental regulation of the spatiotemporal expression of DYRK1A and its role in brain development.
A frequent assumption regarding gene dosage effects in trisomy is that expression of all trisomic genes is upregulated according to the gene copy number (1.5-fold) in every cell in the body throughout the lifespan of the individual, though some have reported dosage compensation for some genes (Ait Yahya-Graison et al., 2007; Hwang et al., 2021; Kahlem et al., 2004; Lyle et al., 2004). Although this view has been challenged by studies of RNA and protein expression, its influence still permeates the interpretation of how DS phenotypes are caused (Antonarakis et al., 2020). Quantification of both mRNA and protein levels in brains of individuals with DS from fetal to adult stages have shown that not all trisomic genes are dysregulated or expressed at 1.5-fold (Lockstone et al., 2007; Mao et al., 2005; Mao et al., 2003). Moreover, studies of a single trisomic gene, including DYRK1A, often report upregulation in a specific tissue for their trisomic gene of interest, but lack systematic evaluation of multiple ages, both sexes, or multiple tissues. An approximate 1.5-fold increase in DYRK1A was found in homogenates from brains from human adults with DS and ~ 1.5-fold increase in DYRK1A from brain homogenates from 15-month-old Ts65Dn mice was observed (Liu et al., 2008). Quantification of DYRK1A protein levels in humans with DS ranging from 1 to 63 years of age found that DYRK1A levels were upregulated 1.3 to 1.5-fold in frontal, temporal, occipital, and cerebellar cortices, and 1.7 to 1.8-fold in the white matter of the corpus callosum and cerebellum (Dowjat et al., 2007). No differences in DYRK1A expression were found in the cerebral cortex of 1 to 3-year-old individuals with DS, but significantly increased expression of DYRK1A in 10 to 30 and ≥ 40-year-old individuals with DS was observed. Brains of 20-week-old fetuses with DS showed 1.5-fold upregulation of DYRK1A RNA and brains of adult Ts65Dn mice showed a 2.1-fold increase of Dyrk1a RNA expression (Guimera et al., 1999). DYRK1A protein levels were also significantly increased 1.3 to 1.8-fold in homogenized brains of 7 to 8-month-old Ts65Dn as compared to control mice (Dowjat et al., 2007). Ts65Dn mouse brain/tissue at 5 months of age yielded no difference in Dyrk1a mRNA expression, but at 12 months Dyrk1a expression was elevated in Ts65Dn mice compared to controls (Choi et al., 2009). In 4 to 8-month-old Ts65Dn mice, DYRK1A was upregulated in the cortex, cerebellum, and hippocampus (Ahmed et al., 2012). DYRK1A expression in the cerebellum, hippocampus, and cerebral cortex of 2 to 3-month Ts65Dn and control mice showed that DYRK1A was not overexpressed in the cerebellum or hippocampus but was overexpressed in the cerebral cortex of Ts65Dn mice (Goodlett et al., 2020; Stringer et al., 2017a). Furthermore, we recently showed that DYRK1A was elevated in P15 Ts65Dn male but not female mice (Hawley et al., 2022). These varied but limited results underscore the need for a systematic, longitudinal, tissue and sex-specific quantification of DYRK1A expression, especially at times when DYRK1A is thought to alter brain developmental processes.
This study provides a systematic profile of the ontogeny of DYRK1A expression to elucidate developmental periods when DYRK1A is overexpressed in different brain regions of developing Ts65Dn mice, including assessment of potential sex differences in the ontogenetic profiles. Two main hypotheses were tested: 1) that DYRK1A overexpression in trisomic mice varies over development in sex- and brain region-dependent manners, is not always proportional to Dyrk1a copy number, and may not correlate with Dyrk1a mRNA expression; and 2) that reduction of Dyrk1a copy number from 3 to 2 at conception in otherwise trisomic mice reduces DYRK1A expression at ages when Dyrk1a is overexpressed but not when trisomic Dyrk1a expression is compensated, independent of other trisomic genes.
2. Materials and methods
2.1. Animals and animal husbandry
Female B6EiC3Sn a/A-Ts(1716)65Dn/J (Ts65Dn) mice (Jackson Laboratory stock number 001924) containing a small marker chromosome (trisomic) (Reeves et al., 1995) were crossed with B6C3F1 male mice (Jackson Laboratory stock number 100010), producing both wildtype (euploid) and trisomic offspring with a genetic background of approximately 50% C57BL/6 and 50% C3H/HeJ (B6C3). New female Ts65Dn and male B6C3F1 animals were obtained from the Jackson Laboratory and introduced to the colony approximately every 6 months to limit genetic drift within the population and maintain consistent animal production. B6.Dyrk1atm1Jdc (Dyrk1afl/fl) mice containing loxP sites flanking Dyrk1a exons 5 and 6 were obtained from Dr. Jon Crispino (Thompson et al., 2015) and bred to C3H/HeJ mice (Jackson Laboratory, stock number 000659), resulting in B6C3F1.Dyrk1afl/wt offspring, containing loxP insertions on one Dyrk1a allele. These heterozygous offspring were intercrossed to produce homozygous B6C3.Dyrk1afl/fl mice on a similar B6C3 genetic background as the Ts65Dn model. Although Ts65Dn pups used in the developmental study contained loxP insertions flanking exons 5 and 6 on one Dyrk1a allele, neither the maternal or paternal line contained components necessary to induce a functional reduction of Dyrk1a by “floxing out”, and tissues collected from the heart, lung, liver, thymus, muscle, kidney, spleen, and toe found no evidence of Dyrk1a truncation via PCR (Supplemental Fig. A). The B6.129 Dyrk1a+/− male mice (Fotaki et al., 2002) were obtained from Dr. Mariona Arbonés and were initially crossed to C3H/HeJ mice and subsequent offspring backcrossed to B6C3F1 mice for ≥10 generations in our colony to parallel the B6C3 genetic background of Ts65Dn mice.
For the developmental age study, male and female trisomic and euploid offspring from Ts65Dn × B6C3.Dyrk1afl/fl matings were used. For the study testing the importance of trisomic Dyrk1a at P6 and P15, the breeding protocol to produce a constitutive Dyrk1a copy number reduction used male and female trisomic and euploid offspring from Ts65Dn × B6C3.Dyrk1a+/− matings. The work presented in this manuscript is part of a multifaceted, longitudinal series of experiments from our laboratory. The breeding design was selected to facilitate comparisons between projects.
All animal research complied with the protocols (SC225 and SC298R) approved by the Institutional Animal Care and Use Committee (IACUC) of the Indiana University-Purdue University Indianapolis (IUPUI). Animals were bred and housed in temperature and humidity-controlled rooms with a diurnal standard 12:12 light:dark cycle (lights on at 0700) in the secure AAALAC-accredited Science Animal Resource Center facility in the IUPUI School of Science. Animals had free access to food and water and were provided nesting material for environmental enrichment. Both dam and sire were housed continually through pregnancy, parturition, and pre-weaning. A total of 390 animals were used in these experiments from 127 litters. Tissues were collected from ~January 2017 through June 2022.
2.2. Tissue collection by age
For fetuses obtained on embryonic (E) day 18.5, pregnancies were generated with timed matings with a vaginal plug confirmation. For all postnatal ages, cages of breeding pairs were checked twice daily for new pups, and the day of birth was designated as postnatal day (P)0. For inclusion in the study, each litter was required to contain a minimum of 3 pups with at least one trisomic pup of either sex. On P0, P3, P6, P12, P15, P18, P21, or P24 full litters of male and female, euploid and trisomic offspring from the (Ts65Dn × B6C3.Dyrk1afl/fl) matings that produce Ts65Dn and euploid mice. Additionally on P6 and P15, offspring from the (Ts65Dn × B6C3. Dyrk1a+/−) matings [that produce Ts65Dn mice with 3 copies of Dyrk1a (Ts,Dyrk1a+/+/+) or two 2 copies of Dyrk1a (Ts65Dn,Dyrk1a+/+/)−, euploid mice with 2 copies of Dyrk1a (Eu,Dyrk1a+/+) or 1 copy of Dyrk1a (Eu,Dyrk1a+/−) were removed from the whelping cage and euthanized with isoflurane, followed by cervical dislocation. From E18.5 offspring, the whole developing brain (WB) was dissected in full. For P0 and P3 offspring, the cerebellum (CB) and forebrain (FB) were collected. At P6-P24, the hippocampus (HIPP), cerebral cortex (CTX), and CB were collected. Brain tissues were rapidly removed, separated, snap frozen in liquid nitrogen, and stored at −80 °C until further processing. Brain tissues from animals with matched RT-qPCR and Western blot analysis were separated by left and right hemispheres before being snap frozen.
2.3. Genotyping
Offspring were genotyped using the breakpoint PCR (Reinholdt et al., 2011) using forward 5′-GTGGCAAGAGACTCAAATTCAAC-3′ (Chromosome 17) and reverse 5′-TGGCTTATTATTATCAGGGCATTT-3′ (Chromosome 16) primers to amplify a ~275 bp product at the translocation point on the 1716 murine chromosome. Samples were separated on a 1.5% agarose gel using SYBR Safe DNA Gel Stain (ThermoFisher) and imaged with UV excitation. When sex determination via visualization was not possible due to age or ambiguity, sex was determined using PCR (McFarlane et al., 2013). Amplification of Sly and Xly genes, which reside on the Y and X chromosome, respectively, was accomplished using forward 5′-GATGATTTGAGTGGAAATGTGAGGTA-3′ and reverse 5′-CTTATGTTTATAGGCATGCACCATGTA-3′ primers and separated, stained, and imaged as above. Experiments using Dyrk1a+/− mice confirmed the presence of the mutated allele with primers Neo P1 (5′-ATTCGCAGCGCATCGCCTTCTATCGCC-3′), and Dyrk1a primers P2 (5′-CTTATGACAGAGTGGAGCAA-3′) and P3 (5′-CGTGATGAGCCCTTACCTATG-3′) using a previously described protocol (Fotaki et al., 2002). Supplemental Fig. B shows the PCR confirming the presence of the mutated Dyrk1a allele.
2.3.1. Protein isolation
Tissues were isolated for protein by using RIPA Lysis and Extraction Buffer (Thermo Scientific) containing 1× Pierce™ Protease Inhibitor Tablet, EDTA-free (Thermo Scientific) following the manufacturer’s instructions. Protein samples were quantified using the Pierce™ Detergent Compatible Bradford Assay Kit (ThermoFisher) (Bradford, 1976) before Western blot analysis.
2.3.2. DYRK1A protein analysis by Western blot
DYRK1A protein levels were quantified using Western blot procedures as described previously (Hawley et al., 2022). Briefly, 20 μg of protein from the designated brain regions of age and sex matched animals were resolved electrophoretically using Bolt™ reagents on Bolt™ Bis-Tris Plus Mini Protein Gels, 4–12% (ThermoFisher). Proteins were transferred to a .045 μm PVDF Transfer Membrane (ThermoFisher) with conditions set to 20 V for 20 h inside a 4 °C refrigerator. PVDF membranes were then blocked in TBS-T containing 5% Blotting Grade Blocker (Bio-Rad) for one hour. Blocking was followed by an overnight incubation of 1:500 DYRK1A monoclonal antibody (M01), clone 7D10 (Abnova) at 4 °C and Donkey anti-mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 790 (ThermoFisher) for 1 h at room temperature.
