


Abstract
A novel and efficient method has been developed for isolation of correctly digested DNA fragments without the use of classic size-dependent electrophoretic separation methods. To achieve this, DNA fragments are end-labelled by haptens. After specific endonuclease digestion of the hapten-labelled DNA, the DNA is incubated with a protein that specifically binds to the hapten. The incubation mixture is then passed through a cartridge containing a protein-binding membrane that does not bind DNA. Undigested and partly digested DNA are retained on the membrane, while correctly digested DNA is selectively recovered for use in further downstream applications.
INTRODUCTION
Size separation of DNA fragments by gel electrophoresis is the standard technique for separating and isolating nucleic acid fragments (1). Liquid chromatography and capillary electrophoresis methods can also be applied for DNA separation (2,3).
Central to DNA cloning is the isolation and purification of endonuclease treated DNA. The desired DNA fragments from the restriction reaction is isolated, typically by agarose gel electrophoresis followed by excision of agarose slices containing DNA identified by UV irradiation (4). The agarose is dissolved, and DNA recovered by absorption to silica gel particles (5). The recovered DNA is often ligated into plasmid DNA previously digested by endonucleases and isolated by similar electrophoretic methods.
Using restriction treatment of DNA by unique DNA endonucleases, several fragments will be generated as a result of incomplete/or lack of digestion (Fig. 1A). To ensure a high level of DNA digestion, elevated enzyme concentrations and/or prolonged incubation times can be used. However, this will increase the possibility for degradation of the compatible DNA ends by traces of contaminating exonucleases present in most enzyme preparations. Endonuclease star activity is also known to occur under these conditions (6,7). Separating the correctly digested from the partly digested DNA can be difficult, as the fragments often differ by only a few bases in size. In addition, the time-consuming gel-based procedures are likely to reduce DNA quality due to degradation by contaminating nucleases or intercalating reagents. UV irradiation can severely degrade the DNA, reducing its biological activity, quality and yield (8). A method for DNA isolation avoiding UV exposure of the DNA is therefore of considerable interest.
Figure 1.

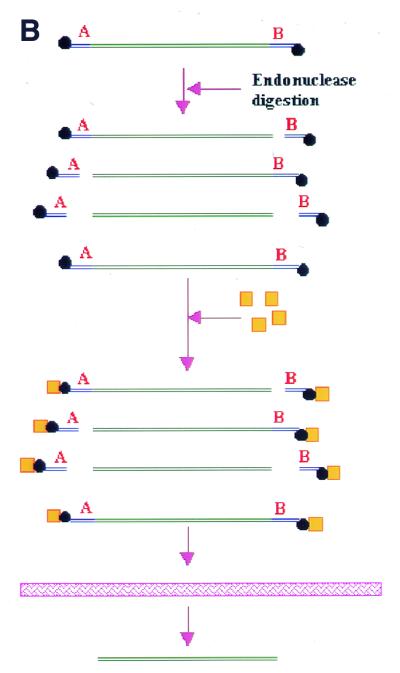
Restriction of DNA product into fragments.(A) Restriction of a DNA product using two unique restriction endonucleases, A and B, will yield four possible fragments, where only one fragment is the result of complete digestion by both endonucleases. (B) By labelling the fragments with a hapten and employing a hapten-binding protein, only the correct fragment will be recovered upon passage of the reaction mixture through the membrane cartridge.
Certain aromatic polymers like poly-p-hydroxystyrene generally bind proteins with a high affinity, while having no affinity for DNA (Seed and Seed, European Patent EP 054800). We will show in this report that by attaching a hapten tag onto the DNA and employing a protein binding partner for the hapten, correctly digested DNA can be selectively isolated with high efficiency by passing the digestion mixture through a protein-binding membrane (Fig. 1B).
