


Abstract
Concentric multilamellar microvesicles, named spherulites™, were evaluated as an oligonucleotide carrier. Up to 80% oligonucleotide was encapsulated in these vesicles. The study was carried out on two different spherulite™ formulations. The spherulite™ size and stability characteristics are presented. Delivery of encapsulated oligonucleotide was performed on a rat hepatocarcinoma and on a lymphoblastoid T cell line, both expressing the luciferase gene. We showed that spherulites™ were able to transfect both adherent and suspension cell lines and deliver the oligonucleotide to the nucleus. Moreover, 48–62% luciferase inhibition was obtained in the rat hepatocarcinoma cell line when the antisense oligonucleotide targeted to the luciferase coding region was encapsulated at 500 nM concentration in spherulites™ of different compositions.
INTRODUCTION
Synthetic oligonucleotides represent an interesting class of compounds for therapeutic development based on their selectivity of interaction with complementary nucleic acid sequences. Their polyanionic nature, though, constitutes a severe drawback for their cellular internalisation. This problem, added to their sensitivity to nuclease degradation, explains their lack of efficiency in cell cultures and in vivo (1,2). Several chemical modifications have been introduced, mostly on the phosphate–sugar backbone, usually leading to analogues that are resistant toward nucleases and with good hybridisation properties (3). One of the main limitations to the development of these molecules remains their intracellular delivery. Different strategies have been designed in order to improve oligonucleotide uptake. Chemical modifications leading to neutral oligonucleotides have been studied (4–7). It was recently shown that temporarily masking the backbone negative charges aided the molecule in passing through the cellular membrane (8). Another strategy to mask the internucleosidic charges consists of forming a neutral or positively charged complex between the oligonucleotide and cationic molecules, such as polymers (9–12) or lipids (13,14). For instance, it had been reported that C-5 propyne phosphorothioates were able to efficiently inhibit gene expression in cell culture when complexed with cytofectin (15). Inhibition of luciferase expression was obtained in the nanomolar range when used on HeLa X1/5 cells that stably express luciferase (16). It has been also shown that antisense oligonucleotides targeted to luciferase or to insulin-like growth factor I (IGF-I) mRNAs were able to inhibit luciferase expression in the nanomolar range in a transient assay using a cationic lipid (17). Cationic lipids are quite efficient vectors for oligonucleotide or gene delivery and they are widely used for in vitro experiments (18). However transfection efficiency of oligonucleotides using cationic lipids is considerably dependent on the lipid and on the oligonucleotide modification (12,19,20). The toxicity of cationic lipids represents an important drawback in vitro, and their application in vivo is further limited due to their unspecific binding to serum proteins and their fast elimination from the circulation (21). Oligonucleotide could also be coupled to transport-enhancing peptides to improve their cellular delivery (22–24). They could also be delivered to the cells using liposomes (9,25), immuno-liposomes (26) or polymers (27,28).
A new system for oligonucleotide delivery was contemplated in order to inhibit the IGF-I gene by an antisense approach. IGF-I and its receptor play a key role in the regulation of normal cell growth and are highly expressed in a wide variety of tumours such as glioblastoma (29), melanoma (30), breast carcinoma (31) and hepatocarcinoma (32). Liposomes represent an interesting way to deliver oligonucleotides inside cells (9,25) and they have been shown to enter cells via endocytosis or membrane disruption (33). But they present a low encapsulation yield, which means that a high ratio of liposome to oligonucleotide must be used to get the appropriate oligonucleotide concentration in the cell. Moreover, their intracellular traffic seems quite limited due to their trapping in endosomal compartments (34,35). pH-sensitive liposomes have been developed to take advantage of the acidic pH in endosomes but they are not very stable in biological fluids (36).
In this context, spherulites™ (37) appeared as potential candidates to overcome some of the limitations of liposomes. As liposomes, they are constituted of phospholipids but their structure is made of concentric bilayers of amphiphiles alternating with layers of aqueous medium, which means that they do not contain a large aqueous core. This structure confers excellent stability in their dispersion medium and a high encapsulation yield, since the oligonucleotide will be located in the aqueous compartments situated between the bilayers (37). Two different spherulite™ formulations were investigated in terms of vesicle stability, encapsulation yield, cell transfection and oligonucleotide release. We showed that spherulites™ could transfect an adherent cell line as well as a suspension cell line. Confocal microscopy on spherulite™-transfected rat hepatocarcinoma cells indicated that spherulites™ accumulated in the cytoplasm while the oligonucleotide was released and reached the nucleus. As shown by radioactivity studies, ∼2.1% of an encapsulated 5′-32P-labelled oligonucleotide were recovered from the spherulite™-transfected cells. Depending on the formulation, 48–62% luciferase inhibition was observed with 500 nM of encapsulated C-5 propyne phosphorothioate targeted to a luciferase coding sequence. One spherulite™ formulation (F1) exhibited some non-specific effect in the absence of any oligonucleotide whereas the other formulation (F2) did not.
MATERIALS AND METHODS
Modified oligonucleotides
Lucif-2 5′-CGU GAU GUU CAC CUC-3′ and Lucif-2MM 5′-CGC UUU CUA UAG CGC-3′, C-5 propyne phosphorothioates were purchased from Eurogentec (Seraing, Belgium). Both U and C were C-5-propynyl derivatives. After precipitation in ethanol with sodium acetate, the oligonucleotides were dissolved in H2O and absorbance at 260 nm was measured using a UV spectrophotometer (Uvikon 860, Kontron Instruments, Bio-Tek, Saint Quentin en Yvelines, France). Oligonucleotide concentrations were determined using molar absorption coefficients calculated as previously described (17).
Spherulite™ preparation
Preparation of spherulites™ was carried out according to the patented procedure (37). F1 contained 5% (w/w) potassium oleate (Fluka, Buchs, Switzerland), 1.5% (w/w) cholesterol sulfate (Aldrich, Saint-Quentin-Fallavier, France), 3.5% (w/w) mannitol oleate (Montanide 80, Seppic, Paris, France), 40% (w/w) lecithin (S100) (Lipoid, Cham, Switzerland) and 50% (w/w) aqueous phase. To prepare 50 mg of lamellar phase, 2.5 mg potassium oleate, 0.75 mg cholesterol sulfate, 1.75 mg montanide and 5 mg aqueous phase were precisely weighed. The mixture was heated at 65°C for 1 h with few stirring intervals. When these constituents were melted, 20 mg phospholipid was added as well as 20 mg H2O (or the same amount of an aqueous 2 mM oligonucleotide solution). The mixture was left overnight at 37°C to allow the phospholipid to hydrate. The next morning, a shearing (10 min) and centrifugation (2 min) were applied to the paste. The phase homogeneity was checked by optical microscopy between crossed polariser and analyser. Dispersion of the paste in 10 times its volume of H2O led to the formation of spherulites™, at a 40 mg/ml concentration of lipid and 80 µM oligonucleotide. The ratio (w/w) between the oligonucleotide and the lipid was 1/100.