Fluorescence was detected using a LI-COR® CLx Odyssey® Imaging System. After imaging the PVDF membrane for DYRK1A fluorescence, it was then stained with Coomassie Brilliant Blue R-250, air dried, and scanned using a Scanjet 5300C Flatbed Scanner (HP, Palo Alto, CA). Alternate mRNA splicing events produce multiple Dyrk1a isoforms of varying protein sizes; the five most abundantly found in brain tissues range from ~520 amino acids (aa) to 763 aa (Guimera et al., 1999) and conserve the c-terminal domain targeted by our chosen primary antibody. The Western blot resolution provided three bands with DYRK1A, one with the 763 aa isoform and two other bands with two isoforms each that were not resolved. We quantified the three DYRK1A isoform bands separately and found no significant differences in the expression of these isoforms for each age, ploidy, and sex.
2.3.3. DYRK1A protein quantification in the developmental age study by relative intensity ratios
A representative blot (Supplemental Fig. C) shows the quantification approach for the developmental analysis (E18.5, P0, P3, P6, P12, P15, P18, P21, and P24) of DYRK1A. DYRK1A protein was quantified from the PVDF membrane fluorescence image using Image Studio™ Lite Software following instructions posted by the manufacturer, producing a numeric value of DYRK1A optical density for each sample. A densiometric measurement of total protein for each sample was then quantified from the same membrane subsequently stained with Coomassie blue using ImageJ software (Schneider et al., 2012), selecting a uniform segment between 15 kDa and 35 kDa to avoid inclusion of areas labeled with exogenous DYRK1A antibodies. A normalized DYRK1A value was produced for each sample by dividing the optical density of the DYRK1A fluorescence signal (from Image Studio™ Lite Software) by the Coomassie blue densitometric measurement of total protein (from ImageJ), as previously described (Aldridge et al., 2008; Nie et al., 2017; Welinder and Ekblad, 2011). The choice to use total protein as a loading control was informed by demonstrations that it can provide a more consistent measure for sample normalization than use of a single housekeeping protein, as it may show better linearity with variation in protein loading (Eaton et al., 2013) and it controls for differences in protein transfer to the membrane during the Western blot procedure (Welinder and Ekblad, 2011).
For quantification of the blots (described here for P6 and older), the hippocampus, cerebral cortex, and cerebellum of four age- and sex-matched animals were assayed on each blot (two euploid and two trisomic) resulting in 3 normalized values for each animal on each blot (one for each brain region). Within each blot, relative DYRK1A expression was calculated separately for each brain region. Trisomic expression is presented as a fold-change relative to the mean of the two euploid samples of the same brain region from each blot. First, an average was taken of the normalized values of both euploid samples of the given brain region. Each normalized trisomic value of the same brain region on the blot was divided by the averaged euploid value, producing a relative intensity ratio for each trisomic sample (to the euploid control mean). The relative intensity ratio of euploid samples were likewise generated by dividing each normalized euploid value by the mean of the two normalized euploid values of the same brain region. The analyses of E18.5 to P3 involved either one or two regions, so more matched cases could be included on those blots. These within-blot relative intensity ratios provide a measure of trisomic expression relative to the matched euploid regions of the same blot; the average values of the euploid brain regions, by definition, equals 1. The number of blots per sex and brain region are shown in Supplemental Table A. Note that for E18.5, whole brain samples used one blot per sex (6–8 mice per genotype). The P0 samples used one blot per sex per brain region (forebrain or cerebellum) with 8 euploid and 8 trisomic mice on each blot. The P3 samples used two blots per sex with both brain regions from four euploid and four trisomic mice on each blot. For P6 to P24, each blot had samples from all three brain regions of two euploid and two trisomic mice of the same sex.
2.3.4. DYRK1A protein quantification in the Dyrk1a copy number reduction study by standardization to pooled homogenate
For the Western blot analysis of the effects of Dyrk1a copy number reduction at P6 and P15, the blot design was modified to incorporate samples of a pooled brain protein homogenate on each blot to serve as a standard across blots. This allowed direct comparison of relative intensity results between blots involving subjects of different ages and sexes. The homogenate was prepared separately in a single batch following the protein isolation protocol described above and consisted of isolated protein from the hippocampus, cerebral cortex, and cerebellum of six male and six female euploid animals and stored as frozen aliquots. These standard homogenates were stored and prepared for analysis in parallel with unknown samples and used in up to three replicate lanes on each blot. Homogenate values normalized to total protein were first calculated and averaged within blot. Then, regardless of brain region, each normalized euploid and trisomic value from a given blot was compared to the mean homogenate value of that blot, resulting in a relative intensity ratio to the homogenate standard [calculated within blot as follows: (normalized value of euploid or trisomic unknown) / (average of the normalized homogenate values)]. A representative blot in Supplemental Fig. D shows the quantification approach for the analysis of Dyrk1a copy number reductions at P6 and P15.
2.3.5. Statistical analysis of protein expression levels
For each age and sex in the developmental analysis, we tested the a priori directional hypothesis that DYRK1A expression is higher in trisomic as compared to euploid mice, comparing DYRK1A relative intensity ratios for each brain region using one-tailed independent groups t-tests (alpha = 0.05) corrected for unequal variances when Levene’s test was significant. Data are presented as scatterplots and means for each group and effect sizes are reported as Cohen’s d. To determine whether the extent of DYRK1A overexpression in trisomic brains differed significantly as a function of brain region, the relative intensity ratios of the three regions of trisomic animals for a given age and sex were compared using within-subjects post hoc tests with Bonferroni corrections.
For the Dyrk1a gene reduction analyses at P6 and P15, three independent a priori directional hypotheses (alpha = 0.05 for each) were tested at each age for each brain region for these DYRK1A relative ratios: a) Ts,Dyrk1a+/+/+ > Eu,Dyrk1a+/+; b) Ts,Dyrk1a+/+/+> Ts,Dyrk1a+/+/−; and c) Eu,Dyrk1a+/+> Eu,Dyrk1a+/−.
2.3.6. RNA isolation
RNA was isolated and purified from each tissue sample using TRIzol™ Reagent (ThermoFisher), following the solubilization and extraction method described previously (Rio et al., 2010). In short, samples were homogenized in TRIzol reagent, centrifuged, and the supernatant removed to new tubes. Phases were separated with choloroform, and RNA precipitated from the aqueous phase using ice-cold isopropanol. The RNA pellet was washed using 75% ethanol, allowed to dry at room temperature, and rehydrated in 30 μL RNase/DNase free water. Samples were quantified for RNA using a NanoDrop 2000 (Thermo Scientific).
2.3.7. mRNA quantification
Isolated and quantified RNA samples were converted to cDNA with a final volume of 20 μL using TaqMan™ Reverse Transcription Reagents following the recommended protocol. The resulting product was diluted 1:5 with sterile milliQ water and stored at −20 °C until analysis. RT-qPCR was performed using a LightCycler® 480 Thermal Block Cycle Unit with LightCycler Software (Roche Diagnostics) on 384-well plates containing age- and sex-matched animals. Reagents consisted of TaqMan™ Gene Expression Master Mix (Roche Diagnostics) and two probes were used to identify different regions of the Dyrk1a transcript, the first probe targeting a region spanning Dyrk1a exons 5–6 (Roche Diagnostics, Mm00432929_m1) and the second targeting Dyrk1a exons 10–11 (Roche Diagnostics, Mm00432934_m1).
2.3.8. RT-qPCR statistical anayses
For the qPCR mRNA analyses, the triplicates of each sample were averaged (arithmetic means for the two Dyrk1a probes; geometric mean the Rn18S probe) for each brain region for each subject. Relative expression was quantified using the 2−ΔCT comparative CT method (Schmittgen and Livak, 2008) and trisomic expression calculated as a fold change relative to the euploid control group, as a function of age, sex, and brain region. We first calculated 2−ΔCT for each sample [as 2−(CTDyrk1a – CTRn18s)sample], then converted that value for each sample to fold change relative to euploid by dividing the sample value by the mean euploid value of subjects from the same sex, tissue, and age obtained from the same plate . Data for each brain region of each group are presented as scatterplots and means for each group and plotted on a log 2 scale. For the statistical analysis of relative expression (fold change) data of euploid and Ts65Dn mice at P0, P3, and P6, the directional hypothesis that trisomic mice had higher Dyrk1a mRNA relative expression than euploid mice was tested separately for each brain region using one-tailed independent groups t-tests (alpha = 0.05), and effect sizes were calculated using Cohen’s d. For the statistical analysis of relative expression (fold change) data from the three groups of P6 male mice from the (Ts65Dn × Dyrk1a+/−) matings, two directional hypotheses were tested separately for each brain region: a) Ts,Dyrk1a+/+/+ > Eu,Dyrk1a+/+ and b) Ts,Dyrk1a+/+/+ > Ts,Dyrk1a+/+/−, using one-tailed independent groups t-tests (alpha = 0.05), and effect sizes were calculated using Cohen’s d.
3. Results
3.1. Ontogeny of DYRK1A overexpression in female and male Ts65Dn mice
To characterize the influence of three copies of Dyrk1a on expression of DYRK1A protein during brain development in the Ts65Dn DS mouse model, DYRK1A protein was quantified at designated ages from embryonic (E) day 18.5 to postnatal (P) day 24. On E18.5, female trisomic mice, but not male trisomic mice, showed significant elevations in whole brain DYRK1A compared to euploid controls (p = 0.018; Cohen’s effect size d = 1.27, Fig. 1A). Sex differences in DYRK1A overexpression were also evident on the day of birth in which DYRK1A was significantly upregulated in female Ts65Dn as compared to control mice in the forebrain (p < 0.001; d = 2.33) and cerebellum (p = 0.009; d = 1.34), but male Ts65Dn mice showed no significant difference in forebrain or cerebellar DYRK1A expression compared to euploid controls (Fig. 1B). On P3, trisomic females continued to show elevated DYRK1A that was significant in the forebrain (p = 0.028; d = 1.04) and approached significance in the cerebellum (p = 0.051; d = 0.88), whereas the males continued to show no significant differences in either region (Fig. 1C). Taken together, these data suggest that during the perinatal period DYRK1A is upregulated approximately 1.5-fold in female developing brain, but not male Ts65Dn as compared to control mice.
Fig. 1.
