

Abstract
Purpose:
To evaluate the retinal histopathology in donor eyes from patients with autosomal recessive retinitis pigmentosa (arRP) caused by EYS mutations.
Methods:
Eyes from a 72 year-old female (donor 1, family 1), a 91 year-old female (donor 2, family 2), and her 97 year-old sister (donor 3, family 2) were evaluated with macroscopic, scanning laser ophthalmoscopy (SLO) and optical coherence tomography (OCT) imaging. Age- normal eyes and an eye donated by donor 1’s asymptomatic mother (donor 4, family 1) were used as controls. The perifovea and peripheral retina were processed for microscopy and immunocytochemistry with markers for cone and rod photoreceptor cells.
Results:
DNA analysis revealed EYS mutations c.2259+1G>A and c.2620C>T (p.Q874X) in family 1 and c.4350_4356del (p.I1451Pfs*3) and c.2739-?_3244+?del in family 2. Imaging studies revealed the presence of bone spicule pigment in arRP donor retinas. Histology of all three affected donor eyes showed very thin retinas with little evidence of stratified nuclear layers in the periphery. In contrast, the perifovea displayed a prominent inner nuclear layer. Immunocytochemistry analysis demonstrated advanced retinal degenerative changes in all eyes with near-total absence of rod photoreceptors. In addition, we found that the perifoveal cones were more preserved in retinas from the donor with the midsize genomic rearrangement (c.4350_4356del (p.I1451Pfs*3) and c.2739-?_3244+?del) than the retinas from the donors with the truncating (c.2259+1G>A and c.2620C>T (p.Q874X) mutations.
Conclusions:
Advanced retinal degenerative changes with near-total absence of rods and preservation of some perifoveal cones are observed in arRP donor retinas with EYS mutations.
Keywords: Recessive retinitis pigmentosa, EYS mutations, histopathology, immunohistochemistry
Introduction
The inherited retinal diseases referred to as retinitis pigmentosa (RP) exhibit a wide variety of genetic heterogeneity, variable expressivity, allelic heterogeneity and phenotypic variability. RP is the most common form of inherited retinal degeneration, affecting 1 in 3,500 people, with more than 1 million patients worldwide [1,2]. It is characterized by progressive rod and cone photoreceptor cell dysfunction and results in the clinical appearance of optic nerve pallor, retinal vascular attenuation and peripheral pigmentary and atrophic changes. Visual deficits in affected individuals initially include night blindness, due to rod photoreceptor loss, followed by progressive loss of peripheral vision. Ultimately, nearly all patients lose central vision between the ages of 50 and 80 years [3].
Families with RP demonstrate all known patterns of Mendelian inheritance, including autosomal dominant, autosomal recessive, and X-linked. Non-Mendelian inheritance patterns such as digenic and maternal inheritance have also been reported. Sixty-percent of all RP cases are autosomal recessive (arRP) [4]. Currently, mutations have been identified in 35 different genes in arRP patients (http://www.sph.uth.tmc.edu/Retnet/). Together, mutations in these genes account for approximately 50% of cases. The genes that are associated with arRP encode proteins that exert their function in different pathways within the retina, including the phototransduction cascade (CNGA1, CNGB1, PDE6A, PDE6B, RGR, RHO, SAG), vitamin A metabolism (ABCA4, LRAT, RLBP1, RPE65), structural or signaling functions (CRB1, RP1, TULP1, USH2A), transcriptional regulation (NR2E3, NRL), and retinal pigment epithelium (RPE) phagocytosis (MERTK), or have unknown functions (CERKL, PRCD, PROM1) [5].
Approximately 5-16% of arRP cases [6] result from mutations in the eyes shut homolog (EYS) gene, identified at the RP25 locus [5,7]. Spanning over 2,000 bp (6p12.1-6q15), EYS is one of the largest genes expressed in the human eye to date [7]. The longest isoform of EYS encodes a protein of 3,165 amino acids whose function remains to be elucidated. The only characterized EYS homologue, Drosophila’s spacemaker or SPAM, is involved in the assembly of the light sensitive rhabdomere, the insect equivalent of vertebrate photoreceptor outer segments [8]. Indeed, the human EYS protein is localized to the photoreceptor outer segments [7]. Considering the evolutionary data and the known function of the Drosophila ortholog, EYS is likely to play a role in the modeling and integrity of retinal architecture [8].
Mutations in more then 60 genes are known to cause RP. Early histopathologic studies of human RP retinas were performed in tissue from donors with unknown genetic mutations and involved only descriptive studies [9–18]. The next group of manuscripts utilized immunocytochemistry assays to analyze the effects of the disease on photoreceptors and the other retinal cells [19–24]. A few recent studies reported histopathologic findings from RP patients with different known gene mutations, including rhodopsin (RHO) [21,25–29], pre-mRNA processing factor 8 (PRPC8) [30], retinitis pigmentosa GTPase regulator (RPGR) [31,32], ATP-binding cassette, sub-family A, member 4 (ABCA4) [33]. Such types of studies are crucial to understand how these genetic mutations lead to dysfunction and photoreceptor cell death in RP patients.
Here we report the ocular histopathology in eyes from three arRP donors with mutations in EYS. We focus on retinal pathology changes and the effect of the disease on the distribution of photoreceptors and other retinal cells. Eye donor 1 carries two heterozygous novel truncating EYS gene mutations and the related eye donors 2 and 3 have two heterozygous EYS deletion mutations. Given the limited molecular characterization of affected individuals whose eyes have been made available for postmortem examination, the opportunity to combine genetic and phenotypic findings is unprecedented.
METHODS
Tissue acquisition and fixation:
Donor eyes were obtained through the Foundation Fighting Blindness (FFB) Eye Donor Program (Columbia, MD). Immunocytochemical analysis was performed with the approval of the Cleveland Clinic Institutional Review Board (IRB #14-057). The research adhered to the tenets of the Declaration of Helsinki. The analyzed tissue included FFB donations #228, 649, 696, 789, 870, 923 and 937. Eyes were obtained from a 72 year-old female (donor 1, family 1, II-4, FFB #923), a 97 year-old female (donor 2, family 2, II-1, FFB #937) and her 91 year-old sister (donor 3, family 2, II-2, FFB #870) (Fig. 1a and 1b). The asymptomatic carrier mother (donor 4, family 1, I-2, FFB #228) of donor 1 was also evaluated (Figure 1a and 1b). Eyes were enucleated 6 hours postmortem and fixed in 4% paraformaldehyde (PF) and 0.5% glutaraldehyde in phosphate buffer. The globes were stored in 2% paraformaldehyde in PBS. Postmortem eyes from 61, 65 and 88 year-old donors without a history of retinal disease were used as controls.
Fig. 1. Mutational Analysis of individuals with arRP due to EYS mutations.
