

Structural variations (SVs) are common in haematological neoplasms. 1 Although most SVs have canonical breakpoints, virtually all have atypical rearrangements that can be of difficult diagnosis. High‐throughput sequencing (HTS) has improved the study of atypical SVs, complementing cytogenetics. However, the short reads (<150 bp) generated by HTS might lead to inaccurate mapping. 2 This may not suppose a mere technical pitfall, but a problem to be faced, to guarantee the monitoring and guided‐treatment of patients bearing atypical SVs when standard procedures are ‘blind’ to them.
Inversion of chromosome 16, inv(16)(p13q22)/t(16;16)(p13;q22), found in 5%–7% of de novo acute myeloid leukaemia (AML), joins CBFB (16q22) and MYH11 (16p13) genes to form a chimeric oncoprotein that sequesters RUNX1. Up to 95% of cases with AML‐inv(16) carry three recurrent breakpoints involving CBFB exon5, and MYH11 exon33, exon29 or exon28, resulting in type A, D or E transcripts, respectively. 3 To the best of our knowledge, at least 10 additional CBFB::MYH11 transcripts with non‐canonical breakpoints have been reported (Table 1). 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 However, direct characterization of inv(16) at the genomic level has rarely been performed. In this context, third‐generation HTS based on long reads, such as nanopore sequencing, could be excellent tools for the identification and comprehensive analysis of atypical SVs. 2
TABLE 1.
Summary of reported CBFB::MYH11 transcripts with exonic/cDNA breakpoints and estimated protein fusion length.
| Transcript type | Breakpoints (Exon) | Breakpoints (cDNA) | Inserted nucleotides at breakpoint | Fusion protein length (residues) | Reference | ||
|---|---|---|---|---|---|---|---|
| CBFB a | MYH11 b | CBFB a | MYH11 b | ||||
| A | 5 | 33 | 495 | 4579 | No | 625 | [4] |
| B | 5 | 32 | 495 | 4366 | No | 696 | [5] |
| C | 5 | 31 | 495 | 4186 | No | 756 | [6] |
| D | 5 | 29 | 495 | 3858 | No | 865 | [4] |
| E | 5 | 28 | 495 | 3652 | No | 934 | [4] |
| F | 4 | 33 | 399 | 4579 | No | 593 | [7] |
| G | 4 | 29 | 399 | 3858 | No | 833 | [7] |
| H | 4 | 28 | 399 | 3746 | 5 bp | 856 | [7] |
| I | 4 | 34 | 399 | 4792 | No | 522 | [8] |
| J | 5 | 30 | 495 | 3977 | No | 830 | [9] |
| K | 6 | 28 | 534 | 3804 | 40 bp (exon 6) | 549 | [10] |
|
Rowe et al. |
5 | 32 | 495 | 4454 | 7 bp | 655 | [11] |
|
Albano et al. |
4 | 33 | 399 | 4708 | 21 bp (399 + 3979_4000ins) | 543 | [12] |
|
Kurata et al. |
5 | 26 | 495 | 3443 | 13 bp | 994 | [13] |
| This study | 4 | 26 | 399 + 5751 | 3581 | 62 bp (399 + 5689_5751ins) | 981 | — |
CBFB reference sequence: NM_022845.3.
MYH11 reference sequence: NM_002474.3.
In this study, we characterize by nanopore sequencing a novel inv(16) in AML. It was detected in a 25‐year‐old woman admitted to our centre because mucosal bleeding. The hemogram revealed thrombocytopenia (18.0 × 109/L) and monocytosis (5.4 × 109/L), with 38% blasts in the blood smear. Bone marrow (BM) aspirate showed the presence of 58% blasts with morphologic and immunophenotypic features of myelomonocytic differentiation. In addition, 15% of atypical eosinophils and their precursors were identified (Figure 1A).
FIGURE 1.
