

Abstract
The mechanistic target of rapamycin (mTOR) is evolutionarily conserved from yeast to humans and is one of the most fundamental pathways of living organisms. Since its discovery three decades ago, mTOR has been recognized as the center of nutrient sensing and growth, homeostasis, metabolism, life span, and aging. The role of dysregulated mTOR in common diseases, especially cancer, has been extensively studied and reported. Emerging evidence supports that mTOR critically regulates innate immune responses that govern the pathogenesis of various cardiovascular diseases. This review discusses the regulatory role of mTOR in macrophage functions in acute inflammation triggered by ischemia and in atherosclerotic cardiovascular disease (ASCVD) and heart failure with preserved ejection fraction (HFpEF), in which chronic inflammation plays critical roles. Specifically, we discuss the role of mTOR in trained immunity, immune senescence, and clonal hematopoiesis. In addition, this review includes a discussion on the architecture of mTOR, the function of its regulatory complexes, and the dual-arm signals required for mTOR activation to reflect the current knowledge state. We emphasize future research directions necessary to understand better the powerful pathway to take advantage of the mTOR inhibitors for innovative applications in patients with cardiovascular diseases associated with aging and inflammation.
Keywords: Ischemia, Inflammation, mTOR, Macrophage, Atherosclerosis
1. Introduction
The mechanistic target of rapamycin (mTOR) is a member of the phosphoinositide 3-kinase (PI3K)-related serine/threonine protein kinase family. As the fundamental mechanism that regulates growth and coordinates growth with the availability of nutrients in the environment, mTOR plays critical roles in homeostasis, metabolism, lifespan, and aging [1]. Dysregulated mTOR contributes to common diseases such as cancer, metabolic disorders, inflammation, neurodegenerative diseases, and epilepsy [2]. Although less studied, emerging evidence demonstrates that mTOR regulates innate immune responses to injuries, threats, and endogenous stimuli, contributing to the pathogenesis of common cardiovascular conditions, including atherosclerosis and heart failure.
mTOR functions through two distinct multi-protein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is better characterized and has well-studied functions of regulating cell growth and metabolism in response to nutrient availability, growth factors, and energy status. On the other hand, mTORC2 is less understood and is known to regulate cell survival, cytoskeletal organization, and metabolism. The majority of work reviewed here is related to mTORC1. Activation of mTORC1 switches on biomass production to increase the biosynthesis of proteins, lipids, and nucleic acids and turns off these macromolecules’ breakdown. These processes are essential to various aspects of macrophage functions. In myocardial infarction (MI), circulating monocytes migrate to the ischemic region of the heart, where they are activated and differentiate into macrophages [4,187,188]. During the differentiation, macrophages undergo remarkable morphological changes and gain many intracellular organelles, especially lysosomes, to support a more complex function profile. Differentiated macrophages also have a higher endocytotic capacity and produce cytokines, chemokines, and lipid mediators such as leukotrienes and prostaglandins at a higher level [4–6]. These processes are metabolically demanding, and the involvement of mTORC1 appears self-evident. In addition, mTORC1 is the node that integrates environment signals with cellular functions, a pathway that can facilitate macrophages to adjust to the environment. Indeed, mTORC1 activation in cardiac macrophages after MI has been recently described [4], although the protective effect of rapamycin or S6 (a downstream target of mTOR complex 1) inhibitor to acute ischemia has been reported earlier [7]. These data suggest a role for mTORC1 in macrophages in acute ischemia, but much remains unknown. For example, mTORC1 activation in the infarct tissue, which lacks traditional nutrients, energy, and oxygen, contradicts the conventional environment necessary for mTORC1 activation.
Emerging evidence also supports mTOR signaling in sustaining chronic inflammation, the underpinning of atherosclerotic cardiovascular disease (ASCVD). Many traditional risk factors and lifestyles, including the Western-style diet (WD), psychological stress, sedentary lifestyle, and sleeping irregularity, promote vascular wall inflammation and atherosclerosis by trained immunity [8–13]. Trained immunity is a form of immune memory for innate immune cells in which activation of these cells can result in enhanced responsiveness to subsequent triggers unrelated to the initial challenge. Metabolic reprogramming to aerobic glycolysis is required for trained immunity, and ample evidence suggests mTOR activation promoted such a type of metabolism in cancer cells and innate and adaptive immune cells [14–16]. In addition, mTOR regulates epigenetic modifications and hematopoiesis, both of which are critical for trained immunity. Senescent monocytes and macrophages were essential contributors to inflamm-aging [17], a persistent low-grade inflammation that occurs with increasing age, which can perpetuate age-related diseases such as ASCVD. Remarkably, a senescent immune system led to the aging of other solid organs, such as the liver and kidney [18]. Rapamycin rejuvenated the aged immune system [18], suggesting immune senescence can be modified, alleviating its impact on the cardiovascular system.
This review aims to examine the role of the mTOR in governing macrophages’ function and its implications in acute ischemia, in chronic inflammation that drives atherosclerotic cardiovascular disease (ASCVD), and in heart failure with preserved ejection function (HFpEF). We discuss the regulatory function of mTOR in trained immunity, immune senescence, clonal hematopoiesis, and CHIP (Clonal hematopoiesis of indeterminate potential), which are biological processes contributing to atherosclerosis and HFpEF. We depict the key aspects of the discussion in Figure 1 and Figure 2.
Figure 1.
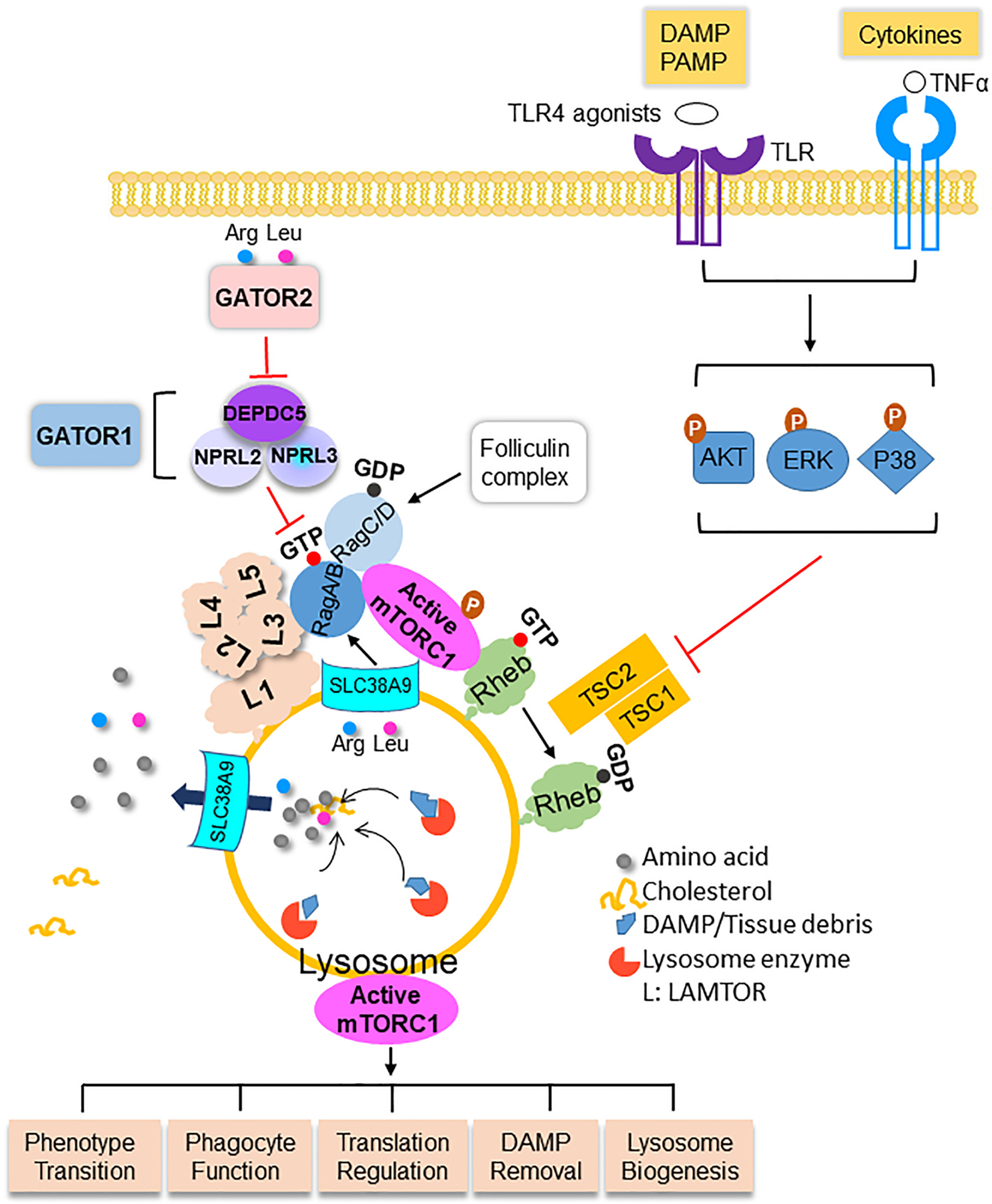
The architecture of mTORC1, the activation and the role of mTORC1 in macrophages in myocardial ischemia. Macrophages digest DAMP/tissue debris to generate amino acids. Amino acids are transported out of lysosomes into the cytosol where arginine (Arg) and leucine (Leu) trigger the translocation of mTORC1 to the surface of lysosomes via Rag GTPase. The LAMTOR (L) complex anchors mTORC1-Rag to the lysosome membrane. Arginine inside the lysosome activates mTORC1 after being sensed by SLC38A9. The assembly of mTORC1 on lysosome membrane is inhibited by GATOR1 and promoted by GATOR2. mTORC1 in macrophages in the infarcted myocardium is activated by the interaction between DAMP and PRR, including TLR4 and its ligands, and cytokines such as TNFα. The stimulation of DAMP and cytokine lead to the activation of AKT, MAPK/ERK, and P38, which phosphorylate the TSC complex. Phosphorylated TSC relieves its inhibitory effect on Rheb that causes activation of mTORC1. mTORC1 regulates essential functions of macrophages including phenotype transition, phagocytotic function, translational regulation of cytokines, DAMP removal, and lysosome proliferation and action. More details are discussed in the main text. DAMP, damage-associated molecular pattern; PRR, pattern recognition receptor; PAMP, pathogen-associated molecular pattern
Figure 2.
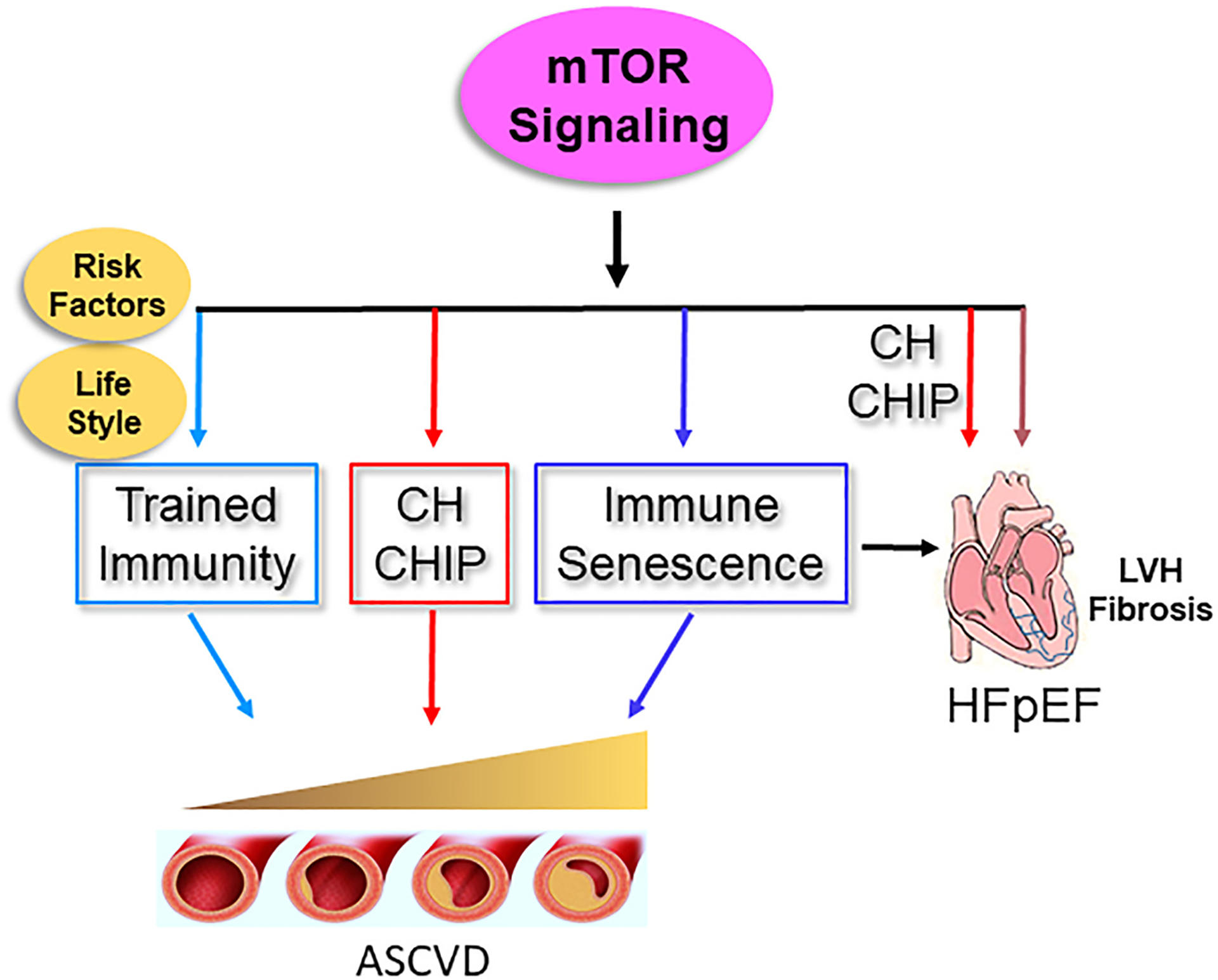
mTOR signaling contributes to atherosclerotic cardiovascular disease (ASCVD) and heart failure with presreved ejection fraction (HFpEF). mTOR activation contributes to atherosclerosis by mediating trained immunity, CH and CHIP, and immune senescence. mTOR activation participates in the pathogenesis of HFpEF by enhancing cardiac fibrosis and promoting inflammation and left ventricle (LV) stiffness mediated by CHIP and immune senescence.
