

SUMMARY
The endocannabinoid (eCB) system is a key modulator of glutamate release within limbic neurocircuitry and thus heavily modulates stress responsivity and adaptation. The ventral hippocampus (vHPC)-basolateral amygdala (BLA) circuit has been implicated in the expression of negative affective states following stress exposure and is modulated by retrograde eCB signaling. However, the mechanisms governing eCB release and the causal relationship between vHPC-BLA eCB signaling and stress-induced behavioral adaptations are not known. Here, we utilized in vivo optogenetic- and biosensor-based approaches to determine the temporal dynamics of activity-dependent and stress-induced eCB release at vHPC-BLA synapses. Furthermore, we demonstrate that genetic deletion of cannabinoid type-1 receptors selectively at vHPC-BLA synapses decreases active stress coping and exacerbates stress-induced avoidance and anhedonia phenotypes. These data establish the in vivo determinants of eCB release at limbic synapses and demonstrate that eCB signaling within vHPC-BLA circuitry serves to counteract adverse behavioral consequences of stress.
Graphical abstract
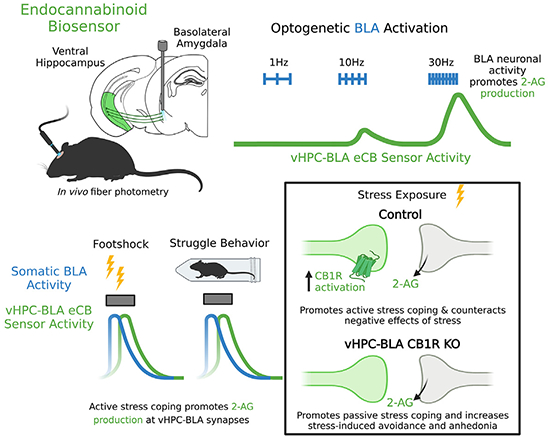
In brief
Kondev et al. define the in vivo determinants of endocannabinoid (eCB) release at ventral hippocampus (vHPC)-basolateral amygdala (BLA) synapses. They also show that stress exposure recruits eCB release at vHPC-BLA synapses, which serves to promote active coping during stress and counteract the subsequent adverse behavioral consequences of stress exposure.
INTRODUCTION
Stress exposure confers risk for the development or exacerbation of psychiatric disorders: from generalized anxiety and major depression to post-traumatic stress disorder (PTSD).1–4 Understanding stress-induced molecular-, cellular-, and circuit-level adaptations could provide critical insight into how stress is translated into affective pathology and may reveal novel therapeutic targets for the treatment of stress-related disorders. One of the leading neuromodulatory signaling systems that has been identified as a prominent drug-development candidate for stress-related psychiatric disorders is the endocannabinoid (eCB) system.4–11
The eCB signaling system, consisting of the cannabinoid receptor type 1 (CB1R) and endogenous ligands including 2-arachidonoylglycerol (2-AG), is a neuromodulatory lipid system heavily implicated in the modulation of the physiological stress response, stress adaptability and susceptibility, and fear responding.12–14 As such, 2-AG signaling specifically within key emotional processing regions, such as the amygdala, has been identified as a critical substrate for mitigating negative affect and stress-induced increases in anxiety-like behavior.15–18 Human and rodent studies have implicated activity of the amygdala and its functionally connected region, the hippocampus, in fear learning and stress-related pathological states, such as PTSD.19–29 The ventral portion of the hippocampus (vHPC) provides strong input to the basolateral amygdala (BLA) that undergoes robust activity-dependent eCB mobilization.19,30 Specifically, we have shown that 2-AG-mediated suppression of vHPC-BLA synapses is associated with resilience to stress-induced behavioral pathology,19 suggesting that 2-AG signaling at these synapses may play an important role in mitigating the adverse consequences of stress exposure.
To directly test the hypothesis that 2-AG signaling at vHPC-BLA synapses confers a protective role against the adverse effects of stress, we utilized a genetically encoded eCB sensor, GRAB-eCB2.0, combined with optogenetics, mouse behavior, and circuit-specific CB1R deletion. Our data establish the in vivo temporal dynamics and neuronal activity determinants of 2-AG release at vHPC-BLA synapses and demonstrate that selective deletion of CB1R from this circuit increases passive coping responses to acute stress and exacerbates subsequent avoidance- and anhedonia-like behaviors. These data suggest that eCB signaling at vHPC-BLA synapses is recruited by stress in an activity-dependent manner and serves to counteract the adverse behavioral consequences of stress exposure.
RESULTS
GRAB-eCB2.0 biosensor elucidates neural determinants of eCB release at vHPC-BLA synapses
To measure eCB release in vivo in a circuit-specific manner, we virally delivered a G protein-coupled receptor (GPCR)-based eCB biosensor (GRAB-eCB2.0 [AAV9-hSyn-GRAB-eCB2.0]) into the vHPC.31 Mice were then ipsilaterally implanted with a fiber optic probe directly above the BLA to measure BLA-dependent eCB production and subsequent sensor activity at vHPC terminals (Figure S1A). Over long experimental time courses, we observed photobleaching of the green fluorescence signal, which was corrected for post hoc (Figure S1B). We found that activation of GRAB-eCB2.0 with the CB receptor agonist CP55,940 (1 mg/kg) increased the fluorescent signal beginning ~20 min after intraperitoneal (i.p.) injection (Figures S1C–S1E). Importantly, this effect was absent in control mice that were injected with the eCB-insensitive mutant virus (GRAB-eCB2.0-mut [AAV9-hSyn-GRABeCB2.0-mutant]) (Figures S1D and S1E). Following the peak fluorescence signal (~45 min after CP55,940 injection), the CB1R inverse agonist rimonabant (10 mg/kg) was injected. Rimonabant significantly reduced the area under the curve (AUC) in GRAB-eCB2.0 mice but not in control, GRAB-eCB2.0-mut mice (Figures S1C, S1F, and S1G). Together, these data confirm the ability to detect GRAB-eCB2.0 activity at vHPC-BLA synapses.