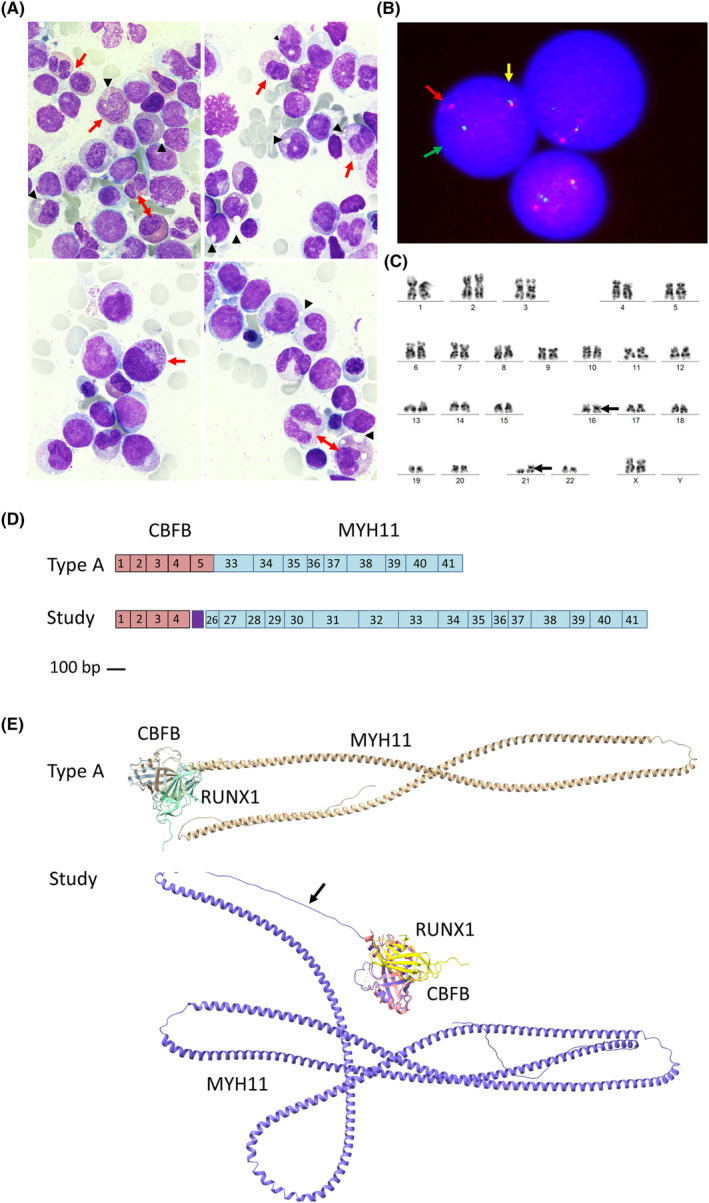
Characterization of atypical inv(16)(p13.1q22), CBFB::MYH11. (A) Bone marrow smear with May Grunwald‐Giemsa (×1000, lower panels are additionally magnified) showing infiltration by myelomonocytic blasts and 15% abnormal eosinophils and their precursors, most of them with abnormal proeosinophilic granules with a characteristic barely pinkish‐orange staining (red arrows). In addition, a remarkable cytoplasmic vacuolization was observed, which was not stained (black arrowheads). (B) FISH of bone marrow cells using the CBFB break apart probe (LSI CBFB Dual Color Break Apart Probe; MetaSystems, Germany) showing one fusion signal (unsplit CBFB, 16q21‐22, yellow arrow), one green signal (split CBFB, 5′ end, green arrow) and one red signal (split CBFB, 3′ end, red arrow). (C) Giemsa‐banded karyotype of bone marrow cells at diagnosis: 46,XX,inv(16)(p13q22),add(21)(p10)[14]/46,XX[6]. (D) Graphical representation of the common type A and the new atypical transcript found in this study type A. Scale representation (scale bar 100pb) of the contribution of the exons of CBFB (red boxes) and MYH11 (blue boxes) to each transcript. The linker peptide of the atypical transcript not belonging to exonic sequences is shown (purple boxes). (E) AlphaFold in silico modelling of the CBFB::MYH11 fusion proteins, and RUNX1 interaction, of the common type A and of the new aberrant inversion found in this study. The linker peptide is indicated with an arrow. Results were visualized with UCSF ChimeraX v1.4.
FISH showed inv(16)(p13q22). Karyotype confirmed inv(16) and add21(p10) (Figure 1B,C). However, RT‐PCR in BM RNA with commercial primers designed to detect the canonical CBFB::MYH11 transcripts (QuantiTec probe RT‐PCR kit, Qiagen, Germany, Table S1) gave negative results. A myeloid HTS panel (Oncomine Myeloid Research Assay‐Chef Ready, Thermofisher, MA, USA) using Ion S5 identified a CBFB::MYH11 rearrangement in 192/24685 RNA reads involving CBFB exon4 and MYH11 exon28. In addition, two pathogenic variants were identified in ZRSR2 (c.1147C > G p.Pro383Ala) and KRAS (c.38G > A p.Gly13Asp).