Formulation 2 (F2) consisted of 45% lecithin (phospholipon P90, Natterman, GmbH, Köln, Germany), 1% dioleoylphosphatidylethanolamine (DOPE, Sigma, Saint-Quentin-Fallavier, France), 20% macrogol oleate (Simulsol 2599, Seppic) and 35% aqueous phase (+/– oligonucleotide). In this case, all the constituents were precisely weighed to obtain 50 mg of paste, mixed and left overnight at 37°C with stirring intervals followed by shearing and dispersion as described above. In this formulation, the ratio (w/w) between the oligonucleotide and the lipid was 1/130.
Spherulite™ size measurement
The size of the particles was determined using a granulometer (Malvern Multisizer). As a diffraction figure corresponds to each vesicle, the granulometric profile obtained gave the volume distribution of the sample objects. The refractive index chosen for the particle was 1.435.
Encapsulation yield and leakage
Amaranth dye (Aldrich) was encapsulated as a control of encapsulation and leakage of the two formulations used. Centrifugation (40 min, 15 300 r.p.m., 4°C) eliminated most of the vesicles. The supernatant containing the amaranth and the smallest vesicles was microfiltrated on a 3 ml cell Amicon under pressure (4 bars). Absorbance of the filtered supernatant was measured at 522 nm.
5′-fluorescein-C-5-propyne phosphorothioate oligonucleotides were also encapsulated. The free fluorescent oligonucleotide was eliminated by ultracentrifugation (15 min, 40 000 r.p.m.) on a Beckman LE-70 equipped with an NVT 65.2 rotor. Measurement of the fluorescence in the supernatant and inside the vesicles was performed on a fluorimeter (Multilabel counter 1420 Victor2, EG and G Wallac, Evry, France) equipped with 485 nm excitation filter and 535 nm emission filter.
To be able to quickly check the encapsulation yield and the oligonucleotide concentration inside the spherulites™ before cell transfection, we used Oligreen® (Molecular Probes, Eugene, OR), a probe emitting fluorescence when associated with a single-stranded nucleic acid. This molecule exhibited a 15 times higher sensitivity than ethidium bromide and could be used as a marker for agarose gels. The gel was analysed by UV transillumination using a standard UV table (312 nm). Gel images were obtained with a Bioprint apparatus (Vilber Lourmat, Marne la Vallée, France) taking care not to saturate the camera. Images were analysed with Image Quant software (APBiotech, Orsay, France). As the spherulites™ could not enter through agarose gels and Oligogreen® could not cross lipid bilayers, only the non-encapsulated oligonucleotides could be visualized. Thus, loading of 10 µl of the encapsulated oligonucleotide solution (1 µM, 0.5 mg/ml lipid for F1, 0.56 mg/ml lipid for F2) on a 1.5% agarose gel as well as the same solution with 10% Triton X-100 to disrupt the spherulites™, gave the ratio between the non-encapsulated oligonucleotide and the oligonucleotide associated with the spherulites™.
Another way to follow the leakage of the oligonucleotide from the spherulites™ was to encapsulate the oligonucleotide 5′-CGTCACCACGACTTCAACGTCC-3′ 5′-32P-radiolabelled with T4 polynucleotide kinase and [γ-32P]ATP. The radiolabelling procedure was done as previously described (12). Analysis was then performed with non-denaturating polyacrylamide gel electrophoresis (10%, w/v) in a TBE running buffer.
Cell lines and cell cultures
The LFCLI1 rat hepatocarcinoma cell line was used for the transfection of antisense oligonucleotides encapsulated in spherulites™. This cell line stably expresses luciferase under the control of the IGF-I promoter. Briefly, this cell clone was obtained by cotransfection of pSV2Neo (Clontech, Palo Alto, USA) and pIGF-1711/luc kindly provided by Dr Peter Rotwein (Seattle, USA) in the LFCl2A rat hepatocarcinoma cell line (32,38). The pIGF-1711/luc contains the promoter and the 5′UTR region of rat IGF-I upstream the coding sequence of luciferase (for construct details see 39). Note that this plasmid could not self-replicate in rodent cells as it lacks eukaryotic replication origin. The LFCLI1 clone was selected on the basis of its geneticin resistance. Using the polymerase chain reaction (PCR), we showed that the promoter, the 5′UTR and the luciferase genes were contiguous in rat chromosomes. This PCR was carried out with two primers : GL1 primer 5′-CAATGTATCTTATGGTACTGTAACTGAGC-3′ which is a complementary sequence upstream the IGF-I promoter of the pGL2 vector, and 25 luc AUG: 5′-TTGGCGTCTTCCATTTTACCAAC-3′ complementary to the ATG codon of luciferase cDNA. PCR cycles were as follows: 1 cycle of 1 min at 95°C, 35 cycles of 30 s at 95°C, 1 min at 62°C, 3 min at 72°C and terminated by 10 min at 72°C. Using primers at 0.5 µM, the Advantage HF PCR kit (Clontech) and genomic DNA from LFCLI1, we found a PCR fragment of 2150 bp in accordance with a complete integration of the IGF-I promoter and luciferase gene from the pIGF-1711/luc plasmid into rat chromosomes. The sequence of this PCR fragment was confirmed using the restriction enzyme SacI, which cleaved the PCR fragment according to the plasmid map of pIGF-1711/luc. No PCR fragment was obtained when using genomic DNA prepared from the parental LFCL2A that did not express luciferase. Supplementary material is available at NAR Online. The LFCLI1 cell line was maintained in minimal essential medium (Sigma) containing 10% (v/v) foetal calf serum (heat inactivated for 30 min at 60°C) supplemented with 0.2 mg/ml G418 (Sigma).
A human lymphoblastoid T cell line, called J.Jhan 5.1, was also used in this study. This cell line, kindly provided by Dr Jean-Louis Virelizier (Institut Pasteur, Paris, France), was derived from a Jurkatt cell line and contained the luciferase reporter gene under the control of the HIV1 Long Terminal Repeat. The luciferase expression was inducible by TNF-α treatment (for more details, see 40).
