

Abstract
Tissue repair responses in metazoans are highly coordinated by different cell types over space and time. However, comprehensive single-cell based characterization covering this coordination is lacking. Here, we captured transcriptional states of single cells over space and time during skin wound closure, revealing choreographed gene-expression profiles. We identified shared space-time patterns of cellular and gene program enrichment: which we call multicellular ‘movements’ spanning multiple cell types. We validated some of the discovered space-time movements using large volume imaging of cleared wounds and demonstrated the value of this analysis to predict ‘sender’ and ‘receiver’ gene programs in macrophages and fibroblasts. Finally, we tested the hypothesis that tumors are like ‘wounds that never heal’ finding conserved wound healing movements in mouse melanoma and colorectal tumor models, as well as human tumor samples revealing fundamental multicellular units of tissue biology for integrative studies.
Graphical Abstract
eTOC
Hu et al. spatially and temporally dissected cell-cell interactions in the mouse skin wound. Mechanisms of fibroblast-macrophage crosstalk were verified through in vitro experiments and imaging. These mechanisms could also be found in tumor samples from mouse models and patients, suggesting they are conserved units of tissue biology.
Introduction
Metazoans rely on intricate networks of cell-cell crosstalk (CCC) for maintenance of tissue homeostasis, repair, and regenerative processes after damage1–3. Given the diversity of cell types within a tissue, all possible ligand-receptor pairings and their signaling dynamics, a formalized method for interrogating CCC over space and time in the tissue remains a daunting task4. Even a minimal two-actor system can exhibit robustness and return to a stable state following perturbation5. This same adaptation to perturbation can be seen when increasing the number of cellular actors and, thus, the number of possible ‘edges’ (i.e. CCC axes), such as the combination of stellate cells, hepatocytes, endothelial cells, and Kupffer cells in liver niches6.
The advent of single-cell technologies allows profiling of cells on the transcriptional level at resolutions previously unattainable, generating rich datasets identifying highly-resolved cell subsets and subtle variations in gene expression and activation states7–9. Several computational approaches seek to infer CCC via paired ligand-receptor and target gene expression10–12,12. These inferences are strengthened by applying spatial and temporal context to single cell transcriptomics which has revealed gene groupings with similar spatiotemporal profiles, shedding light on the spatial segregation of cell functions, the dynamics of cell migration, and tissue zonation13,14. Similarly, describing gene expression in terms of spatiotemporal patterns revealed signaling pathways and co-regulation of genes in sub-compartments of liver and pancreas15,16,15. Concordantly, we were motivated to describe the healing skin wound in terms of spatiotemporal multicellular patterns and gene expression programs as skin wound healing naturally displays well-defined spatial and temporal dimensions16. This process has canonically been segmented into major phases with an initial inflammatory response followed by repair/growth and resolution17,18. Interspersed are coordinated changes in gene expression patterns in diverse cell types from monocyte/macrophages, neutrophils, fibroblasts, endothelial cells, keratinocytes and beyond3,19. Diverse crosstalk mechanisms between these cell types have been identified for regulating the duration of and transition between phases3,19–22. Disruption of these mechanisms often results in aberrant healing, demonstrating the interdependent structure of the wound healing cellular network23,24. Charting the progression of gene expression in single cells over space and time in the wound would yield information on the coordinated behaviors of myriad cell types in an unbiased manner, and how they drive transitions between healing phases.
With macrophages and fibroblasts representing cell types occupying a continuum of gene expression states25,26, as opposed to harboring discrete cell subtypes, clustering approaches are insufficient to capture transitions between states. For example, the M1/M2 ‘binning’ of macrophages may represent too reductive a model, as wound healing macrophages express combinations of canonical M1/M2 genes during all wound phases27. 26,27,29,28Therefore, a method for reframing cellular heterogeneity using overlays of gene programs (i.e. collections of co-expressed genes) in the healing wound may better capture the biology underlying the progression of cellular transcriptional heterogeneity.
An additional important rational for studying space-time progression of multicellular networks relates to chronic disease, where healthy resolution is not achieved. This is exemplified by cancer, where malignant tumor growth co-opts wound healing programs sans resolution, conceptualized in the idea that tumors are ‘wounds that never heal’28–30. This idea motivated us to develop a framework based on conserved gene programs to identify if crosstalk elements of the wound healing cellular network are ‘borrowed’ by tumors. The heterogeneity of a given cell type in different contexts may represent a convolution of conserved differentiation, functional, and tissue-specific expression patterns, as seen in resident immune cells scattered across all tissues31,32. Describing the common biology between two single cell datasets may again require going beyond clustering based approaches which may obscure the identification of overlaid gene programs in a continuum of cell states33.
Using skin wound healing as a well-defined spatial process in tissue repair, we mapped changes in, both, CD45+ and CD45− cell identity that co-occurred in similar space-time patterns. Layered on top of cell identity, we identified spatiotemporally expressed gene programs – or factors – that can be grouped based on their unique space-time profile. Because we found these factors based on their shared space-time patterns across multiple cell types, we refer to these shared factors across distinct cell types as multicellular ‘movements’. Informed by spatiotemporal profiles of gene program expression, we predicted stromal-macrophage CCC over the time-course of wound closure, which we then verified using orthogonal experimental approaches. Finally, we derived a framework for how to identify movements across tissue contexts and identify conservation of correlated immune and non-immune gene program pairs in mouse tumor models as well as human tumors. We then validate our predictions to demonstrate the utility of studying conserved gene program groupings.
Results
Separate waves of immune cell infiltration during wound closure
To establish the compositional changes of immune cells during skin repair, we immunophenotyped cells derived form 4 mm full thickness circular wound on the mouse back via CyTOF. (Figure S1A). This provided an overview of immune cell populations infiltrating the wound engaging in dynamic remodeling (Figures S1B–S1F). In the dimensional space of our CyTOF panel, the wound temporarily reaches the pre-wound composition at around day 7 to 10 post-wounding, and eventually assumes a state that is lymphocyte-rich at day 21 after wounding (Figure S1F and Mendeley Figure S1). For a deeper dive in space and time, we thus focused upon the first 14 days which encompassed the most dramatic changes.
Transcriptional space-time analysis of single cells unveils unique cell patterns during skin repair
Involvement of both immune and non-immune cells are crucial for skin repair34–36. To develop understanding of how gene expression programs in the different phases of skin repair are linked amongst cell types, we chose a spatial study across the radial dimensions of a wound. Thus, we radially sampled wounds using successively larger punches: wound center (2 mm center diameter), wound edge (2–4 mm diameter), wound proximal (4–6 mm diameter), and wound distal (6–8 mm diameter) (Figure 1A). Given the original wound diameter of 4 mm, we captured two distinct areas within the wound bed plus two distinct areas in skin beyond the wound and into uninjured tissue. These spatial subdomains were collected at timepoints suggested by the CyTOF study: early inflammatory (D1), and then at successively longer periods of the resolution as suggested by Figure S1E: transition (D3), return-to-unwounded (D7), and ‘late wound’ (D14) (Figure 1A). Upon digestion of the individual rings of tissue, CD45+ immune and CD45− non-immune cells were sorted separately, barcoded using MULTI-seq37 based on their collected timepoint and area, and then their transcriptional state assessed by using the 10x Genomics single-cell RNA sequencing (scRNAseq) platform (Figure 1A). This provided us with a space-time resolved scRNAseq dataset by which to query linked gene expression.
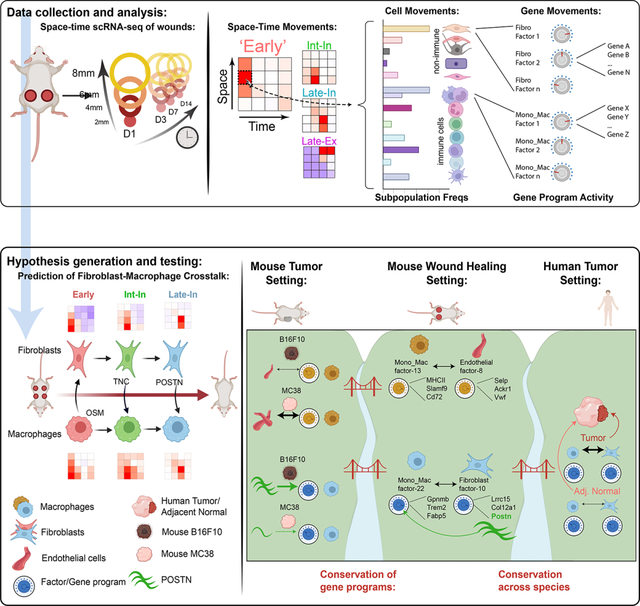
Figure 1. Transcriptional space-time analysis of single cells unveils unique patterns of monocyte/macrophage populations during skin repair.
(A) Experimental layout of transcriptional space-time analysis in wounded skin. Cells analyzed from each wound area and timepoint were pooled from 4 separate wounds. Image generated with Biorender.
(B) UMAP plot of monocyte/macrophage (Mono_Mac) subset from CD45+ object. Dotted line separates eight MHCIIlo and three MHCIIhi Mono_Mac clusters.
(C) UMAP projection of all Mono_Mac cells from (B) in gray and cells highlighted in red by timepoint of wound sampling.
(D) UMAP of MHCIIlo Mono_Mac subset with eight distinct clusters.
(E) Pseudotime trajectory using Monocle 3 on MHCIIlo Mono_Mac subset starting at cluster Mono_1, progressing through all cell clusters and ending at cluster Mono_Mac_6.
(F) Violin plot of MHCIIlo Mono_Mac subpopulations plotted according to their distribution in pseudotime.
(G) Violin plot of MHCIIlo Mono_Mac cells by day post-wounding and plotted according to their presence in pseudotime. D00 equals unwounded skin.
(H) Outline of Space-Time tile plot. The 4×4 grid depicts relative abundance of a cell cluster across radial wound area (y-axis) and time post wounding (x-axis). Each tile is one space-timepoint. Number in tile is the percentage of a subpopulation among all Mono_Mac cells at that specific space-timepoint. Background color indicates relative change compared to unwounded (UW) state: red indicates increase, blue indicates decrease, white indicates no change in subpopulation compared to UW. Exemplary data is depicted.
(I) Space-time tile plots representing Mono_Mac subpopulations.
(J) Phenotypic earth mover’s distance (PhEMD) diffusion map embedding of all space-timepoints. Each dot represents all CD45+ immune cells captured by scRNAseq within that space-timepoint. Dots are color-coded by day and width of band corresponds to area sampled. DC, diffusion coefficient.
See also S1.
Graph-based clustering and differential gene expression analysis of 10,492 CD45+ immune cells revealed heterogenous monocyte and macrophage subpopulations, neutrophil subpopulations, mast cells, dendritic cells, B cells, T cells, and Natural Killer (NK) cells (Figure S1G and S1H). After re-clustering the monocyte/macrophage (Mono_Mac) populations, we identified three MHCIIhi and eight MHCIIlo subpopulations (Figures 1B and S1I) and noted that MHCIIhi cells expressed markers associated with monocyte-derived dendritic cells (moDCs), such as Cd209a38 and Nr4a339 (Figure S1I and S1J). With both MHCIIlo and MHCIhi Mono_Macs emerging early during wound healing (Figure 2C, Day1), we reclustered them separately prior to further analysis (Figures 1D and S1K, respectively).
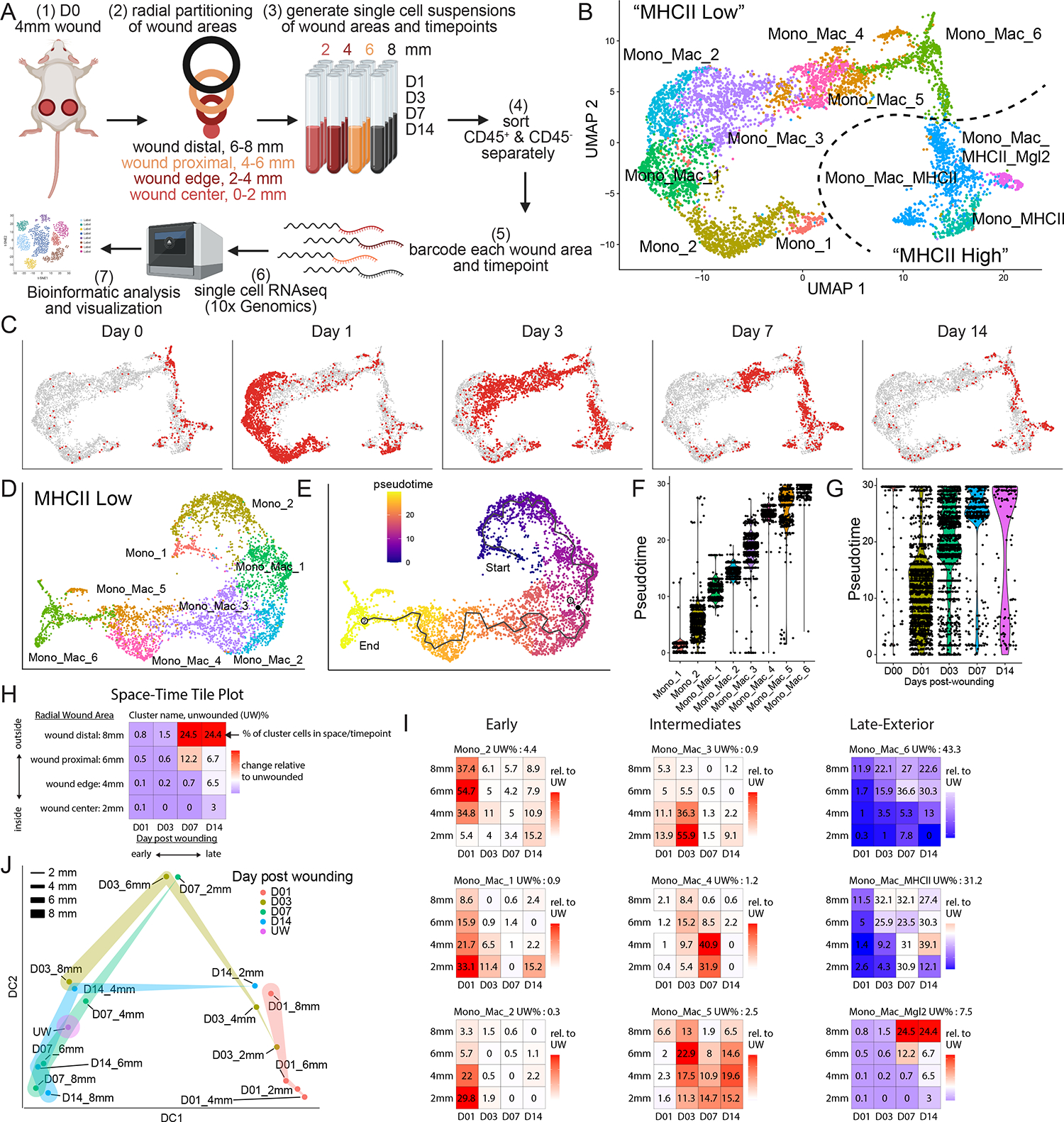
Figure 2. Large volume imaging visualizes spatial distribution of Mono_Mac subsets in whole wounds during skin repair.
(A) Workflow for large volume imaging. A rectangular cuboid covering the wound and surrounding unwounded skin tissue is acquired on a scanning confocal microscope. Acquired images are stitched together and processed for image analysis using Imaris.
(B) ViolinPlot of (left) Arg1 and (right) Mrc1 natural log-normalized mRNA expression level within all Mono_Mac subpopulations.
(C) Space-time tile plot of (left) Arg1 and (right) Mrc1 mRNA expression (normalized to depth) within all Mono_Mac cells. Tiles are color-coded relative to unwounded (UW) state. Red, high. Blue, low.
(D) Top-down view of processed image from Arg1-reporter mouse on days 3, 7, and 14 post-wounding. Colored dots indicate cell location of Arg1− CD11b+ Mono_Mac (green), Arg1+ CD11b− non-myeloid cells (red), and Arg1+ CD11b+ Mono_Mac_3 (yellow). Dotted line represents original 4 mm wound diameter. Bar, 500 μm. Representative of 2 independent experimental replicates is shown.
(E) Quantification of CD11b+ Arg1+ cells in (D) relative to distance from the center of the wound. Percentage of CD11b+ Arg1+ of all CD11b+ cells is plotted by day post wounding, representing an ‘Early Pattern’.
(F) Top-down view of processed image on days 3, 7, and 14 post-wounding. Colored dots indicate cell location of CD206− CD11b+ Mono_Macs (green), CD206+ CD11b− non-myeloid cells (red), and CD206+ CD11b+ Mono_Mac_6 (yellow). Dotted line represents original 4 mm wound diameter Bar, 500 μm. Representative of 2 independent experimental replicates is shown.
(G) Quantification of CD11b+ CD206+ cells in (F) relative to distance from the center of the wound. Percentage of CD11b+ CD206+ of all CD11b+ cells is plotted by day post wounding, representing a ‘Late Exterior Pattern’.
See also S2.
MHCIIlo Mono_Macs consisted of two subsets expressing Ly6c2, annotated as Mono_1 and Mono_2 (Figure S1I). MHCIIlo Mono_Mac_1+2+3 were identified by the transcription factor Klf2, which regulates pro-inflammatory cues in monocytes40 and at least one of the chemokines Cxcl3, Ccl2, Ccl6, Ccl7, and Ccl24. Finally, Mono_Mac_4+5+6 shared Ms4a7 and Apoe expression, two markers associated with microglia and brain-border macrophages41,42. Using pseudotime analysis43, we anchored a trajectory on Mono_1+2 populations, representing cells most similar to Ly6Chi blood-circulating monocytes (Figure S1I), which then proposed a linked transcriptional progression through all clusters successively, eventually ending at Mono_Mac_6, putatively the most terminal Mono_Mac cluster (Figures 1E and 1F). Consistent with this interpretation, Mono_Mac MHCIIlo cells from different timepoints map along the calculated pseudotime (Figure 1G) with the exception that cells collected on day 14 were split between early and late pseudotime, highlighting the status of the ‘late wound’ as a distinct state from unwounded and ‘early wound’ states that was also found in the CyTOF data (Figure S1F). Leveraging this trajectory, we also found trends of gene expression of known myeloid cell states: early expression of inflammatory gene Ly6c244, mid-point expression of Slpi and Mmp12, with Mmp12 being linked to antagonizing further monocyte recruitment45, and late expression of Mrc1, associated with tissue-resident macrophages (Figure S1L).
To search for patterns of space-time distribution of myeloid states, we created tile plots in which each tile shows the prevalence of a cell population—defined from the clustering, above—relative to the unwounded state, both over time (x axis) and space (y axis) (Figure 1H). We discovered greater spatial complexity in how specific immune populations emerge through wound healing phases (Figure S1M), exemplified by the Mono_Macs (Figure 1I). Three subpopulations were characterized as ‘Early’ (i.e. peak at day 1)—represented by Mono_2, Mono_Mac_1 and Mono_Mac_2 and varied from 6 mm to 2 mm in the peak location of the amplified population. Three others had middle-originating waves (D3 or D7): denoted as ‘Intermediates’ and represented by Mono_Mac_3+4+5. Each of these again had specific space-time patterns of recruitment, with Mono_Mac_2 exhibiting an ‘Intermediate Interior (Int-In)’ and Mono_Mac_4 a ‘Late Interior (Late-In)’ pattern. Three others had ‘Late Exterior (Late-Ex)’ distributions, namely Mono_Mac_6, Mono_Mac_MHCII, and Mono_Mac_MHCII_Mgl2 and these were noted as ‘return’ populations, namely that the frequencies dipped and then returned to levels in the unwounded state (Figure 1I). Notably, the earliest progenitor Mono_1, as well Mono_MHCII, did not fit into those patterns (Figure S1N).
Our spatial scRNAseq dataset also captured the space-time accumulation of additional immune cells beyond monocytes and macrophages (Figure S1M). Using PhEMD analysis, the samples occupied an arc-like trajectory in reduced dimensions (Figure 1J), similar superficially to that seen in CyTOF bulk analysis in Figure S1. In this analysis, late wound outside areas (D07_6mm, D07_8mm, D14_6mm, D14_8mm) are more like each other than to the unwounded state, again supporting the previous observation that the immune composition reaches a ‘late wound’ state after skin repair, dissimilar to pre-injury (Figures S1F and 1J).
