

Abstract
Glutamate (Glu) is the most abundant excitatory neurotransmitter in the central nervous system (CNS) and is involved in the pathophysiology of Alzheimer’s disease (AD) in which there is an increased excitotoxicity. Biochemical composition of living tissues including the levels of Glu was analyzed by magnetic resonance spectroscopy (MRS). Previous reports point to decreased levels of Glu in AD. As Glu plays an important role in memory, we hypothesize that Glu levels are decreased in patients with AD when compared with controls. A consecutive sample of 30 patients with mild-to-moderate AD underwent H-MRS with the voxel placed in the bilateral posterior cingulate gyrus. For comparison purposes, we carried out the same technique in 68 patients with mild cognitive impairment (MCI) and in 26 controls. The healthy controls had higher metabolite levels of N-acetyl-aspartate (NAA) than patients with MCI and AD. In turn, patients with MCI and the controls had higher levels of Glu than in patients with AD. The differences were significant in the analysis of variance (ANOVA) test model corrected for age. In the post hoc analysis, the most remarkable differences were seen between patients with AD and the rest (patients with MCI and the controls). In AD, the levels of Glu and NAA are decreased in comparison with MCI and normality, which reflects loss of neurons.
Keywords: 1H-MR spectroscopy, Alzheimer’s disease, mild cognitive impairment, glutamate
Introduction
Previous reports point to an excess of excitatory glutamatergic stimulation in the brain of patients with Alzheimer’s disease (AD), which could lead to neuronal death. 1–4 This fact gave rise to treatment of patients with memantine, a glutamate (Glu) receptor antagonist. The experience with memantine is mostly limited to moderate-to-advanced AD and the results are equally modest, 5,6 but 2 placebo-controlled randomized trials also point to a benefit in mild stages. 7,8
Magnetic resonance spectroscopy (MRS) enables us to measure the concentration of Glu in the brain and other metabolites as well, but there is a paucity of studies measuring such a metabolite. Previous reports point to a decreased levels of Glu in AD in transgenic mice 9 and in humans but the studies were small. 10–12 The main findings described in patients with AD who underwent MRS with a single-voxel spectroscopy areas are as follows: decrease in N-acetyl-aspartate (NAA) in the mesial temporal lobe, posterior cingulate gyrus, occipital lobes, temporal lobes, parietal lobes, and hippocampi; decrease in white matter being smaller than the gray matter 13–16 ; increased myoinositol (mI) in the mesial temporal lobes, anterior and posterior cingulate gyrus, and parietal lobes, with major affectation of the gray matter 13 ; decrease in choline (Cho)/creatine (Cr) levels in patients with mild cognitive impairment (MCI) who did not progress to AD with respect to MCI converters and the controls. 17
The posterior cingulate region of the brain is also a key component of the default mode network (DMN), a constellation of brain regions activated during self-referential thinking. The medial temporal lobe subsystem provides information from previous experiences in the form of memories and associations that are the building blocks of mental simulation. The medial prefrontal subsystem facilitates the flexible use of this information during the construction of self-relevant mental simulations. These 2 subsystems converge on important nodes of integration including the posterior cingulate cortex (PCC). 18
On the basis of the previous reports and the progressive nature in AD, we hypothesize that there is a continuum between normality, MCI, and AD. The patients with AD should have lower levels of Glu than the controls and MCI group in the posterior cingulate gyrus.
Patients and Methods
In this cross-sectional study, we included a consecutive sample of 26 healthy volunteers, 68 patients with amnestic MCI, and 30 patients with mild-to-moderate AD. The patients with MCI were referred by family physicians because of memory complaints corroborated by an informant. They met the criteria of amnestic MCI according to the criteria of Petersen et al. 19 At baseline, the patients underwent neuropsychological analysis encompassing the Memory Impairment Screen (MIS), 20 Mini-Mental State Examination test (Spanish version; with a maximum possible score of 35 points), 21 Blessed Dementia Rating scale, clock drawing test, Geriatric Depression scale (GDS), and the Rey Auditory Verbal Learning Test (RAVLT) delayed recall. The patients with MCI included in this study must score 5 or lower in the MIS, 0.5 in the Clinical Dementia Rating scale (CDR), and a score higher than 21 points in the Mini-Mental State Examination.