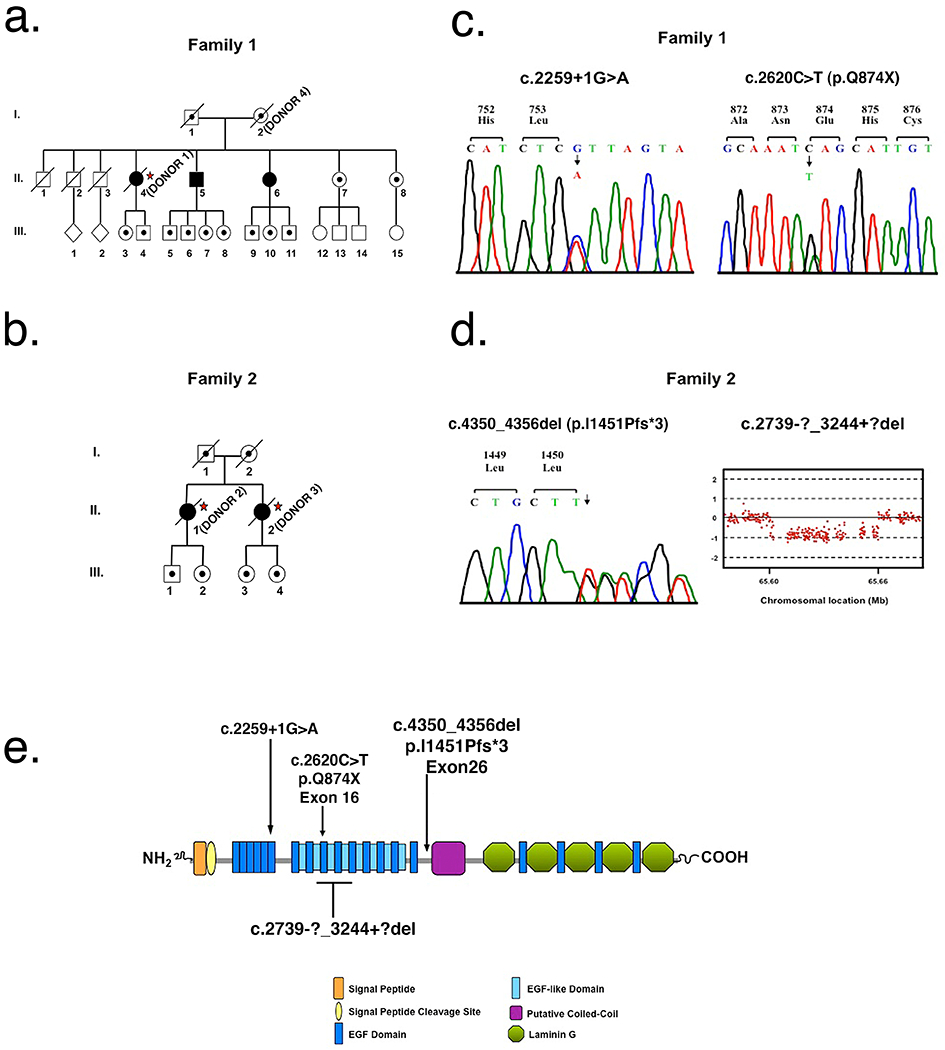
a Pedigree of family 1. Slashed symbols reflect deceased family members. Affected family members are shown with filled symbols, unaffected family members are shown with unfilled symbols and unaffected carrier family members are shown with unfilled symbols with a black dot inside. Postmortem analysis was done on affected member II-4 referred to as donor 1 (*) and in their unaffected mother (I-2) referred to as donor 4 (*). b Pedigree of family 2. Postmortem analysis was done on affected members II-1 and II-2 referred to as donors 2 (*) and 3 (*), respectively. c Sequence analysis of family 1 identified two heterozygous EYS mutations, c.2259+1G>A and p.Q874X. DNA analysis was performed on all three affected members (II-4, II-5 and II-6), their two unaffected living sisters (II-7 and II-8), and their mother (I-2). d Sequence analysis of family 2 identified a heterozygous 7 base pair deletion in EYS, p.I1451Pfs*3. Comparative genomic hybridization identified a heterozygous deletion of exons 15 – 18, c.2739-?_3244+?del. DNA analysis was performed in two affected family members, II-1 and II-2. e Predicted domain structure and distribution of the four EYS mutations identified in this study.
Genetic analysis:
Approximately 10 ml of peripheral blood was collected from donor 1 (family member II-4) and family members II-5, II-6, II-7, II-8 from family 1. Blood was also collected from donor 2 (II-1) of family 2. DNA was extracted and purified from leukocytes by means of the Gentra Systems PUREGENE DNA Purification Kit (Qiagen). DNA for donors 3 (II-2, family 2) and 4 (I-2, family 1) was extracted from fixed or frozen retinal tissue samples using the Gentra Systems PUREGENE DNA Purification Kit (Qiagen).
In family 1, direct testing for mutations in 13 arRP genes (VMD2, CNGA1, CNGB1, CRB1, EYS, LRAT, NR2E3, NRL, PDE6A, PDE6B, RHO, RPE65, USH2A) was performed on patient II-5 by PCR amplification and DNA sequencing in two directions of all coding exons and exon/intron boundaries including at least 50 nucleotides into the intron. Two novel heterozygous mutations were identified in EYS (GenBank RefSeq NM_001142800.1), c.2259+1G>A and c.2620C>T (p.Q874X). Segregation analysis was performed using sequence analysis in all available family members.
In family 2, DNA from patient II-1 (donor 2) was evaluated by Asper Ophthalmics using an APEX (arrayed primer extension) based test. 710 mutations in 28 arRP genes (CERKL, CNGA1, CNGB1, MERTK, PDE6A, PDE6B, PNR, RDH12, RGR, RLBP1, SAG, TULP1, CRB, RPE65, USH2A, USH3A, LRAT, PROML1, PBP3, EYS, ABCA4, AIPL1, CNGA3, CNGB3, GRK1, IMPG2, RHO, and RP1) were evaluated. One heterozygous mutation in EYS was identified, a 7 base pair deletion notated as c.4350_4356del (p.I1451Pfs*3). In an attempt to identify the second pathogenic mutation, PCR products corresponding to the complete EYS coding sequence were amplified from genomic DNA and evaluated by sequence analysis. No mutation was identified. To further search for the second pathogenic variation, the DNA was analyzed using a comparative genomic hybridization array (Oxford Gene Technology; Eye gene array v2). Array data was analyzed by using CytoSure software. A heterozygous midsize genomic rearrangement, c.2739-?_3244+?del, was identified. To further search for the second pathogenic variation, the DNA was analyzed using a comparative genomic hybridization array (Oxford Gene Technology; Eye gene array v2). Array data was analyzed by using CytoSure software. A heterozygous midsize genomic rearrangement, c.2739-?_3244+?del, was identified. The four exon deletion was confirmed by qPCR analysis using primers Hs06795327 and Hs06137405 (Life Technologies).
Fundus Imaging:
Macroscopic fundus images were collected using a Zeiss AxioCam MRC5 camera equipped with a macro zoom lens (Zoom 700 Navitar TV Lens, Navitar, Inc, Rochester, NY) and AxioVision AC Software (Carl Zeiss MicroImaging, Inc.). Prior to imaging, the anterior segment of each eye was removed using a custom-made eye globe holder and microtome blade (AccuEdge 4685, Sakura Finetek USA, Inc. Torrence, CA). The remaining posterior segment was transferred to a custom-made plexiglass chamber, which permitted containment of both the sample and PBS immersion fluids required for imaging. Within the plexiglass chamber, the sample was placed with the posterior pole oriented downward and the opened anterior segment directed upward. The plexiglass chamber was then filled with PBS to the level of the anterior segment opening. In order to eliminate specular reflections from vitreous interface several drops of PBS were dispensed into the open eye cup using a pipette. Angled illumination from a bifurcated fiber optic light source (Fostec EJA, Schott North America, Inc., Elmsford, NY) was used to illuminate the sample for image acquisition.
Scanning Laser Ophthalmoscope (SLO):
SLO images were collected using a model HRA2 confocal scanning laser ophthalmoscope (Heidelberg Engineering, Inc.) in the same manner as the macroscopic fundus images. The HRA2 was rotated 90° so that the scan direction was perpendicular to the table surface. The system was operated in high-resolution mode, which provides an image pixel format of 1536 x 1536 when used with a 55° wide-field objective lens. SLO images of the posterior pole were collected using infrared reflectance (SLO-IR), infrared dark field (SLO-IRDF), autofluorescence (SLO-AF), and red free dark field (SLO-RFDF) imaging modes at field of view (FOV) settings of 55°, 35°, and 25°. Online algorithms within the HRA2 system software enabled automatic real-time averaging and tracking of sequentially collected images. This parameter was preset for averaging 25 image frames, which further enhanced signal-to-noise ratio and image contrast, especially for autofluorescence (SLO-AF) mode.
Spectral Domain Optical Coherence Tomography (SD-OCT):
SD-OCT images were collected using an SD-OCT system (Model SDOIS, Bioptigen, Inc.) in a manner similar to the aforementioned macroscopic and SLO imaging modalities. A single telecentric objective lens was employed to collect 5x5mm and 10x10mm FOV of the posterior pole. The SDOIS system has a center operating wavelength of ~840nm, spectral bandwidth of 65nm and axial, in-depth resolution of ~6-7μm (in air). A 1mm ruby sphere was placed on the optic nerve head to provide a reference scale. SD-OCT imaging was performed using the following scan parameters (1) 5mm linear scan of the horizontal meridian through the optic nerve and fovea @ 1000 A-scans/B-scan, (2) 10mm linear scan of the horizontal meridian through the optic nerve and fovea @ 1000 A-scans/B-scan, (3) 5mm2 volume scan of the posterior pole @ 500 B-scans/volume x 250 A-scans/B-scan, and (4) 10mm2 volume scan of the posterior pole @ 500 B-scans/volume x 250 A-scans/B-scan. Post-acquisition images were averaged with a line and frame filter settings of 3 and 3 respectively via the Bioptigen InVivoVue SDOIS Software version 1.7.