2-AG is canonically produced by postsynaptic neurons in an activity-dependent manner by diacylglycerol-lipase alpha (DAGLα) and activates presynaptic CB1 receptors to reduce neurotransmitter release probability.32–34 To test whether BLA activity could promote 2-AG production at BLA-vHPC synapses, we combined in vivo optogenetics with fiber photometry (Figure 1). A virus expressing the GRABeCB-2.0 was delivered to the vHPC, and the red-shifted excitatory opsin Chrimson (AAV5-hSyn-Chrimson-TdT) was delivered to the BLA; mice were then implanted with a fiber optic above the BLA (Figure 1A). Red light (615nm) was pulsed at different frequencies to stimulate somatic BLA activity, and we simultaneously recorded changes in green fluorescence to measure changes in GRAB-eCB2.0 sensor activity at vHPC terminals in the BLA.
Figure 1. BLA activity promotes increase in GRAB-eCB2.0 biosensor and endogenous CB1R activation at vHPC-BLA synapses.
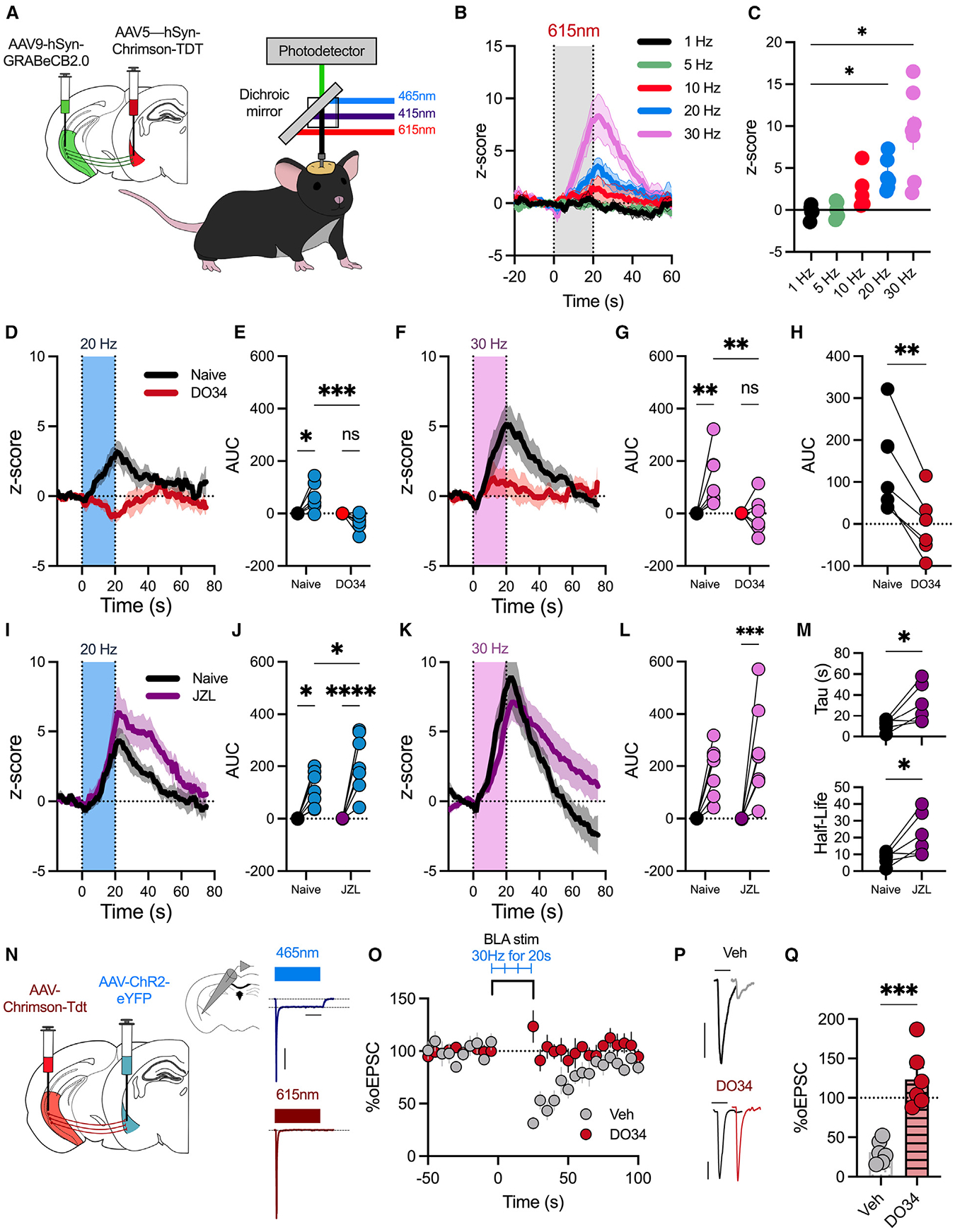
(A) A virus expressing GRAB-eCB-2.0 was delivered to the vHPC, and Chrimson was delivered to the ipsilateral BLA. A fiber optic was implanted above the BLA. In vivo fiber photometry recordings were conducted to simultaneously record changes in GFP fluorescence and stimulate BLA neurons (n = 7 male mice).
(B) Z score of ΔF/F following 20 s of Chrimson stimulation at different frequencies.
(C) Average change in Z score 5 s after stimulation (0–20 s) stops.
(D) Z score of ΔF/F following BLA stimulation (20 Hz, 20 s) in naive mice or mice pretreated with the DAGL inhibitor DO34.
(E) Quantification of area under the curve (AUC) from (D).
(F) Z score of ΔF/F following BLA stimulation (30 Hz, 20 s) in naive mice or DO34-pretreated mice.