MATERIALS AND METHODS
Bacterial strains
XL10 GOLD™ (Stratagene, La Jolla, CA) {TetrD (mcrA)183 D(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac Hte [F′ proAB lacIqZDM15 Tn10 (Tetr) Amy Camr]} supercompetent Escherichia coli bacterial cells were used for all DNA transformations according to the manufacturer’s recommendations.
Plasmids
Plasmid construction. Plasmid pCantab5EApaI/AscI was constructed by insertion of annealed 5′-phosphorylated oligonucleotides CT5E3-5 and CT5E5-3 between SfiI and NotI sites of plasmid pCantab5E (Amersham Pharmacia Biotech, Uppsala, Sweden), introducing unique AscI and ApaI restriction sites.
Sequences of the 5′-phosphorylated primers CT5E3-5 and CT5E5-3 were: CT5E3-5, 5′-GGCCGCGGTACCTACCACCGGCGCGCCGGGCCCACCACCATGGCCCGCT; CT5E5-3, 5′-GGGCCATGGGGTGGTGGGCCCGGCGCGCCGGTGGTAGGTACCGC.
Plasmid isolation. Plasmids were isolated according to the procedure described by Qiagen™ (Hilden, Germany) for use with their Maxiprep™ kit. The final eluate containing plasmid DNA was passed through a HR400 MicroSpin™ (Amersham Pharmacia Biotech) column to remove residual alcohols and salts.
Digestion and labelling of DNA. The pCantab5EApaI/AscI plasmid was digested either with AscI, SfiI or NotI restriction enzymes (Roche, Basel, Switzerland) using 5 µg of plasmid DNA with 30 U of enzyme in the appropriate reaction buffer giving a total volume of 100 µl. The mixture was incubated for 1 h and depleted from salts and proteins by passing it through a HR 400 MicroSpin™ column. PCR products were treated accordingly.
For labelling the restricted plasmid using Klenow fragment fill-in, 5 µg plasmid was added to 30 U of restriction enzyme AscI and appropriate enzyme buffer. To this reaction mixture was added; 40 U of E.coli DNA polymerase I large Klenow fragment (New England Biolabs, Hertfordshire, UK), 2 nmol of 14-modified-dCTP (biotin or fluorescein) (Gibco BRL, Germany) and 10 nmol of dGTP (Gibco BRL) giving a total volume of 100 µl. The reaction mixture was incubated at 37°C for 2 h, and depleted from salts and most proteins by passing it through a HR 400 MicroSpin™ column.
Membrane applications
For all the membrane applications, the Centriflex™ membrane cartridges (EdgeBio Systems, USA) were used.
Biotin-labelled DNA
Biotin-labelled DNA samples were passed through a MicroSpin™ column and subsequently added to at least 2 µg streptavidin (Promega, UK) in a 1:2 molar ratio, giving a total volume of 100 µl. The reaction mixture was vortexed, centrifuged for 1 min and incubated for 10 min at room temperature. The reaction mixture was then added to the membrane cartridge and spun at 13 000 g for 1 min. The membrane cartridge was turned around its axis and 10 µl of water added, before being spun at 13 000 g a second time in order to recover any residual fluid remaining in the cartridge. The supernatant passing through the membrane cartridge was collected for subsequent analysis and downstream application. In the case of excess biotinylated primers or nucleotides remaining in the sample, the supernatant was added another 2 µg streptavidin and incubated for 5 min, before repeating the procedure using a new cartridge.
Fluorescein-labelled DNA
Fluorescein labelled DNA samples were passed through a MicroSpin™ cartridge, and subsequently added at least 4 µg anti-fluorescein antibody (Molecular Probes, USA) in a 1:5 molar ratio. Salts were added to the reaction mixture, to give a buffer composition equal to 1× PBS buffer (150 mM NaCl, 20 mM PO4, pH 7.4). A total volume of 100 µl was used. The reaction mixture was vortexed, centrifuged for 1 min and incubated for 20 min at room temperature. The reaction mixture was then added to the membrane cartridge as for the biotinylated DNA. In the case of excess DNA, fluorescein-labelled primers or nucleotides present in the sample, the supernatant was added another 2 µg antibody and incubated for 15 min, before repeating the procedure on a new cartridge.