The patients with probable AD fulfilled the criteria of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) work group 22 and were included in a randomized trial previously done to compare memantine with donepezil. 15 The patients with AD were reevaluated 6 months after randomization, but a second complete MRS study was only possible in 20 patients (10 under donepezil 10 mg/d and 10 under memantine 20 mg/d).
All patients underwent the neuroimaging techniques of magnetic resonance imaging (MRI). Data were acquired using a 1.5-T Signa HD clinical scanner (GE Healthcare Diagnostic Imaging, Milwaukee, Wisconsin). All images were acquired using an 8-channel phased-array head coil (neurovascular head array [NVHEAD A]). Magnetic resonance spectroscopy was carried out as follows. A midsagital T1-weighted image (repetition time [TR] = 560 ms, echo time [TE] = 12 ms, 90° flip angle, number of excitations = 1, matrix size = 256 × 160, field of view = 24 × 24 cm2, slice thickness/gap = 5/0 mm) was obtained to locate a voxel (2x2c2 cm) including the right and left PCC and inferior precunei (Figure 1 ). 1H-MRS was carried out by a short TE of 35 ms and a TR of 2000 ms and 128 accumulations using a single voxel with a spin echo technique that uses selective excitation with gradient spoiling for water suppression. The mode of spectral acquisition was proton brain spectroscopy–point resolved spectroscopy (PROBE-P) technique. For the quantification of absolute concentrations of brain metabolites, expressed in millimoles per liter per kilogram wet weight, we used the user-independent frequency domain-fitting program LCModel (version 6.2-0), 23 applying an eddy current correction and using internal water signal reference to calculate absolute metabolite concentrations. Apart from the individual analysis of the Glu, Cr, NAA, and mI compounds, we studied the summed concentrations of glutamate + glutamine referred to as Glx. Absolute metabolite values were only considered when the Cramér–Rao lower bound was below 20%, thus indicating that these metabolites could be reliably estimated. Chemical concentrations can now be automatically extracted from magnetic resonance (MR) spectra using sophisticated and well-documented time domain and spectral frequency fitting software packages such as LCModel, 23 a user-independent fitting routine based on a library of model spectra of all individual metabolites. 23 Concentration values are expressed as arbitrary institutional units and are not corrected for contributions by cerebral spinal fluid (CSF) and a small reduction in the numerical values by residual T1 and T2 relaxation effects. We also obtained the ratios of the peak amplitude of the metabolites relative to Cr. In clinical practice, metabolic ratios are assessed using Cr, which is considered as the most stable metabolite and as an internal reference. 24 In Figure 2 , an example of spectrum is given.
Figure 1.
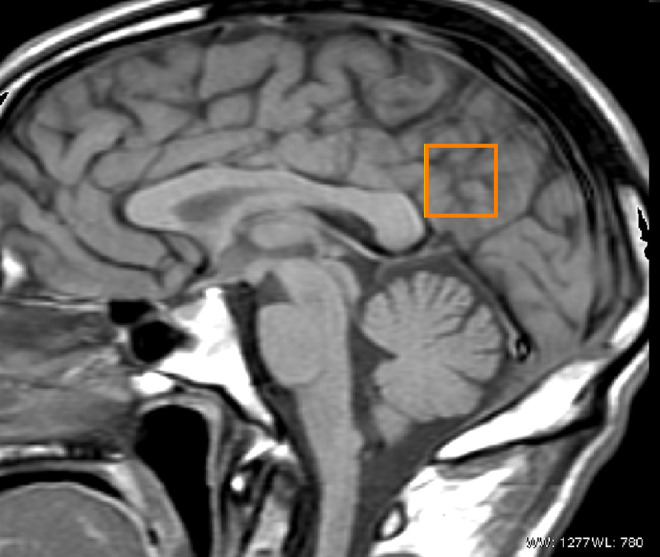
Sagittal T1-weigthed magnetic resonance imaging. Voxel placed in the bilateral posterior cingulate gyrus.
Figure 2.

Example of spectrum obtained with the LC Model. NAA indicates N-acetyl-aspartate; Glx, glutamate + glutamine; Cr, creatine; Cho, choline; mI, myoinositol.
Before starting this study, we studied the test–retest reliability of metabolite measurements in every area in a sample of patients with other pathologies, with 2 consecutive studies without removing the patient from the scanner. According to the resulting α coefficients, we must assume a mean random variation of approximately 8% for mI/Cr and approximately 10% for NAA/Cr and Cho/Cr. 25 We also carried out a second immediate MRS in 16 patients without being removed from the scanner to check the reproducibility of Glu and Glx. The intraclass correlation coefficients were .80 for PCC.