(G) Quantification of AUC from (F).
(H) Within-subject comparison of data in (G).
(I) Z score of ΔF/F following BLA stimulation (20 Hz, 20 s) in naive mice or mice pretreated with the MAGL inhibitor JZL184.
(J) Quantification of AUC from (I).
(K) Z score of ΔF/F following BLA stimulation (30 Hz, 20 s) in naive mice or mice pretreated with the MAGL inhibitor JZL184.
(L) Quantification of AUC from (K).
(M) Decay constant, tau, following JZL184 pretreatment (top); half-life of Z score (bottom).
(N) Schematic of electrophysiological experiments. AAV-ChR2 was delivered to the BLA, and AAV-Chrimson was delivered to the vHPC. Electrophysiological recordings were performed from BLA pyramidal neurons. Representative traces demonstrating that 465 nm light induces depolarization indicative of somatic ChR2, while 615 nm light induces exclusively vHPC-BLA optically evoked excitatory postsynaptic currents (oEPSCs) with no somatic current. Scale bar represents 200 ms and 200 pA. Each data point represents one cell from 5 mice.
(O) Time course of percentage of oEPSC amplitude following stimulation of BLA neurons (30 Hz, 20 s). Slices were incubated in vehicle or DO34 (2.5 μM).
(P) Representative traces of oEPSC amplitude in slices incubated in vehicle (top; scale bar represents 25 ms and 100 pA) or DO34 (bottom; scale bar represents 25 ms and 200 pA).
(Q) Average percentage of oEPSC depression following BLA stimulation.
Data were analyzed by one-way ANOVA (C), two-way ANOVA (E, G, J, and L), Student’s paired two-tailed t test (H and M), or unpaired t test (Q). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent SEM.
Using this approach, we found a frequency-dependent increase in GRAB-eCB2.0 signal with a significant increase in the Z score at 20 and 30 Hz BLA neuron stimulation frequencies (Figures 1B and 1C). To confirm that this effect was driven by activity-dependent BLA production of 2-AG, mice were pretreated with the DAGL inhibitor DO34 (50 mg/kg). The activity-dependent increase in fluorescence at 20 and 30 Hz was blocked by pretreatment with DO34 (Figures 1D–1G). Within-subject comparison demonstrated that DO34 significantly decreased sensor activity following BLA stimulation (Figure 1H). To further confirm activity-dependent release of 2-AG, we used JZL184 (20 mg/kg) to inhibit the 2-AG degradation enzyme MAGL.8,35 JZL184 pretreatment significantly increased activity-dependent sensor activity, evident as an increase in the AUC after both 20 and 30 Hz stimulations (Figures 1I–1L). As expected, JZL184 also increased the decay time constant tau and increased the half-life of the Z score, providing further support for 2-AG remaining in the synapse longer after MAGL inhibition (Figure 1M). Given that activation of Gq-coupled GPCRs is a well-established trigger of 2-AG release, we next examined this receptor-driven form of 2-AG release using chemogenetics via injection of AAV-Gq-coupled DREADDs (AAV5-CaMKII-hM3Dq-mCherry) into the BLA (Figure S1H). Ex vivo, clozapine-N-oxide (CNO) application depolarized BLA neurons (Figure S1I), while in vivo administration of CNO (5 mg/kg) resulted in an increase in GRABeCB-2.0 activity, which was significantly reduced by pretreatment with DO34 (Figures S1J and S1K). These data demonstrate that BLA neuronal firing as well as Gq-coupled GPCR activity can drive 2-AG production and release, and they confirm 2-AG as the major activity-dependent eCB ligand released at vHPC-BLA synapses under these conditions.
These data also suggest that stimulation of BLA neurons can trigger 2-AG production and activation of endogenous CB1Rs on vHPC terminals; we directly tested this hypothesis using ex vivo electrophysiology and optogenetics. Mice were injected with channelrhodopsin (ChR2) (AAV5-CaMKII-ChR2(H134R)-EYFP-WPRE) into the BLA and with Chrimson in the vHPC to allow us to distinctly modulate either BLA somatic or vHPC terminal activity using ex vivo whole-cell electrophysiological recordings of BLA pyramidal neurons (Figure 2N). Blue light stimulation evoked somatic ChR2 activity, while red light stimulation induced monosynaptic vHPC-originating optically evoked excitatory postsynaptic currents (oEPSCs) (Figure 1N). We replicated our in vivo fiber photometry stimulation paradigm ex vivo to analyze a modified form of depolarization-induced suppression of excitation (DSE), a well-characterized form of 2-AG-mediated short-term depression.32 Low blue light power activated BLA neurons (30 Hz, 20 s) and produced ~70% depression of oEPSC amplitude at vHPC-BLA synapses (Figures 2O–2Q). Importantly, our optogenetic protocol did not induce any form of long-term depression or potentiation, evident as no significant change in the percentage of oEPSC amplitude following 3 sequential stimulation events (Figures S1L–S1N). Furthermore, this form of DSE was blocked by pharmacological DAGL inhibition with DO34 (2.5 μM) (Figures 2O–2Q), confirming that our BLA stimulation at the same frequency that resulted in GRAB-eCB2.0 signals in vivo also induces 2-AG production and subsequent synaptic depression of vHPC-BLA synapses.
Figure 2. Footshock and active stress coping recruit BLA activity and eCB production.