Lambda fragment labelling
Two micrograms of lambda HindIII digested DNA (Gibco BRL) was added 40 U of E.coli DNA polymerase I large Klenow fragment (New England Biolabs), 2 nmol of 14-modified-dCTP (biotin or fluorescein) (Gibco BRL) and 10 nmol of dGTP (Gibco BRL) giving a total volume of 100 µl. The reaction mixture was incubated at 37°C for 1 h, and depleted from salts and most proteins by passing it through a HR 400 MicroSpin™ column.
PCR
Template. The template for all PCR reactions was an 800 bp DNA fragment coding for a scFv antibody fragment constructed and isolated according to the RAPS protocol by Pharmacia-Amersham.
Oligonucleotides. For the amplification of the DNA fragment, the 5′-end biotin-labelled oligonucleotides SfiAMP and NotAMP were used: SfiAMP, 5′-GTC CTC GCA ACT GCG GCC CAG CCG GCC ATG GCC GAG; NotAMP, 5′-GAG TCA TTC TGC GGC CGC CCG TTT TAT TTC CAG CTT (SfiI and NotI restriction sites underlined).
For the PCR, using partly restriction site overlapping primers, the 5′-end biotin-labelled primers SfiAMPdel and NotAMPdel were used: SfiAMPdel, 5′-GTC CTC GCA ACT GCGG; NotAMPdel, 5′-GAG TCA TTC TGCGGC C, (nucleotides participating in SfiI and NotI restriction sites are underlined).
PCR reaction conditions. A mixture of 20 ng template DNA, 20 mmol nucleotidemix (Finnzyme, Finland), 25 pmol of each primer, water and 10 µl Expand High Fidelity™ buffer 2 (Roche), was used in the PCR, giving a total volume of 100 µl. The reaction was covered with oil. Three units Expand High Fidelity™ polymerase (Roche) were added using hot-start technique. The reaction mix was hot started at 96°C for 2 min and added enzyme before being cycled 25 rounds at: 96°C 30 s, 60°C 30 s, 72°C 50 s, followed by 2 min extension at 72°C. The PCR product was purified from salts and proteins and most of residual nucleotides by passing it through a HR 200 MicroSpin™ column.
Gel electrophoresis
Agarose gels were made using SeaKem agarose (FMC, UK), and stained by adding ethidium bromide 0.5 µg/ml (Sigma, Germany) to the melted agarose before moulding. Isolation and purification of DNA was performed using Qiagen QuickPCR purification columns (Qiagen™) as directed by the manufacturer. The final eluate containing isolated DNA was passed through a HR 400 MicroSpin™ column to remove residual isopropanol and salts.
Comparing quality of DNA
Preparation of DNA. Plasmid Cantab5EAscI/ApaI (5 µg) was restricted and biotin-labelled using AscI restriction enzyme and the fill-in reaction as previously described. The HR 400 MicroSpin™ treated reaction mixture was added 30 U of the restriction enzyme SfiI together with the SfiI restriction enzyme buffer, incubated 1 h at 50°C. Salts, proteins and most of residual nucleotides were depleted from the mixture by passing it through a HR 400 MicroSpin™ column. This was repeated using the NotI restriction enzyme at 37°C. The mixture was divided in two aliquots. One aliquot was added streptavidin and purified using a Centriflex™ membrane cartridge, while the other aliquot was purified using agarose gel electrophoresis. Equally a biotin-labelled PCR product was digested with SfiI and NotI enzymes and divided into two aliquots for isolation by membrane cartridge or electrophoresis.