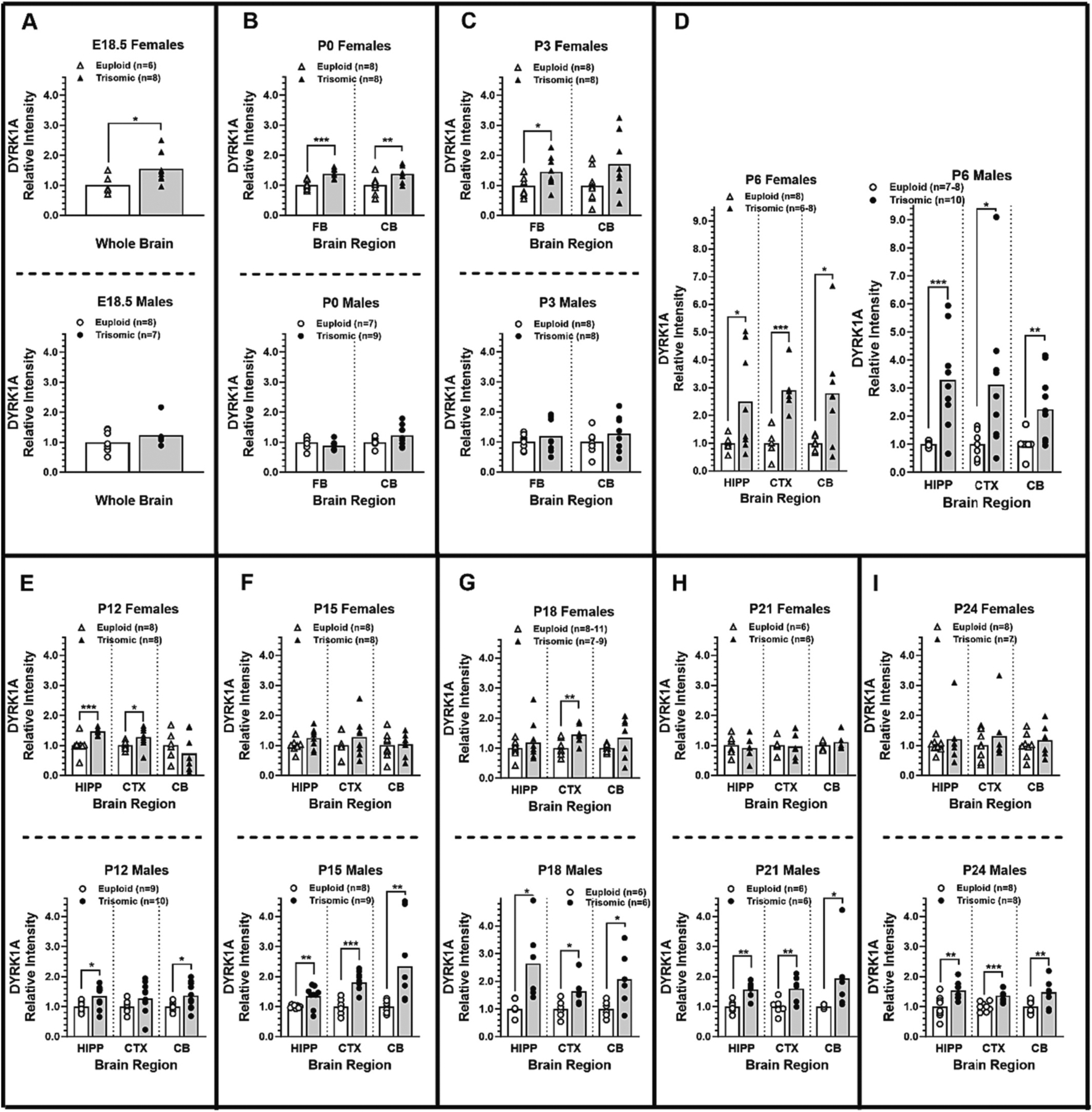
Ontogeny of DYRK1A overexpression in female and male offspring from Ts65Dn × B6C3F1 matings, showing DYRK1A protein expression as relative intensity ratios at E18.5 to P24 (scatterplots and group mean). For each age and sex, the directional hypothesis that trisomic mice had higher DYRK1A ratios as compared to euploid mice was tested separately for each brain region, using one-tailed independent groups t-tests (alpha = 0.05).*p < 0.05; **p < 0.01; ***p < 0.001 (a priori 1-tailed test): The trisomic group (solid bars) was significantly greater than the respective euploid group (open bars) of the same age, sex, and brain region.
In P6-P24 animals, DYRK1A expression was quantified in the hippocampus (HIPP), cerebral cortex (CTX), and cerebellum (CB) (see Supplemental Fig. C for P6 example). At P6, there were large elevations in mean DYRK1A expression in all three brain regions in Ts65Dn as compared to controls in both male and female mice (Fig. 1D). In the P6 hippocampus, DYRK1A in Ts65Dn female mice was upregulated ~2.5-fold (p = 0.028) and in Ts65Dn male mice it was upregulated ~3.3-fold (p < 0.001), and effect sizes were large for both sexes (d = 1.14 and 1.95). In the P6 cerebral cortex, DYRK1A was upregulated ~2.8-fold in Ts65Dn females (p = 0.006) and ~2.3-fold in Ts65Dn males (p = 0.001), again with large effect sizes in both sexes (d = 1.53 and 1.52). Likewise, in the P6 cerebellum DYRK1A was upregulated ~2.8-fold in Ts65Dn females (p = 0.016) and ~2.3-fold in Ts65Dn males (p = 0.008) with both sexes showing large effect sizes (d = 1.33 and 1.14). At P12 (Fig. 1E), DYRK1A was upregulated in female Ts65Dn as compared to euploid littermates in the hippocampus (1.5-fold, p = 0.001) and cerebral cortex (1.3-fold, p = 0.026), with no significant differences in cerebellum. In males, DYRK1A expression was significantly upregulated in the hippocampus (1.2-fold, p = 0.010) and in cerebellum (1.2-fold, p = 0.014), but did not reach significance in the cerebral cortex.
Sex differences in trisomic brain DYRK1A overexpression were evident on P15, data we previously reported (Hawley et al., 2022) but now are included in several new analyses in the current report. In females, there were no significant differences between trisomic mice and their euploid littermates for any brain region. In contrast, P15 Ts65Dn males showed significant overexpression as compared to euploid littermates in all three brain regions, with increases in the hippocampus (1.4-fold, p = 0.009), cerebral cortex (1.9-fold, p < 0.001), and cerebellum (2.1-fold, p = 0.007) (Fig. 1F). For trisomic males, DYRK1A overexpression in both the cerebral cortex and the cerebellum were significantly greater than in the hippocampus (Bonferroni comparisons, p < 0.001 and p = 0.014, respectively). Notably, the effect sizes for P15 males were large (d = 1.34, 2.67, and 1.40, respectively, for hippocampus, cerebral cortex, and cerebellum), whereas for P15 females the effect sizes were much smaller (d = 0.86, 0.49, and 0.09, respectively). Our previously reported finding of sexually dimorphic trisomic DYRK1A overexpression on P15 can now be placed in a larger developmental context in view of the additional findings at P18, P21, and P24 (Fig. 1G, H, and I). The significant overexpression of DYRK1A evident in Ts65Dn male mice at P15 was also seen at P18, P21, and P24, where significant overexpression was found in all brain regions (fold changes ranging from 1.4- to 2.6-fold) and the effect sizes were all large and ranged from 1.14 to 2.67. Furthermore, the lack of significant differences between female Ts65Dn as compared to euploid mice at P15 also extended to P18, P21, and P24 females. The only significant difference between trisomic and euploid females across those ages was the 1.3-fold DYRK1A overexpression in the cerebral cortex at P18 (p = 0.001). All other outcomes failed to reach significance, and for those outcomes the effect sizes ranged from 0.22 to 0.86.
3.2. Developmental- and sex-dependent changes in trisomic DYRK1A overexpression
The presence of significant DYRK1A overexpression in trisomic mice varied across development but the particular ages showing overexpression depended on sex (Fig. 1). For female Ts65Dn mice, overexpression was significant in all regions from E18.5 to P12 with the exception of the cerebellum on P3 and P12. After P12, female group differences failed to reach significance for any region except for P18 cortex. Notably, for trisomic females the largest mean increase in DYRK1A expression relative to euploid littermates was on P6 (mean fold increases of 2.49, 2.90, 2.79 for hippocampus, cortex, and cerebellum, respectively) and P6 also showed the most variable expression levels (CVs = 0.73, 0.28, 0.68, respectively). For male Ts65Dn mice, the first day of significant DYRK1A overexpression was on P6 (Fig. 1D, mean fold increases of 3.28, 3.12, 2.25, and CVs of 0.49, 0.77, 0.54, respectively) and DYRK1A remained significantly elevated at all subsequent ages except for P12 cortex. The cerebellum demonstrated a conspicuous sex difference in trisomic overexpression. In females, significant elevations in cerebellar DYRK1A were present on P0 and P6 but not at any subsequent age. In contrast, males first showed significant elevations in cerebellar DYRK1A on P6 and those remained elevated for all subsequent ages. These age-dependent sex differences in DYRK1A overexpression suggest that differences in the developmental regulation of cerebellar DYRK1A expression in female and male trisomic mice may emerge in the second and third postnatal week in this DS mouse model.
3.3. Dyrk1a copy number reduction normalizes DYRK1A expression in developing Ts65Dn mice
Given the amplified DYRK1A overexpression in male and female Ts65Dn at P6 and the sexually dimorphic overexpression evident at P15, we examined DYRK1A expression in offspring from (Ts65Dn × Dyrk1a+/−) matings producing Ts65Dn (Ts,Dyrk1a+/+/+), Ts65Dn,Dyrk1a+/+/−, Euploid (Eu,Dyrk1a+/+) and Eu,Dyrk1a+/− mice at P6 and P15 to test the hypothesis that these outcomes at P6 and P15 were dependent on Dyrk1a gene dosage and not dependent on other trisomic genes. For female mice on P6 (Fig. 2A), there was significant overexpression of DYRK1A in the Ts65Dn (Ts,Dyrk1a+/+/+) as compared to euploid (Eu, Dyrk1a+/+) offspring in the cerebellum (p = 0.032; d = 0.85) and trends for overexpression in the hippocampus (p = 0.093; d = 0.59) and cerebral cortex (p = 0.076; d = 0.64). These group differences and medium-to-large effect sizes partially replicated the findings of P6 female in the developmental study (compare with Fig. 1D). In the female Ts65Dn,Dyrk1a+/+/− offspring with two copies of Dyrk1a on an otherwise trisomic mouse, DYRK1A levels were significantly reduced relative to Ts,Dyrk1a+/+/+ mice in the cerebellum (p = 0.024; d = 1.21), and the Ts,Dyrk1a+/+/− levels were comparable to Eu,Dyrk1a+/+ littermates. Differences in the hippocampus and cerebral cortex in the Ts,Dyrk1a+/+/− females relative to Ts,Dyrk1a+/+/+ females did not reach significance. Reduction of Dyrk1a copy number in female euploid mice (Eu, Dyrk1a+/−) significantly reduced DYRK1A in all three regions as compared to Eu,Dyrk1a+/+ mice, down to 43% of control in hippocampus (p = 0.004, d = 1.59), 58% of control in cerebral cortex (p = 0.030, d = 0.78), and 58% of control in cerebellum (p = 0.047, d = 0.93).
Fig. 2.