Large volume imaging visualizes spatial distribution of Mono_Mac subsets in wounded skin
We aimed to validate our gene expression patterns via large volume tissue imaging to probe for the localization of individual cell states (Figure 2A). The clearing-enhanced 3D (Ce3D)46 protocol allowed detection of immune and non-immune cells within wounded skin, highlighting their relative position to the wound edge as marked by integrin alpha-6 (ITGA6) staining of the re-established epithelial basement membrane (Figure S2A and Movie S1, white). Of note, ITGA6 staining also highlighted vasculature structures in non-wounded skin, hair follicles, fascia, and severed nerve bundles lying outside of the closing wound (Figure S2B and Movie S2, white).
We focused on two markers, Arg1 and CD206/Mrc1 that, though often grouped together as a combined signature of ‘M2’ macrophages in vitro47, show clear cell subset (Figure 2B) and space-time distinct (Figure 2C) patterns in wound healing, further confirming results from tumors48 that they are not obligately part of the same gene network. By scRNAseq data, ‘early’ inflammatory Mono_Mac subpopulations Mono_Mac_1+2+3 express high levels of Arg1 (Figures 2B and S2C). We validated this via cleared wound images using an Arg1 reporter mouse49, finding CD11b+ Arg1+ myeloid cells were predominantly found at the center of wounds and at early but not late timepoints after wounding (Figures 2D, 2E, S2D, S2E, and S2F; Movies S3 and S4), consistent with the space-time tile plot (Figure 2C). Confirming previous data50, we also detected Arg1+ CD11b− hair follicles in unwounded skin (Figure S2E and S2G). Conversely, Mrc1 expression was highest in the Mono_Mac_6 subpopulation and predominated in exterior wound regions late (Figures 2B, 2C and S2H). Large volume imaging probing for Mrc1-encoded protein CD206 confirmed the absence of CD11b+ CD206+ cells in the center of the wound on day 3 post-wounding (Figure 2F) and their increased abundance on the exterior non-wounded skin area across all time points measured (Figures 2G, S2I, S2J, and S2K; Movies S1 and S2, proving the veracity of our space-time scRNA-Seq mapping.
Unique space-time patterns of fibroblast subpopulations have matching Mono_Mac patterns
Building on these space-time patterns, we focused on other cell types with matching patterns. Unsupervised clustering of 6944 CD45− cells identified 19 different clusters, separated into endothelial cells, fibroblasts, melanocytes, muscle cells, keratinocytes, and dermal sheath papilla cells (Figure 3A and S3A). Akin to immune populations, fibroblasts (Figures 3B–3D) and other skin-resident cells (Figure S3B) displayed distinct space-time distributions. To seek matched patterns, we primarily focused on fibroblasts as their interaction with macrophages is well-documented51. Here, we identified 5 separate fibroblast clusters by distinct gene expression (Figures 3B and S3C) and initially numbered them in accordance with their accumulation in time during skin wound healing (Figures 3C and 3D).
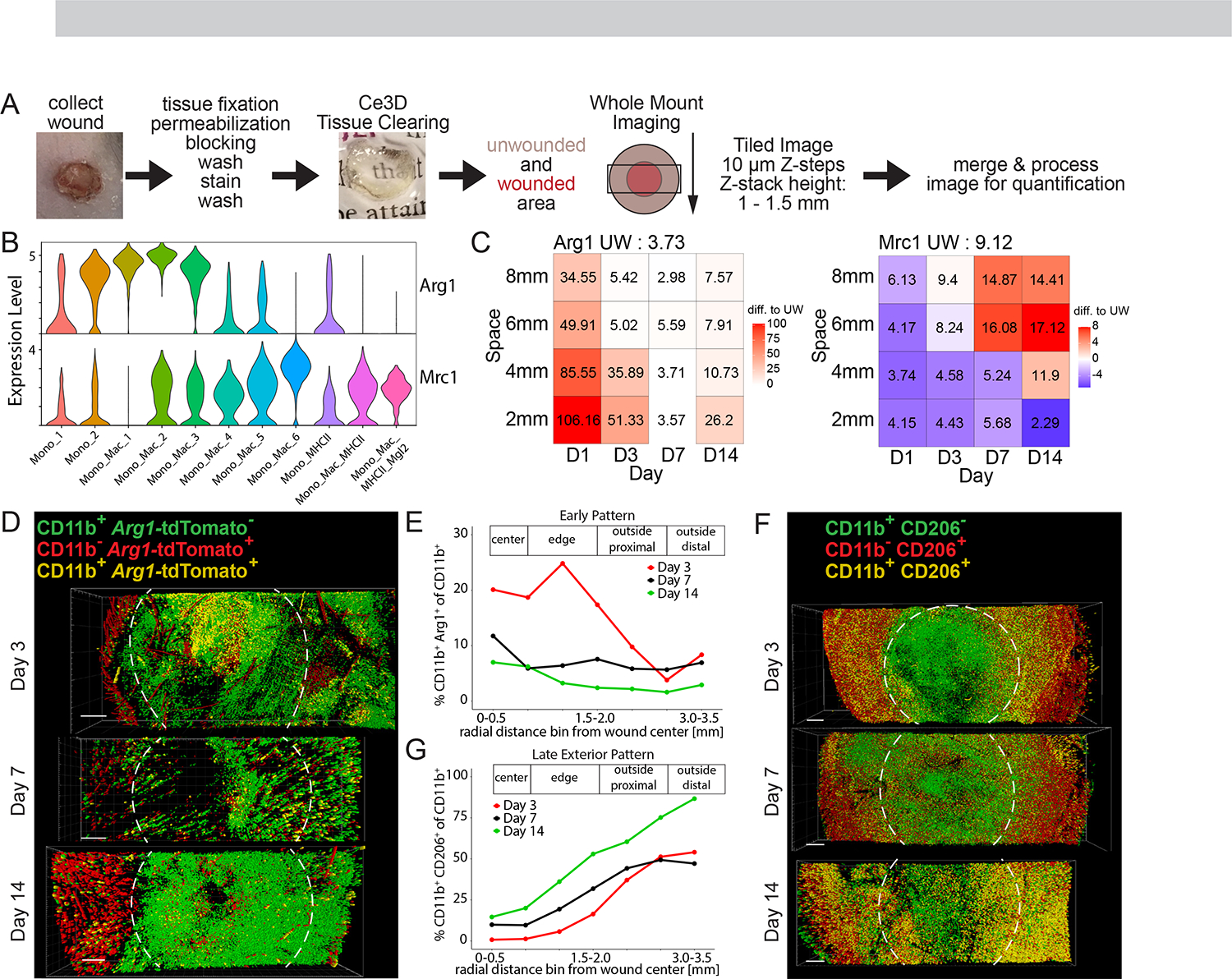
Figure 3. Unique space-time patterns of fibroblast subpopulations have matching Mono_Mac patterns.
(A) UMAP plot of CD45− non-immune cells during skin repair. Endo, endothelial cells. KT, keratinocytes. vSM, vascular smooth muscle.
(B) UMAP plot of the fibroblasts during skin repair.
(C) UMAP projection of all fibroblasts from (B) in gray and cells highlighted in red by timepoint of wound sampling.
(D) Left: Line plots of fibroblast subpopulations identified in the scRNAseq dataset during skin repair. Percentage of each subpopulation within all fibroblasts plotted by day post-wounding. Right: Space-time tile plot representing fibroblast subpopulations. Each tile is one space-timepoint. Number in tile is percentage of subpopulation among all fibroblasts at that specific space-timepoint. Color indicates relative change compared to unwounded (UW) state. Red indicates increase, blue indicates decrease in subpopulation compared to UW.
(E) Schematic depicting wound ‘en face’ imaging. A 250 μm thick cross-section of fixed wound tissue is collected, stained, cleared using Ce3D, and the whole volume is imaged by scanning confocal microscopy.
(F) Space-time tile plot of Postn mRNA expression (normalized to depth) within fibroblasts. Tiles color-coded relative to unwounded (UW) state. Red, high. Blue, low.
(G) 3D-views of dorsal skin wound cross-sections from Pdgfra-reporter mice collected at days 3, 7, and 14 post-wounding. Periostin (POSTN) protein staining shown in red. Dotted vertical line, day 0 wound center. Scale bar, 500 μm.
(H) Quantification of Pdgfra-H2B-EGFP+ spots proximal to POSTN+ staining as a percentage of all Pdgfra-H2B-EGFP+ spots in (G) relative to distance from the center of the wound. Percentage of POSTN-proximal Pdgfra-H2B-EGFP+ spots are plotted by day post wounding representing a ‘Late-Interior Pattern’. Two independent experiments shown overlaid with line dashes representing each replicate.
(I) Outline of Space-Time Correlation Analysis (STCA).
(J) Pearson correlation matrix output of STCA comparing fibroblast and Mono_Mac subpopulations. Correlation was calculated using Pearson correlation and significance adjusted for multiple comparisons using Bonferroni-Hochberg (BH) correction. +p-value<0.05, ++p-value<0.005, +++p-value<0.0005.
(K) Correlation xy-plots of select fibroblast-Mono_Mac subpopulation pairs displaying high Pearson correlation in occurrence in space-time during wound skin repair, as identified in (J). Each dot represents one space-timepoint, i.e. one tile from the space-time tile plot and the unwounded state. The percentage of each paired fibroblast and Mono_Mac cluster within the whole fibroblast or Mono_Mac population, respectively, is plotted for each space-timepoint. Pearson correlation test used to calculate correlation coefficient R and p-value. WH-CM, wound healing cell movement. Int-In, intermediate interior. Late-In, late interior. Late-Ex, late exterior.
See also S3.
The Fibro_1 cluster was annotated as ‘inflammatory’ due to high expression of inflammatory mediators (Figures S3C and S3D)52,53 and their emergence on day 1, representing more than 50% of all wound fibroblasts at that moment (Figure 3D). The Fibro_3 and Fibro_4 clusters expressed genes associated with collagen production and cell contraction (Figures S3C and S3D)53–55 and were annotated as ‘early myofibroblasts’ and ‘myofibroblasts’, respectively. Their space-time profile revealed they make up about 50% of all fibroblasts on day 3 and 7 in the inner regions of the wound (Figure 3D). By day 14, the myofibroblasts have returned to unwounded levels (Figure 3D), highlighting their resolution upon completed skin healing.
With the Fibro_2 cluster showing transcriptional similarity to previously described ‘universal’ and ‘immunomodulating’ fibroblasts and the Fibro_5 cluster displaying similarity to ‘homeostatic/universal’ fibroblasts (Figures S3C–S3F)56,57, they were annotated as such. Of all identified fibroblast subsets, the ‘immunomodulating/universal’ Fibro_2 remained the steadiest in both space and time (Figure 3D), whereas the ‘homeostatic/universal’ Fibro_5 constitute 23.5% of all fibroblasts in unwounded skin and increase over time to reach baseline levels by D7/14 (Figure 3D). Finally, the Fibro_5 dominated the fibroblast population outside of the wounded area (6–8 mm), making up 55–77% of all fibroblast cells there (Figure 3D). This highlights the effect of local wound repair on neighboring unwounded skin areas, where cell compositions are affected, despite the tissue not being subject to direct physical injury.
To visually validate the space-time patterns of fibroblast subsets, we collected wounds at different timepoints post-wounding and generated 250 micron thick wound sections spanning the wound for staining and cleared via Ce3D (Figure 3E) after staining for periostin (POSTN) and α-smooth muscle actin (αSMA), as markers for the myofibroblast-like Fibro_4 and Fibro_3 subsets respectively (Figures S3C and S3D). In our scRNAseq data set, Postn and Acta2 (the gene encoding αSMA) have distinct space-time expression patterns within fibroblasts, with Postn being most prevalent in the center of the wound at day 7 (Figure 3F) and Acta2 emerging on day 3 and then being more focused in the center of the wound at later timepoints (Figure S3G). Using Pdgfra-reporter mice to mark fibroblasts58, we validated the scRNAseq space-time pattern of Postn on the protein level present in the center of the wound on day 7 and markedly reduced D3 and D14 (Figures 3G and 3H). The position of αSMA+ fibroblasts cells not associated with vascular smooth muscle (example in Figure S3H) in the wound over time mirrored the scRNAseq space-time pattern of Acta2: prevalent on the outside on day 3 and then detectable in the center on day 7 and 14 (Figures S3I and S3J). This corroborates the space-time tile plot patterns of the myofibroblast-like Fibro_3+4 subsets and their dynamic appearance and disappearance during the wound healing process.
With the cell states of two key populations (fibroblasts and Mono_Macs) mapped across wound healing space-time (Figures 1 and 2), we asked whether they co-occurred during the wound healing process, highlighting groups of heterotypic cell types putatively engaged in CCC. We performed a Space-Time Correlation Analysis (STCA) of the fibroblast and Mono_Mac subpopulations across the tile plot (Figure 3I, see Methods for further details). This uncovered four co-occurring Mono_Mac/fibroblast pairs with very high degrees of space-time correlation, namely: inflammatory Fibro_1 and Mono_Mac_1, early myofibroblast Fibro_3 and Mono_Mac_3, myofibroblast Fibro_4 and Mono_Mac_4, homeostatic/universal Fibro_5 and Mono_Mac_MHCII_Mgl2 (Figure 3J). To visualize this in another way, we plotted cell percentages for each of these subpopulations over all samples and found high cross-correlations throughout the wound healing process (Figure 3K), with the Fibro_1 and Mono_Mac_1 pair coinciding as an ‘Early’ Wound Healing Cell Movement 1 (henceforth, WH-CM1), Fibro_3 and Mono_Mac_3 representing the ‘Int-In’ pattern (WH-CM2), Fibro_4 and Mono_Mac_4 representing the ‘Late-In’ pattern (WH-CM3), and Fibro_5 and Mono_Mac_MHCII_Mgl2 representing the ‘Late-Ex’ pattern (WH-CM4) (Figures 1I and 3D).
A larger STCA analysis, comparing all identified CD45− non-immune and CD45+ immune cell subpopulations in our spatiotemporal scRNAseq dataset, demonstrated a 5th cluster of correlation (Figures S3K and Supplemental Table 1). This block is only represented by endothelial cells, DCs, and NK cells and represents an ‘Edge’ cell movement (WH-CM5) (Figure S3K). The distinct meanings for some of these additional cell types will await further in-depth analysis as, in this study, we next sought to dive deeper into the gene expression programs that might link two candidate cell types and underlie the pattern of their CCC.
Gene program analysis identifies movements of gene expression across diverse cell types and predicts cell-cell interactions between macrophage and fibroblasts
Moving beyond cluster-based analysis, we applied Non-negative matrix factorization (NMF). This analysis seeks to decompose a cell by gene matrix into the product of two smaller matrices with non-negative components 61,62 and can reveal layers of heterogeneity beyond clustering, especially in a population of cells without clearly demarcated subpopulations33,63,64. Here, collections of genes with similar expression patterns across the cell type contribute to a ‘factor’ with their gene weight denoting the strength of its contribution to that factor (Supplementary Table 2). Each cell is also then assigned a loading value for each factor (e.g. feature plots in Figures 4A and 4B) which can also be visualized in space-time (tile plots in Figures 4A and 4B). We henceforth use the terms ‘factor’ and ‘gene program’ interchangeably. 63,65
Figure 4. Gene program analysis identifies modules of gene expression across diverse cell types and predicts cell-cell interactions between macrophages and fibroblasts.
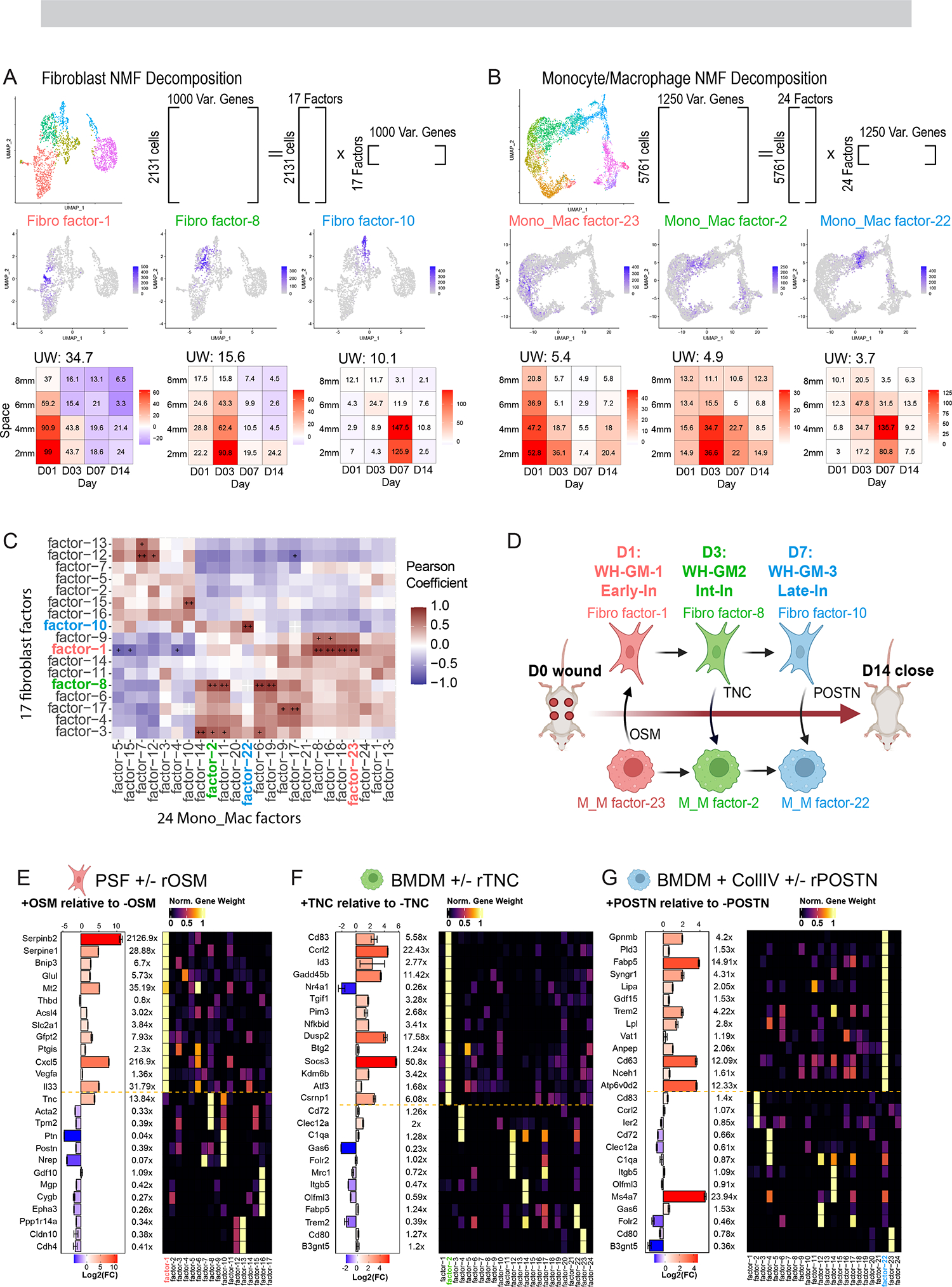
(A and B) Schematic showing strategy for NMF-based decomposition of the (A) fibroblast and (B) monocyte/macrophage populations. NMF decomposition of the fibroblast and Monocyte/Macrophage populations yielded 17/24 factors respectively. Shown are three example FeaturePlots for factor ‘expression’ and Tile Plots describing the average loading of the factor as a function of space-time, with colors indicating change of average expression relative to unwounded as in Fig. 1I
(C) Space-time correlation matrix for average factor expression profiles. Correlation was calculated using Pearson correlation and significance adjusted for multiple comparisons using BH Correction. + (alpha <0.05), ++ (alpha <0.005).
(D) Cartoon schematic of hypothetical fibroblast-macrophage crosstalk and progression over the timespan of wound healing. Three putative interactions that we investigate in vitro are labeled. WH-GM, wound healing gene movement. OSM, oncostatin M. TNC, tenascin-C. POSTN, periostin. Early-In, early interior. Int-In, intermediate interior. Late-In, late interior.
(E) RT-qPCR quantification of gene transcripts in PSF’s predicted from gene program analysis (see S4B) to contribute most to Fibroblast factor-1 as well as genes contributing to other factors as a negative control. Bar chart and color scale denote the Log2 fold-change of relative expression between the OSM treated and untreated PSF’s. Error bars denote standard error of the mean from technical triplicates. Data is representative of two independent experiments. Right heatmap shows the normalized gene weight contribution to all 17 identified fibroblast factors for the genes being probed.