2. The architecture of mTORC1
mTORC1 has three subunits: the regulatory-associated protein of mTOR (Raptor), the positive mTOR regulator mLST8 (mammalian lethal with SEC13 protein 8), and the serine/threonine kinase mTOR (Figure 1). Raptor functions as a scaffold for mTOR kinase to recruit and phosphorylate specific substrates such as the ribosomal protein S6 kinase (S6K) and the eukaryotic translation initiation factor 4E binding proteins (4EBPs) [19]. mLST8 enables protein serine/threonine kinase activator activity [20]. Amino acids signal to the mTORC1 by the Rag GTPase with four isoforms, RagA, RagB, RagC, and RagD. When amino acids are sufficient, Rag GTPases form the heterodimer of GTP-loaded RagA/RagB (RagAGTP/RagBGTP) and GDP-loaded RagC/D (RagCGDP/RagDGDP) that recruits mTORC1 to the lysosomal surface [21,22]. Upon withdrawal of amino acids, the Rag GTPases release mTORC1 into the cytosol to become inactivated [22]. The pentameric Ragulator complex anchors the mTORC1-Rag complex on the lysosome membrane. Ragulator contains two heterodimers: Lamtor2 (late endosomal/lysosomal adaptor and MAPK and mTOR activator 2)-Lamtor3 and Lamtor4-Lamtor5, and these heterodimers are held together by Lamtor1 [23]. Another complex that interacts with Rag GTPases is GAP (GTPase-activating protein) activity toward the Rags (Gator1), which consists of three components: Nprl2 (nitrogen permease regulator-like 2), Nprl3, and Depdc5 (Dishevelled Egl-10 and Pleckstrin domain-containing protein 5). Gator1 functions as a GAP toward RagA and RagB [24] and is a negative regulator of mTORC1 [24]. Depdc5 is the only subunit of Gator1 that binds the Rag GTPases, and the interaction between Depdc5 and Rag GTPases confers the inhibitory mode of Gator1 [25]. Deletion of Depdc5 leads to constitutively elevated mTORC1 activation regardless of amino acid abundance [26]. The third complex that potentially interacts with Rag GTPase is Gator2, which is composed of MIOS (meiosis regulator for oocyte development), WDR59 (WD repeat domain 59), WDR24 (WD Repeat Domain 24), Seh1L (SEH1-like nucleoporin), and SEC13 (SEC13 homolog, nuclear pore and COPII coat complex component). Gator2 directly responds to leucine and arginine and suppresses Gator1, releasing Gator1’s inhibition to mTORC1 [24]. The fourth complex interacting with Rag GTPase is the folliculin complex (FLCN with FNIP1 (Folliculin-interacting protein 1) or FNIP2). FLCN/FNIP has GAP activity on Rag C/D, promoting Rag GTPase transition from inactive to active isoforms [27]. Detailed mechanisms on the fifth complex that interacts with Rag, the vacuolar H+-ATPase (VATPase), remain to be fully elucidated and will not be a focus for this review.
3. Nutrient sensing and mTORC1 activation
Amino acids such as leucine and arginine are the strongest in stimulating mTORC1 activation [2,28]. Leucine binds to its sensor Sestrin2 [29], and the interaction relieves Sestrin2’s inhibitory effect on Gator2 that suppresses Gator1, the negative regulator of mTORC1. Similarly, Castor1 (cytosolic arginine sensor for mTORC1 subunit 1) is an arginine sensor, and arginine-bound Castor1 disrupts the inhibitory complex of Castor1-Gator2, leading to Gator2-mediated Gator1 inhibition and mTORC1 activation [30]. Interestingly, different groups have reported SLC38A9 (solute carrier 38A9) as a unique sensor of arginine and transporter of essential amino acids [31–34]. SLC38A9 is a lysosomal membrane protein integral to the Ragulator-Rag GTPase complex [31,33]. As a particular sensor, SLC38A9 detects arginine inside the lysosomal lumen and in the cytosol and stimulates mTORC1 activation [31,32,34]. In addition, SLC38A9 transports amino acids generated by the degradation of proteins out of lysosome into the cytosol, where they induce mTORC1 activation [34]. Besides leucine and arginine, S-adenosylmethionine (SAM), a metabolite of amino acid methionine, activates mTORC1 by the sensor Samtor (S-adenosylmethionine sensor upstream of mTORC1) [35]. SAM-infused Samtor dissociates from Gator1, which releases the inhibition of Gator1 to mTORC1. As SAM is the universal methyl donor in methylating RNA, DNA, and proteins such as histones [36], it connects mTORC1 with potential epigenetic regulation via one-carbon metabolism. Besides amino acids, recent evidence suggests cholesterol derived from low density lipoprotein (LDL) processing in the lysosomes activated mTORC1 by the transporter SLC38A9 [37,182]. In addition, loss of function of Niemann-Pick C1 (NPC1), the cholesterol exporter and the mutation of which results lipid accumulation, fatal metabolic derangement, and a neurodegenerative diseases, constititutively activated mTORC1 [37,182], further supporting cholesterol as a form of nutrient sensed by mTORC1. The consummate effect of nutrient abundance is for mTORC1 to assemble on the outer membrane surface of lysosomes, where mTORC1 is activated by kinases downstream of growth factors. Importantly, cholesterol-derived from LDL as an activator of mTORC1 provides the mechanistic basis for mTOR’s role in atherosclerosis, which will be discussed later.
4. Growth factors and mTORC1 activation
Once the mTORC1 complex localizes on the lysosome membrane, growth factors and cellular stresses activate the mTOR kinase by the Rheb (Ras homolog enriched in brain) GTPases. An earlier study reported that Rheb bound to FKBP38 (FK506-binding protein), an endogenous mTOR inhibitor, dissociated it from mTOR in a GTP-dependent fashion [38]. However, these findings were not substantiated by others [39]. Recent data obtained by cryo-electron microscopy suggested that binding of Rheb to the mTOR kinase allosterically realigned active-site residues and relieved the auto-inhibition [40]. Activated mTOR kinase phosphorylates its downstream substrates, including S6K, 4EBP1, and TFEB (Transcription factor EB). These events lead to synthesizing proteins, lipids, and nucleotides and suppressing lysosome degradation. The tuberous sclerosis complex (TSC) takes center stage to transduce signals from the growth factor arm to mTORC1. The TSC complex suppresses mTORC1 by functioning as a GAP to the Rheb GTPases that hydrolyze GTP loaded on Rheb to GDP-Rheb, which loses its ability to stimulate the mTORC1 kinase activity [41]. The TSC complex is composed of TSC1, TSC2, and TBC1D7 [42–44]. While TSC2 of the TSC complex confers the GAP function, TSC1 enhances its GAP activity and stabilizes TBC1D7 within the trimeric complex [45]. The role of TBC1D7 is not entirely clear but appears to maintain the proper function of the complex [44]. Growth factors such as insulin and insulin-like GF (IGF) activate the PI3K/AKT, which phosphorylates the Ser939, Ser1086, Ser1088, and Thr1422 residues on TSC2 [42,46]. Phosphorylation of TSC2 releases the inhibitory effect of the TSC complex on Rheb, leading to mTORC1 activation.
Besides insulin and IGFs, receptor tyrosine kinase-dependent Ras signaling activates mTORC1 via the MAP Kinase and its effector p90RSK by phosphorylating/inhibiting TSC2 [47,48]. EGF and phorbol esters (PMA), ligands of Ras, and constitutively activated Ras mutants phosphorylate TSC and relieve its inhibition of mTORC1 [48,49]. Additionally, TSC2 becomes heavily phosphorylated in response to various cellular stress signals [42,46], therefore highlighting the crucial role of TSC in stress-induced mTORC1 activation. GTP-loaded Ras can also directly bind and allosterically activate PI3K [50], initiating the PI3K/AKT-activated mTORC1 cascade. Interestingly, agonists of Ras/MAPK/ERK (EGF and PMA) induce direct phosphorylation of Raptor at Ser8, Ser696, and Ser863 and promote mTORC1 signaling by phosphorylating 4EBP1 [51]. Although this observation was made using multiple cell lines initially, defective phosphorylation at Ser696, Thr706, and S863 impaired mTORC1 activation in skeletal muscle cells [52]. These findings imply that the Ras/MAPK stimulation can bypass TSC and directly affect the subunit of the mTORC1 complex. It also suggests a nutrient-Ras/MAPK arm in mTORC1 activation in addition to the nutrient and growth factor branch. Alternatively, as Ras is an oncogene and an essential mediator of inflammatory diseases [53], it raises the possibility of Ras-induced aberrant mTORC1 activation that escapes the regulation from nutrients in these conditions.
5. mTORC1 in regulating macrophage function
Macrophages rely on stimulation from the environment for activation [3,4]. Deletion of mTOR and Raptor using the LysM-Cre, a recombinase that causes conditional gene deletions in monocytes, macrophages, and neutrophils, enhanced macrophages’ response to M1 polarization agents LPS and IFNγ in vitro [54]. Similarly, mice with conditional knockout of mTOR and Raptor by LysM-Cre also had an over-exuberant reaction to LPS injection with a more significant drop in body temperature and producing more TNF-α and IL-6 [54]. mTORC1 inhibitor rapamycin increased the production of IL-12p40, IL6, and TNFα in peripheral monocytes after LPS stimulation in an NFkb-dependent manner [55,56]. The detailed mechanism of the suppressive role of mTORC1 to the inflammatory response from monocytes and macrophages remains to be determined. Some evidence suggests that mTORC1 inhibition by rapamycin suppressed the translation of low abundance cytokine (IL10), thus biasing the production of cytokines with high levels of transcripts, which include many proinflammatory cytokines (Il1b, Tnf, Il6) [57]. The same study further demonstrated that mTORC1 inhibition led to 4EBP1 and 4EBP2 binding to eIF4E (elongation initiation factor 4E), a subunit of the initiation complex, and subsequent suppression of cap-dependent translation. The translational bias with mTORC1 inhibition is an intriguing mechanism. However, it cannot explain the increased cytokine transcripts by the same mTORC1 inhibitor [56]. Additionally, others have demonstrated that 4EBP1 was insensitive to rapamycin treatment [58]. A possible explanation is that the translation bias may occur in selective conditions, as it was initially observed in virulent pathogen-infected macrophages. More studies are needed to characterize context-, substrate-, and inflammatory stimuli-dependent regulations from mTORC1.
In contrast to the immune suppressive effect discussed above, most evidence supports the immune stimulatory action of mTORC1. mTORC1 was constitutively active in macrophages deficient in TSC1 [59], and the persistent mTORC1 activation enhanced macrophages’ response to inflammatory stimuli such as LPS but resisted M2 polarization (anti-inflammatory) agents such as IL4 [59]. Macrophage activation syndrome (MAS) is a potentially life-threatening complication of systemic inflammatory disorders manifested mainly as systemic juvenile idiopathic arthritis (also known as Stills Disease) and systemic lupus erythematosus [60]. IL1, one of the cytokines activating MAS’s macrophages, induced mTORC1 activation [61]. The activated mTORC1 promoted the expansion of inflammatory monocytes and inflammation. Suppressing mTORC1 by rapamycin alleviated systemic inflammatory response [61]. In addition, mice deficient in TSC2 in hematopoietic cells, achieved by using the Mx-Cre, spontaneously developed arthritis and a MAS-like syndrome [61], further supporting the pro-inflammation role of mTORC1. Interestingly, TSC2 deletion using the same Mx-Cre enhanced myeloid cell development and maturation via the Myc gene [62]. The activated mTORC1 induced hypertrophy and proliferation of macrophages, which led to granuloma formation in vivo [63]. These data consistently suggest that mTORC1 activation promotes monocyte/macrophage proliferation to enhance inflammation. It is unclear if TSC deficiency-induced inflammation is primarily driven by monocytes/macrophage proliferation and if intrinsic elevated inflammation contributes to the phenotype. Although the enhanced myeloid proliferation by mTORC1 activation via TSC2 deletion is an extreme example, it may nonetheless shed light on the role of mTORC1 in promoting myelopoiesis, a key feature of trained immunity for ASCVD, which will be further discussed later.