Cellular uptake of the spherulites™ loaded with radiolabelled oligonucleotide
Cells were seeded in a 12-well plate at a 105/ml cell density and treated with 12 000 c.p.m./µl vesicle dilution in 300 µl optiMEM (Life Technologies, Cergy-Pontoise, France) over 5 and 24 h. The medium was removed and the associated radioactivity counted with a liquid scintillation counter (1219 rack beta LKB Wallac). The following PBS washes were also counted. Cells were lysed in the following lysis buffer : 10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 0.5% SDS, 0.01% proteinase K. Phenol extraction of the cell extract, followed by an ethanol precipitation (–20°C, overnight) allowed us to recover the nucleic acids associated with the cells.
Cellular uptake of the spherulites™ by FACS analysis
Cells were seeded in a 12-well plate at a 105/ml cell density and treated with 200 nM encapsulated or non-encapsulated 5′ fluorescein C5-propyne oligophosphorothioate in 300 µl optiMEM. Treated cells were trypsinized and re-suspended in MEM supplemented with 10% FCS. After centrifugation at 3000 r.p.m. for 5 min, pellets were washed twice with PBS and submitted to centrifugation in order to minimise detection of cell surface adsorption during FACS analysis. The supernatant was discarded and cells re-suspended in 500 µl PBS. Fluorescence from 5 × 104 individual cells was analysed by cytometry (FACSort, Becton Dickinson, San Jose, CA) excluding dead cells. Cytotoxicity induced by the spherulite™ composition was checked using cell counting, LDH and MTS colorimetric tests (Promega). Thus, we showed that a lipid concentration of empty or loaded spherulites™ as high as 1 mg/ml (formulations F1 and F2) did not exhibit any cytotoxicity. Fluorescein diacetate (Aldrich) was also added to the empty spherulite™-treated cells to confirm by epifluorescence microscopy that we measured fluorescence inside viable cells.
Laser scanning confocal microscopy
LFCLI1 cells were seeded at a density of 2 × 104 cells/chamber into an 8-chamber glass slide (Lab-Tek, Nalge Nunc International, Roskilde, Denmark) and the spherulite™ dilution was added corresponding to 100 nM 5′-fluorescein C-5 propyne phosphorothioate, 36–58 µg/ml of lipid including or not 1% rhodamine phosphoethanolamine (N-Rh-PE, L-1392, Molecular Probes). The slides were observed either with live cells or with fixed cells. In this case, two washes with PBS were applied and cells were fixed with 2% paraformaldehyde (Sigma) for 30 min, then mounted with Mowiol 4-88 (Hoechst F., Frankfurt, Germany). Analyses were performed with a confocal imaging system (MRC-600, Bio-Rad, Hercules, CA) equipped with a Nikon Optiphot epifluorescence microscope (Nikon, Tokyo, Japan) and a 50× and 100× Planapo objective (numerical aperture 1.4). A krypton/argon laser tuned to produce 488 nm fluorescein excitation and 568 nm rhodamine excitation allowed simultaneous reading of both fluorescent signals and image merging. Diaphragm and fluorescence detection levels were adjusted to eliminate any interference between fluorescein and rhodamine channels. Pictures were recorded with a Kalman filter (average of five images).
Cell transfection and measurement of luciferase activity
A typical transfection experiment was performed as follows. Cells were trypsinized, counted and mixed with spherulites™ with or without an oligonucleotide, then set as quadruplicates in a 24-well plate (50 000 cells/well; 50–500 nM oligonucleotide; 18–290 µg/ml lipid) in 300 µl optiMEM and left at 37°C, 5% CO2, for 24–48 hours. Photinus pyralis luciferase activity was measured in cell extracts by using the luciferase assay kit (Promega). Typically, treated LFCLI1 cells were passively lysed with 100 µl/well of passive lysis buffer (Promega) for 30 min at room temperature. Protein concentration of cell extracts was determined using the Bradford protein assay kit (Bio-Rad). Luminescence was measured using a luminometer (Multilabel counter 1420 Victor2, EG and G Wallac) equipped with a co-injector, that delivered 80 µl of luciferase substrate into 40 µl of cell extracts. The indicated luciferase activity corresponds to the ratio between the detected light unit and the protein amount. Typically, the relative light unit per mg of protein for the untreated LFCLI1 cell line was found to be 63 000 R.L.U./mg, whereas the parental cell line LFCL2A that did not express luciferase, exhibited a background emission of 3700 R.L.U./mg. Results were normalised using the R.L.U. per mg of protein of untreated cells taken as 100% of luciferase expression.
RESULTS AND DISCUSSION
Oligonucleotide-carrier uptake in living cells involves three main steps. The carrier should interact with the cells, be internalised and then release the oligonucleotide, which might reach the nucleus or distribute between the cytosol and the nucleus. Two different spherulite™ formulations were evaluated for their ability to meet these criteria. Formulation compositions are indicated in Materials and Methods. The first formulation (F1) had been optimised and applied with success to a previous study on macrophage cells infected with leishmania parasites (C.Bourget and J.J.Toulmé, unpublished results). The second formulation (F2) was optimised during this work according to the results of transfection experiments. This included the necessity to ease the preparation on small amounts and to obtain a formulation that would lead to small particles with a desired stability. These particles have to be stable enough to be kept in an extra-cellular medium but not too stable in cells in order to release the encapsulated oligonucleotide.
Physical chemistry
Spherulite™ preparation. The originality of these particles compared to liposomes comes from the manner in which they are prepared (41). The mixture of phospholipids with an optimised amount of water leads to the formation of a homogeneous liquid-crystal lamellar phase. The shearing and then the dispersion into an aqueous medium of this lamellar phase induce the formation of the spherulites™ (see Materials and Methods). The preparation process eliminates the use of any organic solvent or high shear, therefore minimising the risk of decomposition or externalisation of the oligonucleotide. Observation of a homogeneous phase by optical microscopy between crossed polariser and analyser is a good indicator of spherulite™ formation. The lamellar phase should present a homogeneous birefringent texture. After dispersion, the vesicles may be observed with the same technique, since spherulites™ are birefringent. However, their small size often prevents the observation of the birefringence. Obtaining a homogeneous phase for F1 requires a heating step in order to melt all the constituents, and the phospholipids are added afterwards. In this context, F2 vesicle preparation is easier. It only requires the mixing of all constituents and incubation of the paste obtained at 37°C to allow the phospholipids to hydrate.