Ligation and transformation. Enzyme digested plasmid DNA (1 µg) isolated by either the standard or membane method, was added 200 ng of enzyme digested PCR product isolated with a corresponding method. The ligation mixture was added ligation buffer, 5 U of T4 DNA ligase (Roche) and water to a total of 50 µl. The ligation reaction was incubated for 1 h at 20°C. The ligation reactions were digested with AscI enzyme to linearise religated plasmid. The reaction was purified from salts and proteins by passing it through a HR 400 MicroSpin™ column. Digested ligation mixture (5 µl) was added to 100 µl XL10 GOLD competent cells. The cells were treated as described by Stratagene, and spread on agar for colony growth overnight. Colonies were counted the next day. The experiment was repeated five times.
RESULTS
Retainment of biotin-labelled DNA on a protein binding membrane
Lambda HindIII digested DNA fragments were labelled with biotin using Klenow fragment and biotin-labelled nucleotides. The Klenow fill-in reaction will result in each lambda DNA fragment having a maximum of two biotins attached, except for the two lambda fragments containing cos-sites, which could have a maximum of six biotins attached. As shown in Figure 2A, biotin-labelled DNA is retained on the membrane when added streptavidin and passed through the membrane cartridge. Unlabelled DNA passes unhindered, demonstrating a high efficiency and selectivity in the biotin-labelling and subsequent removal of the labelled DNA.
Figure 2.
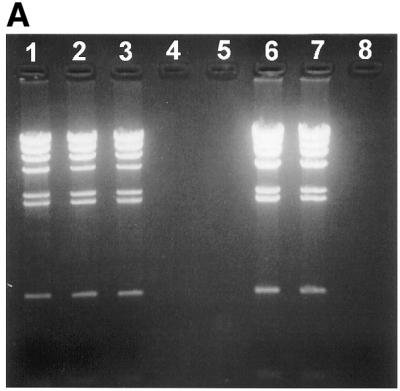
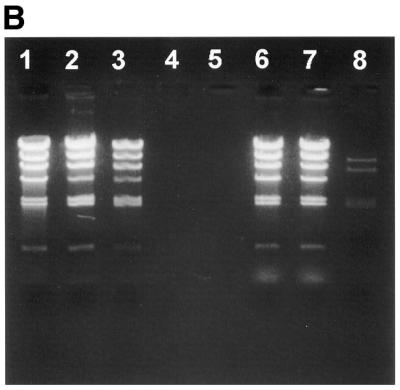
Retainment of labelled lambda HindIII restricted DNA fragments by the membrane cartridge. Lambda HindIII DNA fragments are labelled with streptavidin in (A), and with fluorescein in (B). An aliquot of 1 µg lambda HindIII digested DNA fragments were used in each lane. Lane 1, unlabelled lambda HindIII fragments; lane 2, unlabelled lambda HindIII fragments incubated with 5 µg BSA and passed through the membrane cartridge; lane 3, unlabelled lambda HindIII fragments incubated with 5 µg streptavidin and passed through the membrane cartridge; lanes 4 and 5, empty; lane 6, labelled lambda HindIII fragments; lane 7, labelled lambda HindIII fragments incubated with 5 µg BSA and passed through the membrane cartridge; lane 8, labelled lambda HindIII fragments incubated with 5 µg streptavidin (A) or anti-fluorescein antibody (B) and passed through the membrane cartridge.
Retainment of fluorescein-labelled DNA on a protein binding membrane
Lambda HindIII DNA fragments were labelled with fluorescein and added anti-fluorescein antibodies. Although not as efficient as the biotin–streptavidin interaction in Figure 2A, the interaction between the fluorescein-labelled DNA and the antibody seem efficient enough to retain most of the labelled DNA on the membrane (Fig. 2B). Note that the fragments containing the cos-sites are most efficiently retained on the membrane. Experiments performed confirm that one biotin is sufficient for DNA retainment on the membrane, while 5–10 fluorescein molecules are needed to get an equally efficient retainment (data not shown). Biotin/streptavidin therefore seems the preferred system for use with the membrane-method.