(F) RT-qPCR quantification of gene transcripts in BMDM’s predicted from gene program analysis (see S4A) to contribute most to Mono_Mac factor-2 as well as genes contributing to other factors as a negative control. Bar chart and color scale denote the Log2 fold-change of relative expression between the TNC treated and untreated BMDM’s. Error bars denote standard error of the mean from technical triplicates. Data is representative of two independent experiments. Right heatmap shows the normalized gene weight contribution to all 17 identified Mono_Mac factors for the genes being probed.
(G) RT-qPCR quantification of gene transcripts in BMDM’s predicted from gene program analysis (see S4A) to contribute most to Mono_Mac factor-22 as well as genes contributing to other factors as a negative control. Bar chart and color scale denote the Log2 fold-change of relative expression between the POSTN treated and untreated BMDM’s. Error bars denote standard error of the mean from technical triplicates. Data is representative of two independent experiments. Right heatmap shows the normalized gene weight contribution to all 17 identified Mono_Mac factors for the genes being probed.
We applied a variation of NMF analysis, termed non-smooth NMF (nsNMF)66 to our fibroblast and macrophage subset because of its sparser output during factorization as compared to the base NMF algorithm67,68. We applied a cophenetic metric to identify the optimal number of factors as in Figure S4C69. This resulted in 24 factors for all Mono_Mac populations and 17 factors for the fibroblasts, each with a largely unique and specific collection of top-contributing genes (Figures S4A and S4B). Some genes are shared strongly between factors. For example, MHCII associated genes (e.g. H2-Aa, H2-Ab1) are shared between Mono_Mac factor-4 and factor-7 (Figure S4A), however such overlaps remain rare.
Like the STCA above with cell subpopulation fractions, we asked if the space time patterns of certain factors in the Mono_Mac population were correlated with factors in the fibroblasts. Using a similar approach to Figure 3I, we identified pairs of strongly correlated Mono_Mac and Fibroblast factors. We focused on three sets of correlated factors, henceforth gene movements (GM): WH-GM-1 through 3. These respectively exhibit space-time patterns broadly similar to WH-CM1 (‘Early’), WH-CM2 (‘Int-In’) and WH-CM-3 (‘Late-In)’ patterns described above (Figure 4C and tile plots in Figures 4A and 4B).
Based on the space-time coincidence of these factors, we explored the hypothesis that such correlations could reveal cell-cell signaling between fibroblasts and Mono_Macs that drive the emergence of reciprocal gene programs over the time course of wound closure (Figure 4D). We began by examining fibroblast/Mono_Mac movements WH-GM-1 through 3 using CellChatDB10 to help identify putative ligands that are products of genes contributing to a ‘sender’ factor (Figure S5D, and Mendeley Figure S4). We then experimentally queried if the set of top contributing genes of the correlated ‘target’ factor, in the opposite cell type, might be upregulated by the ligand of interest. We explored three such relationships between macrophages and fibroblasts:
WH-GM-1/Early-In:
The Oncostatin M (OSM) pathway was predicted by CellChat to be most prevalent at D1 (Figure S4D) with Osm being a major contributing gene to Mono_Mac factor-23 (Figure S4A). That, in turn was defined to be in a space-time gene movement with fibroblast factor-1 (Figure 4C). We then used real-time quantitative RT-PCR to test how in vitro stimulation with OSM would affect gene expression in primary skin fibroblasts (PSFs), finding that genes shown in Figure S4B that contribute most heavily to fibroblast factor-1 (e.g. Serpinb2, Serpine1, Bnip3, Glul) were significantly upregulated by OSM treatment (Figure 4E). Importantly, top contributing genes to other factors (e.g. Acta2, Cygb, Postn) were not similarly upregulated indicating the specificity of the predictions generated from NMF analysis.
WH-GM-2/Int-In:
We noted the emergence of Mono_Mac factor-2 at D3 in the interior of the wound, coinciding with fibroblast factor-8 (Figure 4C). Tnc encoding tenascin-C was a major contributing gene to fibro factor-8 and is known to skew macrophage polarization70. We predicted exposure of BMDMs to tenascin-C would upregulate the suite of top contributing genes to Mono_mac factor-2. We found that genes that strongly and specifically contributed to M_M factor-2 (e.g. Cd83, Ccrl2, Id3, Gadd45b, Tgif1 among others) were upregulated following tenascin-C treatment (Figure 4F).
WH-GM-3/Late-In:
CellChat predicted the Periostin (POSTN) pathway for D3/7 could signal from fibroblasts to macrophages (Figure S4D and Mendeley Figure S4) with predicted signaling from Fibro_3/4 to Mono_Mac_3/4, specifically through interaction with Integrins alpha-V and beta-3 (Itgav, Itgb3)71. Postn was a top contributing gene to fibroblast factor-10 (Figure S4B), which correlated well in space-time with Mono_Mac factor-22 to make up WH-GM-3 (Figure 4C). Using an in vitro stimulation of BMDMs, we confirmed that recombinant POSTN, in combination with plate-bound collagen specifically induced expression of top contributing members of Mono_Mac factor-22 (e.g. Gpnmb, Pld3, Fabp5) but not those from other factors (Figure 4G). We then examined if the paired signal and response genes were found co-localized in the healing wound. Imaging sections of D7 wounds, we were able to simultaneously stain for the protein product of Postn (sender) and a response gene in Macrophages, Gpnmb (Figure S4E). We found GPNMB+, CD11b+ cells in the vicinity of thick POSTN deposition (Figure S4F). We also observed a gradient in POSTN signal moving outwards from the center of the wound, matching our scRNA-Seq tileplots as in Figure 3. Concurrently, mean GPNMB signal within CD11b+ cells decreased beyond the original wound edge at around 2mm distance from the center (Figure S4G), matching the Mono_Mac factor 22 profile (Figure 4B). When examining the distance of CD11b+ cells to the nearest POSTN surface, we also found that GPNMB+CD11b+ cells were significantly closer to POSTN signal than their GPNMB-CD11b+ counterparts, with a median distance of 5 microns vs 55 microns (Figure S4H).
Extending NMF to all the other cell types, we identified 114 gene programs scattered across our broad cell type definitions (top contributing genes and weights found in Supplementary Table 1); analysis of their correlated space-time profiles revealed ‘blocks’ of shared space-time patterns including but also extending beyond the Early, Edge, Int-In, Late-In, and Late-Ex patterns described above (Figures S4I–J and Mendeley Figure S4).
Identification of conserved gene programs between wound healing and cancer
The paradigm of the tumor as a ‘wound that never heals’ has been hypothesized28,29 and we sought to quantify the degree the TME could be described by wound healing factors. We hypothesized that NMF analysis could reveal conserved gene programs between the wound healing and tumor tissue contexts. We first generated an integrated myeloid-subset scRNA-Seq dataset from two different mouse tumor models—B16F10 mouse melanoma and MC38 mouse colorectal (Figure 5A and S5A) and applied our nsNMF workflow, identifying 25 factors (Figures S5B and S5C) We then sought to quantify the degree of factor similarity between the two contexts; we prioritized that a conserved pair of factors exhibits significant overlap in the top contributing genes; therefore, we applied a Jaccard distance metric based on the top 20 genes by weight or distance. Most factor pairings displayed little to no overlap in their top 20 contributing genes, but a few rose prominently from the background (Figure 5B).
Figure 5. Identification of conserved gene programs in Mono_Mac between wound healing and mouse tumor models.
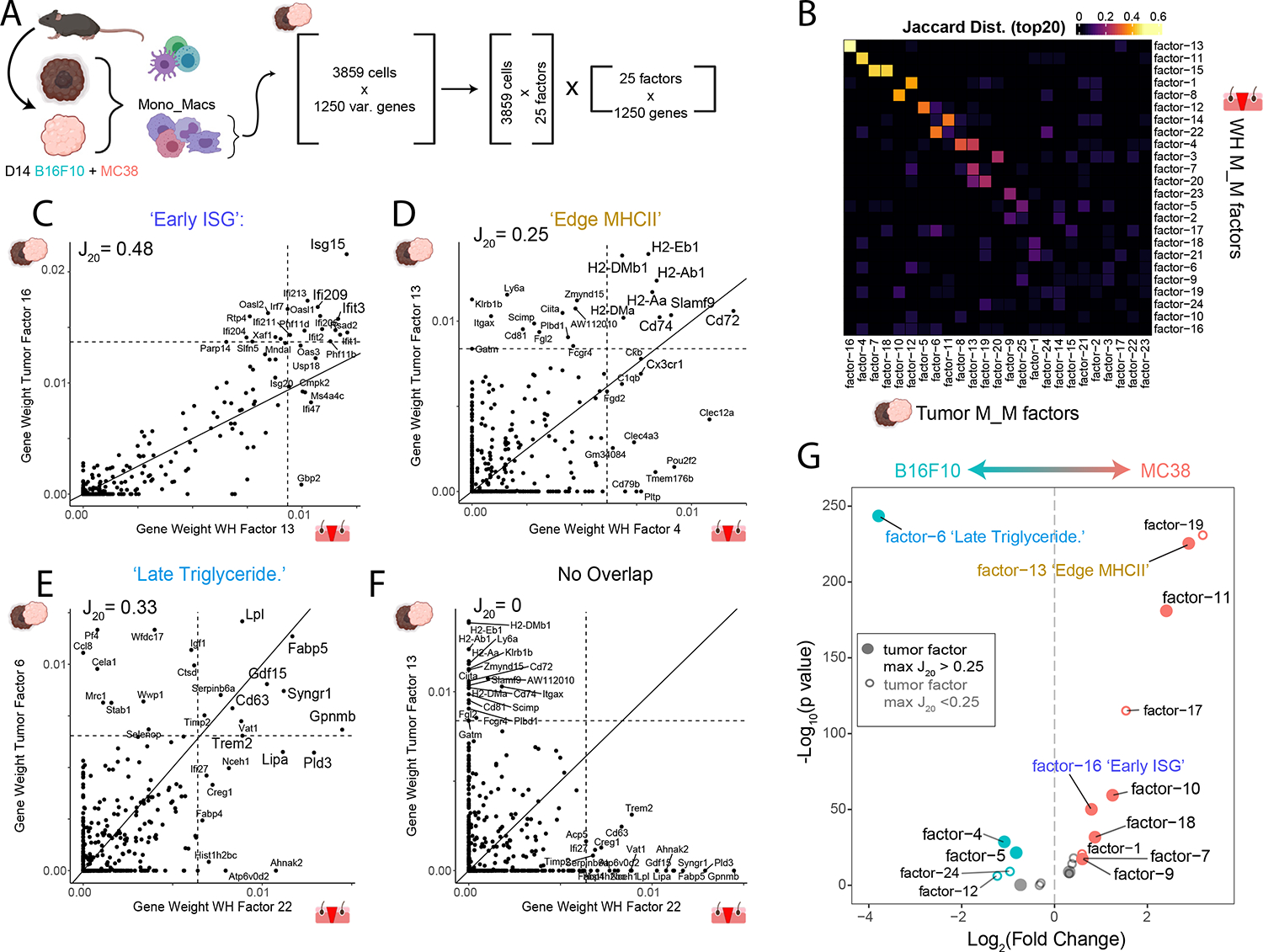
(A) Strategy for generation of a multi-tumor model Mono_Mac CD45+ scRNA-Seq dataset. Following integration, Mono_Mac populations were selected for NMF decomposition, starting with 3859 cells and the top 1250 variable genes expressed in at least 2% of cells. This resulted in 25 factors of interest based on the cophenetic metric (seen in Figure S5C)
(B) Heatmap showing the Jaccard20 distance (defined in STAR Methods) between all 25 M_M tumor and 24 M_M WH factors based on top contributing gene weights.
(C-F) Scatter plots for selected tumor/WH factor pairs for (C) Tumor factor-16 vs WH factor-13, (D) Tumor factor-13 vs WH factor-4, (E) Tumor factor-6 vs WH factor-22, and (F) Tumor factor-13 and WH factor-22 with the gene weight contributions plotted as calculated from the basis matrix in the NMF output (see Figure S4A for WH factors and S5B for tumor factors). Slope represents x=y line and dotted lines represent the weight for the 20th highest gene contribution in either factor. The Jaccard20 index is shown and thus reflects the frequency of points in quadrant I over quadrants I, II and IV. For pairings in C-E, top shared genes in the upper right quadrant were put through Enrichr to find overrepresented cellular processes with the top result by p-value listed. Full Enrichr output can be found in the extended data (Table S2).
(G) Volcano plot showing differential loading of factors between MC38 and B16F10 Mono_Mac. Datasets for the 25 identified factors. Y-axis denotes log10 of unadjusted p-value. Labelled points have adjusted p-value < 0.05 (Bonferroni correction) and absolute log2 fold-change greater than 0.5. Colored points have absolute log2 fold change greater than 0.5.
We generated scatterplots of gene weights to examine shared gene contributions more closely between pairs of WH and Tumor factors (examples in Figures 5C–5F and S5D–S5I). While most factor pairings resembled Figure 5F with little to no overlap in gene contributions, we did identify nine strong pairings (Figures 5C–5E and S5D–S5I). Gene Ontology (GO) analysis on the shared genes in the upper right quadrant revealed unique cellular processes associated with each pairing which could, at least partially, reflect the functional output of those gene programs (full list of GO terms in Supplementary Table 3). We focus on three of these pairings below:
‘Early ISG’ Program:
Of the stronger pairings, the WH M_M-13-to-Tumor M_M-16 factors was highly characterized by a collection of well-described Interferon stimulated genes72 (ISG) (Figure 5C). GO analysis yielded the term ‘Type I interferon signaling pathway’. In the wound, this WH M/M factor-13 followed an ‘Early’ WH-GM1 spacetime profile (Figure S4J).
‘Edge MHCII program:
Another factor pairing, (WH M_M-4-to-Tumor M_M-13; Figure 5D), involved genes associated with antigen presentation through MHCII. In this case, there were highly correlated expression levels of H2-Ab1, H2-Eb1 and other genes associated with antigen presentation (e.g. Cd74). GO analysis indicated antigen processing and presentation via MHCII. WH M_M factor-4 followed an ‘Edge’ WH-GM5 space time profile which seemed to follow the closure of the wound (Figure S4I–J).
‘Late Triglyceride’:
The WH M_M factor-22 to tumor M_M factor-6 pairing, was marked by genes including Gpnmb, Fabp5, Syngr1, Cd63, Trem2, and Lipa among others (Figure 5E). GO analysis revealed an enrichment for ‘Triglyceride Sequestration’ suggesting a functional output associated with intracellular vesicle trafficking and exocytosis. This factor also followed the space time pattern we termed ‘Late-In’ or WH-GM3 (Figure S4J).
Finally, we sought to determine whether such programs are used equivalently in the two model tumor systems. The MC38 dataset displayed significant enrichment of factors including tumor factors 11 and 13 which had strong correspondence to a WH factor. Meanwhile, B16F10 tumors were uniquely marked by a separate and very strong enrichment of factor-6, corresponding to WH factor-22 (Figure 5G). Together this suggests that while tumors may indeed borrow factors from wound healing, individual tumors may do so uniquely.
We were also curious how different timepoints of the tumor might resemble different space-time coordinates of wound healing. We used two published scRNA-Seq datasets on fibroblasts collected at different stages of tumor progression in either a B16F10 melanoma model73 (Figure S5J) or tumors arising in KPP GEMM animals54 (Figure S5K). We used our fibroblast factors to generate signature scores for each program in these tumor datasets to relate their similarity to space-time coordinates of wound healing. Strikingly, we found both tumor models showed similar trajectories over tumor stage, moving towards the D7_2mm and D7_4mm coordinates and away from the UW and late edge coordinates (Figure S5L–N). This was driven by gain of factors-8,10 and loss of factors 12,13, suggesting that as the tumor progresses in these models, the fibroblast compartment comes to resemble a late resolving wound.
We next sought to generalize our findings to a more clinically relevant setting. Using a merged scRNA-Seq dataset collected from patient tumor resections (from lung adenocarcinomas ‘LUNG’ and head and neck squamous cell carcinoma ‘HNSC’) with some matched adjacent normal tissue (Figure 6A), we extracted the fibroblast and Mono_Mac subsets (Figures 6B, 6C and S6A–S6D). We applied NMF decomposition and compared gene weight contributions as above, with conversion of mouse gene symbols to human ones for comparison. Despite the difficulties in comparing gene programs across species and tissue type, we observed several overlapping mouse wound healing (mWH) and human tumor (HuTumor) factors (Figure S6G, S6H). mWH Mono_Mac factor-22 overlapped well with human tumor (HuTumor) Mono_Mac factor-7 with many overlapping genes as seen in a gene weight scatterplot (Figure 6D). Meanwhile mWH Fibroblast factor-10 was conserved well with HuTumor Fibroblast factor-5 (Figure 6E). This suggested that these D7 dominant gene programs (mWH Fibroblast factor-10 and mWH Mono_Mac factor-22) represented a cross-species conserved state in human tissues. When we plotted the mean loading of HuTumor Fibro factor-5 vs. the mean loading of HuTumor M_M factor-7 across samples, we found a correlated increase in these factors going from adjacent normal to tumor tissue in HNSC and LUNG (Figure 6F, 6G). When considering the HNSC and LUNG indications separately, we found a significant correlation between the levels of this HuTumor fibro factor-5 and HuTumor M_M factor-7. Thus, not only are these factors conserved across species, but also their cooccurrence.
Figure 6. The wound healing gene movement-3 (WH-GM3) is conserved in human tumors.
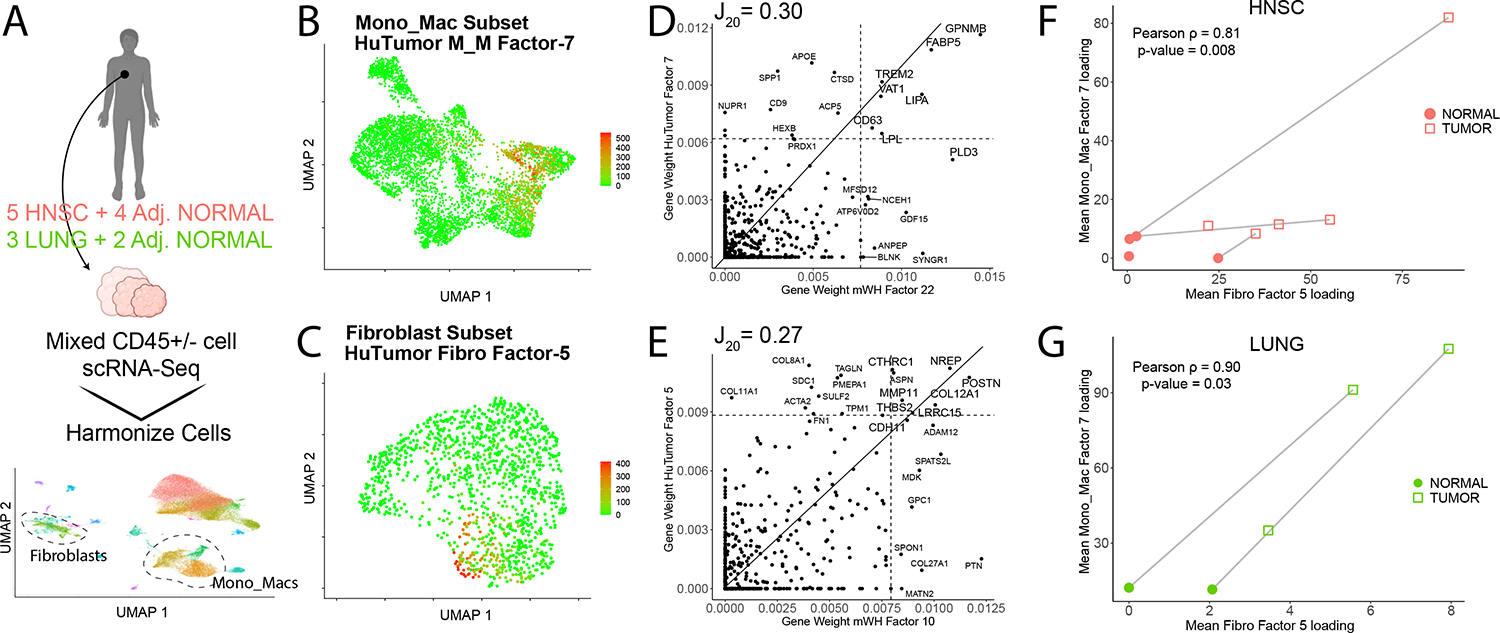
(A) scRNA-seq datasets of both CD45+ and CD45− compartments from patient tumor resections and adjacent normal samples were collected from lung adenocarcinomas (LUNG) and head and neck squamous cell carcinomas (HNSC) as described in STAR Methods (n = 77,270 cells).
