

SUMMARY
Cellular signaling involves a large repertoire of membrane receptors operating in overlapping spatiotemporal regimes and targeting many common intracellular effectors. However, both the molecular mechanisms and the physiological roles of crosstalk between receptors, especially those from different superfamilies, are poorly understood. We find that the receptor tyrosine kinase (RTK) TrkB and the G-protein-coupled receptor (GPCR) metabotropic glutamate receptor 5 (mGluR5) together mediate hippocampal synaptic plasticity in response to brain-derived neurotrophic factor (BDNF). Activated TrkB enhances constitutive mGluR5 activity to initiate a mode switch that drives BDNF-dependent sustained, oscillatory Ca2+ signaling and enhanced MAP kinase activation. This crosstalk is mediated, in part, by synergy between Gβγ, released by TrkB, and Gαq-GTP, released by mGluR5, to enable physiologically relevant RTK/GPCR crosstalk.
Graphical Abstract
In brief
Lao-Peregrin et al. find that BDNF-driven hippocampal synaptic plasticity is dependent on functional crosstalk with mGluR5. Mechanistic studies reveal a general mode of RTK/GPCR crosstalk that is driven by G-protein synergy.
INTRODUCTION
Cell signaling is based on a complex interplay between ensembles of receptors that sense extracellular signal dynamics and convert them into intracellular cascades that shape a multitude of functions.1-3 In the central nervous system, rapid synaptic transmission and its plasticity are mediated by ion-channel-linked and G-protein-coupled receptors (GPCRs) that sense neurotransmitters, such as glutamate.4 In contrast, growth factors like brain-derived neurotrophic factor (BDNF) typically signal via receptor tyrosine kinases (RTKs) on slower time scales to regulate the induction and expression of synaptic plasticity.5 In addition to their roles in normal brain function, both glutamate and BDNF signaling underlie many aspects of the pathophysiology and treatment of neurodevelopmental and neurodegenerative disorders.6-8 Despite both forms of signaling likely occurring coincidentally at excitatory synapses, little is known about how neurotrophin and neurotransmitter receptor classes work in concert to tune synaptic plasticity.
Tropomyosin-related kinase B (TrkB) and metabotropic glutamate receptor 5 (mGluR5) represent two of the most abundant, widely expressed membrane receptors in the brain with overlapping synaptic roles. TrkB is an RTK that senses BDNF, which drives autophosphorylation of intracellular tyrosine residues on the kinase domain to initiate intracellular signaling cascades through the binding of adaptor proteins.9 For example, the Shc adaptor protein links activated TrkB to two separate pathways: PI-3 kinase and MAP kinase signaling.10 In addition, phospholipase C-γ (PLC-γ) binds to phosphorylated TrkB and initiates IP3 production to drive release of intracellular Ca2+ stores. mGluR5 is a Gq-coupled family C GPCR that uses heterotrimeric G proteins to initiate signaling cascades. Most prominently, mGluR5 activation leads to distinctive Ca2+ oscillations due to reversible protein kinase C (PKC)-dependent phosphorylation of intracellular residues.11-14 In addition to its glutamate-evoked activity, under many circumstances, mGluR5 produces tonic glutamate-independent signaling.15,16
In the hippocampus, BDNF-TrkB signaling contributes to tetanus-induced long-term potentiation (LTP),17-19 and exogenous BDNF application alone produces a chemical form of LTP (“BDNF-LTP”).20-23 In contrast, hippocampal mGluR5 can contribute to electrical and chemical forms of either LTP or long-term depression (LTD), depending on the context.4,24-29 Both mGluR5 and TrkB signal via biochemical cascades and activate common downstream signaling effectors strongly implicated in synaptic plasticity, such as intracellular Ca2+ release and MAP kinase activation.28,30 These overlapping synaptic roles and signaling properties together raise the possibility that crosstalk between TrkB and mGluR5 may serve as a link between rapid neurotransmission and long-term plasticity.
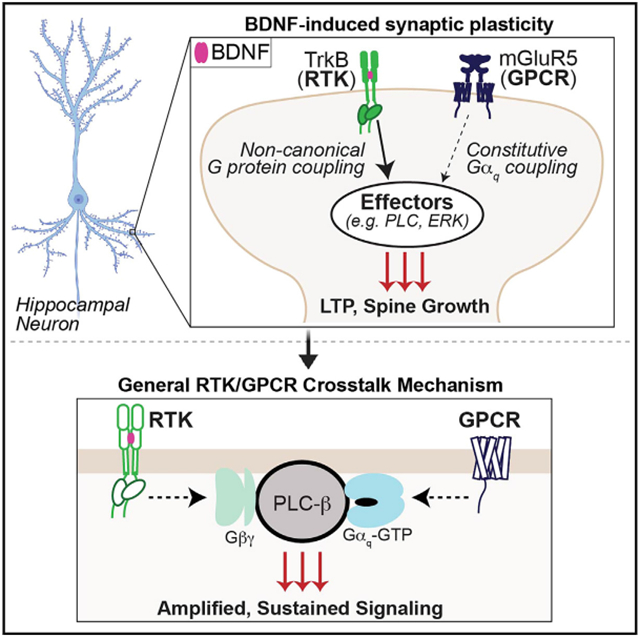
It has previously been shown that GPCR activation can elicit signaling through transactivation of RTKs, such as the epidermal growth factor receptor (EGFR) or TrkB, through a variety of mechanisms, including regulation of RTK ligand release or RTK phosphorylation following GPCR activation of the tyrosine kinase Src.31-38 However, minimal work has addressed the ability of a GPCR to alter the signaling response of an RTK to its native ligand.39,40 Using acute slice electrophysiology and cultured neuron imaging, we find that BDNF-TrkB-induced synaptic plasticity is dependent on constitutive, coincident dendritic mGluR5 signaling. Using a battery of assays in cultured cells, we reveal that mGluR5 co-expression or positive allosteric modulation dramatically boosts the BDNF sensitivity and response duration of TrkB in terms of both intracellular Ca2+ release and MAP kinase pathway activation. This crosstalk is mediated by constitutive G-protein activation by mGluR5 and non-canonical activation of G-protein signaling by TrkB, which, together, enable cooperative activation of downstream effectors, such as phospholipase C-β (PLC-β). This G-protein-dependent crosstalk mechanism underlies the mGluR5 dependence of BDNF-induced synaptic plasticity and occurs for a range of RTKs and GPCRs, indicating that this is a general mode of RTK/GPCR synergy that is relevant across physiological systems.
RESULTS
mGluR5 activity is required for BDNF-induced synaptic plasticity
Given that both TrkB and mGluR5 can drive certain forms of LTP in the hippocampal CA3-CA1 synapse, we asked if these receptors functionally interact in this context. We performed field recordings from adult mouse hippocampal slices with exogenous BDNF application, as done previously.20-23 Application of BDNF for 30 min produced a dose-dependent increase in field excitatory post-synaptic potential (fEPSP) slope that persisted for an hour after washout (Figure S1A). This effect was blocked by the selective TrkB antagonist ANA-1241 (Figure S1B). Throughout this study, we refer to this chemical form of long-term synaptic plasticity as BDNF-LTP. As a first test for a potential contribution of mGluR5 to this form of plasticity, we asked if pharmacological inhibition of mGluR5 would alter BDNF-LTP induction or expression. Strikingly, when slices were pre-treated and maintained in the mGluR5 negative allosteric modulator MPEP (40 μM) during BDNF (100 ng/mL) application, induction of BDNF-LTP was largely diminished (Figures 1A and 1B). Application of MPEP in the absence of BDNF had no effect on fEPSP slope (Figure S1C). MPEP treatment following BDNF-induced potentiation also had no effect (Figure S1D), indicating that mGluR5 activation is required for BDNF-LTP induction, but not for expression or maintenance. Furthermore, BDNF-LTP was not associated with changes in paired pulse ratio (Figure S1E), as shown previously,22 in line with a post-synaptic mechanism. Consistent with pharmacological blockade of mGluR5, post-development conditional knockout (KO) of mGluR5 in CA1 neurons via virally delivered Cre-recombinase (Figure S1F) prevented BDNF-LTP (Figures 1C and 1D). Importantly, CA1 KO of mGluR5 also strongly diminished sensitivity to the group I mGluR agonist, dihydroxyphenylglycine (DHPG) (Figures S1G and S1 H), but did not alter basal synaptic strength as assessed with a titration of stimulus intensity (Figure S1I).
Figure 1. BDNF-induced LTP is dependent on mGluR5 and enhanced by an mGluR5 allosteric modulator.

(A) fEPSP slope time course showing that blockade of mGluR5 with MPEP prevents BDNF-induced LTP. Gray bars show regions averaged for baseline and post-BDNF values in (B). Right, representative fEPSP traces recorded during basal and after 60 min of BDNF perfusion.
(B) Summary bar graph showing a lack of BDNF-induced potentiation in the presence of MPEP.
(C and D) Conditional KO of mGluR5 in CA1 pyramidal neurons impairs BDNF-LTP compared with control slices.
(E and F) Co-application of the mGluR5 PAM VU-29 enhances LTP induced by low-dose (50 ng/mL) or high-dose (100 ng/mL) BDNF.
(G and H) BDNF-induced LTP is blocked by an ERK inhibitor (PD98059, 50 μM) or a PLC inhibitor (U-73122, 5 μM), but not an inactive PLC inhibitor analog (U-73343, 5 μM). For (B), (D), (F), (H): individual points represent independent slices taken from distinct mice. For (B) and (D), unpaired t test is used. For (F) and (H), one-way ANOVA with Sidak’s multiple comparisons is used. All data are shown as the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S1.
Given that mGluR5 activity is required for BDNF-LTP induction, we hypothesized that positive allosteric modulators (PAMs) of mGluR5 may enhance BDNF-induced potentiation. A prior study showed that the mGluR5 PAM VU-29 can potentiate tetanus-induced LTP,26 which also involves BDNF-TrkB signaling.30 Consistent with this study, VU-29 did not alter baseline synaptic transmission (Figure S1J). However, co-application of VU-29 with a lower dose of BDNF (50 ng/mL) dramatically enhanced LTP (Figures 1E and 1F). VU-29 was also able to modestly enhance the LTP induced by a higher BDNF dose (100 ng/mL) (Figure 1F). The effects of VU-29 further indicate that synergistic interplay between TrkB and mGluR5 can drive synaptic strengthening. For further insight into the signaling mechanisms underlying mGluR5-dependent BDNF-LTP, we assessed the contributions of extracellularly regulated kinase (ERK) and phospholipase C (PLC), two key signaling molecules previously implicated in BDNF-mediated plasticity.42,43 Application of either PD98059, an ERK1/2 inhibitor, or U73122, a PLC inhibitor, had no effect on basal fEPSP slope (Figures S1K and S1L). However, co-application of PD98059 or U73122 with BDNF prevented BDNF-LTP (Figures 1G and 1H), suggesting that both ERK and PLC signaling are required for the induction of BDNF-LTP.
In addition to its ability to tune the electrical properties of glutamatergic synapses, BDNF can also produce morphological changes in dendritic spines.44-46 This raises the possibility that mGluR5 also contributes to structural forms of BDNF-induced synaptic plasticity. To assess whether TrkB and mGluR5 are co-localized in such a way that they may co-regulate spine growth, we visualized both endogenous receptors in primary cultured hippocampal neurons using immunocytochemistry and scanning confocal microscopy (Figure 2A). Both TrkB and mGluR5 puncta were observed along the dendritic shaft and spines, including a substantial co-localized population within the same spine or shaft region (Figure 2A). In addition, mGluR5 was detected in TrkB immunoprecipitates from hippocampal neurons, as assessed via western blot (Figure S2A). These results provide evidence that mGluR5 and TrkB compartmentalize together within signaling microdomains, providing a basis for functional crosstalk via access to an overlapping pool of signaling targets.
Figure 2. BDNF-induced dendritic spine growth and calcium signaling are dependent on mGluR5.

(A) Confocal image of fixed hippocampal neuron showing anti-TrkB and anti-mGluR5, including co-localization in dendrites (scale bars, 1 μm dendrite; 0.1 μm spine and shaft). Of the spines, 49.3% ± 6.6% showed co-expression of TrkB and mGluR5; Pearson’s correlation co-efficient between TrkB and mGluR5 in dendritic shaft (top 10% of pixels) = 0.49 ± 0.05 (n = 12 neurons).