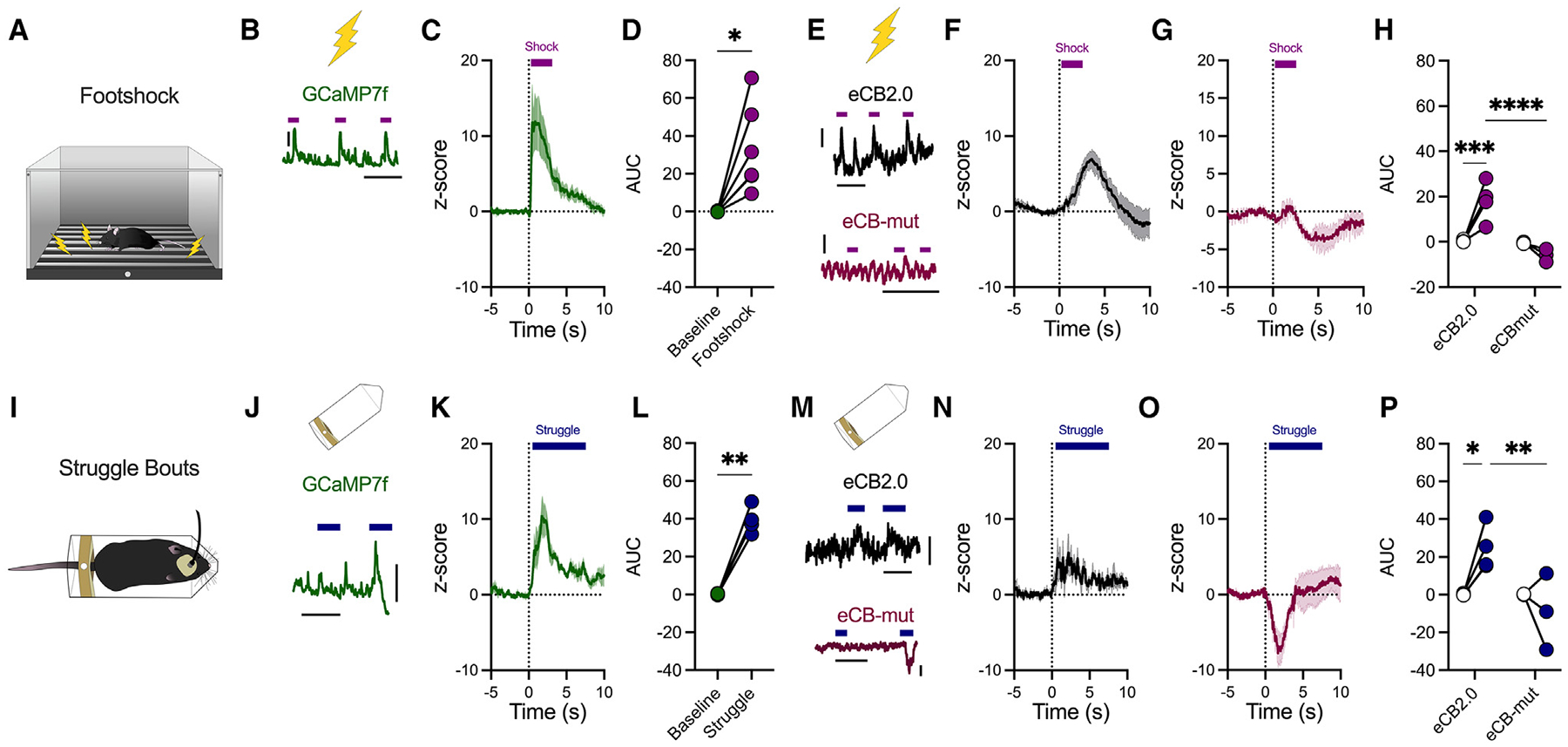
(A) Footshock stress diagram.
(B) Representative ΔF/F trace of mice injected with a virus expressing GCaMP7f (AAVrg-hSyn-jGCaMP7f-WPRE) into the BLA and photometry recordings of somatic BLA activity were conducted during footshock exposure (purple bars) (n = 6 male mice; scale bar represents 0.1 ΔF/F and 50 ms).
(C) Z score of ΔF/F following footshock in GCaMP7f-injected mice; baseline was −3 s to −1 s before footshock onset (0 s).
(D) AUC of (C).
(E) Representative trace of ΔF/F during footshock (purple bars) in mice injected with a virus expressing the GRAB-eCB2.0 (top) or eCB-mut (bottom). Scale bar represents 0.005 ΔF/F and 50 ms.
(F) Z score of ΔF/F following footshock in GRAB-eCB2.0 mice (n = 5 male mice).
(G) Z score of ΔF/F following footshock in GRAB-eCB-mut mice (n = 3 male mice).
(H) Quantification of AUC following footshock in GRAB-eCB2.0 or GRAB-eCB-mut mice.
(I) Struggle behavior during restraint stress.
(J) Representative ΔF/F trace of mice injected with a virus expressing GCaMP7f into the BLA and recording of BLA activity during struggle (blue bars). Scale bar represents 0.05 ΔF/F and 20 ms.
(K) Z score of ΔF/F following struggle behavior in GCaMP7f mice (n = 4 male mice).
(L) AUC quantification from (K).
(M) Representative traces of ΔF/F during struggle behavior (blue bars) in GRAB-eCB2.0 (top) or GRAB-eCB-mut (bottom) mice. Scale bar represents 0.005 ΔF/F and 20 ms.
(N) Z score of ΔF/F following struggle behavior in GRAB-eCB2.0 mice (n = 5 male mice).
(O) Z score of ΔF/F following struggle behavior in GRAB-eCB-mut mice (n = 3 male mice).
(P) Quantification of AUC following struggle onset.
Data were analyzed via paired Student’s two-tailed t test (D and L) or two-way ANOVA (H and P). *p < 0.05, **p < 0.01, ***p <0.001, ****p < 0.0001. Error bars represent SEM.
