

Abstract
Potent anti-tumor T cell response and efficient intratumoral T cell infiltration are the major challenges for therapeutic cancer vaccines. To address these issues, a nano-vaccine system has been designed to promote anti-tumor T cell responses, and intratumoral infiltration was examined in various murine tumor models. Subcutaneous vaccination with nanodiscs carrying human papillomavirus (HPV)-16 E7 antigen elicits as high as ~32% E7-specific CD8 α + T cell responses in circulation, representing a 29-fold improvement over the soluble peptide vaccination. Importantly, nanodisc vaccination also promotes robust intratumoral T cell infiltration and eliminates HPV16 E6/E7-expressing TC-1 tumors at mucosal sites, including lungs, inner lip, and intravaginal tissues. In a benchmark study with a live Listeria vaccine combined with anti-PD-1 IgG, nanodiscs plus anti-PD-1 immune checkpoint blockade elicits comparable levels of T cell responses with anti-tumor efficacy. Furthermore, compared with Complete Freund’s Adjuvant combined with tetanus toxoid, nanodisc vaccination in HLA-A02 mice generates >200-fold stronger IFN-γ+ T cell responses against a neoantigen from an HLA-A02 melanoma patient. Overall, these results show that the nanodisc system is a promising cancer vaccine platform for inducing anti-tumor T cell responses.
Keywords: nanoparticles, cancer vaccine, papillomavirus, neoantigen
Graphical Abstract
Efficient infiltration of T cells in solid cancer is a major challenge for cancer immunotherapy. A nanoparticle vaccine system has been developed to promote T cell infiltration into peripheral mucosal tissues and eliminate disseminated tumors. Nanodiscs are broadly applicable with a wide range of tumor antigens, thus providing a versatile and potent vaccine platform for eliciting T cell immunity.
1. INTRODUCTION
Induction of anti-tumor T cell responses with vaccination is an attractive therapeutic strategy against multiple types of cancer,[1–4] and various cancer vaccine platforms have been reported to induce tumor-specific T cell responses.[5–12] Cancer vaccines can be generally classified into 2 categories: live vector-based vaccines[6,9,10] and subunit vaccines.[7,11,12] The inherent pathogen-like properties of live vectors allow for the induction of strong innate and adaptive immune responses.[10,13] For example, several clinical trials have examined TA-HPV,[9] a live recombinant vaccinia virus-based human papillomavirus (HPV) vaccine encoding E6 and E7 antigen of HPV 16 and 18 as well as Lm-LLO-E7,[6,10] a live attenuated Listeria monocytogenes vector expressing E7 and listeriolysin O. Despite their ability to induce cytotoxic T lymphocyte (CTL) responses in humans,[4,6,9,14,15] live vector-based vaccines need to overcome many challenges.[4,16] First, their therapeutic effects are hindered by pre-existing immunity against the vector itself as well as neutralizing anti-vector antibodies generated after multiple immunizations.[4,16,17] Second, safety concerns and adverse effects associated with live vectors pose additional challenges.[4] For instance, 40% of patients experienced severe grade 3 side effects in a phase I clinical trial with Lm-LLO-E7.[6]
On the other hand, subunit vaccines, composed of defined tumor antigens and immunostimulatory agents, offer safer alternatives.[5,7,12] However, weak T cell responses and inefficient intratumoral infiltration of T cells are the major hurdles to overcome.[4] Here, we sought to address these issues with a potent subunit vaccine platform based on nanodiscs. “Blank” nanodiscs, composed of phospholipids and Apolipoprotein-mimetic peptide, have been previously manufactured in Kg scales and shown to be safe in humans for cardiovascular applications, thus providing a promising platform for drug delivery applications.[18] We have reported that nanodiscs carrying peptide antigens and adjuvant molecules efficiently deliver them to dendritic cells (DCs) in lymph nodes (LNs), leading to strong anti-tumor T cell responses in combination with immune checkpoint blockade (ICB).[5,19,20] Therefore, nanodiscs with demonstrated large scale manufacturability, safety, and potency for immune activation offer an attractive platform for cancer vaccination.
Using the nanodisc technology, here we aimed to answer the following questions: (1) What is the optimal route of nanodisc vaccination for promoting antigen-specific T cell responses and T cell infiltration into the tumor microenvironment (TME)? (2) How does the therapeutic efficacy of nanodiscs compare with other leading vaccine technologies, such as live Listeria vaccine? (3) Can we demonstrate the wide applicability of nanodisc technology with clinically relevant human HLA-restricted antigens? To address these questions, we have compared the subcutaneous (s.c.) versus the intranasal (i.n.) route of nanodisc vaccination using HPV16 E7 antigen and assessed their anti-tumor efficacy in multiple mucosal tumor models (Figure 1). Although prophylactic vaccines have been highly effective against HPV infection,[21–23] development of successful therapeutic vaccines against established HPV+ cancer,[4,11,24,25] such as in head & neck and cervical cancer,[21,22,26,27] has been elusive due to inefficient T cell induction and infiltration into mucosal TME.[28]
Figure 1.

Schematic illustration of nanodisc vaccination and immune monitoring in HPV16 mucosal tumor models.
Here, using TC-1 cells expressing HPV16 E6/E7 oncoprotein, we have demonstrated that s.c. nanodisc vaccination in mice induced as high as ~32% E7-specific CD8+ T cell response among all CD8+ T cells in circulation, promoting robust T cell infiltration into peripheral mucosal tissues. In TC-1 models of HPV-associated lung metastasis, head & neck,[29] and cervical cancer,[27,30] we show that s.c. nanodisc vaccination generated superior T cell responses than i.n. nanodisc vaccination and eliminated TC-1 tumors from the lungs, inner lip, and reproductive tract. Furthermore, we performed a head-to-head comparison study between a nanodisc vaccine and a Listeria-based live vector vaccine, a representative cancer vaccine in the late stage of clinical development.[6] While both vaccine platforms combined with ICB achieved comparable levels of T cell responses and tumor regression rates, nanodisc s.c. vaccination offers a convenient off-the-shelf product and a safer alternative to intravenous vaccination with live attenuated Listeria vaccines. Lastly, HLA-A02 transgenic mice immunized with nanodiscs elicited strong T cell responses against HLA-A02-restricted antigens, including a neoantigen from a melanoma patient and M2 flu antigen, thus demonstrating the versatility of the nanodisc platform for a wide range of peptide antigens.
2. RESULTS AND DISCUSSION
2.1. Subcutaneous nanodisc vaccination induces strong E7-specific CD8+ T cell responses.
Recruitment of CD8+ T cells into the TME is critical for successful cancer immunotherapy, especially for tumors located in mucosal tissues characterized by a low frequency of T cells.[28,31] Previously, i.n. vaccination has been shown to promote T cell infiltration in mucosal tumors, such as lung tumors and head & neck tumors by targeting lung-associated mediastinal LNs; however, it remains unclear whether i.n. vaccination is effective against distal mucosal tumors, such as intravaginal tumors.[28,30–33] Here, we set out to examine whether potent systemic T cell responses elicited by parenteral vaccination with a potent vaccine platform can lead to T cell infiltration into local as well as disseminated mucosal tumors (Figure 1).