(B) Representative images showing BDNF-induced (100 ng/mL) spine density increases in DIV 21 hippocampal neurons. Arrowheads indicate spine heads enriched with F-actin.
(C and D) Bar graphs summarizing the BDNF-induced increases in spine density. MPEP blocks 100 ng/mL BDNF-induced spine density increase (C), while VU-29 potentiates the increase in spine density induced by low-dose 50 ng/mL BDNF (D).
(E) Representative image of GCaMP8m-expressing hippocampal neuron (left, scale bar, 10 μm), with snapshots of dendrites (white rectangles) taken in the presence of 100 ng/mL BDNF (middle, scale bar, 5 μm). Right, representative trace from 10 μm regions of interest (ROIs) with Ca2+ response (yellow rectangles) or without Ca2+ response (red rectangle) from each dendrite.
(F) Bar graph showing that 1 μM MPEP co-application significantly decreases the percentage of total dendritic area with BDNF-induced Ca2+ responses.
(G) Bar graph showing that co-application of low-dose (50 ng/mL) BDNF with 500 nM VU-29 leads to an increased percentage of total dendritic area with Ca2+ responses. For (C), (D), (F), and (G), individual points represent separate neurons, taken from at least three separate culture preparations. For (C) and (D), one-way ANOVA with Tukey’s multiple comparisons is used. For (F) and (G), unpaired t test is used. All data are shown as the mean ± SEM, *p < 0.05, ***p < 0.001. See also Figure S2.
To assess BDNF-induced spine growth, we acutely added BDNF for 30 min in mature primary hippocampal neurons. We observed a dose-dependent 20%–30% increase in spine density (Figures 2B, 2C, and S2B), which was prevented by pre-treatment with ANA-12 (Figure S2C). As was seen with field recordings in acute slices, BDNF-induced spine density increase was not observed in the presence of MPEP (Figure 2C), indicating that BDNF-induced structural plasticity is also dependent on mGluR5. Furthermore, VU-29 enhanced BDNF-induced structural plasticity (Figure 2D), while PLC or ERK inhibition prevented BDNF-induced spine growth (Figures S2D and S2E). Together, these data show that electrical and structural forms of BDNF-driven plasticity share common mGluR5-dependent properties.
mGluR5 co-expression and allosteric modulation control BDNF-TrkB signaling dynamics
Based on the PLC dependence of BDNF-LTP in our study and prior studies implicating intracellular Ca2+ stores in BDNF-LTP,47 we assessed the dendritic Ca2+ responses to BDNF application using the genetically encoded fluorescent sensor GCaMP8m in primary hippocampal neuronal cultures. In the presence of tetrodotoxin (TTX) to prevent action potential firing and associated neurotransmitter release, sporadic Ca2+ events were observed in dendrites. Application of BDNF led to a substantial increase in the amount of dendritic Ca2+ events, which showed a wide variety of temporal dynamics and were confined within ~5–30 μm dendritic patches (Figure 2E). Pre-application of MPEP substantially decreased the proportion of dendritic area showing BDNF responses (Figures 2F and S2F), in line with its inhibitory effect on BDNF-LTP and spine growth. Furthermore, application of VU-29 enhanced the response to a lower dose of BDNF (50 ng/mL) (Figures 2G and S2G). These results show that mGluR5 activity contributes to the initial dendritic response to BDNF, motivating a subsequent focus on how mGluR5 shapes acute BDNF-TrkB Ca2+ signaling dynamics.
To further probe the signaling that underlies the mGluR5 dependence of BDNF-TrkB synaptic plasticity, we turned to a simplified HEK 293 cell context. In a stable cell line expressing TrkB (“HEK 293-TrkB”),48 BDNF produced dose-dependent Ca2+ responses in the form of a single transient of 50–200 s duration, consistent with the established ability of TrkB to activate PLC-γ49 (Figures 3A and S3A-S3E). In contrast to RTKs, Gαq-coupled GPCRs, such as mGluR5, signal via the PLC-β family.50 HEK 293 cells transiently transfected with mGluR5 responded to glutamate application with oscillatory Ca2+ responses with event durations of ~30 s and were insensitive to BDNF (Figures 3B, S3F, and S3G). mGluR5-induced Ca2+ oscillations were due to reversible PKC-mediated phosphorylation of receptor C-terminal-domain residues.11,13,14 Based on initial event duration (Figure S3G), responses were classified as “slow Ca2+ wave” or “Ca2+ oscillations,” with the former associated with TrkB and the latter with mGluR5.
Figure 3. BDNF-mediated TrkB activation produces mGluR5-dependent Ca2+ oscillations in HEK 293 cells.

(A–C) Representative traces showing intracellular Ca2+ responses to TrkB activation by BDNF (A), mGluR5 activation by glutamate (B), and TrkB activation by BDNF in mGluR5 co-expressing cells (C).
(D) Distribution of BDNF responses in the absence or presence of mGluR5 co-expression. Only cells showing a response to glutamate are analyzed in the TrkB + mGluR5 condition.
(E–G) Representative traces showing Ca2+ responses to extended 30 min BDNF application in cells expressing TrkB (E) or TrkB and mGluR5 (F), with summary histogram (G) showing the distribution of response durations.
(H and I) Co-application of the mGluR5 PAM VU-29 enhances the response to low-dose BDNF, as seen in a representative cell (H) and a summary bar graph of the percentage of cells responding to BDNF (I). Only cells responding to glutamate were included in the bar graph in (I).
(J and K) Representative western blot (J) and quantification (K) of BDNF-induced ERK activation (p-ERK/ERK normalized ratio at 15 min) in HEK293-TrkB cells co-expressing mGluR5, showing an enhanced response in the presence of 500 nM VU-29. For (D) and (I), points represent values from individual movies taken from distinct coverslips. For (K), individual points represent values from individual blots. For all conditions, the data come from at least three separate cell preparations. Unpaired t test for (I); one-way ANOVA with Tukey’s multiple comparisons for (K). All data are shown as the mean ± SEM; **p < 0.01, ***p < 0.001. See also Figures S3 and S4.
Strikingly, upon co-expression of mGluR5 in HEK 293-TrkB cells, BDNF elicited Ca2+oscillations in ~60% of glutamate-sensitive cells, while only ~20% produced a slow Ca2+ wave response (Figures 3C and 3D). This suggests a mode switch in BDNF-induced Ca2+ signaling upon mGluR5 co-expression. In addition to BDNF, neurotrophin-3 (NT-3) and neurotrophin-4 (NT-4), which also activate TrkB, produced mGluR5-dependent Ca2+ oscillations (Figures S4A-S4D). BDNF-induced Ca2+ oscillations were observed with application of low-dose (25 ng/mL) (Figures S4E and S4F) and high-dose (100 ng/mL) BDNF (Figures 3C and 3D). Notably, mGluR5 co-expression slightly decreased the amplitude (Figure S4G) and substantially decreased the latency of BDNF responses (Figure S4H), demonstrating a robust acceleration of TrkB signaling. When BDNF was applied continuously for 30 min, as was done for synaptic plasticity induction (Figures 1 and 2), TrkB alone produced a single slow Ca2+ wave (Figure 3E), while mGluR5 co-expression enabled Ca2+ oscillations that persisted for the entire BDNF application time (Figures 3F and 3G). Similarly, mGluR5 co-expression increased the amplitude and duration of ERK phosphorylation following BDNF treatment in HEK 293-TrkB cells (Figures S4I and S4J). We also found that application of the mGluR5 PAM VU-29 did not directly activate mGluR5 Ca2+ signaling (Figures S4K and S4 L) but dramatically increased the proportion of cells responding to low-dose BDNF (25 ng/mL) with Ca2+ oscillations (Figures 3H, 3I, and S4M), showing that mGluR5 can effectively sensitize TrkB to its endogenous ligand. With a high dose of BDNF, VU-29 enhanced the proportion of cells responding to >90% (Figure S4N). VU-29 also increased the amplitude (Figure S4G), decreased the latency (Figure S4H), and increased the Ca2+ oscillation frequency of BDNF responses (Figure S4O). Last, co-application of VU-29 increased the extent of ERK phosphorylation following low-dose (25 ng/mL) BDNF application in HEK 293-TrkB cells co-expressing mGluR5 (Figures 3J and 3K). Together, these data indicate that mGluR5 co-expression and positive allosteric modulation can amplify the TrkB response to BDNF, which may underlie the mGluR5 dependence of BDNF-LTP induction.
Molecular mechanisms of TrkB/mGluR5 signaling crosstalk
We next investigated the molecular mechanisms that mediate TrkB/mGluR5 signaling crosstalk. Like many GPCRs, mGluR5 has previously been shown to display constitutive activity,15 which may explain its contribution to the BDNF response in the absence of glutamate application. However, the relatively high affinity of mGluR5 for glutamate makes it hard to rule out a role for ambient glutamate or for BDNF-induced glutamate release as a driver of Ca2+ oscillations. To clarify this, we removed the extracellular domains of mGluR5, including the glutamate binding domain, to produce “mGluR5-ΔECD.”51 Co-expressing mGluR5-ΔECD with TrkB did not prevent BDNF-induced Ca2+ oscillations (Figures 4A and 4B). Importantly, prior work has shown that mGluR5-ΔECD still can couple to G proteins and shows constitutive activity.51 Indeed, VU0360172, an mGluR5 allosteric agonist, still elicited robust Ca2+ oscillations with this construct (Figure 4A). We then probed if mGluR5 activation is required for BDNF-induced Ca2+ oscillations by using MPEP, which fully abolished BDNF-induced Ca2+ oscillations, indicating that mGluR5 activation is required (Figures 4B and S5A). Importantly, MPEP did not alter BDNF Ca2+ responses in cells expressing TrkB but not mGluR5 (Figures S5B and S5C). MPEP treatment also blocked slow Ca2+ wave responses (Figure S5A) and decreased the ERK phosphorylation response to BDNF in cells co-expressing TrkB and mGluR5 (Figures S5D and S5E). Furthermore, introduction of a mutation into a conserved intracellular loop 3 residue52 (F768D) to prevent mGluR5 from coupling with G proteins also prevented BDNF-induced Ca2+ oscillations (Figures 4B, S5F, and S5G) without altering mGluR5 surface levels (Figure S5H). Together, these experiments show that TrkB/mGluR5 signaling crosstalk is dependent on mGluR5/G protein coupling but is independent of glutamate binding to mGluR5.
Figure 4. Molecular determinants of BDNF-induced Ca2+ oscillations.

(A) Representative trace showing BDNF-induced Ca2+ oscillations in cells co-expressing TrkB and mGluR5-ΔECD. Ca2+ oscillations are also produced with VU0360172, an mGluR5 allosteric agonist.
(B) Summary bar graph showing lack of BDNF-induced Ca2+ oscillations when mGluR5 is blocked by MPEP or when the F768D mutation is introduced, but not when the ECD is removed. Only cells responding to glutamate or VU0360172 (for mGluR5-ΔECD) were analyzed.
(C) Representative trace showing BDNF-induced Ca2+ oscillations in cells co-expressing mGluR5 and TrkB-ΔFrs-ΔShc-ΔPLCγ.
(D) Summary bar graph showing lack of BDNF-induced Ca2+ oscillations with kinase-dead TrkB (K571N), but clear oscillations for TrkB-ΔPLCγ and TrkB-ΔFrs-ΔShc-ΔPLCγ. Only cells showing a response to glutamate were analyzed. Points in (B) and (D) represent values from individual movies taken from distinct coverslips. For all conditions, data come from at least three separate cell preparations. All data are shown as the mean ± SEM. See also Figure S5.
To probe the determinants of TrkB/mGluR5 crosstalk at the level of TrkB via perturbations directly to TrkB, we turned to transient co-transfection of TrkB and mGluR5, which also enabled BDNF-induced, mGluR5-dependent Ca2+ oscillations (Figure 4D). To determine if TrkB kinase activity is required for Ca2+ oscillations, we tested the TrkB-K571N “kinase-dead” mutant.53 Introduction of this mutation prevented BDNF-induced Ca2+ responses in the presence or absence of mGluR5 (Figures 4D and S5I-S5K). In contrast, removal of the PLC-γ recruitment site alone (“TrkB-ΔPLCγ”) or in combination with the Shc and FRS2 sites (“TrkB-ΔFrs-ΔShc-ΔPLCγ”) did not prevent BDNF-induced Ca2+ oscillations in cells co-expressing mGluR5 (Figures 4C, 4D, and S5J), despite preventing Ca2+ responses in cells expressing only TrkB (Figure S5K). As a key control, we showed that both the TrkB kinase-dead and the TrkB-ΔFrs-ΔShc-ΔPLCγ mutants are unable to activate the ERK pathway, while the TrkB-ΔPLCγ mutant maintained a weak BDNF-induced ERK response (Figures S5L and S5M). Furthermore, all TrkB mutants showed clear surface expression, with TrkB-ΔFrs-ΔShc-ΔPLCγ showing increased surface expression compared with wild-type TrkB (Figure S5N). These results indicate that BDNF-induced Ca2+ oscillations are not dependent on canonical TrkB signaling mechanisms via the Shc, FRS2, or PLC-γ pathways, but they do require tyrosine kinase activity.