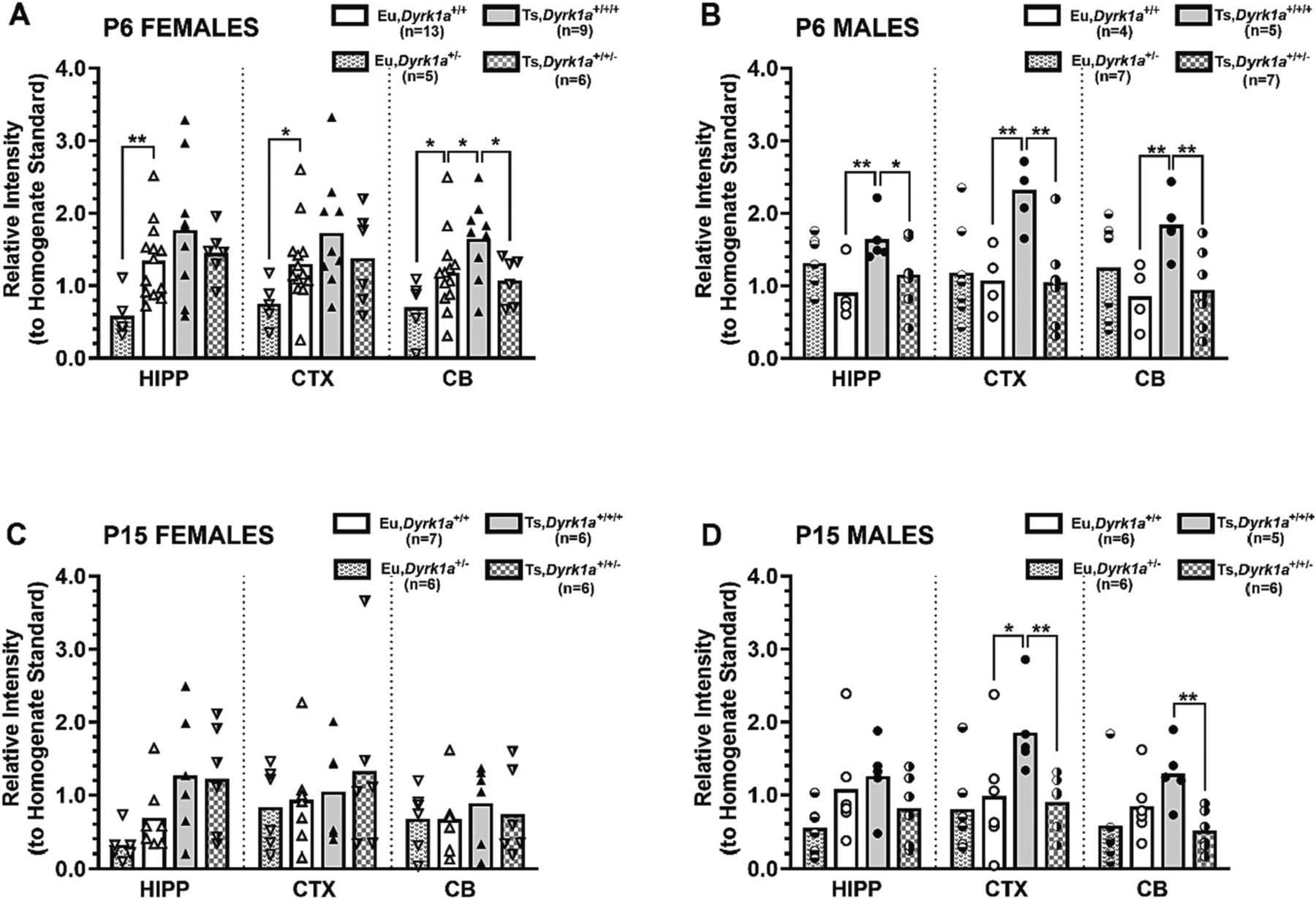
Dyrk1a copy number reduction normalizes DYRK1A expression in developing Ts65Dn mice. DYRK1A expression in P6 and P15 offspring from Ts65Dn × Dyrk1a+/− matings, with data shown as scatterplot with group mean. Three a priori directional hypotheses were tested for each brain region at each age: a) Ts, Dyrk1a+/+/+ > Eu,Dyrk1a+/+; b) Ts,Dyrk1a+/+/+ > Ts,Dyrk1a+/+/−; and c) Eu,Dyrk1a+/+ > Eu,Dyrk1a+/−, alpha = 0.05 for each region.
For P6 male mice from the (Ts65Dn × Dyrk1a+/−) matings, there was significant DYRK1A overexpression in Ts,Dyrk1a+/+/+ as compared to Eu,Dyrk1a+/+ offspring in the hippocampus (p = 0.010), cerebral cortex (p = 0.002), and cerebellum (p = 0.005) in offspring (Fig. 2B). These group differences and large effect sizes in male P6 mice replicated the outcomes of the developmental DYRK1A protein expression study. Reduction of Dyrk1a copy number in the otherwise trisomic males (Ts, Dyrk1a+/+/−) significantly reduced DYRK1A levels relative to Ts, Dyrk1a+/+/+ males in all three regions (p = 0.035 for hippocampus; p = 0.002 for cerebral cortex; p = 0.006 for cerebellum), and the Ts, Dyrk1a+/+/− levels were comparable to levels found in Eu,Dyrk1a+/+ littermates. Reduction of Dyrk1a copy number in male euploid mice (Eu, Dyrk1a+/−) did not significantly reduce DYRK1A relative to Eu,Dyrk1a+/+ males in any brain region.
At P15, an age when elevated DYRK1A levels were present in male but not female trisomic mice in the developmental study, male offspring of (Ts65Dn × Dyrk1a+/−) matings (Fig. 2D) showed DYRK1A expression that was significantly higher in male Ts,Dyrk1a+/+/+ compared to Eu, Dyrk1a+/+ littermates in the cerebral cortex (p = 0.037; d = 1.23) and non-significant increases in the cerebellum (p = 0.113; d = 1.06). These results partially replicated the P6 male findings in the developmental study (compare Figs. 1F and 2D). Functional reduction of one copy of Dyrk1a (Ts,Dyrk1a+/+/−) significantly reduced DYRK1A as compared to male Ts,Dyrk1a+/+/+ mice in the cerebral cortex (p = 0.005) and cerebellum (p = 0.002), yielding large effect sizes (d = 1.96 and 2.24, respectively) and trending in the hippocampus (0.082; d = 0.92), reaching levels approximating those of euploid males. Dyrk1a copy number reduction in the male euploid mice (Eu,Dyrk1a+/−) did not significantly affect DYRK1A expression in the cerebral cortex or cerebellum, but there was a trend toward significance in the hippocampus (p = 0.062; d = 0.97). For the females at P15 (Fig. 2C), there were no significant group differences in any brain region, though there were non-significant trends toward increased expression in the hippocampus in Ts, Dyrk1a+/+/+ as compared to Eu,Dyrk1a+/+ females (p = 0.073; d = 0.87) and for the euploid knockdown to reduce hippocampal DYRK1A relative to Eu,Dyrk1a+/+ (p = 0.054; d = 0.98). The lack of significant differences between Eu,Dyrk1a+/+ and Ts,Dyrk1a+/+/+ females and the relatively small effect sizes in the cerebral cortex and cerebellum mirrored the P15 female findings in the developmental study (compare Figs. 1F and 2C). In addition, for the euploid control groups there appeared to be a trend for the P15 females to have lower DYRK1A expression in all three brain regions than the P6 females (compare Eu, Dyrk1a+/+ groups in Fig. 2A and C), whereas the euploid male levels appeared to be similar across ages. However, a post hoc 2 × 2 factorial ANOVA on the Eu,Dyrk1a+/+ data with Age and Sex as grouping factors failed to yield any significant main or interactive effects for any brain region (p’s > 0.064).
3.4. Dyrk1a mRNA expression in developing Ts65Dn mice and effects of Dyrk1a copy number reduction
Dyrk1a mRNA relative expression was quantified with qPCR at P0, P3, and P6 (Fig. 3) following the 2−ΔCT comparative CT method (Schmittgen and Livak, 2008). For P0 and P3, RNA and protein were isolated from brain tissue of the same animals at each age, but for P6 the RNA and protein had to be isolated from different animals. The relative overexpression of Dyrk1a mRNA in trisomic mice depended on age. At P0, Ts65Dn females (Fig. 3A) had significant elevations both in the forebrain (1.6-fold; p = 0.029, d = 0.86) and the cerebellum (1.9-fold; p = 0.0096, d = 1.12). Ts65Dn males at P0 (Fig. 3D) showed significant and large elevations as compared to euploid littermates in both the forebrain (3.3-fold; p = 0.004, d = 1.56) and the cerebellum (4.3-fold; p = 0.005,d = 1.48). At P3, there were no significant group differences in mRNA expression in either brain region for either sex (Fig. 3B and E) and effect sizes were smaller (d’s between 0.08 and 0.75). At P6, significantly higher Dyrk1a expression was again evident in trisomic mice; females (Fig. 3C) showed significant increases in all three regions [hippocampus (1.8-fold; p = 0.030, d = 1.38), cerebral cortex (3.0-fold, p = 0.016, d = 1.00) and cerebellum (2.3-fold, p = 0.009, d = 2.02) and males (Fig. 3F) showed significant increases in the cerebral cortex (1.7-fold, p = 0.018, d = 1.20) and cerebellum (1.9-fold, p = 0.003, d = 1.75). We replicated these data with a different mRNA probe that recognized the exons corresponding with the C terminal end of Dyrk1a and the data from the two different probes were highly correlated in both sexes at all three ages (r’s ranging from 0.925 to 0.998; see Supplemental Fig. E). Both probes confirmed the greater and more variable overexpression outcomes in the P0 males. The large increase in mRNA expression in trisomic P0 males in both forebrain and cerebellum (Fig. 3D) contrasts with the lack of significant protein overexpression in the same animals in either region (Fig. 1B), and this mismatch was confirmed by the lack of significant correlation between mRNA and protein in either brain region (r = +0.18 for forebrain; r = 0.0 for cerebellum) in trisomic P0 males. Protein and mRNA expression in trisomic P0 females was likewise not significantly correlated (r = −0.08 for forebrain; r = −0.28 for cerebellum). The lack of group differences in mRNA expression at P3 further highlights the developmental change in mRNA expression at P6, showing increases in trisomic mice of both sexes—generally consistent with the parallel emergence of the large increase in protein expression seen in trisomic mice at P6.
Fig. 3.
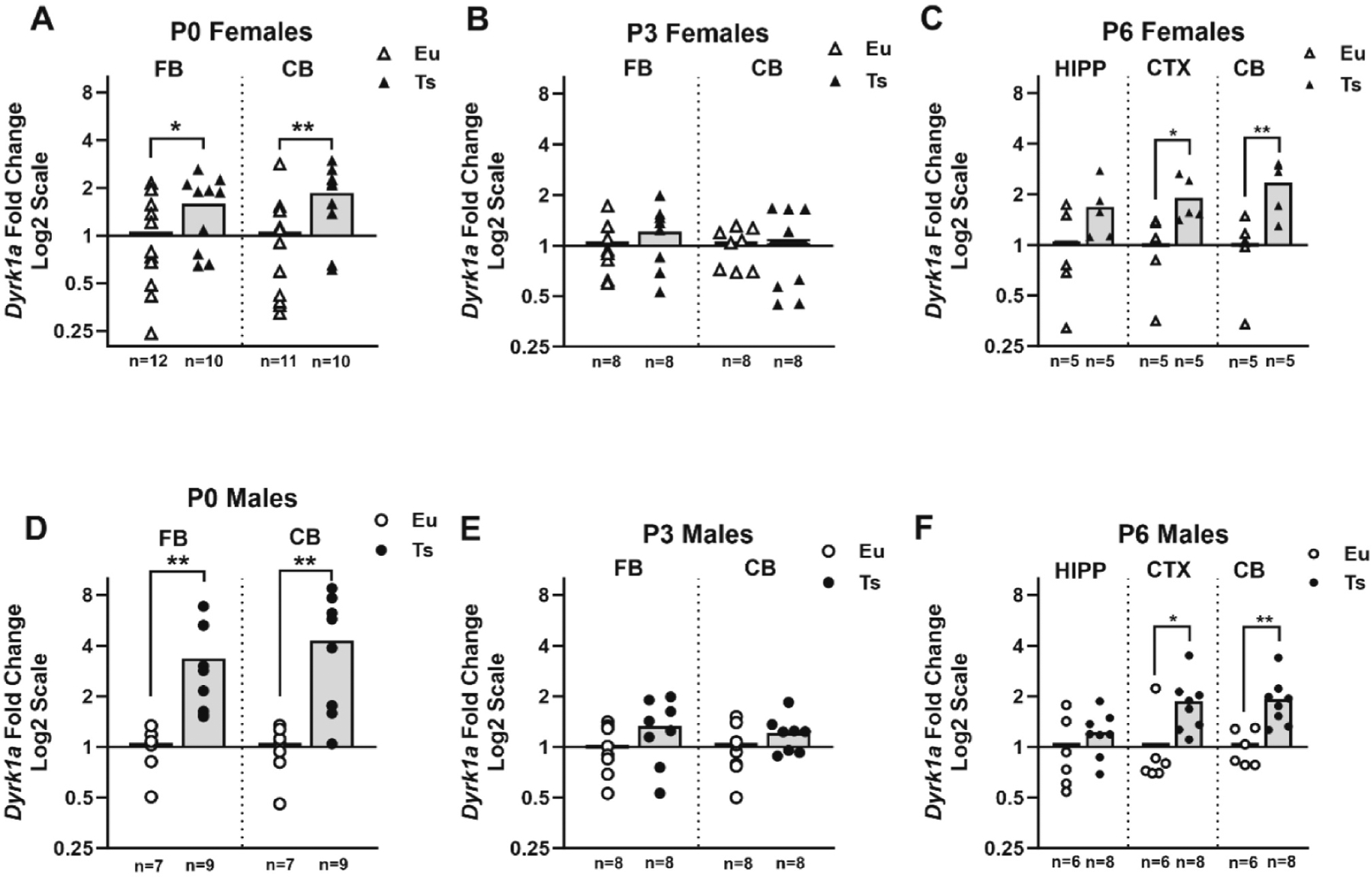
Dyrk1a mRNA expression in developing Ts65Dn mice. Quantitative polymerase chain reaction (PCR) analysis of Dyrk1a mRNA expression was performed in mice at P0, P3, and P6. Relative fold change was calculated using the 2−ΔCT comparative method relative to within-plate euploid means, and data are shown as scatterplots with group means and plotted using a log 2 scale (euploid means =1). For each age and sex, the directional hypothesis that trisomic mice had higher Dyrk1a mRNA relative expression than euploid mice was tested separately for each brain region, using one-tailed independent groups t-tests (alpha = 0.05). *p < 0.05; **p < 0.01 (a priori 1-tailed t-test), the trisomic group (dark bars and symbols) was significantly greater than the respective euploid group (open symbols) of the same age, sex, and brain region.