Semi-thin Epon sections and morphological analysis:
A small area of the retina/RPE/choroid complex from both the perifoveal and peripheral regions of the arRP donors, asymptomatic mother and controls were fixed in 2.5% glutaraldehyde in 0.1M cacodylate buffer, sequentially dehydrated in ethanol and embedded in Epon. Plastic sections (1 μm) of all samples were stained with toluidine blue and photographed by light microscopy with a Zeiss Imager Z.1 microcroscope equipped with a Zeiss AxioCam MRC5 camera.
Immunohistochemistry:
Small areas from the perifovea and peripheral eye wall were cut and infused with 10% and 20% sucrose in PBS, and embedded in Tissue-Tek “4583” (Miles Inc., Elkhart, IN). 10 μm cryosections were cut on a HM 505E cryostat (Microm, Walldorf, Germany) equipped with a CryoJane Tape-Transfer system (Instrumedics, Inc., Hackensack, NJ). Prior to labeling, embedding medium was removed through two consecutive PBS incubations for 20 min. The tissue was then processed for immunofluorescence labeling. Sections were blocked in PBS supplemented with 1% BSA for 30 min and incubated with monoclonal antibodies 1D4 to rhodopsin (ab5417, 1:1000, from Abcam), 7G6 to cone arrestin (1:100, from Dr. P. MacLeish, Morehouse School of Medicine, Atlanta, GA), and polyclonal antibodies to red/green cone opsin (AB5405, 1:1200, Chemicon) in PBS/BSA overnight at 4°C. Cell nuclei were labeled with TO-PRO®-3 iodide (blue, 1mg/ml, Molecular Probes, Eugene, OR). Sections were then labeled with secondary antibodies goat anti-mouse IgG Alexa Fluor 488 (1:1000; Molecular Probes), goat anti-rabbit IgG Alexa Fluor 594 (1:1000; Molecular Probes) and goat anti-rabbit IgG Alexa Fluor 488 (1:1000; Molecular Probes) for 1h at room temperature. Sections were analyzed using a Leica laser scanning confocal microscope (TCS-SP2, Leica, Exton, PA). A series of 1 μm xy (en face) sections were collected. Each individual xy image of the retinas stained represents a three-dimensional projection of the entire cryosection (sum of all images in the stack). Microscopic panels were composed using AdobePhotoshop CS3 (Adobe, San Jose, CA). Two different commercially available EYS antibodies were tested but filed to react with our tissue probably due to the presence of glutaraldehyde in the fixative buffer.
Results
Clinical Findings
Two unrelated families were evaluated. Family 1 reported Irish and French ancestry whereas family 2 is Scandinavian. Donor 1 (family 1, II-4) was most recently examined at age 70, two years before her death (Fig. 1a, star). At that time, her visual acuity was hand motion OD and 20/400 OS. Dilated fundus examination of both eyes showed a pale optic nerve head, attenuated vessels, and bone spicule pigment in the periphery with pavingstone degeneration inferiorly. She also had some mottled RPE pigmentation in her left eye. No other clinical or imaging information was available. Donor 2 (family 2, II-1) was last examined at age 96, one year before her death (Fig. 1b). At that time her visual acuity was 20/400 OU. Dilated fundus exam showed optic disc atrophy, choroidal atrophy, vascular attenuation, and chorioretinal degeneration. Donor 3 (family 2, II-2) was last examined at age 88, three years prior to her death (Fig. 1b). She had light perception vision in both eyes. Her previous fundus examinations showed the typical triad of characteristics indicative of RP – optic nerve head pallor, attenuated retinal vessels and bone spicule pigmentation in the periphery. Clinical records were not available for donor 4 (family 1, I-2) (Fig. 1a).
Molecular Genetic Analysis
In family 1, genetic analysis was performed on the affected donor 1 (II-4), her unaffected mother (donor 4, I-2), two affected siblings (II-5 and II-6) and two unaffected siblings (II-7 and II-8) (Fig. 1a). DNA sequence analysis in comparison to GenBank entry NM_001142800 revealed two novel heterozygous mutations in EYS, c.2259+1G>A and c.2620C>T (p.Q874X), in patient II-5 (Fig. 1c). Segregation analysis revealed that both affected sisters, II-4 (donor 1) and II-6, carried both mutations; while the two unaffected sisters were both heterozygous for the c.2259+1G>A mutation.
In family 2, genetic analysis was performed on affected donor 2 (II-1) (Fig. 1b). DNA analysis revealed a heterozygous 7 bp deletion in EYS, c.4350_4356del (p.I1451Pfs*3), and comparative genomic hybridization revealed an additional deletion of exons 15 – 18, c.2739-?_3244+?del (Fig. 1d). Targeted sequence analysis in patient II-2 (donor 3) revealed that she was also heterozygous for p.I1451Pfs*3 (Fig. 1b). Insufficient DNA from donor 3 prevented the performance of midsize deletion analysis. The locations of these identified mutations relative to the predicted EYS protein domain structure are shown in Fig. 1e.
Ex-vivo Imaging of Donor Eyes
The combination of fundus, confocal scanning laser ophthalmoscopy (SLO) and spectral-domain optical coherence tomography (SD-OCT) imaging systems can provide comprehensive characterization of retinal lesions prior to histopathology 34. Therefore, these imaging techniques were performed on all arRP and control eye donors and qualitatively compared (Figs. 2 and 3). Anatomical landmarks, such as the optic nerve and fovea, were clearly identified in each donor eye using all three imaging modalities.
Fig. 2. Ex-vivo imaging of arRP donor eyes with EYS mutations.
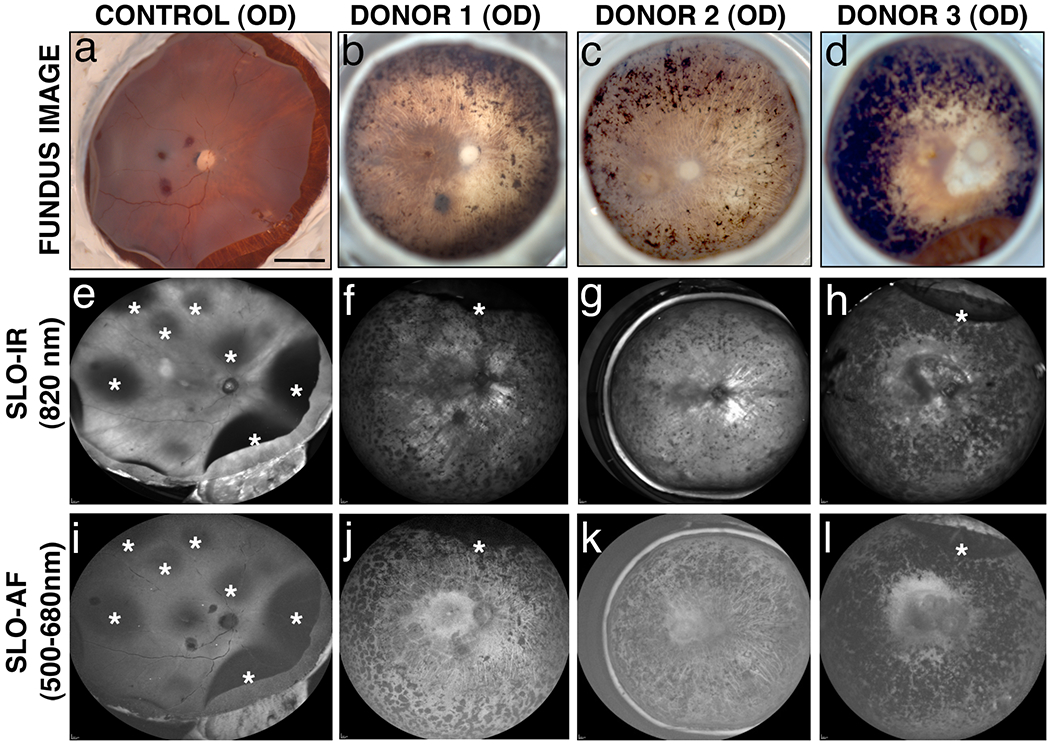
Fundus images (a-d) and SLO images (e-l) were collected from donor 1, 2 and 3 and an age-similar control. In the control eye, detached retinas are apparent with all imaging modalities (a, e, i, *). In all three arRP eyes, fundus images reveal bone spicule pigment in mid-peripheral and peripheral areas to varying degrees (b-d). SLO-IR imaging identified degeneration in the entire posterior pole region including the macula, perimacula and areas surrounding the optic nerve due to focal loss of RPE in donors 2 (g) and 3 (h) as compared to an age-matched control eye (e). SLO-AF imaging identified hypofluorescence in one contiguous region involving the macula and area surrounding the optic disk of donor 3 (l) as opposed to the individually demarcated and isolated regions seen with both donors 1 (j) and 2 (k) and the control eye (i). Scale bars in fundus image = 0.5 cm.