The ability of BDNF to produce mGluR5-dependent Ca2+ oscillations via the TrkB-ΔFrs-ΔShc-ΔPLCγ construct, which lacks canonical mechanisms for downstream signaling, motivated us to investigate alternative pathways. A variety of RTKs have been shown to couple to heterotrimeric G proteins via non-canonical mechanisms that nonetheless rely on kinase activity.54-58 Furthermore, a recent study showed that TrkB is highly sensitive to KO of Gαi1 and Gαi3 G proteins, which dramatically reduced BDNF-induced signaling in hippocampal neurons.59 We hypothesized that upon BDNF binding, TrkB can promote Gi/o signaling, which then synergizes with Gq/11 proteins tonically activated by mGluR5 (Figure 5A). Informed by a model proposed to explain the crosstalk between Gαi/o-coupled GPCRs and Gq/11-coupled GPCRs,60,61 we reasoned that Gβγ subunits released by Gαi/o under the control of TrkB and Gαq-GTP promoted by mGluR5 would synergize by simultaneously binding to PLC-β to produce Ca2+ oscillations. Consistent with this idea and with prior work showing that group I mGluR signaling can be promoted by Gαi/o-coupled receptors,62,63 we found that activation of Gαi/o signaling upon stimulation of different GPCRs (mu-opioid receptor [MOR] or gamma-aminobutyric acid B receptor [GABAβR]) led to mGluR5-dependent Ca2+ oscillations in the absence of glutamate (Figures S6A and S6B). The similarities of these responses with those elicited by BDNF (Figure 3) motivated us to test the effect of G-protein perturbations on the interplay between TrkB and mGluR5.
Figure 5. G-protein dependence of TrkB/mGluR5 crosstalk.

(A) Schematic of the proposed “G-protein synergy” mechanism underlying TrkB/mGluR5 crosstalk.
(B) Summary bar graph showing effects of G-protein perturbations on the efficiency of 100 ng/mL BDNF-induced Ca2+ oscillations in cells expressing TrkB and mGluR5. PTX, pertussis toxin; DN-Gαi3, Gαi3-G203A; GRK2-CAAX, membrane-tethered GRK2; R587Q impairs Gβγ binding; D110A impairs Gαq binding; GRK2-CT, isolated, membrane-tethered PH domain of GRK2; WT, wild type; YM-254890 (20 μM), Gαq blocker.
(C–E) Schematic of cAMP signaling (C) with average traces (D) and summary bar graph (E) showing that TrkB activation by 100 ng/mL BDNF inhibits cAMP production in a G-protein-dependent manner.
(F–H) Schematic of GBAi inhibition of G-protein coupling (F) with summary bar graphs showing that GBAi overexpression decreases TrkB/mGluR5 crosstalk (G) but not MOR/mGluR5 crosstalk (H). For (B) and (F) through (H), only cells showing a response to 100 μM glutamate were analyzed, except for the YM-254890 condition, in which only cells responding to BDNF were included. Points represent values from individual movies taken from distinct coverslips (≥3 separate cell preparations per condition). All conditions were compared with the control group using one-way ANOVA with Dunnett’s multiple comparisons. All data are shown as the mean ± SEM; **p < 0.01; ***p < 0.001. See also Figure S6.
First, we found that co-expression of a dominant-negative Gαi3 mutant, G203A (“DN-Gαi3”), that prevents the dissociation of Gβγ from Gα64-66 dramatically reduced the proportion of cells showing mGluR5-dependent Ca2+ oscillations in response to BDNF in HEK 293-TrkB cells (Figure 5B). Importantly, DN-Gαi3 did not alter BDNF responses in cells expressing TrkB alone or mGluR5-mediated glutamate responses (Figures S6C and S6D). This dependence on Gi/o signaling was also corroborated by the observation of a large reduction in DAMGO-induced Ca2+ oscillations in mGluR5 and MOR-expressing cells upon expression of DN-Gαi3 (Figure S6E). However, BDNF and DAMGO were affected differently by pertussis toxin (PTX), which modifies the C terminus of Gα subunits in the Gi/o family67. While PTX completely blunted DAMGO-induced mGluR5-dependent Ca2+ oscillations (Figure S6E), as expected for a GPCR-mediated mechanism, it had no effect on BDNF-induced oscillations (Figure 5B). This lends confidence to the idea that BDNF exerts its action through a non-canonical, RTK-dependent Gi/o-Gβγ signaling that is PTX insensitive57 rather than through the activation of a GPCR intermediate.
We then co-expressed a membrane-tethered version of GPCR kinase 2 (“GRK2-CAAX”), which can bind and sequester both Gβγ and Gαq-GTP.68,69 Indeed, GRK2-CAAX co-expression reduced the proportion of cells responding to BDNF with Ca2+ oscillations (Figure 5B) without altering BDNF responses in cells expressing TrkB alone (Figure S6C) or mGluR5 glutamate responses (Figure S6D). Mutation of both the Gβγ (R587Q) and the Gαq (D110A) binding sites on GRK2 was required to rescue efficient TrkB/mGluR5 crosstalk (Figure 5B). Notably, the isolated GRK2 C terminus (“GRK2-CT”), which contains only the Gβγ binding site, was also able to blunt both TrkB/mGluR5 (Figure 5B) and MOR/mGluR5 crosstalk (Figure S6E) without altering BDNF responses in cells expressing TrkB alone (Figure S6C) or mGluR5 glutamate responses (Figure S6D). The Gαq inhibitor YM-254890 was able to block TrkB/mGluR5 crosstalk (Figure 5B) and mGluR5 glutamate responses (Figure S6D), but not slow-wave Ca2+ responses from TrkB (Figure S6C). Together, these data are consistent with a working “G-protein synergy” model that is sensitive to blockade of either TrkB-induced Gβγ release or mGluR5-induced Gαq activation (Figure 5A).
Based on these results, we further probed the potential Gαi/o coupling of TrkB. First, we tested if BDNF-mediated activation of TrkB can lead to similar downstream effects as Gαi/o-coupled GPCRs. We turned to cAMP imaging using the “cAMPr” sensor.70 Upon forskolin application to stimulate adenylate cyclase activity, Gαi/o-coupled receptor agonism typically decreases the enhancement of cAMP levels, as seen with MOR activation by DAMGO (Figures S6F and S6G). BDNF application decreased the forskolin response in TrkB-expressing cells (Figures 5C-5E). Consistent with a G-protein-dependent mechanism, BDNF-induced cAMP inhibition was not seen upon co-expression of DN-Gαi3 or GRK2-CT (Figure 5E). However, it is worth noting that the inhibition of cAMP levels by TrkB (~15%) was substantially smaller than that by MOR (~50%), indicating that TrkB is a much less efficient activator of G proteins. Consistent with this apparent functional interaction between TrkB and Gαi/o proteins, TrkB was able to co-immunoprecipitate Gαi3 both in HEK 293 cells and in neurons (Figure S6H), as has previously been shown.59
We then asked if the family of G-protein regulators that contain a G-protein-binding-and-activating (GBA) motif mediate the effects of TrkB on the crosstalk with mGluR5. This family of broadly expressed guanine nucleotide exchange factors (GEFs), including proteins like Girdin/GIV and DAPLE, has been shown to drive nucleotide exchange and Gβγ release from G-protein heterotrimers,57,71-76 including in response to RTK activation.56,57,72-74,76,77 To determine the necessity of direct involvement of G proteins for TrkB to crosstalk with mGluR5, we turned to a previously reported tool, termed GBAi.78,79 This is an engineered synthetic protein based on Gα that binds with high affinity to GBA motifs, but not to other known Gα interactors (i.e., Gβγ, GPCRs, effectors, RGS proteins, Ric-8A, GoLoco motifs), to specifically block GBA-dependent G-protein signaling without interference in canonical signaling via GPCRs (Figure 5F). Expression of GBAi reduced the efficiency of TrkB/mGluR5 crosstalk as assessed by Ca2+ imaging (Figure 5G). This inhibitory effect was enhanced by a mutation (S252A) that increases affinity for GBA motifs but was abolished by a different mutation (W211A) that prevents GBA motif binding.76,79 Crucially, GBAi-S252A had no effect on the response to BDNF in cells expressing only TrkB (Figure S6I), on the response to glutamate in cells co-expressing TrkB and mGluR5 (Figure S6J), or on the MOR/mGluR5 crosstalk (Figure 5H). Taken together with other results presented above, these data support a model in which Gβγ released upon the action of GBA proteins is responsible for the contribution of TrkB responses to the crosstalk with mGluR5 in regulating Ca2+ dynamics.
G-protein synergy underlies the mGluR5 dependence of BDNF-induced synaptic plasticity
To test if the G-protein synergy model of TrkB/mGluR5 crosstalk is consistent with BDNF-LTP, we used two perturbations to block either Gβγ or Gαq-GTP. First, to blunt the contribution of Gβγ, we injected a Cre-dependent adeno-associated virus (AAV) containing the membrane-tethered GRK2-CT construct into the CA1 with or without co-injection of a Cre construct under the CaMKII promoter to target expression to the CA1 pyramidal neurons (Figure S7A). After 5–6 weeks, strong expression of GRK2-CT was observed via immunohistochemistry, including clear dendritic localization (Figure S7A). Consistent with our model, GRK2-CT expression blunted BDNF-LTP to a similar degree compared with mGluR5 blockade or knockdown (Figures 6A-6C), without altering basal synaptic properties (Figure S7B) or DHPG responses (Figures S7C and S7D). GRK2-CT expression in cultured hippocampal neurons (Figure S7E) also impaired the BDNF-induced increase in spine density (Figure 6D). To test the contribution of Gαq-GTP, we applied YM-254890, which produced a small potentiation of basal fEPSP amplitude (Figure S7F) and fully blocked BDNF-induced LTP (Figures 6E-6G). YM-254890 pre-incubation also blocked BDNF-induced spine growth in cultured hippocampal neurons (Figure 6H), further supporting the G-protein synergy model.
Figure 6. mGluR5-dependent BDNF-LTP and BDNF-induced dendritic spine growth are mediated by G-protein crosstalk.

(A) Schematic of GRK2-CT mechanism.
(B and C) fEPSP slope time course (B) showing that overexpression of GRK2-CT leads to attenuation of BDNF-LTP. In (B), gray bars show regions averaged for baseline and post-BDNF values in (C).
(D) Bar graph summarizing the partial suppression of BDNF-induced increases in spine density with overexpression of GRK2-CT.
(E) Schematic of YM-254890 mechanism.
(F and G) fEPSP slope time course (F) showing that pre-incubation with YM-254890 leads to attenuation of BDNF-LTP. In (F), gray bars show regions averaged for baseline and post-BDNF values in (G).
(H) Pre-incubation with YM-254890 inhibits BDNF-induced spine density increase.
(I) Schematic of GBAi-S252A mechanism.
(J and K) fEPSP slope time course (J) showing that overexpression of GBAi-S252A leads to attenuation of BDNF-LTP. Overexpression of the non-binding GBAi-W211A does not affect BDNF-LTP. In (J), gray bars show regions averaged for baseline and post-BDNF values in (K).
(L) Bar graph summarizing the suppression of BDNF-induced increases in spine density with overexpression of GBAi-S252A, but not with overexpression of GBAi-W211A.
(M) Schematic of RTK/GPCR synergy model that drives BDNF-dependent LTP and spine growth. Points in (D) and (G) represent independent slices from separate mice. Points in (D), (H), and (L) represent independent neurons from three or four separate preparations. Unpaired t test is used for (C), (G), and (K). One-way ANOVA with Tukey’s multiple comparisons is used for (D), (H), and (L). All data are shown as the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S7.