Expression of Dyrk1a mRNA was also assessed in P6 male mice from the (Ts65Dn × Dyrk1a+/−) matings using the Eu,Dyrk1a+/+, Ts, Dyrk1a+/+/+, and Ts,Dyrk1a+/+/− genotypes; female brain tissues were not analyzed due to insufficient numbers of females produced. As shown in Fig. 4, The Ts,Dyrk1a+/+/+ male mice showed significant increases in mRNA expression relative to the Eu,Dyrk1a+/+ mice in all three regions: hippocampus [2.6-fold; p ≤0.001, d = 2.6]; cerebral cortex [2.0-fold; p = 0.017, d = 1.4]; cerebellum [2.6-fold; p = 0.021, d = 1.5]. The functional reduction of one copy of Dyrk1a in trisomic mice (Ts, Dyrk1a+/+/−) failed to significantly reduce mRNA expression in any brain region.
Fig. 4.
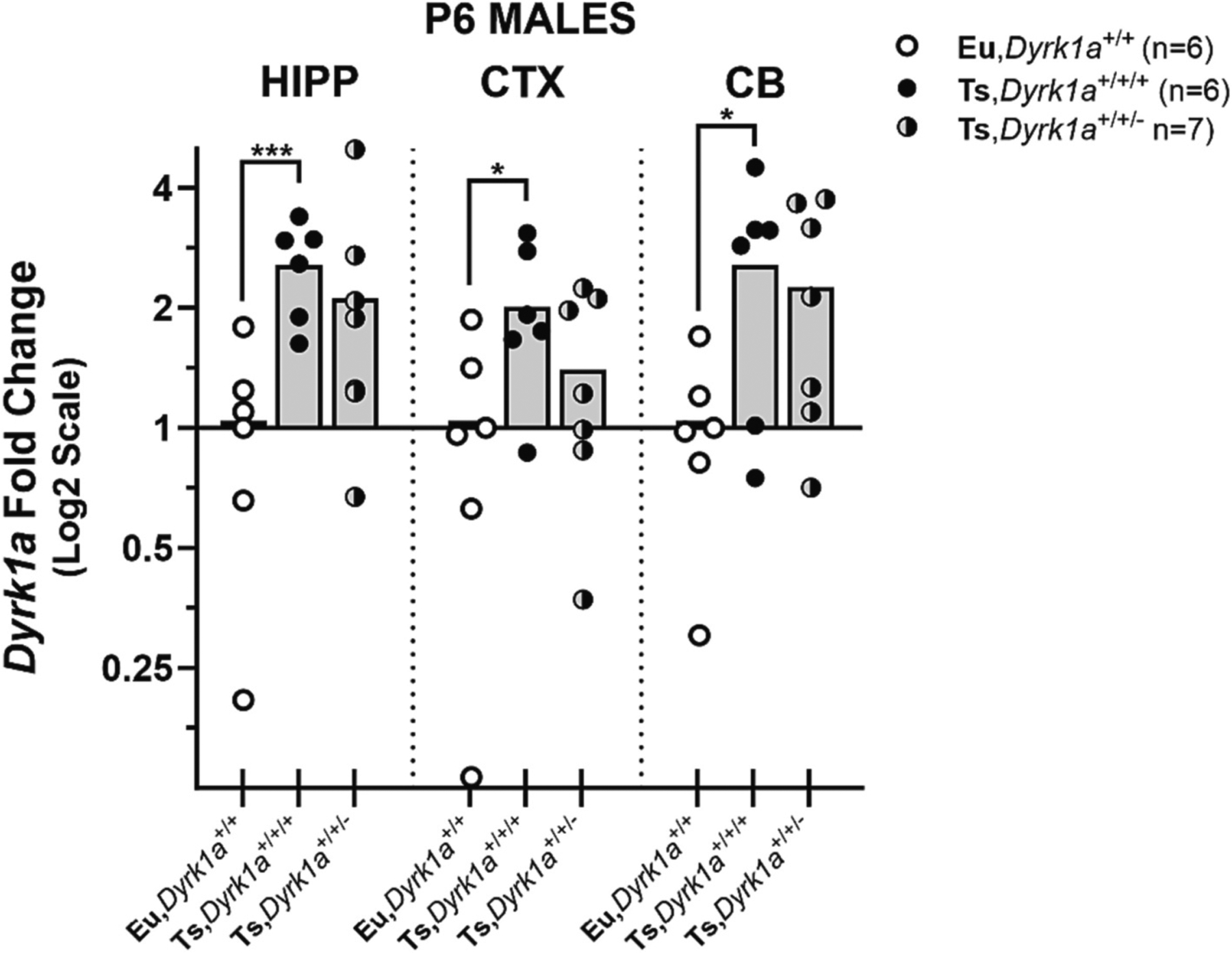
Effects of Dyrk1a copy number reduction on Dyrk1a mRNA expression in P6 male Ts65Dn mice. Quantitative polymerase chain reaction (qPCR) analysis was performed in mice of each genotype: euploid (Eu,Dyrk1a+/+), Trisomic (Ts,Dyrk1a+/+/+), and trisomic with 2 copies of Dyrk1a Ts,Dyrk1a+/+/−). Relative fold change was calculated using the 2−ΔCT comparative method relative to within-plate euploid means. Two directional hypotheses regarding the expression of Dyrk1a mRNA were tested separately for each brain region: a) Ts,Dyrk1a+/+/+ > Eu,Dyrk1a+/+ and b) Ts,Dyrk1a+/+/+ > Ts,Dyrk1a+/+/−, using one-tailed independent groups t-tests (alpha = 0.05). Data are shown as scatterplots with group means and plotted using a log 2 scale (euploid means = 1).
4. Discussion
4.1. Spatial, temporal, and sex-specific developmental patterns of trisomic DYRK1A expression levels
Results from this developmental study of trisomic Dyrk1a overexpression in the Ts65Dn DS mouse model demonstrate spatial, temporal, and sex-specific expression of DYRK1A in a trisomic system. As we and others have previously described, DYRK1A expression in the trisomic system is not consistently upregulated 1.5-fold that of normal gene expression (Cheon et al., 2003; Goodlett et al., 2020; Stringer et al., 2017a). The current study confirms that relative expression varies depending on the age of developing trisomic mice, suggesting that expression of triplicated Dyrk1a is controlled in a temporally specific manner—being overexpressed at some developmental ages and not at others. These new data extend the range of our previous report of DYRK1A expression in brain tissues of P15 and young adult Ts65Dn brain tissue (Goodlett et al., 2020; Hawley et al., 2022) and contribute more systematic evidence of ontogenetic changes in DYRK1A expression across early development.
The current data show that P6 is a prominent developmental time-point of amplified DYRK1A overexpression in brain tissues of both male and female Ts65Dn mice. These significant and large increases in DYRK1A expression suggest that P6—roughly equivalent to the period of human brain development in the early third trimester (Workman et al., 2013)— may be a time of excessive DYRK1A kinase activity that alters the trajectory of the trisomic brain at a critical period of its development. Significant cerebellar deficits have been found in Ts65Dn mice at P6 and abnormal trisomic cerebellar development has been a therapeutic target at this stage (Das et al., 2013; Roper et al., 2006a); the hippocampus is also altered at P6 in Ts65Dn mice (Lorenzi and Reeves, 2006). When treatment was given at birth to correct the cerebellar phenotype via a Sonic Hedgehog agonist, cerebellar structure was normalized and hippocampal-dependent learning, memory, and function were also improved (Das et al., 2013).
Sex-specific effects in trisomic DYRK1A overexpression were identified (in opposite directions) during two developmental periods, one in the perinatal period (E18.5-P3) and another in the third postnatal week (P15-P24). During the perinatal period, female trisomic mice showed significant increases in DYRK1A relative to euploid littermates (~1.5-fold) whereas males did not. Notably, a study of protein expression in human female fetuses with and without trisomy (n = 4 each) at gestational week 18–19, a stage of human brain development that is roughly equivalent to the late fetal period in mouse brain development (Workman et al., 2013), found no differences in protein expression (Cheon et al., 2003). Although DYRK1A expression in the cerebral cortex did not differ between fetuses with and without trisomy, the specificity of the DYRK1A antibody used was unclear because the size of the major protein variant identified (200 kDa) was larger than typically reported. Future studies need better alignment of the mouse model and human data to compare DYRK1A expression; results from female and male fetal mice at early ages would be useful for better temporal resolution of periods of sex differences in trisomic DYRK1A overexpression. In addition, a comparative assessment of male and female human fetuses with and without trisomy at multiple gestational ages, but especially at week 18–19, could identify whether sex differences in DYRK1A expression are present (as predicted by these mouse model data). Sex differences in the opposite direction were evident on and after P15, with only male trisomic mice showing significant DYRK1A overexpression. The P15 data were included in our previous report (Hawley et al., 2022), and the new data show that similar sex differences extended to P18 (except for cerebral cortex), P21, and P24, implicating important sex differences in DYRK1A levels in Ts65Dn mice that emerge during the third postnatal week.