The patient was diagnosed with AML‐inv(16). BM re‐evaluation after 3 + 7 induction showed persistence of 29% blasts. Salvage chemotherapy with FLAG‐IDA (PETHEMA‐CBF‐2016) was then administered. Subsequently, haematologic and cytogenetic complete remission (CR) was achieved. However, flow cytometry revealed positive minimal residual disease (MRD 0.20%). The patient then underwent allogeneic haploidentical haematopoietic stem cell transplantation (HSCT) from a sibling. Since day +28 post‐transplant and to date (4 years later), she is in CR with negative MRD by flow cytometry and has complete donor chimerism.
The patient fortunately achieved a durable remission. However, at the diagnostic level, this scenario revealed a problem to be solved: the impossibility of monitoring the atypical CBFB::MYH11 transcript using standard diagnostic systems, including RT‐PCR kits. The root of this difficulty was the incomplete molecular characterization of the inv(16). Cytogenetics and Oncomine RNA sequencing identified an inv(16) and a CBFB::MYH11 transcript, but did not accurately characterized them.
Therefore, we performed genomic long‐read sequencing using nanopore technology. The study was performed on a MinION instrument (Oxford Nanopore Technologies [ONT], UK) using adaptive sampling, a computationally driven method for targeted sequencing of individual DNA molecules based on the identity of an initial set of approximately 450 bp, which are real‐time compared to a reference sequence to decide whether a given molecule should be sequenced further or rejected. The genome coordinates for enrichment were chr16:66297445–67,900,545 and chr16:14968145–16,559,989, which contain the CBFB and MYH11 genes, respectively. The library was prepared using the kit SQK‐LSK109 (ONT, UK). The run (20 h) yielded 10Gb. Mean coverage in the region of interest was 3.6X, while it was 1.3X in the rest of the genome. Basecalling was done with Guppy 6.4.2 using FAST. An in‐house multimodal pipeline including Sniffles software, 2 found four reads supporting a 51 Mb inversion (Figure S1). De novo assembly of these reads described an atypical breakpoint affecting c.399 + 5751 of CBFB intron4 and c.3581 MYH11 exon26, results discordant with the data initially provided by Oncomine. Genomic PCR covering the inversion (Table S2) and Sanger sequencing confirmed the breakpoint observed by nanopore: seq[GRCh38] inv(16)(p13q22) chr16:g.[15,735,438–67,072,549 inv] (Figure S1).
HSF v3.1 and NetGene2 v.2.42 were used for prediction of splicing. A very strong acceptor signal was identified in CBFB intron4, surrounded by silencer splicing signals (Figure S2). However, in the fusion gene, the MYH11 exon26, located only 62 bp from this intronic acceptor site, provided counteracting enhancer splicing signals (Figure S2). Furthermore, the use of this cryptic intronic acceptor site would preserve the MYH11 open reading frame. To test our prediction, RT‐PCR was performed on BM RNA at diagnosis using the primers and conditions listed in Table S2. This assay yielded only the expected 235 bp amplicon in the patient, and Sanger sequencing confirmed the predicted fusion. A nested RT‐PCR was also designed to increase sensitivity and evaluate MRD in patient's serial samples during treatment. This analysis did not detect the fusion transcript after HSCT, confirming the favourable evolution of the case (Figure S2).
The resulting chimeric protein might contain 981 residues: 133 from CBFB (exon 1–4), 20 from the retained region of CBFB intron 4, and 829 from MYH11 (exon 26–41). Notably, it represents one of the longest CBFB::MYH11 described to date, and the one with the highest number of residues at the fusion point not corresponding to CBFB or MYH11 exons (Table 1, Figure 1D).
Finally, we used Alphafold for in silico modelling of the atypical oncoprotein, 14 and compared it with the common type A form. As shown in Figure 1E, in both fusion proteins, the N‐terminus encoded by CBFB was able to bind RUNX1, while the C‐terminus encoded by MYH11 formed a long α‐helix. However, the length of the α‐helix was extremely different: the one resulting from the new transcript was almost twice as long as the canonical oncoprotein. In addition, CBFB intron4 encoded a long, unstructured loop connecting the CBFB globular domain to the MYH11 helix.