Throughout our studies, we synthesized nanodiscs as described previously[5,19,20] and observed efficient loading of peptide antigens and cholesterol-CpG (Table S1, Supporting Information). We compared nanodisc vaccination given via the s.c. or i.n. route of administration. C57BL/6 mice were vaccinated either at the s.c. tail base area or both nostrils on days 0 and 14 with nanodiscs containing 20 μg E7 peptide and 10 μg CpG. The control groups included the same doses of E7 peptide and CpG formulated in a soluble form or emulsified in Montanide. On day 21, we examined the frequency of antigen-specific CD8+ T cells among PBMCs with the tetramer staining assay. Nanodisc vaccination induced ~32% E7-specific CD8+ T cells among PBMCs, representing a 29-fold increase compared with the soluble vaccine or a 15-fold increase compared with the Montanide control (Figure 2A–B). Interestingly, nanodiscs administered via the i.n. route induced only ~3.8 % E7-specific CD8+ T cells among PBMCs (Figure 2A–B). These results indicated that nanodisc vaccination administered via the s.c. route elicited more potent E7-specific CD8+ T cell responses in the systemic compartment, compared with conventional soluble peptide vaccines or intranasal nanodiscs vaccination.
Figure 2. Subcutaneous nanodisc vaccination induced effective cancer antigen-specific T cell response via efficient lymph node draining.

A-B) C57BL/6 mice were vaccinated on days 0 and 14 with 20 μg E7 peptide and 10 μg CpG in the indicated formulations. Vaccines were given via either the subcutaneous (s.c.) route at tail base or intranasal (i.n.) route. On day 21, the frequency of E7-specific CD8+ T cells among PBMCs was measured by the tetramer staining assay. Shown are A) the representative flow cytometry scatter plots and B) the average values. C) Serial PET images of C57/BL mice at various time points post-injection of 64Cu-NOTA-E7 or 64Cu-NOTA-nanodisc-E7. D-E) Time−radioactivity curves of Injection site, axillary LNs, inguinal LNs, intestine, liver, blood, and muscle after s.c. injection. F) Biodistribution of 64Cu-NOTA-E7 and 64Cu-NOTA-nanodisc-E7 at 46 h post-injection. Data are presented as mean ± s.e.m. from a representative experiment from 2 independent experiments (n = 4–5). *p < 0.05, **p < 0.01, ****p < 0.0001 analyzed by (B) one-way ANOVA or (F) two-way ANOVA, with Tukey’s HSD multiple comparison post hoc test.
We examined the biodistribution profiles of nanodiscs with positron emission tomography (PET) imaging. Nanodisc vaccination given via the s.c. route resulted in a significant amount of 64Cu-tagged E7 antigen accumulating in multiple draining LNs (dLNs) even within 1 h of injection (Figure 2C,E). After 46 h, we detected ~20% injection dose per gram of tissue in proximal inguinal LNs as well as in distal axillary LNs (Figure 2E). On the other hand, free E7 peptide administered s.c. resulted in rapid systemic dissemination of antigen with minimal signal in dLNs (~4% and ~11% ID/g for axillary and inguinal dLNs, respectively) (Figure 2C,D). To validate the results, we isolated various tissues at 46 h and quantified radioactivity of 64Cu with gamma counter. Ex vivo measurement indicated that s.c. nanodisc vaccination increased delivery of E7 antigen to axillary and inguinal LNs by 12-fold and 2.3-fold, respectively, compared with free soluble vaccination (Figure 2F). In contrast, i.n. vaccination resulted in the accumulation of nanodiscs in the lungs, cervical LNs, and GI tract (data not shown).
2.2. Therapeutic vaccination against lung metastasis
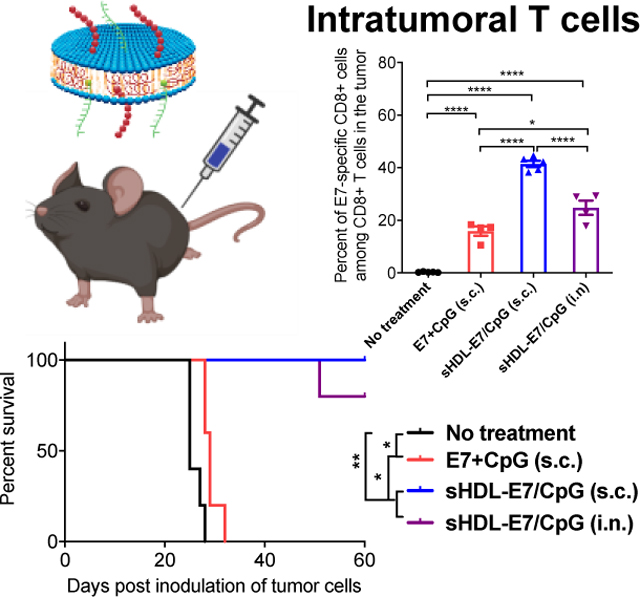
Next, we examined whether strong systemic T cell responses induced by s.c. nanodisc vaccination can inhibit tumor growth in mucosal tissues. We first evaluated the therapeutic effect of nanodiscs in a lung metastasis model. C57BL/6 mice were administered intravenously with TC-1 tumor cells expressing HPV16 E6/E7. Non-treated animals died within 25 days of tumor inoculation due to tumor burden and difficulty in breathing (Figure 3A–B). Subcutaneous vaccination with a soluble mixture of 20 μg E7 peptide and 10 μg CpG had only a moderate effect, with all animals succumbing to the tumor burden within 30 days. In stark contrast, s.c. vaccination with nanodiscs carrying the same dose of E7 peptide and CpG (sHDL-E7/CpG) eliminated lung metastases within 2 weeks after treatment without any sign of tumor for 60 days (Figure 3A–B). Nanodisc vaccination via the i.n. route also potently inhibited lung metastasis and prolonged the animal survival (Figure 3A–B). T cell responses examined on day 3 after the second vaccination revealed that s.c. nanodisc vaccination induced ~18% circulating E7-specific CD8+ T cells, representing 5.7-fold stronger response than s.c. vaccination of soluble vaccines (P < 0.0001, Figure 3C–D). Robust CD8+ T cell response in circulation correlated with the high frequency of intratumoral T cells, with nanodisc-immunized animals harboring ~2.6-fold higher frequency of E7-specific CD8+ T cells within the TME, compared with the soluble vaccine group (P < 0.0001, Figure 3E–F). Notably, mice immunized with nanodisc via the i.n. route generated weak E7-specific CD8+ T cell responses in the systemic compartment, but they had a higher frequency of E7-specific CD8+ T cells in the lung tissues, compared with s.c. soluble vaccination (Figure 3C–F).
Figure 3. Nanodisc vaccination in the TC-1 lung metastasis model.