It is intriguing that suppressing or activating mTORC1 by genetically deleting components of the mTORC1 (mTOR or Raptor) or TSC from the growth factor responding arm, respectively, enhances inflammation in macrophages. Negative feedback via the mTORC1 complex is a possible explanation, supported by the recent evidence of mTORC1 regulating the IFNγ-activated inhibitor of translation (GAIT) system. GAIT inhibited the translation of inflammatory mediators and promoted inflammation resolution [64]. S6K1 (also known as P70S6 kinase (P70S6K)) is a substrate of mTORC1 that phosphorylated the Thr389 residue of S6K1 in monocytes [65,66]. The C terminus of S6K1 was phosphorylated by cyclin-dependent kinase 5 (CDK5) at Ser424 and Ser429. The cooperative phosphorylation by the two kinases was required to induce glutamylprolyl tRNA synthetase (EPRS) activation, a component of the GAIT complex [64]. Deletion of mTOR or Raptor, therefore, interrupts the negative feedback by inhibiting GAIT complex formation. We should also recognize that the regulatory function of TSC to mTORC1 is complex. For example, TSC deletion paradoxically activated MiT/TFE [67], which enhanced catabolism by increasing lysosome gene expression and biogenesis. Similar complexity could also be extended to other inflammatory pathways in the TSC loss-of-function model. More studies are necessary to delineate the multifacet regulation at different levels of the mTORC1 pathway in macrophages.
At the metabolic level, mTORC1 is reported to enhance macrophage inflammation through glycolysis, the preferred metabolic mechanism for M1 inflammatory macrophages in vitro [68]. mTORC1 promoted glycolysis by increasing the translation of glycolytic enzymes and their transcriptional regulators in bone marrow-derived and peritoneal macrophages [69]. The enhanced glycolysis subsequently promoted the activation of NLRP3 inflammasome [69]. Inflammasome activation leads to the release of IL1 family cytokines in their active isoforms via membrane pores formed by GSDMD (Gasdermin D). A recent study identified that the Ragulator-Rag complex was required for GSDMD pore formation by a genome-wide genetic screen [70]. The study suggested that Ragulator-Rag-mTOR increased reactive oxygen species (ROS) production, which promoted the oligomerization of GSDMD in macrophages [70]. This process depended on mTORC1 activity but was only suppressed by Torin 1, not rapamycin. It remains unclear what contributed to the differential impact. The different mechanisms by which the two agents inhibit mTORC1 could play a role. Rapamycin inhibits mTORC1 by forming a complex with FKBP12, which creates a ternary complex with mTOR to interfere with substrate recruitment. In contrast, Torin 1 is an ATP-competitive inhibitor suppressing the phosphorylation/activation of mTORC1. Additionally, rapamycin has a limited inhibitory effect on some mTORC1 substrates, such as 4EBP1 [71], which may contribute to the lack of impact on GSDMD by rapamycin.
Besides enhancing inflammation by the abovementioned mechanisms, mTORC1 regulated pathogen clearance through autophagy [72]. Although the understanding is that mTORC1 inhibits autophagy, a recent study reported an unexpected role of Lamtor5 in degrading TLR4, the receptor for LPS, by enhancing autophagy in macrophages [73]. Lamtor5 is one of the subunits of Ragulator that anchors mTORC1 to the lysosome membrane. Gain- or loss-of-function of Lamtor5 suppressed or enhanced the LPS-TLR4-mediated signaling, respectively, which includes the activation of NFkb, IRF3, and TBK1. The loss of Lamtor5 led to sustained inflammation and increased mortality during endotoxic shock [73]. Although not characterized in detail, Torin 1 treatment suppressed the exuberant LPS-TLR4 signaling in Lamtor5-deficient macrophages [73].
In summary, mTORC1 plays a complex role in governing macrophage function. While there is some evidence of mTORC1 being immune-inhibitory, most data support its’ pro-inflammatory role in macrophages. The dichotomy is created by suppressing or enhancing mTORC1 activity via deleting different components of the mTOR complexes. As most clinical diseases related to the mTOR signaling demonstrate enhanced mTORC1 activation, the model reflecting such may be more relevant in this setting.
6. AMP-activated protein kinase in regulating macrophage function in hypoxia
AMP-activated protein kinase (AMPK) senses the energy levels in the cells and is activated by high AMP/ATP and ADP/ATP ratio. In contrast to mTORC1, which detects and responds to nutrient and energy abundance, AMPK is activated by energy deficiency. AMPK inhibits mTORC1 activity to conserve energy and promote cell survival in energy crises. In acute and complete ischemia, the infarct tissue is hypoxic and lacks nutrients in regular isoforms that serve as substrates to generate the fuel. Therefore, AMPK is expected to activate in macrophages in this context. Evidence suggests Liver kinase B1 (LBK1) and calcium release-activated calcium channels (CaMKKβ) stimulated AMPK in hypoxia [74]. However, the activity state and pathways responsible for AMPK activation in macrophages in acute injury and inflammation remain largely undetermined. We refer readers to recent reviews for discussions on pathways hypoxia employs to trigger AMPK activation [75] and the regulatory role of AMPK to mTOR pathway in hypoxia[75,76], which summarized evidence from ranges of cells and experimental models, including cancer cells, smooth muscle cells, epithelial cells, and cardiomyocytes. These studies provide valuable insights into the two opposing pathways in hypoxia. However, the response of resident cells, such as cardiomyocytes and smooth muscle cells, to hypoxia is likely different from that of cells migrating into the injury sites, such as monocytes that evolve into macrophages in the hypoxia tissue.
Although the evidence is limited, there are interesting observations about the function of AMPK in macrophages. It was reported that macrophages expressed predominantly the AMPKα1 subunit, the activation of which polarized bone marrow-derived macrophages toward anti-inflammatory phenotype [77]. This in vitro observation was substantiated by findings from an in vivo model of acute limb ischemia concerning angiogenesis, a process facilitated by anti-inflammatory/alternatively activated macrophages. The authors uncovered that deleting AMPKα1, not AMPKα2, impaired arteriogenesis [78]. Interestingly, the total macrophages accumulated in the ischemic limb was also reduced in AMPKα1 knockout mice [78]. As macrophages in the acute inflammatory sites evolve in a continuum from the inflammatory to the anti-inflammatory phenotype, it is possible that the reduced number of macrophages initially, thus decreasing the total anti-inflammatory macrophages, also contributed to the impaired angiogenesis when AMPKα1 was suppressed. The data also suggest that AMPK regulates a specific subtype of anti-inflammatory macrophages with primary functions of generating pro-angiogenic factors, extracellular matrix remodeling, and neovascular sprouting.
7. mTORC1 in regulating macrophages function in acute myocardial ischemia
Acute ischemia of the myocardium induces a robust inflammatory response in which macrophages are key players. The localized tissue injury differs from systemic inflammation by LPS injection or in vitro experimental settings many studies discussed so far employed. Because the interaction between macrophages and the ischemic environment dictates macrophage functions, mTORC1 could affect a broader effector function of these cells. The macrophage activation in the infarct tissue depends on recognizing damage-associated molecular patterns (DAMP) by pattern-recognizing receptors (PRR). There are more than 40 different PRRs [79]. Toll-like receptors (TLR), especially TLR4, are the most studied DAMP-PRR that induces mTORC1 activation [5,80]. TLR4 recognizes a variety of exogenous and endogenous ligands. DAMP signals released from necrotic myocardium that are recognized by TLR4 included S100 calcium-binding protein A1 (S100A1), S100A8/A9, HMGB1 (High mobility group box 1 protein), galectin-3, S100β, and IL-1α [81,82]. Injecting the mixture of HMGB1, galectin-3, S100β, and IL-1α or HMGB1 alone provoked cardiac inflammation in the wild type but not in TLR4-deficient mice [81]. Knocking down TLR4 reduced TNFα and IL6 in the infarct tissue and improved cardiac function four weeks after MI [83]. Although TLR-mediated signaling in acute ischemia was not directly assessed in macrophages, knocking-down TLR4 by injecting shRNA into the infarct region immediately after the MI procedure strongly suggests the contribution of macrophages to the phenotype. More work is needed to delineate if TLRs activate mTORC1 in macrophages after MI using macrophage conditional knockout models or chimeric mice generated by bone marrow transplant.
TLR-ligands interactions activate mTORC1 in multiple ways. Stimulating TLR4 by LPS activated MAP3K that phosphorylated P70S6K [84], which directly phosphorylates/activates S6. At the same time, LPS-TLR4 interaction induced AKT phosphorylation that relieved the inhibitory effect of the TSC complex on mTORC1 [84,85]. As there are numerous endogenous TLR4 ligands, and some of them are abundantly present in the infarct tissue (fibronectin, HMGB1, for example), the LPS-TLR4 is used here as an example of TLR4 activation. Stimulation of TLRs, TNFαR, IL1R, and some GPCRs activated MAP3K8, the kinase that initiated the phosphorylation sequence of MEK, ERK, and 4EBP1, leading to the release of translation suppression of inflammatory cytokines [86]. Ligand-bound TLR4 also recruited a small GTPase Rab8a as an adaptor protein to engage PI3Kγ, leading to mTORC1 activation via the PI3K/AKT path [87]. Furthermore, TLR3 and TLR4 activated TBK1 (TANK-binding kinase 1) that phosphorylated mTOR at Ser2159 residue [88]. These data suggest TLR activation can stimulate mTORC1 by directly increasing the phosphorylation of the mTORC1 substrates, mTOR itself, or by relieving the suppression from the TSC complex. It is essential to realize that the signaling pathways discussed above have not been explicitly studied in the myocardial ischemia setting and therefore serve as indirect evidence.
Inflammatory cytokines, including IL-1β, TNFα, and IFNγ and their receptors, are known DAMPs-PRRs that activated mTORC1 [65,89]. These cytokines are produced abundantly in the infarct tissue [90] and likely are the primary sources of signals for the growth factor/stress arm of mTORC1 activation. In macrophages, DAMP-PRR interactions activate mTORC1 by the MAP3K/ERK and PI3K/AKT pathway. Although these pathways have not been studied explicitly as the activating signals for mTORC1 in myocardial ischemia, the available evidence supports the active roles of MAP3K/ERK and PI3K/AKT in post-infarct repair. The Ras-ERK pathway promotes chronic inflammation in obesity [91], rheumatoid arthritis, psoriatic arthritis, and the progressive form of osteoarthritis [92]. In myocardial ischemia/reperfusion injury, studies have reported protective [93] and detrimental [94] roles of ERK activation. Activated ERK was detected predominately in non-cardiomyocytes in the infarct region, suggesting macrophages are a source of ERK activation after MI [95]. Macrophages from the MI tissue displayed a strong activation of ERK1/2 (unpublished observation), which is associated with robust activation of mTORC1 [4].
The PI3K/AKT is the archetypical pathway that activates mTORC1. In a murine model of MI, global AKT1 deficiency increased cardiomyocyte apoptosis 24 h after MI. Still, it reduced fibrosis and infarct size and improved cardiac function four weeks after MI [96], pointing to an immune cell-mediated process. Interestingly, mortality caused predominantly by left ventricle rupture from severe inflammation in experimental MI models was lower in AKT1 null mice [96]. This finding suggests that AKT1 activation contributed to the acute inflammation via immune cells such as macrophages. Additionally, global AKT1 deficiency in the background of double deletion of ApoE and SR-BI (scavenger receptor class B type I) reduced inflammatory macrophage and neutrophil infiltration in spontaneous MI developed in these mice and improved survival and function [97]. In contrast, PI3K/AKT activation was associated with the inflammatory phenotype of macrophages after MI [98]. The evidence suggests a pro-inflammatory and detrimental role of AKT1-mTORC1 activation macrophages in acute ischemia. These observations are in line with recent findings that robust mTORC1 activation in macrophages from the infarct tissue is detrimental to myocardial repair and remodeling [4]. There also appears distinct activation patterns of substrates of mTORC1 in inflammatory and reparative macrophages, suggesting that the individual substrate of mTORC1 regulates molecular events responsible for switching macrophages from inflammatory to reparative phenotype.
7.1. Perspectives and future studies
Most research about mTORC1 in cardiac diseases focuses on muscle biology and myocyte response to stress, especially hypertrophic stimuli [99]. This is not surprising given mTORC1 is the fundamental pathway by which eukaryotic cells maintain growth, which is essential for cardiomyocytes to respond to stress. The robust activation of mTORC1 in macrophages in MI brings new research interests to an important yet poorly understood area. As we begin to understand the role of mTORC1 in macrophages in myocardial ischemia, many questions remain to be answered before we can fully appreciate this pathway and take advantage of the clinically available mTORC1 inhibitors for bedside translation. mTORC1 activation strictly requires dual input from growth/stress signals and nutrients. Although the DAMP-PRR interaction serves as the input from the growth factor/stress arm, it is unclear what sources supply nutrients. The infarct tissue lacks classical nutrients such as monomeric amino acids and is a hypoxic environment without energy; all suppress mTORC1 activation contrary to our findings [4]. One possibility is that macrophages, as professional phagocytes, take advantage of the massive dead myocardium and turn it into nutrients. The nutrients generated in this manner regulate cellular function by activating mTORC1 and serve as the substrates for building the repairing structure. Often, the primary focus when macrophages are studied is their ability to stimulate or inhibit inflammation. Their role as professional phagocytes to remove dead tissue is much less investigated. Macrophages as phagocytes are expected to function at their peak in MI to remove the tissue debris and initiate repair. To delineate how macrophages “extract” valuable nutrients from dead myocardium and use them to regulate their function by mTORC1 activation provides a different vantage point and helps our understanding of a critical aspect of the functional repertoire of macrophages that has been largely missing. As the function of phagocytes is closely tied with lysosomes, future research that answers questions such as how mTORC1 regulates macrophage differentiation and phenotypes by affecting lysosome proliferation/function is also essential to a better understanding of macrophage biology in ischemia.