(B) Feature plot shown for the Mono_Mac object for HuTumor Mono_Mac factor-7.
(C) Feature plot shown for the fibroblast object for HuTumor fibro factor-5.
(D) Scatter plot for gene weights from factor pair (human) HuTumor Mono_Mac factor-7 vs. (mouse) mWH Mono_Mac factor-22 (right). Mouse gene symbols were converted to their ortholog for comparison purposes with the gene weight contributions plotted as calculated from the basis matrix in the NMF output. Slope represents x = y line, and dotted lines represent the weight for the 20th highest gene contribution in either factor. The Jaccard20 index is shown.
(E) Scatter plots for selected human (hu) tumor/murine (m) WH factor pairs huTumor Fibroblast Factor 5 vs. mouse WH Fibroblast factor-10. Mouse gene symbols were converted to their ortholog for comparison purposes with the gene weight contributions plotted as calculated from the basis matrix in the NMF output. Slope represents x = y line and dotted lines represent the weight for the 20th highest gene contribution in either factor. The Jaccard20 index is shown.
(F and G) Scatter plot showing mean factor levels calculated for huTumor Mono_Mac factor-7 and huTumor fibroblast factor-5 across HNSC samples (F) and LUNG (G). Point shapes denote adjacent normal vs. tumor samples and lines between points represent paired adjacent normal and tumor samples. Pearson’s rho denoted and p value calculated via Pearson’s method.
Conservation of Mono_Mac factors predicts increased POSTN density and decreased Selectin-P+ vessel density in B16F10 vs. MC38 tumor models
We finally sought to study the conserved gene programs spatially and confirm the differential usage of wound healing factors in different tumor models. To this end, we used our Mono_Mac factor translation matrix (Figure 5B) and our wound healing movement identification, to make and then test predictions about the state of the tumor microenvironment in either tumor model. As observed in Figure 5G, the M_M tumor factor-6 was more highly expressed in B16F10 vs MC38. This tumor macrophage factor corresponded to M_M WH factor-22 (Figure 5B). We used the observation that the latter factor was paired in a space-time movement with fibroblast factor-10 in our wound healing dataset (Figure 4C), to form the prediction that the sender signal POSTN, would be more prevalent in B16F10 vs. MC38 model (Figure 7A). Immunofluorescence staining of both tumors identified indeed profoundly larger density of POSTN fibers within B16F10 tumors as compared to MC38 (Figures 7B and 7C). In addition, a considerably larger fraction of CD11b+ cells were in close contact with POSTN fibers in the B16F10 model vs. MC38 (Figures 7D, 7E, and S7A).
Figure 7. Conservation of Mono_Mac factors predicts specific differential features of microenvironments in B16F10 vs MC38 tumor models.
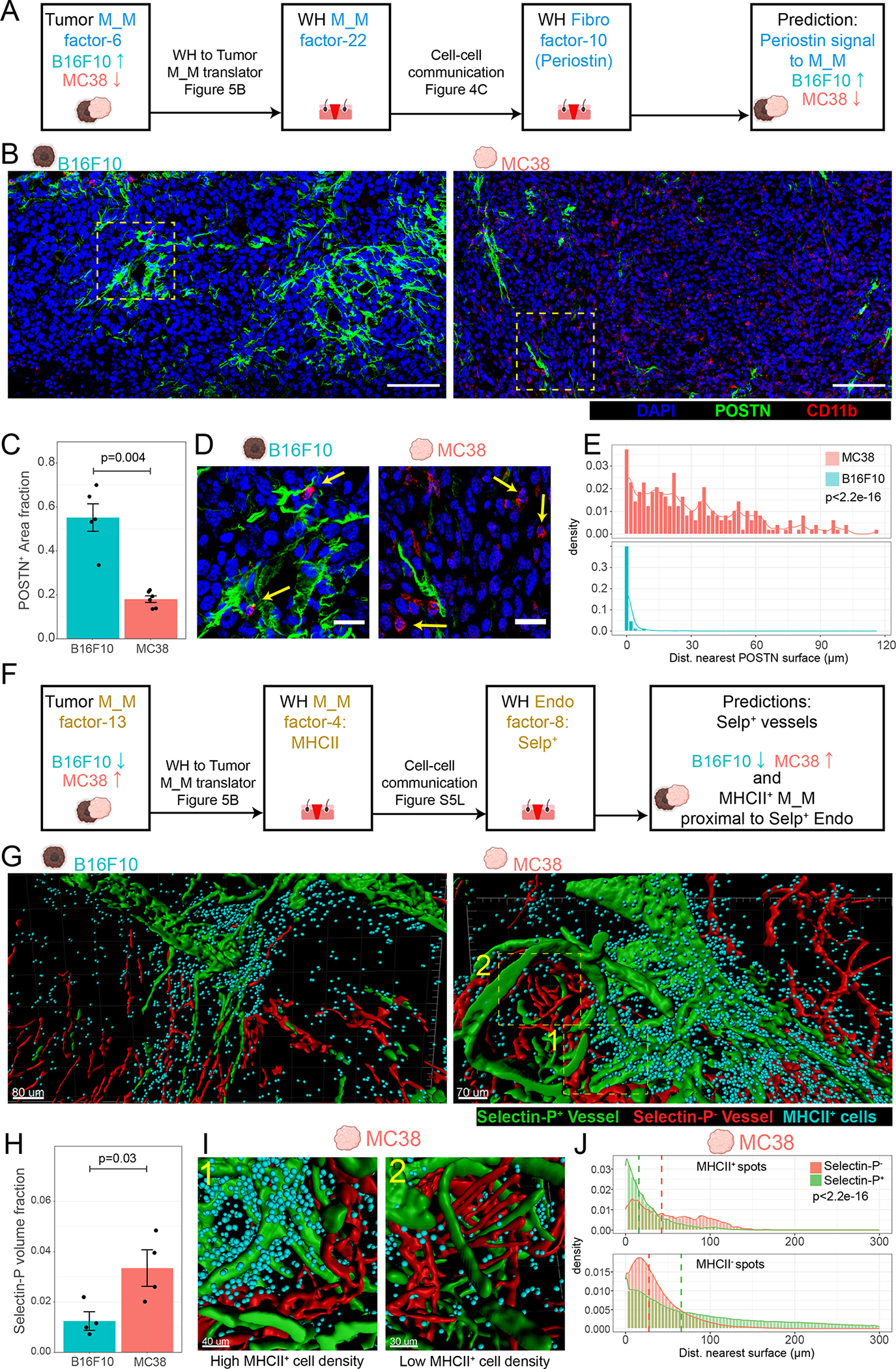
(A) Schematic for hypothesis generation in the tumor setting. Translation of the tumor M_M factor-6 to the WH M_M factor-22 allows prediction that the same stimuli (POSTN from WH Fibro factor-10) might underlie tumor M_M factor-6 and thus be more prevalent in the B16F10 vs. MC38 tumor model.
(B) Representative immunofluorescent images of 10 μm sections of D14 B16F10 and MC38 tumors stained for DAPI (blue), POSTN (green), and CD11b (red). Scale bar = 100 μm. Representative of 2 independent replicates consisting of total of 6 and 5 samples for MC38 and B16F10 respectively.
(C) Barchart denoting fraction area of POSTN+ surfaces as a fraction of total imaged tissue area. One-sided Wilcoxon rank-sum test used. Each point represents a scanned area from 6/5 separate tumor samples MC38/B16F10 respectively.
(D) Insets from denoted regions of interest in Figure 6B. Arrows denote CD11b+ cells in close contact with POSTN+ surfaces (B16F10) or not (MC38). Scale bar = 25 μm.
(E) Histograms showing distribution of distance of CD11b+ cells to nearest POSTN surface. Representative of 3 independent replicates (3 tumors). Bin-width = 2 μm. Kolmogorov-Smirnov (KS) test used to test for distribution similarity.
(F) Schematic for hypothesis generation in the tumor setting. Translation of the tumor M_M factor-13 to the WH M_M factor-4 allows prediction that the same association between WH M_M factor-4 with endothelial WH factor-8 might be found in the tumor setting and that endothelial factor-8 might be more prevalent in MC38 vs. B16F10 tumor model.
(G) Processed 3D images of cleared 250 μm thick tumor slices from a (left) B16F10 and (right) MC38 tumor. Generated surfaces based on Selectin-P+ staining (green) and CD31+/Selectin-P− signal (red). Dots (cyan) denote MHCII+ cells. Representative of 4 independent replicates (4 separate tumors).
(H) Bar chart showing comparison of the cumulative Selectin-P+ surfaces volume normalized to total imaged tissue volume between MC38 and B16F10. One-sided Wilcoxon rank sum test used.
(I) Zoomed in and rotated insets from MC38 tumor in Figure 6G exemplifying (1) dense accumulation of MHCII+ cells proximal to Selectin-P+ vessels and (2) sparse MHCII+ cell accumulation proximal to Selectin-P−.
(J) Histograms indicating the distances of MHCII+ spots and MHCII− spots to the nearest Selectin-P+ or Selectin-P− surface in the MC38 model. Dashed line indicates the median. Histograms representative of 4 independent replicates (4 separate tumors). P-value calculated via KS test.
See also S7.
Conversely, the shared ‘Edge MHCII’ factor (tumor M_M factor-13 corresponding to WH M_M factor-4 Figure 5B) was more dominant in MC38 (Figure 5G). Going back to the space-time correlations in Figure S4J, WH M_M factor-4, as an ‘Edge’ pattern WH-GM5, grouped with WH endothelial factor-8, which comprised genes including Selp, Vwf, and Ackr1. Following a similar line of inference as before with POSTN signaling to macrophages, we predicted a higher density of Selectin-P+ vasculature in MC38 vs. B16F10 tumors (Figure 7F).
Using 3D imaging of cleared thick tumor sections (250 μm) (Figures 7G, S7B), we found a markedly increased density of Selectin-P+ vasculature in the MC38 tumor relative to B16F10 tumor (Figure 7H), consistent with our prediction in Figure 7F. Analysis also revealed clear physical proximity of MHCII+ cells (cyan points) with Selectin-P+ vasculature (green) vs Selectin-P− vasculature (red) in both tumor models (Figures 7I, 7J, and S7C). We also note this preference for Selectin-P vessels was not found in MHCII− cells (Figure 7J, and S7C). These lines of evidence provide examples of how the conceptual framework of conserved gene programs and multicellular movements can inform hypothesis generation spanning tissue contexts.
Discussion
Characterizing how diverse cell types are spatially and temporally organized within the tissue will help us understand the underlying dynamic nature of tissues. Here, we established a spatiotemporal framework to study pairing of cell types during the physiologically complex process of wound repair. In this setting, the concept that spatiotemporal correlation may indicated paired biology drove the identification of groups of cell types and gene programs that together form larger cellular movements, and partially conserved in both skin repair and tumor growth.
Several fibroblast-macrophage interactions have been described in health and disease, such as fibrosis and cancer22. The use of space time correlation analysis (STCA) takes this systematically one step further and identified three distinct fibroblast-macrophage cellular pairings during wound repair (Figure 3J), each characterized by a distinct space-time pattern during the repair process. The earliest pattern consisted of inflammatory Fibro_1, expressing the neutrophil-attracting chemokine Cxcl574 and the early inflammatory gene Ptx375. Accordingly, this ‘Early’ pattern was accompanied by neutrophils and monocytes (Figure S3K) whose accumulation in early wounds had been previously identified3 but not tied to this fibroblast population. Conversely, the ‘Late-Ex’ pattern ties together Fibro_5, mast cells, MHCIIhi M_M, T cells, and keratinocyte subsets. These multicellular patterns are akin to the described collection of cell types that coordinate monocyte differentiation in the liver6.
By combining NMF-based decomposition with spatiotemporal data, we identified co-occurring gene programs that provide candidates for reciprocal interaction between cell types. We present this approach as a framework for identifying CCC pathways and their downstream effects on gene expression. In our framework, factors or gene programs represent a functional module activated in a cell type due to response to external stimuli which in turn may be derived from programs activated in other cell types. One example is OSM from M_M to Fibroblasts, previously known to induce Serpine1 and Il33 upregulation76,77. Using NMF analysis combined with spatiotemporal correlations across cell types, we identified a host of other genes (e.g. Glul, Mt2, Acsl4) (Figure 4E) not previously described to be OSM induced.
Reciprocally, we predicted TNC to induce a host of genes (e.g. Cd83, Ccrl2, Id3) in M_M factor-2 in D3 wound interior. Testing TNC treatment on BMDM’s revealed a specific upregulation of these predicted genes in a specific manner, failing to upregulate genes contributing to other gene programs (Figure 4F). Recent work has discovered a TNC-TLR4 signaling axis in macrophages that promotes a pro-metastatic perivascular niche70; we note several of these target genes we verified are known to sit downstream of TLR4 activation78,79.
Similarly, our movement identification suggested POSTN, which emerges later in wound healing in fibroblasts, as a candidate to induce the genes defined in factor-22 in M_M’s (Figure 4G) in D7 wound interiors. This factor includes Gpnmb, Lipa, and Trem2, implicated as a marker for a tumor associated macrophage population that can be targeted for immunotherapy80–82. Previous reports have mainly described the ability of POSTN to promote adhesion and migration of macrophages in tumor settings83,84. 80–82 85
Our dataset suggests a multitude of major groupings of spatiotemporally correlated gene programs in disparate cell types, with each grouping displaying a unique space-time pattern (Figures S4I, S4J and Mendeley Figure S4). Similar to the idea of ‘hubs’ of gene programs, these movements of gene programs could represent spatially co-localized, functional units of cell organization in tissue64. Additionally, such groupings could underlie the findings that tumor microenvironments tend to adopt defined compositional ‘Archetypes’86. The gene programs identified here, and their shared spatiotemporal profiles, will inform future studies to identify which correlations are indicative of true CCC87.12
We posit that gene program analysis can serve as a powerful tool for integrative studies across tissue types and disease models. 33We identified conserved gene programs in the M_M populations from two tumor models and our wound healing dataset. We also found conserved gene programs in both macrophages and fibroblasts from human tumor samples, demonstrating that these programs may represent fundamental cross-species biology (Figure 6A–6E). We also note that factor pairings are not perfect, with some genes showing little contribution in one setting versus major contribution in another (Figure 5B). We theorize this could indicate either a purely coincidental co-expression in one setting, gene dropout differences, or an additional layer of epigenetic regulation. We also note the presence of many factors without a good counterpart which could represent artifacts introduced in the processing pipeline or be representative of context-specific programs. This methodology could also be applied to models of fibrosis88, wherein many of the same CCC’s that drive regeneration become dysregulated, e.g. persisting inappropriately22,89–91. Thus, identifying the space-time profiles of conserved gene programs and between fibroses, wound healing, and tumorigenesis could identify where the dynamics of pathologies begin to diverge in state space from normal29.
We further demonstrated that testable hypotheses can be generated from our correlative studies by proceeding in a two-step fashion, first translating tumor Mono_Mac factors to wound healing Mono_Mac factors (Figure 5B), then using identified multicellular movements to translate WH M/M factors to correlated WH factors in other cell types (Figure S4J). This analysis allowed us to predict increased POSTN signaling to macrophages in B16F10 versus MC38 tumors which we verified downstream (Figure 7A–7E). Of note, this pair of macrophage/fibroblast programs (M_M factor-22 and Fibroblast factor-10) that dominated in the D7 wound interior was found conserved in human tumor samples from HNSC and lung adenocarcinoma resections (Figure 6A–6E) with a paired enrichment in tumor tissue vs. adjacent normal (Figure 6F, 6G). We suggest that different tumor archetypes in humans and mouse models can be described in terms of wound healing space-time coordinates; in this instance, the B16F10 model and some human tumors can be said to resemble a D7 wound interior in the resolution phase. Altogether, our in-silico analysis suggests that as tumors progress they become enriched in features found in the late wound interior while losing features associated with the unwounded and late wound exterior coordinates (Figure S5L–S5N). Notably, our tumor datasets lack spatial resolution and future work could illuminate neighborhoods in the tumor microenvironment with similarity to distinct wound space-time points using spatial transcriptomics92–94. Conversely, we also predicted an enrichment for P-selectin-expressing endothelial cells (endo factor-8) in MC38 versus B16F10 tumor (Figure 7F). The close association of MHCIIhi macrophages with CD31+ vasculature has been reported previously95 but not specifically Selectin-P+ vasculature. Of interest, the endothelial factor-8 genes including Selp, Sele, and Ackr1 also define a tumor associated high endothelial venule network found to be correlated with patient response to checkpoint blockade immunotherapy96, suggesting an etiology for the unresponsiveness of the B16-F10 melanoma model versus the MC38 model to ICB90,97
Limitations of study:
Our scRNA-seq study identified several immune and non-immune cells and their dynamic changes during the skin repair process. However, we noticed no capture of adipocytes, neurons, or glial cells. Presumably our workflow of cell sorting and 10x Genomics encapsulation led to their disproportionate loss. This precludes analysis of their potential involvement in the movements we describe. In addition, we noted reduced representation of Neutrophils in our scRNA-Seq study compared to our initial CyTOF study, possibly due to the sensitivity of these cells to the scRNA-Seq workflow. Previous studies have highlighted the importance of adipocytes in skin repair34, as well as the role of macrophage-neuron crosstalk skin homeostasis98.
While we show multiple lines of evidence for POSTN-mediated factor-22 upregulation in macrophages several questions remain. Firstly, POSTN is known to mediate several effects on TGFβ signaling99 and macrophage recruitment100. The signaling events leading directly from POSTN binding via theoretical receptors (ITGAV, ITGAM) to upregulation of Mono/Mac factor-22 genes remains unclear. Secondly, in our tumor settings, it remains unknown if the POSTN observed is derived from fibroblasts or tumor cells themselves.
The skin is colonized by a microbiome that breaches skin barrier upon injury which are sensed by local immune cells. In our otherwise sterile tumor models, pathogen sensing would be less likely to influence CCC. Thus, commonalities discovered here using subcutaneous tumor models and skin wounding are conserved in both absence and presence of pathogen detection while programs during wound healing without a counterpart in tumor models may represent a response downstream of pathogen sensing.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew F. Krummel (matthew.krummel@ucsf.edu).