Next, to test if GBA-dependent G-protein signaling (Figure 6I) underlies BDNF-induced plasticity, we used Cre-dependent AAVs for GBAi-S252A or the inactive GBAi-W211A construct. Both constructs expressed well after 5–6 weeks and showed dendritic localization (Figure S7G). Following transduction with GBAi-W211A AAV, normal BDNF-LTP was observed in hippocampal slices (Figures 6J and 6K). In contrast, transduction with GBAi-S252A led to a substantial decrease in BDNF-LTP (Figures 6J and 6K). There was no difference in basal synaptic strength between slices expressing GBAi-W211A and slices expressing GBAi-S252A (Figure S7H) and no difference in DHPG-induced depression (Figures S7I and S7J). Last, expression of GBAi-S252A (Figure S7L), but not GBAi-W211A (Figure S7K), impaired BDNF-induced spine growth in cultured hippocampal neurons (Figure 6L). Together, these results support a model in which non-canonical G-protein signaling activation by GBA proteins mediates TrkB synergistic contribution to mGluR5-dependent synaptic plasticity (Figure 6M).
TrkB/mGluR5 crosstalk mechanism is generalizable across receptor subtypes
As TrkB/mGluR5 synergy merely requires simultaneous BDNF-mediated activation of TrkB and constitutive Gαq activation by mGluR5, we asked if such crosstalk could proceed independent of the identity of the Gαq-coupled receptor. We first tested if overexpressing Gαq would be sufficient to boost TrkB-induced Ca2+ responses. Indeed, low-dose (25 ng/mL) BDNF responses were strongly enhanced in terms of both the percentage of cells responding (Figure 7A) and the response amplitude (Figure S8A) upon Gαq co-expression in HEK 293 cells. This suggests that elevated levels of Gαq-GTP sensitize cells to the response to TrkB activation. Consistent with this interpretation, YM25 abolished the boost in Ca2+ responses seen with Gαq overexpression (Figure 7A). We then asked if Gαq-coupled receptors other than mGluR5 can undergo crosstalk with TrkB. We used the TrkB-ΔPLCγ construct to abolish canonical PLC-γ-mediated TrkB responses and co-expressed either mGluR1 or 5-HT2AR, which are both primarily coupled to Gαq.4,80 mGluR1 co-expression allowed clear responses to BDNF in ~20%–40% of cells (Figures 7C and S8B), but 5-HT2AR co-expression enabled responses in only ~5%–10% of cells (Figure 7C). Based on our prior work,69 we reasoned that 5-HT2AR may not produce sufficient tonic Gαq activation and added a subthreshold level of 5-HT (1 nM). This boosted the proportion of cells responding to BDNF to 20%–30% (Figures 7B and 7C). As an important control, the Gαi/o-coupled mGluR2 did not enable BDNF responses either under basal conditions or in the presence of glutamate (Figure 7C), confirming the need for a Gαq-coupled GPCR to enable crosstalk with TrkB.
Figure 7. RTK/GPCR crosstalk is observed across a panel of receptors.

(A) Summary bar graph showing that co-expression of Gαq enhances Ca2+ responses upon low-dose BDNF-induced TrkB activation in HEK 293-TrkB cells.
(B) Representative trace showing Ca2+ response upon co-application of BDNF and subthreshold dose of 5-HT in HEK 293 cells co-expressing TrkB-ΔPLC and 5-HT2AR.
(C) Summary bar graph showing percentage of cells responding to BDNF in HEK293 cells co-expressing TrkBΔPLC and mGluR1 or 5-HT2AR or mGluR2 with or without co-application of 5-HT or glutamate.
(D and E) Representative traces showing intracellular Ca2+ responses to endogenous EGFR activation by EGF in the absence (D) or presence (E) of mGluR5.
(F) Distribution of EGF responses in the absence or presence of mGluR5 co-expression.
(G and H) Representative traces showing intracellular Ca2+ responses to TrkA activation by NGF in the absence (G) or presence (H) of mGluR5.
(I) Distribution of NGF responses in the absence or presence of mGluR5 co-expression.
(J and K) Representative traces showing intracellular Ca2+ responses to IGF1R activation by IGF in the absence (J) or presence (K) of mGluR5.
(L) Distribution of IGF responses in the absence or presence of mGluR5 co-expression. For (A) (C), (F), (I), and (L), points represent values from individual movies taken from distinct coverslips. For all conditions, data come from at least three separate cell preparations. Only cells showing a response to glutamate are analyzed in the conditions with mGluR5. One-way ANOVA with Tukey’s multiple comparisons is used for (A) and (C). All data are shown as the mean ± SEM; **p < 0.01, ***p < 0.001. See also Figure S8.
Given that RTKs other than TrkB have been shown to rely on heterotrimeric G proteins and/or GBA-motif-mediated mechanisms to propagate signaling,54,56,58,59,81-87 we asked if the cross-talk observed between TrkB and Gαq-coupled GPCRs could be observed with other RTKs. In HEK 293 cells, we detected slow-wave Ca2+ responses upon EGF application, indicating signaling functionality of endogenously expressed EGFR (Figure 7D). Upon mGluR5 co-expression, nearly half of the cells showed oscillatory Ca2+ responses to EGF (Figures 7E and 7F), suggesting a mechanism of crosstalk analogous to that characterized for TrkB/mGluR5. Similarly, exogenous expression of TrkA, another neurotrophin receptor in the same family as TrkB,88 enabled nerve growth factor (NGF)-induced slow-wave Ca2+ responses that converted to oscillatory responses upon mGluR5 co-expression (Figures 7G-7I). Finally, we tested the insulin growth factor 1 receptor (IGF1R) which, in contrast to other RTKs tested, showed no Ca2+ response to its native ligand, IGF-I (Figure 7J). Strikingly, mGluR5 co-expression enabled robust IGF-I responses that were primarily oscillatory (Figures 7K and 7L). Co-expression of IGF1R with mGluR1 or with 5-HT2AR both also enabled IGF responses in cells expressing IGF1R (Figures S8C and S8D). This further supports the finding that Gαq tone can re-shape the acute response to RTK activation. Finally, we tested if EGF- and IGF-induced, Gαq-dependent Ca2+ responses were mediated by the same G-protein synergy mechanism as observed with TrkB. Indeed, both EGF- and IGF-induced Ca2+ oscillatory responses upon co-expression of mGluR5 were decreased by co-expression of DN-Gαi3, GRK2-CT, or GBAi-S252A (Figures S8E and S8F), suggesting a mechanism similar to that of TrkB/mGluR5 crosstalk. Together, these data reveal that RTK activation can synergize with tonic GPCR activation to re-shape downstream signaling dynamics.
DISCUSSION
Our work identifies a mode of RTK/GPCR crosstalk by which mGluR5 acts as a critical mediator of TrkB effects by amplifying and altering the spatiotemporal dynamics of downstream signaling to drive a form of BDNF-induced synaptic plasticity. We show that TrkB enhances constitutive mGluR5 activity to initiate a mode switch leading to sustained, oscillatory Ca2+ signaling as well as enhanced MAP kinase pathway activation. This crosstalk is mediated by cooperativity between Gαq-GTP, released by mGluR5, and Gβγ, released by non-canonical TrkB signaling. This molecular mechanism appears to be conserved across different RTKs and Gαq-coupled GPCRs. Our findings thus support a G-protein synergy model (Figures 5A and 6M), in which simultaneous activation of both an RTK and a GPCR drives non-linear signal summation to re-shape the long-term consequences of receptor activation.
Given their extensive co-expression throughout the nervous system and the key roles of both TrkB and mGluR5 in synaptic plasticity, this neurotrophin/neurotransmitter receptor signaling crosstalk may contribute to a wide range of phenomena that have been previously attributed to either individual receptor system. For example, modulating TrkB or mGluR5 signaling has been proposed for treatment of many neurodegenerative and neuropsychiatric diseases. Recently, both TrkB and mGluR5 signaling have been shown to mediate the synaptic adaptations associated with rapid antidepressant action,6,89,90 which may employ a form of synaptic potentiation similar to that observed in this study. In this context, our findings that TrkB-driven synaptic plasticity is significantly enhanced by mGluR5 PAMs (Figures 1E and 2D) has potential therapeutic implications for next-generation antidepressant treatment strategies that would target this form of neuromodulatory crosstalk.
Furthermore, the G-protein synergy model proposed here represents a paradigm of RTK/GPCR crosstalk with relevance beyond the TrkB/mGluR5 combination and outside of the brain. We show that such crosstalk is observed with EGFR, TrkA, and IGF1R, critical RTKs with major relevance in cancer, development, and metabolism. For example, many cancers are associated with constitutively active Gαq mutations,91-93 which may provide a means of amplifying the pro-cancer signaling of RTKs94,95 and serve as promising therapeutic targets.96 In contrast to prior work that uncovered modes of GPCR transactivation of RTKs,31,32,37,38,97-101 our study represents an example of an RTK engaging constitutive GPCR signaling to mediate its effects by expanding its signaling repertoire. In our model, a downstream effector (i.e., PLC) serves as a coincidence detector to amplify the response to simultaneous RTK and GPCR activation. Notably, in line with this model, a recent study found that RTK-initiated downstream signaling can be blunted by Gαq inhibition.102 Importantly, this crosstalk mode does not require direct heteromerization between RTK and GPCR (e.g., TrkB and mGluR5), although the relative localization of the two receptors is likely a critical determinant of when and where such crosstalk may occur in native systems. In principle, this form of crosstalk could apply to many RTK/Gαq-coupled GPCR pairs and is likely subject to further layers of tuning and regulation, motivating future studies across biological contexts.
Limitations of the study
While we present a multitude of evidence for a G-protein synergy model (Figure 5A), it is important to note that many of our perturbations, such as expression of GRK2-CT, may affect G-protein tone broadly and, thus, perturb cells and synapses in ways that are difficult to interpret. Furthermore, other modes of crosstalk likely occur simultaneously to contribute to the complex signaling response to BDNF. For example, given that both TrkB and mGluR5 stimulate release of intracellular Ca2+ stores, Ca2+-induced Ca2+ release103 may contribute to crosstalk in the dendritic spines and shafts where we observe complex Ca2+ signaling dynamics. There may also be more indirect downstream modes of RTK/Gαq signaling pathway crosstalk at play, including feedback excitation and inhibition. This may explain why MPEP application blocks both BDNF-induced Ca2+ oscillations and slow-wave Ca2+ responses (Figures 4 and S5). Furthermore, it is worth noting that both TrkB and mGluR5 undergo complex modes of endocytosis, trafficking, and endosomal signaling,104,105 contributing to the functional interactions between receptors. Overall, the relative contributions of different modes of crosstalk likely shape the degree and spatiotemporal dynamics of crosstalk between TrkB and mGluR5 in different contexts, including different types of synapses and in different plasticity induction paradigms. This is also likely the case for tuning the crosstalk between other RTKs and other Gαq-coupled receptors that we propose as a general mode of receptor-receptor interaction (Figure 7).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dr. Francis S. Lee (fslee@med.cornell.edu).
Materials availability
All materials can be provided pending a completed material transfer agreement. Requests should be submitted to the lead contact Dr. Francis S. Lee (fslee@med.cornell.edu).
Data and code availability
All data are available in the main text or the supplementary materials.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Animal care was in accordance with Weill Cornell Institutional Animal Care and Use Committee. Mice were maintained with a 12-hour light/dark cycle at 18°C - 22°C and had ad libitum access to water and food. 8–10-week-old wild-type C57BL/6J male mice (Jackson laboratory) were used for electrophysiology. The mGluR5FL/FL mouse was purchased from Jackson Laboratory (B6.129-Grm5tm1.1Jixu/J, JAX stock #028626), and genotyping was performed per Jackson Laboratory’s protocol. All litters were weaned and sex segregated at P21. For primary hippocampal neuronal cultures, pregnant wild-type C57BL/6N mice were purchased from Charles River.
Human cell lines
HEK 293 cells were purchased from ATCC (CRL-1573) and authenticated by Bio-Synthesis, Inc. HEK 293-TrkB stable cell line is as previously described.48 All cells were tested for mycoplasma every 8 weeks with ATCC Universal Mycoplasma Detection Kit to ensure no contamination. HEK 293 cells were maintained in HEK media, consisting of DMEM (Gibco) supplemented with 10% fetal bovine serum, 100 mM sodium pyruvate, and 100 U/mL penicillin-streptomycin. HEK 293-TrkB stable cells were maintained in HEK media with 200 mg/ml G418 selection antibiotic to sustain a pure population of HEK 293-TrkB cells. Cells were passaged every 3-4 days when they reached to 90% confluency.