Recent work demonstrated that mTORC1 inhibited nutrient generation from protein scavenging via macropinocytosis, a process of uptaking extracellular soluble components and small debris (<5 μm) into large vacuoles (>250 nm). Macropinocytosis is an actin- and PI3K-dependent process active at baseline or triggered by cytokines. Macropinocytosis improves cell survival in a harsh environment, like cancer cells in the center of a solid tumor. mTORC1 directly suppressed the degradation of the proteins by suppressing V-ATPase-mediated acidification of lysosomes [100]. V-ATPase suppression was mediated by phosphorylating STK11IP (serine/threonine kinase 11 interacting protein), a new substrate of mTORC1 identified by quantitative phosphoproteomic analyses [101]. Macrophages from different immune phenotypes have striking differences in their ability to perform macropinocytosis [102]. Human inflammatory macrophages differentiated from monocytes followed by treatment of IFNγ and LPS possessed 5% of the macropinocytosis capacity as that of the anti-inflammatory macrophages polarized by IL4 [102]. The underlying mechanisms for macropinocytosis also differed: the inflammatory macrophages depended on Rho GTPases or Rac1, and anti-inflammatory macrophages relied on PI3K, Ca++ and CaSR (Ca sensing receptor). These results have important implications for the phagocytotic function of macrophages in the ischemic myocardium. Our observation (unpublished) demonstrates only soluble DAMPs (sDAMP) from the infarct tissue trigger inflammation, suggesting macropinocytosis can serve as mechanism to remove DAMPs. Targetting mTORC1 to boost macropinocytosis is a novel way to increase the efficiency of clearing DAMPs and therefore potentially reducing inflammation and inhibiting remodeling and heart failure after MI.
In Figure 1, we summarize the potential mTORC1 activation signals in acute ischemia and the implications of mTORC1 activation based on current available evidence.
8. mTOR Signaling and ASCVD
Atherosclerosis is the culprit responsible for most acute and chronic ischemia. The initial event of atherosclerosis is an intra-mural accumulation of apolipoprotein B-containing lipoproteins in the susceptible regions of the vasculature. The sequestered lipoproteins trigger inflammatory changes in the overlaying endothelium, promoting monocyte adhesion and infiltration into the vascular wall. Monocytes recruited engulf the lipoprotein and its by-products from oxidation and degradation and evolve into “foam cells”, the signature macrophages of the atherosclerosis lesion. Macrophages contribute to the initiation, progression, and remodeling of atherosclerotic lesions [103,185,186] via their versatile effector functions that include producing chemokines/cytokines/ROS, secreting matrix-degrading proteases, endocytosis and degrading engulfed material, and by inflammatory-stimulated cell death. This section focuses on how trained immunity links risk factors to atherosclerosis and the role of mTOR therein.
8.1. Trained immunity and ASCVD
Trained immunity is a process by which innate immune cells gain memories after exposure to certain stimuli and augment their response to subsequent insults. Classical stimuli that lead to trained immunity include β-glucan (a primary cell wall component of fungi), the BCG vaccine, oxidized LDL (oxLDL), low-level LPS, and aldosterone. Three biological processes must be in place for trained immunity: metabolic rewiring, including glycolysis and glutaminolysis, epigenetic reprogramming, and enhanced myelopoiesis [104]. Although the precise sequence of the three events remains unclear, some evidence suggests the metabolites from the metabolic reprogramming cause epigenetic changes, suggesting the interconnection between these two processes.
The role of trained immunity in ASCVD has recently gained attention. Although the term “trained immunity” was introduced in 2011, a study from 1999 has already demonstrated BCG-immunized rabbits fed with a Western-style diet (WD) had increased atherosclerosis [105], therefore likely the first report linking trained immunity with atherosclerotic disease. Since then, studies have uncovered that trained immunity in human monocytes can be induced by oxLDL [106–108], acetylated LDL [107], glucose [109], Lp(a) [110], and aldosterone [111], all of which are risk factors of human ASCVD. More importantly, intriguing evidence also connects lifestyle risk factors as triggers of trained immunity. Psychological stress is a known risk factor for ASCVD [112]. When mice were exposed to variable stressors such as crowding, isolation, cage tilt, and frequent light-dark cycle changes, monocytes from these mice demonstrated transcriptomic and epigenomic changes that biased these cells to a hyper-inflammatory phenotype and enhanced myeloipoiesis [12]. Monocytes from human subjects with elevated stress levels, assessed by the Perceived Stress Scale, or from stressed mice had exacerbated responses to inflammatory stimuli such as TLR ligands [12], consistent with prior priming events similar to those in trained immunity. These monocytes exhibited enhanced activities in PI3K/AKT, mTOR, and Hif-1α and decreased oxidative phosphorylation [12], indicating metabolic reprogramming. Intriguingly, stress activated all three processes, including metabolic and epigenetic reprogramming and myelopoiesis [12], required for trained immunity. The molecular triggers are yet to be defined and could be a composite of metabolites and neurohormonal mediators.
Western-style diet (WD), a diet that is high in saturated fats, refined carbohydrates, and salt, and low in complex carbohydrates, fiber, vitamins and minerals, induced inflammation [113]. Feeding WD to Ldlr−/− mice caused systemic inflammation manifested by elevations of more than 20 cytokines and chemokines, most of which are pro-inflammatory [11]. Myeloid progenitor cells from WD-fed mice had increased proliferation, leading to more mature monocytes and granulocytes while not impacting lymphocytes. The enhanced myelopoiesis was a result of transcriptomic and epigenomic reprogramming of the progenitors. Significantly, the responses of monocytes from WD-fed mice to TLR agonists (TLR2, 4, 7/8, and 9) were augmented compared to cells from chow-fed mice, suggesting a possible role of WD in trained immunity [11]. Replacing WD with a regular chow diet normalized serum inflammation markers, but it failed to reverse the augmented responses of myeloid cells to inflammatory stimuli [11]. Similarly, epigenetic reprogramming of adipose tissue macrophages and retinal myeloid cells and the augmented inflammatory response persisted after weight reduction in high-fat diet-induced obesity [9]. The lasting priming effect of WD on myeloid cells was further supported by the evidence that bone marrow cells from WD-fed mice retained the ability to promote atherosclerosis after being transplanted to Ldlr−/− mice on a regular chow diet [10]. Interestingly, the donor Hematopoietic stem cells (HSCs) from WD-fed mice maintained the hypomethylated promoters for Pu.1 and IRF8 [10], key transcription factors that could explain the enhanced myelopoiesis. These data suggest that epigenetic changes induced by WD have a long lasting effect in promoting the production of monocytes and granulocytes.
While the priming factors from WD to induce trained immunity have yet to be fully defined, one candidate is LPS. LPS can be detected in healthy subjects who consumed WD [114], a phenomenon known as metabolic endotoxemia. Changes in the gut microbiota by WD can compromise the gastrointestinal barrier and allow LPS to enter the circulation [115]. Additionally, the consumption of sucralose or sucrose modified gut microbiota and enriched the bacterial genes involved in the synthesis of LPS [116]. These events likely contribute to the low level of LPS in the blood stream. Unlike high concentrations that cause tolerance, low concentration of LPS is known to induce trained immunity. Research has found that low-dose LPS injection (5ng per kg body weight twice a week for two months) in ApoE−/− mice, an attempt to mimick metabolic endotoxemia, aggravated atherosclerosis [117]. Therefore, WD may have incurred trained immunity by the products of metabolic derangement, byproducts from the diet itself, or the consequential impact of WD on the gut microbiota and mucosal barrier.
Poor sleeping quality, abnormal quantity that is either too short or too long, and fragmented sleep are associated with ASCVD and ASCVD-related mortality [118]. Even in healthy young volunteers, slight sleep restriction (from 7.4 h to 6.1 h) for six weeks expanded HSPC (Hematopoietic Stem and Progenitor Cells) population and increased circulating classical and non-classical monocytes in the evening [119]. In addition, the sleep restriction (SR) augmented the activities of histone acetyltransferase (HAT) and deacetylase (HDAC). However, only the changes in HDAC reached statistical significance [119], suggesting possible epigenomic changes in HSPCs by SR. Sleep fragmentation (SF) in ApoE−/− mice fed with a high-fat diet accelerated atherosclerosis [8]. The proliferation of Lin− Sca1+ cKit+ (LSK) cells (a type of HSC) in the BM was enhanced, and inflammatory monocytes (Ly6Chi) in the circulation were increased by SF [8], both contributing to the phenotype. Interestingly, restoring to regular sleep for ten weeks only partially reversed the epigenomic changes induced by SF (69%), leaving the remaining 31% unchanged. The 31% epigenomic loci alterations may determine the augmented inflammatory response in trained immunity. Similarly, restoring to a regular sleep schedule did not reverse the SF-induced hyper-inflammatory response to cecal ligation and puncture, which included higher monocytosis, augmented BM hematopoiesis, and increased plasma cytokine levels, resulting in a worse clinical score [119]. These data suggest that trained immunity connects sleep disruption and ASCVD. However, the sleep fragmentation, performed in the preclinical model using a sweep bar moved along the bottom of the cage every 2 min during the light cycle, could represent severe disruption of sleep. Therefore, the clinical relevance is still unclear.
While the current evidence supports lifestyle risk factors such as diet and sleep disruption induced trained immunity that led to the exacerbated inflammatory response that cannot be reversed by switching back to a regular diet, by weight loss, or restoring normal sleep, exercise has been shown to protect mice and humans with atherosclerosis from chronic leukocytosis [120]. Physical inactivity is associated with a higher risk of developing ASCVD. Exercise, on the other hand, mitigated the proinflammatory state in human subjects [121]. In patients with existing coronary artery disease, exercise 4 times or more per week was associated with lower blood leukocyte count [120]. While the guideline recommends exercise regularly for primary prevention of ASCVD [122], the mechanism is not fully understood. In ApoE−/−mice fed with WD, voluntary exercise inhibited leukocytosis, lowered immune cell accumulation in the aorta and decreased the size of atherosclerotic plaques. The benefit of exercise was attributed to decreased leptin levels from the visceral fat tissue, which promoted hematopoietic quiescence [120]. Although exercise-induced changes, including the decrease in leukocyte production and the epigenomic and transcriptomic modifications, remained in place after stopping exercise for several weeks, they were not permanent, which supports the current recommendation regarding daily activity at the molecular level. It is unclear if exercise reverses trained immunity based on the current data from a specific experimental setup. More work is needed to answer this question.
8.2. mTOR and trained immunity
Innate immune cells re-wire their metabolic pathway to aerobic glycolysis to sustain the trained immunity. This is not surprising as glycolysis supplies metabolic intermediates to enhance macrophage inflammatory response. Human primary monocytes trained with β-glucan preferred aerobic glycolysis with high glucose consumption, high lactate production, a high NAD+/NADH ratio, and a reduced basal respiration rate [123]. Trained immunity was inhibited by mTORC1 inhibitor rapamycin, AMPK activator metformin (which also inhibits mTOR), and deletion of Hif1α [123]. Similarly, oxLDL-induced trained immunity in human peripheral blood monocytes depended on glycolysis [106]. The enhanced glycolysis by mTOR activation feeds pyruvate to the TCA cycle, producing acetyl-CoA, which is further metabolized into mevalonate, a precursor metabolite for cholesterol synthesis [108]. Mevalonate activated PI3K-mTOR, forming a loop of mTOR-glycolysis-Mevalonate that induced long-lasting trained immunity. It is interesting to note that rapamycin consistently inhibited the augmented inflammatory response in trained immunity [123,124]. However, the direct metabolic impact of mTOR inhibition on monocytes or macrophages is less established and sometimes inferred from indirect mTOR suppression by AMPK activation from metformin [125]. Nonetheless, coupled with the ample evidence supporting mTOR’s role in promoting aerobic glycolysis in inflammatory macrophages [15], T cells [14], and cancer cells [16], mTOR signaling likely serves as a crucial pathway for trained immunity.
Rapamycin and its derivatives, the so-called rapalog-coated stents, are widely used to treat coronary artery stenosis in patients and deliver better outcomes compared to bare metal stents or stents coated with Paclitaxel that suppressed coronary artery smooth muscle cell proliferation by inducing mitotic arrest [126]. In a recent study, myeloidspecific nanobiologics that targeted mTOR or S6K1, one of the downstream kinases activated by mTOR, reduced lesion size and inhibited inflammation in the ApoE-deficient mice [127]. With the evidence discussed above linking risk factors and lifestyles with trained immunity through mTOR activation, there could be a value of systemic administered immune modulators targeting the mTOR pathway in earlier stages of the ASCVD or even for prevention purposes in selected patients. The challenge is that mTOR signaling regulates a wide range of biological processes with growing downstream targets, and many questions regarding mTOR’s role in trained immunity have yet to be answered. For example, is mTORC1 the predominant complex in inducing trained immunity, and what are the specific substrates of mTORC1 involved, and is the regulation transcriptional or translational or both, and does mTOR direct or indirectly induce epigenetic reprogramming?
In summary, accumulating evidence points to trained immunity as an underlying mechanism linking risk factors and life styles to the pathogenesis of ASCVD. There appears to be a long-lasting effect of the trained immunity-mediated augmented inflammatory response by these factors, which cannot be reversed by simply “discontinuing” the life style practice. Exercise has a temporary effect on this process. In this context, pharmacological interventions may provide some options. Immune modulators targeting mTOR are promising in reversing trained immunity in ASCVD. However, more research is warranted to address outstanding questions.