Fig. 3. Ex-vivo OCT imaging of arRP donor eyes with EYS mutations.
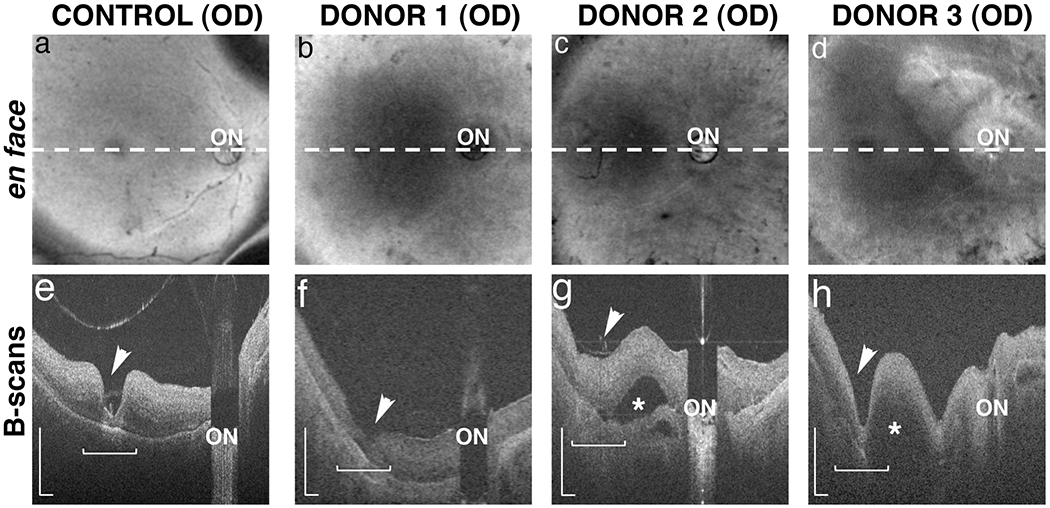
OCT images were collected from donors 1, 2 and 3 and an age-matched control. En face images reveal the location (dashed lines) of the in-depth, B-scan images of control (a) and arRP donors 1 (b), 2 (c) and 3 (d). The fovea (arrowhead) and optic nerve (ON) were identified in all donor eyes. In-depth B-scans from the control eye (e) revealed a normal appearing retina with clearly defined fovea, some evidence of laminar architecture, and no appreciable evidence of retinal thinning or degeneration. Images from the arRP donors (f-h) revealed retinas of appreciable thickness but with less organized architecture and integrity in the macular region (horizontal bracket). In contrast, the perimacular regions showed some evidence of thinned retina relative to the control retina suggesting degeneration. Donor’s 2 (g) and 3 (h) had macular (*) and choroidal (arrow) detachments as indicated in the B-scans. B-scan scale is 0.5mm.
Visible light fundus imaging showed inherent differences between the control and arRP eyes that included the presence or absence of peripheral bone spicule pigment and inherent differences in the overall color hue of the fundus. Bone spicule pigment density was more pronounced in donor 3 (Fig. 2d) than in donors 1 and 2 (Fig. 2b and 2c). The fundi of arRP donor eyes had hues that were similar in appearance, but were more “whitish” in color (Fig. 2b to 2d) than the control eye (Fig. 2a). Optic disc pallor, attenuated retinal vessels, and profound atrophy of the RPE and choriocapillaris were only observed in arRP eyes.
Examination by SLO revealed areas of detached retina in the control eye (Fig. 2e and 2i, asterisks) and in the majority (2/3) of arRP eyes (Figs. 2f, 2h, 2j, 2l, asterisks); the retinal detachments were attributed to post-mortem enucleation and removal of anterior segments in preparation for imaging. In arRP eyes, both SLO infrared (SLO-IR) and autofluorescence (SLO-AF) showed bone spicule pigment in agreement with fundus macroscopic imaging. SLO-IR imaging revealed loss of RPE in the posterior pole including the macula, perimacula and areas surrounding the optic nerve in the eyes of donors 2 and 3 (Fig. 2g and 2h) as compared to control (Fig. 2e). SLO-AF imaging identified hypofluorescence in one contiguous region involving the macula and area surrounding the optic disk of donor 3 (Fig. 2l) as opposed to the focal loss of RPE in demarcated regions in eyes from donors 1 and 2 (Fig. 2j and 2k), and an absence of both abnormal findings in the control retina (Fig. 2i). These data suggest that different EYS mutations in arRP donors lead to retinal changes that can be observed using multiple imaging modalities.
SD-OCT imaging was performed on each of the arRP and control eyes (Fig. 3). In-depth B-scans from the control eye (Fig. 3a and 3e) revealed a normal-appearing retina with clearly defined fovea, some evidence of laminar architecture, and no appreciable evidence of retinal thinning and/or degeneration. Images from arRP donors revealed retinas of appreciable thickness but with less organized architecture and integrity in the macular region (Fig. 3b–d and 3f–h, bracket). In contrast, the peripheral regions showed a retina that was thinner than control eyes, suggesting loss of cells and axons (data not shown). Eyes from donors 2 and 3 had macular detachments between the optic nerve and fovea (Fig. 3g and 3h, asterisk) that were not previously identified by either fundus macroscopic imaging or SLO techniques. Donor 2’s eye also showed what appeared to be a choroidal detachment adjacent to the optic nerve and below the region of retinal detachment (Fig. 3g, arrow). These data suggest that arRP EYS mutations lead to the atrophy of the retinas in addition to substantial disorganization of the cellular layers in the macular and perifoveal regions.
Histopathology of Donor Retinas
To evaluate the effect of EYS mutations on retinal structure, toluidine blue-stained plastic sections of arRP and control donor eyes were examined. A schematic drawing depicts the regions harvested and processed for observation in both the morphological and immunohistological assays (Fig. 4a). These include: the periphery (Fig. 4a, region 1) and perifovea (Fig. 4a, region 2). Histology of control retinas in the periphery (Fig. 4b) and perifovea (Fig. 4f) showed normal lamination patterns. In all three arRP donors, a highly degenerated retina with disorganization of the retinal laminae and gliosis was evident in all peripheral areas analyzed (Fig. 4c–e). Moreover, photoreceptor outer segments were also absent in all areas analyzed. Intraretinal bone spicule pigment was visible in the retinas of donors 2 (Fig. 4d, arrow) and 3 (Fig. 4e, arrow). The perifovea displayed a prominent inner nuclear layer. Eyes from donors 2 (Fig. 4h) and 3 (Fig. 4i) displayed localized areas of RPE atrophy in the perifovea whereas the RPE was uniformly thin in the perifovea of donor 1 (Fig. 4g). Patchy, disorganized cones were observed subjacent to the RPE in the perifovea of donors 2 (Fig. 4h, arrowheads) and 3 (Fig. 4i, arrowheads). These data suggest that the EYS mutations cause profound changes in the retina leading to the loss of several retinal cell layers and the appearance of gliosis.
Fig. 4. Histology of arRP donor eyes with EYS mutations.