Digested PCR products are easily purified by the membrane-method
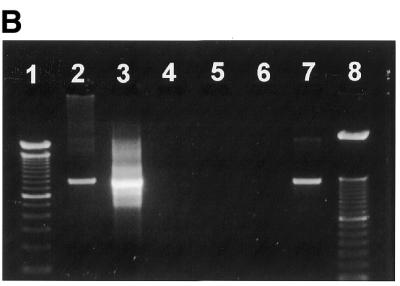
To investigate the recovery of purified PCR products by the membrane-method, an 800 bp DNA fragment was amplified by PCR using 5′-end biotinylated primers. The PCR product was digested with enzymes SfiI and NotI corresponding to restriction sites found at each flank of the amplified PCR product. Figure 3A shows that the PCR product only when digested by both enzymes is allowed to pass through the membrane cartridge. PCR product digested with only one or none of the enzymes are retained on the membrane.
Figure 3.


Purification of digested DNA. An aliquot of 200 ng of DNA was used for each lane. (A) Lane 1, 100 bp DNA size standard; lane 2, empty; lane 3, PCR product; lane 4, PCR product added streptavidin and passed through the membrane cartridge; lane 5, empty; lane 6, PCR product restricted by SfiI endonuclease; lane 7, PCR product restricted by SfiI endonuclease added streptavidin and passed through the membrane cartridge; lane 8, empty; lane 9, PCR product restricted by NotI endonuclease; lane 10, PCR product restricted by NotI endonuclease added streptavidin and passed through the membrane cartridge; lane 11, empty; lane 12, PCR product restricted by NotI and SfiI endonucleases; lane 13, PCR product restricted by NotI and SfiI endonucleases added streptavidin and passed through the membrane cartridge; lane 14, empty; lane 15, 100 bp DNA size standard. (B) Lane 1, Lambda HindIII size standard; lane 2, empty; lane 3, AscI digested and biotin labelled plasmid DNA; lane 4, AscI digested and biotin labelled plasmid DNA added streptavidin and passed through the membrane cartridge; lane 5, empty; lane 6, AscI digested and biotin labelled plasmid DNA restricted by SfiI endonuclease; lane 7, AscI digested and biotin labelled plasmid DNA restricted by SfiI endonuclease added streptavidin and passed through the membrane cartridge; lane 8, empty; lane 9, AscI digested and biotin labelled plasmid DNA restricted by NotI endonuclease; lane 10, AscI digested and biotin labelled plasmid DNA restricted by NotI endonuclease added streptavidin and passed through the membrane cartridge; lane 11, empty; lane 12, AscI digested and biotin labelled plasmid DNA restricted by NotI and SfiI endonucleases; lane 13, AscI digested and biotin labelled plasmid DNA restricted by NotI and SfiI endonucleases added streptavidin and passed through the membrane cartridge; lane 14, empty; lane 15, Lambda HindIII size standard.
Digestion of plasmid DNA for subsequent cloning
To investigate the ability of the method to purify digested plasmid DNA, the pCantab5EApaI/AscI plasmid was initially digested by AscI restriction enzyme, located within the DNA fragment to be removed, and biotin-labelled by a Klenow fill-in reaction. The biotin-labelled plasmid was then digested with enzymes SfiI and NotI. Figure 3B shows that the plasmid DNA, only when digested by both enzymes, is allowed to pass through the membrane cartridge. Plasmid DNA digested with only one or none of the enzymes are retained on the membrane.
Certain PCR products can be purified directly by the membrane-method without the need of further purification steps
Erroneous primer annealing during the PCR will give unwanted by-products. Performing the PCR using primers partly overlapping unique restriction sites flanking the template eliminates this problem (Fig. 4A). Amplifying the 800 bp insert with primers partly overlapping the unique SfiI and NotI restriction sites will subsequently enable the removal of any by-products made by erroneous annealing of primers. The resulting by-products will not contain both SfiI and NotI sites. Consequently, all by-products will be removed upon digestion with both SfiI and NotI, followed by the addition of streptavidin and passage through the membrane cartridge.
Figure 4.