Materials Availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNAseq data have been deposited at GEO and Mendeley Data under and are publicly available as of the date of publication. Accession numbers are listed in the key resource table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD16/32 (clone 2.4G2) | Tonbo Biosciences | 70-0161-U500 |
| anti-mouse CD45 Alexa Fluor 647 (clone 30-F11) | Biolegend | 103124 |
| anti-mouse GPNMB eFluor™ 660 (clone CTSREVL) | eBioscience | 50-5708-80 |
| anti-h/mPeriostin (clone 345613) | R&D Systems | MAB3548 |
| anti-alpha smooth muscle actin antibody (polyclonal) | Abcam | AB5694 |
| anti-mouse P-selectin (polyclonal) | R&D Systems | AF737 |
| anti-CD31 AlexaFluor647 (clone 390) | BioLegend | 102416 |
| anti-CD11b AlexaFluor594 (clone M1/70) | BioLegend | 101254 |
| anti-CD206-AlexaFluor647 (clone C068C2) | BioLegend | 141712 |
| anti-I-A/I-E AlexaFluor594 (clone M5/114.15.2) | BioLegend | 107650 |
| anti-I-A/I-E AlexaFluor488 (clone M5/114.15.2) | BioLegend | 107616 |
| goat anti-rat IgG (H+L) AlexaFluor488 | Thermo Fisher Scientific | A11006 |
| donkey anti-goat IgG (H+L) AlexaFluor488 | Thermo Fisher Scientific | A11055 |
| F(ab')2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 555 | Thermo Fisher Scientific | A48283 |
| Alexa Fluor® 488 AffiniPure Fab Fragment Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch | 112-547-003 |
| Alexa Fluor® 647 AffiniPure Fab Fragment Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch | 112-607-003 |
| anti-mouse B220 (clone RA3–6B2) | Biolegend | 103202 |
| anti-mouse CCR7 (clone 4B12) | Biolegend | 120101 |
| anti-mouse CD103 (clone 2e7) | Biolegend | 121402 |
| anti-mouse CD11b (clone M1/70) | Biolegend | 101202 |
| anti-mouse CD11c (clone N418) | Biolegend | 117302 |
| anti-mouse CD16/32 (clone 2.4G2) | BD | 553142 |
| anti-mouse CD19 (clone 6D5) | Biolegend | 115501 |
| anti-mouse CD206 (clone C068C2) | Biolegend | 141702 |
| anti-mouse CD207 (clone 4C7) | Biolegend | 144202 |
| anti-mouse CD24 (M1/69) | Biolegend | 101802 |
| anti-mouse CD3e (clone 17A2) | Biolegend | 100202 |
| anti-mouse CD38 (clone 90) | Biolegend | 102702 |
| anti-mouse CD4 (clone RM4-5) | Biolegend | 100506 |
| anti-mouse CD44 (clone IM7) | Biolegend | 103002 |
| anti-mouse CD45 (clone 30-F11) | Biolegend | 103102 |
| anti-mouse CD49b (clone HMa2) | Biolegend | 103513 |
| anti-mouse CD62L (clone MAB5671) | R&D | MAB5761 |
| anti-mouse CD64 (clone (X54-5/7.1) | Biolegend | 139302 |
| anti-mouse CD69 (polyclonal) | R&D | AF2386 |
| anti-mouse CD8a (clone 53-6.7) | Biolegend | 100702 |
| anti-mouse CD86 (clone GL-1) | Biolegend | 105002 |
| anti-mouse CD90 (clone G7) | Biolegend | 105202 |
| anti-mouse c-Kit (clone 2B8) | Biolegend | 105802 |
| anti-mouse CTLA-4 (clone UC10-4B9) | Biolegend | 106302 |
| anti-mouse F4/80 (clone BM8) | Biolegend | 123102 |
| anti-mouse FceR1a (clone MAR-1) | Biolegend | 134302 |
| anti-mouse Foxp3 (clone NRRF-30) | eBiosciences | 14-4771-80 |
| anti-mouse GATA3 (clone 16E10A23) | Biolegend | 653802 |
| anti-mouse Ki67 (clone SolA15) | eBiosciences | 14-5698-82 |
| anti-mouse Ly6C (clone HK1.4) | Biolegend | 128002 |
| anti-mouse Ly6G (clone 1A8) | Biolegend | 127602 |
| anti-mouse MHC-II (clone M5/114.15.2) | Biolegend | 107602 |
| anti-mouse PD-1 (clone 29F.1A12) | Biolegend | 135202 |
| anti-mouse PD-L1 (clone 10F.9G2) | Biolegend | 124302 |
| anti-mouse PDCA1 (clone 927) | Biolegend | 127002 |
| anti-mouse RORgt (clone B2D) | eBiosciences | 14-6981-82 |
| anti-mouse Siglec F (clone E50-2440) | BD Biosciences | 552125 |
| anti-mouse SIRPa (clone P84) | Biolegend | 144002 |
| anti-mouse T-bet (clone 4B10) | Biolegend | 644802 |
| anti-mouse TCRgd (clone GL3) | Biolegend | 118101 |
| anti-mouse Ter119 (clone Ter119) | Biolegend | 116202 |
| anti-mouse TIM-3 (clone B8.2C12) | Biolegend | 134002 |
| anti-human CD45 APC/e780 (clone HI30) | Thermo Fisher | 47-0459-42 |
| anti-human CD3e PerCP/e710 (clone OKT3) | Thermo Fisher | 46-0037-42 |
| anti-human HLA-DR BUV395 (clone G46-6) | BD Biosciences | 564040 |
| Biological samples | ||
| Fetal Bovine Serum, Benchmark | GeminiBio | 100-106 |
| Chemicals, peptides, and recombinant proteins | ||
| Methanol | Fisher Scientific | A412-4 |
| recombinant human Tenascin | Merck Millipore | CC-065 |
| recombinant mouse Periostin | R&D Systems | 2955-F2-050 |
| recombinant mouse Oncostatin M | BioLegend | 762802 |
| Albumin, Bovine (BSA), nuclease free | VWR | 332 |
| Albumin, Bovine (BSA) | Sigma | A7906 |
| EDTA | Teknova | E0306 |
| Maxpar PBS | Fluidigm | 201058 |
| Maxpar Cell Staining Buffer | Fluidigm | 201068 |
| Maxpar Water | Fluidigm | 201069 |
| EQ Four Element Calibration Beads | Fluidigm | 201078 |
| Saponin | Sigma | S-7900 |
| Cisplatin | Enzo Life Sciences | ALX-400-040 |
| NaN3 | Sigma | S-8032 |
| Cell-ID 20-Plex Pd Barcoding Kit | Fluidigm | 201060 |
| Cell-ID 125 μm Iridium Intercalator | Fluidigm | 201192A |
| 16% paraformaldehyde | Electron Microscopy Sciences | 15710 |
| DNase I | Millipore Sigma | 10104159001 |
| Collagenase IX | Millipore Sigma | C7657 |
| Hyaluronidase | Worthington Biochemical Corp | LS005477 |
| buprenorphine hydrochloride | Hospira | 0409-2012-32 |
| bupivicaine | AuroMedica Pharma LLC | 55150-167-10 |
| OCT | Sakura | 4583 |
| Triton X-100 | Sigma | T8787 |
| Mouse serum | Jackson ImmunoResearch | 015-000-120 |
| Normal rat serum | Stem Cell Technologies | 13551 |
| Goat serum | Sigma | G9023 |
| Histodenz | Sigma | D-2158 |
| N-methylacetamide | Sigma | M26305-100G |
| 1-Thioglycerol | Sigma | M1753-100ML |
| 5X iScript RT Supermix | Bio-Rad | L001404B |
| SsoFast EvaGreen Supermix | Bio-Rad | L000915B |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1, 16 rxns | 10X Genomics | PN1000121 |
| Chromium Next GEM Chip G Single Cell Kit, 48 rxns | 10X Genomics | PN-1000120 |
| Chromium Next GEM Single Cell 5’ Library & Gel Bead Kit v1.1, 16 rxns | 10x Genomics | PN-1000165 |
| 10X Chromium Controller | 10x Genomics | N/A |
| Deposited data | ||
| Mouse spatiotemporal scRNAseq wound data set | This paper | GEO: GSE204777 |
| Human tumor scRNAseq data set | This paper | 10.17632/nnmb6m5p5j.1 |
| Mouse melanoma fibroblast data set | Davidson et al. (PMID: 32433953)73 | ArrayExpress: E-MTAB-7427 |
| Mouse pan-tissue fibroblast scRNAseq atlas | Buechler et al. (PMID: 33981032)22 | ArrayExpress:E-MTAB-10315 |
| Mouse pancreatic cancer fibroblast scRNAseq data set | Dominguez et al. (PMID: 31699795)54 | ArrayExpress: E-MTAB-8483 |
| Mouse wound scRNAseq data set | Guerrero-Juarez et al. (PMID: 31699795)26 | GEO: GSE113854 |
| Supplementary Mendeley Figures & Tables | Mendeley Data | 10.17632/kmmw43j2z6.2 |
| Experimental models: Cell lines | ||
| B16-F10 (mouse melanoma cell line) | ATCC | CRL-6475 |
| MC38 (mouse colorectal tumor cell line) | Kerafast | ENH204-FP |
| Experimental models: Organisms/strains | ||
| Mouse: wild-type C57BL/6 | The Jackson Laboratory | Stock# 000664 |
| Mouse: Arg1-tdTomato-CreERT2 × R26R-EYFP | H.-E. Liang & R. Locksley (UCSF)49 | https://www.sciencedirect.com/science/article/pii/S1074761319301992 |
| Mouse: Pdgfra-H2B-EGFP | A. Molofsky (UCSF)58 | Stock# 007669, |
| Oligonucleotides | ||
| See Supplementary Table S2 for qPCR primers | IDT | N/A |
| Software and algorithms | ||
| Normalizer | Finck et al, 20131103; https://onlinelibrary.wiley.com/doi/10.1002/cyto.a.22271 | https://rdrr.io/bioc/CATALYST/man/normCytof.html |
| Rphenograph | Levine et al, 2015104; https://www.sciencedirect.com/science/article/pii/S0092867415006376 | https://github.com/JinmiaoChenLab/Rphenograph |
| R environment | R Development Core Team | https://cran.r-project.org/bin/windows/base/ |
| Premessa | Parker ICI | https://github.com/ParkerICI/premessa |
| Flow Jo v10.8.1 | Tree Star | https://www.flowjo.com/ |
| Imaris 9.2.1 | Bitplane | https://imaris.oxinst.com/products/imaris-for-cell-biologists?gad=1&gclid=Cj0KCQjw3a2iBhCFARIsAD4jQB2RkztA4vyhmDUQ3_eo2WwjysDt-NhFrH-evt8PxW5s5oKpw_WYJhcaAuhZEALw_wcB |
| MATLAB | The MathWorks, Inc. | https://www.mathworks.com/products/matlab.html |
| LASX | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| CellChat (R package) | Jin et al. 202110; https://www.nature.com/articles/s41467-021-21246-9 | https://github.com/sqjin/CellChat |
| Seurat v3 (R package) | Stuart and Butler et al.105; Comprehensive Integration of Single-Cell Data. Cell (2019) [Seurat V3] | https://satijalab.org/seurat/ |
| NMF (R package) | Gaujoux R, Seoighe C (2010)110. “A flexible R package for nonnegative matrix factorization.” BMC Bioinformatics, 11(1), 367. ISSN 1471-2105, doi: 10.1186/1471-2105-11-367, http://www.biomedcentral.com/1471-2105/11/367. | https://cran.r-project.org/web/packages/NMF/index.html |
| Corrplot (R package) | https://cran.r-project.org/web/packages/corrplot/vignettes/corrplot-intro.html | https://cran.r-project.org/web/packages/corrplot/vignettes/corrplot-intro.html |
| Ggpubr (R package) | https://cran.r-project.org/web/packages/ggpubr/index.html | https://cran.r-project.org/web/packages/ggpubr/index.html |
| PhEMD | Chen et al., 2020106; https://www.nature.com/articles/s41592-019-0689-z | https://www.bioconductor.org/packages/release/bioc/html/phemd.html |
| Monocle 3 | Trapnell et al., 201443; http://dx.doi.org/10.1038/nbt.2859 | https://github.com/cole-trapnell-lab/monocle3 |
| CellRanger v 4.0.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Biorender | https://www.biorender.com | https://www.biorender.com/ |
| Other | ||
| lipid-modified oligonucleotides (LMO) | McGinnis et al., 201937; https://www.nature.com/articles/s41592-019-0433-8 | N/A |
| MaxPAR antibody conjugation kit | Fluidigm | 201146B |
| Dermal biopsy punch, 2 mm | Acuderm inc. | 69038-02 |
| Dermal biopsy punch, 4 mm | Integra | 33-34 |
| Dermal biopsy punch, 6 mm | Integra | 33-36 |
| Dermal biopsy punch, 8 mm | Integra | 33-37 |
| RNeasy Mini Kit | Qiagen | 74104 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All mice were housed in an American Association for the Accreditation of Laboratory Animal Care (AALAC)-accredited animal facility and maintained in specific pathogen-free conditions. All animal experiments were approved and performed in accordance with the Institutional Animal Care and Use Program protocol number AN184232. Wild-type female C57BL/6 mice between 6–12 weeks old were purchased from The Jackson Laboratory, Arg1-tdTomatoCreERT2 × R26R-EYFP mice were a kind gift from Drs. Hong-Erh Liang and Richard Locksley (UCSF), Pdgfra-H2B-EGFP+/wt mice were a kind gift from Dr. Ari Molofsky (UCSF). All mice were housed at the University of California, San Francisco (UCSF) animal facility with typical light/dark cycles and standard chow. For tumor growth studies, MC38 colon cancer (5×105 cells / 50 μl) or B16-F10 melanoma cancer cells (1×105 cells / 50 ul) were transplanted into the subcutaneous region of the mouse flank. On day 14 after tumor challenge, when tumors reached a size/volume of approximately 0.5 cm3, mice were sacrificed, tumors were excised and processed for downstream analysis.
Cell lines
B16-F10 and MC38 cells were purchased from ATCC and Kerafast, respectively, and cultured at 37°C in 5% CO2 in DMEM supplemented with 10% FBS (Benchmark), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Human tumor collection of the UCSF Immunoprofiler Initiative (IPI)
Tumor samples for the Immunoprofiler was transported from various cancer operating rooms (ORs). All patients consented by the UCSF IPI clinical coordinator group for tissue collection under a UCSF IRB approved protocol (UCSF IRB# 20-31740). Samples were obtained after surgical excision with biopsies taken by Pathology Assistants to confirm the presence of tumor cells. Patients were selected without regard to prior treatment. Freshly resected samples were placed in ice-cold PBS or Leibovitz’s L-15 medium in a 50 mL conical tube and immediately transported to the laboratory where they were chopped in small pieces (2–3mm2) and placed in a cryovial filled with 1mL of freezing media (10% DMSO, 90% FBS) and stored at −80 in a Polycarbonate container, blue high-density polyethylene closure, white high-density polyethylene vial holder overnight. The cryovials were then moved to a nitrogen tank for long-term storage.
Human tissue digestion and cell sorting for single cell RNA sequencing analysis
Tumor tissue contained in a cryovial was rapidly thawed in a 37C water bath and then the tissue was rinsed 3 times in complete media (RPMI with 10% FBS) before being thoroughly chopped with surgical scissors and transferred to GentleMACs C Tubes (Miltenyi Biotec) containing 20 uL/mL Liberase TL (5 mg/ml, Roche) and 50 U/ml DNAse I (Roche) in RPMI 1640 per 0.3 g tissue. GentleMACs C Tubes were then installed onto the GentleMACs Octo Dissociator (Miltenyi Biotec) and incubated for up to 45min according to the manufacturer’s instructions. Samples were then quenched with 15 mL of sort buffer (PBS/2% FBS/2mM EDTA), filtered through 100 μm filters and spun down. Red blood cell lysis was performed with 175 mM ammonium chloride if needed. Cells were then incubated with Human FcX (BioLegend) to prevent non-specific antibody binding. Cells were then washed in DPBS and incubated with Zombie Aqua Fixable Viability Dye (Thermo). Following viability dye incubation, cells were washed with sort buffer and incubated with cell surface antibodies mix diluted (containing anti-human CD45, anti-human CD3e, and anti-human HLA-DR) in the BV stain buffer (BD Biosciences) following manufacturer instruction for 30 minutes on ice in the dark. Cells were then washed three times and resuspended in sort buffer (PBS/2% FBS/2mM EDTA) after filtering through a 100 μm filter prior to flow sorting. Each sample was then sorted for viable immune cell (CD45+ viability dye−) and viable non-immune cells (CD45− viability dye−). Each sample was enriched for immune cells at a ratio of 80% immune and 20% non-immune cells. After sorting, cells were pelleted and resuspended at 103 cells/ul in 0.04%BSA/PBA and loaded onto the Chromium Controller (10X Genomics). Samples were processed for single-cell encapsulation and cDNA library generation using the Chromium Single Cell 5’ v1.1 Reagent Kits (10X Genomics). The library was subsequently sequenced on an Illumina Novaseq (Illumina). All samples were sequenced at 25,000 reads per cell.
Method details
Full-thickness wounding
All mice used for wounding experiments were between 7–10 weeks old (second telogen hair follicle phase). Two to four days before wounding, back skin was shaved and residual hair was removed using NAIR (Walgreens). On the day of wounding, mice were anesthetized with 3% isofluorane and subcutaneously injected with 50 μl of 0.25% bupivacaine and 50 μl of 50 μg/ml buprenorphine for analgesia. The back skin was then sterilized using a Betadine Solution swab stick (for scRNAseq and imaging experiments) or ChloraPrep (BD Biosciences) (for Mass Cytometry experiments to avoid iodine contamination during sample acquisition). Four full-thickness wounds were generated with a 4 mm biopsy punch. Wound diameter was measured with calipers.
Tissue Processing
At time of analysis, mice were euthanized, back skin was dissected from mice, excess scapular and inguinal fat was removed, and wounds plus adjacent tissue was excised using an 8 mm biopsy punch. An 8 mm biopsy punch was used to collect unwounded back skin as a day 0 or unwounded control. Four to six 8 mm biopsy punches were pooled from unwounded back skin to collect sufficient cells for downstream analysis. On ice, tissue was finely minced with scissors and then placed in a 2 ml tube containing 1 ml of digestion medium (2 mg/ml collagenase XI, 0.5 mg/ml hyaluronidase, 0.1 mg/ml DNase in RPMI with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM beta-mercaptoethanol). The tube was placed horizontally in a bacterial shaker for 45 min at 37°C and 225 rpm. The sample was then filtered through a 100 μm filter and washed with 10 ml cold RPMI. The generated single cell suspension was then processed for further depending on analysis method.
Mass Cytometry
Antibody conjugate generation
All mass cytometry antibodies are listed in the Key Resource Table. Primary conjugates of mass cytometry antibodies were prepared using the MaxPAR antibody conjugation kit (Fluidigm) according to the manufacturer’s instructions. After labeling, antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) containing 0.02% NaN3 at 0.1–0.3 mg/ml. Antibody conjugates were stored at 4°C. Each antibody clone and lot was titrated to optimal staining concentrations.
Cell Preparation
After single cell generation, cells were resuspended at 1×106 cells / ml PBS with 5 mM EDTA. An equal volume of PBS with 5 mM EDTA plus 50 μM Cisplatin (Enzo Life Sciences, Farmingdale, NY) was added and incubated for 60 s before quenching with an equal volume of PBS with 5 mM EDTA plus 0.5% BSA. Cells were centrifuged at 500 g for 5 min at 4°C. Washed cells were resuspended in PBS with 5 mM EDTA and fixed with 2.67% PFA for 10 min at RT. Fixation was quenched by adding 10x volume of PBS with 5 mM EDTA plus 0.5% BSA. Cells were centrifuged at 500 g for 5 min at 4°C and resuspended in PBS with 0.5% BSA and 10% DMSO before frozen and stored at −80°C until barcoding.
Mass-Tag Cellular Barcoding
After thawing stored samples at RT, up to 1×106 cells from each mouse were barcoded with distinct combinations of stable Pd isotopes in PBS with 0.02% saponin as described before101. After incubation at RT for 15 min on a shaker at 90 rpm, cells were pelleted at 600 g for 5 min at 4°C and washed two more times with cell staining media (CSM: PBS with 0.5% BSA and 0.02% NaN3). After the last wash, all samples were pooled into a single 15 ml tube.
Mass Cytometry Staining and Acquisition
After barcoding and pooling of samples, all cells were pelleted at 600 g for 5 min at 4°C and resuspended in CSM containing a metal-labeled anti-CD16/32 antibody for 5 min at RT on shaker at 90 rpm to block Fc receptors. Extracellular cell markers were stained by adding a master mix of metal-labeled antibodies listed in the Key Resource Table. After incubation for 30 min at RT on a shaker at 90 rpm, cells were washed with 5 ml of CSM. For cell permeabilization prior to intracellular stain, pelleted cells were resuspended in 1 ml of pre-chilled 99% MeOH (Sigma Aldrich) and incubated for 10 min at 4°C. Cells were washed twice in 5 ml of CSM prior to intracellular stain. For intracellular staining, pelleted cells were resuspended in the master mix of metal-labeled antibodies for intracellular targets and incubated for 30 min at RT on a shaker 90 rpm. Cells were washed with 5 ml of CSM and then resuspended in 2 ml PBS with 1.6% PFA and 0.55 μl 191/193Ir DNA Intercalator (Fluidigm). Cells were incubated overnight at 4°C. Cells were then washed once with 12 ml CSM, once with 12 ml PBS, and once with 12 ml of ddH2O prior to dilution in H2O at about 3×106 cells/ml containing normalization beads (see below), filtered through a 70 μm cell strainer and then analyzed on a Helios mass cytometer (Fluidigm, San Francisco, CA). We analyzed 3×104–2×105 cells per sample.