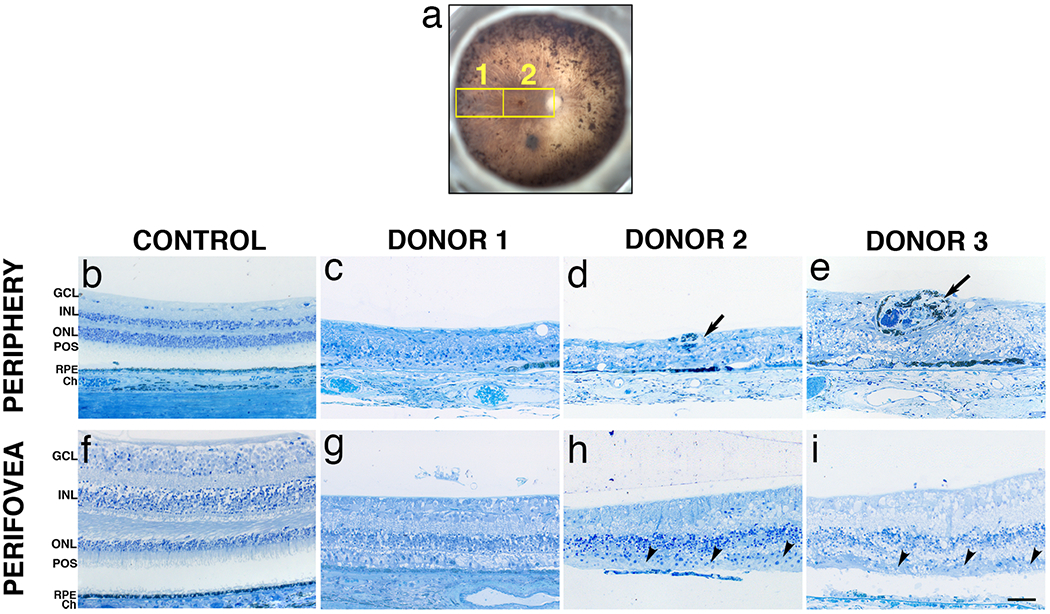
A fundus image of the studied eye (OS) with a schematic drawing of the regions cut and processed for cryosectioning and immunolabeling is shown (a). Toluidine blue-stained plastic sections (1μm) of retinas from donors 1, 2 and 3 and an age-matched control. Morphology of the control retina in the periphery (b) and perifoveal (f) regions displayed typical retinal lamina. Histology of the periphery of all three donors revealed a highly degenerated retina with disorganization of the lamina and cellular layers and gliosis in all areas analyzed (c-e). Photoreceptor outer segments were also absent in all areas analyzed. Intraretinal bone spicule pigments were visible in the retinas of donors 2 (d, arrow) and 3 (e, arrow). In contrast, the perifovea of all arRP donors displayed a prominent inner nuclear layer. Donors 2 (h) and 3 (i) displayed localized areas of RPE atrophy whereas the RPE was thin in the perifovea of donor 1 (g) as compared to the control donor (f). Patchy, disorganized cone remnants were observed on top of the RPE in the perifovea of donors 2 (h, arrowhead) and 3 (i, arrowhead). GCL= ganglion cell layer; INL= inner nuclear layer; ONL= outer nuclear layer; POS = photoreceptor outer segments; RPE= retinal pigment epithelium; Ch = Choroid. Scale bar =50μm.
Immunohistochemistry of Donor Retinas
Human EYS protein is localized to the photoreceptor outer segments and is believed to play a role in the modeling of retinal architecture 8. In order to investigate if mutations in EYS affect the retinal architecture, immunofluorescent studies with antibodies to photoreceptor outer segment proteins were performed. Immunostaining of control retinas with rhodopsin antibodies revealed that rhodopsin distribution was restricted to the rod outer segments in the retinal periphery (Fig. 5a) and perifovea (Fig. 5e). Rhodopsin-labeled cells were rarely encountered in the retinal periphery of donor 1 (Fig. 5b) while some were still present in the periphery of donors 2 (Fig. 5c) and 3 (Fig. 5d). Rhodopsin-positive cells were also observed along the outer retina directly abutting the choroid in some areas where the RPE was absent (Fig. 5c and 5d, arrows). In addition, rhodopsin was not restricted to the outer segments in all the arRP donor retinas and rhodopsin-positive cells had lost their slim, rod-shaped morphology. In the perifovea of donors 1 (Fig. 5f) and 2 (Fig. 5g), no rhodopsin-labeled cells were found while donor 3 (Fig. 5h) displayed several disorganized rhodopsin-labeled cells. Although retinas have reached an advanced stage of degeneration, our data suggest that the EYS mutations identified in arRP donors have an effect on the subcellular localization of rhodopsin and the distribution of rhodopsin-positive cells retinas.
Fig. 5. Immunocytochemistry of arRP retinal sections with EYS mutations stained with rhodopsin antibodies.
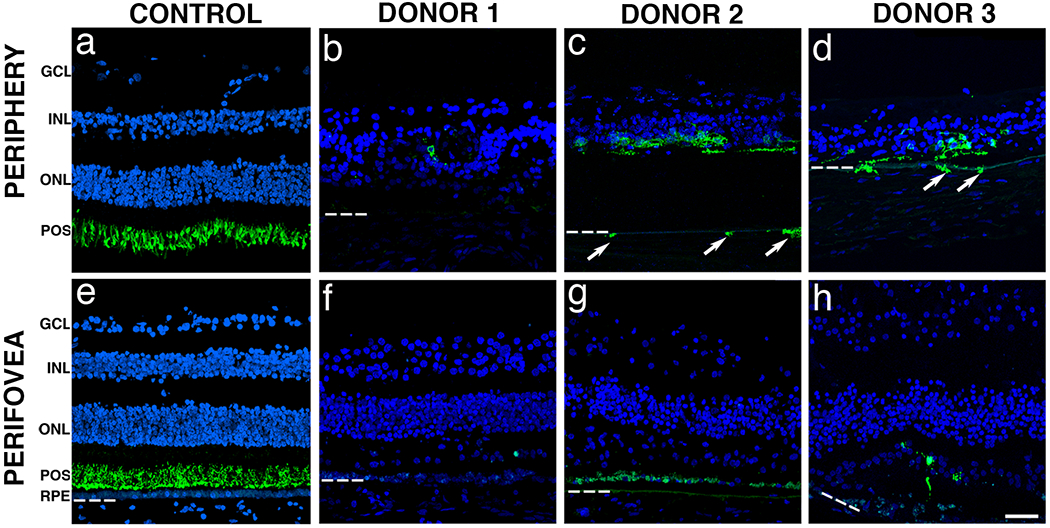
Immunofluorescence of arRP retinal sections labeled with antibodies to rhodopsin (green) showed significantly decreased staining when compared to control. The control retina showed that rhodopsin was restricted to the rod outer segments in both the periphery (a) and perifovea (e). Rhodopsin-labeled cells were essentially absent from the periphery of donor 1 (e). However, rhodopsin-labeled cells were still present in the periphery of donors 2 (c) and 3 (d). Of interest, some rhodopsin-labeled cells were observed in the choroid of these eyes (arrows). In the perifovea, donors 1 (f) and 2 (g) displayed no rods while donor 3 (h) displayed a few disorganized rods. Bruch’s membrane is indicated by hashed white line. Nuclei were labeled with TO-PRO-3. GCL= ganglion cell layer; INL= inner nuclear layer; ONL= outer nuclear layer; POS = photoreceptor outer segments; RPE= retinal pigment epithelium. Scale bar = 40μm.
Next we investigated the distribution of cone photoreceptors in retinas harboring EYS mutations. Immunostaining of control retinas with cone arrestin antibodies revealed that the protein was distributed along the entire conical-shaped plasma membrane, from the tip of the outer segment to the synaptic base both in the retinal periphery (Fig. 6a, green) and in the perifovea (Fig. 6e, green). Immunostaining of control retinas with antibodies against red-green cone opsin revealed that this protein is restricted to cone outer segments both in the periphery (Fig. 6a, red) and perifovea (Fig. 6e, red). Strikingly, cones were mostly absent from the periphery of all the arRP donor retinas (Fig. 6b–d). However, in the perifovea of all arRP donors, cone arrestin staining displayed the typical conical-shaped cellular distribution with lack of synaptic terminals and outer segments (Fig. 6e–h). These cone arrestin-positive cells were also characterized by the redistribution of red-green opsin to the inner segments and cell bodies. In the perifovea of donor 1, red-green opsin labeling was significantly decreased and the cone arrestin distribution was mostly confined to a row of shortened cells (Fig. 6f). Interestingly, the perifovea from both donors 2 (Fig. 6g) and 3 (Fig. 6h) displayed cone arrestin-labeled cells concentrated in areas that still had some RPE cells. These cones displayed a distribution of red-green opsin through the entire cell body. These data suggest that EYS mutations cause a profound loss of peripheral cones and can affect the subcellular localization of cone opsin- and cone arrestin-positive cells in the few perifoveal cones that survive.
Fig. 6. Immunocytochemistry of arRP retinal sections with EYS mutations stained with cone-specific antibodies.