Active stress coping promotes BLA activity and 2-AG release at vHPC-BLA synapses
Having gained insight into the neural activity determinants of GRAB-eCB2.0 activation at vHPC-BLA synapses, we next evaluated how stressful stimuli may drive changes in BLA activity and eCB production at vHPC-BLA synapses (Figure 2). First, using the calcium indicator GCaMP7f (AAVrg-hSyn-jGCaMP7f-WPRE), we show that footshock delivery (2 s, 0.7 mA) promotes BLA neuron activity (Figures 2A–2D). A separate cohort of mice expressed GRAB-eCB2.0 or GRAB-eCB2.0-mut in the vHPC, with a fiber optic implanted above the BLA (Figures 2E–2H). Footshock promoted an increase in GRAB-eCB2.0 sensor activity at vHPC terminals in the BLA, indicating stress-induced eCB release at vHPC-BLA synapses (Figures 2F and 2H). Importantly, this increase in GRAB-eCB2.0 fluorescent signal was absent in mice expressing GRAB-eCB2.0-mut (Figures 2G and 2H).
We confirmed this stress-induced increase in BLA activity and eCB release by exposing mice to a restraint stress session (Figure S2A). Acute restraint stress had no significant effect on overall BLA activity, and restraint stress-induced changes in overall fluorescence did not differ between GRAB-eCB2.0- and GRAB-eCB2.0-mut-injected mice (Figures S2A–S2E). We hypothesized that perhaps changes in BLA eCB signaling are related to active bouts of coping behavior during stress rather than the stress exposure itself. To investigate this, we assessed struggle behavior during restraint (Figure 2I). Whole-body struggle behavior elicited a significant increase in BLA activity, as well as an increase in GRAB-eCB2.0 sensor activity, that was not present in GRAB-eCB2.0-mut mice (Figures 2J–2P). We confirmed that changes in GRAB-eCB2.0-mut activity were due to eCB-independent signaling by comparing stress-coping-induced changes in fluorescence to another eCB-independent fluorophore, GFP (AAV5-CMV-PI-EGFP-WPRE) (Figure S2F). Indeed, we observed that neither footshock nor struggle bouts during restraint increases fluorescence in GFP- or GRAB-eCB2.0-mut-injected mice (Figures S2G–S2J).
To validate that this increase in eCB2.0 sensor activity was due to stress-induced 2-AG release, we pretreated mice with the DAGL inhibitor DO34 (50 mg/kg) (Figure S2K). Pharmacological inhibition of DAGL significantly attenuated both footshock- and struggle-induced sensor activity, demonstrating that active struggle behavior and footshock exposure elicit 2-AG production and release in the BLA (Figures S2L–S2O). Given our optogenetic data above indicating that increases in BLA neuron activity can drive 2-AG release at vHPC-BLA synapses and the association between stress-induced increases in BLA activity and 2-AG release, we hypothesized that neuronal activity contributes to stress-induced 2-AG signaling. Supporting this notion, we found a significant lag in sensor activity compared with somatic BLA activity (Figure S2P). We thus used inhibitory a Gi-DREADD (AAV5-hSyn-hM4Di-mCherry) to decrease BLA activity during stress and to test whether BLA activity is necessary for eCB production (Figure S2Q). Chemogenetic BLA inhibition significantly attenuated stress-induced increases in eCB2.0 sensor activity, evident as a decrease in AUC following footshock and struggle behavior (Figures S2R–S2U). Given that eCB sensor activity peaked between 2 and 5 s, we confirmed that BLA stimulation at a frequency that was previously shown to induce 2-AG release (Figures 1B and 1C) could still promote sensor activity at the same temporal scale as external stimuli; indeed, 5 s of BLA stimulation at 30 Hz was sufficient to induce eCB sensor activity at vHPC-BLA synapses (Figure S2V). These data ultimately demonstrate that active stress coping is associated with enhanced BLA activity that can drive 2-AG release at vHPC-BLA synapses.
Deletion of vHPC-BLACB1R reduces active coping and worsens behavioral consequences of stress
We have thus far demonstrated that stress promotes activity-dependent phasic 2-AG signaling at vHPC-BLA synapses. We next wanted to establish the role for the endogenous CB1R at vHPC-BLA synapses in the regulation of stress-induced behavioral adaptations. eCB signaling along vHPC-BLA synapses constrains activity of this circuit, and it has been proposed that 2-AG signaling within the BLA serves to counteract the negative effects of stress.8,19 We thus hypothesized that eCB signaling along vHPC-BLA circuitry facilitates physiological stress-coping responses and prevents adverse behavioral effects of stress.
To test this hypothesis, we deleted CB1R from the vHPC-BLA circuit and assessed changes in stress-coping and behavioral sequalae (Figure 3A). Combination injection of retrograde Flp recombinase (rgAAV2-Ef1a-mCherry-IRES-Flpo) in the BLA and Flp-dependent Cre (AAV5-CMV-fDIO-Cre-mNeonGreen-WPRE) in the vHPC of CB1Rfl/fl resulted in functional deletion of the CB1R specifically from BLA-projecting vHPC neurons but not from nucleus accumbens (NAc)-projecting vHPC neurons (Figure S3A). This circuit-specific deletion was validated by electrophysiology, demonstrating a lack of CP55–940-induced synaptic depression at vHPC-BLA, but not vHPC-NAc, synapses (Figures 3B–3D and S3B–S3E).
Figure 3. Deletion of vHPCCB1R-BLA decreases active stress coping, exacerbates stress-induced anhedonia and avoidance, and increases stress susceptibility.