Statistical Analysis
The differences of mean metabolite values and ratios were analyzed with the ANOVA test and Bonferroni correction. However, as these values and ratios correlated with age significantly (P = .003 or lower) and as the mean age of controls was lower than that of patients, we made a multivariate analysis to correct the differences in age. We used the t test for paired samples to compare the changes in metabolite levels after treatment using donepezil and memantine separately, and for the whole sample of treated patients.
Results
We included 124 patients in the study: 68 patients with MCI, 30 patients with AD, and 26 controls (healthy volunteers). The baseline variables and the results of memory test and scales are included in Table 1 .
Table 1.
Baseline Variables for MCI, Patients With AD, and Controls
| Variables in MCI patients (n = 68) |
| Age: 74.2 year (SD: 6.9; range: 58-88) |
| Sex: 42 were female |
| MEC: 28.3 (SD: 3.1; range: 21-34) |
| BDRS: 2.7 (SD: 0.8; range: 1-4) |
| MIS: 2.4 (SD: 1.6; range: 0-5) |
| RAVLT: 3.2 (SD: 2.5; range: 0-6) in delayed recall. |
| Hypertension: 24 (34%) |
| APOE4 genotype: 21 had 1 or 2 alleles |
| Variables in the group of patients with AD (n = 30) |
| Age: 78.8 (SD: 4.65; range: 65-86) |
| Sex: 22 were female |
| MEC: 23.3 (SD: 3.5; range: 16-30) |
| BDRS: 7.8 (SD: 2.7; range: 4-14) |
| ADAS-cog: 22.7 (SD: 8.7; range: 13-48) |
| NPI: 10.8 (SD: 8.8; range: 2-40) |
| DAD: 74% (SD: 15; range: 20%-100%) |
| Hypertension: 10 (33%) |
| APOE4 genotype: 15 had 1 or 2 alleles |
| Control group (N = 26) |
| 12 were male; 14 were female |
| Age: 69.96 (SD: 17.29; range: 60-90) |
Abbreviations: MIS, Memory Impairment Screen; RAVLT, Rey Auditory Verbal Learning Test; SD, standard deviation; MCI, mild cognitive impairment; AD, Alzheimer’s diease; MEC, Mini-Examen Cognoscitivo; BDRS, Blessed Dementia Rating Scale; ADAS-cog, Alzheimer Disease Assessment Scale-cognitive part; NPI, neuropsychiatric inventory; DAD, disability assessment for dementia.
We observed that controls were somewhat younger than the patients with MCI and AD (mean age: 69.9 vs 74 and 78, respectively). Female predominance was noted in all the groups—MCI group (61%), AD group (73%), and control group (53.8%). Hypertension was described in MCI (34%) and AD (33%) groups. The presence of 1 or 2 APOE4 alleles in MCI and AD groups was 30.8% and 50%, respectively (Table 1).
Table 2 lists the metabolite levels and ratio of Cr in all the 3 groups of patients. When we compared the absolute values of NAA in the posterior cingulate gyrus, we found significant differences (F = 6.3; P = .002), with lower levels in AD (6.9 mmol/L) and MCI (7.3) groups than in the control group (7.58).
Table 2.
Metabolite Values and Ratios of Creatine in Each Group a
| Metabolite | Patients With AD | Patients With MCI | Controls | Significance P Value |
|---|---|---|---|---|
| NAA | 6.9 (0.88) | 7.3 (0.67) | 7.82 (0.61) b | .008 |
| Cho | 1.12 (0.18) | 1.1 (0.13) | 1.09 (0.14) | |
| mI | 4.99 (0.92) | 5.1 (0.65) | 4.77 (0.56) | |
| Glu | 6.63 (0.96) | 7.25 (0.82) | 7.52 (1.05) b | .036 |
| GLX | 9.02 (1.26) | 9.74 (1.24) | 9.69 (1.38) b | .019 |
| NAA/Cr | 1.23 (0.1) | 1.27 (0.1) | 1.38 (0.12) | |
| Cho/Cr | 0.2 (0.1) | 0.19 (0.22) | 0.19 (0.18) | |
| mI/Cr | 0.89 (0.13) | 0.9 (0.12) | 0.84 (0.02) | |
| Glu/Cr | 1.19 (0.14) | 1.26 (0.14) | 1.33 (0.19) | |
| GLX/Cr | 1.63 (0.26) | 1.7 (0.22) | 1.71 (0.25) b | .04 |
Abbreviations: NAA, N-acetyl-aspartate; GLX, glutamate + glutamine; Cr, creatine; Cho, choline; mI, myoinositol; Glu, glutamate; MCI, mild cognitive impairment; AD, Alzheimer’s disease; ANOVA, analysis of variance; SD, standard deviation.
a Mean and SD are expressed in mmol/L.
b Significant in the ANOVA test corrected for age.