Purification of PCR products without the need for size-separation methods. (A) A DNA template for use in a PCR has two unique flanking restriction sites A and B. For PCR amplification two biotinylated primers are used, each partly overlapping the restriction sites of the template (bold). However, within the template are sequences related to the restriction sites, where the primers could anneal, giving erroneous PCR products (C and D). The PCR reaction now produces a mixture of DNA products, with only one product being wanted. Annealing of the primers to C or D do not create new restriction sites, so that only annealing of primers to A or B will give products with the correct restriction sites A and B. By digesting the PCR products with restriction enzymes A and B, and subsequently adding streptavidin to the DNA fragments, all by-products and incompletely digested PCR products are tagged by the steptavidin. By passing the reaction through the membrane cartridge only the wanted PCR product is recovered. (B) Lane 1, 100 bp size standard; lane 2, PCR template; lane 3, suboptimal PCR products; lane 4, PCR products with added streptavidin and passed through the membrane cartridge; lane 5, NotI restricted PCR products added streptavidin and passed through the membrane cartridge; lane 6, SfiI restricted PCR products added streptavidin and passed through the membrane cartridge; lane 7, NotI and SfiI restricted PCR products added streptavidin and passed through the membrane cartridge; lane 8, 50 bp size marker.
The 800 bp DNA fragment was amplified using sub-optimal PCR conditions (annealing temperature of 55°C) using partly overlapping 5′-biotinylated primers. As shown in Figure 4B, all unwanted by-products are efficiently removed from the PCR upon enzyme digestion and passage through the membrane cartridge.
The quality of DNA prepared by the membrane-method is better than DNA prepared by standard gel-electrophoretic methods
In order to investigate the quality of the DNA obtained, plasmid DNA and PCR products were isolated in parallel using both the standard and the membrane-method. Equal amounts of the DNA fragments obtained from each method were ligated to give circular ligation products. After digesting the ligation products with AscI to remove religated products, they were transformed into bacteria to measure the ligation efficiency. Comparing the number of ampicillin-resistant bacterial colonies obtained from each method reflects the quality of the DNA (Table 1). Twenty times more colonies were seen using the membrane-method than standard methods, demonstrating a substantial improvement in DNA quality.
Table 1. Comparing DNA quality.
| Experiment | Mean | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Standard method, c.f.u.a | 35 | 62 | 40 | 21 | 39 | 39 |
| Membrane-method, c.f.u. | 720 | 685 | 832 | 625 | 852 | 743 |
Ligation products resulting from DNA isolated by the membrane-method perform 20-fold better in bacterial transformation than ligation products resulting from equal amounts of DNA isolated by standard method. aColony forming units.
DISCUSSION
We have developed a new strategy for purification of DNA fragments, which have several advantages over standard size separating gel electrophoretic methods. The method described here involves the use of a matrix in the form of a membrane. The membrane specifically binds proteins and not DNA. These properties are used to separate protein-tagged DNA from untagged by retaining the protein-tagged DNA on the membrane. The untagged DNA elutes unhindered through the membrane.
The main advantages of the membrane-method include fast purification, high DNA separating capacity and high purity of the DNA. Furthermore, UV-irradiation of the DNA is avoided. The low hands-on time and the low-priced reagents used are also interesting aspects of the membrane-method.
The standard gel electrophoresis has several disadvantages compared to our membrane-method. Gel electrophoresis and isolation of the gel-imbedded DNA is time consuming. It also involves the use of ethidium bromide and UV irradiation that is potentially harmful for the operator. Furthermore, DNA exposed to UV irradiation is seriously damaged, leading to a reduced biological activity (8,9). The lengthy purification process could also impair DNA quality by exposure for potential DNases and intercalacing dyes like ethidium bromide.