Spherulite™ size. Determination of the particle size was carried out to evaluate the effects of the shearing time, the dispersion time and the dispersion medium. Different samples were prepared with the modification of one parameter at a time, and an analysis of the size of the population(s) was performed. We showed that the shearing process was optimal after 10–15 min, no change in the population repartition being observed if shearing was performed for a longer period of time. As shown in Figure 1, formulation F1 led, after 10 min shearing, to two populations with a mean size ∼0.7 and 4.0 µm when dispersed in H2O or in phosphate buffer, respectively. The F2 formulation which is an alternative of formulation F1 led to one population with a smaller mean size ∼0.3 µm after 15 min shearing when dispersed either in H2O or in phosphate buffer (Fig. 1).
Figure 1.

Analysis of formulations 1 and 2 by granulometry. Population repartition was determined for the two formulations investigated. F1 gave two populations centred at 0.7 and 4 µm, F2 gave one population centred at 0.3 µm.
Encapsulation yield and spherulite™ stability. Optimisation of a formulation requires the inclusion of the exact amount of water that will lead to a high encapsulation yield. A precise study was carried out for each new formulation to get the best ratio of water compared to the lipid.
Oligreen“ binding to oligonucleotides was used as a tool to measure the small amounts of oligonucleotide released outside the vesicles and the encapsulated oligonucleotide concentration. First, using agarose gel electrophoresis, a standard curve was obtained from known concentrations of free oligonucleotide in order to estimate the encapsulated oligonucleotide amount based on the encapsulation yield, after lysis of the capsules (Fig. 2, lane 1). Triton X-100 (10%) was added to a non-encapsulated oligonucleotide solution to make sure that Triton X-100 did not interfere with the fluorescence intensity (Fig. 2, lane 2). The loading of a dilution of non-purified spherulite™ dispersion allowed us to calculate the encapsulation yield, comparing lanes with and without Triton X-100 (Fig. 2, lanes 5 and 6). In the experiment shown on Figure 2, a 45% encapsulation yield was found, showing that this spherulite preparation was not as successful as the preparation mentioned below. The same type of loading after elimination of the free oligonucleotide by ultracentrifugation indicated no signal outside the spherulites™ (lane 3). Therefore, ultracentrifugation represents a valid way to eliminate the non-encapsulated oligonucleotide. Moreover, the absolute concentration within the spherulites™ could be estimated using known concentration of oligonucleotides (comparison of lanes 2 and 4).
Figure 2.

Analysis of spherulites™ by gel electrophoresis. In lane 1, the free oligonucleotide (49 ng) was loaded on the gel. In lane 2, the free oligonucleotide (49 ng) was treated with Triton X-100 (10%). In lanes 3–6, the oligonucleotide was associated with spherulites™ and treated (lanes 4 and 6) or not treated (lanes 3 and 5) with Triton X-100 (10%). Spherulites™ purified by ultracentrifugation (40 000 r.p.m., 15 min) were loaded on lanes 3 and 4. Oligreen® (0.5% v/v) was included in the agarose gel (1.5 % w/v). S represents the position of the wells.
The stability of the two microvesicle compositions (F1 and F2) was evaluated using amaranth as a leakage marker. Extraction of the amaranth released in the extra spherulite™ medium and UV measurement indicated that these formulations were stable over a period of 34 days, when kept at 4°C, if the media inside and outside the spherulite™ was at the same salt concentration. However, we observed that a 150 mM NaCl difference between the intra- and extra-spherulite™ media was enough to induce a leakage. Encapsulation yields of amaranth in F1 and F2 formulations were found to be 75 and 72%, respectively. Stability and encapsulation yields were also checked using radiolabelling. A 32P-labelled 22mer phosphodiester was encapsulated and the non-encapsulated oligonucleotide was eliminated by ultracentrifugation. The encapsulation yields obtained after analysis on a non-denaturing polyacrylamide gel were as high as 80 and 85% for F1 and F2, respectively. The time-dependent stability of the vesicle dispersion at different temperatures was evaluated. The acrylamide gel presented in Figure 3 shows that after 10 days most of the oligonucleotide was released from the F2 vesicles if the intra- and extra-vesicle salt concentrations were not identical. Indeed, in this experiment the oligonucleotide was dissolved in H2O while the spherulites™ were dispersed in phosphate buffer (PBS). Lane 1 (Fig. 3) corresponds to the free oligonucleotide and lane 2 to the total amount of oligonucleotide obtained by lysing the spherulites™ with 10% Triton X-100 during 15 min. We can see that a 150 mM difference in salt concentration was enough to induce a leakage of the vesicles which was significant for F2 since 61% free oligonucleotide was found in the PBS (lanes 1 and 2). The same experiment performed with an identical buffer, water or PBS, inside and outside the vesicles showed that the dispersion was stable at 4°C. The spherulites™ (F2) were then prepared and dispersed in H2O in order to obtain stable vesicles under storage conditions at 4°C. Oligonucleotide leakage from F1 was also checked. Figure 3 shows the leakage measured when oligonucleotide was dissolved in H2O and the spherulites™ dispersed in phosphate buffer, after 10 days at 4°C (lanes 3 and 4). 13% oligonucleotide was found to be released. This formulation was more stable than F2, which was not surprising since F1 contains components aimed at stabilising the capsule lipid layers.
Figure 3.
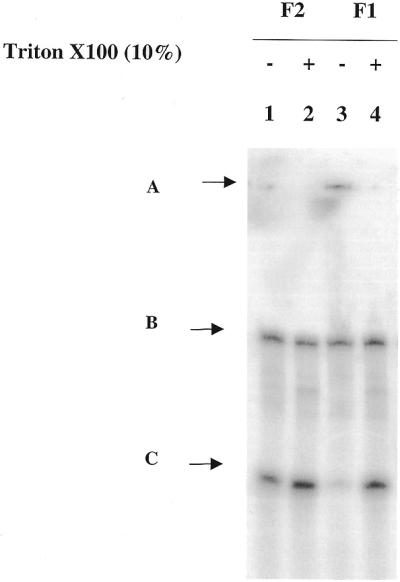
Spherulite™ stability analysed by gel electrophoresis. The two formulations F1 and F2 were prepared with 5′-radiolabelled oligonucleotide dissolved in H2O. The spherulites™ were dispersed in PBS. Vesicle leakage was followed by gel electrophoresis after 10 days at 4°C. On this 10% non-denaturating polyacrylamide gel was loaded the oligonucleotide contained in F2 (lanes 1 and 2) and oligonucleotide contained in F1 (lanes 3 and 4). 10% Triton X-100 was added to both formulations (lanes 2 and 4) to disrupt the vesicles and give the 100% oligonucleotide concentration. (A) Top of the gel, (B) internal marker (59mer) to control the homogeneity of the loading, (C) 5′-radiolabelled encapsulated 22mer PO.