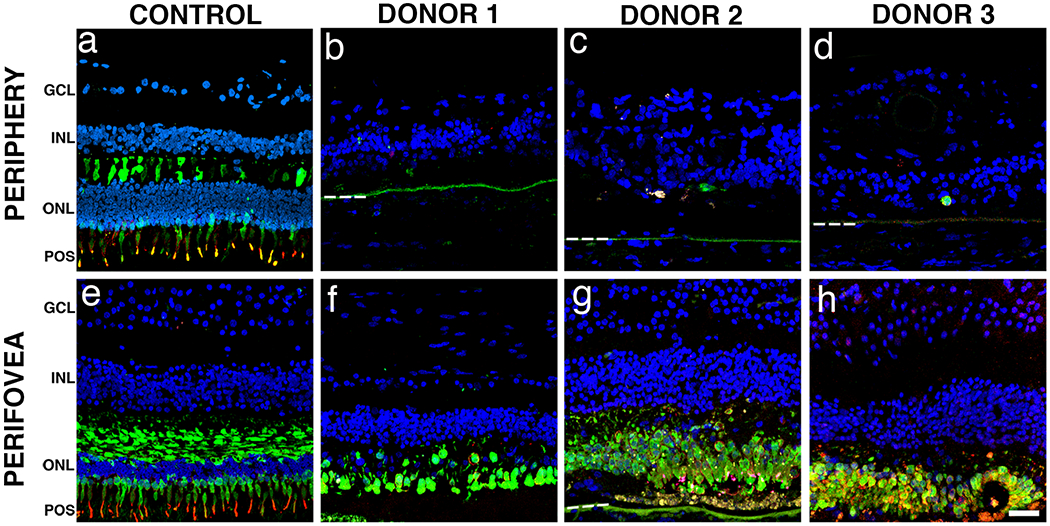
Immunofluorescence of arRP retinal sections labeled with antibodies to cone arrestin (green) and red/green cone opsin (red) showed significantly decreased staining in the periphery when compared to control. In the peripheral (a) and perifoveal (e) regions of the control retina, cone arrestin was distributed along the entire plasma membrane, from the tip of the outer segment to the synaptic base, while the red/green cone opsin was restricted to the outer segments. Cone-specific labeled cells were mostly absent from the periphery of all three arRP donor retinas (b-d). In contrast, cone-specific labeled cells were present but highly disorganized in the perifovea of donor 1 (f). Interestingly, the perifovea from both donor 2 (g) and 3 (h) displayed disorganized cone-specific labeled cells concentrated in the areas that still maintained RPE. Bruch’s membrane is indicated by hashed white line. Nuclei were labeled with TO-PRO-3. GCL= ganglion cell layer; INL= inner nuclear layer; ONL= outer nuclear layer; POS = photoreceptor outer segments. Scale bar = 40μm.
Histology and Immunocytochemistry of the Unaffected Eye from the Mother of Donor 1
The retinal periphery of the unaffected mother of donor 1 (donor 4) revealed typical retinal lamination similar to an age-matched control; however, a significant decrease in the number of nuclei of the inner and outer nuclear layers was noted (Fig. 7b and 7d). In addition, donor 4 displayed both rhodopsin (Fig. 7b, green) and red-green cone opsin (Fig. 7d, red) distribution restricted to the photoreceptor outer segments as observed in the control retinas (Fig. 7a and 7c). Cone arrestin was also distributed throughout the entire cell in donor 4 (Fig. 7d, green) similar to control retinas (Fig. 7c). These data suggest that the unnafected mother of the arRP donor 1, carrying an obligatory heterozygous EYS mutation, does not display any significant changes in retinal structure and is similar to age-similar normal controls.
Fig. 7. Immunocytochemistry of retinal sections of an unaffected carrier with a heterozygous EYS mutation.
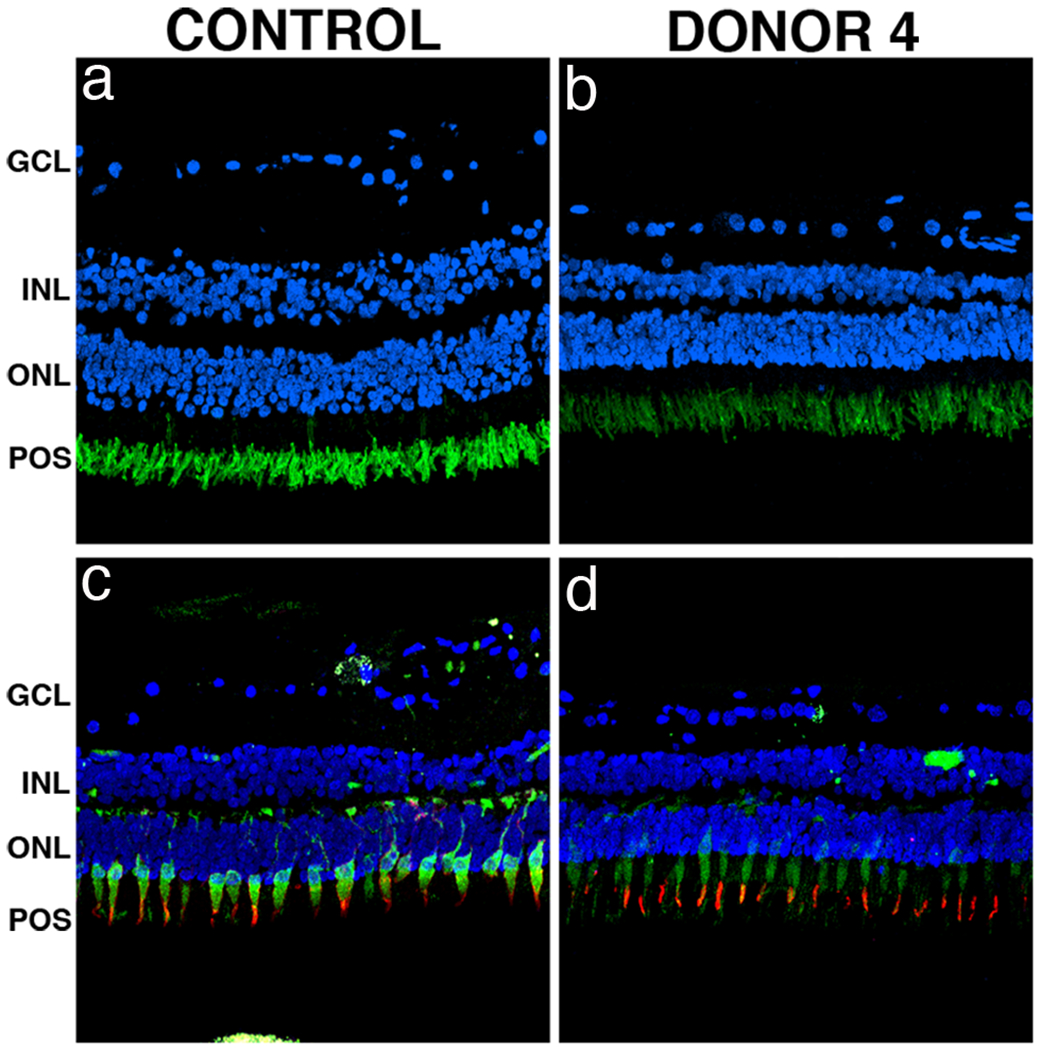
Immunofluorescence of peripheral retina sections of the unaffected mother of donor 1 (donor 4) labeled with antibodies to rhodopsin, cone arrestin and red/green cone opsin showed no significant changes in staining of these proteins as compared to control. The unaffected carrier donor retina displayed both rhodopsin (b, green) and red/green cone opsin (d, red) restricted to the photoreceptor outer segments as observed in control retinas (a, c). Cone arrestin labeling in the unaffected carrier donor retina (d, green) displayed similar distribution when compared to the control retina (c). Nuclei were labeled with TO-PRO-3. GCL= ganglion cell layer; INL= inner nuclear layer; ONL= outer nuclear layer; POS = photoreceptor outer segments. Scale bar = 40μm.
DISCUSSION
The examination of postmortem eye tissues obtained from individuals in whom the disease- causing gene has been identified offers a unique opportunity to study the relationship between genotype and disease pathogenesis and the effects of the genetic defect on retinal structures. There continues to be a relative lack of the combination of genetic information with clinical and histopathological data from the same individual. We present for the first time the ocular histopathological changes in two arRP families with different, novel mutations in the EYS gene. We compare the relationship between genotype and disease phenotype in these eyes.