Another finding was the lower levels of Glu in AD (6.63 mmol/L) and MCI (7.25) groups in comparison with the control group (7.19) with statistical significance (F = 4.68; P = .01). Lower levels of Glx were also found in patients with AD (9.02) in comparison with patients with MCI (9.7) and the controls ([9.45] F = 3.13; P = .047). The differences found in NAA and Glu were confirmed in a multivariate ANOVA test model corrected for age. In the post hoc analysis (Bonferroni test), the most remarkable differences were seen between patients with AD and the rest (patients with MCI and the controls), but not between patients with MCI and the controls. Figure 3 shows the box plots for NAA and Glu levels in each group.
Figure 3.

Box plots for the NAA and Glu levels in the 3 groups. 1 indicates AD group; 2, MCI group; 3, controls; GLU, glutamate; NAA, N-acetyl-aspartate.
With regard to the patients with AD, 20 of them underwent a second MRS after 6 months of treatment; 10 patients were on donepezil and 10 on memantine. In general, we did not see significant global changes after treatment with either donepezil or memantine in the t test for paired data. There was a trend of increased Glu levels, but the difference was only significant for the Glx/Cr ratio (means: 1.63 at baseline and 1.86 in the second scan; t = −3.01, P = .007). Despite treatment, the NAA levels decreased significantly (6.8 vs 6.2; t = 2.2; P = .04), which is consistent with a progressive degeneration and neuronal loss (P = .008). The NAA/Cr ratios did not change significantly. When the differences were analyzed separately for donepezil/memantine (Table 3 ), we only saw significant changes in the variable Glx/Cr ratio (.36 for donepezil vs .06 for memantine; t = −2.3; P = .03).
Table 3.
Ratios Before and After Treatment in the Donepezil and Memantine Groups a
| Donepezil Group, n = 10 | Memantine Group, n = 10 | |||
|---|---|---|---|---|
| Metabolite | Baseline | After Treatment | Baseline | After Treatment |
| NAA | 6.61 (1.24) | 5.74 (1.36) | 6.99 (0.5) | 6.68 (0.83) |
| NAA/cr | 1.21 (0.13) | 1.24 (0.18) | 1.22 (0.02) | 1.22 (0.13) |
| Glu | 6.57 (1.07) | 5.99 (1.49) | 6.65 (0.58) | 6.51 (0.83) |
| Glu/Cr | 1.21 (0.15) | 1.29 (0.15) | 1.16 (0.14) | 1.19 (0.14) |
| GLX | 9.12 (1.45) | 9.37 (1.07) | 8.87 (1.1) | 9 (0.94) |
| GLX/cr | 1.7 (0.3) | 2.06 (0.22) b | 1.55 (0.23) | 1.63 (0.3) |
Abbreviations: NAA, N-acetyl-aspartate; GLX, glutamate + glutamine; Cr, creatine; Glu, glutamate; SD, standard deviation.
a Mean (and SD) metabolite values in mmol/L.
b Indicates significance.
Discussion
As expected, decreased levels of NAA and NAA/Cr in posterior cingulate gyrus were found in the patients with AD and MCI compared with controls. Additionally, absolute Glu and Glx levels and Glx/Cr ratio were reduced in patients with AD versus MCI group. Patients with MCI showed NAA and NAA/Cr levels that were intermediate between controls and patients with AD. There were no changes detected in the absolute levels of Cho and mI in patients with MCI or AD compared with the control group.