A previous study of DYRK1A protein levels in the forebrain of Ts65Dn mice at P5, P10, P15, P20, P25, P30, and P35 in Ts65Dn mice (sex not specified) found that trisomic mice showed significantly elevated levels of DYRK1A protein in the forebrain at all seven ages (Yin et al., 2017). There are several important methodological differences between that study and ours that may account for the discrepancies beyond just the specific regions used for DYRK1A quantification and the specific primary antibody used. The sex of the mice utilized in Yin et al. was not disclosed, but the significant DYRK1A elevations we found in male mice from P6 through P24 may be relatively similar to what Yin et al. reported for P5 to P25 (if those were male). In addition, the current study normalized DYRK1A to total protein staining as compared to β-actin in the Yin et al. study. Total protein provides a stable reference measure that accounts for variation in protein loading and in the amount of protein transferred to the membrane (Welinder and Ekblad, 2011), and can have superior linearity over a range of concentrations when compared to using a single reference protein (Aldridge et al., 2008; Eaton et al., 2013). This may be particularly relevant if the reference protein has differential expression across regions, ages, or genotypes. Additionally, the current study used larger sample sizes (ranging from 6 to 11 mice per group as compared to 3–5 mice per group) which may be necessary to achieve reliable estimates given the high phenotypic variability in Ts65Dn mice (Roper et al., 2020).
4.2. Variability in DYRK1A expression between brain regions
The majority of previous studies of DYRK1A expression in DS mouse models have used adults and have reported significant elevations in DYRK1A protein levels, but many of these examined either whole brain homogenate or a single brain region (reviewed in Stringer et al., 2017b). This study is the first systematic developmental assessment of levels of DYRK1A protein in three major brain regions of Ts65Dn male and female mice. In general, when DYRK1A was upregulated in one brain region of trisomic mice of a given age and sex, it typically was upregulated in other regions as well, particularly when comparing the cerebral cortex and hippocampus. This suggests a common mechanism for DYRK1A regulation in these two forebrain regions. Notable exceptions of multi-region upregulation included the lack of overexpression in the P12 female cerebellum despite upregulation in hippocampus and cerebral cortex, and significant overexpression in female P18 cerebral cortex but not the other regions. Although brain regions tended to show similar patterns of DYRK1A overexpression across regions at a given age and sex, other data from our lab assessing DYRK1A levels in young adult Ts65Dn mice showed instances of regional discordance. At P68, Ts65Dn male mice maintained in individual cages after P25 showed no differences in DYRK1A in cerebral cortex and hippocampus but had significantly lower levels in the cerebellum as compared to euploid controls (Stringer et al., 2017a). In contrast, Ts65Dn male mice group housed and given chronic daily gavage of EGCG or vehicle for three weeks starting on P42 showed increased DYRK1A expression at P68 only in the cerebral cortex and not in the cerebellum or hippocampus (Goodlett et al., 2020). Future studies need to compare DYRK1A protein levels across multiple brain regions, as well as rates of synthesis and degradation of DYRK1A, as this will be a necessary step toward understanding the regional landscape of DYRK1A function in DS model mice.
4.3. Sex-specific expression of Dyrk1a
Sex is recognized as a contributing factor in brain development and differences between male and female gene expression are being reported more frequently in DS research. A study of the circulating proteome did not test for differences in DYRK1A protein expression specifically but did find differences in protein expression in general between males and females (Sullivan et al., 2017). In a study of expression in gene pathways of brain development in the Dp1Yey mouse model, DYRK1A was differently expressed in the hippocampi of male and female control animals aged 7 to 9 months old (Block et al., 2015). At P15, DYRK1A was significantly overexpressed in the hippocampus, cerebral cortex, and cerebellum of male Ts65Dn mice compared to euploid littermate controls; expression in P15 females was not significantly higher in any brain region tested (Hawley et al., 2022). To date, these data are the only reports of sex-specific DYRK1A expression in DS mouse models, emphasizing the limited analysis of effects sex has on protein or transcript quantities in trisomy and the importance of the demonstrated sex differences in DYRK1A expression in the developing brain in the current study. Sex differences in DYRK1A protein expression in Ts65Dn pups may result from the general, genome-wide transcriptional dysregulation in triplicated and non-triplicated genes caused by abnormal gene dosage of Hsa21 genes combined with differences in sex chromosome complement (Saran et al., 2003; Xing et al., 2023). The observed sex differences in DYRK1A expression in the developing Ts65Dn brain occur against the backdrop of the developmental cascades of sex differentiation of the typical brain. Classically, the organizational effects of fetal sex steroids were produced during time-limited critical periods to shape enduring differences in male and female brain structure and function (Morris et al., 2004). Recent evidence suggests that sex chromosome complement may play an important role in sex differences in brain (beyond the classic organizational model), including large adult sex differences in brain regional transcriptome expression profiles and in expression of neuroinflammatory mediators of immune signaling (McCarthy, 2020). This suggests that brain transcriptome analyses in the Ts65Dn model at key developmental ages (P6 and P15) may help identify the extent to which the combination of sex chromosome complement and trisomy results in sex differences in gene expression profiles across a large set of genes in various tissues.
4.4. Genetic alterations of Dyrk1a and resultant DYRK1A levels
The DYRK1A expression of the P6 and P15 offspring from the (Ts65Dn × Dyrk1a+/−) mating to reduce Dyrk1a copy number in some animals confirms several important aspects of developmental regulation of brain DYRK1A expression together with emergent sex differences and the role of trisomic Dyrk1a in the Ts65Dn mouse model. First, having three copies of Dyrk1a was shown to be necessary for DYRK1A overexpression on P6 (seen in all three brain regions in males and in cerebellum in females) in Ts65Dn relative to euploid control mice from (Ts65Dn × Dyrk1a+/−) matings, independent of the effects of other trisomic genes. Reduction of one copy of Dyrk1a from conception in otherwise trisomic mice reduced DYRK1A to levels that approximated those of euploid controls in both sexes at P6. Second, sex-dependent differences in DYRK1A expression that emerged by P15 in the developmental study were partially replicated at P15 in this constitutive knockdown study, such that trisomic males showed significant DYRK1A overexpression in the cerebral cortex in both studies whereas no significant differences in any brain region were evident in P15 females in either study. Third, when there was no increase in trisomic DYRK1A levels with three copies of Dyrk1a, there was no reduction of DYRK1A levels when Dyrk1a copy number was normalized to two copies on an otherwise trisomic background. Fourth, an unexpected significant sex difference was seen on P6 in the effects of functional reduction of Dyrk1a in euploid mice (from two to one copy), in which females showed significant reductions in DYRK1A expression in all three brain regions whereas males showed no significant changes in any region. Collectively, these contrasting outcomes both at P6 (trisomic overexpression in both sexes that requires three functional copies, but differences between sexes in euploid mice with one functional copy in which only females showed haploinsufficiency effects), and at P15 (trisomic overexpression only in males that also requires three functional copies) all suggest the presence of emerging sex differences in developmental regulation of brain DYRK1A expression in a manner that depends on Dyrk1a gene copy number.
4.5. Comparison of trisomic Dyrk1a mRNA and protein expression
In the developmental analyses at P0, P3, and P6, different patterns of Dyrk1a mRNA and protein levels were evident across age and sex. There was a general concordance of overexpression of Dyrk1a mRNA and protein at P6, and the changes in magnitude were more similar in females than males. In contrast, at P0 and P3 large differences between the amount of relative expression of Dyrk1a mRNA and protein were evident in the trisomic groups. Note that the mRNA and protein samples came from different mice at P6 (due to technical problems) but from the same brain samples at P0 and P3; the lack of correlation of mRNA and protein in the trisomic P0 and P3 brains was evident even though the tissues were matched by individual animal. Also note the stark contrast in outcomes of mRNA and protein levels in the comparisons of Ts,Dyrk1a+/+/+ and Ts,Dyrk1a+/+/− P6 males: no significant differences in mRNA expression were found between the two in any brain region, but levels of DYRK1A were significantly reduced (to near euploid levels) in all three regions in the Ts,Dyrk1a+/+/− mice. Discordant outcomes between Dyrk1a RNA and protein expression in brain have been reported in older male Ts65Dn mice, although these comparisons came from different experiments (Ahmed et al., 2012; Choi et al., 2009; Sultan et al., 2007). Transcript and protein fold changes only correlated weakly both when assessing individuals with DS as compared to those without DS and when comparing a discordant trisomic twin pair; experimental and technical noise was acknowledged as a potential contributor to this lack of correlation (Liu et al., 2017). Post transcriptional regulation and turnover, especially of trisomic proteins, was shown to account for differences between transcriptome and proteome products. Others have shown in trisomic fibroblasts that some trisomic transcripts were upregulated but the corresponding proteins were not (or vice versa), and hypothesize those differences reflect post transcriptional regulation, maintenance of stoichiometry of protein complexes, or buffering (Liu et al., 2017). Overexpression of DYRK1A protein may or may not follow overexpression of Dyrk1a mRNA due to posttranscriptional regulation including a lag in translation. As expression levels of trisomic genes are quantified, mRNA and protein levels could be different in any tissue at any developmental time.
4.6. DYRK1A expression and molecular mechanisms
DYRK1A is expressed in non-trisomic mice at several embryonic stages in cortical layers, forebrain neurons, and the nucleus of cells from the ventricular zone in ICR mice (Hammerle et al., 2008). At P5, DYRK1A was localized in the nucleus of cerebellar Purkinje cells (Hammerle et al., 2008); At P14, significant DYRK1A expression was detected in the external granular, molecular layer, and internal granule cell layer of the cerebral cortex (Marti et al., 2003). DYRK1A expression studies have not been extensively conducted in Ts65Dn mice, but Ts65Dn mice do exhibit deficient neurological structural phenotypes in brain regions where we found DYRK1A overexpression including during perinatal or early postnatal ages (Baxter et al., 2000; Belichenko et al., 2004; Bianchi et al., 2010; Chakrabarti et al., 2007; Roper et al., 2006b; Stagni et al., 2016). Many of these histological neurological deficits are thought to be the result of cell cycle dysfunction, to which DYRK1A overexpression has been strongly linked. For example, an overexpression of DYRK1A (observed in the current study) is hypothesized to cause cell cycle dysregulation in mouse models of DS (Haydar and Reeves, 2012) including the G1/S phase, a crucial time when cells will either leave the cell cycle in order to differentiate, or pass through the G1 checkpoint and commit to undergo another round of cell division (Ohnuma and Harris, 2003). Expression of MNB/DYRK1A or Dyrk1a induces neuronal differentiation in neural cell lines (Kelly and Rahmani, 2005; Yang et al., 2001); however, its stable overexpression appears to impair normal neuronal differentiation (Park et al., 2007). Thus, the transient expression of Dyrk1a could promote cell cycle exit, and its subsequent down regulation allows cells to undergo neuronal differentiation (Hammerle et al., 2008; Hammerle et al., 2011). This mechanism of action could be attributed to the ability of DYRK1A to phosphorylate and possibly regulate Cyclin D1 (G1-to-S phase transition and cell proliferation) and p27 (neuronal differentiation and cell cycle exit) (Frank and Tsai, 2009; Hindley and Philpott, 2012; Najas et al., 2015; Tejedor and Hammerle, 2011).
Overexpression of DYRK1A/Dyrk1a both in vivo and in vitro results in the nuclear export of Cyclin D1, resulting in reductions in Cyclin D1 protein levels at multiple developmental ages in TgDyrk1a mice (E11.5 & E14.5) (Najas et al., 2015; Park et al., 2010; Soppa et al., 2014; Yabut et al., 2010). In addition, DYRK1A/Dyrk1a overexpression results in more cells in the G1/0 phase versus S or G2/M phase, leading to cell proliferation deficits, premature cell exit and differentiation, and the nuclear export of Cyclin D1 (Najas et al., 2015; Soppa et al., 2014). Ts65Dn E11.5 embryos contain 1.5-fold more DYRK1A protein and display significant decreases in Cyclin D1 levels. Normalization of Dyrk1a copy number restored levels of Cyclin D1, the number of intermediate progenitors, as well as the production of neurons at multiple developmental time points (Najas et al., 2015).