A-F) To establish a lung metastasis model, C57BL/6 mice were inoculated intravenously with 1×105 TC1-luc cells on day 0. On days 10 and 16, animals were vaccinated with 20 μg E7 peptide and 10 μg CpG formulated as a soluble vaccine or sHDL vaccine. Vaccines were given via either s.c. at the tail base or intranasal (i.n.) route. A) Tumor burden was monitored over time using in vivo whole animal imaging (IVIS). B) Animal survival was measured over 60 days. C-F) Three days after the second vaccination, the frequency of E7-specific CD8a+ T cells was measured among C-D) PBMCs or E-F) lung tissues by the tetramer staining assay. Shown are C, E) the representative flow cytometry scatter plots and D, F) the average values of E7-tetramer+ CD8⍺+ T-cells. Data are presented as mean ± s.e.m. from a representative experiment from 2 independent experiments (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 analyzed by (D, F) one-way ANOVA with Tukey’s HSD multiple comparison post hoc test or by (B) log-rank (Mantel−Cox) test.
2.3. Therapeutic vaccination against inner lip tumors.
We evaluated the therapeutic effect of nanodisc vaccination against sublingual inner lip tumor - a widely used orthotopic model for HPV-associated head and neck cancer.[22,28,29] We established the model by inoculating TC-1 tumor cells directly in the inner lip of mice and initiated vaccination on day 6. Non-treated animals died within 20 days of tumor inoculation. Whereas s.c. vaccination with a soluble mixture of 20 μg E7 peptide and 10 μg CpG led to ~40% of animals eliminating tumor cells, we observed 100% tumor eradication in animals vaccinated s.c. with nanodiscs (Figure 4A–B). In contrast, i.n. nanodisc vaccination produced a moderate response with ~60% survival rate. Mice bearing TC-1 inner lip tumors generated ~22% circulating E7-specific CD8+ T cells after s.c. nanodisc vaccination, representing a 13-fold improvement over the soluble vaccine given via the same route (P < 0.0001, Figure 4C–D). Strong systemic T cell responses correlated with robust CD8+ T-cell infiltration into inner lip tumors, with the s.c. nanodisc group having ~3.6-fold higher frequency of E7-specific CD8+ T cells in the TME, compared with the s.c. soluble group (P < 0.0001, Figure 4E–F). In contrast, i.n. vaccination with nanodiscs induced significantly lower frequency of E7-specific CD8+ T cells in circulation as well as within the TME, comparable to the s.c. soluble vaccination (Figure 4C–F).
Figure 4. The therapeutic effect of nanodisc vaccination in TC-1 head and neck cancer model.

To establish a head and neck model, C57BL/6 mice were inoculated with 50,000 TC1-luc cells in the inner lip on day 0. On days 6 and 12, animals were vaccinated with 20 μg E7 peptide and 10 μg CpG formulated as a soluble vaccine or sHDL vaccine. The route of vaccination was either s.c. at the tail base or intranasal (i.n.) vaccination as indicated. A) Tumor burden was monitored over time using in vivo whole animal imaging (IVIS). B) Animal survival was measured over 60 days. C-F) Three days after the second vaccination, the frequency of E7-specific CD8⍺+ T cells was measured among C-D) PBMCs or E-F) tumor tissues by the tetramer assay. Shown are C, E) the representative flow cytometry scatter plots and D, F) the average values of E7-tetramer+ CD8⍺+ T-cells. Data are presented as mean ± s.e.m. from a representative experiment from 2 independent experiments (n = 5). *p < 0.05, ****p < 0.0001 analyzed by (D, F) one-way ANOVA with Tukey’s HSD multiple comparison post hoc test or by (B) log-rank (Mantel−Cox) test.
2.4. Therapeutic vaccination against intravaginal tumors.
We also assessed nanodisc vaccination against intravaginal TC-1 tumors, an aggressive model with clinical features of HPV+ cervical cancer.[27] C57BL/6 mice were inoculated with TC-1 cells into the vagina after diestrus synchronization as previously reported[27,30,34] and vaccinated starting day 6 post tumor inoculation. Non-treated animals died within 20 days, while s.c. vaccination with a soluble mixture of E7 peptide and CpG had a modest anti-tumor effect (Figure 5A–B). In stark contrast, s.c. vaccination with nanodiscs significantly extended the animal survival, with ~40% of animals eliminating tumors (Figure 5A–B). Notably, unlike in the case of lung and inner lip tumor models (Figure 3A–B, 4A–B), i.n. nanodisc vaccination had a minimal impact on the animal survival (Figure 5A–B). T cell analysis revealed that s.c. nanodisc vaccination in mice bearing intravaginal TC-1 tumors elicited ~23% circulating E7-specific CD8+ T cells, representing 11-fold and 13-fold improvement over s.c. soluble vaccination and i.n. nanodisc vaccination, respectively (P < 0.0001, Figure 5C–D). Similarly, the s.c. nanodisc vaccine group had 2.3-fold and 2-fold higher frequency of E7-specific CD8+ T cells in the intravaginal TME, compared with s.c. soluble vaccination or i.n. nanodisc vaccination, respectively (P < 0.001, Figure 5E–F). Overall, these results suggest that s.c. nanodisc vaccination elicits robust E7-specific CD8+ T cell responses in circulation, leading to efficient T cell infiltration into peripheral mucosal tumors, whereas i.n. nanodisc vaccination was only effective against tumors proximal to the site of vaccination (e.g. lungs and sublingual tissues).
Figure 5. The therapeutic effect of nanodisc vaccination in TC-1 cervical cancer model.

To establish an HPV-associated cervical cancer model, C57BL/6 mice were inoculated with 4×104 TC1-luc cells in the vaginal tract on day 0. On days 6 and 12, animals were vaccinated with 20 μg E7 peptide and 10 μg CpG formulated as a soluble vaccine or sHDL vaccine. The route of vaccination was either s.c. at the tail base or intranasal (i.n.) vaccination as indicated. A) Tumor burden was monitored over time using in vivo whole animal imaging (IVIS). B) Animal survival was measured over 60 days. C-F) Three days after the second vaccination, the frequency of E7-specific CD8a+ T cells was measured among C-D) PBMCs or E-F) tumor tissues by the tetramer staining assay. Shown are C, E) the representative flow cytometry scatter plots and D, F) the average values of E7-tetramer+ CD8a+ T-cells. Data are presented as mean ± s.e.m. from a representative experiment from 2 independent experiments (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 analyzed by (D, F) one-way ANOVA with Tukey’s HSD multiple comparison post hoc test or by (B) log-rank (Mantel−Cox) test.