9. mTOR and immune senescence
Cellular senescence is a relatively well-characterized phenotype [128]. Senescent cells have a distinct morphology (enlarged and flattened), dysfunctional mitochondria with enhanced ROS release, resistance to apoptosis, exited cell cycle, protein modifications, accumulation of lipofuscin granules, and expression of SA-β-galactosidase. Senescence is also associated with changes in the epigenome and the transcriptome. Functionally, senescent cells develop senescence-associated secretory phenotype (SASP) that contains hundreds of molecules with comprehensive ranges of functions, including inflammatory cytokines (IL6, IL8), chemokines (MCP1, MIPs), growth factors (TGFβ), proteases, hemostatic factors, bradykinins, extracellular matrix components, metalloproteinases, miRNAs, ROS, metabolites, and DAMPs [129]. SASP induces and/or reinforces senescent changes in the surrounding cells and attracts immune cells to remove the senescent cells. As a result, small proportions of senescent cells, for example, 2%, can have a significant impact.
By definition, immune senescence is a process of immune dysfunction that occurs with age leading to changes in the immune functions that increase vulnerability to infections and chronic diseases and results in a lack of response to vaccine. However, senescence in immune cells is difficult to define because of the diverse immune cells of innate and adaptive immune systems, and a broad spectrum of phenotypes developed by individual immune cell types in response to the environment, for example, the macrophages. However, the cardinal features of immune senescence have been described to include immune cell function changes, altered cell type composition such as the loss of naïve T cells and an increase in highly differentiated CD28- memory T cells, defective response to new antigen, impaired calcium-mediated signaling, thymic atrophy, enhanced auto-immunity, and inflamm-aging [130]. In addition, many senescent immune cells expressed elevated p16 and p21 and generated SASP [131]. The focus of this review is the senescent monocyte and macrophage phenotype.
In humans, the atypical monocytes (CD14lowCD16hi) accumulated in elderly patients were considered senescent cells as they produced SASP and were proinflammatory [132]. Additionally, a subset of bone marrow monocyte-macrophages that were enriched in pathways critical for inflammation, senescence, and tumorigenesis increased with aging, consistent with the senescent phenotype [133]. Aged monocytes-macrophages-produced grancalcin induced marrow adipogenesis and reduced dynamic bone formation rates, characteristics of bone aging [133]. These data suggest that the senescent monocyte-macrophage population produces unique SASPs to cause aging of the organ they reside in, and it is possible specific SASPs made by aged macrophages in the heart could contribute to age-related hypertrophy and fibrosis. In addition, macrophages in the bone fracture site of old mice (24 months) had an increased pro-inflammatory profile compared to young mice (3 months) and delayed fracture healing [134]. In an animal model of immune senescence induced by selectively deleting the DNA repair protein, Ercc1, in mouse hematopoietic cells, premature aging in solid organs such as the liver and kidney occurred [18], suggesting immune senescence can lead to aging at the systemic level. Although vascular aging was not characterized in this study, macrophages, among other immune cells (T cells, B cell, NK cells), significantly increased expression of senescent markers p16 and p21 and produced SASP in this model [18]. Therefore, these mice could develop early onset atherosclerosis in experimental settings such as using WD and in mice with ApoE or Ldlr deletion.
mTOR plays a central role in regulating aging-related biological processes. Inhibition of mTOR by rapamycin extends the life span in various species. For immune senescence, mTOR activation was essential for the secretory phenotype and the increased production of IL6 and IL8 in myeloid cells after being stimulated with the senescent signal Ras [135]. mTOR enhanced the stability of SASP mRNA by inhibiting the RNA-binding protein ZFP36L1 [136]. mTOR inhibition by rapamycin suppressed the secretion of inflammatory cytokines by senescent cells and impaired SASP production [136,137]. Furthermore, rapamycin rejuvenated the immune system in an immune senescence model induced by deleting a DNA repair mechanism in hemopoietic cells [18]. However, it is unclear if rapamycin also reversed the premature aging of the solid organs by immune senescence, as the study did not report it.
Both preclinical and clinical studies examined if mTOR pathway inhibitors can mitigate the sequelae of immune senescence. Inhibition of PI3K/mTOR activity by BEZ235 upregulated IFN-induced antiviral immune responses and rescued mice from lethal influenza infection [138]. In a double-blind, randomized, placebo-controlled trial using the same inhibitor BEZ235 (name changed to RTB101), RTB101 upregulated IFN-induced antiviral responses in older adults (age ≥ 65). Interestingly, RTB101 reduced the incidents of respiratory tract infection in the phase 2B trial but did not demonstrate a similar impact in the phase 3 trial [139]. The authors proposed to refine endpoints for future studies to determine the effectiveness of RTB101 further. The data nonetheless suggested that daily administering RTB101 for 16 weeks was safe and well-tolerated in this patient population. Additionally, mTOR inhibitor RAD001 boosted influenza vaccine responses in elderly patients [140]. The evidence in aggregates suggested that immune senescence can be modified, and mTOR pathway inhibitors can be considered immune modulators to reverse immune senescence, at least to a certain degree.
The driving factors for immune senescence and ASCVD overlap significantly, and metabolic derangement (diabetes, hypercholesterolemia), genetics, and smoking are just a few examples. The persistent, low-grade inflammation, also known as inflamm-aging and a hallmark of immune senescence, is a critical driving force of atherosclerosis. Therefore, it is very likely that immune senescence plays an essential role in the pathogenesis of ASCVD. The lipid-laden macrophages (foam cells) with senescence markers produced more atherogenic and inflammatory cytokines, chemokines, and metalloprotease, promoting lesion progression and instability [141]. Senolytics that target the aged macrophages blunted the atherosclerotic lesion progression [142]. Moreover, rapamycin consistently reduced the burden of atherosclerosis in animal models of accelerated atherosclerosis (ApoE−/− and Ldlr−/−fed with high-fat diet), which is reviewed elsewhere [143], suggesting the possible role of mTOR in immune senescence in ASCVD. There is limited data on the molecular mechanisms of monocyte/macrophage senescence in atherosclerosis. miR-216a was reported to promote atherosclerosis partly due to its role in macrophage aging [144].
10. mTOR and clonal hematopoiesis
Clonal hematopoiesis of indeterminate potential (CHIP) is an age-related phenomenon in which HSCs acquire leukemogenic mutations, leading to a clonally expanded HSC. These HSCs have survival advantages and continue to expand with aging. As a result, selective expansion of a subpopulation of blood cells occurs. The individuals with CHIP do not have evident hematologic malignancy, dysplasia, or cytopenia [145]. The known mutations of CHIP are typically epigenetic regulators such as DNMT3A, TET2, and ASXL1, DNA damage repair genes including PPM1D, TP53, the regulatory tyrosine kinase JAK2, or mRNA spliceo-some components SF3B1 and SRSF2. The prevalence of CHIP increases with aging, detectable in up to 10% of adults at age 70 or greater and nearly 20% of adults older than 90 years old using the next-generation sequencing of peripheral blood cells [146–148]. CHIP is associated with increased risk for coronary artery disease [146,149] and stroke [150]. Individuals with TET2, JAK2, and ASXL1 CHIP had a higher incidence of heart failure, and ASXL1 CHIP was associated with decreased LV ejection fraction [151]. Interestingly, evidence from 50 years ago suggested the monoclonality in human atherosclerotic lesions [152]. Besides CHIP, mosaic chromosomal alterations (mCAs) are another clonal hematopoiesis (CH) form. Compared to the specific gene mutations in CHIP, mCA-mediated CH has larger (often >1 megabase in length) structural alterations that can be insertions or deletions or copy number neutral loss of heterozygosity [153]. While CHIP is associated with more myeloid malignancies, lymphoid malignancies are more common for mCAs [148,153]. As monocytes and monocyte-derived macrophages are drivers of chronic inflammation in the atherosclerotic lesions, the differentially affected cell populations by CHIP and mCA may explain the certain and uncertain associations of CHIP and mCA [153–155] with coronary artery disease, respectively. Even though the association of mCA-mediated CH with atherosclerosis is not yet defined, evidence suggested mCA increased cardiovascular mortality [154], and a recently published analysis of 10,070 cancer survivors demonstrated carriers of any mCA were at an increased risk of CAD death compared with noncarriers [156].
We are beginning to understand the role of mTOR in CH. mTOR was reported to promote ASXL1 mutation-induced CH [157]. ASXL1 is one of three human homologs of the Drosophila Asx gene and was involved in epigenetic regulation [158]. There are also unique differences in ASXL1 CHIP and CHIPs from other epigenetic modulators such as DNMT3A and TET2. DNMT3A and TET2 mutations confer selection advantages, and the corresponding HSCs have enhanced self-renewal ability. In contrast, HSCs from ASXL1 mutation knock-in mice had decreased self-renewal ability leading to a competitive disadvantage after transplantation [157,159]. However, in the native environment, the ASXL1 mutant conferred a clonal advantage on LT-HSCs (Multipotent long-term HSCs) in hematopoiesis. ASXL1 mutation induced aberrant expansion of the LT-HSC compartment through activation of the AKT/mTOR pathway. Mechanistically, mutant ASXL1 interacted with and stabilized BAP1 (BRCA1 Associated Protein 1), a nuclear ubiquitin carboxy-terminal hydrolase, resulting in the deubiquitination and activation of AKT, leading to the subsequent mTOR stimulation [157]. Notably, the expanded LT-HSCs lost the quiescence feature as a large proportion of the LT-HSCs were at the advanced stage of the cell cycle. mTORC1 inhibitor rapamycin reversed the aberrant proliferation of LT-HSCs [157].
There appears to be more evidence supporting mTOR’s role in promoting the cell cycle in CH. The DNMT3A gene was frequently mutated in hematopoietic malignancies, and DNMT3AR878H was the most common mutant. Conditional knock-in DNMT3AR878H in LSK progenitor cells using Mx1-Cre and the Cre-inducing agent plpC increased cell proliferation [160]. These mutant LSK progenitors exhibited higher mTOR activity that drove cell proliferation through activating CDK1 [160]. The increased mTOR activation was associated with mTOR gene hypomethylation. Additionally, HSCs harboring both DNMT3AR878H and nucleophosmin (NPM1) mutations gained accessibility to promoters enriched in cell cycle and PI3K/Akt/mTOR signaling [161]. The combination of these two mutations resulted in the most potent transformation to hematologic malignancy, highlighting the importance of the mTOR pathway in sustaining rapid cell proliferation.
Overall, these data support the role of mTOR in the cell cycle and proliferation during CH, including both benign CH and malignant transformation. Significantly, mTORC1 inhibition by rapamycin attenuated the clonal expansion in both settings. These data also corroborate the finding of enhanced myelopoiesis-induced by mTOR activation from TSC2 deletion [62]. Although it remains to be determined if the mTOR pathway actively contributes to CHIP-mediated atherosclerosis, the evidence of mTOR’s involvement in CH and CHIP as a driver of myeloid cell proliferation suggests such a potential. There are interesting and clinically relevant questions. For example, it is a known phenomenon that some patients develop accelerated atherosclerosis after acute ischemia. It is tempting to speculate that ischemia-triggered emergent hematopoiesis may selectively expand the CHIP population, and mTOR inhibition in such a patient population may confer protection.
11. mTOR and HFpEF
By AHA/ACC and ESC definition, heart failure with preserved ejection fraction (HFpEF) is defined as signs and symptoms of HF with the left ventricular ejection fraction (LVEF) ≥50% [162,163]. Historically, a patient population with typical heart failure symptoms and preserved LVEF was reported in 1982 [164]. We now understand that HFpEF accounts for 50% of all HF patients and 47% of all HF hospital admissions [165]. The comorbidities of HFpEF include obesity, hypertension, type 2 diabetes, coronary artery disease, atrial fibrillation, chronic kidney disease, obstructive sleep apnea, and chronic obstructive pulmonary disease (COPD). Aging and female sex are the additional important risk factors for developing HFpEF, and almost all patients older than 90 develop HFpEF [166]. Although HFpEF as a disease entity is much better characterized, the mechanisms driving the pathogenesis of HFpEF remain to be fully determined.
Evidence supporting the interplay between the immune-mediated processes and HFpEF has begun to emerge. In HFpEF patients, inflammatory cytokines such as TNFα, MCP1/CCL2, IL6, IL12, and CXCL10 were significantly elevated in the peripheral circulation compared with asymptomatic patients of hypertension or LV diastolic dysfunction [167]; patients with worsening HFpEF had higher TNF-α, IL-6, and CCL2 than stable patients [168]. Interestingly, the cytokines from the alternatively activated macrophages (anti-inflammatory macrophages) were also increased in HFpEF patients [167]. Correlating to the overall elevated immune/inflammatory profile, there was more myeloid cell infiltration in the myocardium and 2–4-fold increases in classical, intermediate, and nonclassical monocyte in the circulation of HFpEF patients [167,169]. This clinical observation suggests the association of an enhanced inflammatory profile with HFpEF. In an experimental model of diastolic dysfunction generated by salty drinking water, unilateral nephrectomy, and chronic exposure to aldosterone (SAUNA), the production of neutrophils, monocytes, and B cells from the spleen were elevated, and the density of macrophages in the myocardium were increased [170]. Deletion of IL10, an anti-inflammatory and profibrotic cytokine from macrophages, improved diastolic function in animals exposed to SAUNA [170]. Stress triggered the influx of CCR2 + Ly6Chigh monocytes that evolve into macrophages in the myocardium, contributing to the majority of macrophages and the inflammatory response [171]. Preventing the recruitment of CCR2+ monocytes to the heart by deleting CCR2 or MCP1, thereby blocking the CCR2+ macrophage expansion in the myocardium, remarkably decreased fibrosis and improved diastolic function [172]. These pre-clinical data support that macrophages can drive cardiac fibrosis and stiffness, leading to the hemodynamic features of HFpEF.