The DMN of brain is a recently described brain system that has been identified using neuroimaging methods. The posterior cingulate region of the brain is also a key component of the DMN. 18
Patients with AD showed decreased resting-state activity in the posterior cingulate and hippocampus, suggesting that disrupted connectivity between these regions accounts for the posterior cingulate hypometabolism and hypoperfusion commonly detected in positron emission tomography studies of early AD. 26 The activity in the DMN may prove a sensitive and specific biomarker for cognitive decline. 27
Previous spectroscopy studies in patients with AD and MCI have found decreased levels of NAA, NAA/Cr, and NAA/mI. 16,28–30 The PCC is an area involved in memory and many times studied in MCI and AD. With regard to the best area to be explored, it seems that the posteromedial parietal cortex is particularly useful in terms of reproducibility. 25
N-acetyl-aspartate is a marker of neuronal viability and axonal density. Studies of MRS found decreased NAA and increased mI in the occipital, temporal, parietal, and frontal lobes in patients with AD even in the early stages of the disease. 31 N-acetyl-aspartate is believed to be located predominantly in neural bodies, dendrites, and axons. Cortical NAA concentrations provide information on neuronal and synaptic growth and pruning, while white matter NAA levels reflect development of axonal system. 32 Spectral abnormalities found in the elderly individuals suggest that there is a reduction in the number of neurons and of neuron viability (diminished NAA) associated with a more degradation of membranes and/or increase in the number of glial cells (elevated Cho and Cho/Cr ratio). Magnetic resonance spectroscopy in posterior cingulate discriminate AD and MCI according to the differences in NAA/Cr ratio. 30 The NAA/Cr ratio in the posterior cingulate gyrus predicted conversion from MCI to probable AD with a sensitivity of 82% and specificity of 72% 16 and correlate closely with clinical severity scales. 33 The ratio of NAA/Cr ratio ≤1.43 in the posteromedial bilateral parietal cortex predicted conversion to probable AD with 74.1% sensitivity and 83.7% specificity. The cross-validated accuracy 28 of classification was 82%.
In the neurotransmission process, Glu is released into the synapse from neuronal cells, then taken up by astroglial cells and transformed into glutamine, and afterward transported back to the neuron to be recycled to the Glu pool. As part of its neurotransmitters role, Glx is an excitatory aminoacid, and excessive Glx neurotransmission has been implicated in excitotoxic neuronal damage. 34 Some studies found that the persistence of elevated Ca2+ levels in hippocampal neurons exposed to Glu correlated with the extent of neuronal death 35 and that a large increase in Ca2+ in cultured hippocampal neurons after Glu application predicted cell death. 36 We have metabotropic receptors (mGLUr 1-8) and ionotropic receptors (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA], kainic acid [KA], and N-methyl-d-aspartate [NMDA]), each one of them with its own characteristics. 37 In humans, as in animals, an acute hypofunctional NMDA receptor (NMDAR) state is associated with increased glutamatergic activity. 38
The control group and MCI have significantly higher Glu levels than patients with AD, since many of the metabolite alterations that occur in AD processes including reductions in Glu are also known to occur with normal aging. 39 Amyloid-β (Aβ) can induce a number of biochemical changes in cells including an increase in the levels of cytosolic calcium and the stimulation of kinases. Recently, it has been reported that Aβ deposits can impair default network function in older individuals even without clinical dementia symptoms. 40 Thus, Aβ may impair the function of neurons at an early stage in the development of AD. Synaptic plasticity is a key component of memory and cognition, which regulates the strength of signaling between neurons. 41 Synaptic plasticity can be mediated by changes in the level of receptors on both presynaptic and postsynaptic membranes (postsynaptic density). 41 α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) are key regulators of synaptic function and cognition. In AD, cell surface AMPARs are downregulated; however, the reason for this downregulation is not clear; but recently, Liu et al have described that the Aβ protein induces a number of biochemical changes in cells including an increase in cytosolic calcium, and this contributes to a downregulation in the expression of Glu receptors in postsynaptic membrane. 42 This decrease in AMPARs may contribute to the decline in cognitive function seen in AD. The affection of the synaptic plasticity could explain the low levels of Glu found in our research. Finally, it has been reported that the Aβ protein inhibits presynaptic function due to the depletion of calcium-dependent vesicules, 43 and this also could explain the lower levels of Glu found in AD.
Limitations
The 1H-MRS signal arises from gray and white matter and is an ensemble average of multiple different cell types. Levels of Glu are often contaminated in part by glutamine, and Glu exists in the neurotransmitter pool as well as a metabolic pool.
Conclusions
This study points Glu to be a marker of neuronal loss and cognitive deterioration. The metabolite levels of Glu in patients with MCI were generally intermediate to control group and AD values, supporting the hypothesis of a pathological continuum. This study places Glu, a key neurotransmitter, in the forefront of our thinking behind the molecular processes involved in MCI and AD. We propose that decreased levels of Glu in the PCC, a key zone in the default network hypothesis, cause cellular damage and disruptions in circuits involved in cognition and behavior.