2.5. Benchmarking nanodiscs to Listeria-based vaccine.
Given the strong therapeutic efficacy of nanodiscs, we sought to directly compare nanodiscs to a leading T cell vaccine technology, namely live attenuated Listeria vaccine, which has reached but ultimately failed in phase III trials.[6,10] First, we examined antigen-specific T-cell responses in non-tumor bearing mice that received either vaccine carrying a model antigen, Gp33 peptide, which is an immunodominant epitope derived from lymphocytic choriomeningitis virus.[35] C57BL/6 mice were vaccinated s.c. at tail base on days 0 and 30 with nanodiscs carrying Gp33 and CpG. In parallel, mice were vaccinated on days 0 and 30 with Listeria vector encoding Gp33 administered via the i.v. route - the traditional route employed in phase III trials.[6,10] On day 7 after priming vaccination, nanodiscs generated stronger Gp33-specific, polyfunctional IFN-γ+TNF-⍺+ CD8+ T cell responses than Listeria vaccination (Figure 6A). After the boost vaccination, both vaccine groups achieved similar levels of Gp33-specific CD8+ T cell responses. Furthermore, we also compared nanodiscs to Listeria vaccine using a neoantigen, Adpgk, derived from MC-38 colon carcinoma model.[36] For this study, mice were also given anti-PD-1 IgG or isotype IgG via intraperitoneal (i.p.) administration on day 4 after each vaccination. While the prime vaccination resulted in similar levels of neoantigen-specific CD8+ T cell responses between the nanodisc and Listeria groups, boost vaccination with nanodiscs further improved Adpgk-specific CD8+ T cell responses, compared with Listeria vaccination, regardless of anti-PD-1 IgG co-treatment (Figure 6B).
Figure 6. CD8+ T cell responses induced by nanodiscs and Listeria vaccine.

A) C57BL/6 mice were vaccinated on days 0 and 30 with 107/100 μl/mouse Listeria-Gp33 (i.v. route) or sHDL nanodiscs carrying Gp33 and CpG (s.c. route). On days 7 and 35, splenocytes were re-stimulated with Gp33 for intracellular cytokine staining (ICS) for IFN-γ and TNF-⍺. B) C57BL/6 mice were vaccinated on days 0 and 30 with 107/100 μl/mouse Listeria-Adpgk (i.v. route) or sHDL nanodiscs carrying Adpgk and CpG (s.c. route). Anti-PD-1 IgG was given i.p. on days 4 and 34. On days 7 and 35, splenocytes were re-stimulated with Adpgk for ICS. Data are presented as mean ± s.e.m. from a representative experiment from 2 independent experiments (n = 7). *p < 0.05, **p < 0.01, analyzed by one-way ANOVA with Tukey’s HSD multiple comparison post hoc test.
To examine whether the strong systemic antigen-specific T cell response could induce better T cell homing into tumor and eradicate the tumor, we tested the therapeutic effect of nanodiscs and Listeria vaccine in a subcutaneous MC38 tumor model. As shown in Figure 7A, C57BL/6 mice were inoculated on day 0 with 5 × 105 MC-38 cells at s.c. flank. On day 10 and 17, MC-38 tumor-bearing mice were immunized with nanodiscs delivering 20 μg Adpgk peptide and 15 μg CpG or 1×107 CFU Listeria- Adpgk-multiepitope. Nanodisc vaccines were administered at s.c. tail base as above, whereas Listeria vaccines were given via i.v. route. Anti-PD-1 IgG or isotype IgG was given i.p. on days 11, 14, 18, and 21. Nanodisc vaccination alone did not lead to tumor regression, whereas Listeria vaccine alone eliminated tumors in 4 out of 10 animals (Figure 7B, Figure S1, Supporting Information). In contrast, when combined with anti-PD-1 IgG, nanodisc vaccines eradicated tumors in 4 out of 10 animals, which was statistically comparable to Listeria vaccine plus anti-PD-1 IgG therapy (Figure 7B). We also evaluated antigen-specific T cell responses in circulation as well as in the TME. Listeria vaccine alone group exhibited an increased trend of Adpgk-tetramer+, IFN-γ+TNF-⍺+ polyfunctional CD8+ T cells, compared with nanodisc vaccine group (Figure 7C–D). However, nanodiscs co-treated with anti-PD-1 IgG amplified antigen-specific T cell responses in circulation and TME for the nanodisc group, reaching comparable levels as in the Listeria vaccine + anti-PD-1 IgG group (Figure 7C–D). Note that we administered live attenuated Listeria vaccine via i.v. route since s.c. vaccination of Listeria vectors has been reported to induce much weaker immune response.[37] Overall, these studies showed that s.c. nanodisc vaccination is a promising platform for cancer vaccination.
Figure 7. Comparison of therapeutic effect and T cell infiltration in TME after Nanodiscs and Listeria vaccine.

A) C57BL/6 mice were inoculated at s.c. flank with MC38 cancer cells on day 0. On days 10 and 17, animals were vaccinated with 107/100 μl/mouse Listeria-Adpgk intravenously (i.v.) or nanodisc-Adpgk/CpG (s.c.). Anti-PD-1 IgG was injected i.p. on days 11, 14, 18, and 21. For antitumor immune evaluation, the spleens were harvested on day 28 and processed for peptide stimulation and intracellular cytokine staining (ICS). B) Tumor growth was monitored. C) Adpgk-specific CD8a+ T cells were quantified by the tetramer staining among PBMCs, spleen, and tumor tissues. D) PBMCs, spleen, and tumor tissues were isolated and re-stimulated with Adpgk neoepitope, followed by intracellular cytokine staining. Data are presented as mean ± s.e.m. n = 10 for B) and n = 4–7 for C-D).
2.6. Nanodisc vaccination with human HLA-A02 epitopes.
Lastly, we examined the nanodisc platform for eliciting T cells against a human neo-antigen derived from a HLA-A02 melanoma patient.[38] For the control group, HLA-A02 transgenic mice were vaccinated with 10 μg/dose HLA-A02 neoantigen peptide and 2 μg tetanus toxoid emulsified in Complete Freund’s Adjuvant (CFA), which is a potent yet toxic adjuvant system.[39,40] Nevertheless, HLA-A02 transgenic mice that received prime-boost-boost immunizations with CFA-TT adjuvant system generated only a basal level of IFN-γ+ T cell response (Figure 8A–B). Strikingly, switching the last boost immunization with sHDL nanodiscs achieved >200-fold stronger neoantigen-specific IFN-γ+ T cell responses (P < 0.001, Figure 8A–B). We also employed an HLA-A02-restricted influenza peptide, GILGFVFTL (M158–66) as a positive control. After HLA-A02 transgenic mice were prime-boost vaccinated with nanodiscs carrying either HLA-A02-restricted melanoma neoantigen or flu antigen, we observed robust IFN-γ+ CD8+ T cell responses among PBMCs, as shown by intracellular cytokine staining (Figure 8C–E). Overall, these results suggest that the nanodisc platform is compatible with other vaccine technologies in heterologous immunization regimens and generates robust T cell responses to a wide range of antigens, including HLA-restricted antigens.
Figure 8.