The CHIP phenomenon further illustrated the role of monocytes/macrophages in developing HFpEF. In a case-controlled study, CHIP mutations were significantly associated with HFpEF in patients younger than 65 [173]. Similarly, a recent study (pre-print) suggested CHIP was significantly associated with a 42% increased risk of HFpEF in post-menopausal women (n = 5214) [174]. Specifically, the study showed a stronger association of TET2 mutation with HFpEF than DNMT3A or ASXL1. Interestingly, there was no evidence of the association between CHIP and the risk of incidents of HFrEF in this patient population. In mice infused with Ang II, inactivation of TET2 and DNMT3 in HSCs exacerbated cardiac hypertrophy and fibrosis of the heart and kidney and decreased the fraction shortening by 20 to 25% [175]. A similar observation was made in the hypertrophy model generated by pressure overload by trans-aortic constriction (TAC). In the TAC model, including 10% TET2-deficient bone marrow cells at the time of bone marrow transplantation enhanced cardiac hypertrophy, fibrosis, and a modest reduction of cardiac function [176]. Although the endpoints of these studies demonstrated a modest decrease in cardiac function, hypertrophy, fibrosis, and diastolic function likely preceded systolic dysfunction. Furthermore, a recent study suggested a high prevalence of CHIP of TET2 mutation in patients with HFpEF [177]. These data in aggregates point to a connection of TET2 mutation with HFpEF. Introducing TET2 mutation in HSCs in the HFpEF model induced by a high-fat diet and nitric oxide synthase inhibition [178] may provide further evidence of CHIP in HFpEF.
The only class 1 recommended therapy for HFpEF is diuretics, which reduces congestion and relieves symptoms [162]. The Sodium-Glucose Cotransporter-2 (SGLT2) inhibitors are recently added as a class 2a recommendation for HFpEF [162]. The disease-modulating treatment of HFpEF is lacking. In preclinical models, treating 25 months old female mice with ten weeks of calorie restriction or rapamycin at 2.24 mg/kg/day reversed aging-associated diastolic function [179]. At the molecular level, these treatments reversed the aging-related decrease in proteins involved in mitochondrial function, electron transport chain, citric acid cycle, and fatty acid metabolism, as well as increased proteins of glycolysis and oxidative stress response [179]. Similar benefits of rapamycin on diastolic function and stiffness of the myocardium were extended to both aged male and female mice [180]. A proof-of-concept study is ongoing to determine if rapamycin improves patients of HFpEF with frailty (NCT04996719). These studies revealed the global effect of mTOR inhibition by rapamycin. Still, the data is consistent with the role of mTOR in promoting glycolysis, the critical metabolism for inflammatory macrophages. The inflammatory (CCR2+) monocytes become MHChi macrophages in HFpEF, which are highly enriched in profibrotic IL10, TGF-β, and osteopontin [170], in contrast to the pro-inflammatory macrophages when the same monocytes are recruited to the ischemic tissue early on after MI. Given the strong suppression of fibrogenesis mTOR inhibition in various organs, it is possible that mTOR inhibition inhibits the pro-fibrotic macrophages in HFpEF, likely by metabolic re-configuration. At systemic level, mTOR inhibition can mitigate enhanced hematopoiesis and myeloid cell proliferation in HFpEF, therefore offering novel strategies for managing this complex form of heart failure.
In summary and as depicted in Figures 1 and 2, this review focuses on the role of mTOR in monocytes and macrophages in various cardiovascular diseases that include acute ischemia, ASCVD, and HFpEF. We discuss the vital function of mTOR in trained immunity, immune senescence, clonal expansion, and CHIP, which contribute to the pathogenesis of ASCVD and HFpEF. Many cardiovascular diseases are age-related and share common risk factors. Therefore, it is not surprising that mTOR regulates biological processes contributing to diverse cardiovascular conditions, which attests to mTOR as a powerful pathway that can be tapped into. There are substantial translation potentials for targeting mTOR-mediated signaling to modulate acute and chronic inflammation and mitigate the detrimental effects of mTOR on cardiovascular health. Further research is warranted to unravel the intricacies of the mTOR-macrophage axis and identify potential therapeutic interventions for these conditions.
Acknowledgement
M.C.M. is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health by award number T32HL125247. J. W and D.J.C are supported by National Heart, Lung, and Blood Institute award R01HL145298. D. J. C is supported by the VA MERIT Review award 1 I01 BX004562.
References
- [1].Saxton RA, Sabatini DM, mTOR Signaling in growth, metabolism, and disease, Cell. 168 (6) (2017) 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu GY, Sabatini DM, mTOR at the nexus of nutrition, growth, ageing and disease, Nat. Rev. Mol. Cell Biol 21 (4) (2020) 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Desalegn G, Pabst O, Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine, Nature Communications 10 (1) (2019) 3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen G, Phan V, Luo X, Cao DJ, The mechanistic target of rapamycin complex 1 critically regulates the function of mononuclear phagocytes and promotes cardiac remodeling in acute ischemia, J. Mol. Cell. Cardiol 159 (2021) 62–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weichhart T, Hengstschläger M, Linke M, Regulation of innate immune cell function by mTOR, Nat. Rev. Immunol 15 (10) (2015) 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G, Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes, Proc. Natl. Acad. Sci 107 (17) (2010) 7817–7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, et al. , Beneficial effects of mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction, J. Am. College Cardiol 54 (25) (2009) 2435–2446. [DOI] [PubMed] [Google Scholar]
- [8].McAlpine CS, Kiss MG, Rattik S, He S, Vassalli A, Valet C, et al. , Sleep modulates haematopoiesis and protects against atherosclerosis, Nature. 566 (7744) (2019) 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hata M, Andriessen EMMA, Hata M, Diaz-Marin R, Fournier F, Crespo-Garcia S, et al. , Past history of obesity triggers persistent epigenetic changes in innate immunity and exacerbates neuroinflammation, Science. 379 (6627) (2023) 45–62. [DOI] [PubMed] [Google Scholar]
- [10].van Kampen E, Jaminon A, van Berkel TJC, Van Eck M, Diet-induced (epigenetic) changes in bone marrow augment atherosclerosis, J. Leukoc. Biol 96 (5) (2014) 833–841. [DOI] [PubMed] [Google Scholar]
- [11].Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. , Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming, Cell. 172 (1) (2018), 162–75.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barrett TJ, Corr EM, van Solingen C, Schlamp F, Brown EJ, Koelwyn GJ, et al. , Chronic stress primes innate immune responses in mice and humans, Cell Rep. 36 (10) (2021), 109595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Seijkens T, Hoeksema MA, Beckers L, Smeets E, Meiler S, Levels J, et al. , Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis, FASEB J. 28 (5) (2014) 2202–2213. [DOI] [PubMed] [Google Scholar]
- [14].DiToro D, Harbour SN, Bando JK, Benavides G, Witte S, Laufer VA, et al. , Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity, Immunity. 52 (4) (2020), 650–67.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].O’Neill LA, A broken Krebs cycle in macrophages, Immunity. 42 (3) (2015) 393–394. [DOI] [PubMed] [Google Scholar]
- [16].Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH, Kong HK, et al. , Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1α Axis and restores tamoxifen sensitivity in Antiestrogen-resistant breast cancer cells, PLoS One 10 (7) (2015), e0132285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yarbro JR, Emmons RS, Pence BD, Macrophage immunometabolism and inflammaging: roles of mitochondrial dysfunction, cellular senescence, CD38, and NAD, Immunometabolism. 2 (3) (2020), e200026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. , An aged immune system drives senescence and ageing of solid organs, Nature. 594 (7861) (2021) 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hara K, Maruki Y, Long X, Yoshino K-I, Oshiro N, Hidayat S, et al. , Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action, Cell 110 (2) (2002) 177–189. [DOI] [PubMed] [Google Scholar]
- [20].Kim D-H, Sarbassov DD, Ali SM, Latek RR, Guntur KVP, Erdjument-Bromage H, et al. , GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR, Mol. Cell 11 (4) (2003) 895–904. [DOI] [PubMed] [Google Scholar]
- [21].Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. , The rag GTPases bind raptor and mediate amino acid Signaling to mTORC1, Science 320 (5882) (2008) 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM, Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids, Cell. 141 (2) (2010) 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].de Araujo MEG, Naschberger A, Fürnrohr BG, Stasyk T, Dunzendorfer-Matt T, Lechner S, et al. , Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling, Science. 358 (6361) (2017) 377–381. [DOI] [PubMed] [Google Scholar]
- [24].Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. , A tumor suppressor complex with GAP activity for the rag GTPases that signal amino acid sufficiency to mTORC1, Science. 340 (6136) (2013) 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shen K, Huang RK, Brignole EJ, Condon KJ, Valenstein ML, Chantranupong L, et al. , Architecture of the human GATOR1 and GATOR1–rag GTPases complexes, Nature. 556 (7699) (2018) 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yuskaitis CJ, Modasia JB, Schrotter S, Rossitto LA, Groff KJ, Morici C, et al. , DEPDC5-dependent mTORC1 signaling mechanisms are critical for the anti-seizure effects of acute fasting, Cell Rep. 40 (9) (2022) 111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ramirez Reyes JMJ, Cuesta R, Pause A, Folliculin: a regulator of transcription through AMPK and mTOR Signaling pathways, Frontiers in Cell and Developmental Biology. (2021) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Condon KJ, Sabatini DM, Nutrient regulation of mTORC1 at a glance, J. Cell Sci 132 (21) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. , Sestrin2 is a leucine sensor for the mTORC1 pathway, Science. 351 (6268) (2016) 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. , The CASTOR proteins are arginine sensors for the mTORC1 pathway, Cell. 165 (1) (2016) 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jung J, Genau HM, Behrends C, Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9, Mol. Cell. Biol 35 (14) (2015) 2479–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Metabolism., et al. , Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1, Science. 347 (6218) (2015) 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rebsamen M, Pochini L, Stasyk T, de Araújo MEG, Galluccio M, Kandasamy RK, et al. , SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1, Nature. 519 (7544) (2015) 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. , mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient, Cell. 171 (3) (2017), 642–54. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, et al. , SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway, Science. 358 (6364) (2017) 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mentch SJ, Locasale JW, One-carbon metabolism and epigenetics: understanding the specificity, Ann. N. Y. Acad. Sci 1363 (1) (2016) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lim C-Y, Davis OB, Shin HR, Zhang J, Berdan CA, Jiang X, et al. , ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann–pick type C, Nature Cell Biology 21 (10) (2019) 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, et al. , Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38, Science. 318 (5852) (2007) 977–980. [DOI] [PubMed] [Google Scholar]
- [39].Uhlenbrock K, Weiwad M, Wetzker R, Fischer G, Wittinghofer A, Rubio I, Reassessment of the role of FKBP38 in the Rheb/mTORC1 pathway, FEBS Lett. 583 (6) (2009) 965–970. [DOI] [PubMed] [Google Scholar]
- [40].Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, et al. , Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40, Nature. 552 (7685) (2017) 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, et al. , Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat cell biol. 5 (6) (2003) 559–565. [DOI] [PubMed] [Google Scholar]
- [42].Li Y, Corradetti MN, Inoki K, Guan K-L, TSC2: filling the GAP in the mTOR signaling pathway, Trends Biochem. Sci 29 (1) (2004) 32–38. [DOI] [PubMed] [Google Scholar]
- [43].McManus EJ, Alessi DR, TSC1–TSC2: a complex tale of PKB-mediated S6K regulation, Nat. Cell Biol 4 (9) (2002). E214–E6. [DOI] [PubMed] [Google Scholar]
- [44].Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, et al. , TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1, Mol. Cell 47 (4) (2012) 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D, Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins, Nature Cell Biology 5 (6) (2003) 578–581. [DOI] [PubMed] [Google Scholar]
- [46].Inoki K, Li Y, Zhu T, Wu J, Guan K-L, TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling, Nat. Cell Biol 4 (9) (2002) 648–657. [DOI] [PubMed] [Google Scholar]
- [47].Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP, Phosphorylation and functional inactivation of TSC2 by Erk: implications for tuberous sclerosisand cancer pathogenesis, Cell. 121 (2) (2005) 179–193. [DOI] [PubMed] [Google Scholar]
- [48].Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J, Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase, Proc. Natl. Acad. Sci 101 (37) (2004) 13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ballif BA, Roux PP, Gerber SA, MacKeigan JP, Blenis J, Gygi SP, Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors, Proc. Natl. Acad. Sci 102 (3) (2005) 667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Suire S, Hawkins P, Stephens L, Activation of phosphoinositide 3-kinase γ by Ras, Current Biology 12 (13) (2002) 1068–1075 (Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase direct target of Ras. Nature. 1994;370(6490):527–32). [DOI] [PubMed] [Google Scholar]