Although the deficits in cortical, hippocampal, and cerebellar formation in Ts65Dn mice are well established, connections between these deficits and observable behavioral phenotypes at a specific age are not well understood. The current study discovered that DYRK1A protein is significantly elevated at P6, which could be directly affecting the observed hippocampal neurogenesis deficit via hyperphosphorylation of cell cycle proteins (i.e., Cyclin D1). However, future studies will need to determine in what cell types (i.e., pyramidal cells, neurons, Purkinje cells) and where DYRK1A localizes (nucleus, cytosol) to pursue hypotheses concerning which specific substrates and intracellular processes DYRK1A could be regulating. The substrates regulated by DYRK1A phosphorylation are located in both the nucleus and cytosol, and this regulation is likely to be tissue and time-specific (Arron et al., 2006; Chen-Hwang et al., 2002; de Graaf et al., 2004; Kaczmarski et al., 2014; Marti et al., 2003; Woods et al., 2001). Assessing protein levels in a brain region is a fundamental first step toward understanding the influence of trisomic DYRK1A on DS-related phenotypes.
4.7. Potential for treatment guided by sex-specific spatiotemporal expression patterns
The majority of therapeutics to date administered to Ts65Dn mice across ages have targeted specific neurotransmitter systems or aberrant neural pathways, based on their contribution to cognitive and behavioral phenotypes (Lorenzon et al., 2023). Alternatively, a targeted gene or set of genes hypothesized to have a major influence on a phenotype due to overexpression in DS may be a therapeutic focus (Korbel et al., 2009; Lyle et al., 2009). However, the presence of a triplicated gene does not mean that it will alter development and function in every cell across development. Thus, it is critical to identify the spatial and temporal expression of a trisomic gene in conjunction with specific phenotypes, in order to understand when the trisomic gene(s) cause(s) divergence from normal patterns of development (Roper and Reeves, 2006; Stringer et al., 2017b). Because the overexpression of DYRK1A is hypothesized to alter developmental trajectories and cause cognitive, skeletal, and other DS-related phenotypes (Duchon and Herault, 2016), and the presumed elevation of DYRK1A may be a target of therapy, we reasoned that a developmental timeline of DYRK1A expression would be important (Stringer et al., 2017b). As confirmed by the current results with DYRK1A, expression of trisomic genes may not be as simple as the “1.5-fold upregulation rule”; compensation, or different classes or levels of expression of trisomic genes may influence DS phenotypic development (Ait Yahya-Graison et al., 2007). DYRK1A may also self-regulate by autophosphorylation which may enhance its own kinase activity while reducing its degradation (Atas-Ozcan et al., 2021). Altered expression of DYRK1A at different points in development may lead to tissue- and sex-specific phenotypic alterations, necessitating differential gene specific pharmacological treatments.
4.8. Limitations of the study
Ts65Dn mice are one of many DS mouse models and have been the primary model used in DS preclinical neurobiological and neuro-behavioral research, including extensive use in testing therapies designed to normalize DS phenotypes (Garcia-Cerro et al., 2017; Gardiner, 2015; Reeves et al., 1995). They are the only DS mouse model currently characterized that exhibit early postnatal gene expression, neuroanatomical, and developmental behavioral deficits like those observed in DS. Limitations of the Ts65Dn model have been identified, the most notable being that the trisomic mice also contain ~35 protein coding genes found on mouse chromosome 17 that are not homologous to Hsa21 (Duchon et al., 2011; Reinholdt et al., 2011). With the development of the Ts66Yah and other DS mouse models, the contributions of these non-Hsa21 genes are becoming understood (Duchon et al., 2022; Xing et al., 2023). These newer models may have more accurate face validity but may still lack many Hsa21 homologous genes. More research with additional models is needed to find the genetic interactions of Dyrk1a and other trisomic genes.
In the developmental analysis of DYRK1A, the lack of inclusion of a protein homogenate standard in common across all blots—needed to provide a means to standardize expression levels of samples across blots—is a methodological limitation. This limits our analyses to within-blot comparisons of trisomic samples relative to euploid samples (same sex, age, and brain region), precluding direct quantitative comparisons of changes in trisomic and euploid groups across age and sex. Without across-blot standardization, the extent to which DYRK1A expression changed as a function of age or sex in trisomic and euploid mice in the developmental study cannot be determined. This study also is limited by its focus on tissue levels of DYRK1A; neither the cellular and sub-cellular localization of DYRK1A expression nor the kinase activity of DYRK1A was assessed, so the cellular localization and functional activity throughout development remain to be characterized. Given that the developmental changes in DYRK1A overexpression were demonstrated to be gene-dosage dependent on P6 and P15, it will be important to determine whether differences in DYRK1A protein expression are accompanied by proportional differences in kinase activity.
Supplementary Material
Acknowledgements
The authors thank Dr. Jon Crispino for the Dyrk1a conditional knockout mice and Dr. Mariona Arbonés for the Dyrk1a knockout mice. This project was supported by Beaty/C-Tech Fund—Blue River Community Foundation; the Indiana Clinical and Translational Sciences Institute and funded in part by Grant Number UL1TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award; and by the National Institues of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR078663 to RJR and CRG.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
CRediT authorship contribution statement
Laura E. Hawley: Conceptualization, Methodology, Validation, Investigation, Data curation, Formal analysis, Writing – original draft, Writing – review & editing, Visualization, Supervision, Project administration. Megan Stringer: Conceptualization, Methodology, Investigation, Supervision, Project administration. Abigail J. Deal: Investigation, Formal analysis, Data curation, Writing – review & editing. Andrew Folz: Investigation, Validation, Writing – review & editing. Charles R. Goodlett: Conceptualization, Validation, Formal analysis, Resources, Writing – original draft, Writing – review & editing, Visualization, Supervision, Funding acquisition. Randall J. Roper: Conceptualization, Validation, Resources, Writing – original draft, Writing – review & editing, Visualization, Supervision, Funding acquisition.
Declaration of Competing Interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2023.106359.
Data availability
All data related to this study have been deposited at www.datadryad.org and are publicly available as of the date of publication.
References
- Ahmed MM, et al. , 2012. Loss of correlations among proteins in brains of the Ts65Dn mouse model of down syndrome. J. Proteome Res 11, 1251–1263. [DOI] [PubMed] [Google Scholar]
- Ahn KJ, et al. , 2006. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis 22, 463–472. [DOI] [PubMed] [Google Scholar]
- Ait Yahya-Graison E, et al. , 2007. Classification of human chromosome 21 gene-expression variations in down syndrome: impact on disease phenotypes. Am. J. Hum. Genet 81, 475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge GM, et al. , 2008. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 172, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altafaj X, et al. , 2013. Normalization of Dyrk1A expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of down syndrome. Neurobiol. Dis 52, 117–127. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, et al. , 2020. Down syndrome. Nat. Rev. Dis. Primers 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arron JR, et al. , 2006. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 441, 595–600. [DOI] [PubMed] [Google Scholar]
- Atas-Ozcan H, et al. , 2021. Dyrk1a from gene function in development and physiology to dosage correction across life span in down syndrome. Genes (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, et al. , 1999. MRI volumes of the hippocampus and amygdala in adults with Down’s syndrome with and without dementia. Am. J. Psychiatry 156, 564–568. [DOI] [PubMed] [Google Scholar]
- Aziz NM, et al. , 2018. Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of down syndrome. Dis. Model. Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter LL, et al. , 2000. Discovery and genetic localization of down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum. Mol. Genet 9, 195–202. [DOI] [PubMed] [Google Scholar]
- Becker W, et al. , 2014. DYRK1A: a potential drug target for multiple down syndrome neuropathologies. CNS Neurol. Disord. Drug Targets 13, 26–33. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, et al. , 2004. Synaptic structural abnormalities in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol 480, 281–298. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, et al. , 2007. Synaptic and cognitive abnormalities in mouse models of down syndrome: exploring genotype-phenotype relationships. J. Comp. Neurol 504, 329–345. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, et al. , 2015. Down syndrome cognitive phenotypes modeled in mice Trisomic for all HSA 21 homologues. PLoS One 10, e0134861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi P, et al. , 2010. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for down syndrome. J. Neurosci 30, 8769–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block A, et al. , 2015. Sex differences in protein expression in the mouse brain and their perturbations in a model of down syndrome. Biol. Sex Differ 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM, 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Branchi I, et al. , 2004. Transgenic mouse in vivo library of human down syndrome critical region 1: association between DYRK1A overexpression, brain development abnormalities, and cell cycle protein alteration. J. Neuropathol. Exp. Neurol 63, 429–440. [DOI] [PubMed] [Google Scholar]
- Canzonetta C, et al. , 2008. DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in down syndrome. Am. J. Hum. Genet 83, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, et al. , 2007. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of down syndrome. J. Neurosci 27, 11483–11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Hwang MC, et al. , 2002. Dynamin is a minibrain kinase/dual specificity Yak1-related kinase 1A substrate. J. Biol. Chem 277, 17597–17604. [DOI] [PubMed] [Google Scholar]
- Cheon MS, et al. , 2003. Protein levels of genes encoded on chromosome 21 in fetal down syndrome brain: challenging the gene dosage effect hypothesis (part II). Amino Acids 24, 119–125. [DOI] [PubMed] [Google Scholar]
- Choi JH, et al. , 2009. Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn down syndrome mouse model. J. Neurochem 110, 1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, et al. , 2007. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus. 17, 665–678. [DOI] [PubMed] [Google Scholar]
- Contestabile A, et al. , 2017. The GABAergic hypothesis for cognitive disabilities in down syndrome. Front. Cell. Neurosci 11, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, et al. , 2013. Hedgehog agonist therapy corrects structural and cognitive deficits in a down syndrome mouse model. Sci. Transl. Med 5, 201ra120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Torre R, Dierssen M, 2012. Therapeutic approaches in the improvement of cognitive performance in down syndrome: past, present, and future. Prog. Brain Res 197, 1–14. [DOI] [PubMed] [Google Scholar]
- Dowjat WK, et al. , 2007. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with down syndrome. Neurosci. Lett 413, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes EC, et al. , 2008. Loss of synaptophysin and synaptosomal-associated protein 25-kDa (SNAP-25) in elderly down syndrome individuals. Neuropathol. Appl. Neurobiol 34, 12–22. [DOI] [PubMed] [Google Scholar]
- Duchon A, Herault Y, 2016. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in down syndrome. Front. Behav. Neurosci 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchon A, et al. , 2011. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mamm. Genome 22, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchon A, et al. , 2021. Multi-influential genetic interactions alter behaviour and cognition through six main biological cascades in down syndrome mouse models. Hum. Mol. Genet 30, 771–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchon A, et al. , 2022. Ts66Yah, a mouse model of down syndrome with improved construct and face validity. Dis. Model. Mech 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton SL, et al. , 2013. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS One 8, e72457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgin JO, et al. , 2012. Human and mouse model cognitive phenotypes in down syndrome: implications for assessment. Prog. Brain Res 197, 123–151. [DOI] [PubMed] [Google Scholar]
- Fotaki V, et al. , 2002. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol 22, 6636–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CL, Tsai LH, 2009. Alternative functions of core cell cycle regulators in neuronal migration, neuronal maturation, and synaptic plasticity. Neuron. 62, 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gameren-Oosterom HB, et al. , 2011. Development, problem behavior, and quality of life in a population based sample of eight-year-old children with down syndrome. PLoS One 6, e21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gameren-Oosterom HB, et al. , 2013. Problem behavior of individuals with down syndrome in a nationwide cohort assessed in late adolescence. J. Pediatr 163, 1396–1401. [DOI] [PubMed] [Google Scholar]