Mass Cytometry Bead Standard Data Normalization
Data normalization was performed as previously described102. All mass cytometry files were normalized together using the mass cytometry data normalization algorithm103, which uses the intensity values of a sliding window of the bead standards to correct for instrument fluctuations over time and between samples.
Data Analysis
After bead normalization and debarcoding using the Premessa package (https://github.com/ParkerICI/premessa), singlets were gated by Event Length and DNA content. Live cells were identified as Cisplatin-negative cells. Immune cells were identified as CD45+ cells and downsampled to 3.000–10.000 cells per sample prior to export and concatenation in FlowJo (Treestar). After concatenation of all cells from each timepoint, cell clusters were identified using the Phenograph algorithm104 using k=190 nearest neighbors. Mean intensity values of each marker per cluster were exported from FlowJo and imported into Morpheus (https://software.broadinstitute.org/morpheus/) to generate heatmaps. The identity of each cluster was determined based on expression of stained markers. Cell counts ber sample and CyTOF run can be found in Mendeley Table S1.
Single cell RNA sequencing of skin wound-associated cells
For spatiotemporal scRNAseq analysis, skin wounds were processed as described above to generate single cell suspensions, except that after excising the skin wound and adjacent tissue with an 8 mm biopsy punch, the excised skin wounds were further partitioned by successively using a 2 mm, 4 mm, and 6 mm biopsy punch, resulting in three rings and one round center sample, all with different diameters (Figure 1A). Digestion of tissue proceeded as described above, except that no DNase I was used in the digestion media to avoid potential downstream cleaving of nucleotide barcodes. After washing and pelleting of cells at 500 g for 5 min at 4, samples were each resuspended in 100 μl of staining buffer (PBS plus 2% FBS and 2 mM EDTA) and 2 μl of purified anti-mouse CD16/32 (Fc Shield, clone 2.4G2, Tonbo Biosciences, 2 mg/ml) was added to the sample. Samples were incubated for 15’ on ice to block FC receptors. Following this incubation, anti-mouse CD45 Alexa Fluor 647 (clone 30-F11, Biolegend, 1:1000) was added and cells were incubated for an additional 20’ on ice. Cells were washed using staining buffer, pelleted at 500 g for 5 min, and resuspended in staining buffer plus DAPI (1 μM). Cells were filtered on a 40 μm cell strainer right before being sorted on a BD FACSAria II cell sorter (BD Biosciences). CD45+ and CD45− cells were collected separately in ice cold collection buffer (colorless RPMI supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM beta-mercaptoethanol). Across all 17 samples (= 4 timepoints multiplied by 4 areas plus one unwounded sample), 206 × 103 CD45+ and 263.7 × 103 CD45− cells were collected. Cells of individual samples were pelleted at 500 g for 5 min at 4°C. Supernatant was removed and cells were resuspended in 160 μl of colorless RPMI. Samples were then barcoded using lipid modified oligonucleotides (LMO) as in McGinnis et al.37 before pooling. The pool was then split over 5 lanes of a 10X 3’ NEXTGEM chip before encapsulation with a targeted cell number of 12,000 per lane.
Following the MULTI-Seq library construction protocol, additive primer was spiked in during the cDNA amplification step as described in McGinnis et al37, the supernatant was reserved following SPRI cleanup and separately amplified. Finally, libraries were pooled for sequencing using a 1:10 molar ratio of LMO : gene expression (GEX) libraries with approximately equal representation from each lane. Pooled samples were then sequenced using 1 lane of a S4 flowcell for a target of ~3B reads. GEX libraries had total read counts of (493M, 515M, 495M, 452M, 403M) for lanes (1,2,3,4,5) respectively, showing similar order of magnitude representation in read counts. The estimated sequencing saturation ranged from 58–64% (calculated via CellRanger), indicating sequencing depth did not vary appreciably between lanes. LMO libraries had total read counts of (37M, 39M, 38M, 36M, 32M) also showing stable representation from all 5 lanes.
Each individual well was demultiplexed using deMULTIplex package37 to remove doublets and unlabeled cells. Confidently hashed cells were then carried forward for integration in Seurat v3105. From there, cells with percent mitochondrial reads >25 percent and number of genes <200 were filtered out. Following an initial high-level clustering, we removed several clusters composed mostly of high mitochondrial % cells or low nUMI and a immune/non-immune doublet cluster. An initial high-level clustering and dimensional reduction was used to define the CD45+ and CD45− subsets. Once subsetted, each group was then further re-clustered to generate the ‘final’ CD45+/− datasets.
Candidate identification of ligand-receptor interactions using CellChat
For CellChat analysis10, the M/M subset and fibroblast subsets were merged into a single Seurat object, then split into 5 Seurat objects based on the ‘Day’ metadata. Each Seurat object was then imported into a CellChat object using the ‘RNA’ assay. Signaling network likelihoods were calculated using CellChat’s computeCommunProb function using the trimean method, using raw data, and with population size scaling. Stacked barchart was calculated using the rankNet function with genes ordered by day weighted average.
Embedding a low-dimensional representation of samples using PhEMD
For mass cytometry data, PhEMD was employed to generate a two- or three-dimensional embedding of all samples split by timepoint based on their immune cell profiles106. For scRNAseq data, a two-dimensional embedding of CD45+ samples split by all space/timepoints based on their immune cell profiles was generated. Briefly, PhEMD first generates a reference map of cell subtypes, then uses Earth Mover’s Distance (EMD) to compute pairwise dissimilarities between samples (incorporating sample-to-sample differences in cell fractions of each cell subtype as well as intrinsic dissimilarities between subtypes based on the cell subtype reference map), and finally applies a dimensionality reduction technique to the sample-to-sample distance matrix to generate a final embedding of samples. The Seurat implementation of 3D Uniform Manifold Approximation and Projection (UMAP) was used to map the cell-subtype space. Dissimilarity between each pair of cell subtypes was defined as the distance between the centroids (in UMAP space) of all cells assigned to the two respective subtypes. PHATE was applied to the EMD patient-to-patient distance matrix to generate the final 3D embedding of samples107. For PhEMD application to CyTOF data, the fully concatenated clean datasets were converted into feature-cell sparse matrices that Seurat could import, analogous to a scRNA-Seq dataset. The data was then transformed using the arcsinh transform and processed as described above.
Pseudotime Analysis in scRNAseq data set
Monocle analysis
Raw counts from the individual cell-specific object were used to create a Monocle343,108,109 cell_data_set object, and the PCA and UMAP embeddings were imported directly from the Seurat object. Each cell-specific trajectory was inferred by reverse embedding the UMAP coordinates using the DDRTree method. The root cell states for the trajectory in the MHCIIlow and MHCIIhi Mono_Mac objects were chosen based on which cell cluster was present on day 1 post-wounding. Relative pseudotime was obtained through a linear transformation relative to the cells with the lowest and highest pseudotimes ((1 − min_pseudotime) / max_pseudotime).
Space/Time Tileplot of pseudotime
Similar to the Space/Time Tileplot of cell frequencies described above, a tileplot for the MHCIIlow and MHCIIhi Mono_Macs was created. The numbers in each tile represent the average pseudotime value of all cells found at that particular space/timepoint. ‘D00’ represents cells found in the unwounded skin and, therefore, is not split into different wound regions.
Ce3D Tissue Clearing and Whole Mount Imaging
Tissue staining and clearing
Wounds and surrounding tissue were excised from back skin using an 8 mm biopsy punch. Wound samples were cleared using the Ce3D clearing protocol46 with minor modifications. First, wound samples were fixed in 4% PFA (16% PFA, Electron Microscopy Sciences, diluted in PBS) at 4°C overnight on a horizontal shaker at 100 rpm by sandwiching them between two layers of cell strainer mesh to prevent sample curling. Subsequent steps were all performed protected from light. One wound sample was then transferred into a 2 ml tube and washed three times 30–60 min in 1 ml of wash buffer (PBS, 0.3% Triton X-100, 0.5% 1-thioglycerol) on a horizontal shaker at RT at 100 rpm. The inside of the 2 ml tube lid was plugged with a polydimethylsiloxane (PDMS) cut out to prevent the sample from being stuck during all subsequent washing, blocking, staining, and clearing steps. After washing, the sample was immersed in filtered blocking solution (PBS, 0.3% Triton X-100), 1% BSA, 1% normal mouse serum, and 1% normal serum of host species of used staining antibodies) overnight at 37°C on a horizontal shaker at 100 rpm. The next day, the sample was stained in 1 ml blocking buffer supplemented with DAPI (1 ug/ml) for nuclear staining, anti-CD49f/ITGA6 AF488 (clone GoH3; 1:100, stock 200 μg/ml), anti-CD11b AF647 (clone M1/70; 1:100, stock 500 μg/ml), anti-CD11b AF594 (clone M1/70; 1:100, stock 500 μg/ml), and/or anti-CD206 AF647 (clone C068C2; 1:200, stock 500 μg/ml) antibodies. The sample was incubated for three days at 37°C on a horizontal shaker at 100 rpm. After the staining incubation, the sample was transferred into a new 2 ml tube and washed once using 1 ml wash buffer for 8–12 h at 37°C on a horizontal shaker at 100 rpm, and then three more times with 1 ml wash buffer for 8–12 h at RT on a horizontal shaker at 100 rpm. After washing, the sample was transferred into a new 2 ml tube for clearing by subsequently incubating the sample in 1 ml of 33%, 50%, 80%, or 100% Ce3D clearing solution (2.75 ml 40% N-methylacetamide, 4 g Histodenz, 5 μl Triton X-100, 25 μl 1-thioglycerol) diluted in wash buffer at RT on a horizontal shaker at 100 rpm. The 33%, 50%, and 80% Ce3D clearing steps were done for 1 h each and the 100% clearing step was done overnight. After clearing, the sample was mounted epidermal side facing down on a PDMS chamber that fit the sample in the middle. Ce3D clearing solution was used as the mounting medium.
‘Thick’ section clearing:
For ‘thick’ section (250–300 μm) clearing and imaging for both wounds and tumor samples, we started with whole tissue (wound or tumor) and fixed with 4% PFA at 4C overnight. This was followed by a progressive 15% to 30% w/v sucrose gradient then embedding and freezing in OCT. Thick sections were then made using a cryostat and placed into PBS to wash OCT residue away. These sections were then carried through a generalized Ce3D workflow12. Blocking and wash buffers same as above. For the POSTN and αSMA staining in Figure 3 and S3, we used the following primary antibodies for 1 day at 37C followed by 1 day at RT in Ce3D blocking buffer supplemented with 1% Normal Goat Serum: anti-aSMA (1:200 Abcam stock at 0.2mg/mL) and anti-POSTN (1:200 R&D stock 500 μg/ml). Following a 1 day wash, we stained with the following secondaries for 2 days at RT: Goat anti-rabbit F(ab’)2 AF555 (1:1000, Thermo stock 2 mg/mL) and Goat anti rabbit F(ab) AF647 (1:1000, Jackson Immuno stock 1.25 mg/mL) and DAPI at 2 ug/mL. Samples were then washed for another day before clearing and mounting as above.
For imaging of Selectin-P and CD31 vasculature in thick tumor sections in Figure 7, sections were blocked in Ce3D blocking buffer with 1% NDS and NRS, then stained with primary anti mouse P-selectin (1:200, R&D stock 0.2 mg/mL) and anti-CD31 (1:100) and. After washing, we applied to following stains: donkey anti-goat AF488 (1:1000, Thermo stock 2mg/mL), anti-CD31 (1:100, Biolegend 0.5 mg/mL) and anti-I-A/I-E AlexaFluor594 (1:200, Biolegend stock 0.5mg/mL) and DAPI at 2 ug/mL. Following staining, thick sections were cleared as above and mounted in additional clearing solution in cavity slides, then cover slipped.
‘Thin’ cryosection imaging:
For thin (10 μm) sections, samples were embedded and frozen in OCT, then sectioned on the cryostat and transferred to slides. Samples were then fixed 2 hr 4C in 4% PFA before permeabilization for 15 min in 0.2% Triton X-100. Samples were then blocked using (PBS, 0.1% Triton X-100), 1% BSA, and 1% normal serum of host species of used staining antibodies) and stained in the same buffer. All staining steps were performed at 4C overnight. For thin sections of wounds in Supplementary Figure 4, we first stained for POSTN using rat-derived anti-human/mouse periostin antibody (clone 345624, R&D Systems, 1:200, stock 500 μg/ml) and secondary AF488 goat anti-rat F(ab) (1:1000, stock 2 mg/mL). Following washing and blocking with Normal Rat Serum, we stained with anti-mouse GPNMB eFluor660 (clone CTSREVL, ThermoFisher, 1:200, 200 μg/ml) as well as 1 ug/mL DAPI. For thin tumor sections in Figure 7, we first stained for POSTN using rat-derived anti-human/mouse periostin antibody (clone 345624, R&D Systems, 1:200, stock 500 μg/ml) and secondary AF488 goat anti-rat F(ab) (1:1000, stock 2 mg/mL) as above Following washing and blocking with Normal Rat Serum, we stained with anti-mouse CD11b AF594 (Biolegend clone M1/70; 1:100, stock 500 μg/ml) as well as 1 ug/mL DAPI. Slides were washed and coverslipped using VectaShield.
Image acquisition and analysis
All samples were imaged using a Leica SP8 laser scanning confocal microscope with a white light laser and 405 nm diode. For whole cleared wounds, a 16X 0.6NA (HC Fluotar L 16x/0.6 IMM CORR DLS, Leica) objective was used during acquisition. ‘Thick’ (250–300 μm) and ‘thin’ (10 μm) cryosections were imaged using a 20X 0.75NA (HC PL APO 20x/0.75 IMM CORR CS2, Leica) objective. After acquisition, individual tiled images were stitched together using the LAS X software (Leica) and then analyzed using the Imaris software suite (Bitplane).
Imaris processing
For large volume images, spots were created using the Imaris Spots function and inputting the DAPI nuclear stain signal. Parameters for Spots generation are listed in Mendeley Table S2. This first step is done to locate and identify individual cells within the imaged sample. Spherical spots here are created based on signal above a user-defined threshold value in the DAPI channel and based on the size of the DAPI object. Next, Surfaces for channels CD11b, Arg1-tdTomato, and CD206 were created based on the parameters listed in Mendeley Table S2 using the Imaris Surfaces function. Surface marker-positive cells were identified by their vicinity to a previously identified DAPI spot. Depending on surface marker (see Mendeley Table S2 for details), DAPI spots 5–20 μm close to the Surface were identified as CD11b+, Arg1+, and/or CD206+ using the Find Spots Close to Surface function in Imaris. To identify CD11b+ Arg1+ or CD11b+ CD206+ double positive cells, the Colocalize Spots function selecting CD11b -and Arg1-positive spots or CD11b- and CD206-positive spots, respectively, with a threshold of =<1 μm was used in Imaris.
Ex vivo experiments of primary skin fibroblast and bone marrow-derived macrophages (BMDMs)
Primary skin fibroblast isolation
6–12 week old C57BL/6 mice were euthanized and their ventral skin covering the thorax was shaved and residual hair were removed by NAIR application. The skin was excised and remaining subcutaneous fat was removed before the tissue was finely minced with scissors. The tissue was then transferred into a 2 ml tube containing 1 ml of digestion medium (2 mg/ml collagenase XI, 0.5 mg/ml hyaluronidase, 0.1 mg/ml DNase in RPMI with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM beta-mercaptoethanol). Tissue from one skin excision was split into 3 tubes. The tubes were placed horizontally in a bacterial shaker for 90 min at 37°C and 225 rpm. After digestion, the samples were filtered on a 100 μm cell strainer and the digestion was quenched by adding 20 ml of ice cold RPMI. The single cell suspension was then seeded at a concentration of 2×104 cells / cm2 in fibroblast media (DMEM supplemented with 10% FBS, 2 mM L-glutamine, 50 μM beta-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B) at 37°C in 5% CO2. After 3 days or when culture reached 80–90% confluency, cells were collected, aliquoted, and stored in liquid nitrogen (passage 1). For subsequent co-culture experiments, cells were thawed and cultured in fibroblast media for one more passage before use in experiments.
Generation of bone marrow-derived macrophages (BMDMs)
6–12 week old C57BL/6 mice were euthanized and their femurs and tibiae were excised. Using PBS, the bone marrow was flushed from the long bones after cutting off the ends. After pelleting the bone marrow, the red blood cells were lysed using RBC lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0.1 mM EDTA) for 5 min at RT. Cells were washed with DMEM and filtered through a 40 μm cell strainer before being seeded at 1×106 cells / ml on a low-adherent cell culture dish in BMDM media (DMEM supplemented with 10% FBS (Benchmark), 2 mM L-glutamine, 50 μM beta-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml M-CSF). After 3 days in culture, cells were split 1:2 into new low-adherent cell culture dishes with fresh BMDM media. On day 6–7 of culture, BMDMs were harvested and used for experiments.
For Oncostatin M stimulation, PSF’s were grown to 80% confluency, before switching to DMEM+1% FBS with 50 ng/mL of recombinant OSM (Biolegend) and incubated 24hrs at 37C before harvest for RNA extraction.
Conditioned BMDM media from Grn−/− was generated as follows: BMDM’s were generated as described above from bone marrow from Grn−/− mice. On D7 following BMDM isolation and differentiation, the media was replaced with DMEM+1%FBS and harvested 24 hours following incubation. PSF’s were grown till 80% confluent then media was replaced with Grn−/− BMDM CM with or without 1 ug/mL recombinant Progranulin (R&D). PSF’s were incubated at 37C for 24hrs before harvest for RNA extraction. For Periostin treatment, Collagen IV coated 6 well plates (Corning 354428) were coated with 1 ug/mL recombinant mouse Periostin (R&D 2955-F2-050) overnight at 4C. Plates were then washed twice with PBS before cell plating. BMDM’s D8 post-isolation and differentiation were then plated at a density of 750k cells per well and briefly spun down at 200g for 15 seconds to force them into contact with the plate. BMDM’s were then incubated 24 hrs at 37C before RNA extraction.
Reverse transcription real-time quantitative PCR
Following cell harvest and washing 1X cold PBS, cells were lysed and RNA extraction performed using the RNeasy kit from Qiagen. Roughly equal mass of RNA was then used for reverse transcription using the iScript Master Mix. The reaction mix was directly used for RT-qPCR with the SsoFast EvaGreen kit and read out on a Bio-Rad thermocycler with the following cycling protocol: [95C for 30s, 95C for 5s, 60C for 2s, repeat from step 2 44x, 65C to 95C melt curve with 0.5C increments at 5s each]. Primer sequences can be found in the key resources table. We confirmed that melt curves were consistent across samples and the non-template controls has no appreciable amplification. Cq was calculated at roughly ¼ the maximum signal and then corrected against 18s Cq before conversion to relative expression. Shown bar charts are representative of the mean+SE of 3 technical replicate wells.
Quantification and statistical analysis
Statistical details of individual experiments can be found in their respective figure legends.
Quantification of cells in microscopy images
Quantification in whole-mount wounds
To quantify the number of cells per area of wound in the Imaris software, first, a user-defined ‘center’ spot was selected in the wound that represents the center of the wound. This spot was duplicated to create a new spot object. Then, the duplicated ‘center’ spot was selected and using the ‘Distance Transformation’ tool under ‘Tools -> Create new Channel’ a new channel was generated, wherein the channel intensity across the sample reflects the distance from the previously selected center spot. Areas close to the center spot have a low intensity and areas distant from the center have a high intensity in the ‘Distance Transformation’ channel. To calculate how many cells/spots are located at what distance from the center of the wound, the 8 mm wound cutout used for imaging was subdivided into 8 circular bins (0–0.5 mm distance from center of wound; 0.5–1.0; 1.0–1.5; 1.5–2.0; 2.0–2.5; 2.5–3.0; 3.0–3.5; 3.5–4.0). Cells/spots with a cell marker of interest was selected, for example ‘CD11b+’ spots, and a new filter was created called ‘Intensity Mean’ of the ‘Distance Transformation’ channel. Selection of the maximum and minimum filter settings reflect which of the 8 circular bins all ‘CD11b+’ spots are counted. The same was done for all ‘CD11b+ Arg1-tdTomato+’ spots and the percentage of ‘CD11b+ Arg-tdTomato+ of CD11b+’ spots was calculated for each of the 8 circular bins and plotted. The same analysis was done for ‘CD11b+ CD206+’ spots.