METHOD DETAILS
Reagents and antibodies
Recombinant human BDNF (Cat # 450-02), NT-3 (Cat # 450-03), NT-4 (Cat # 450-04), EGF (Cat #AF-100-15), IGF-I (Cat #100-11), NGF (Cat #450-01) were purchased from Peprotech and reconstituted in Invitrogen Ultra-Pure DNase/RNase-Free water. Picrotoxin (Cat #1128), tetrodotoxin (TTX, Cat # 1069), DHPG (Cat # 0805), MPEP (Cat # 1212), U73122 (Cat # 1268), U73343 (Cat # 4133), PD98059 (Cat # 1213), DAMGO (Cat # 1171) and baclofen (Cat # 0417), were purchased from Tocris Bioscience. YM254890 was purchased from Cayman Chemical. VU-29 was purchased from HelloBio (Cat # HB0642). Antibodies for western blots and co-immunoprecipitation assays were anti-phospho-p44/42 MAPK (Cell Signaling, Technology, 9101), anti-p44/42 MAPK (Cell Signaling Technology, 9102), anti-β-actin (Sigma, A1978), anti-TrkB (Millipore Sigma, 07-225), and anti-Gαi3 (Santa Cruz, H-7). Antibodies for immunocytochemistry were TrkB (R&D, AF1494), mGluR5 (Alomone, AGC-007), Alexa Fluor 546 Phalloidin (Invitrogen, A22283). Antibody for immunohistochemistry was anti-HA-Tag (Cell Signaling, C29F4, 3724) and anti-myc-Tag (Cell Signaling, 71D10, 2278).
Electrophysiology
Coronal brain slices (300 μm thick) containing hippocampus were prepared from male mice (C57BL/6J wildtype or mGluR5FL/FL). Following isoflurane anesthesia, mice were transcardially perfused, followed by brain extraction. Slices were cut with a vibratome (VT1200S, Leica) in ice-cold, oxygenated NMDG-HEPES aCSF112 containing (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2·2H2O, and 10 MgSO4·7H2O, pH to 7.3–7.4. Slices were incubated for 15-20 min on NMDG-HEPEs aCSF at 37°C then transferred to an incubation chamber at room temperature for at least 1.5 hrs containing HEPES aCSF (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2·2H2O, and 2 MgSO4·7H2O, pH to 7.3–7.4. Individual slices were transferred to an immersion recording chamber, where they were submerged in oxygenated aCSF containing (in mM): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 12.5 glucose, 5 HEPES, 2 CaCl2·2H2O, and 2 MgSO4,·7 H2O, and 10 μM picrotoxin, pH to 7.3-7.4, flowing 3 mL/min at room temperature. Field Excitatory Postsynaptic Potentials (fEPSP) were recorded through a carbon fiber electrode (Carbostar-1, Kation Scientific) placed in the stratum radiatum of the CA1 region. Evoked fEPSPs were elicited by Schaffer collateral stimulation with an extracellular bipolar tungsten electrode via a constant current stimulator (DS3, Constant Current Isolated Stimulator, Digitimer Ltd.) using monophasic currents of 200 μs duration. Baseline recording was obtained by stimulating the slice every 60 sec until the signal was stable using a current that elicited a 30–40% maximal response measured as the initial slope. fEPSPs were recorded in Axon Clampex 9.2 (Molecular Devices) using a Multiclamp 700b amplifier (Molecular Devices), a Digidata 1324 (Molecular Devices) with 10 kHz sampling and analyzed using Clampfit 9.2 (Molecular Devices). Following a stable baseline of ≥30 min, BDNF was applied for 30 min and recording proceeded for at least another 60 min. ANA-12 (10 μM), MPEP (40 μM), VU-29 (500 nM) were perfused for 20 min, while U73343 (5 μM), U73122 (5 μM), PD-98059 (50 μM), YM254890 (20 μM) for 60 min before and during BDNF perfusion. For DHPG-induced depression, stable baseline was established for at least 20 minutes before DHPG (50 μM) was perfused at 3 mL/min at room temperature. For paired-pulse analysis, paired stimuli with variable interstimulus intervals (25-500 ms) were applied to the Schaffer collaterals.
Stereotaxic viral injections for ex vivo slice electrophysiology
Mice were anesthetized with a ketamine and xylazine cocktail (0.1 mL per 10 g of body weight) and mounted on a stereotaxic frame (David Kopf Instruments). Viral injections were all done in the dorsal CA1 region of the hippocampus (2.2 mm AP, −1.5 mm ML, −1.25 mm DV from bregma), using a Nanoject II Auto-Nanoliter Injector (Drummond Scientific Company) equipped with a Nanofil syringe (World Precision Instruments). For ex vivo mGluR5 genetic knockout slices, 7-week-old mGluR5FL/FL male mice were injected with 150 nL of AAV8-hSyn-mCherry-Cre (UNC Vector Core) or control AAV8-hSyn-mCherry (UNC Vector Core). Electrophysiology experiments were performed 5-6 weeks after injections to allow sufficient time for knockdown. To express HA-GRK2-CT, Cre-dependent AAV8-HA-GRK2-CT (UNC Vector Core, viral titer 6 x 1012) was pre-mixed with AAV5-CaMKII-mCherry-Cre (UNC Vector Core, viral titer 3.5 x 1012) at 2:1 volume ratio before injection into wildtype C57BL/6J 7-week-old male mice, at a final volume of 150 nL. For control, AAV5-CaMKII-mCherry (UNC Vector Core, viral titer 3.5 x 1012) was used. To express GBAi-W211A and GBAi-S252A, Cre-dependent AAV8-DIO-GBAi-W211A (UNC Vector Core, viral titer 1.5 x 1012) or AAV8-DIO-GBAi-S252A (UNC Vector Core, viral titer 1 x 1012) was pre-mixed with AAV5-CaMKII-mCherry-Cre (UNC Vector Core, viral titer 3.5 x 1012) at 2:1 volume ratio before injection into wildtype C57BL/6J 7-week-old male mice, at a final volume of 150 nL. Electrophysiology experiments were performed 5-6 weeks after. To validate viral expression, mice were perfused and coronal sections from fixed brains were prepared, stained for anti-HA (for HA-GRK2-CT) or anti-myc (for myc-GBAi-W211A and myc-GBAi-S252A), and imaged on the Zeiss LSM 880 confocal (refer to “Immunohistochemistry and confocal imaging” section). Confocal imaging to show representative slices of the CA1 injections and expression of the viruses were performed at the Weill Cornell Medicine CLC Imaging Core Facility using Zeiss LSM 880 equipped with Zeiss Plan-Apochromat 20x / NA 0.8. Z-stack tiled image was acquired and stitched together with a 10% overlay, leading to the composite image of the whole hippocampus.
Primary hippocampal neuronal cultures
Primary hippocampal neurons were prepared from E18 mice, as previously described,113 with some modifications. Briefly, bilateral hippocampi were collected and digested with 0.22 μm PES membrane-filtered custom digestion solution containing 5 mg/mL deoxyribonuclease I (Sigma, D4527/10KU), 1.5 mM CaCl2, 0.75 mM EDTA, 200 units of papain (Worthington, LS003127), and 2.5 mM L-cysteine (Sigma, C7352) for 5 min at 37°C. The cells were plated on autoclaved, nitric acid-washed, poly-L-lysine (Sigma) coated glass cover slips (Electron Microscopy Sciences) in Neuronal Plating Medium for 2-4 hours. They were maintained in modified Neurobasal/B27 Medium, which also had 1 mM sodium pyruvate (Gibco, 11360070) and 100 U/mL penicillin-streptomycin (Gibco, 15070063) in addition to the B27 and GlutaMAX-I supplements. On days in vitro (DIV) 0, 4 μM cytosine-1-β-D-arabinofuranoside (Sigma, 251010) was added to limit glial proliferation.
Immunocytochemistry and confocal imaging of dendritic spines
DIV 21-24 primary hippocampal neurons were pre-treated with 1 μM TTX and the drug of interest (1 μM MPEP for 20 min, 500 nM VU-29 for 20 min, 10 μM ANA-12 for 1 hour, 5 μM U73122 for 1 hour, 50 μM PD98059 for 1 hour) before BDNF (50 ng/mL or 100 ng/mL) addition for 30 min at 37°C. The neurons were then washed twice with pre-warmed Neurobasal media and fixed with 4% paraformaldehyde/4% sucrose solution for 10 min at room temperature. Permeabilization and blocking were achieved with 0.1% Triton X-100 in 3% donkey serum and 3% BSA PBS solution for 30 min at room temperature. Primary antibodies were applied for 1 hour at room temperature or 4° overnight, per each antibody’s manufacturer protocols. Subtype-specific fluorophore-conjugated secondary antibodies at 1:500 dilutions were added for 30 min at room temperature. Coverslips were mounted with ProLong Gold antifade reagent (Invitrogen, P10144). Confocal imaging of the fixed coverslips was performed within 1 week of staining. Confocal imaging was performed at the Weill Cornell Medicine CLC Imaging Core Facility using Zeiss LSM 880 equipped with 32-element AiryScan detector for super-resolution imaging and 32-channel GaAsP array for spectral imaging. For spine imaging, AiryScan imaging was performed using Zeiss Plan-Apochromat 63x/1.4 Oil DIC M27 with NA 1.4 at with zoom 3.0x. Z-stack images were collected at 0.22 μm intervals and AiryScan deconvolution (Zeiss ZEN Black) was performed for each image. Spine density analysis was done by linearizing discrete secondary dendrites on ImageJ followed by manual counting of protruding dendritic spines performed blind to the experimental conditions. Approximately 40-70 μm of secondary dendrite was analyzed per neuron.
Calcium imaging in primary hippocampal neurons
For neuronal Ca2+ imaging experiments, pGP-AAV1-Syn-GCaMP8m-WPRE (Addgene, Cat# 162375, viral titer per well 1×108vg/mL) was added to the cultures at DIV 14-17. At DIV 18-23 hippocampal neuron coverslips were placed in a perfusion chamber at 33°C with a continual gravity-based perfusion of neuronal extracellular solution containing 1 μM TTX, 138 mM NaCl, 1.5 mM KCl, 5 mM HEPES, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM glucose, pH 7.4. Imaging was performed on an Olympus IX83 microscope equipped with 488 nm laser illumination and a Hamamatsu ORCA-Flash4.0 V2 sCMOS camera imaging at 10 Hz with 42 ms exposure time. For the BDNF condition, baseline recording with neuronal extracellular solution perfusion was done for 5 minutes, followed by 100 ng/mL BDNF for 10 min, then washed out for 5 min. For the MPEP condition, baseline recording was done for 5 min, followed by 1 μM MPEP for 5 min, 1 μM MPEP and 100 ng/mL BDNF for 10 min, and washout for 5 min. For VU-29 condition, baseline recording was done for 5 minutes, followed by 500 nM VU-29 for 5 min, 500 nM + 50 ng/mL BDNF for 10 min, and washout for 5 min. Uniform 10 μm dendritic ROIs were manually drawn along each dendrite on ImageJ. Time series mean intensities were extracted for each ROI, and average background fluorescence subtracted. Photobleaching was corrected with a fitted exponential curve and intensities were normalized to a 1 min sliding median window as dF/F. To calculate the proportion of dendritic area with a BDNF response, ROIs were manually categorized as ROIs with or without signal based on the presence or absence of clear Ca2+ elevations in the presence of BDNF.
Plasmids and molecular cloning
Previously described106,114 HA-SNAP-mGluR5 plasmids were used and modified via site-directed mutagenesis. For heterologous expression of TrkB, a SNAP-TrkB clone was made via Gibson assembly and contains an N-terminal signal sequence followed by an HA-tag and a SNAP-tag prior to full length rat TrkB. Point mutations (“kinase dead” K571N) and deletions (PLC-γ site: Δ806-821; FRS site: Δ510-513, Shc site: Δ806-821). GRK2-CAAX (# 166224) and cAMPr (# 99143) were purchased from Addgene. The PM-GRK2-CT plasmid107 and PTX-S1 constructs were previously described.108 The GRK2-CT ORF was subcloned into a Cre-dependent AAV plasmid by restriction enzyme ligation and an HA-tag was added to the N-terminus. The G203A mutation was introduced into a rat Gαi3 clone (gift from D. Logothetis). An HA tag followed by a SNAP tag was introduced via Gibson assembly at the N-terminus of the rat MOR (gift from J. Broichhagen). The rat GABABR clones were as previously described.109 All constructs were verified by sequencing. The pLIC-myc-GBAi (# 171753) and pLIC-myc-GBAi W211A (# 171754) plasmids were purchased from Addgene. The S252A mutation was introduced into the pLIC-myc-GBAi clone.78 The GBAi S252A and GBAi W211A were subcloned into a Cre-dependent AAV plasmid by restriction enzyme ligation.