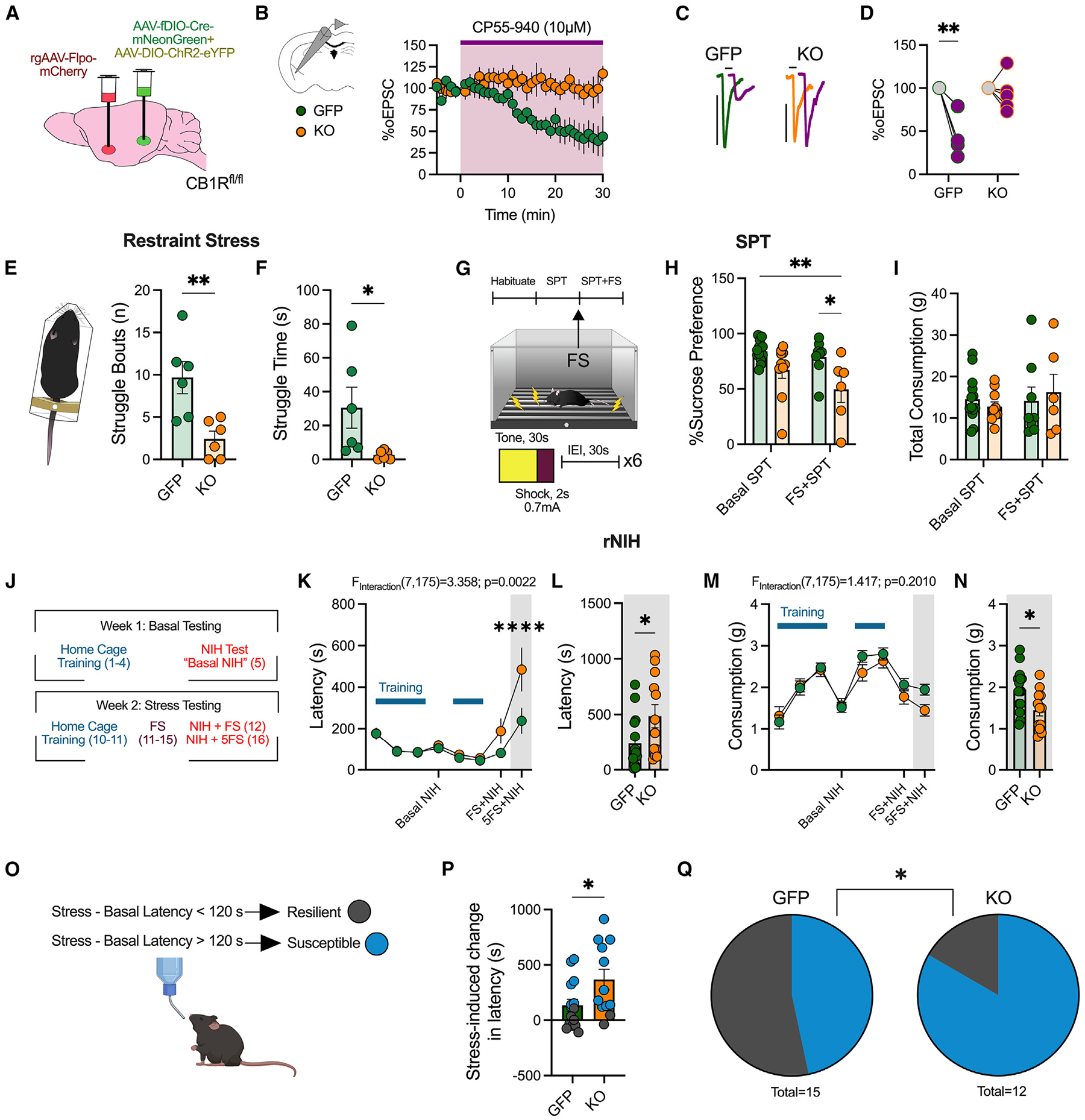
(A) Schematic of experimental design and viral surgeries to selectively delete the CB1R from vHPC-BLA circuit (KO) or control (GFP).
(B) Ex vivo electrophysiological recordings from BLA pyramidal neurons. Time course of vHPC-mediated oEPSCs following bath application of CP55,940 (10 μM) (n = 3 male mice/group).
(C) Representative traces before and after CP55,940 wash-on in control mice (GFP) and vHPCCB1R KO-BLA mice (KO).
(D) Quantification of average percentage of oEPSC depression following CP55,940 wash-on.
(E) Average number of struggle bouts during 4 min restraint stress exposure (n = 6 male and female mice/group).
(F) Quantification of total time spent struggling during restraint.
(G) Experimental timeline for sucrose preference test (SPT). Basal SPT was assessed 24 h after sucrose was provided. Mice were then exposed to an acute footshock exposure, consisting of 6 shocks. 24 h after footshock, sucrose consumption was assessed again (FS + SPT).
(H) Percentage of sucrose preference basally and after one acute footshock session (GFP: n = 13, KO: n = 10 basal and GFP: n = 8 and KO: n = 6 post-stress, male and female mice).
(I) Total consumption of liquid (water and sucrose solution).
(J) Experimental timeline for repeated novelty induced hypophagia (rNIH) testing (GFP: n = 15 and KO: n = 12 male and female mice).
(K) Latency to drink Ensure over time.
(L) Feeding latency during NIH after 5 days of footshock stress.
(M) Consumption of Ensure over time.
(N) Consumption during NIH after 5 days of footshock stress.
(O) Criteria used for determining stress-susceptible versus-resilient mice.
(P) Quantification of stress-induced change in latency, comparing latency after 5 days of footshock with basal latency.
(Q) Proportion of susceptible versus resilient mice following vHPCCB1R KO-BLA.
Data were analyzed via two-way ANOVA (D, H, I, K, and M), unpaired Student’s t test (E, F, L, N, and P), or chi-squared test (Q) performed as analysis. *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars represent SEM.
We first examined changes in stress coping via assessment of struggling behavior during restraint stress in vHPC-BLACB1R−KO (knockout) mice (Figures 3E and 3F). Selective KO of the CB1R from vHPC-BLA synapses resulted in a decrease in the number of whole-body struggle bouts during restraint exposure, as well as a decrease in the total time spent struggling (Figures 3E and 3F). We confirmed this deficit in active stress coping by testing mice in the tail suspension test (TST) and the forced swim test (FST); vHPC-BLACB1R−KO mice exhibited an increase in immobility in both measures, further suggesting a deficit in active stress-coping behavior upon deletion of CB1R from the vHPC-BLA circuit (Figures S3F and S3G). To confirm that this phenotype was not due to general changes in locomotion, we exposed mice to open field testing and found no change in distance traveled (Figure S3H).