The gene responsible for arRP at the RP25 locus was identified several years ago as EYS (an ortholog of Drosophila eyes shut, eys), and encodes an ortholog of the Drosophila protein SPAM [7], a multi-domain protein containing 3,165 amino acids and comprised of several protein motifs commonly found in extracellular molecules (Fig. 1e). EYS consists of 21 epidermal growth factor (EGF)-like domains in its N-terminal region followed by five C-terminal laminin G domains that are interspersed with additional EGF repeats. The protein is also predicted to contain a signal peptide for secretion into the extracellular environment. Drosophila spam is expressed in the eye of several insect species with an open rhabdom system, wherein the rhabdomeres or photoreceptor cells of each ommatidium in the compound eye are separated from each other allowing each photoreceptor to act as an independent unit. Loss of EYS switches an open rhabdom system to a more primitive closed one found in insects such as ants and bees [7]. Considering the evolutionary data and the known function of the Drosophila ortholog, human EYS is likely to have a role in the integrity of photoreceptor architecture. Indeed, immunohistochemical studies confirmed the localization of this protein to the photoreceptor outer segments [7]. Interestingly, despite the reported mutations and the presumed loss of function leading to arRP, EYS is not present in rodent eyes making this gene the fourth mendelian disease–associated human gene whose orthologs are disrupted or absent from rodent genomes [7].
EYS is a major arRP disease-causing gene. Mutations have been identified in families of different ancestral origin and are estimated to account for 5 – 16% of arRP cases [6]. The types of mutations identified in patients include missense, nonsense, microdeletions and insertions, 5’UTR variations and copy-number variations such as midsize genomic rearrangements. In the present study, we performed a comprehensive comparison of four eye donors (3 affected and one carrier) from two unrelated families with EYS mutations, including clinical, genetic and morphologic analyses. The heterozygous EYS mutations c.2259+1G>A and c.2620C>T were identified in family 1, whereas the heterozygous mutations p.I1451Pfs*3 and c.2739-?_3244+?del were identified in family 2. The two mutations in family 1 are novel and have not been reported in previous studies in patients of different ethnicities [6,35–43]. On the other hand, the deletions identified in family 2 have been previously reported [6,35,36,41,42]. All four mutations described in the present report are considered pathogenic. Two of them (c.2259+1G>A and p.Q874X) are located within EGF domains and are predicted to have a truncating effect on the protein. p.I1451Pfs*3 is predicted to lead to a premature stop codon three amino acids downstream and likely truncates the last half of the protein including the coiled coil domain and all laminin G domains. c.2739-?_3244+?del is predicted to lead to a loss of four exons which deletes a portion of the EGF domains. In family 1, it is likely that the altered mRNA transcripts would be degraded through nonsense mediated decay. In family 2, it is possible that the mRNA from the midsize four-exon deletion is used for protein translation and leads to an abnormal yet partially functional protein product.
Clinically, patients carrying pathogenic EYS mutations have night blindness as the initial symptom, as well as retinal bone spicule pigmentation and attenuated retinal vessels, followed by progressive visual field constriction. On average, visual acuity begins to decrease at around 30 years of age and continues to deteriorate over the next few decades [6,36,43]. The phenotype in patients with RP due to EYS mutations has been described previously and is relatively homogeneous [6,38,44,45]. Generally, they exhibit markedly reduced scotopic and photopic responses on ERG; have retinal thinning with macular involvement on OCT; and a fundus appearance typical for RP. Overall, patients with EYS mutations have a more severe clinical course compared to some of the other RP genotypes. Central vision is relatively preserved until late in the disease course.
In the present study of eyes from older individuals with EYS mutations, both imaging and histological examinations revealed highly degenerated retinas with little evidence of stratified nuclear layers in the periphery. In contrast, the perifoveal region maintained a prominent inner nuclear layer. Donors 2 (age 97) and 3 (age 91) from family 2 had a few areas of preserved RPE in both the periphery and perifovea, whereas the RPE was thin in the perifovea and absent in the periphery of donor 1 (age 72). Rods were mostly absent in all arRP donors, except in the periphery where the cells appeared morphologically abnormal. Cones were also mostly absent from the periphery of all arRP donors; however, patches of cones were observed in the perifovea of donors 2 and 3. This preservation of cones in the perifovea is likely responsible for the central vision that some patients maintain until late in the course of the disease.
It is important to note that the donor ages differed by 20 – 25 years, suggesting that different stages of the disease may be represented in the tissues studied. Interestingly, the disease pathology of the older aged donors from family 2 appeared to be less severe. We speculate that the retinas from donors 2 and 3 would have had better morphology than the retina from donor 1 if they would have been evaluated at a similar age. It is possible that the two different types of mutations represented in these donors may show specific differences in the rate of retinal degeneration but this is beyond what can be inferred by the end-stage pathology of limited specimens.
The data presented here provides new insights into the pathology and disease manifestation caused by EYS mutations. Previous studies have shown that EYS mutations lead to severe defects in inter-rhabdomeral space formation in Drosophila eyes [8]. As a result, the human EYS protein has been hypothesized to be essential for the formation and integrity of the photoreceptor outer segment and overall retinal architecture. We show that combinations of four different EYS mutations lead to advanced retinal degeneration with near-total loss of rods. The preservation of macular cones in arRP donor retinas with a midsize exonic deletion may indicate an ability for individuals with this type of mutation to maintain some central vision for a longer duration than individuals with truncating mutations and despite severe visual dysfunction, suggests an opportunity for cell rescue if therapy becomes available. This observation is significant due to the estimation that midsize genomic rearrangements in EYS are responsible for the disease in ~4% of arRP cases in the absence of other mutations, and constitute the second pathogenic variation in ~15% of cases where a second mutation is detected by sequence analysis [41]. Further experimental research will be required to corroborate this possible association. The present study demonstrates that the identification of disease-causing mutations combined with the histopathological analyses of donor eyes provides a more comprehensive platform for the study of the natural history of retinal dystrophies, and possible predictions of the clinical course of the disease. These observations undoubtedly help genetic counseling as well as future targeted therapies.
ACKNOWLEDGEMENTS
The authors thank Dr. Peter MacLeish (Morehouse School of Medicine, Atlanta, GA) for providing us with the antibody to cone arrestin (7G6) and Xiaoping Yang for expert technical assistance. Supported by The Foundation Fighting Blindness Histopathology Grant F-OH01-1102-0231 (JGH), Research Center Grants from The Foundation Fighting Blindness (JGH), Research to Prevent Blindness Unrestricted Grant and National Institutes of Health grant R01EY014240-08 (JGH).