HEK 293 cell transfection
For transfection and Ca2+ imaging and cAMP imaging, cells were plated on poly-L-lysine coated glass coverslips and transfected using Lipofectamine 2000 or 3000 (Thermo Fisher Scientific). For all transfections, 0.5 μg of DNA was used for each plasmid, except 0.3 μg of GCaMP6f and 0.7 μg of mGluR5-ΔECD. With experiments involving heterologous expression of mGluR5, cells were maintained in MPEP (1 μM) post-transfection to maintain cell health.
Calcium imaging in HEK 293 cells
GCaMP6f imaging was performed 24-48 hours after transfection at room temperature on either an inverted fluorescent Nikon Eclipse Ti2-E microscope equipped with an Andor Zyla 5.5 sCMOS camera or an Olympus IX83 microscope equipped with a Hamamatsu ORCA-Flash4.0 V2 sCMOS camera. All imaging was done with a 20x objective using either a 488 nm LED (Nikon microscope) or 488 nm laser (Olympus microscope). Movies were the acquired with 100 ms exposures at 0.5-1.0 Hz. Cells were continuously super-fused with extracellular solution containing (in mM): 135 NaCl, 5.4 KCl, 10 HEPES, 2 CaCl2, 1 MgCl2, pH=7.4). Drugs were applied using a gravity driven perfusion system. Analysis was performed using ImageJ and NIS-Elements Advance Research 5.2.6 software. After selecting single-cell regions of interest, fluorescence intensities were quantified as dF/F by subtracting each ROI’s average resting baseline fluorescence from the first 2 min of recording before ligand addition (F0) from the fluorescence from any given time point (Ft), divided by F0. Response latency was quantified as time from initial drug application to the first rise of response above baseline and response duration was defined as the amount of time required for fluorescence intensity to return to baseline levels after the initial rise (baseline-to-baseline). Response amplitude is the peak dF/F value and response frequency was defined as the reciprocal of averaged interval between peaks. Responses were identified as either “oscillatory” or “slow wave” based on response duration (>50 s for slow wave) and if multiple peaks were observed. At least 2 biological replicates (i.e. separate transfections) were included for all experiments (see Figure legends for details). All analyses were manually performed on Microsoft Excel, with statistical analysis performed on GraphPad Prism. Analysis of surface expression of SNAP-tagged constructs was performed as previously described.115
cAMP imaging in HEK 293 cells
cAMPr imaging was performed 24-48 hours after transfection at room temperature on an inverted fluorescent Nikon Eclipse Ti2-E microscope equipped with an Andor Zyla 5.5 sCMOS camera. Briefly, movies were the acquired with 100 ms exposures at 0.25-0.5Hz with 20x objective. Cells were continuously perfused with the same extracellular solution same as used in HEK 293 calcium imaging experiments. BDNF or DAMGO was applied to cells 2 min prior to Forskolin co-application. Fluorescence intensity was analyzed in similarly, as previously described for HEK 293 calcium imaging. Area under the curve (AUC) of the normalized traces was used to quantify the cAMP response. Analysis was performed using ImageJ and NIS-Elements Advance Research 5.2.6 software. All analyses were manually performed on Microsoft Excel, with statistical analysis performed on GraphPad Prism
Western blot and immunoprecipitation assays
For western blot lysate preparation, HEK 293-TrkB cells were treated with drugs prior to cell lysis with cold Pierce RIPA lysis buffer (Thermo Scientific, 89901) supplemented with protease inhibitor set I (Sigma, 539131) and phosphatase inhibitor set II (Sigma, 524625). Protein extracts were quantified using Pierce BCA protein assay kit (Thermo Scientific, 23225). 20 μg (HEK 293-TrkB) of protein was reduced in NuPAGE LDS sample buffer (Invitrogen, NP0007) with NuPAGE sample reducing agent (Invitrogen, NP0009) and boiled at 65°C for 10 min. Proteins were then run on 4-12% Bolt-Tris Plus mini gels for separation and transferred onto Bio-Rad Immun-Blot PVDF membrane using Bio-Rad Trans-Blot Turbo Transfer System. Membranes were blocked for at least 1 hour at room temperature prior to incubation with primary antibodies 4°C overnight or 1 hour at room temperature, depending on each antibody’s manufacturer recommendations. Following 3 washes in TBST, membranes were incubated with appropriate HRP secondary antibodies (Jackson laboratories) at room temperature for 30 minutes prior to visualization by ECL detection (Thermo Scientific, 32106) and Bio-Rad ChemiDoc XRS+ imaging system with Image Lab Software. Membranes were stripped with Restore stripping buffer (Thermo Scientific, 21063), rinsed 4 times with TBST, before blocking and probing again with other antibodies. Each experiment was repeated at least three times. Blots were analyzed on ImageJ by measuring intensity of phosphorylated ERK1/2 bands with corresponding ERK1/2 bands for normalization. Total ERK1/2 was first normalized to actin-loading control bands to accommodate for any variability in amount of protein loaded into each well. For all immunoprecipitation assays, cells were lysed with immunoprecipitation lysis buffer (20 mM HEPES, 5 mM Mg-acetate, 125 mM K-acetate, 0.4% Triton X-100, 1mM dithiothreitol, 100 μM sodium orthovanadate, and phosphatase/protease inhibitors from Millipore Sigma, as noted previously). For the TrkB and mGluR5 co-immunoprecipitation experiment in DIV 19 primary hippocampal neurons, no ligand was added prior to cell lysis. For the TrkB and Gαi3 co-immunoprecipitation experiments in HEK 293 cells and primary hippocampal neurons, BDNF 100 ng/mL was added at 37°C for 15 minutes prior to cell lysis. Sepharose protein G beads were pre-incubated with antibodies to form antibody-bead complexes overnight at 4°C overnight. These complexes were then added to protein lysates for 2-4 hours at room temperature to allow for binding of desired protein to the beads. The beads were then washed 4 times in immunoprecipitation lysis buffer, then similarly reduced in NuPAGE LDS sample buffer and NuPAGE sample reducing agent prior to boiling at 65°C for 10 min to allow elution of the proteins off the beads. The immunoprecipitated proteins were then resolved as per western blot protocol above.
Immunohistochemistry and confocal imaging
Whole brain coronal sections (40 μm) were prepared using a sliding microtome. Serial sections were washed in TBS, incubated for 30 min in a blocking solution containing 4% normal horse serum (vol/vol), 1% BSA in TBS with 0.2% Triton X-100, and incubated overnight at 4°C with primary antibodies diluted in blocking solution. Sections were then washed in TBS and incubated for 2 hours with subtype-specific Alexa-488 conjugated secondary antibody at room temperature. After washing three times for 10 min, sections were mounted, coverslipped with water soluble glycerol-based mounting medium containing DAPI. Confocal imaging was performed on a Zeiss LSM 880 with Zeiss Plan-Apochromat 20x/0.8 DIC-II. Tiled Z-stack images were collected at 0.94 μm intervals.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as means ±SEM and analyzed with GraphPad Prism 9.0 software. Statistical significances were calculated via unpaired student t test (for two group comparisons), one-way ANOVA with Šidák and Tukey’s post hoc tests to control for multiple comparisons (for three or more group comparisons), or two-way ANOVA with Sidak’s multiple comparison tests (to assess statistical significance between means), as indicated within individual figure legends. For frequency distribution histogram statistics, exact sum-of-squares F-test was used. In figures, asterisks denote statistical significance marked by * P < 0.05, ** P < 0.01, *** P < 0.001, and “n.s.” indicates no statistical significance.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-phospho-p44/42 MAPK | Cell Signaling Technology | Cat # 9101 RRID: AB_331646 |
| anti-p44/42 MAPK | Cell Signaling Technology | Cat # 9102 RRID: AB_330744 |
| anti-β-actin | Sigma-Aldrich | Cat # A1978, RRID: AB_476692 |
| anti-TrkB | R and D Systems | Cat # AF1494 RRID:AB_2155264 |
| anti-TrkB | Millipore Sigma | Cat #07-225, RRID: AB_310445 |
| anti-Gαi3 (H-7) | Santa Cruz Biotechnology | Cat #sc-365422, RRID: AB_10847081 |
| anti-mGluR5 | Alomone Labs | Cat # AGC-007, RRID:AB_2039991 |
| anti-HA-Tag (C29F4) | Cell Signaling Technology | Cat # 3724, RRID: AB_1549585 |
| anti-myc-Tag (71D10) | Cell Signaling Technology | Cat #2278, RRID: AB_490778 |
| Alexa 546 Phalloidin | Invitrogen | Cat # A22283 |
| Bacterial and virus strains | ||
| AAV8-hSyn-mCherry-Cre | UNC Vector Core | N/A |
| AAV8-hSyn-mCherry | UNC Vector Core | N/A |
| AAV8-HA-GRK2-CT | UNC Vector Core | N/A |
| AAV8-DIO-GBAi-W211A-myc | UNC Vector Core | N/A |
| AAV8-DIO-GBAi-S252A-myc | UNC Vector Core | N/A |
| AAV5-CaMKII-mCherry-Cre | UNC Vector Core | N/A |
| AAV5-CaMKII-mCherry | UNC Vector Core | N/A |
| AAV1-Syn-GCaMP8m-WPRE | Addgene | Cat # 162375 |
| Chemicals, peptides, and recombinant proteins | ||
| Picrotoxin | Tocris | Cat #1128 |
| Tetrodotoxin (TTX) | Tocris | Cat # 1069 |
| DHPG | Tocris | Cat # 0805 |
| MPEP | Tocris | Cat # 1212 |
| U73122 | Tocris | Cat # 1268 |
| U73343 | Tocris | Cat # 4133 |
| PD98059 | Tocris | Cat # 1213 |
| DAMGO | Tocris | Cat # 1171 |
| Baclofen | Tocris | Cat # 0417 |
| YM254890 | Cayman | Cat # 29735 |
| Forskolin | Tocris | Cat # 1099 |
| VU-29 | HelloBio | Cat # HB0642 |
| ANA-12 | Tocris | Cat # 4781 |
| Recombinant human BDNF | Peprotech | Cat # 450-02 |
| Recombinant human NT-3 | Peprotech | Cat # 450-03 |
| Recombinant human NT-4 | Peprotech | Cat # 450-04 |
| Recombinant human EGF | Peprotech | Cat # AF-100-15 |
| Recombinant human IGF-I | Peprotech | Cat # 100-11 |
| Recombinant human NGF | Peprotech | Cat # 450-01 |
| SNAP-Surface Alexa Fluor 546 | New England Biolabs | Cat #S9132S |
| Critical commercial assays | ||
| Universal Mycoplasma Detection Kit | ATCC | Cat #30-1012K |
| Experimental models: Cell lines | ||
| HEK 293 | ATCC | CRL-1573 |
| HEK293-TrkB | Narisawa-Saito et al.48 | |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| B6.129-Grm5tm1.1Jixu/J | The Jackson Laboratory | JAX: 028626 |
| C57BL/6N | Charles River | Strain 027 |
| Recombinant DNA | ||
| HA-SNAP-mGluR5 | Gutzeit et al. 106 | |
| HA-SNAP-mGluR5 ΔECD | This paper | |
| HA-SNAP-mGluR5 F768D | This paper, based on Francesconi and Duvoisin 52 | |
| HA-SNAP-TrkB | This paper | |
| HA-SNAP-TrkB-K571N (“kinase dead”) | This paper | |
| HA-SNAP-TrkB-Δ806-821 (“TrkB-ΔPLC-γ” ) | This paper | |
| HA-SNAP-TrkB-Δ510-513-Y515F-Δ806-821 (“TrkB-ΔFRS2-ΔShc-ΔPLC-γ”) | This paper | |
| GRK2-CAAX | Addgene | Cat. # 166224 |
| PM-HA-GRK2-CT | Irannejad and Wedegaertner 107 | |
| PTX-S1 | Vivaudou et al. 108 | |
| rat GABABR | Zheng et al. 109 | |
| Gαi3- G203A | gift from D. Logothetis | |
| MOR | gift from J. Broichhagen | |
| myc-GBAi | Addgene | Cat # 171753 78 |
| myc-GBAi-W211A | Addgene | Cat #171754 78 |
| myc-GBAi-S252A | de Opakua et al. 79 | |
| IGF1R | Sino Biological | Cat.# HG10164-NY |
| 5-HT2A | Morsetin et al. 110 | |
| cAMPr | Addgene | Cat. # 99143 |
| Software and algorithms | ||
| pCLAMP 9.2 (Clampex and Clampfit) | Molecular Devices | RRID:SCR_011323 |
| ImageJ | Schneider et al.111 | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
| NIS-Elements Advance Research 5.2.6 | Nikon | https://nikon.com; RRID: SCR_014329 |
| Olympus cellSens | Olympus | https://olympus-lifescience.com/cellsens, RRID: SCR_016238 |
| Zeiss Zen Black | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html, RRID:SCR_013672 |
| Microsoft Excel | Microsoft Office | https://products.office.com/en-us/excel, RRID: SCR_016137 |
| ChemiDoc Image Lab software | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software, RRID: SCR_104210 |
| GraphPad Prism 9 | GraphPad | https://graphpad.com, RRID:SCR_002798 |
| Adobe Illustrator | Adobe | https://adobe.com, RRID: SCR_010279 |
Highlights.