- Garcia-Cerro S, et al. , 2014. Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and Neuromorphological alterations found in a mouse model of down syndrome. PLoS One 9, e106572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cerro S, et al. , 2017. Normalizing the gene dosage of Dyrk1A in a mouse model of down syndrome rescues several Alzheimer’s disease phenotypes. Neurobiol. Dis 106, 76–88. [DOI] [PubMed] [Google Scholar]
- Gardiner KJ, 2015. Pharmacological approaches to improving cognitive function in down syndrome: current status and considerations. Drug Des. Devel. Ther 9, 103–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett CR, et al. , 2020. Evaluation of the therapeutic potential of Epigallocatechin-3-gallate (EGCG) via oral gavage in young adult down syndrome mice. Sci. Rep 10, 10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodliffe JW, et al. , 2016. Absence of prenatal forebrain defects in the Dp(16)1Yey/+ mouse model of down syndrome. J. Neurosci 36, 2926–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf K, et al. , 2004. Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: phosphorylation by DYRK1A and colocalization with splicing factors. J. Biol. Chem 279, 4612–4624. [DOI] [PubMed] [Google Scholar]
- Guidi S, et al. , 2008. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with down syndrome. Brain Pathol. 18, 180–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimera J, et al. , 1999. Human minibrain homologue (MNBH/DYRK1): characterization, alternative splicing, differential tissue expression, and overexpression in down syndrome. Genomics. 57, 407–418. [DOI] [PubMed] [Google Scholar]
- Hammerle B, et al. , 2008. The spatio-temporal and subcellular expression of the candidate down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur. J. Neurosci 27, 1061–1074. [DOI] [PubMed] [Google Scholar]
- Hammerle B, et al. , 2011. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development. 138, 2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley LE, et al. , 2022. Sexually dimorphic DYRK1A overexpression on postnatal day 15 in the Ts65Dn mouse model of down syndrome: effects of pharmacological targeting on behavioral phenotypes. Pharmacol. Biochem. Behav 217, 173404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar TF, Reeves RH, 2012. Trisomy 21 and early brain development. Trends Neurosci. 35, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herault Y, et al. , 2017. Rodent models in down syndrome research: impact and future opportunities. Dis. Model. Mech 10, 1165–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindley C, Philpott A, 2012. Co-ordination of cell cycle and differentiation in the developing nervous system. Biochem. J 444, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, et al. , 2021. Consequences of aneuploidy in human fibroblasts with trisomy 21. Proc. Natl. Acad. Sci. U. S. A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, et al. , 2015. Genetic dissection of the down syndrome critical region. Hum. Mol. Genet 24, 6540–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarski W, et al. , 2014. Intracellular distribution of differentially phosphorylated dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A). J. Neurosci. Res 92, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlem P, et al. , 2004. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome Res. 14, 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, et al. , 2022. A transchromosomic rat model with human chromosome 21 shows robust down syndrome features. Am. J. Hum. Genet 109, 328–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PA, Rahmani Z, 2005. DYRK1A enhances the mitogen-activated protein kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol. Biol. Cell 16, 3562–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesslak JP, et al. , 1994. Magnetic resonance imaging analysis of age-related changes in the brains of individuals with Down’s syndrome. Neurology. 44, 1039–1045. [DOI] [PubMed] [Google Scholar]
- Kojima S, Cimini D, 2019. Aneuploidy and gene expression: is there dosage compensation? Epigenomics. 11, 1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, et al. , 2009. The genetic architecture of down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U. S. A 106, 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, et al. , 2008. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in down syndrome. FASEB J. 22, 3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. , 2017. Systematic proteome and proteostasis profiling in human trisomy 21 fibroblast cells. Nat. Commun 8, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockstone HE, et al. , 2007. Gene expression profiling in the adult down syndrome brain. Genomics. 90, 647–660. [DOI] [PubMed] [Google Scholar]
- Lorenzi HA, Reeves RH, 2006. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 1104, 153–159. [DOI] [PubMed] [Google Scholar]
- Lorenzon N, et al. , 2023. State-of-the-art therapy for down syndrome. Dev. Med. Child Neurol 65, 870–884. [DOI] [PubMed] [Google Scholar]
- Lyle R, et al. , 2004. Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res. 14, 1268–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, et al. , 2009. Genotype-phenotype correlations in down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur. J. Hum. Genet 17, 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maatta T, et al. , 2006. Mental health, behaviour and intellectual abilities of people with down syndrome. Downs Syndr. Res. Pract 11, 37–43. [DOI] [PubMed] [Google Scholar]
- Mao R, et al. , 2003. Global up-regulation of chromosome 21 gene expression in the developing down syndrome brain. Genomics. 81, 457–467. [DOI] [PubMed] [Google Scholar]
- Mao R, et al. , 2005. Primary and secondary transcriptional effects in the developing human down syndrome brain and heart. Genome Biol. 6, R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal JP, et al. , 2016. Growing up with down syndrome: development from 6 months to 10.7 years. Res. Dev. Disabil 59, 437–450. [DOI] [PubMed] [Google Scholar]
- Marti E, et al. , 2003. Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res. 964, 250–263. [DOI] [PubMed] [Google Scholar]
- Martinez de Lagran M, et al. , 2004. Motor phenotypic alterations in TgDyrk1a transgenic mice implicate DYRK1A in down syndrome motor dysfunction. Neurobiol. Dis 15, 132–142. [DOI] [PubMed] [Google Scholar]
- Martinez-Cue C, et al. , 2002. Differential effects of environmental enrichment on behavior and learning of male and female Ts65Dn mice, a model for down syndrome. Behav. Brain Res 134, 185–200. [DOI] [PubMed] [Google Scholar]
- Martinez-Cue C, et al. , 2006. Anxiety and panic responses to a predator in male and female Ts65Dn mice, a model for down syndrome. Genes Brain Behav. 5, 413–422. [DOI] [PubMed] [Google Scholar]
- McCarthy MM, 2020. A new view of sexual differentiation of mammalian brain. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol 206, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane L, et al. , 2013. Novel PCR assay for determining the genetic sex of mice. Sex. Dev 7, 207–211. [DOI] [PubMed] [Google Scholar]
- Morris JA, et al. , 2004. Sexual differentiation of the vertebrate nervous system. Nat. Neurosci 7, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Moyer AJ, et al. , 2021. All creatures great and small: new approaches for understanding down syndrome genetics. Trends Genet. 37, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najas S, et al. , 2015. DYRK1A-mediated cyclin D1 degradation in neural stem cells contributes to the neurogenic cortical defects in down syndrome. EBioMedicine. 2, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, et al. , 2017. An appropriate loading control for western blot analysis in animal models of myocardial ischemic infarction. Biochem. Biophys. Rep 12, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma S-I, Harris WA, 2003. Neurogenesis and the cell cycle. Neuron. 40, 199–208. [DOI] [PubMed] [Google Scholar]
- Park J, Chung KC, 2013. New perspectives of Dyrk1A role in neurogenesis and Neuropathologic features of down syndrome. Exp. Neurobiol 22, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, et al. , 2007. Dyrk1A overexpression in immortalized hippocampal cells produces the neuropathological features of down syndrome. Mol. Cell. Neurosci 36, 270–279. [DOI] [PubMed] [Google Scholar]
- Park J, et al. , 2010. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J. Biol. Chem 285, 31895–31906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter JD, et al. , 2001. Neuroanatomy of Down’s syndrome: a high-resolution MRI study. Am. J. Psychiatry 158, 1659–1665. [DOI] [PubMed] [Google Scholar]
- Raz N, et al. , 1995. Selective neuroanatomic abnormalities in Down’s syndrome and their cognitive correlates: evidence from MRI morphometry. Neurology. 45, 356–366. [DOI] [PubMed] [Google Scholar]
- Reeves RH, et al. , 1995. A mouse model for down syndrome exhibits learning and behaviour deficits. Nat. Genet 11, 177–184. [DOI] [PubMed] [Google Scholar]
- Reinholdt LG, et al. , 2011. Molecular characterization of the translocation breakpoints in the down syndrome mouse model Ts65Dn. Mamm. Genome 22, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC, et al. , 2010. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc., 2010, pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- Roper RJ, Reeves RH, 2006. Understanding the basis for down syndrome phenotypes. PLoS Genet. 2, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, et al. , 2006a. Defective cerebellar response to mitogenic hedgehog signaling in down [corrected] syndrome mice. Proc. Natl. Acad. Sci. U. S. A 103, 1452–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, et al. , 2006b. Perinatal loss of Ts65Dn down syndrome mice. Genetics. 172, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, et al. , 2020. Influence of allelic differences in down syndrome. Prog. Brain Res 251, 29–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saran NG, et al. , 2003. Global disruption of the cerebellar transcriptome in a down syndrome mouse model. Hum. Mol. Genet 12, 2013–2019. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ, 2008. Analyzing real-time PCR data by the comparative C (T) method. Nat. Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schneider CA, et al. , 2012. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppa U, et al. , 2014. The down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 13, 2084–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagni F, et al. , 2016. Short- and long-term effects of neonatal pharmacotherapy with epigallocatechin-3-gallate on hippocampal development in the Ts65dn mouse model of down syndrome. Neuroscience. 333, 277–301. [DOI] [PubMed] [Google Scholar]
- Stagni F, et al. , 2018. Neurogenesis impairment: an early developmental defect in down syndrome. Free Radic. Biol. Med 114, 15–32. [DOI] [PubMed] [Google Scholar]
- Stringer M, et al. , 2017a. Epigallocatechin-3-gallate (EGCG) consumption in the Ts65Dn model of down syndrome fails to improve behavioral deficits and is detrimental to skeletal phenotypes. Physiol. Behav 177, 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer M, et al. , 2017b. Targeting trisomic treatments: optimizing Dyrk1a inhibition to improve down syndrome deficits. Mol. Genet. Genomic. Med 5, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, et al. , 2017. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep 7, 14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, et al. , 2007. Gene expression variation in Down’s syndrome mice allows prioritization of candidate genes. Genome Biol. 8, R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor FJ, Hammerle B, 2011. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 278, 223–235. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, et al. , 2015. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing cyclin D3. J. Exp. Med 212, 953–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis S, et al. , 1991. Down syndrome: MR quantification of brain structures and comparison with normal control subjects. AJNR Am. J. Neuroradiol 12, 1207–1211. [PMC free article] [PubMed] [Google Scholar]
- Welinder C, Ekblad L, 2011. Coomassie staining as loading control in Western blot analysis. J. Proteome Res 10, 1416–1419. [DOI] [PubMed] [Google Scholar]
- Woods YL, et al. , 2001. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J 355, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AD, et al. , 2013. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci 33, 7368–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, et al. , 2023. Dissection of a down syndrome-associated trisomy to separate the gene dosage-dependent and -independent effects of an extra chromosome. Hum. Mol. Genet 32, 2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabut O, et al. , 2010. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci 30, 4004–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang EJ, et al. , 2001. Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem 276, 39819–39824. [DOI] [PubMed] [Google Scholar]
- Yin X, et al. , 2017. Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn down syndrome mice. Sci. Rep 7, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data related to this study have been deposited at www.datadryad.org and are publicly available as of the date of publication.