Quantification in thick and thin wound and tumor cryosections
To quantify the distance of cells to POSTN or Selectin-P or CD31 surfaces, we used surfaces generated using parameters listed in the Imaris parameters table (see Mendeley Data) and created a distance transform channel from the outside of these surfaces. The value of this channel at the center of the cells/spots was used as the distance value in histograms and distributions for KS testing.
Space/Time Tileplot of cell frequencies
To depict the distribution of identified cell subsets in the scRNAseq data set within the wound over time, we devised a Space/Time Tileplot that is a 4×4 grid, where the x-axis is split into the four time points of sampling (day 1, day 3, day 7, and day 14) and the y-axis is split into the four areas of the sampled wound (2 mm wound center, 4 mm ring, 6 mm ring, 8 mm ring). This generates 16 space/timepoints. To plot the tileplot, we used the ‘geom_tile’ function from the ggplot2 R package. First, each cell cluster’s percentage within a larger object (for example the ‘Mono_Mac_1’ cluster within the larger ‘Mono_Mac’ object) is calculated for each space/timepoint. These are then plotted on the 4×4 grid and color-coded based on their relative change compared to the unwounded state. Red indicates increase in subpopulation, white indicates the same percentage, blue indicates decrease in subpopulation compared to UW. Code is deposited and provided.
Space/Time Correlation Analysis (STCA)
To identify cell subsets in the scRNAseq data set that shared similar space/time profiles, we calculated the Pearson correlation coefficient, R, using the percentage for every space/timepoint between pairs of cell subsets. A Pearson correlation coefficient of R = 1 between a pair of cell subsets would indicate that they have the same space/time profile across the wound healing process. A Pearson correlation coefficient of R = −1 would indicate an negative correlation between the cell subsets, i.e. that the presence of the two cell subsets are inversely correlated across the wound. P-values were calculated using either Pearson or Spearman method (noted in legends). The Benjamini-Hochberg correction was applied for false discovery rate correction.
NMF Decomposition
The CD45−/+ objects were subsetted according to broad cell type definitions (M/M, Neutrophils, DC’s, Mast Cells, T, TNK, B and Fibroblast, Keratinocyte, Endothelial, Melanocyte, Dermal sheath papilla, vSM). The top 1250 most variable genes (depending on diversity of subset expression patterns) were selected using the ‘vst’ method in Seurat. Using the scaled RNA assay (non-centered) and subsetting out the most variable genes, we thus created a 1250xN (N = # of cells) expression matrix, then applied the non-smooth NMF algorithm (as described in NMF package110). We used a parameter sweep combined with the cophenetic metric to find the most stable number of factors using 50 iterations. For gene weights per factor, we used the basis matrix output from the nsNMF and sorted based on the highest contributing genes. For plotting purposes, we normalized gene contributions across all factors to understand how specific a given gene was for a factor. We used the coefficient matrix as the ‘expression’ value for each cell for that factor. This value was used directly for average factor expression and feature plots.
When translating factors between tumor and wound healing, we used a Jaccard distance metric based on the top 20 contributing genes for each factor.
Supplementary Material
Clearing-enhanced (Ce3D) skin wound D3 post-wounding of wild-type C57BL/6 mouse. Original wound size, 4 mm. Movie starts with internal-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and CD206 (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis. ITGA6/CD49b staining (white) highlights re-established epithelial basement membrane, vasculature structures in non-wounded skin, hair follicles, and fascia.
Clearing-enhanced (Ce3D) skin wound D7 post-wounding of wild-type C57BL/6 mouse. Original wound size, 4 mm. Movie starts with internal-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and CD206 (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis. ITGA6/CD49b staining (white) highlights re-established epithelial basement membrane, vasculature structures in non-wounded skin, hair follicles, fascia, and severed nerve bundles.
Clearing-enhanced (Ce3D) skin wound D3 post-wounding of Arg1-tdTomato reporter mouse. Original wound size, 4 mm. Movie starts with internal-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and Arg1-tdTomato (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis. ITGA6/CD49b staining (white) highlights reestablished epithelial basement membrane, vasculature structures in non-wounded skin, hair follicles, and fascia.
Clearing-enhanced (Ce3D) skin wound D7 post-wounding of Arg1-tdTomato reporter mouse. Original wound size, 4 mm. Movie starts with external-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and Arg1-tdTomato (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis.
Top 20 contributing genes by gene weight from basis matrix shown for each of the 114 identified factors along with their gene weight towards that factor.
Genes in the upper right quadrant (I) of the tumor by wound healing M_M factor scatterplots (Figure 6C–E and Supplementary Figure SD–I) were used for GO analysis. Table shows a truncated output from each query for each pair with
Highlights:
Space-time scRNA-Seq of healing skin wounds across immune and non-immune cells
Gene program analysis reveals coordinated multicellular movements
Prediction of macrophage-fibroblast crosstalk involving OSM, TNC, and POSTN signals
Translation of gene program pairs to mouse and human tumor settings
Acknowledgements:
We would like to thank Drs. Hong-Erh Liang and Richard Locksley for the generous gift of the Arg1-reporter mouse. We also thank Drs. Chris McGinnis and Zev Gartner for the LMO’s and comments on manuscript and Dr. Ian Boothby for advice on the mouse wound healing model. Additionally, we thank members of the Krummel lab for comments on the manuscript. This work was supported by funds from NIH R01CA197363. K.H.H. is supported by the American Cancer Society Postdoctoral Fellowship (#133078-PF-19-222-01-LIB) and N.F.K. is supported by NIH T32 5T32CA108462-17. Flow cytometry was performed at the UCSF Parnassus Flow CoLab, RRID:SCR_018206, and supported in part by the DRC Center Grant NIH P30 DK063720. We also thank the Data Science CoLab for bioinformatic support and the Center for Advanced Technology at UCSF for sequencing support. Finally, we thank all patients and their families, for placing their trust in us.
Footnotes
Declarations of interest:
M.F.K. is a founder and shareholder of PIONYR immunotherapeutic and FOUNDERY innovations. All other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wertheimer T, Velardi E, Tsai J, Cooper K, Xiao S, Kloss CC, Ottmüller KJ, Mokhtari Z, Brede C, deRoos P, et al. (2018). Production of BMP4 by endothelial cells is crucial for endogenous thymic regeneration. Sci Immunol 3. 10.1126/sciimmunol.aal2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walesky CM, Kolb KE, Winston CL, Henderson J, Kruft B, Fleming I, Ko S, Monga SP, Mueller F, Apte U, et al. (2020). Functional compensation precedes recovery of tissue mass following acute liver injury. Nat Commun 11. 10.1038/s41467-020-19558-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodrigues M, Kosaric N, Bonham CA, and Gurtner GC (2019). Wound healing: A cellular perspective. Physiol Rev. 10.1152/physrev.00067.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li P, and Elowitz MB (2019). Communication codes in developmental signaling pathways. Development (Cambridge) 146. 10.1242/dev.170977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, Flavell RA, Mayo A, Alon U, and Medzhitov R (2018). Circuit Design Features of a Stable Two-Cell System. Cell 172. 10.1016/j.cell.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, Vanneste B, De Prijck S, Nedospasov SA, Kremer A, et al. (2019). Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 51. 10.1016/j.immuni.2019.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33. 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, Ni B, Sklar J, Przytycka TM, Childs R, et al. (2015). Genome-wide detection of DNase i hypersensitive sites in single cells and FFPE tissue samples. Nature 528. 10.1038/nature15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523. 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat Commun 12. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc 15. 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 12.Browaeys R, Saelens W, and Saeys Y (2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 17. 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 13.Moor AE, Harnik Y, Ben-Moshe S, Massasa EE, Rozenberg M, Eilam R, Bahar Halpern K, and Itzkovitz S (2018). Spatial Reconstruction of Single Enterocytes Uncovers Broad Zonation along the Intestinal Villus Axis. Cell 175. 10.1016/j.cell.2018.08.063. [DOI] [PubMed] [Google Scholar]
- 14.Manco R, Averbukh I, Porat Z, Bahar Halpern K, Amit I, and Itzkovitz S (2021). Clump sequencing exposes the spatial expression programs of intestinal secretory cells. Nat Commun 12. 10.1038/s41467-021-23245-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egozi A, Bahar Halpern K, Farack L, Rotem H, and Itzkovitz S (2020). Zonation of Pancreatic Acinar Cells in Diabetic Mice. Cell Rep 32. 10.1016/j.celrep.2020.108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S, Gonzalez DG, Guirao B, Boucher JD, Cockburn K, Marsh ED, Mesa KR, Brown S, Rompolas P, Haberman AM, et al. (2017). Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nat Cell Biol 19. 10.1038/ncb3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin P (1997). Wound healing - Aiming for perfect skin regeneration. Science (1979) 276. 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 18.Eming SA, Wynn TA, and Martin P (2017). Inflammation and metabolism in tissue repair and regeneration. Science (1979) 356. 10.1126/science.aam7928. [DOI] [PubMed] [Google Scholar]
- 19.Brazil JC, Quiros M, Nusrat A, and Parkos CA (2019). Innate immune cell–epithelial crosstalk during wound repair. Journal of Clinical Investigation 129. 10.1172/JCI124618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eming SA, Martin P, and Tomic-Canic M (2014). Wound repair and regeneration: Mechanisms, signaling, and translation. Sci Transl Med 6. 10.1126/scitranslmed.3009337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson HN, and Hardman MJ (2020). Wound healing: cellular mechanisms and pathological outcomes: Cellular Mechanisms of Wound Repair. Open Biol 10. 10.1098/rsob.200223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buechler MB, Fu W, and Turley SJ (2021). Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 54. 10.1016/j.immuni.2021.04.021. [DOI] [PubMed] [Google Scholar]
- 23.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, Roers A, and Eming SA (2010). Differential Roles of Macrophages in Diverse Phases of Skin Repair. The Journal of Immunology. 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 24.Shook BA, Wasko RR, Rivera-Gonzalez GC, Salazar-Gatzimas E, López-Giráldez F, Dash BC, Muñoz-Rojas AR, Aultman KD, Zwick RK, Lei V, et al. (2018). Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science (1979). 10.1126/science.aar2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu SX, Gustafson HH, Jackson DL, Pun SH, and Trapnell C (2020). Trajectory analysis quantifies transcriptional plasticity during macrophage polarization. Sci Rep 10. 10.1038/s41598-020-68766-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerrero-Juarez CF, Dedhia PH, Jin S, Ruiz-Vega R, Ma D, Liu Y, Yamaga K, Shestova O, Gay DL, Yang Z, et al. (2019). Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat Commun 10. 10.1038/s41467-018-08247-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brancato SK, and Albina JE (2011). Wound macrophages as key regulators of repair: Origin, phenotype, and function. American Journal of Pathology 178. 10.1016/j.ajpath.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dvorak HF (2015). Tumors: Wounds that do not heal-redux. Cancer Immunol Res 3. 10.1158/2326-6066.CIR-14-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacCarthy-Morrogh L, and Martin P (2020). The hallmarks of cancer are also the hallmarks of wound healing. Sci Signal 13. 10.1126/SCISIGNAL.AAY8690. [DOI] [PubMed] [Google Scholar]
- 30.Nathan C, and Ding A (2010). Nonresolving Inflammation. Cell 140. 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 31.Rosato PC, Wijeyesinghe S, Stolley JM, and Masopust D (2020). Integrating resident memory into T cell differentiation models. Curr Opin Immunol 63. 10.1016/j.coi.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159. 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, Warren A, Rodman C, Roth JA, Bender SA, et al. (2020). Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 52. 10.1038/s41588-020-00726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shook BA, Wasko RR, Mano O, Rutenberg-Schoenberg M, Rudolph MC, Zirak B, Rivera-Gonzalez GC, López-Giráldez F, Zarini S, Rezza A, et al. (2020). Dermal Adipocyte Lipolysis and Myofibroblast Conversion Are Required for Efficient Skin Repair. Cell Stem Cell 26. 10.1016/j.stem.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson-Smith AC, et al. (2013). Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504. 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mascharak S, des Jardins-Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL, Chen K, Duoto B, Chinta M, Foster DS, et al. (2021). Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science (1979) 372. 10.1126/science.aba2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS, Werb Z, et al. (2019). MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat Methods 16. 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, Patel R, Gautier EL, Hugues S, Longhi MP, et al. (2016). The Heterogeneity of Ly6Chi Monocytes Controls Their Differentiation into iNOS+ Macrophages or Monocyte-Derived Dendritic Cells. Immunity 45. 10.1016/j.immuni.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boulet S, Daudelin JF, Odagiu L, Pelletier AN, Yun TJ, Lesage S, Cheong C, and Labrecque N (2019). The orphan nuclear receptor NR4A3 controls the differentiation of monocyte-derived dendritic cells following microbial stimulation. Proc Natl Acad Sci U S A 116. 10.1073/pnas.1821296116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, and Jain MK (2006). Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci U S A 103. 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, and Barres BA (2018). A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98. 10.1016/j.neuron.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50. 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blecher-Gonen R, Bost P, Hilligan KL, David E, Salame TM, Roussel E, Connor LM, Mayer JU, Bahar Halpern K, Tóth B, et al. (2019). Single-Cell Analysis of Diverse Pathogen Responses Defines a Molecular Roadmap for Generating Antigen-Specific Immunity. Cell Syst 8. 10.1016/j.cels.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, and Overall CM (2008). Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR + CXC chemokines and generates CCL2, −7, −8, and −13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112. 10.1182/blood-2007-12-129080. [DOI] [PubMed] [Google Scholar]
- 46.Li W, Germain RN, and Gerner MY (2019). High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat Protoc. 10.1038/s41596-019-0156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado JDD, Popovich PG, Partida-Sanchez S, and Guerau-De-arellano M (2015). Novel markers to delineate murine M1 and M2 macrophages. PLoS One 10. 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mujal AM, Combes AJ, Rao AA, Binnewies M, Samad B, Tsui J, Boissonnas A, Pollack JL, Argüello RJ, Meng MV, et al. (2022). Holistic Characterization of Tumor Monocyte-to-Macrophage Differentiation Integrates Distinct Immune Phenotypes in Kidney Cancer. Cancer Immunol Res 10, 403–419. 10.1158/2326-6066.CIR-21-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider C, Lee J, Koga S, Ricardo-Gonzalez RR, Nussbaum JC, Smith LK, Villeda SA, Liang HE, and Locksley RM (2019). Tissue-Resident Group 2 Innate Lymphoid Cells Differentiate by Layered Ontogeny and In Situ Perinatal Priming. Immunity 50. 10.1016/j.immuni.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu H, Yoo PK, Aguirre CC, Tsoa RW, Kern RM, Grody WW, Cederbaum SD, and Iyer RK (2003). Widespread expression of arginase I in mouse tissues: Biochemical and physiological implications. Journal of Histochemistry and Cytochemistry 51. 10.1177/002215540305100905. [DOI] [PubMed] [Google Scholar]
- 51.Franklin RA (2021). Fibroblasts and macrophages: Collaborators in tissue homeostasis. Immunol Rev 302. 10.1111/imr.12989. [DOI] [PubMed] [Google Scholar]
- 52.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214. 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 9. 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov 10. 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 55.Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, et al. (2020). Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun 11. 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, Yang YA, Tam L, Caothien R, Roose-Girma M, et al. (2021). Cross-tissue organization of the fibroblast lineage. Nature 593. 10.1038/s41586-021-03549-5. [DOI] [PubMed] [Google Scholar]
- 57.Vu R, Jin S, Sun P, Haensel D, Nguyen QH, Dragan M, Kessenbrock K, Nie Q, and Dai X (2022). Wound healing in aged skin exhibits systems-level alterations in cellular composition and cell-cell communication. Cell Rep 40, 111155. 10.1016/j.celrep.2022.111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamilton TG, Klinghoffer RA, Corrin PD, and Soriano P (2003). Evolutionary Divergence of Platelet-Derived Growth Factor Alpha Receptor Signaling Mechanisms. Mol Cell Biol 23, 4013–4025. 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Droin C, Kholtei J. El, Bahar Halpern K, Hurni C, Rozenberg M, Muvkadi S, Itzkovitz S, and Naef F. (2021). Space-time logic of liver gene expression at sub-lobular scale. Nat Metab 3, 43–58. 10.1038/s42255-020-00323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, Burcham AR, Salhotra A, Ransom RC, Henn D, et al. (2021). Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2110025118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee DD, and Seung HS (1999). Learning the parts of objects by non-negative matrix factorization. Nature 401. 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- 62.Stein-O’Brien GL, Arora R, Culhane AC, Favorov AV, Garmire LX, Greene CS, Goff LA, Li Y, Ngom A, Ochs MF, et al. (2018). Enter the Matrix: Factorization Uncovers Knowledge from Omics. Trends in Genetics 34. 10.1016/j.tig.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kotliar D, Veres A, Nagy MA, Tabrizi S, Hodis E, Melton DA, and Sabeti PC (2019). Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife 8. 10.7554/eLife.43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184. 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeBruine ZJ, Melcher K, and Triche TJ Jr (2021). Fast and robust non-negative matrix factorization for single-cell experiments. bioRxiv. 10.1101/2021.09.01.458620. [DOI] [Google Scholar]
- 66.Pascual-Montano A, Carazo JM, Kochi K, Lehmann D, and Pascual-Marqui RD (2006). Nonsmooth nonnegative matrix factorization (nsNMF). IEEE Trans Pattern Anal Mach Intell 28. 10.1109/TPAMI.2006.60. [DOI] [PubMed] [Google Scholar]
- 67.Elosua-Bayes M, Nieto P, Mereu E, Gut I, and Heyn H (2021). SPOTlight: Seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res 49. 10.1093/nar/gkab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pascual-Montano A, Carazo JM, Kochi K, Lehmann D, and Pascual-Marqui RD (2006). Nonsmooth nonnegative matrix factorization (nsNMF). IEEE Trans Pattern Anal Mach Intell 28. 10.1109/TPAMI.2006.60. [DOI] [PubMed] [Google Scholar]
- 69.Brunet JP, Tamayo P, Golub TR, and Mesirov JP (2004). Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A 101. 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hongu T, Pein M, Insua-Rodríguez J, Gutjahr E, Mattavelli G, Meier J, Decker K, Descot A, Bozza M, Harbottle R, et al. (2022). Perivascular tenascin C triggers sequential activation of macrophages and endothelial cells to generate a pro-metastatic vascular niche in the lungs. Nat Cancer 3, 486–504. 10.1038/s43018-022-00353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, and Chang DD (2002). Periostin secreted by epithelial ovarian carcinoma is a ligand for αVβ3 and αVβ5 integrins and promotes cell motility. Cancer Res 62. [PubMed] [Google Scholar]
- 72.Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: A complex web of host defenses. Annu Rev Immunol 32. 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, Kar G, Vento-Tormo R, Hagai T, Chen X, et al. (2020). Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep 31. 10.1016/j.celrep.2020.107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Walz A, Burgener R, Car B, Baggiolini M, Kunkel SL, and Strieter RM (1991). Structure and Neutrophil-activating Properties of a Novel Inflammatory Peptide (ENA-78) with Homology to Interleukin 8. Journal of Experimental Medicine 174. 10.1084/jem.174.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doni A, Stravalaci M, Inforzato A, Magrini E, Mantovani A, Garlanda C, and Bottazzi B (2019). The long pentraxin PTX3 as a link between innate immunity, tissue remodeling, and cancer. Front Immunol 10. 10.3389/fimmu.2019.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richards CD, Izakelian L, Dubey A, Zhang G, Wong S, Kwofie K, Qureshi A, and Botelho F (2016). Regulation of IL-33 by Oncostatin M in Mouse Lung Epithelial Cells. Mediators Inflamm 2016. 10.1155/2016/9858374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Junk DJ, Bryson BL, Smigiel JM, Parameswaran N, Bartel CA, and Jackson MW (2017). Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 36. 10.1038/onc.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yin W, Li Y, Song Y, Zhang J, Wu C, Chen Y, Miao Y, Lin C, Lin Y, Yan D, et al. (2021). CCRL2 promotes antitumor T-cell immunity via amplifying TLR4-mediated immunostimulatory macrophage activation. Proceedings of the National Academy of Sciences 118. 10.1073/pnas.2024171118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu X, Zhang Y, Yu Y, Yang X, and Cao X (2008). SOCS3 promotes TLR4 response in macrophages by feedback inhibiting TGF-β1/Smad3 signaling. Mol Immunol 45, 1405–1413. 10.1016/j.molimm.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 80.Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T, Jahchan NS, Canaday P, Lu E, Norng M, et al. (2021). Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep 37. 10.1016/j.celrep.2021.109844. [DOI] [PubMed] [Google Scholar]
- 81.Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, Bornstein C, Moshe A, Keren-Shaul H, Cohen M, et al. (2020). Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 182. 10.1016/j.cell.2020.06.032. [DOI] [PubMed] [Google Scholar]
- 82.Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, et al. (2020). TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182. 10.1016/j.cell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. (2015). Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17. 10.1038/ncb3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kormann R, Kavvadas P, Placier S, Vandermeersch S, Dorison A, Dussaule JC, Chadjichristos CE, Prakoura N, and Chatziantoniou C (2020). Periostin promotes cell proliferation and macrophage polarization to drive repair after AKI. Journal of the American Society of Nephrology 31. 10.1681/ASN.2019020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.González-González L, and Alonso J (2018). Periostin: A matricellular protein with multiple functions in cancer development and progression. Front Oncol 8. 10.3389/fonc.2018.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Combes AJ, Samad B, Tsui J, Chew NW, Yan P, Reeder GC, Kushnoor D, Shen A, Davidson B, Barczak AJ, et al. (2022). Discovering dominant tumor immune archetypes in a pan-cancer census. Cell 185, 184–203.e19. 10.1016/j.cell.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harnik Y, Buchauer L, Ben-Moshe S, Averbukh I, Levin Y, Savidor A, Eilam R, Moor AE, and Itzkovitz S (2021). Spatial discordances between mRNAs and proteins in the intestinal epithelium. Nat Metab 3, 1680–1693. 10.1038/s42255-021-00504-6. [DOI] [PubMed] [Google Scholar]
- 88.Henderson NC, Rieder F, and Wynn TA (2020). Fibrosis: from mechanisms to medicines. Nature 587. 10.1038/s41586-020-2938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eming SA, Murray PJ, and Pearce EJ (2021). Metabolic orchestration of the wound healing response. Cell Metab 33. 10.1016/j.cmet.2021.07.017. [DOI] [PubMed] [Google Scholar]
- 90.Wynn TA, and Vannella KM (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44. 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adler M, Mayo A, Zhou X, Franklin RA, Meizlish ML, Medzhitov R, Kallenberger SM, and Alon U (2020). Principles of Cell Circuits for Tissue Repair and Fibrosis. iScience 23. 10.1016/j.isci.2020.100841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu KH, Eichorst JP, McGinnis CS, Patterson DM, Chow ED, Kersten K, Jameson SC, Gartner ZJ, Rao AA, and Krummel MF (2020). ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat Methods. 10.1038/s41592-020-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee Y, Bogdanoff D, Wang Y, Hartoularos GC, Woo JM, Mowery CT, Nisonoff HM, Lee DS, Sun Y, Lee J, et al. (2021). Xyzeq: Spatially resolved single-cell RNA sequencing reveals expression heterogeneity in the tumor microenvironment. Sci Adv 7. 10.1126/sciadv.abg4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, and Macosko EZ (2019). Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science (1979) 363. 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stoltzfus CR, Sivakumar R, Kunz L, Olin Pope BE, Menietti E, Speziale D, Adelfio R, Bacac M, Colombetti S, Perro M, et al. (2021). Multi-Parameter Quantitative Imaging of Tumor Microenvironments Reveals Perivascular Immune Niches Associated With Anti-Tumor Immunity. Front Immunol 12. 10.3389/fimmu.2021.726492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Asrir A, Tardiveau C, Coudert J, Laffont R, Blanchard L, Bellard E, Veerman K, Bettini S, Lafouresse F, Vina E, et al. (2022). Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell 40, 318–334.e9. 10.1016/j.ccell.2022.01.002. [DOI] [PubMed] [Google Scholar]
- 97.Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, et al. (2016). Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 6, 71–79. 10.1158/2159-8290.CD15-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kolter J, Feuerstein R, Zeis P, Hagemeyer N, Paterson N, d’Errico P, Baasch S, Amann L, Masuda T, Lösslein A, et al. (2019). A Subset of Skin Macrophages Contributes to the Surveillance and Regeneration of Local Nerves. Immunity 50. 10.1016/j.immuni.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 99.Lorts A, Schwanekamp JA, Baudino TA, McNally EM, and Molkentin JD (2012). Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-β pathway. Proc Natl Acad Sci U S A 109. 10.1073/pnas.1204708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Allard DE, Wang Y, Li JJ, Conley B, Xu EW, Sailer D, Kimpston C, Notini R, Smith CJ, Koseoglu E, et al. (2018). Schwann cell-derived periostin promotes autoimmune peripheral polyneuropathy via macrophage recruitment. Journal of Clinical Investigation 128. 10.1172/JCI99308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zunder ER, Finck R, Behbehani GK, Amir EAD, Krishnaswamy S, Gonzalez VD, Lorang CG, Bjornson Z, Spitzer MH, Bodenmiller B, et al. (2015). Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc. 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP, and Bendall SC (2013). Normalization of mass cytometry data with bead standards. Cytometry Part A. 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162. 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stuart T, Butler A, Hoffman P, Stoeckius M, Smibert P, Satija R, Hafemeister C, Papalexi E, Mauck Iii WM, and Hao Y (2019). Comprehensive Integration of Single-Cell Data Resource Comprehensive Integration of Single-Cell Data. Cell 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen WS, Zivanovic N, van Dijk D, Wolf G, Bodenmiller B, and Krishnaswamy S (2020). Uncovering axes of variation among single-cell cancer specimens. Nat Methods 17. 10.1038/s41592-019-0689-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, Elzen A, van den, Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol 37. 10.1038/s41587-019-0336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14. 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566. 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gaujoux R, and Seoighe C (2010). A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11. 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clearing-enhanced (Ce3D) skin wound D3 post-wounding of wild-type C57BL/6 mouse. Original wound size, 4 mm. Movie starts with internal-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and CD206 (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis. ITGA6/CD49b staining (white) highlights re-established epithelial basement membrane, vasculature structures in non-wounded skin, hair follicles, and fascia.