BDNF-driven synaptic plasticity is dependent on mGluR5
mGluR5 enables a mode switch in BDNF-TrkB signaling
Non-canonical G-protein coupling drives BDNF-induced plasticity
G-protein-dependent crosstalk is seen between multiple RTKs and Gq-coupled GPCRs
ACKNOWLEDGMENTS
We are grateful to David Simon (Weill Cornell Medicine) for access to his Nikon Ti2-E microscope and valuable discussions. We thank Hermany Munguba (Weill Cornell Medicine) for setting up the electrophysiology rig. We thank Rasmus Herlo Jensen (Columbia University) and Logan Grosenick (Weill Cornell Medicine) for their insights on neuronal calcium signaling analysis. Last, we thank research technicians RoshelleSmith (Weill Cornell Medicine), Chienchun (Ted) Huang (Weill Cornell Medicine), Nikhil Kothari (Weill Cornell Medicine), and Rui Rong Yang (Weill Cornell Medicine) for assistance with preparation of cultures, mouse colony management, heterologous cell maintenance, and immunohistochemistry. Funding sources for this work are National Institutes of Health grants R01NS126590 (J.L. and F.S.L.), R35GM124731 (J.L.), R01MH123154 (F.S.L.), and R01GM136132 (M.G.-M.); a Rohr Family Research Scholar Award (J.L.); a Monique Weill-Caulier award (J.L.); the Pritzker Neuro-psychiatric Disorders Research Consortium (F.S.L.); the New York – Presbyterian Center for Youth Mental Health (F.S.L.); National Research Foundation (NRF) of Korea grant NRF-2021R1I1A3055750 (M.S.); and a Burroughs Well-come Fund – Weill Cornell Physician-Scientist Academy award (J.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113595.
DECLARATION OF INTERESTS
The authors declare that they have no competing interests.
REFERENCES
- 1.Grundmann M, and Kostenis E (2017). Temporal Bias: Time-Encoded Dynamic GPCR Signaling. Trends Pharmacol. Sci 38, 1110–1124. [DOI] [PubMed] [Google Scholar]
- 2.Lemmon MA, and Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pierce KL, Premont RT, and Lefkowitz RJ (2002). Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol 3, 639–650. [DOI] [PubMed] [Google Scholar]
- 4.Reiner A, and Levitz J (2018). Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 98, 1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee FS, and Hempstead BL (2018). New Roles for an Ancient Factor. Trends Neurosci. 41, 765–767. [DOI] [PubMed] [Google Scholar]
- 6.Castrén E, and Monteggia LM (2021). Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 90, 128–136. [DOI] [PubMed] [Google Scholar]
- 7.Javitt DC (2004). Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 9, 984–997. [DOI] [PubMed] [Google Scholar]
- 8.Volk L, Chiu SL, Sharma K, and Huganir RL (2015). Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci 38, 127–149. [DOI] [PubMed] [Google Scholar]
- 9.Huang EJ, and Reichardt LF (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem 72, 609–642. [DOI] [PubMed] [Google Scholar]
- 10.Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, and Kaplan DR (1994).Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron 12, 691–705. [DOI] [PubMed] [Google Scholar]
- 11.Flint AC, Dammerman RS, and Kriegstein AR (1999). Endogenous activation of metabotropic glutamate receptors in neocortical development causes neuronal calcium oscillations. Proc. Natl. Acad. Sci. USA 96, 12144–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Codazzi F, Teruel MN, and Meyer T (2001). Control of astrocyte Ca(2+) oscillations and waves by oscillating translocation and activation of protein kinase C. Curr. Biol 11, 1089–1097. [DOI] [PubMed] [Google Scholar]
- 13.Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Nakanishi S, and Okada M (1996). Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature 383, 89–92. [DOI] [PubMed] [Google Scholar]
- 14.Kim CH, Braud S, Isaac JTR, and Roche KW (2005). Protein kinase C phosphorylation of the metabotropic glutamate receptor mGluR5 on Serine 839 regulates Ca2+ oscillations. J. Biol. Chem 280, 25409–25415. [DOI] [PubMed] [Google Scholar]
- 15.Ango F, Prézeau L, Muller T, Tu JC, Xiao B, Worley PF, Pin JP, Bockaert J, and Fagni L (2001). Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature 411, 962–965. [DOI] [PubMed] [Google Scholar]
- 16.Kubo Y, Miyashita T, and Murata Y (1998). Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science 279, 1722–1725. [DOI] [PubMed] [Google Scholar]
- 17.Minichiello L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci 10, 850–860. [DOI] [PubMed] [Google Scholar]
- 18.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, and Lu B (1996). Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381, 706–709. [DOI] [PubMed] [Google Scholar]
- 19.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, and Bonhoeffer T (1996). Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc. Natl. Acad. Sci. USA 93, 12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang H, and Schuman EM (1995). Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662. [DOI] [PubMed] [Google Scholar]
- 21.Kang H, and Schuman EM (1996). A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406. [DOI] [PubMed] [Google Scholar]
- 22.Ji Y, Lu Y, Yang F, Shen W, Tang TTT, Feng L, Duan S, and Lu B (2010). Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci 13, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomassoni-Ardori F, Fulgenzi G, Becker J, Barrick C, Palko ME, Kuhn S, Koparde V, Cam M, Yanpallewar S, Oberdoerffer S, and Tessarollo L (2019). Rbfox1 up-regulation impairs BDNF-dependent hippocampal LTP by dysregulating TrkB isoform expression levels. Elife 8, e49673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, and Collingridge GL (1993). Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature 363, 347–350. [DOI] [PubMed] [Google Scholar]
- 25.Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, and Roder JC (1997). Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J. Neurosci 17, 5196–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, et al. (2009). mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34, 2057–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber KM, Roder JC, and Bear MF (2001). Chemical induction of mGluR5- and protein synthesis–dependent long-term depression in hippocampal area CA1. J. Neurophysiol 86, 321–325. [DOI] [PubMed] [Google Scholar]
- 28.Lüscher C, and Huber KM (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dogra S, and Conn PJ (2021). Targeting metabotropic glutamate receptors for the treatment of depression and other stress-related disorders. Neuropharmacology 196, 108687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park H, and Poo MM (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci 14, 7–23. [DOI] [PubMed] [Google Scholar]
- 31.Luttrell LM, Daaka Y, and Lefkowitz RJ (1999). Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr. Opin. Cell Biol 11, 177–183. [DOI] [PubMed] [Google Scholar]
- 32.Di Liberto V, Mudò G, and Belluardo N (2019). Crosstalk between receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCR) in the brain: Focus on heteroreceptor complexes and related functional neurotrophic effects. Neuropharmacology 152, 67–77. [DOI] [PubMed] [Google Scholar]
- 33.Shah BH, and Catt KJ (2004). GPCR-mediated transactivation of RTKs in the CNS: mechanisms and consequences. Trends Neurosci. 27, 48–53. [DOI] [PubMed] [Google Scholar]
- 34.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, and Ullrich A (1999). EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402, 884–888. [DOI] [PubMed] [Google Scholar]
- 35.Cussac D, Schaak S, Denis C, and Paris H (2002). alpha 2B-adrenergic receptor activates MAPK via a pathway involving arachidonic acid metabolism, matrix metalloproteinases, and epidermal growth factor receptor transactivation. J. Biol. Chem 277, 19882–19888. [DOI] [PubMed] [Google Scholar]
- 36.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, and Rockman HA (2007). Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest 117, 2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, Qiao Y, and Li Z (2018). New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci 39, 367–386. [DOI] [PubMed] [Google Scholar]
- 38.Lee FS, and Chao MV (2001). Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. USA 98, 3555–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mira E, Lacalle RA, González MA, Gómez-Moutón C, Abad JL, Bernad A, Martínez-A C, and Mañes S (2001). A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep. 2, 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delcourt N, Thouvenot E, Chanrion B, Galéotti N, Jouin P, Bockaert J, and Marin P (2007). PACAP type I receptor transactivation is essential for IGF-1 receptor signalling and antiapoptotic activity in neurons. EMBO J. 26, 1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, and Rognan D (2011). Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest 121, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TVP, and Bramham CR (2002). Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci 22, 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, and Korte M (2002). Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36, 121–137. [DOI] [PubMed] [Google Scholar]
- 44.Ji Y, Pang PT, Feng L, and Lu B (2005). Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat. Neurosci 8, 164–172. [DOI] [PubMed] [Google Scholar]
- 45.Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, and McNamara JO (2016). Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka JI, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GCR, and Kasai H (2008). Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319, 1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang H, and Schuman EM (2000). Intracellular Ca(2+) signaling is required for neurotrophin-induced potentiation in the adult rat hippocampus. Neurosci. Lett 282, 141–144. [DOI] [PubMed] [Google Scholar]
- 48.Narisawa-Saito M, Iwakura Y, Kawamura M, Araki K, Kozaki S, Takei N, and Nawa H (2002). Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. J. Biol. Chem 277, 40901–40910. [DOI] [PubMed] [Google Scholar]
- 49.Middlemas DS, Meisenhelder J, and Hunter T (1994). Identification of TrkB autophosphorylation sites and evidence that phospholipase C-gamma 1 is a substrate of the TrkB receptor. J. Biol. Chem 269, 5458–5466. [PubMed] [Google Scholar]
- 50.Kadamur G, and Ross EM (2013). Mammalian phospholipase C. Annu. Rev. Physiol 75, 127–154. [DOI] [PubMed] [Google Scholar]
- 51.Goudet C, Gaven F, Kniazeff J, Vol C, Liu J, Cohen-Gonsaud M, Acher F, Prézeau L, and Pin JP (2004). Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc. Natl. Acad. Sci. USA 101, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Francesconi A, and Duvoisin RM (1998). Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J. Biol. Chem 273, 5615–5624. [DOI] [PubMed] [Google Scholar]
- 53.Atwal JK, Massie B, Miller FD, and Kaplan DR (2000).The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron 27, 265–277. [DOI] [PubMed] [Google Scholar]
- 54.Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, Ear J, Jung BH, Cabrera B, Carethers JM, and Farquhar MG (2010). A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol. Biol. Cell 21, 2338–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin C, Ear J, Midde K, Lopez-Sanchez I, Aznar N, Garcia-Marcos M, Kufareva I, Abagyan R, and Ghosh P (2014). Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol. Biol. Cell 25, 3654–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalogriopoulos NA, Lopez-Sanchez I, Lin C, Ngo T, Midde KK, Roy S, Aznar N, Murray F, Garcia-Marcos M, Kufareva I, et al. (2020). Receptor tyrosine kinases activate heterotrimeric G proteins via phosphorylation within the interdomain cleft of Galphai. Proc. Natl. Acad. Sci. USA 117, 28763–28774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garcia-Marcos M. (2021). Complementary biosensors reveal different G-protein signaling modes triggered by GPCRs and non-receptor activators. Elife 10, e65620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS, Marshall J, Jiang M, and Chu WM (2009). Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci. Signal 2, ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marshall J, Zhou XZ, Chen G, Yang SQ, Li Y, Wang Y, Zhang ZQ, Jiang Q, Birnbaumer L, and Cao C (2018). Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc. Natl. Acad. Sci. USA 115, E3549–E3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfeil EM, Brands J, Merten N, Vögtle T, Vescovo M, Rick U, Albrecht IM, Heycke N, Kawakami K, Ono Y, et al. (2020). Heterotrimeric G Protein Subunit Galphaq Is a Master Switch for Gbetagamma-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 80, 940–954.e6. [DOI] [PubMed] [Google Scholar]