Footnotes
This study was approved by our regional ethical committee. Informed consent was acquired according to the Declaration of Helsinki.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study has been conducted with the financial support of the Instituto Aragones de Ciencias de la Salud (Spain). Project P-100/2008.
References
- 1. Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8(1):185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23(9):1261–1276. [DOI] [PubMed] [Google Scholar]
- 3. Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist: a review of preclinical data. Neuropharmacology. 1999;38(6):735–767. [DOI] [PubMed] [Google Scholar]
- 4. Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRX. 2004;1(1):101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reisberg B, Doody R, Stoffler A. Memantine in moderate to severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. [DOI] [PubMed] [Google Scholar]
- 6. Areosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev. 2005;18(2):CD003154. [DOI] [PubMed] [Google Scholar]
- 7. Peskind ER, Potkin SG, Pomara N. For the memantine MEM-MD-10 study group. Memantine treatment in mild to moderate Alzheimer disease: A 24-week randomized, controlled trial. Am J Geriatr Psychiatry. 2006;14(8):704–715. [DOI] [PubMed] [Google Scholar]
- 8. Backchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results of a randomized, duble-blind, placebo-controlled 6-month study. J Alzheimers Dis. 2007;11(4):471–479. [DOI] [PubMed] [Google Scholar]
- 9. Salek RM, Xia J, Innes A, et al. A metabolic study of the CRND8 transgenic mouse model of Alzheimer's disease. Neurochem Int. 2010;56(8):937–947. [DOI] [PubMed] [Google Scholar]
- 10. Antuono PG, Jones JL, Wang Y, Li SJ. Decreased glutamate + glutamine in Alzheimer's disease detected in vivo with (1)H-MRS at 0.5 T. Neurology. 2002;56(6):737–742. [DOI] [PubMed] [Google Scholar]
- 11. Hattori N, Abe K, Sakoda S, Sawada T. Proton MR spectroscopic study at 3 Tesla on glutamate/glutamine in Alzheimer's disease. Neuroreport. 2002;13(1):183–186. [DOI] [PubMed] [Google Scholar]
- 12. Rupsingh R, Borrie M, Smith M, Wells JL, Bartha R. Reduced hippocampal glutamate in Alzheimer disease. Neurobiol Aging. 2009;32(5):802–810. [DOI] [PubMed] [Google Scholar]
- 13. Soher BJ, Doraiswamy PM, Charles HC. A review of 1H MR spectroscopy findings in Alzheimer’s disease. Neuroimaging Clin N Am. 2005;15(4):847–852. [DOI] [PubMed] [Google Scholar]
- 14. Modrego PJ, Fayed N, Pina MA. Conversion from mild cognitive impairment to probable Alzheimer's disease predicted by brain magnetic resonance spectroscopy. Am J Psychiatry. 2005;162(4):667–675. [DOI] [PubMed] [Google Scholar]
- 15. Modrego PJ, Fayed N, Errea JM, Rios C, Pina MA, Sarasa M. Memantine versus donepezil in Alzheimer’s disease. A randomized trial with magnetic resonance spectroscopy. Eur J Neurol. 2010;17(3):405–412. [DOI] [PubMed] [Google Scholar]
- 16. Fayed N, Dávila J, Oliveros A, Castillo J, Medrano JJ. Utility of different MR modalities in mild cognitive impairment and its use as a predictor of conversion to probable dementia. Acad Radiol. 2008;15(9):1089–1098. [DOI] [PubMed] [Google Scholar]
- 17. Kantarci K, Weigand SD, Petersen RC. Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2007;28(9):1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1 124:1–38. [DOI] [PubMed] [Google Scholar]
- 19. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment. Clinical characterization and outcome. Arch Neurol. 1999;56(3):303–308. [DOI] [PubMed] [Google Scholar]
- 20. Buschke H, Kuslansky G, Katz M, et al. Screening for dementia with the memory impairment screen. Neurology. 1999;52(2):231–238. [DOI] [PubMed] [Google Scholar]
- 21. Lobo A, Ezquerra J, Gomez Burgada F, Sala JM, Seva Diaz A. Cognocitive mini-test (a simple practical test to detect intellectual changes in medical patients [in Spanish]. Actas Luso Esp Neurol Psiquiatr Cienc Afines. 1979;7(3):189–202. [PubMed] [Google Scholar]
- 22. McKhan G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group. Neurology. 1984;34(7):939–944. [DOI] [PubMed] [Google Scholar]