Biological studies
Cell system. IGF-I, a 70-residue peptide that is structurally related to insulin, plays a fundamental role in growth and development. It seems to mediate many of the post-natal effects of growth hormone. It is also expressed in many foetal tissues and in various tumours, and has been recently identified in tumour cell lines such as glioblastoma and hepatocarcinoma. The RNA antisense strategy was used to disrupt IGF-I expression in tumorigenic cell lines (29,32). IGF-I inhibition led to a suppression of glioblastoma and hepatocarcinoma growth in a rat model (32). We chose to target IGF-I using an antisense oligonucleotide in the hepatocarcinoma cell line LFCL2A since the important role in tumorigenesis of this growth factor has previously been demonstrated (32). This cell line produces voluminous tumours when injected into syngenic Commentry rats providing a model to perform further in vivo studies with spherulite™-encapsulated antisense oligonucleotides. In order to screen easily a variety of spherulite™ formulations, a stable cell line derived from LFCL2A and expressing the luciferase reporter gene under the control of the IGF-I promoter was selected.
Cell uptake. Use of 5′-radiolabelled oligonucleotide to quantitate the amount of oligonucleotide in the cells when carried by the spherulites™. After 5 and 24 h treatment of LFCLI1 cells with encapsulated 32P-labelled phosphodiester oligonucleotide, the radioactivity associated with the supernatant and the cells was counted. After 24 h, 97 and 94% were found in the transfection medium for formulations 1 and 2, respectively. The radioactivity associated with the cells was 0.8 and 2.1% for formulations 1 and 2, respectively. No detectable signal was observed when the cells were treated with the non-encapsulated oligonucleotide. Thus, oligonucleotide encapsulation induces an increase in its internalisation even if it remains low. Usually, in vitro transfections, with oligonucleotide carriers are performed with cationic lipids because it is easier for a cationic compound to be taken up via electrostatic interactions with the cell membrane. In our case, incorporation of cationic lipids in the spherulite™ formulation would lead to a better uptake of the complex but the ultimate goal being the in vivo experiments, we chose to keep these non-cationic formulations. A lot of difficulties have indeed been reported when trying to extrapolate from in vitro to in vivo situations using cationic lipids, some groups did even skip the in vitro studies (35). It is noteworthy that F2 contains less negative charges than F1 and this may explain the difference observed in cell uptake. Moreover, F2 does contain DOPE, a zwitterionic lipid that exhibits fusiogenic properties (42).
Flow cytometry studies of cells transfected with spherulite™ containing 5′-fluorescein oligonucleotide. Despite the low amount of 5′-32P oligonucleotide recovered from the cells, we were interested in determining the oligonucleotide localisation after release from the vesicles. For this purpose, we used a 5′-fluorescein-labelled C-5-propyne phosphorothioate oligonucleotide and studied cellular uptake by fluorescence using cytofluorimetry and confocal microscopy (described below). 5′-fluorescein propynyl Lucif-2 was encapsulated in F1 and F2 vesicles, the non-encapsulated oligonucleotide was eliminated from the supernatant by ultracentrifugation as described in Materials and Methods. We demonstrated by flow cytometry that the fluorescence intensity associated with LFCLI1 cells exposed to 200 nM fluorescent oligonucleotide was increased 2.0- and 3.2-fold compared to untreated cells when the oligonucleotide was encapsulated in spherulite™ formulations F1 and F2, respectively (Fig. 4). Note that the measurement of fluorescence associated with the cells did not demonstrate that spherulites™ were inside the cells or associated with the cell membrane. For this purpose, further microscopy studies were performed (see below). The same FACS experiment carried out on J.Jhan 5.1 suspension cell line showed a fluorescence intensity increased 25- to 400-fold with F1 and F2 formulations, respectively (Fig. 4). To demonstrate that cells treated with spherulites™ were alive, double labelling with fluorescein diacetate and encapsulated 5′-rhodamine phosphodiester oligonucleotide was performed. Co-localisation of live (green coloured) and transfected cells (red coloured) was observed by microscopy, confirming that suspension cells were still alive after treatment with spherulites™. All cells in suspension were fluorescent after 17 h treatment while adherent cells were not all transfected yet (see below, Fig. 6). Therefore, spherulites™ appear as an interesting tool for transfection of cells in suspension, also shown by confocal microscopy (see below).
Figure 4.

Cell uptake studied by flow cytometry. Two different cell lines, LFCLI1 (left) and J.Jhan5.1 (right), were transfected during 24 h with 5′-fluorescein C-5 propyne phosphorothioate oligonucleotide encapsulated in either formulations F1 and F2. Dashed lines represent the untreated cells, the broad line corresponds to the F1 formulation and the filled spectrum to the F2 formulation.
Figure 6.

Transfection of J.Jhan 5.1 cells in suspension with F2-encapsulated 5′-fluorescein C-5 propyne phosphorothioate oligonucleotide. Observation by confocal microscopy 17 h after transfection. Left panel represents lymphocytes in phase contrast. Right panel shows the cell fluorescence when transfected with encapsulated 5′-fluorescein oligonucleotide.