- 61.Philip F, Kadamur G, Silos RG, Woodson J, and Ross EM (2010). Synergistic activation of phospholipase C-beta3 by Galpha(q) and Gbetagamma describes a simple two-state coincidence detector. Curr. Biol 20, 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Menna L, Joffe ME, Iacovelli L, Orlando R, Lindsley CW, Mairesse J, Gressèns P, Cannella M, Caraci F, Copani A, et al. (2018). Functional partnership between mGlu3 and mGlu5 metabotropic glutamate receptors in the central nervous system. Neuropharmacology 128, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rives ML, Vol C, Fukazawa Y, Tinel N, Trinquet E, Ayoub MA, Shigemoto R, Pin JP, and Prézeau L (2009). Crosstalk between GABAB and mGlu1a receptors reveals new insight into GPCR signal integration. EMBO J. 28, 2195–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Knight KM, Ghosh S, Campbell SL, Lefevre TJ, Olsen RHJ, Smrcka AV, Valentin NH, Yin G, Vaidehi N, and Dohlman HG (2021). A universal allosteric mechanism for G protein activation. Mol. Cell 81, 1384–1396.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee E, Taussig R, and Gilman AG (1992). The G226A mutant of Gs alpha highlights the requirement for dissociation of G protein subunits. J. Biol. Chem 267, 1212–1218. [PubMed] [Google Scholar]
- 66.Berghuis AM, Lee E, Raw AS, Gilman AG, and Sprang SR (1996). Structure of the GDP-Pi complex of Gly203–>Ala gialpha1: a mimic of the ternary product complex of galpha-catalyzed GTP hydrolysis. Structure 4, 1277–1290. [DOI] [PubMed] [Google Scholar]
- 67.Burns DL (1988). Subunit structure and enzymic activity of pertussis toxin. Microbiol. Sci 5, 285–287. [PubMed] [Google Scholar]
- 68.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, and Tesmer JJG (2005). Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science 310, 1686–1690. [DOI] [PubMed] [Google Scholar]
- 69.Xiang G, Acosta-Ruiz A, Radoux-Mergault A, Kristt M, Kim J, Moon JD, Broichhagen J, Inoue A, Lee FS, Stoeber M, et al. (2022). Control of Galpha(q) signaling dynamics and GPCR cross-talk by GRKs. Sci. Adv 8, eabq3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hackley CR, Mazzoni EO, and Blau J (2018). cAMPr: A single-wave-length fluorescent sensor for cyclic AMP. Sci. Signal 11, eaah3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Marcos M, Ghosh P, and Farquhar MG (2009). GIV is a non-receptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. USA 106, 3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aznar N, Midde KK, Dunkel Y, Lopez-Sanchez I, Pavlova Y, Marivin A, Barbazan J, Murray F, Nitsche U, Janssen KP, et al. (2015). Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. Elife 4, e07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garcia-Marcos M, Ghosh P, Ear J, and Farquhar MG (2010). A structural determinant that renders G alpha(i) sensitive to activation by GIV/girdin is required to promote cell migration. J. Biol. Chem 285, 12765–12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia-Marcos M, Kietrsunthorn PS, Wang H, Ghosh P, and Farquhar MG (2011). G Protein binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of G(alpha)i-regulatory motifs. J. Biol. Chem 286, 28138–28149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coleman BD, Marivin A, Parag-Sharma K, DiGiacomo V, Kim S, Pepper JS, Casler J, Nguyen LT, Koelle MR, and Garcia-Marcos M (2016). Evolutionary Conservation of a GPCR-Independent Mechanism of Trimeric G Protein Activation. Mol Biol E 33, 820–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maziarz M, Broselid S, DiGiacomo V, Park JC, Luebbers A, Garcia-Navarrete L, Blanco-Canosa JB, Baillie GS, and Garcia-Marcos M (2018). A biochemical and genetic discovery pipeline identifies PLCdelta4b as a nonreceptor activator of heterotrimeric G-proteins. J. Biol. Chem 293, 16964–16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao J, DiGiacomo V, Ferreras-Gutierrez M, Dastjerdi S, Ibáñez de Opakua A, Park JC, Luebbers A, Chen Q, Beeler A, Blanco FJ, and Garcia-Marcos M (2023). Small-molecule targeting of GPCR-independent noncanonical G-protein signaling in cancer. Proc. Natl. Acad. Sci. USA 120. e2213140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leyme A, Marivin A, Maziarz M, DiGiacomo V, Papakonstantinou MP, Patel PP, Blanco-Canosa JB, Walawalkar IA, Rodriguez-Davila G, Dominguez I, and Garcia-Marcos M (2017). Specific inhibition of GPCR-independent G protein signaling by a rationally engineered protein. Proc. Natl. Acad. Sci. USA 114, E10319–E10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Opakua AI, Parag-Sharma K, DiGiacomo V, Merino N, Leyme A, Marivin A, Villate M, Nguyen LT, de la Cruz-Morcillo MA, Blanco-Canosa JB, et al. (2017). Molecular mechanism of Galphai activation by non-GPCR proteins with a Galpha-Binding and Activating motif. Nat. Commun 8, 15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hauser AS, Avet C, Normand C, Mancini A, Inoue A, Bouvier M, and Gloriam DE (2022). Common coupling map advances GPCR-G protein selectivity. Elife 11, e74107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luttrell LM, van Biesen T, Hawes BE, Koch WJ, Touhara K, and Lefkowitz RJ (1995). G beta gamma subunits mediate mitogen-activated protein kinase activation by the tyrosine kinase insulin-like growth factor 1 receptor. J. Biol. Chem 270, 16495–16498. [DOI] [PubMed] [Google Scholar]
- 82.Dalle S, Ricketts W, Imamura T, Vollenweider P, and Olefsky JM (2001). Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J. Biol. Chem 276, 15688–15695. [DOI] [PubMed] [Google Scholar]
- 83.Conway AM, Rakhit S, Pyne S, and Pyne NJ (1999). Platelet-derived-growth-factor stimulation of the p42/p44 mitogen-activated protein kinase pathway in airway smooth muscle: role of pertussis-toxin-sensitive G-proteins, c-Src tyrosine kinases and phosphoinositide 3-kinase. Biochem. J 337, 171–177. [PMC free article] [PubMed] [Google Scholar]
- 84.Fedorov YV, Jones NC, and Olwin BB (1998). Regulation of myogenesis by fibroblast growth factors requires beta-gamma subunits of pertussis toxin-sensitive G proteins. Mol. Cell Biol 18, 5780–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Swaney DL, Ramms DJ, Wang Z, Park J, Goto Y, Soucheray M, Bhola N, Kim K, Zheng F, Zeng Y, et al. (2021). A protein network map of head and neck cancer reveals PIK3CA mutant drug sensitivity. Science 374, eabf2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.El-Shewy HM, Johnson KR, Lee MH, Jaffa AA, Obeid LM, and Luttrell LM (2006). Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine 1-phosphate receptors. J. Biol. Chem 281, 31399–31407. [DOI] [PubMed] [Google Scholar]
- 87.Aznar N, Ear J, Dunkel Y, Sun N, Satterfield K, He F, Kalogriopoulos NA, Lopez-Sanchez I, Ghassemian M, Sahoo D, et al. (2018). Convergence of Wnt, growth factor, and heterotrimeric G protein signals on the guanine nucleotide exchange factor Daple. Sci. Signal 11, eaao4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee FS, Kim AH, Khursigara G, and Chao MV (2001). The uniqueness of being a neurotrophin receptor. Curr. Opin. Neurobiol 11, 281–286. [DOI] [PubMed] [Google Scholar]
- 89.Holz A, Mülsch F, Schwarz MK, Hollmann M, Döbróssy MD, Coenen VA, Bartos M, Normann C, Biber K, van Calker D, and Serchov T (2019). Enhanced mGlu5 Signaling in Excitatory Neurons Promotes Rapid Antidepressant Effects via AMPA Receptor Activation. Neuron 104, 338–352.e7. [DOI] [PubMed] [Google Scholar]
- 90.Parekh PK, Johnson SB, and Liston C (2022). Synaptic Mechanisms Regulating Mood State Transitions in Depression. Annu. Rev. Neurosci 45, 581–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Annala S, Feng X, Shridhar N, Eryilmaz F, Patt J, Yang J, Pfeil EM, Cervantes-Villagrana RD, Inoue A, Häberlein F, Reher R, Kehraus S, Monteleone S, Schrage R, Heycke N, Rick U, Engel S, Pfeifer A, Kolb P, König G, Bünemann M, Tüting T, Vázquez-Prado J, Gutkind JS, Gaffal E, and Kostenis E (2019). Direct targeting of Galphaq and Galpha11 oncoproteins in cancer cells. Sci. Signal 12, eaau5948. [DOI] [PubMed] [Google Scholar]
- 92.O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, and Gutkind JS (2013). The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, and Bastian BC (2009). Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Regad T. (2015). Targeting RTK Signaling Pathways in Cancer. Cancers 7, 1758–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Du Z, and Lovly CM (2018). Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kostenis E, Pfeil EM, and Annala S (2020). Heterotrimeric Gq proteins as therapeutic targets? J. Biol. Chem 295, 5206–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Daub H, Weiss FU, Wallasch C, and Ullrich A (1996). Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557–560. [DOI] [PubMed] [Google Scholar]
- 98.Daub H, Wallasch C, Lankenau A, Herrlich A, and Ullrich A (1997). Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 16, 7032–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luttrell LM, Della Rocca GJ, van Biesen T, Luttrell DK, and Lefkowitz RJ (1997). Gbetagamma subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J. Biol. Chem 272, 4637–4644. [DOI] [PubMed] [Google Scholar]
- 100.Tsai W, Morielli AD, and Peralta EG (1997). The m1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. EMBO J. 16, 4597–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou R, Han B, Xia C, and Zhuang X (2019). Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science 365, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patt J, Alenfelder J, Pfeil EM, Voss JH, Merten N, Eryilmaz F, Heycke N, Rick U, Inoue A, Kehraus S, et al. (2021). An experimental strategy to probe Gq contribution to signal transduction in living cells. J. Biol. Chem 296, 100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clapham DE (2007). Calcium signaling. Cell 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- 104.Barford K, Deppmann C, and Winckler B (2017). The neurotrophin receptor signaling endosome: Where trafficking meets signaling. Dev. Neurobiol 77, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Purgert CA, Izumi Y, Jong YJI, Kumar V, Zorumski CF, and O’Malley KL (2014). Intracellular mGluR5 can mediate synaptic plasticity in the hippocampus. J. Neurosci 34, 4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gutzeit VA, Thibado J, Stor DS, Zhou Z, Blanchard SC, Andersen OS, and Levitz J (2019). Conformational dynamics between transmembrane domains and allosteric modulation of a metabotropic glutamate receptor. Elife 8, e45116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Irannejad R, and Wedegaertner PB (2010). Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein betagamma subunits. J. Biol. Chem 285, 32393–32404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, and Logothetis DE (1997). Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J. Biol. Chem 272, 31553–31560. [DOI] [PubMed] [Google Scholar]
- 109.Zheng S, Abreu N, Levitz J, and Kruse AC (2019). Structural basis for KCTD-mediated rapid desensitization of GABAB signalling. Nature 567, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Morstein J, Bader T, Cardillo AL, Schackmann J, Ashok S, Hougland JL, Hrycyna CA, Trauner DH, and Distefano MD (2022). Photoswitchable Isoprenoid Lipids Enable Optical Control of Peptide Lipidation. ACS Chem. Biol 17, 2945–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, and Lein E (2018). Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method. J Vis Exp. 10.3791/53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kaech S, and Banker G (2006). Culturing hippocampal neurons. Nat. Protoc 1, 2406–2415. [DOI] [PubMed] [Google Scholar]
- 114.Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, and Pin JP (2011). A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J 25, 66–77. [DOI] [PubMed] [Google Scholar]
- 115.Thibado JK, Tano JY, Lee J, Salas-Estrada L, Provasi D, Strauss A, Marcelo Lamim Ribeiro J, Xiang G, Broichhagen J, Filizola M, et al. (2021). Differences in interactions between transmembrane domains tune the activation of metabotropic glutamate receptors. Elife 10, e67027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.