- 23. Provencher SW. Estimation of metabolite concentrations from localised in vivo proton NMR spectra. Magn Reson Med. 1993;30(6):672–679. [DOI] [PubMed] [Google Scholar]
- 24. Danielsen ER, Ross B. Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases. New York, NY: Marcel Dekker; 1999:5–22. [Google Scholar]
- 25. Fayed N, Modrego PJ, Medrano J. Comparative test-retest reliability of metabolite values assessed with magnetic resonance spectroscopy of the brain. The LCModel versus the manufacturer software. Neurol Res. 2009;31(5):472–477. [DOI] [PubMed] [Google Scholar]
- 26. Matsuda H. Cerebral blood flow and metabolic abnormalities in Alzheimer's disease. Ann Nucl Med. 2001;15(2):85–92. [DOI] [PubMed] [Google Scholar]
- 27. Sperling RA, Dickerson BC, Pihlajamaki M, et al. Functional alterations in memory networks in early Alzheimer's disease. Neuromolecular Med. 2010;12(1):27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Modrego P, Fayed N, Sarasa M. Magnetic resonance spectroscopy in the prediction of early conversion from amnestic mild cognitive impairment to dementia: a prospective cohort. BMJ Open 2011. doi: 10.1136/bmjopen-2010-000007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kantarci K, Knopman DS, Dickson DW, et al. Alzheimer disease: postmortem neuropathologic correlates of antemortem 1H MR Spectroscopy metabolite measurements. Radiology. 2008;248(1):210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kantarci K, Xu Y, Shiung MM, et al. Comparative diagnostic utility of different MR modalities in mild cognitive impairment and Alzheimer's disease. Dement Geriatr Cogn Disord. 2002;14(4):198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valenzuela MJ, Sachdev P. Magnetic resonance spectroscopy in AD. Neurology. 2001;56(5):592–598. [DOI] [PubMed] [Google Scholar]
- 32. Horska A, Kaufmann W, Brant L, Naidu S, Harris J, Barker P. In vivo quantitative proton MRSI study of brain development from childhood to adolescence. J Magn Reson Imaging. 2002;15(2):137–143. [DOI] [PubMed] [Google Scholar]
- 33. Fayed N, Dávila J, Oliveros A, Jr, Medrano J, Castillo J. Correlation of findings in advanced MR techniques with global severity scales in patients with some grade of cognitive impairment. Neurol Res. 2010;32(2):157–165. [DOI] [PubMed] [Google Scholar]
- 34. Bleich S, Römer K, Wiltfang J, Kornhuber J. Glutamate and the glutamate receptor system: a target for drug action. Int J Geriatr Psychiatry. 2003;18(suppl 1):S33–S40. [DOI] [PubMed] [Google Scholar]
- 35. Ogura A, Miyamoto M, Kudo Y. Neuronal death in vitro: parallelism between survivability of hippocampal neurones and sustained elevation of cytosolic Ca2+ after exposure to glutamate receptor agonist. Exp Brain Res. 1988;73(3):447–458. [DOI] [PubMed] [Google Scholar]
- 36. Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9(11):3728–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gazulla J, Cavero-Nagore M. Glutamato y enfermedad de Alzheimer. Rev Neurol. 2006;42(7):427–432. [PubMed] [Google Scholar]
- 38. Rowland LM, Bustillo JR, Mullins PG, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162(2):394–396. [DOI] [PubMed] [Google Scholar]
- 39. Zahr NM, Mayer D, Pfefferbaum A, Sullivan EV. Low striatal glutamate levels underlie cognitive decline in the elderly: evidence from in vivo molecular spectroscopy. Cereb Cortex. 2008;18(10):2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sperling RA, Laviolette PS, O’Keefe K, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63(2):178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kullmann DM, Lamsa KP. Longterm synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8(9):687–699. [DOI] [PubMed] [Google Scholar]
- 42. Liu SJ, Gasperini R, Foa L, Small DH. Amyloid-β decreases cell surface AMPA receptors by increasing intracellular calcium and phosphorylation of GluR2. J Alzheimer’s Dis. 2010;21(2):655–666. [DOI] [PubMed] [Google Scholar]
- 43. Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. Amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol chem. 2010;285(4):2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]