Transfection efficiency studied by confocal microscopy. As it had been reported that the fixation protocol could interfere with confocal microscopy observation (43), we compared first the live and fixed cells 24 h after transfection. No noticeable difference emerged from the two protocols, therefore we usually fixed the cells. After 15 h, only few cells showed a fluorescence intensity when transfected with either free or encapsulated 5′-fluorescein oligonucleotide. But this intensity was higher when the oligonucleotide was encapsulated. Thus, ∼5% fluorescent cells could be counted after 17 h, 20% after 24 h and almost 100% after 48 and 62 h. This could mean that either adherent cells require a long time to be transfected or the release of the oligonucleotide from the spherulites™ is quite slow. Note that during these long incubation times hepatocarcinoma cells divided in the presence of spherulites™. It follows therefrom that cell division could be required for significant uptake of spherulites™. The adsorption of spherulites™ on cell membranes could also be the limiting step. Electron microscopic studies are in progress in order to determine if spherulites™ multilamellar structures can be observed inside the cells. A comparison of the results obtained at 41 h when LFCLI1 cells were transfected with free or encapsulated 5′-fluorescein labelled oligonucleotide is presented in Figure 5A and B. First, we observed that the fluorescence intensity was higher when the oligonucleotide was encapsulated (Fig. 5B) than when it was not (Fig. 5A). Second, the fluorescence was diffused when free oligonucleotide was used (Fig. 5A), while it was localised to the nucleus when transfection was performed in the presence of the carrier (Fig. 5B). Double labelling of F2 allowed us to follow the lipid (PE-Rh) and the oligonucleotide separately. The fluorescence from fluorescein was localised in the nucleus (Fig. 5C) and the rhodamine fluorescence showed up only in the cytoplasm (Fig. 5D). As no punctate fluorescence due to the free fluorescein (44) was observed during the experiment, we assumed that the 5′-labelled oligonucleotide was still intact, probably protected from nuclease degradation by the spherulites™. At earlier times (t = 17 h), we did observe on the few fluorescent cells a yellow coloration in the cytoplasm when both labelling were merged indicating a co-localisation of the two markers. But after 30 h, the rhodamine fluorescence was so intense that co-localisation could not be detected. This may also be explained by the possible traffic of the lipids to the nucleus membrane and then release of the oligonucleotide.
Figure 5.

Transfection of adherent LFCLI1 cells with F2-encapsulated 5′-fluorescein C-5-propyne phosphorothioate oligonucleotide. Observation by confocal microscopy 41 h after transfection.. (A) Non-encapsulated 5′-fluorescein Lucif-2, (B) encapsulated 5′-fluorescein Lucif-2, (C) and (D) double labelling with encapsulated 5′-fluorescein Lucif-2 in F2 containing rhodamine phosphoethanolamine (N-PE-Rh). (C) and (D) show the fluorescein and the rhodamine fluorescence, respectively.
As it took quite a long time to observe fluorescence on adherent cells, we thought that the vesicle-cell interaction could be one of the limiting step. Indeed, most of the spherulites™ remain in the suspension medium. Therefore cells in suspension represented an interesting target for transfection. The J.Jhan 5.1 model was tranfected with the encapsulated 5′-fluorescein oligonucleotide using the same protocol as for LFCLI1 cells. What we observed is presented in Figure 6. All cells were transfected after 17 h when the oligonucleotide was encapsulated in spherulites™ while the free oligonucleotide only accumulated in dead cells. From an independant experiment, we have previously demonstrated that J.Jhan 5.1 cells treated with unloaded spherulites™ or loaded with 5′-rhodamine oligonucleotide remained alive as shown by fluorescein diacetate experiment mentioned in FACS study. These spherulite™ formulations are negatively charged and did not interact easily with LFCLI1 adherent cells. Most cationic lipids do not allow transfection of cells in suspension, therefore it is noteworthy that we were able to transfect the J.Jhan 5.1 cell line in suspension with our spherulite™ carrier.
Luciferase inhibition
The LFCLI1 cells used for the study express the luciferase gene under the control of IGF-I promoter. Luciferase fluorescence eased the screening of different spherulite™ formulations, the objective being to target IGF-I if the screening was successful. We could not perform these experiments on J.Jhan 5.1 cells since they did not express enough luciferase activity in our hands despite an inducible TNF-α luciferase expression system (40). Hepatocarcinoma cells were transfected with different concentrations of spherulites™ containing the anti-luciferase antisense oligonucleotide. A 15mer modified C-5 propyne phosphorothioate, Lucif-2, targeting the coding region (502–516) of the luciferase mRNA was previously described to specifically inhibit luciferase expression (16,17,45). An 8-base mismatch, Lucif-2MM, was chosen as a control sequence according to Flanagan et al. (16). The efficacy of luciferase inhibition with encapsulated oligonucleotides was compared to that of the empty vesicles and of the non-encapsulated Lucif-2 and Lucif-2MM.
At 24 h, 62% inhibition of luciferase was observed using 500 nM oligonucleotide contained in F1 formulation, while the free oligonucleotide exhibited 17% inhibition in the same conditions (Table 1). Some luciferase inhibition was also observed when transfection was carried out with the corresponding concentration of empty spherulites™. When the luciferase inhibition obtained with empty spherulites™ was subtracted from that obtained with antisense-loaded spherulites™ (Formulation F1), a 23% specific inhibition was observed at 24 h. It has been recently shown that VCAM-1 expression could be inhibited unexpectedly by the use of high concentration of cationic lipid in the absence of oligonucleotide (46). In our case, it follows that a higher loading of the spherulites™ would be a good way to reduce these observed non-specific activities, allowing use of the same amount of oligonucleotide and less particles for the transfection experiments. Higher loading should not present any problem since we showed that increasing the loading of spherulites™ by a factor of 10 did not affect the shape of the particles and that we were still below the saturating concentration. In order to load more oligonucleotide into the spherulites™ and maintain an affordable price, the process is under revision to improve the preparation of the particles on a small scale (5 mg). We then compared F1 and F2. Empty spherulites™ with F2 did not induce any unspecific effect (only 5% of luciferase inhibition at 24 h, Table 1) in contrast to what was observed with F1 (37% at 24 h, Table 1). The specific luciferase inhibition obtained with both formulations of antisense-loaded spherulites™ was similar (∼20%) to that obtained with the non-encapsulated antisense oligonucleotide with a 24 h treatment. The time-dependent inhibition of luciferase expression by the antisense oligonucleotide was then measured. Table 1 shows that 24 h after transfection there was not much difference between the encapsulated and free oligonucleotide but 48 h after transfection the non-encapsulated Lucif-2 had no effect while the encapsulated oligonucleotide inhibited luciferase expression by 48%. These results are in agreement with what we observed by epifluorescence microscopy; it takes longer for the cells to be transfected by the encapsulated oligonucleotide than by the free oligonucleotide. Using encapsulation, the oligonucleotide may be slowly delivered in the cells leading to a limited but prolonged inhibition of luciferase expression. It should be noted that in all cases the mismatched oligonucleotide exhibited some inhibitory effects as previously reported (16).