A) HLA-A02 transgenic mice were vaccinated on days 0 and 14 with 10 μg/dose of neoantigen peptide from a HLA-A02 melanoma patient in Complete Freund's Adjuvant (CFA) containing 2 μg/dose of tetanus toxoid. On day 28, the animals were boosted with either the same CFA +TT formulation or nanodiscs containing 15 μg/dose of CpG. B) On day 35, antigen-specific T cell responses were evaluated by ELISPOT after restimulating splenocytes with 0.1, 1, or 10 μg/mL of the antigen peptide. C) HLA-A02 transgenic mice were vaccinated on days 0 and 21 with nanodiscs containing 15 μg/dose of CpG and either neoantigen peptide from an HLA-A02 melanoma patient (Mel-Ag) or HLA-A02-restricted flu antigen peptide, M158–66. D-E) On day 28, PBMCs were analyzed for antigen-specific, IFN-γ+ T-cell responses by intracellular cytokine staining after ex vivo restimulation with 10 μg/mL of each peptide. Data are presented as mean ± s.e.m. from a representative experiment from 2 independent experiments (n = 3). ****p < 0.0001 analyzed by (b) two-way ANOVA with Tukey’s HSD multiple comparison post hoc test.
3. CONCLUSION
In this work, we examined antigen-specific T cell responses generated by nanodisc vaccination and compared the strength of nanodiscs vaccine against other vaccine platforms. We chose HPV16 E7 antigen for our initial studies since HPV16 E7 is one of the most thoroughly studied antigens in the context of HPV-associated cancer.[4,10,14] Among various therapeutic HPV vaccines under development, TA-HPV and Lm-LLO-E7 targeting HPV16 E6/E7 are the leading vaccine candidates. TA-HPV was first evaluated clinically in the 1990s. Three out of eight patients with late-stage cervical cancer generated antigen-specific T cell responses against HPV, and two of them remained tumor-free after 15 and 21 months of vaccination.[9] The subsequent clinical studies showed that TA-HPV induced serological and T cell responses, alleviating HPV-associated lesions.[14,15] Lm-LLO-E7, which is based on Listeria vector expressing E7 antigen fused to a part of virulence factor, listeriolysin O, has been evaluated in phase I-III studies.[11] The first study, published in 2009, showed that i.v. administration of Lm-LLO-E7 induced E7-specific T cell responses, but 40% patients experienced grade 3 side effects.[6] To address the safety issues and regulatory challenges associated with live vectors, peptide-based subunit vaccines with HPV16 E6/E7 antigens have been studied extensively.[7,41,42] However, peptide-based subunit vaccines generally suffer from limited anti-tumor efficacy due to inefficient antigen delivery to lymphoid tissues and the lack of appropriate innate immune stimulation.[4,12] In our previous work, we have shown that the nanodisc vaccine technology administered s.c. can efficiently drain to LNs and generate potent antigen-specific T cell responses.[5,19,20,43] In this work, we have utilized the nanodisc platform for therapeutic vaccination targeted against HPV16 E7 and shown elicitation of robust E7-specific CD8+ T cells, leading to the elimination of TC-1 tumors inoculated in various mucosal tissues, including intravaginal TC-1 model known for low T cell infiltration and aggressive features of HPV+ cervical cancer.[27,30,34] Importantly, in our head-to-head comparison studies, nanodisc vaccination induced comparable levels of antigen-specific CD8+ T cell responses as Listeria vectors (Figure 6–7) but without any overt sign of toxicity or anti-vector immunity associated with live vector vaccines.
Efficient infiltration of T cells in solid cancer is a major challenge for therapeutic vaccines, especially for HPV-associated tumors in mucosal sites.[44–46] Previous studies have reported that i.n. vaccination targets DCs in proximal draining LNs and promotes T cell infiltration in mucosal tumors, including lungs and head and neck cancer.[28,31] However, it remains unclear whether i.n. vaccination can also promote T cell infiltration into distant mucosal tumors, such as in the reproductive tract, characterized by a low frequency of T cells and unresponsiveness to conventional therapies.[27,32] Here, we have demonstrated that s.c. vaccination with nanodiscs induced up to ~32% E7-specific CD8+ T cells in circulation, leading to efficient intratumoral infiltration of T cells against mucosal tumors in the lungs, inner lip, and intravaginal tissues. In contrast, i.n. nanodisc vaccination failed to induce T cell infiltration into distal mucosal sites (i.e., intravaginal tissues) (Figure 5), whereas we observed modest T cell infiltration into mucosal tissues proximal to the site of immunization (e.g., the lungs and inner lip) (Figure 3–4). While the exact mechanism of action is beyond the scope of this paper, we speculate that s.c. nanodisc vaccination allows for efficient dissemination of nanodiscs from the injection site to multiple LNs, including inguinal and axillary LNs (Figure 2), leading to a high frequency of antigen-specific CD8+ T cells in the circulation and subsequent infiltration of CD8+ T cells into peripheral mucosal tumors that release cytokine/chemokine signals and/or antigens.
To demonstrate the broad applicability of nanodisc vaccine, we evaluated whether the nanodisc platform can elicit T cell responses against HLA-restricted antigens, including a neoantigen from HLA-A02 melanoma patient as well as a widely used influenza epitope M158–66. Interestingly, nanodisc immunization rescued low level of T cell responses observed in HLA-A02 transgenic mice after CFA plus tetanus toxoid vaccination and elicited significantly amplified antigen-specific T cell responses against HLA-A02-restricted neoantigen (Figure 8A–B). We have also demonstrated induction of robust CD8+ T cell responses against M158–66 epitope. These results show that nanodiscs are broadly applicable with a wide range of antigens, including neoantigens, shared tumor antigens, and viral antigens, and are compatible with other vaccine platforms in the context of heterologous vaccination. Overall, nanodiscs offer a versatile and promising vaccine platform for eliciting robust T cell immunity and may provide a new avenue for advancing combination cancer immunotherapy.[47]
4. MATERIALS & METHODS
Materials
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), and 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine-N-[3-(2-pyridyldithio)propionate] (DOPE-PDP) were purchased from Avanti Polar Lipids (Alabaster, AL). S-2-(4-Isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA) was purchased from Macrocyclics, Inc. (Dallas, TX). SEPPIC INC MONTANIDE (Catalog# NC0962946) was purchased from Fisher Scientific. E7 peptide (GQAEPDRAHYNIVTFCCKCD), HLA-A02-restricted flu antigen peptide (CSSGILGFVFTL) and HLA-A02-restricted melanoma patient peptide (CSS-GIPENSFNV) were synthesized by AnaSpec (Fremont, CA). Gp33 peptide (CSSKAVYNFATM) and Adpgk peptide (CSSASMTNMELM) were respectively synthesized by Genemed Synthesis (San Antonio, TX) and RS Synthesis (Louisville, KY). The oligodeoxynucleotide TLR9 ligand CpG 1826 (5’-tccatgacgttcctgacgtt-3’, lower case letters represent phosphorothioate backbone), and CpG 1826 modified with cholesterol (Cho-CpG) were synthesized by Integrated DNA Technologies (Coralville, IA). Anti-mouse CD16/32 was from eBioscience (San Diego, CA). Anti-mouse CD8α-APC (Catalog# 553035) were from BD Biosciences (San Jose, CA). Anti-CD4-FITC (Catalog# 100406), anti-CD8-PerCPCy5.5 (Catalog# 100734), anti-IFNγ-APC (Catalog# 505810), anti-TNF α -PE-Cy7 (Catalog#506324), anti-IL-2-BV421 (Catalog#503825), and anti-CD40L-PE (Catalog# 106506) were from BioLegend. Tetramer H-2Db-RAHYNIVTF-BV421 was kindly provided by the NIH Tetramer Core Facility (Atlanta, GA).