Our membrane-method involves the interaction of a hapten and a protein. The success is dependent on the efficient hapten labelling of all DNA and the efficient binding of the protein to the hapten. For this purpose, the streptavidin/biotin interaction is very suitable. Incorporation of biotin into DNA is efficiently achieved either by use of biotinylated primers in PCR (10) or by a number of enzymatic reactions (11,12). The streptavidin/biotin interaction is extraordinarily strong (13), enabling us to bind biotin-labelled DNA to added streptavidin very efficiently. As a consequence, DNA can be separated in a size independent manner.
Digestion of DNA often requires the subsequent removal of small DNA fragments, making it difficult to isolate and identify correctly digested of DNA by conventional gel electrophoresis. Isolation of a mixture of partially and correctly digested DNA will lower the efficiency of further downstream applications of the digested DNA. Our method eliminates these problems, as only correctly digested DNA passes the membrane cartridge, giving excellent purity of the DNA. In addition, the membrane-method enables us to perform the digestion with minute amounts of restriction endonucleases to further minimise exonuclease attack on the DNA, while still allowing the recovery of only the correctly digested DNA fragments.
Isolation of a high amount of purified DNA is suitable with the membrane-method. By standard methods, the gel loading capacity limits the amount of DNA that can be handled. Using the Centriflex™ cartridges 20 µg DNA can easily be processed by the cartridge (data not shown), while standard methods would be limited to 3–4 µg DNA per lane. Optimising the membrane-method and the membrane cartridge could improve the capacity even further. In contrast to the membrane-method, standard DNA purification methods result in dilution of the DNA samples which in turn must be handled by precipitation before being used in further downstream applications.
Our membrane-method has a number of potential applications in different biotechniques, involving separation of DNA. We have shown that the method is particularly useful in separation of DNA fragments resulting from enzyme digestion reactions, greatly improving the DNA quality. This is shown by elevated transformation efficiencies demonstrating its usefulness in library-constructing approaches, which require high transformation efficiencies for obtaining optimal results.
The very high quality of the DNA obtained by the membrane-method, as well as the rapidity at which it is performed with, suggests that this method should be implemented as a technique in most DNA laboratories, replacing a number of sub-optimal methods for DNA fragment isolation.
Acknowledgments
ACKNOWLEDGEMENT
The study was supported by a grant from the Norwegian Research Council #32680/213.
REFERENCES
- 1.Slater G.W., Kist,T.B., Ren,H. and Drouin,G. (1998) Electrophoresis, 19, 1525–1541. [DOI] [PubMed] [Google Scholar]
- 2.Huber C.G. (1998) J. Chromatogr. A., 806, 3–30. [DOI] [PubMed] [Google Scholar]
- 3.Sunada W.M. and Blanch,H.W. (1997) Electrophoresis, 18, 2243–2254. [DOI] [PubMed] [Google Scholar]
- 4.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY.
- 5.Vogelstein B. and Gillespie,D. (1979) Proc. Natl Acad. Sci. USA, 76, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kriss J., Herrin,G., Forman,L. and Cotton,R. (1990) Nucleic Acids Res., 18, 3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasri M. and Thomas,D. (1986) Nucleic Acids Res., 14, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartman P.S. (1991) Biotechniques, 11, 747–748. [PubMed] [Google Scholar]
- 9.Hardy E., Pupo,E., Casalvilla,R., Sosa,A.E., Trujillo,L.E., Lopez,E. and Castellanos-Serra,L. (1996) Electrophoresis, 17, 1537–1541. [DOI] [PubMed] [Google Scholar]
- 10.Nelson P.S., Kent,M. and Muthini,S. (1992) Nucleic Acids Res., 20, 6253–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klevan L. and Gebeyehu,G. (1990) Methods Enzymol., 184, 561–577. [PubMed] [Google Scholar]
- 12.Flickinger J.L., Gebeyehu,G., Buchman,G., Haces,A. and Rashtchian,A. (1992) Nucleic Acids Res., 20, 2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaiet L.a.W.F.L.1. (1964) Arch. Biochem. Biophys., 106, 1. [DOI] [PubMed] [Google Scholar]