Table 1. Inhibition of luciferase expression in LFCLI1 cells by F1- and F2-encapsulated C-5 propyne phosphorothioate oligonucleotide (500 nM) compared to the free oligonucleotide.
| Spherulite™ formulations | F1 | F2 | |
|---|---|---|---|
| t = 24 h | t = 24 h | t = 48 h | |
| Untreated Cells | 100.0 ± 4.8 | 100.0 ± 5.1 | 100.0 ± 3.2 |
| + Non encapsulated Lucif-2 | 83.1 ± 2.1 | 82.3 ± 1.0 | 92.0 ± 1.1 |
| + Non encapsulated Lucif-2MM | 92.0 ± 2.0 | 91.0 ± 2.2 | 99.9 ± 5.6 |
| + Empty vesicles | 62.3 ± 4.2 | 95.1 ± 0.9 | 96.5 ± 2.1 |
| + Encapsulated Lucif-2 | 38.6 ± 3.0 | 71.4 ± 0.5 | 52.1 ± 1.0 |
| + Encapsulated Lucif-2MM | 69.8 ± 4.5 | 88.7 ± 2.2 | 74.1 ± 3.2 |
The lipid concentration was 0.25 mg/ml for F1 and 0.3 mg/ml for F2 formulations. Normalised relative light units corresponding to luciferase expression of treated cells are given. Luciferase activity of untreated cells was chosen as 100% of luciferase expression. Results are given as means ± SD.
CONCLUSION
Spherulites™ were evaluated as potential carriers for oligonucleotide delivery. The encapsulation yield of oligonucleotides in these concentric multilamellar structures was found to be as high as 80%. Their preparation appears to be quite simple, does not require any organic solvent and the vesicles are stable enough to be stored at 4°C. We showed by radiolabelling, flow cytometry and confocal microscopy that non-cationic spherulites™ were able to transfect 100% of LFCLI1 cells. A fluorescence maximum was observed after 41 h and persisted over 62 h. This fluorescence, corresponding to the labelled oligonucleotide, was localised in the nucleus. We showed that 500 nM antisense oligonucleotide encapsulated with F2 exhibited 50% inhibition of luciferase expression 48 h after the beginning of incubation. Work is in progress to prepare vesicles with higher quantities of oligonucleotide in order to avoid any non-specific luciferase inhibition exhibited by empty spherulites™ and to envision in vivo studies. The ability of spherulites™ to be used in vivo was demonstrated on mice models for antigen delivery (S. Gaubert and R. Laversanne, unpublished results).
We also demonstrated that the J.Jhan 5.1 cell line in suspension was efficiently transfected with spherulite™-encapsulated oligonucleotide which is quite an interesting result since not many transfectants are available for the transfection of cell lines in suspension (47,48). In a study performed in parallel with the present one, plasmids were shown to be incorporated in a very efficient manner into spherulites™ (T. Pott and D. Roux, unpublished results). Therefore spherulites™ might represent a new and efficient way of transfecting both plasmids and oligonucleotides into non-adherent cells.
SUPPLEMENTARY MATERIAL
See Supplementary Material available at NAR Online.
ACKNOWLEDEGMENTS
We thank Dr P. Rotwein for providing us with the IGF-I plasmid, Dr C. Lafarge-Frayssinet and Dr A. Sarasin for providing us with the parental rat hepatocarcinoma cell line LFCL2A, and Dr J.L. Virelizier for the gift of the J.Jhan 5.1 cell line. We are grateful to Y. Hamel, G. Doré and W. Prudhomme for their technical contributions. This work was supported by grants to J.C.F. and R.L. from the Ligue Nationale contre le Cancer and from the French Minister of Research.
REFERENCES
- 1.Agrawal S. and Zhao,Q. (1998) Curr. Opin. Chem. Biol., 2, 519–528. [DOI] [PubMed] [Google Scholar]
- 2.Persidis A. (1999) Nature Biotech., 17, 403–404. [DOI] [PubMed] [Google Scholar]
- 3.Crooke R.M. (1998) In Crooke,S.T. (ed.), Antisense Research and Application, Vol 131. Springer-Verlag, Germany, pp. 103–140.
- 4.Miller P.S., McParland,K.B., Jayaraman,K. and Ts’o,P.O. (1981) Biochemistry, 20, 1874–1880. [DOI] [PubMed] [Google Scholar]
- 5.Letsinger R.L., Singman,C.N., Histand,G. and Salunkhe,M. (1988) J. Am. Chem. Soc., 110, 4470–4471. [Google Scholar]
- 6.Levis J.T., Butler,W.O., Tseng,B.Y. and Ts’o,P.O. (1995) Antisense Res. Dev., 5, 251–259. [DOI] [PubMed] [Google Scholar]
- 7.Mignet N. and Gryaznov,S.M. (1998) Nucleic Acids Res., 26, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vivès E., Dell’Aquilla,C., Bologna,J.C., Morvan,F., Rayner,B. and Imbach,J.L. (1999) Nucleic Acids Res., 27, 4071–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarenc J.P., Degols,G., Leonetti,J.P., Milhaud,P. and Lebleu,B. (1993) Anticancer Drug Des., 8, 81–94. [PubMed] [Google Scholar]
- 10.Boussif O., Lezoualc’h,F., Zanta,M.A., Mergny,M.D., Scherman,D., Demeneix,B. and Behr,J.P. (1995) Proc. Natl Acad. Sci. USA, 92, 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delong R., Stephenson,K., Loftus,T., Fisher,M., Alahari,S., Nolting,A. and Juliano,R.L. (1997) J. Pharm. Sci., 86, 762–764. [DOI] [PubMed] [Google Scholar]
- 12.Dheur S., Dias,N., Van Aerschot,A., Herdewijn,P., Bettinger,T., Rémy,J.S., Hélène,C. and Saison-Behmoaras,T.E. (1999) Antisense Nucleic Acid Drug Dev., 9, 515–525. [DOI] [PubMed] [Google Scholar]
- 13.Behr J.P. (1994) Bioconjug. Chem., 5, 382–389. [DOI] [PubMed] [Google Scholar]
- 14.Hughes J., Astriab,A., Yoo,H., Alahari,S., Liang,E., Sergueev,D., Shaw,B.R. and Juliano,R.L. (1999) Methods Enzymol., 313, 342–358. [DOI] [PubMed] [Google Scholar]
- 15.Flanagan M.W. and Wagner,R.W. (1998) In Stein,C.A. and Krieg,A.M. (eds), Applied Antisense Oligonucleotide Technology, Vol. 9. Wiley-Liss, London, pp. 175–191.