Given that stress exposure can facilitate the development of innate avoidance and anhedonia-like behaviors, we hypothesized that deletion of CB1R from vHPC-BLA synapses would worsen the adverse behavioral consequences of stress exposure. Indeed, vHPC-BLACB1R−KO mice exhibited a robust decrease in sucrose preference following an acute footshock exposure, but not basally, relative to control mice (Figures 3G and 3H). Importantly, there were no differences in total fluid consumption between groups (Figure 3I). Mice were also tested in the repeated novelty-induced hypophagia (rNIH) test, which is modulated by BLA 2-AG levels19 (Figure 3J). We found that deletion of the CB1R from vHPC-BLA synapses had no effect on the latency to drink or the total consumption of the Ensure reward across training or during basal NIH testing. However, exposure to 5 days of footshock stress significantly increased the latency to drink, and decreased the consumption of, Ensure in vHPC-BLACB1R−KO mice relative to controls (Figures 3K–3N). Together, these data suggest that deletion of CB1R within vHPC-BLA circuits exacerbates stress-induced avoidance and anhedonia without affecting basal affective phenotypes.
We have previously demonstrated that BLA 2-AG signaling critically regulates susceptibility to developing avoidance phenotypes in the rNIH test.19 We therefore used this rNIH paradigm to explore whether vHPC-BLACB1R−KO mice exhibited enhanced stress susceptibility (Figure 3O).19 A stress-induced change in latency of greater than 120 s was used to categorize mice as stress susceptible or resilient based on our previous work.19 Importantly, we observed that vHPC-BLACB1R−KO promoted a significant increase in stress-induced feeding latency following 5 days of footshock stress (Figure 3P). This ultimately resulted in an increase in the proportion of stress-susceptible mice (Figure 3Q). These data support a model whereby stress recruits 2-AG signaling at vHPC-BLA synapses to promote active stress-coping responses and to counteract the behavioral sequalae of stress exposure, including anhedonia and avoidance.
To validate that these behavioral effects were not due to general vHPC CB1R deletion, a separate cohort of CB1Rfl/fl mice were injected with a virus expressing Cre (AAV8-Ef1a-mCherry-IRES-Cre) or GFP (AAV5-CMV-PI-EGFP-WPRE) to delete CB1R from all vHPC cells, including projection neurons (Figures S3H–S3L). Ex vivo electrophysiological recordings of CP55,940-induced depression of vHPC-mediated oEPSCs in the NAc were significantly attenuated following Cre injection (Figures S3J–S3L). We exposed vHPCCB1R KO mice to the TST and the FST and observed no significant changes in active stress coping (Figures S3M and S3N). We also assessed changes in stress-induced anhedonia and avoidance via the SPT and rNIH. We found that global vHPCCB1R KO had no effect on sucrose preference or general consumption (Figure S3O). Furthermore, general vHPCCB1R KO did not exhibit a change in the latency to drink Ensure or in the total consumption of Ensure, either basally or following 5 days of footshock stress (Figures S3P–S3S). As such, global vHPCCB1R deletion also had no effect on the stress-induced change in feeding latency, stress susceptibility, or general locomotion as measured during the open field test (Figures S3T–S3V). These experiments confirm circuit specificity of vHPCCB1R-BLA-deletion-mediated effects, suggesting that the collapse of eCB regulation of vHPC-BLA promotes passive stress coping and negative affect following stress.
DISCUSSION
Activity within the amygdala and the vHPC (the anterior hippocampus in humans) has been implicated in the pathogenesis of stress-related neuropsychiatric disorders, and preclinical studies have confirmed that vHPC-amygdala circuits are critical for driving affective behavioral responses to stress.20–24,36–39 We have previously shown that vHPC afferents to the amygdala express CB1 receptors and 2-AG-dependent retrograde synaptic suppression.19 Importantly, the magnitude of eCB synaptic suppression was greatest in mice that exhibited resilience to adverse behavioral consequences of stress, suggesting that eCB signaling at vHPC-BLA synapses could be actively recruited during stress exposure to dampen vHPC afferent excitation and could serve to counteract the adverse behavioral consequences of stress exposure. Here, we directly test these hypotheses using a combination of GPCR-based biosensor recordings, pharmacology, and intersectional genetics and establish a causal role for vHPC-BLA 2-AG-CB1 signaling in the regulation of stress-coping responses and adverse behavioral consequences of stress exposure. These data provide circuit-specific mechanistic insight into the regulation of stress adaptation and a critical role for eCB signaling in the regulation of vHPC-BLA function.