- [51].Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, et al. , ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1), J Biol Chem. 286 (1) (2011) 567–577.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Frey JW, Jacobs BL, Goodman CA, Hornberger TA, A role for raptor phosphorylation in the mechanical activation of mTOR signaling, Cell. Signal 26 (2) (2014) 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sadeghi Shaker M, Rokni M, Mahmoudi M, Farhadi E, Ras family signaling pathway in immunopathogenesis of inflammatory rheumatic diseases, Front. Immunol 14 (2023) 1151246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Collins SL, Oh M-H, Sun I-H, Chan-Li Y, Zhao L, Powell JD, et al. , mTORC1 Signaling regulates proinflammatory macrophage function and Metabolism, J. Immunol 207 (3) (2021) 913–922. [DOI] [PubMed] [Google Scholar]
- [55].Weichhart T, Haidinger M, Katholnig K, Kopecky C, Poglitsch M, Lassnig C, et al. , Inhibition of mTOR blocks the anti-inflammatory effects of glucocorticoids in myeloid immune cells, Blood. 117 (16) (2011) 4273–4283. [DOI] [PubMed] [Google Scholar]
- [56].Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, et al. , The TSC-mTOR signaling pathway regulates the innate inflammatory response, Immunity. 29 (4) (2008) 565–577. [DOI] [PubMed] [Google Scholar]
- [57].Ivanov SS, Roy CR, Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR, Nat. Immunol 14 (12) (2013) 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Woodcock HV, Eley JD, Guillotin D, Platé M, Nanthakumar CB, Martufi M, et al. , The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis, Nat. Commun 10 (1) (2019) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. , The TSC-mTOR pathway regulates macrophage polarization, Nat. Commun 4 (1) (2013) 2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Crayne CB, Albeituni S, Nichols KE, Cron RQ, The immunology of macrophage activation syndrome, Front. Immunol 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Huang Z, You X, Chen L, Du Y, Brodeur K, Jee H, et al. , mTORC1 links pathology in experimental models of Still’s disease and macrophage activation syndrome, Nat. Commun 13 (1) (2022) 6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lee PY, Sykes DB, Ameri S, Kalaitzidis D, Charles JF, Nelson-Maney N, et al. , The metabolic regulator mTORC1 controls terminal myeloid differentiation, Science Immunology. 2 (11) (2017) eaam6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Linke M, Pham HTT, Katholnig K, Schnöller T, Miller A, Demel F, et al. , Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression, Nat. Immunol 18 (3) (2017) 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Arif A, Yao P, Terenzi F, Jia J, Ray PS, Fox PL, The GAIT translational control system, WIREs RNA. 9 (2) (2018), e1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Arif A, Jia J, Moodt RA, DiCorleto PE, Fox PL, Phosphorylation of glutamylprolyl tRNA synthetase by cyclin-dependent kinase 5 dictates transcript-selective translational control, Proc. Natl. Acad. Sci. U. S. A 108 (4) (2011) 1415–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Arif A, Jia J, Willard B, Li X, Fox PL, Multisite phosphorylation of S6K1 directs a kinase Phospho-code that determines substrate selection, Mol. Cell 73 (3) (2019), 446–57.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Alesi N, Akl EW, Khabibullin D, Liu H-J, Nidhiry AS, Garner ER, et al. , TSC2 regulates lysosome biogenesis via a non-canonical RAGC and TFEB-dependent mechanism, Nature Communications 12 (1) (2021) 4245.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. , Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype, J. Biol. Chem 289 (11) (2014) 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Moon J-S, Hisata S, Park M-A, DeNicola Gina M., Ryter Stefan W, Nakahira K, et al. , mTORC1-induced HK1-dependent glycolysis regulates NLRP3 Inflammasome activation, Cell Rep. 12 (1) (2015) 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [70].Evavold CL, Hafner-Bratkovič I, Devant P, D’Andrea JM, Ngwa EM, Boršić E, et al. , Control of gasdermin D oligomerization and pyroptosis by the Ragulator-rag-mTORC1 pathway, Cell. 184 (17) (2021), 4495–511.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. , An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1*, J. Biol. Chem 284 (12) (2009) 8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Joe Y, Chen Y, Park J, Kim HJ, Rah S-Y, Ryu J, et al. , Cross-talk between CD38 and TTP Is Essential for Resolution of Inflammation during Microbial Sepsis, Cell Reports 30 (4) (2020) 1063–1076, e5. [DOI] [PubMed] [Google Scholar]
- [73].Zhang W, Zhuang N, Liu X, He L, He Y, Mahinthichaichan P, et al. , The metabolic regulator Lamtor5 suppresses inflammatory signaling via regulating mTOR-mediated TLR4 degradation, Cellular & Molecular Immunology. 17 (10) (2020) 1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Moral-Sanz J, Lewis SA, MacMillan S, Ross FA, Thomson A, Viollet B, et al. , The LKB1–AMPK-α1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria, Science Signaling 11 (550) (2018) eaau029.. [DOI] [PubMed] [Google Scholar]
- [75].Chun Y, Kim J, AMPK-mTOR Signaling and cellular adaptations in hypoxia, Int. J. Mol. Sci 22 (18) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Dengler F, Activation of AMPK under hypoxia: many roads leading to Rome, Int. J. Mol. Sci 21 (7) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sag D, Carling D, Stout RD, Suttles J, Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype, J. Immunol 181 (12) (2008) 8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhu H, Zhang M, Liu Z, Xing J, Moriasi C, Dai X, et al. , AMP-activated protein kinase α1 in macrophages promotes collateral Remodeling and Arteriogenesis in mice in vivo, Arterioscler. Thromb. Vasc. Biol 36 (9) (2016) 1868–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li D, Wu M, Pattern recognition receptors in health and diseases, Signal Transduction and Targeted Therapy 6 (1) (2021) 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bao W, Wang Y, Fu Y, Jia X, Li J, Vangan N, et al. , mTORC1 Regulates Flagellin-Induced Inflammatory Response in Macrophages, PLoS One 10 (5) (2015) e0125910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang W, Lavine KJ, Epelman S, Evans SA, Weinheimer CJ, Barger PM, et al. , Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo, J. Am. Heart Assoc 4 (6) (2015), e001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rohde D, Schön C, Boerries M, Didrihsone I, Ritterhoff J, Kubatzky KF, et al. , S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via toll-like receptor 4, EMBO Molecular Medicine. 6 (6) (2014) 778–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liu L, Wang Y, Cao Z-Y, Wang M-M, Liu X-M, Gao T, et al. , Up-regulated TLR4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction, J. Cell. Mol. Med 19 (12) (2015) 2728–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gajanayaka N, Dong SXM, Ali H, Iqbal S, Mookerjee A, Lawton DA, et al. , TLR-4 agonist induces IFN-γ production selectively in Proinflammatory human M1 macrophages through the PI3K-mTOR- and JNK-MAPK-activated p70S6K pathway, J. Immunol 207 (9) (2021) 2310–2324. [DOI] [PubMed] [Google Scholar]
- [85].Wang H, Brown J, Gao S, Liang S, Jotwani R, Zhou H, et al. , The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells, J. Immunol 191 (3) (2013) 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].López-Pelaéz M, Fumagalli S, Sanz C, Herrero C, Guerra S, Fernandez M, et al. , Cot/tpl2-MKK1/2-Erk1/2 controls mTORC1-mediated mRNA translation in toll-like receptor-activated macrophages, Mol. Biol. Cell 23 (15) (2012) 2982–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Luo L, Wall AA, Yeo JC, Condon ND, Norwood SJ, Schoenwaelder S, et al. , Rab8a interacts directly with PI3Kγ to modulate TLR4-driven PI3K and mTOR signalling, Nat. Commun 5 (2014) 4407. [DOI] [PubMed] [Google Scholar]
- [88].Bodur C, Kazyken D, Huang K, Ekim Ustunel B, Siroky KA, Tooley AS, et al. , The IKK-related kinase TBK1 activates mTORC1 directly in response to growth factors and innate immune agonists, EMBO J. 37 (1) (2018) 19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Liu Y, Cao G-F, Xue J, Wan J, Wan Y, Jiang Q, et al. , Tumor necrosis factor-alpha (TNF-α)-mediated in vitro human retinal pigment epithelial (RPE) cell migration mainly requires Akt/mTOR complex 1 (mTORC1), but not mTOR complex 2 (mTORC2) signaling, European Journal of Cell Biology 91 (9) (2012) 728–737. [DOI] [PubMed] [Google Scholar]
- [90].Bartekova M, Radosinska J, Jelemensky M, Dhalla NS, Role of cytokines and inflammation in heart function during health and disease, Heart Fail. Rev 23 (5) (2018) 733–758. [DOI] [PubMed] [Google Scholar]
- [91].Ohkura T, Yoshimura T, Fujisawa M, Ohara T, Marutani R, Usami K, et al. , Spred2 regulates high fat diet-induced adipose tissue inflammation, and metabolic abnormalities in mice, Front. Immunol 10 (2019) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lu N, Malemud CJ, Extracellular signal-regulated kinase: a regulator of cell growth, inflammation, chondrocyte and bone cell receptor-mediated gene expression, Int. J. Mol. Sci 20 (15) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Chen Y, Ba L, Huang W, Liu Y, Pan H, Mingyao E, et al. , Role of carvacrol in cardioprotection against myocardial ischemia/reperfusion injury in rats through activation of MAPK/ERK and Akt/eNOS signaling pathways, European Journal of Pharmacology 796 (2017) 90–100. [DOI] [PubMed] [Google Scholar]
- [94].Zhou Q-L, Teng F, Zhang Y-S, Sun Q, Cao Y-X, Meng G-W, FPR1 gene silencing suppresses cardiomyocyte apoptosis and ventricular remodeling in rats with ischemia/reperfusion injury through the inhibition of MAPK signaling pathway, Experimental Cell Research 370 (2) (2018) 506–518. [DOI] [PubMed] [Google Scholar]
- [95].Yeh CC, Li H, Malhotra D, Turcato S, Nicholas S, Tu R, et al. , Distinctive ERK and p38 signaling in remote and infarcted myocardium during post-MI remodeling in the mouse, J. Cell. Biochem 109 (6) (2010) 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ma L, Kerr BA, Naga Prasad SV, Byzova TV, Somanath PR, Differential effects of Akt1 signaling on short- versus long-term consequences of myocardial infarction and reperfusion injury, Lab. Investig 94 (10) (2014) 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kerr BA, Ma L, West XZ, Ding L, Malinin NL, Weber ME, et al. , Interference with Akt Signaling protects against myocardial infarction and death by limiting the consequences of oxidative stress, Sci. Signal 6 (287) (2013) ra67–ra. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Li Z, Liu X, Zhang X, Zhang W, Gong M, Qin X, et al. , TRIM21 aggravates cardiac injury after myocardial infarction by promoting M1 macrophage polarization, Front. Immunol 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Sciarretta S, Forte M, Frati G, Sadoshima J, New insights into the role of mTOR Signaling in the cardiovascular system, Circ. Res 122 (3) (2018) 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ratto E, Chowdhury SR, Siefert NS, Schneider M, Wittmann M, Helm D, et al. , Direct control of lysosomal catabolic activity by mTORC1 through regulation of V-ATPase assembly, Nat. Commun 13 (1) (2022) 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zi Z, Zhang Z, Feng Q, Kim C, Wang X-D, Scherer PE, et al. , Quantitative phosphoproteomic analyses identify STK11IP as a lysosome-specific substrate of mTORC1 that regulates lysosomal acidification, Nat. Commun 13 (1) (2022) 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Redka DyS M Gütschow, S. Grinstein, J. Canton, Differential ability of proinflammatory and anti-inflammatory macrophages to perform macropinocytosis, Mol. Biol. Cell 29 (1) (2018) 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Moore Kathryn J, Tabas I, Macrophages in the pathogenesis of atherosclerosis, Cell 145 (3) (2011) 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Fanucchi S, Domínguez-Andrés J, Joosten LAB, Netea MG, Mhlanga MM, The intersection of epigenetics and Metabolism in trained immunity, Immunity. 54 (1) (2021) 32–43. [DOI] [PubMed] [Google Scholar]
- [105].Lamb DJ, Eales LJ, Ferns GA, Immunization with bacillus Calmette-Guerin vaccine increases aortic atherosclerosis in the cholesterol-fed rabbit, Atherosclerosis. 143 (1) (1999) 105–113. [DOI] [PubMed] [Google Scholar]
- [106].Keating ST, Groh L, Thiem K, Bekkering S, Li Y, Matzaraki V, et al. , Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein, J. Mol. Med 98 (6) (2020) 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP, Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes, Arterioscler. Thromb. Vasc. Biol 34 (8) (2014) 1731–1738. [DOI] [PubMed] [Google Scholar]
- [108].Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. , Metabolic induction of trained immunity through the mevalonate pathway, Cell. 172 (1) (2018), 135–46.e9. [DOI] [PubMed] [Google Scholar]
- [109].Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, et al. , Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis, Circulation. 144 (12) (2021) 961–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. , Oxidized phospholipids on lipoprotein(a) elicit Arterial Wall inflammation and an inflammatory monocyte response in humans, Circulation. 134 (8) (2016) 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].van der Heijden C, Keating ST, Groh L, Joosten LAB, Netea MG, Riksen NP, Aldosterone induces trained immunity: the role of fatty acid synthesis, Cardiovasc. Res 116 (2) (2020) 317–328. [DOI] [PubMed] [Google Scholar]
- [112].Dimsdale JE, Psychological stress and cardiovascular disease, J. Am. Coll. Cardiol 51 (13) (2008) 1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lopez-Garcia E, Schulze MB, Fung TT, Meigs JB, Rifai N, Manson JE, et al. , Major dietary patterns are related to plasma concentrations of markers of inflammation and endothelial dysfunction, Am J Clin Nutr. 80 (4) (2004) 1029–1035. [DOI] [PubMed] [Google Scholar]