Clearing-enhanced (Ce3D) skin wound D7 post-wounding of wild-type C57BL/6 mouse. Original wound size, 4 mm. Movie starts with internal-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and CD206 (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis. ITGA6/CD49b staining (white) highlights re-established epithelial basement membrane, vasculature structures in non-wounded skin, hair follicles, fascia, and severed nerve bundles.
Clearing-enhanced (Ce3D) skin wound D3 post-wounding of Arg1-tdTomato reporter mouse. Original wound size, 4 mm. Movie starts with internal-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and Arg1-tdTomato (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis. ITGA6/CD49b staining (white) highlights reestablished epithelial basement membrane, vasculature structures in non-wounded skin, hair follicles, and fascia.
Clearing-enhanced (Ce3D) skin wound D7 post-wounding of Arg1-tdTomato reporter mouse. Original wound size, 4 mm. Movie starts with external-facing view of wound and DAPI (blue), ITGA6/CD49b (white), CD11b (green), and Arg1-tdTomato (red) channels on. Individual channels are turned on and off as wound image rotates along horizontal axis.
Top 20 contributing genes by gene weight from basis matrix shown for each of the 114 identified factors along with their gene weight towards that factor.
Genes in the upper right quadrant (I) of the tumor by wound healing M_M factor scatterplots (Figure 6C–E and Supplementary Figure SD–I) were used for GO analysis. Table shows a truncated output from each query for each pair with
Data Availability Statement
Single-cell RNAseq data have been deposited at GEO and Mendeley Data under and are publicly available as of the date of publication. Accession numbers are listed in the key resource table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD16/32 (clone 2.4G2) | Tonbo Biosciences | 70-0161-U500 |
| anti-mouse CD45 Alexa Fluor 647 (clone 30-F11) | Biolegend | 103124 |
| anti-mouse GPNMB eFluor™ 660 (clone CTSREVL) | eBioscience | 50-5708-80 |
| anti-h/mPeriostin (clone 345613) | R&D Systems | MAB3548 |
| anti-alpha smooth muscle actin antibody (polyclonal) | Abcam | AB5694 |
| anti-mouse P-selectin (polyclonal) | R&D Systems | AF737 |
| anti-CD31 AlexaFluor647 (clone 390) | BioLegend | 102416 |
| anti-CD11b AlexaFluor594 (clone M1/70) | BioLegend | 101254 |
| anti-CD206-AlexaFluor647 (clone C068C2) | BioLegend | 141712 |
| anti-I-A/I-E AlexaFluor594 (clone M5/114.15.2) | BioLegend | 107650 |
| anti-I-A/I-E AlexaFluor488 (clone M5/114.15.2) | BioLegend | 107616 |
| goat anti-rat IgG (H+L) AlexaFluor488 | Thermo Fisher Scientific | A11006 |
| donkey anti-goat IgG (H+L) AlexaFluor488 | Thermo Fisher Scientific | A11055 |
| F(ab')2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 555 | Thermo Fisher Scientific | A48283 |
| Alexa Fluor® 488 AffiniPure Fab Fragment Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch | 112-547-003 |
| Alexa Fluor® 647 AffiniPure Fab Fragment Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch | 112-607-003 |
| anti-mouse B220 (clone RA3–6B2) | Biolegend | 103202 |
| anti-mouse CCR7 (clone 4B12) | Biolegend | 120101 |
| anti-mouse CD103 (clone 2e7) | Biolegend | 121402 |
| anti-mouse CD11b (clone M1/70) | Biolegend | 101202 |
| anti-mouse CD11c (clone N418) | Biolegend | 117302 |
| anti-mouse CD16/32 (clone 2.4G2) | BD | 553142 |
| anti-mouse CD19 (clone 6D5) | Biolegend | 115501 |
| anti-mouse CD206 (clone C068C2) | Biolegend | 141702 |
| anti-mouse CD207 (clone 4C7) | Biolegend | 144202 |
| anti-mouse CD24 (M1/69) | Biolegend | 101802 |
| anti-mouse CD3e (clone 17A2) | Biolegend | 100202 |
| anti-mouse CD38 (clone 90) | Biolegend | 102702 |
| anti-mouse CD4 (clone RM4-5) | Biolegend | 100506 |
| anti-mouse CD44 (clone IM7) | Biolegend | 103002 |
| anti-mouse CD45 (clone 30-F11) | Biolegend | 103102 |
| anti-mouse CD49b (clone HMa2) | Biolegend | 103513 |
| anti-mouse CD62L (clone MAB5671) | R&D | MAB5761 |
| anti-mouse CD64 (clone (X54-5/7.1) | Biolegend | 139302 |
| anti-mouse CD69 (polyclonal) | R&D | AF2386 |
| anti-mouse CD8a (clone 53-6.7) | Biolegend | 100702 |
| anti-mouse CD86 (clone GL-1) | Biolegend | 105002 |
| anti-mouse CD90 (clone G7) | Biolegend | 105202 |
| anti-mouse c-Kit (clone 2B8) | Biolegend | 105802 |
| anti-mouse CTLA-4 (clone UC10-4B9) | Biolegend | 106302 |
| anti-mouse F4/80 (clone BM8) | Biolegend | 123102 |
| anti-mouse FceR1a (clone MAR-1) | Biolegend | 134302 |
| anti-mouse Foxp3 (clone NRRF-30) | eBiosciences | 14-4771-80 |
| anti-mouse GATA3 (clone 16E10A23) | Biolegend | 653802 |
| anti-mouse Ki67 (clone SolA15) | eBiosciences | 14-5698-82 |
| anti-mouse Ly6C (clone HK1.4) | Biolegend | 128002 |
| anti-mouse Ly6G (clone 1A8) | Biolegend | 127602 |
| anti-mouse MHC-II (clone M5/114.15.2) | Biolegend | 107602 |
| anti-mouse PD-1 (clone 29F.1A12) | Biolegend | 135202 |
| anti-mouse PD-L1 (clone 10F.9G2) | Biolegend | 124302 |
| anti-mouse PDCA1 (clone 927) | Biolegend | 127002 |
| anti-mouse RORgt (clone B2D) | eBiosciences | 14-6981-82 |
| anti-mouse Siglec F (clone E50-2440) | BD Biosciences | 552125 |
| anti-mouse SIRPa (clone P84) | Biolegend | 144002 |
| anti-mouse T-bet (clone 4B10) | Biolegend | 644802 |
| anti-mouse TCRgd (clone GL3) | Biolegend | 118101 |
| anti-mouse Ter119 (clone Ter119) | Biolegend | 116202 |
| anti-mouse TIM-3 (clone B8.2C12) | Biolegend | 134002 |
| anti-human CD45 APC/e780 (clone HI30) | Thermo Fisher | 47-0459-42 |
| anti-human CD3e PerCP/e710 (clone OKT3) | Thermo Fisher | 46-0037-42 |
| anti-human HLA-DR BUV395 (clone G46-6) | BD Biosciences | 564040 |
| Biological samples | ||
| Fetal Bovine Serum, Benchmark | GeminiBio | 100-106 |
| Chemicals, peptides, and recombinant proteins | ||
| Methanol | Fisher Scientific | A412-4 |
| recombinant human Tenascin | Merck Millipore | CC-065 |
| recombinant mouse Periostin | R&D Systems | 2955-F2-050 |
| recombinant mouse Oncostatin M | BioLegend | 762802 |
| Albumin, Bovine (BSA), nuclease free | VWR | 332 |
| Albumin, Bovine (BSA) | Sigma | A7906 |
| EDTA | Teknova | E0306 |
| Maxpar PBS | Fluidigm | 201058 |
| Maxpar Cell Staining Buffer | Fluidigm | 201068 |
| Maxpar Water | Fluidigm | 201069 |
| EQ Four Element Calibration Beads | Fluidigm | 201078 |
| Saponin | Sigma | S-7900 |
| Cisplatin | Enzo Life Sciences | ALX-400-040 |
| NaN3 | Sigma | S-8032 |
| Cell-ID 20-Plex Pd Barcoding Kit | Fluidigm | 201060 |
| Cell-ID 125 μm Iridium Intercalator | Fluidigm | 201192A |
| 16% paraformaldehyde | Electron Microscopy Sciences | 15710 |
| DNase I | Millipore Sigma | 10104159001 |
| Collagenase IX | Millipore Sigma | C7657 |
| Hyaluronidase | Worthington Biochemical Corp | LS005477 |
| buprenorphine hydrochloride | Hospira | 0409-2012-32 |
| bupivicaine | AuroMedica Pharma LLC | 55150-167-10 |
| OCT | Sakura | 4583 |
| Triton X-100 | Sigma | T8787 |
| Mouse serum | Jackson ImmunoResearch | 015-000-120 |
| Normal rat serum | Stem Cell Technologies | 13551 |
| Goat serum | Sigma | G9023 |
| Histodenz | Sigma | D-2158 |
| N-methylacetamide | Sigma | M26305-100G |
| 1-Thioglycerol | Sigma | M1753-100ML |
| 5X iScript RT Supermix | Bio-Rad | L001404B |
| SsoFast EvaGreen Supermix | Bio-Rad | L000915B |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1, 16 rxns | 10X Genomics | PN1000121 |
| Chromium Next GEM Chip G Single Cell Kit, 48 rxns | 10X Genomics | PN-1000120 |
| Chromium Next GEM Single Cell 5’ Library & Gel Bead Kit v1.1, 16 rxns | 10x Genomics | PN-1000165 |
| 10X Chromium Controller | 10x Genomics | N/A |
| Deposited data | ||
| Mouse spatiotemporal scRNAseq wound data set | This paper | GEO: GSE204777 |
| Human tumor scRNAseq data set | This paper | 10.17632/nnmb6m5p5j.1 |
| Mouse melanoma fibroblast data set | Davidson et al. (PMID: 32433953)73 | ArrayExpress: E-MTAB-7427 |
| Mouse pan-tissue fibroblast scRNAseq atlas | Buechler et al. (PMID: 33981032)22 | ArrayExpress:E-MTAB-10315 |
| Mouse pancreatic cancer fibroblast scRNAseq data set | Dominguez et al. (PMID: 31699795)54 | ArrayExpress: E-MTAB-8483 |
| Mouse wound scRNAseq data set | Guerrero-Juarez et al. (PMID: 31699795)26 | GEO: GSE113854 |
| Supplementary Mendeley Figures & Tables | Mendeley Data | 10.17632/kmmw43j2z6.2 |
| Experimental models: Cell lines | ||
| B16-F10 (mouse melanoma cell line) | ATCC | CRL-6475 |
| MC38 (mouse colorectal tumor cell line) | Kerafast | ENH204-FP |
| Experimental models: Organisms/strains | ||
| Mouse: wild-type C57BL/6 | The Jackson Laboratory | Stock# 000664 |
| Mouse: Arg1-tdTomato-CreERT2 × R26R-EYFP | H.-E. Liang & R. Locksley (UCSF)49 | https://www.sciencedirect.com/science/article/pii/S1074761319301992 |
| Mouse: Pdgfra-H2B-EGFP | A. Molofsky (UCSF)58 | Stock# 007669, |
| Oligonucleotides | ||
| See Supplementary Table S2 for qPCR primers | IDT | N/A |
| Software and algorithms | ||
| Normalizer | Finck et al, 20131103; https://onlinelibrary.wiley.com/doi/10.1002/cyto.a.22271 | https://rdrr.io/bioc/CATALYST/man/normCytof.html |
| Rphenograph | Levine et al, 2015104; https://www.sciencedirect.com/science/article/pii/S0092867415006376 | https://github.com/JinmiaoChenLab/Rphenograph |
| R environment | R Development Core Team | https://cran.r-project.org/bin/windows/base/ |
| Premessa | Parker ICI | https://github.com/ParkerICI/premessa |
| Flow Jo v10.8.1 | Tree Star | https://www.flowjo.com/ |
| Imaris 9.2.1 | Bitplane | https://imaris.oxinst.com/products/imaris-for-cell-biologists?gad=1&gclid=Cj0KCQjw3a2iBhCFARIsAD4jQB2RkztA4vyhmDUQ3_eo2WwjysDt-NhFrH-evt8PxW5s5oKpw_WYJhcaAuhZEALw_wcB |
| MATLAB | The MathWorks, Inc. | https://www.mathworks.com/products/matlab.html |
| LASX | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| CellChat (R package) | Jin et al. 202110; https://www.nature.com/articles/s41467-021-21246-9 | https://github.com/sqjin/CellChat |
| Seurat v3 (R package) | Stuart and Butler et al.105; Comprehensive Integration of Single-Cell Data. Cell (2019) [Seurat V3] | https://satijalab.org/seurat/ |
| NMF (R package) | Gaujoux R, Seoighe C (2010)110. “A flexible R package for nonnegative matrix factorization.” BMC Bioinformatics, 11(1), 367. ISSN 1471-2105, doi: 10.1186/1471-2105-11-367, http://www.biomedcentral.com/1471-2105/11/367. | https://cran.r-project.org/web/packages/NMF/index.html |
| Corrplot (R package) | https://cran.r-project.org/web/packages/corrplot/vignettes/corrplot-intro.html | https://cran.r-project.org/web/packages/corrplot/vignettes/corrplot-intro.html |
| Ggpubr (R package) | https://cran.r-project.org/web/packages/ggpubr/index.html | https://cran.r-project.org/web/packages/ggpubr/index.html |
| PhEMD | Chen et al., 2020106; https://www.nature.com/articles/s41592-019-0689-z | https://www.bioconductor.org/packages/release/bioc/html/phemd.html |
| Monocle 3 | Trapnell et al., 201443; http://dx.doi.org/10.1038/nbt.2859 | https://github.com/cole-trapnell-lab/monocle3 |
| CellRanger v 4.0.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Biorender | https://www.biorender.com | https://www.biorender.com/ |
| Other | ||
| lipid-modified oligonucleotides (LMO) | McGinnis et al., 201937; https://www.nature.com/articles/s41592-019-0433-8 | N/A |
| MaxPAR antibody conjugation kit | Fluidigm | 201146B |
| Dermal biopsy punch, 2 mm | Acuderm inc. | 69038-02 |
| Dermal biopsy punch, 4 mm | Integra | 33-34 |
| Dermal biopsy punch, 6 mm | Integra | 33-36 |
| Dermal biopsy punch, 8 mm | Integra | 33-37 |
| RNeasy Mini Kit | Qiagen | 74104 |