REFERENCES
- 1.Boughman JA, Conneally PM, Nance WE (1980) Population genetic studies of retinitis pigmentosa. Am J Hum Genet 32:223–235. [PMC free article] [PubMed] [Google Scholar]
- 2.Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH (1984) Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 97:357–365. [DOI] [PubMed] [Google Scholar]
- 3.Dryja TP, McGee TL, Hahn LB, Cowley GS, Olsson JE, Reichel E, Sandberg MA, Berson EL (1990) Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med 323:1302–1307. [DOI] [PubMed] [Google Scholar]
- 4.Tsang SH, Wolpert K (2010) The Genetics of Retinitis Pigmentosa. Retinal Physician. [Google Scholar]
- 5.Collin RW, Littink KW, Klevering BJ, van den Born LI, Koenekoop RK, Zonneveld MN, Blokland EA, Strom TM, Hoyng CB, den Hollander AI, Cremers FP (2008) Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet 83:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Littink KW, van den Born LI, Koenekoop RK, Collin RW, Zonneveld MN, Blokland EA, Khan H, Theelen T, Hoyng CB, Cremers FP, den Hollander AI, Klevering BJ (2010) Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 117:2026–2033, 2033 e2021-2027. [DOI] [PubMed] [Google Scholar]
- 7.Abd El-Aziz MM, Barragan I, O’Driscoll CA, Goodstadt L, Prigmore E, Borrego S, Mena M, Pieras JI, El-Ashry MF, Safieh LA, Shah A, Cheetham ME, Carter NP, Chakarova C, Ponting CP, Bhattacharya SS, Antinolo G (2008) EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet 40:1285–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zelhof AC, Hardy RW, Becker A, Zuker CS (2006) Transforming the architecture of compound eyes. Nature 443:696–699. [DOI] [PubMed] [Google Scholar]
- 9.Mizuno K, Nishida S (1967) Electron microscopic studies of human retinitis pigmentosa. I. Two cases of advanced retinitis pigmentosa. Am J Ophthalmol 63:791–803. [DOI] [PubMed] [Google Scholar]
- 10.Kolb H, Gouras P (1974) Electron microscopic observations of human retinitis pigmentosa, dominantly inherited. Invest Ophthalmol Vis Sci 13:487–498. [PubMed] [Google Scholar]
- 11.Szamier RB, Berson EL (1977) Retinal ultrastructure in advanced retinitis pigmentosa. Invest Ophthalmol Vis Sci 16:947–962. [PubMed] [Google Scholar]
- 12.Rayborn ME, Moorhead LC, Hollyfield JG (1982) A dominantly inherited chorioretinal degeneration resembling sectoral retinitis pigmentosa. Ophthalmology 89:1441–1454. [DOI] [PubMed] [Google Scholar]
- 13.Szamier RB, Berson EL (1982) Histopathologic study of an unusual form of retinitis pigmentosa. Invest Ophthalmol Vis Sci 22:559–570. [PubMed] [Google Scholar]
- 14.Szamier RB, Berson EL (1985) Retinal histopathology of a carrier of X-chromosome-linked retinitis pigmentosa. Ophthalmology 92:271–278. [DOI] [PubMed] [Google Scholar]
- 15.Szamier RB, Berson EL, Klein R, Meyers S (1979) Sex-linked retinitis pigmentosa: ultrastructure of photoreceptors and pigment epithelium. Invest Ophthalmol Vis Sci 18:145–160. [PubMed] [Google Scholar]
- 16.Farber DB, Flannery JG, Bird AC, Shuster T, Bok D (1987) Histopathological and biochemical studies on donor eyes affected with retinitis pigmentosa. Prog Clin Biol Res 247:53–67. [PubMed] [Google Scholar]
- 17.Tucker GS, Jacobson SG (1988) Morphological findings in retinitis pigmentosa with early diffuse rod dysfunction. Retina 8:30–41. [DOI] [PubMed] [Google Scholar]
- 18.Santos A, Humayun MS, de Juan E Jr., et al. (1997) Preservation of the inner retina in retinitis pigmentosa. A morphometric analysis. Arch Ophthalmol 115:511–515. [DOI] [PubMed] [Google Scholar]
- 19.Li ZY, Kljavin IJ, Milam AH (1995) Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci 15:5429–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li ZY, Possin DE, Milam AH (1995) Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology 102:805–816. [DOI] [PubMed] [Google Scholar]
- 21.Fariss RN, Apte SS, Luthert PJ, Bird AC, Milam AH (1998) Accumulation of tissue inhibitor of metalloproteinases-3 in human eyes with Sorsby’s fundus dystrophy or retinitis pigmentosa. Br J Ophthalmol 82:1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milam AH, Li ZY, Fariss RN (1998) Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res 17:175–205. [DOI] [PubMed] [Google Scholar]
- 23.Tulvatana W, Adamian M, Berson EL, Dryja TP (1999) Photoreceptor rosettes in autosomal dominant retinitis pigmentosa with reduced penetrance. Arch Ophthalmol 117:399–402. [DOI] [PubMed] [Google Scholar]
- 24.Fariss RN, Li ZY, Milam AH (2000) Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol 129:215–223. [DOI] [PubMed] [Google Scholar]
- 25.Li ZY, Jacobson SG, Milam AH (1994) Autosomal dominant retinitis pigmentosa caused by the threonine-17-methionine rhodopsin mutation: retinal histopathology and immunocytochemistry. Exp Eye Res 58:397–408. [DOI] [PubMed] [Google Scholar]
- 26.Milam AH, Li ZY, Cideciyan AV, Jacobson SG (1996) Clinicopathologic effects of the Q64ter rhodopsin mutation in retinitis pigmentosa. Invest Ophthalmol Vis Sci 37:753–765. [PubMed] [Google Scholar]
- 27.To K, Adamian M, Dryja TP, Berson EL (2000) Retinal histopathology of an autopsy eye with advanced retinitis pigmentosa in a family with rhodopsin Glu181Lys. Am J Ophthalmol 130:790–792. [DOI] [PubMed] [Google Scholar]
- 28.John SK, Smith JE, Aguirre GD, Milam AH (2000) Loss of cone molecular markers in rhodopsin-mutant human retinas with retinitis pigmentosa. Mol Vis 6:204–215. [PubMed] [Google Scholar]
- 29.To K, Adamian M, Dryja TP, Berson EL (2002) Histopathologic study of variation in severity of retinitis pigmentosa due to the dominant rhodopsin mutation Pro23His. Am J Ophthalmol 134:290–293. [DOI] [PubMed] [Google Scholar]
- 30.To K, Adamian M, Berson EL (2004) Histologic study of retinitis pigmentosa due to a mutation in the RP13 gene (PRPC8): comparison with rhodopsin Pro23His, Cys110Arg, and Glu181Lys. Am J Ophthalmol 137:946–948. [DOI] [PubMed] [Google Scholar]
- 31.Aguirre GD, Yashar BM, John SK, et al. (2002) Retinal histopathology of an XLRP carrier with a mutation in the RPGR exon ORF15. Exp Eye res 75:431–443. [PubMed] [Google Scholar]
- 32.Jacobson SG, Buraczynska M, Milam AH, et al. (1997) Disease expression in X-linked retinitis pigmentosa caused by a putative null mutation in the RPGR gene. Invest Ophthalmol Vis Sci 38:1983–1997. [PubMed] [Google Scholar]
- 33.Mullins RF, Kuehn MH, Radu RA, et al. (2012) Autosomal recessive retinitis pigmentosa due to ABCA4 mutations: clinical, pathologic, and molecular characterization. Invest Ophthalmol Vis Sci 53:1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagheri N, Bell BA, Bonilha VL, Hollyfield JG (2012) Imaging human postmortem eyes with SLO and OCT. Adv Exp Med Biol 723:479–488. [DOI] [PubMed] [Google Scholar]
- 35.A Abd El-Aziz MM, O’Driscoll CA, Kaye RS, Barragan I, El-Ashry MF, Borrego S, Antinolo G, Pang CP, Webster AR, Bhattacharya SS (2010) Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 51:4266–4272. [DOI] [PubMed] [Google Scholar]
- 36.Audo I, Sahel JA, Mohand-Said S, Lancelot ME, Antonio A, Moskova-Doumanova V, Nandrot EF, Doumanov J, Barragan I, Antinolo G, Bhattacharya SS, Zeitz C (2010) EYS is a major gene for rod-cone dystrophies in France. Hum Mutat 31:E1406–1435. [DOI] [PubMed] [Google Scholar]
- 37.Barragan I, Borrego S, Pieras JI, Gonzalez-del Pozo M, Santoyo J, Ayuso C, Baiget M, Millan JM, Mena M, Abd El-Aziz MM, Audo I, Zeitz C, Littink KW, Dopazo J, Bhattacharya SS, Antinolo G (2010) Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Mutat 31:E1772–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bandah-Rozenfeld D, Littink KW, Ben-Yosef T, et al. (2010) Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest Ophthalmol Vis Sci 51:4387–4394. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Zhang J, Li C, Yang G, Liu M, Wang QK, Tang Z (2010) Identification of a novel homozygous nonsense mutation in EYS in a Chinese family with autosomal recessive retinitis pigmentosa. BMC Med Genet 11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan MI, Collin RW, Arimadyo K, et al. (2010) Missense mutations at homologous positions in the fourth and fifth laminin A G-like domains of eyes shut homolog cause autosomal recessive retinitis pigmentosa. Mol Vis 16:2753–2759. [PMC free article] [PubMed] [Google Scholar]
- 41.Pieras JI, Barragan I, Borrego S, et al. (2011) Copy-number variations in EYS: a significant event in the appearance of arRP. Invest Ophthalmol Vis Sci 52:5625–5631. [DOI] [PubMed] [Google Scholar]
- 42.Hosono K, Ishigami C, Takahashi M, et al. (2012) Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PloS one 7:e31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suto K, Hosono K, Takahashi M, et al. (2014) Clinical Phenotype in Ten Unrelated Japanese Patients with Mutations in the EYS Gene. Ophthalmic Genet 35:25–34. [DOI] [PubMed] [Google Scholar]
- 44.Iwanami M, Oshikawa M, Nishida T, Nakadomari S, Kato S (2012) High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 53:1033–1040. [DOI] [PubMed] [Google Scholar]
- 45.Katagiri S, Akahori M, Hayashi T, et al. (2014) Autosomal recessive cone-rod dystrophy associated with compound heterozygous mutations in the EYS gene. Doc Ophthalmol 128:211–217. [DOI] [PubMed] [Google Scholar]