Cell culture
TC-1 cells expressing luciferase (TC-1-luc) were kindly provided by Dr. T.C. Wu from Johns Hopkins University (Baltimore, MD). The cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, 2 mM glutamine, 1 mM sodium pyruvate, non-essential amino acids, and 400 μg/mL G418. MC38 cells were cultured in Dulbecco's Modified Eagle Media (DMEM) supplemented with 10% v/v heat-inactivated FCS at Bristol Myers Squibb.
Preparation of vaccine nanodiscs
Vaccine nanodiscs were prepared following the previous reports [5]. Briefly, DMPC and 22A in the weight ratio of 2:1 were dissolved in acetic acid, followed by lyophilization and hydration with PBS to form nanodiscs. Each antigen peptide was reacted with DOPE-PDP at a 1.5:1 molar ratio, and the resulting lipid-peptide conjugates were dissolved in DMSO and incubated with pre-formed nanodiscs at room temperature for 30 min. Unreacted antigen peptides were removed by ultrafiltration. Cholesterol modified CpG was incubated with nanodiscs for 30 min. Table S1 Supporting Information shows the conjugation efficiency of tumor antigen peptides as determined by LC-MS, and the loading efficiency of CpG as measured by gel permeation chromatography.
Tumor models and therapy
Mice were cared for following the federal, state, and local guidelines. All work performed on animals was in accordance with and approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan, Ann Arbor, and Bristol Myers Squibb. Female C57BL/6 (5 – 6 weeks) were purchased from Envigo or Jackson Laboratory (USA). For the lung metastasis model, C57BL/6 mice were intravenously injected with 1×105 TC-1-luc cells on day 0. For the inner lip tumors [29], C57BL/6 mice were injected with 50,000 TC-1-luc cells in the inner lip on day 0. For the HPV-associated cervical cancer model [27,30,34], female mice received s.c. injection of medroxyprogesterone (3 mg/mouse) for diestrus synchronization, and after 4 days, the animals were inoculated with 40,000 TC-1-luc cells by intravaginal administration. For each model, animals were vaccinated on indicated days with 20 μg E7 and 10 μg CpG through tail base s.c. vaccination or intranasal vaccination. Bioluminescence from tumor cells was visualized using IVIS after intraperitoneal (i.p.) injection of luciferin.
For comparison of vaccine nanodiscs and Listeria vector vaccine, C57BL/6 mice were injected on days 0 and 30 with nanodiscs at s.c. tail base or 107/100 μl/mouse Listeria-Gp33/Adpgk intravenously (i.v.). Listeria was cultured in sterile Brain Heart Infusion Broth, Modified (Teknova Inc., Hollister, CA) overnight to achieve stationary phase culture of 109 CFU/mL which was further diluted with Hank’s Balanced Salt solution (HBSS) to make 108 CFU/mL for vaccination. On Day 7 post priming and day 5 post boost, spleen was harvested and processed for antigen stimulation and intracellular cytokine staining (ICS). For the therapeutic studies in the MC38 model, C57BL/6 mice were subcutaneously injected with 0.5 million MC38 cells on day 0. On days 10 and 17, animals were vaccinated with nanodiscs at s.c. tail base or i.v. with 107/100 μl/mouse Listeria vaccine. A subset of animals received 100 μg anti-PD-1 i.p. on days 11, 14, 18, and 21.
For evaluation of immune responses of HLA-A02-restricted peptides, HLA-A02 transgenic mice (Jackson Laboratory, Bar Harbor, ME) were immunized on indicated days with vaccine formulations. Mice were vaccinated with 10 μg/dose of neoantigen peptide (SILMHGLVSL) from a HLA-A02 melanoma patient in the form of either CFA containing 2 μg/dose of tetanus toxoid or nanodiscs containing 15 μg/dose of CpG. For a positive control group, HLA-A02 mice were immunized with nanodiscs delivering 10 μg/dose of A02-restricted influenza peptide, M158–66 GILGFVFTL and 15 μg/dose of CpG.
Copper-64 labeling of nanodiscs and PET imaging
Copper-64 (64Cu) was produced with an onsite cyclotron (GE PETtrace). 64CuCl2 (74 MBq) was diluted in 0.3 mL of 0.1 M sodium acetate buffer (pH 5.0) and mixed with 0.5 mg of nanodisc. The reaction was conducted at 37 °C for 30 min with constant shaking. Then 5 μL 0.1 M EDTA (ethylenediaminetetraacetic acid) was added into the solution and shaken for 5 min to remove non-specifically bound 64Cu. The resulting 64Cu-NOTA–nanodisc was purified by PD-10 size exclusion column chromatography using PBS. The radioactive fractions were collected for further in vivo studies. C57BL/6 mice were administered with 5–8 MBq of 64Cu-NOTA–nanodisc via s.c. or intranasal route, and PET imaging was performed over time using a microPET/microCT Inveon rodent model scanner (Siemens Medical Solutions USA, Inc.). Quantitative PET data for the major organs were presented as the percentage injected dose per gram of tissue (%ID g−1). To validate these results, blood and major organs/tissues were collected and weighed at 24 h post-injection, and the samples were measured for radioactivity using a gamma counter (PerkinElmer).
Examination of T cell responses
The frequency of tumor antigen-specific CD8α+ T cells was analyzed using the tetramer staining assay as described previously [5]. Blood was collected from each mouse by submandibular bleeding, and red blood cells were lysed using Ammonium-Chloride-Potassium (ACK) lysis buffer. Tumor tissues harvested on indicated time points were cut into small pieces of 2 to 4 mm, and cells were dissociated in digestion buffer [collagenase type IV (1 mg/ml) and deoxyribonuclease I (100 U/ml) in serum-free RPMI] for 30 min at 37°C with gentle shaking. Cell suspension was passed through a 70-μm nylon strainer and washed with FACS buffer (1% BSA in PBS). Cells were then incubated with CD16/32 for 10 min, incubated with peptide-MHC tetramer (H-2Db-RAHYNIVTF-BV421) for 30 min at room temperature, and stained with antibodies against CD8a (53–6.7) on ice for 20 min. Cells were washed twice with FACS buffer and resuspended in 7AAD solution (0.5 μg/mL) for analysis by flow cytometry (Cyan 5, Beckman Coulter).
For intracellular cytokine staining (ICS) assay, 100–150 μL peripheral blood collected from vaccinated mice was lysed with ACK lysis buffer, washed with PBS, and plated at ~10 million cells/mL in 50 μL T cell media (RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 55 μM β-mercaptoethanol, 1 mM pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin, HEPES, and non-essential amino acids) in 96-well U bottom plates. Cells were pulsed with 10 μg/mL antigen peptides for 6 h, with brefeldin A (BD Biosciences) added during the last 4 h of incubation. Cells were then washed twice with ice-cold FACS buffer, followed by incubation with anti-CD16/32 for 10 minutes and anti-CD8α for 20 min on ice. Cells were then fix/permeabilized for 20 min on ice and then stained with anti-IFN-γ or anti-TNF α for 30 min on ice. After extensive washing, cells were analyzed by flow cytometry.