- 16.Flanagan M.W., Kothavale,A. and Wagner,R. (1996) Nucleic Acids Res., 24, 2936–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamel Y., Lacoste,J., Frayssinet,C., Sarasin,A., Garestier,T., Francois,J.C. and Hélène,C. (1999) Biochem. J., 339, 547–553. [PMC free article] [PubMed] [Google Scholar]
- 18.Byk G., Dubertret,C., Escriou,V., Frederic,M., Jaslin,G., Rangara,R., Pitard,B., Crouzet,J., Wils,P., Schwartz,B. and Scherman,D. (1998) J. Med. Chem., 41, 229–235. [DOI] [PubMed] [Google Scholar]
- 19.Conrad A.H., Behlke,M.A., Jaffredo,T. and Conrad,G.W. (1998) Antisense Nucleic Acid Drug Dev., 8, 427–434. [DOI] [PubMed] [Google Scholar]
- 20.Kang S.H., Zirbes,E.L. and Kole,R. (1999) Antisense Nucleic Acid Drug Dev., 9, 497–505. [DOI] [PubMed] [Google Scholar]
- 21.Plank C., Mechtler,K., Szoka,J. and Wagner,E. (1996) Hum. Gene Ther., 7, 1437–1446. [DOI] [PubMed] [Google Scholar]
- 22.Bongartz J.P., Aubertin,A.M., Milhaud,P.G. and Lebleu,B. (1994) Nucleic Acids Res., 22, 4681–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vives E., Brodin,P. and Lebleu,B. (1997) J. Biol. Chem., 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- 24.Morris M.C., Chaloin,L., Mery,J., Heitz,F. and Divita,G. (1999) Nucleic Acids Res., 27, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juliano R.L. and Akhtar,S. (1992) Antisense Res. Dev., 2, 165–176. [DOI] [PubMed] [Google Scholar]
- 26.Zelphati O., Imbach,J.L., Signoret,N., Zon,G., Rayner,B. and Leserman,L. (1994) Nucleic Acids Res., 22, 4307–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwab G., Chavany,C., Duroux,I., Goubin,G., Lebeau,J., Helene,C. and Saison-Behmoaras,T. (1994) Proc. Natl Acad. Sci. USA, 91, 10460–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis K.J., Irwin,W.J. and Akhtar,S. (1998) J. Drug Target, 5, 291–302. [DOI] [PubMed] [Google Scholar]
- 29.Trojan J., Johnson,T.R., Rudin,S.D., Tykocinski,M.L. and Ilan,J. (1993) Science, 259, 94–97. [DOI] [PubMed] [Google Scholar]
- 30.Resnicoff M., Coppola,D., Sell,C., Rubin,R., Ferrone,S. and Baserga,R. (1994) Cancer Res., 54, 4848–4850. [PubMed] [Google Scholar]
- 31.Neuenschwander S., Roberts,C. and Leroith,D. (1995) Endocrinology, 136, 4298–4303. [DOI] [PubMed] [Google Scholar]
- 32.Lafarge-Frayssinet C., Duc,H., Frayssinet,C., Sarasin,A., Anthony,D., Guo,Y. and Trojan,J. (1997) Cancer Gene Ther., 5, 276–285. [PubMed] [Google Scholar]
- 33.Szoka F.C., Xu,Y. and Zelphati,O. (1996) J. Liposome Res., 6, 567–587. [Google Scholar]
- 34.Miller A.D. (1998) Angew. Chem. Int. Ed. Engl., 37, 1768–1785. [Google Scholar]
- 35.Mahato R.I., Smith,L.C. and Rolland,A. (1999) Adv. Genet., 41, 95–156. [DOI] [PubMed] [Google Scholar]
- 36.Zelphati O. and Szoka,F. (1996) J. Controlled Release, 41, 99–119. [Google Scholar]
- 37.Mahy P., Roux,D., Laversanne,R., Amédée,J. and Freund,O. (1998) World Patent, WO 98/02144.
- 38.Hatzfeld A., Feldmann,G., Guesnon,J., Frayssinet,C. and Shapira,F. (1978) Cancer Res., 38, 16–22. [PubMed] [Google Scholar]
- 39.Hall L.J., Kajimoto,Y., Bichell,D., Kim,S., James,P.L., Counts,D., Nixon,L.J., Tobin,G. and Rotwein,P. (1992) DNA Cell Biol., 11, 301–312. [DOI] [PubMed] [Google Scholar]
- 40.Israel N., Gougerot-Pocidalo,M.A., Aillet,F. and Virelizier,J.L. (1992) J. Immunol., 149, 3386–3393. [PubMed] [Google Scholar]
- 41.Gulik T., Dedieu,J.C., Roux,D., Degert,C. and Laversanne,R. (1996) Langmuir, 12, 4668–4670. [Google Scholar]
- 42.Farhood H., Serbina,N. and Huang,L. (1995) Biochim. Biophys. Acta, 1235, 289–295. [DOI] [PubMed] [Google Scholar]
- 43.Pichon C., Monsigny,M. and Roche,A.C. (1999) Antisense Nucleic Acid Drug Dev., 9, 89–93. [DOI] [PubMed] [Google Scholar]
- 44.Fisher T.L., Terhorst,T., Cao,X. and Wagner,R.W. (1993) Nucleic Acids Res., 21, 3857–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis J., Lin,K.Y., Kothavale,A., Flanagan,W.M., Matteucci,M.D., DePrince,R.B., Mook,R.A., Hendren,R.W. and Wagner,R.W. (1996) Proc. Natl Acad. Sci. USA, 93, 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maus U., Rosseau,S., Mandrakas,N., Schlingensiepen,R., Maus,R., Muth,H., Grimminger,F., Seeger,W. and Lohmeyer,J. (1999) Antisense Nucleic Acid Drug Dev., 9, 71–80. [DOI] [PubMed] [Google Scholar]
- 47.Keller H., Yunxu,C., Marit,G., Pla,M., Reiffers,J., Theze,J. and Froussard,P. (1999) Gene Ther., 6, 931–938. [DOI] [PubMed] [Google Scholar]
- 48.Marit G., Cao,Y., Froussard,P., Ripoche,J., Dupouy,M., Elandaloussi,A., Lacombe,F., Mahon,F.X., Keller,H., Pla,M., Reiffers,J. and Theze,J. (2000) Eur. J. Haematol., 64, 22–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.