- [114].Pendyala S, Walker JM, Holt PR, A high-fat diet is associated with endotoxemia that originates from the gut, Gastroenterology. 142 (5) (2012), 1100–1.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Mohammad S, Thiemermann C, Role of metabolic Endotoxemia in systemic inflammation and potential interventions, Front. Immunol 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Sánchez-Tapia M, Miller AW, Granados-Portillo O, Tovar AR, Torres N, The development of metabolic endotoxemia is dependent on the type of sweetener and the presence of saturated fat in the diet, Gut Microbes 12 (1) (2020) 1801301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Geng S, Chen K, Yuan R, Peng L, Maitra U, Diao N, et al. , The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis, Nat. Commun 7 (1) (2016) 13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].St-Onge M-P, Grandner MA, Brown D, Conroy MB, Jean-Louis G, Coons M, et al. , Sleep duration and quality: impact on lifestyle behaviors and cardiometabolic health: a scientific statement from the American Heart Association, Circulation. 134 (18) (2016) e367–e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].McAlpine CS, Kiss MG, Zuraikat FM, Cheek D, Schiroli G, Amatullah H, et al. , Sleep exerts lasting effects on hematopoietic stem cell function and diversity, J. Exp. Med 219 (11) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Frodermann V, Rohde D, Courties G, Severe N, Schloss MJ, Amatullah H, et al. , Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells, Nat. Med 25 (11) (2019) 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Noz MP, Hartman YAW, Hopman MTE, Willems P, Tack CJ, Joosten LAB, et al. , Sixteen-week physical activity intervention in subjects with increased cardiometabolic risk shifts innate immune function towards a less proinflammatory state, J. Am. Heart Assoc 8 (21) (2019), e013764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. , 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines, Circulation. 140 (11) (2019) e596–e646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. , mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity, Science. 345 (6204) (2014) 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. , Immunometabolic pathways in BCG-induced trained immunity, Cell Reports. 17 (10) (2016) 2562–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Arts RJW, Novakovic B, ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. , Glutaminolysis and fumarate accumulation integrate Immunometabolic and epigenetic programs in trained immunity, Cell Metab. 24 (6) (2016) 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Stone GW, Kedhi E, Kereiakes DJ, Parise H, Fahy M, Serruys PW, et al. , Differential clinical responses to Everolimus-eluting and paclitaxel-eluting coronary stents in patients with and without diabetes mellitus, Circulation. 124 (8) (2011) 893–900. [DOI] [PubMed] [Google Scholar]
- [127].van Leent MMT, Beldman TJ, Toner YC, Lameijer MA, Rother N, Bekkering S, et al. , Prosaposin mediates inflammation in atherosclerosis, Sci. Transl. Med 13 (584) (2021) eabe1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Elder SS, Emmerson E, Senescent cells and macrophages: key players for regeneration? Open Biol. 10 (12) (2020), 200309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Birch J, Gil J, Senescence and the SASP: many therapeutic avenues, Genes Dev. 34 (23–24) (2020) 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Goronzy JJ, Weyand CM, Understanding immunosenescence to improve responses to vaccines, Nature Immunology 14 (5) (2013) 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Vicente R, Mausset-Bonnefont A-L, Jorgensen C, Louis-Plence P, Brondello JM, Cellular senescence impact on immune cell fate and function, Aging Cell 15 (3) (2016) 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ong S-M, Hadadi E, Dang T-M, Yeap W-H, Tan CT-Y, Ng T-P, et al. , The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence, Cell Death Dis. 9 (3) (2018) 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Li C-J, Xiao Y, Sun Y-C, He W-Z, Liu L, Huang M, et al. , Senescent immune cells release grancalcin to promote skeletal aging, Cell Metab. 33 (10) (2021), 1957–73.e6. [DOI] [PubMed] [Google Scholar]
- [134].Clark D, Brazina S, Yang F, Hu D, Hsieh CL, Niemi EC, et al. , Age-related changes to macrophages are detrimental to fracture healing in mice, Aging Cell 19 (3) (2020), e13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. , Spatial coupling of mTOR and autophagy augments secretory phenotypes, Science. 332 (6032) (2011) 966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. , mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype, Nat. Cell Biol 17 (9) (2015) 1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. , MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation, Nat. Cell Biol 17 (8) (2015) 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Smallwood HS, Duan S, Morfouace M, Rezinciuc S, Shulkin BL, Shelat A, et al. , Targeting metabolic reprogramming by influenza infection for therapeutic intervention, Cell Rep. 19 (8) (2017) 1640–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Mannick JB, Teo G, Bernardo P, Quinn D, Russell K, Klickstein L, et al. , Targeting the biology of ageing with mTOR inhibitors to improve immune function in older adults: phase 2b and phase 3 randomised trials, Lancet Healthy Longevity. 2 (5) (2021) e250–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, et al. , mTOR inhibition improves immune function in the elderly, Sci. Transl. Med 6 (268) (2014), 268ra179–268ra179. [DOI] [PubMed] [Google Scholar]
- [141].Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM, Senescent intimal foam cells are deleterious at all stages of atherosclerosis, Science. 354 (6311) (2016) 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Pham LM, Kim E-C, Ou W, Phung CD, Nguyen TT, Pham TT, et al. , Targeting and clearance of senescent foamy macrophages and senescent endothelial cells by antibody-functionalized mesoporous silica nanoparticles for alleviating aorta atherosclerosis, Biomaterials. 269 (2021), 120677. [DOI] [PubMed] [Google Scholar]
- [143].Selvarani R, Mohammed S, Richardson A, Effect of rapamycin on aging and age-related diseases-past and future, Geroscience. 43 (3) (2021) 1135–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Yang S, Li J, Chen Y, Zhang S, Feng C, Hou Z, et al. , MicroRNA-216a promotes M1 macrophages polarization and atherosclerosis progression by activating telomerase via the Smad3/NF-κB pathway, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 1865 (7) (2019) 1772–1781. [DOI] [PubMed] [Google Scholar]
- [145].Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. , Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes, Blood. 126 (1) (2015) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Natarajan P, Jaiswal S, Kathiresan S, Clonal Hematopoiesis, Circul.: Genomic Precis. Med 11 (7) (2018), e001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. , Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence, N. Engl. J. Med 371 (26) (2014) 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. , Age-related clonal hematopoiesis associated with adverse outcomes, N. Engl. J. Med 371 (26) (2014) 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. , Clonal Hematopoiesis and risk of atherosclerotic cardiovascular disease, N. Engl. J. Med 377 (2) (2017) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Bhattacharya R, Zekavat SM, Haessler J, Fornage M, Raffield L, Uddin MM, et al. , Clonal Hematopoiesis is associated with higher risk of stroke, Stroke. 53 (3) (2022) 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, et al. , Association of Clonal Hematopoiesis with Incident Heart Failure, J. Am. Coll. Cardiol 78 (1) (2021) 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Benditt EP, Benditt JM, Evidence for a monoclonal origin of human atherosclerotic plaques, Proc. Natl. Acad. Sci 70 (6) (1973) 1753–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Loh PR, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, et al. , Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations, Nature. 559 (7714) (2018) 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Saiki R, Momozawa Y, Nannya Y, Nakagawa MM, Ochi Y, Yoshizato T, et al. , Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis, Nat. Med 27 (7) (2021) 1239–1249. [DOI] [PubMed] [Google Scholar]
- [155].Zekavat SM, Lin S-H, Bick AG, Liu A, Paruchuri K, Wang C, et al. , Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection, Nat. Med 27 (6) (2021) 1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Sun M, Cyr M-C, Sandoval J, Lemieux Perreault L-P, Busque L, Tardif J-C, et al. , Somatic mosaic chromosomal alterations and death of cardiovascular disease causes among cancer survivors, Cancer Epidemiol. Biomark. Prev 32 (6) (2023) 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Fujino T, Goyama S, Sugiura Y, Inoue D, Asada S, Yamasaki S, et al. , Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway, Nat. Commun 12 (1) (2021) 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Inoue D, Fujino T, Sheridan P, Zhang YZ, Nagase R, Horikawa S, et al. , A novel ASXL1-OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies, Leukemia 32 (6) (2018) 1327–1337. [DOI] [PubMed] [Google Scholar]
- [159].Hsu Y-C, Chiu Y-C, Lin C-C, Kuo Y-Y, Hou H-A, Tzeng Y-S, et al. , The distinct biological implications of Asxl1 mutation and its roles in leukemogenesis revealed by a knock-in mouse model, J. Hematol. Oncol 10 (1) (2017) 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Dai Y-J, Wang Y-Y, Huang J-Y, Xia L, Shi X-D, Xu J, et al. , Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement, Proc. Natl. Acad. Sci 114 (20) (2017) 5237–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].SanMiguel JM, Eudy E, Loberg MA, Miles LA, Stearns T, Mistry JJ, et al. , Cell origin–dependent cooperativity of mutant Dnmt3a and Npm1 in clonal hematopoiesis and myeloid malignancy, Blood Adv. 6 (12) (2022) 3666–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Kittleson MM, Panjrath GS, Amancherla K, Davis LL, Deswal A, Dixon DL, et al. , 2023 ACC expert consensus decision pathway on management of heart failure with preserved ejection fraction, J. Am. Coll. Cardiol 81 (18) (2023) 1835–1878. [DOI] [PubMed] [Google Scholar]
- [163].Webb J, Fovargue L, Tøndel K, Porter B, Sieniewicz B, Gould J, et al. , The emerging role of cardiac magnetic resonance imaging in the evaluation of patients with HFpEF, Curr Heart Fail Rep. 15 (1) (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Luchi RJ, Snow E, Luchi JM, Nelson CL, Pircher FJ, Left ventricular function in hospitalized geriatric patients, J. Am. Geriatr. Soc 30 (11) (1982) 700–705. [DOI] [PubMed] [Google Scholar]
- [165].Chang PP, Chambless LE, Shahar E, Bertoni AG, Russell SD, Ni H, et al. , Incidence and survival of hospitalized acute decompensated heart failure in four US communities (from the atherosclerosis risk in communities study), Am J Cardiol 113 (3) (2014) 504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Upadhya B, Kitzman DW, Heart failure with preserved ejection fraction: new approaches to diagnosis and management, Clin. Cardiol 43 (2) (2020) 145–155 (Li Z, Lefer DJ. Demystifying Cardiac Aging. Circulation Research. 2021;128(4): 508–10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Glezeva N, Voon V, Watson C, Horgan S, McDonald K, Ledwidge M, et al. , Exaggerated inflammation and Monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis, J. Card. Fail 21 (2) (2015) 167–177. [DOI] [PubMed] [Google Scholar]
- [168].Abernethy A, Raza S, Sun JL, Anstrom KJ, Tracy R, Steiner J, et al. , Proinflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction, J. Am. Heart Assoc 7 (8) (2018), e007385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. , Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction, Circul.: Heart Fail 4 (1) (2011) 44–52. [DOI] [PubMed] [Google Scholar]
- [170].Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, et al. , Cardiac macrophages promote diastolic dysfunction, J. Exp. Med 215 (2) (2018) 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. , Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation, Immunity. 40 (1) (2014) 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Xu J, Lin S-C, Chen J, Miao Y, Taffet GE, Entman ML, et al. , CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis, American journal of physiology-heart and circulatory physiology 301 (2) (2011). H538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Shi C, Aboumsallem JP, Suthahar N, de Graaf AO, Jansen JH, van Zeventer IA, et al. , Clonal haematopoiesis of indeterminate potential: associations with heart failure incidence, clinical parameters and biomarkers, Eur. J. Heart Fail 25 (1) (2023) 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Reiner AP, Roberts MB, Honigberg MC, Kooperberg C, Desai P, Bick AG, et al. , Association of clonal hematopoiesis of indeterminate potential with incident heart failure with preserved ejection fraction, medRxiv (2023), 10.1101/2023.06.07.23291038. [DOI] [Google Scholar]
- [175].Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K, CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease, Circ. Res 123 (3) (2018) 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. , Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 Inflammasome, J. Am. Coll. Cardiol 71 (8) (2018) 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Thomas T, Ji Y, Kalkan F, Son A, Pandey A, Subramanian V, et al. , Clonal hematopoiesis and heart failure with preserved ejection fraction, Blood. 140 (Supplement 1) (2022) 8611–8612. [Google Scholar]
- [178].Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. , Nitrosative stress drives heart failure with preserved ejection fraction, Nature. 568 (7752) (2019) 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Dai D-F, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, et al. , Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart, Aging Cell 13 (3) (2014) 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Quarles E, Basisty N, Chiao YA, Merrihew G, Gu H, Sweetwyne MT, et al. , Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment, Aging Cell 19 (2) (2020), e13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, et al. , Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex, Science 355 (6331) (2017) 1306–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Moore KJ, Sheedy FJ, Fisher EA, Macrophages in atherosclerosis: a dynamic balance, Nat. Rev. Immunol 13 (2013) 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Tabas I, Lichtman AH, Monocyte-Macrophages and T Cells in Atherosclerosis, Immunity 47 (2017) 621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. , The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions, J. Exp. Med 204 (12) (2007) 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Hilgendorf I, Gerhardt LM, C Tan T, Winter C, Holderried TA, Chousterman BG, et al. , Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium, Circ. Res 114 (10) (2014) 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]