For ELISPOT assays, spleens from immunized mice were harvested, processed into single cell suspensions for each mouse, and seeded in 96-well PVDF plates (EMD Millipore) pre-incubated overnight with IFN-γ coating Ab (R&D Systems). Splenocytes were co-incubated with antigen peptides (2 μg/ml) or controls for 24 h. Assays were completed using sequential incubations with biotinylated-secondary Ab, streptavidin-alkaline phosphatase (Sigma Chemical), and NBT/BCIP substrate (Surmodics). Spots developed were analyzed using an AID iSpot Reader (Autoimmun Diagnostika GmbH, Germany).
Statistical analysis
For animal studies, mice were randomized to match the similar average tumor burden before the initiation of any treatments. All procedures were performed in a non-blinded fashion. Statistical analysis was performed with Prism 6.0 software (GraphPad Software) by one-way or two ANOVA with Tukey’s HSD multiple comparison post hoc test. Statistical significance for the survival curve was calculated by the Mantel-Cox log-rank test. Statistical significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data were approximately normally distributed, and variance was similar between the groups. Data are presented as mean ± s.e.m. and sample sizes are reported in each figure legend.
Supplementary Material
Acknowledgements
This work was supported in part by NIH (R01EB022563, J.J.M.; R01CA210273, J.J.M.; R21NS091555, A.S.; R01HL134569, A.S.; R01CA155010, C.J.W.; R21CA216772, D.B.K.; NCI-SPORE-2P50CA101942, D.B.K.), MTRAC for Life Sciences Hub, UM Forbes Institute for Cancer Discovery Pilot Grant, and Emerald Foundation. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369), and NSF CAREER Award (1553831). R.K. is supported by the Broomfield International Student Fellowship and the AHA Predoctoral Fellowship (15PRE25090050). W.Y. is supported by AHA Postdoctoral Fellowship (16POST27760002). L.S. was supported in part by T32 GM07767. C.J.W. is a scholar of the Leukemia and Lymphoma Society, and is supported in part by the Parker Institute for Cancer Immunotherapy. Opinions interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for the provision of MHC-I tetramers.
Disclosure of Potential Conflicts of Interest
A patent application for nanodisc vaccines has been filed, with J.J.M., A.S., and R.K. as inventors. J.J.M. and A.S. are co-founders of EVOQ Therapeutics, LLC. that develops the nanodisc technology for vaccine applications. P.B.S. and R.J. are employees of Bristol Myers Squibb. C.J.W. is a co-founder, equity holder, and SAB member of Neon Therapeutics, Inc. D.B.K. has previously advised Neon Therapeutics, and has received consulting fees from Gerson Lehrman Group, Guidepoint, Neon Therapeutics, System analytic Ltd and The Science Advisory Board. D.B.K. owns equity in Aduro Biotech, Agenus Inc., Armata Pharmaceuticals, Biomarin Pharmaceutical Inc., Breakbio Corp., Bristol Myers Squibb Co., Celldex Therapeutics Inc., Editas Medicine Inc., Exelixis Inc., Gilead Sciences Inc., IMV Inc., Lexicon Pharmaceuticals Inc., and Stemline Therapeutics Inc.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library
Contributor Information
Dr. Rui Kuai, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA.; Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Priti B. Singh, Bristol Myers Squibb, Redwood City, CA, USA..
Xiaoqi Sun, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA.; Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Dr. Cheng Xu, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA. Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Alireza Hassani Najafabadi, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA.; Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Dr. Lindsay Scheetz, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA. Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Dr. Wenmin Yuan, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA. Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Yao Xu, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, USA.; Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
Hao Hong, State Key Laboratory of Pharmaceutical Biotechnology, Medical School of Nanjing University, Nanjing University, Nanjing, 210093, China.
Derin B. Keskin, Broad Institute of MIT and Harvard, Cambridge, MA, USA. Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts, USA. The Translational Immunogenomics Lab (TIGL), Dana-Farber Cancer Institute, Boston, Massachusetts, USA.
Catherine J. Wu, Broad Institute of MIT and Harvard, Cambridge, MA, USA. The Translational Immunogenomics Lab (TIGL), Dana-Farber Cancer Institute, Boston, Massachusetts, USA. Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA
Renu Jain, Bristol Myers Squibb, Redwood City, CA, USA..
Anna Schwendeman, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, 48109, USA.; Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
James J. Moon, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, MI, 48109, USA.; Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI, USA. Biointerfaces Institute, University of Michigan, Ann Arbor, MI, USA.
References
- [1].Sahin U, Tureci O, Science. 2018, 359, 1355. [DOI] [PubMed] [Google Scholar]
- [2].Hu Z, Ott PA, Wu CJ, Nat Rev Immunol. 2018, 18, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, Gilboa E, Hammond SA, Hershberg R, Korman AJ, Kvistborg P, Melief C, Mellman I, Palucka AK, Redchenko I, Robins H, Sallusto F, Schenkelberg T, Schoenberger S, Sosman J, Tureci O, Van den Eynde B, Koff W, Coukos G, Sci Transl Med. 2016, 8, 334ps9. [DOI] [PubMed] [Google Scholar]
- [4].Yang A, Farmer E, Wu TC, Hung CF, J Biomed Sci. 2016, 23, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ, Nat Mater. 2017, 16, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maciag PC, Radulovic S, Rothman J, Vaccine. 2009, 27, 3975. [DOI] [PubMed] [Google Scholar]
- [7].de Vos van Steenwijk PJ, Ramwadhdoebe TH, Lowik MJ, van der Minne CE, Berends-van der Meer DM, Fathers LM, Valentijn AR, Oostendorp J, Fleuren GJ, Hellebrekers BW, Welters MJ, van Poelgeest MI, Melief CJ, Kenter GG, van der Burg SH, Cancer Immunol Immunother. 2012, 61, 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Santin AD, Bellone S, Palmieri M, Zanolini A, Ravaggi A, Siegel ER, Roman JJ, Pecorelli S, Cannon MJ, J Virol. 2008, 82, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Borysiewicz LK, Fiander A, Nimako M, Man S, Wilkinson GW, Westmoreland D, Evans AS, Adams M, Stacey SN, Boursnell ME, Rutherford E, Hickling JK, Inglis SC, Lancet. 1996, 347, 1523. [DOI] [PubMed] [Google Scholar]