Using nanopore sequencing and a novel methodology, adaptive sampling, which does not require expensive equipment, in a fast and economical process, we characterized a new atypical inv(16) with nucleotide resolution in a patient with de novo AML. It remains to be determined whether this oversized oncoprotein might influence AML‐inv(16) pathogenesis. Unlike most cases with non‐A type transcripts that have been reported, and despite having one of the most genuine CBFB::MYH11 fusions, the patient showed many of the features that can be expected of a bona fide AML‐inv(16). Interestingly, transcripts with an alternative 5′ breakpoint at CBFB exon4 (i.e. lacking CBFB exon5) have been associated with AML‐inv(16) without myelo‐monocytic differentiation. 3 , 4 , 5 Shigesada et al. suggested that longer transcripts at the expense of the MYH11 N‐terminus might have uncoiled, flexible regions N‐terminal to the coiled‐coil polymerization domains, which might help CBFB bind RUNX1 with higher affinity. 3 In this sense, we could speculate that, in contrast to other transcripts lacking CBFB exon5, the oversized N‐terminal region of MYH11 in our case, separated from CBFB by an unfolded linker of 20 residues, could lead to the type A‐like leukaemic phenotype by (1) cooperation with the truncated CBFB to overcome the exon5 deletion and bind RUNX1 and (2) straight fibre formation by an extra‐long coiled‐coil domain.
It is even more difficult to speculate on the clinical implications of this new aberrant inversion in an isolated patient. Case reports and small series suggest that non‐A type transcripts are associated with worse clinical outcomes than A type transcripts. However, this assumption has been challenged by others. In addition, the level of complexity may be increased by concomitant chromosomal or gene abnormalities. 3 , 4
Finally, our findings may have therapeutic implications. Genomic mapping of the inversion allowed the discovery of the mechanism leading to the fusion protein: the use of an intronic cryptic acceptor splice site. Our findings provide the rationale to test novel therapeutic approaches targeting splicing in preclinical models of AML.
In conclusion, this study reveals new evidence for the diagnostic potential of long‐read sequencing to offer a ‘whole picture’ of SVs that may not be adequately represented by the ‘sum of parts’ of short‐read HTS methods. 15 Moreover, it illustrates how the complete characterization of the driver defect of any haematological neoplasm could not only allow us to refine disease monitoring and patient management, thereby solving clinical problems, but also provide deeper insights into the underlying pathogenic mechanisms. Ultimately, new and specific therapies may be identified based on these findings.
AUTHOR CONTRIBUTIONS
Carlos Bravo‐Perez: Conceptualization (equal); data curation (equal); formal analysis (lead); investigation (equal); methodology (equal); visualization (equal); writing – original draft (lead); writing – review and editing (lead). Rosa Cifuentes‐Riquelme: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (lead); visualization (equal); writing – original draft (equal). José Padilla: Data curation (equal); investigation (equal); methodology (equal); software (equal); writing – original draft (supporting); writing – review and editing (supporting). Maria Eugenia de la Morena‐Barrio: Conceptualization (equal); investigation (equal); methodology (equal); software (equal); validation (equal); visualization (equal); writing – original draft (supporting); writing – review and editing (supporting). Francisco Ortuño: Investigation (equal); methodology (equal); resources (equal); visualization (equal); writing – original draft (supporting); writing – review and editing (supporting). Pedro Garrido‐Rodríguez: Data curation (equal); investigation (equal); methodology (equal); software (equal); visualization (equal); writing – original draft (supporting); writing – review and editing (supporting). MariLuz Amigo: Data curation (equal); resources (equal); writing – original draft (supporting); writing – review and editing (supporting). Inmaculada Heras: Data curation (equal); resources (equal); writing – original draft (supporting); writing – review and editing (supporting). Vicente Vicente: Conceptualization (equal); supervision (equal); writing – original draft (supporting); writing – review and editing (supporting). María Luisa Lozano: Conceptualization (equal); supervision (equal); writing – original draft (supporting); writing – review and editing (supporting). Raul Teruel‐Montoya: Data curation (equal); investigation (equal); methodology (equal); resources (equal); writing – original draft (supporting); writing – review and editing (supporting). Belen de la Morena‐Barrio: Data curation (equal); methodology (equal); resources (equal); writing – original draft (supporting); writing – review and editing (supporting). Javier Corral: Conceptualization (lead); funding acquisition (lead); project administration (lead); supervision (lead); writing – original draft (lead); writing – review and editing (lead).
FUNDING INFORMATION
The study was funded by Instituto de Salud Carlos III (ISCIII) through the project ‘PMP21/00052’, which was co‐funded by the European Union‐Next Generation EU. Cifuentes‐Riquelme R has a PFIS contract. de la Morena‐Barrio ME has a Ramon y Cajal (RYC2021‐031000‐I) from Ministerio de Ciencia e Innovación. Bravo‐Pérez C has a Juan Rodés fellowship (JR22/00041). de la Morena‐Barrio B has a postdoctoral contract from CIBERER. Garrido‐Rodríguez P has a predoctoral contract from CIBERER.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Data S1.