- [10].Sewell DA, Shahabi V, Gunn GR 3rd, Pan ZK, Dominiecki ME, Paterson Y, Cancer Res. 2004, 64, 8821. [DOI] [PubMed] [Google Scholar]
- [11].Chabeda A, Yanez RJR, Lamprecht R, Meyers AE, Rybicki EP, Hitzeroth II, Papillomavirus Res. 2018, 5, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ, Nature. 2014, 507, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nascimento IP, Leite LC, Braz J Med Biol Res. 2012, 45, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Baldwin PJ, van der Burg SH, Boswell CM, Offringa R, Hickling JK, Dobson J, Roberts JS, Latimer JA, Moseley RP, Coleman N, Stanley MA, Sterling JC, Clin Cancer Res. 2003, 9, 5205. [PubMed] [Google Scholar]
- [15].Kaufmann AM, Stern PL, Rankin EM, Sommer H, Nuessler V, Schneider A, Adams M, Onon TS, Bauknecht T, Wagner U, Kroon K, Hickling J, Boswell CM, Stacey SN, Kitchener HC, Gillard J, Wanders J, Roberts JS, Zwierzina H, Clin Cancer Res. 2002, 8, 3676. [PubMed] [Google Scholar]
- [16].Medina E, Guzman CA, Vaccine. 2001, 19, 1573. [DOI] [PubMed] [Google Scholar]
- [17].Ura T, Okuda K, Shimada M, Vaccines (Basel). 2014, 2, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A, ACS Nano. 2016, 10, 3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kuai R, Sun X, Yuan W, Xu Y, Schwendeman A, Moon JJ, Bioconjugate Chemistry. 2018, 29, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kuai R, Sun X, Yuan W, Ochyl LJ, Xu Y, Najafabadi AH, Scheetz L, Yu M-Z, Balwani I, Schwendeman A, Moon JJ, Journal of Controlled Release. 2018, 282, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hildesheim A, Herrero R, Wacholder S, Rodriguez AC, Solomon D, Bratti MC, Schiller JT, Gonzalez P, Dubin G, Porras C, Jimenez SE, Lowy DR, Costa Rican HPVVTG, JAMA. 2007, 298, 743. [DOI] [PubMed] [Google Scholar]
- [22].Mork J, Lie AK, Glattre E, Hallmans G, Jellum E, Koskela P, Moller B, Pukkala E, Schiller JT, Youngman L, Lehtinen M, Dillner J, N Engl J Med. 2001, 344, 1125. [DOI] [PubMed] [Google Scholar]
- [23].Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT, Proc Natl Acad Sci U S A. 1992, 89, 12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Trimble CL, Frazer IH, Lancet Oncol. 2009, 10, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Melief CJ, van der Burg SH, Nat Rev Cancer. 2008, 8, 351. [DOI] [PubMed] [Google Scholar]
- [26].Peng S, Wang JW, Karanam B, Wang C, Huh WK, Alvarez RD, Pai SI, Hung CF, Wu TC, Roden RB, PLoS One. 2015, 10, e116389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Decrausaz L, Goncalves AR, Domingos-Pereira S, Pythoud C, Stehle JC, Schiller J, Jichlinski P, Nardelli-Haefliger D, Int J Cancer. 2011, 128, 2105. [DOI] [PubMed] [Google Scholar]
- [28].Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, Bouguin C, Merillon N, Dransart E, Tran T, Quintin-Colonna F, Autret G, Thiebaud M, Suleman M, Riffault S, Wu TC, Launay O, Danel C, Taieb J, Richardson J, Zitvogel L, Fridman WH, Johannes L, Tartour E, Sci Transl Med. 2013, 5, 172ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mondini M, Nizard M, Tran T, Mauge L, Loi M, Clemenson C, Dugue D, Maroun P, Louvet E, Adam J, Badoual C, Helley D, Dransart E, Johannes L, Vozenin MC, Perfettini JL, Tartour E, Deutsch E, Mol Cancer Ther. 2015, 14, 1336. [DOI] [PubMed] [Google Scholar]
- [30].Domingos-Pereira S, Decrausaz L, Derre L, Bobst M, Romero P, Schiller JT, Jichlinski P, Nardelli-Haefliger D, Mucosal Immunol. 2013, 6, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nardelli-Haefliger D, Dudda JC, Romero P, Sci Transl Med. 2013, 5, 172fs4. [DOI] [PubMed] [Google Scholar]
- [32].Decrausaz L, Domingos-Pereira S, Duc M, Bobst M, Romero P, Schiller JT, Jichlinski P, Nardelli-Haefliger D, Int J Cancer. 2011, 129, 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bialkowski L, van Weijnen A, Van der Jeught K, Renmans D, Daszkiewicz L, Heirman C, Stange G, Breckpot K, Aerts JL, Thielemans K, Sci Rep. 2016, 6, 22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].van Poelgeest MI, Welters MJ, Vermeij R, Stynenbosch LF, Loof NM, Berends-van der Meer DM, Lowik MJ, Hamming IL, van Esch EM, Hellebrekers BW, van Beurden M, Schreuder HW, Kagie MJ, Trimbos JB, Fathers LM, Daemen T, Hollema H, Valentijn AR, Oostendorp J, Oude Elberink JH, Fleuren GJ, Bosse T, Kenter GG, Stijnen T, Nijman HW, Melief CJ, van der Burg SH, Clin Cancer Res. 2016, 22, 2342. [DOI] [PubMed] [Google Scholar]
- [35].Liu Y, Xu L, Jiang Y, Sun J, He X, Cell Mol Immunol. 2007, 4, 431. [PubMed] [Google Scholar]
- [36].Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L, Nature. 2014, 515, 572. [DOI] [PubMed] [Google Scholar]
- [37].Qiu J, Yan L, Chen J, Chen CY, Shen L, Letvin NL, Haynes BF, Freitag N, Rong L, Frencher JT, Huang D, Wang X, Chen ZW, Clin Vaccine Immunol. 2011, 18, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, Nature. 2017, 547, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shapira M, Jolivet M, Arnon R, International journal of immunopharmacology. 1985, 7, 719. [DOI] [PubMed] [Google Scholar]
- [40].Fairweather NF, Lyness VA, Maskell DJ, Infect Immun. 1987, 55, 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].van Poelgeest MI, Welters MJ, van Esch EM, Stynenbosch LF, Kerpershoek G, van EL van Meerten Persijn, van den Hende M, Lowik MJ, Berends-van der Meer DM, Fathers LM, Valentijn AR, Oostendorp J, Fleuren GJ, Melief CJ, Kenter GG, van der Burg SH, J Transl Med. 2013, 11, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kruse S, Buchler M, Uhl P, Sauter M, Scherer P, Lan TCT, Zottnick S, Klevenz A, Yang R, Rosl F, Mier W, Riemer AB, Oncoimmunology. 2019, 8, e1524694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Scheetz L, Kadiyala P, Sun X, Son S, Najafabadi AH, Aikins M, Lowenstein PR, Schwendeman A, Castro MG, Moon JJ, Clin Cancer Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Riley RS, June CH, Langer R, Mitchell MJ, Nat Rev Drug Discov. 2019, 18, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stevanovic S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, Somerville RPT, Klebanoff CA, Kammula US, Sherry RM, Yang JC, Rosenberg SA, Hinrichs CS, Clin Cancer Res. 2019, 25, 1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Maskey N, Thapa N, Maharjan M, Shrestha G, Maharjan N, Cai H, Liu S, Cancer Manag Res. 2019, 11, 7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nam J, Son S, Park KS, Zou W, Shea LD, Moon JJ, Nature Reviews Materials. 2019, 4, 398. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.