Bravo‐Perez C, Cifuentes‐Riquelme R, Padilla J, et al. The whole is greater than the sum of its parts: Long‐read sequencing for solving clinical problems in haematology. J Cell Mol Med. 2024;28:e17961. doi: 10.1111/jcmm.17961
Carlos Bravo‐Perez and Rosa Cifuentes‐Riquelme these authors contributed equally.
DATA AVAILABILITY STATEMENT
For original data, please contact javier.corral@carm.es.
REFERENCES
- 1. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703‐1719. doi: 10.1038/s41375-022-01613-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single‐molecule sequencing. Nat Methods. 2018;15(6):461‐468. doi: 10.1038/s41592-018-0001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shigesada K, van de Sluis B, Liu PP. Mechanism of leukemogenesis by the inv(16) chimeric gene CBFB/PEBP2B‐MHY11. Oncogene. 2004;23(24):4297‐4307. doi: 10.1038/sj.onc.1207748 [DOI] [PubMed] [Google Scholar]
- 4. Liu PP, Hajra A, Wijmenga C, Collins FS. Molecular pathogenesis of the chromosome 16 inversion in the M4Eo subtype of acute myeloid leukemia. Blood. 1995;85(9):2289‐2302. [PubMed] [Google Scholar]
- 5. Schnittger S, Bacher U, Haferlach C, Kern W, Haferlach T. Rare CBFB‐MYH11 fusion transcripts in AML with inv(16)/t(16;16) are associated with therapy‐related AML M4eo, atypical cytomorphology, atypical immunophenotype, atypical additional chromosomal rearrangements and low white blood cell count: a study on 162. Leukemia. 2007;21(4):725‐731. doi: 10.1038/sj.leu.2404531 [DOI] [PubMed] [Google Scholar]
- 6. Claxton DF, Liu P, Hsu HB, et al. Detection of fusion transcripts generated by the inversion 16 chromosome in acute myelogenous leukemia. Blood. 1994;83(7):1750‐1756. [PubMed] [Google Scholar]
- 7. Shurtleff SA, Meyers S, Hiebert SW, et al. Heterogeneity in CBF beta/MYH11 fusion messages encoded by the inv(16)(p13q22) and the t(16;16)(p13;q22) in acute myelogenous leukemia. Blood. 1995;85(12):3695‐3703. [PubMed] [Google Scholar]
- 8. Zhang W, Wang H, Zhang P, Li H, Ma X, Liu H. Rare type I CBFβ/MYH11 fusion transcript in primary acute myeloid leukemia with inv(16)(p13.1q22): a case report. Braz J Med Biol Res. 2021;54(12):e11605. doi: 10.1590/1414-431X2021e11605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trnková Z, Peková S, Bedrlíková R, et al. Type J CBFbeta/MYH11 transcript in the M4Eo subtype of acute myeloid leukemia. Hematology. 2003;8(2):115‐117. doi: 10.1080/1024533031000084259 [DOI] [PubMed] [Google Scholar]
- 10. Park TS, Lee ST, Song J, et al. Detection of a novel CBFB/MYH11 variant fusion transcript (K‐type) showing partial insertion of exon 6 of CBFB gene using two commercially available multiplex RT‐PCR kits. Cancer Genet Cytogenet. 2009;189(2):87‐92. doi: 10.1016/j.cancergencyto.2008.10.012 [DOI] [PubMed] [Google Scholar]
- 11. Rowe D, Strain L, Lowe C, Jones G. A case of acute myeloid leukemia with inv(16)(p13q22) reveals a novel MYH11 breakpoint and a new CBF beta‐MYH11 transcript variant. Haematologica. 2007;92(10):1433‐1434. doi: 10.3324/haematol.11536 [DOI] [PubMed] [Google Scholar]
- 12. Albano F, Anelli L, Zagaria A, et al. Acute myeloid leukemia with t(16;16) (p13;q22) showing a new CBFB‐MYH11 fusion transcript associated with an atypical leukemic blasts morphology. Hum Pathol. 2014;45(3):643‐647. doi: 10.1016/j.humpath.2013.09.013 [DOI] [PubMed] [Google Scholar]
- 13. Kurata K, Yamamoto K, Okazaki Y, et al. Detection of a novel CBFB‐MYH11 fusion transcript in acute myeloid leukemia M1 with inv(16)(p13q22). Cancer Genet. 2020;241:72‐76. doi: 10.1016/j.cancergen.2019.07.005 [DOI] [PubMed] [Google Scholar]
- 14. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583‐589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Minervini CF, Cumbo C, Orsini P, et al. Nanopore sequencing in blood diseases: a wide range of opportunities. Front Genet. 2020;11:76. doi: 10.3389/fgene.2020.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
For original data, please contact javier.corral@carm.es.