We first utilized the GPCR-based biosensor to evaluate the in vivo neural activity determinants of eCB release at vHPC-BLA synapses. Using a combination of optogenetics, pharmacology, and GRAB-eCB2.0 fiber photometry, we found that neuronal stimulation at 20–30 Hz resulted in robust retrograde release of 2-AG at vHPC-BLA synapses. Importantly, postsynaptic stimulation at these frequencies resulted in the engagement of endogenous CB1R at vHPC terminals, as revealed by our ex vivo electrophysiological experiments demonstrating 2-AG-mediated synaptic depression after 30 Hz stimulation of BLA neurons. Ultimately, these studies provide compelling evidence for activity-dependent release of 2-AG within a defined neural circuit and provide a new insight into the neural activity frequencies sufficient for in vivo 2-AG release. Indeed, photostimulation of BLA neurons at 20 Hz during an auditory and visual cue that predicts water delivery has been previously shown to promote approach behavior, suggesting that induction of these high-frequency stimulation patterns could be important for modulating behavior and recruitment of eCB signaling.40
Acute and repeated homotypic stress have been demonstrated to increase tissue levels of 2-AG in the amygdala, and it has been suggested that this increase serves as part of the endogenous stress adaptation response.9,12,15,19,41,42 Our data here refine the temporal specificity for stress-induced elevations in BLA eCBs by revealing phasic increases in eCB production and release at vHPC-BLA synapses that closely parallel increases in BLA activity. Specifically, eCB release was associated with increases in intracellular calcium in BLA neurons and behavioral responses including fleeing/darting (to footshock) and struggling (to restraint stress). Interestingly, GRAB-eCB2.0 responses to stress were blocked by the DAGL inhibitor DO34, confirming 2-AG, rather than other ligands, as the primary eCB released at vHPC-BLA synapses under these conditions. These data are consistent with bulk tissue analyses, which demonstrate that stress decreases levels of AEA in the amygdala and other limbic brain regions.12 Furthermore, reductions in neuronal activity using DREADD-based approaches reduced stress-induced increases in GRAB-eCB2.0 signal, supporting neuronal activity-dependent release of 2-AG in response to stress. These data provide high-temporal-resolution analysis of 2-AG release within defined neural circuits critical for stress reactivity and affective behavior and in vivo activity-dependent 2-AG release in response to salient environmental stimuli.
A key question arises from our previous data demonstrating relatively enhanced DSE at vHPC-BLA synapses in stress-resilient mice19 and from our current data revealing 2-AG release at vHPC-BLA synapses in response to stress exposure: is 2-AG-CB1R signaling at vHPC-BLA synapses causally linked to stress-induced behavioral reactivity and adaptation? Our data address this question in two separate, but related, ways. First, we found that 2-AG release occurred during bouts of active stress coping that were preceded by increases in BLA neuron activity. Combined with previous data suggesting that BLA activity (or activity of a subset of BLA output neurons) is associated with active behavioral responses to stress,43 this suggests a possible scenario whereby increases in BLA activity drive 2-AG release to dampen vHPC afferent excitation via activation of presynaptic CB1R. That deletion of CB1R from vHPC-BLA terminals reduced active stress coping in a variety of assays (restraint stress, TST, FST) suggests that vHPC inputs to the BLA promote passive stress coping. This hypothesis is consistent with data demonstrating that vHPC-BLA circuits are critical for driving freezing responses to contextual threat.44 How vHPC inputs to the BLA selectively drive “passive” output circuits or inhibit “active” output circuits from the BLA is not known but represents an important open question. Second, we found that deletion of CB1R from vHPC-BLA synapses promotes anhedonia and avoidance selectively after stress exposure and, more robustly, after repeated stress exposure. These data suggest that 2-AG released within the synapse during stress exposure plays an important role in buffering against subsequent adverse behavioral consequences of stress, consistent with previous studies.5–10,12 Taken together, these data indicate that 2-AG-CB1 signaling at vHPC-BLA synapses plays an important role in regulating behavioral responses during and after stress exposure and provides further support for the contention that 2-AG signaling within defined limbic circuits is a critical mechanism buffering against the adverse effects of stress exposure.
Our data and previous studies provide accumulating support for the notion that impairments in 2-AG-CB1R signaling could contribute to the pathogenesis of stress-related neuropsychiatric disorders. Indeed, recent identification of 2-AG signaling within BLA-PFC circuits as a key mechanism regulating stress-induced avoidance behaviors suggests a widely distributed role for 2-AG signaling within interconnected limbic nodes in the regulation of stress reactivity and adaptation.16 These studies also provide support for the continued investigation of pharmacological 2-AG augmentation strategies, including MAGL inhibitors, for the treatment of stress-related neuropsychiatric disorders.
Limitations of the study
One limitation of the present work is the apparent discrepancy between optogenetic BLA stimulation requirements for eCB release detection and physiological activity patterns of BLA neurons. Specifically, while the BLA is comprised of pyramidal cells that basally fire at low frequencies (<6 Hz), as well as interneuron populations that fire at higher frequencies (10–30 Hz),45 neuronal firing at 20–30 Hz was required for clear eCB detection using optogenetic approaches. How does this relate to stress-induced increases in GRAB-eCB2.0 signal if it is unlikely that BLA pyramidal neurons fire at such frequencies? First, many other factors contribute to eCB release beyond just cell firing that are not present during isolated optogenetic stimulation. For example, activation of Gq-coupled GPCRs facilitates eCB release, as we have shown here using a chemogenetic approach; it is possible that other neurotransmitters and neuromodulators like acetyl-choline, serotonin, or neuropeptides are released during stress and that these signals combined with lower levels of neuronal firing are sufficient to release eCBs at these synapses. Indeed, stress increases GRAB-eCB2.0 signals demonstrating that eCBs are released, indicating that there are likely to be unknown additional factors that align with neural firing to coordinate synaptic 2-AG release in vivo. Future studies will be required to identify these factors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources, reagents, and code should be directed to and will be fulfilled by the Lead Contact, Sachin Patel (sachin.patel@northwestern.edu).
Materials availability
All materials and code will be freely available upon request to the lead contact upon completion of materials transfer agreement, if relevant.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request
This paper does not contain original code
Access to any additional information can be requested from the lead contact
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committees and were conducted in accordance with the National Institute of Health guidelines for the Care and Use of Laboratory Animals. Adult C57BL/6J wild-type (Figures 1 and 2) or CB1Rfl/fl (Figure 3) mice were used as outlined in the figure legends. Mice were housed on a 12-hourlight/12-h dark cycle with lights on at 6:00 h; food and water were provided ad libitum. Mice that underwent surgery were 5–10 weeks old, and behavior was assessed at least 5 weeks following viral injection.
Male and female CB1Rfl/fl mice were bred in house on a homozygous x homozygous breeding scheme, and littermate-matched controls were used for all behavioral experiments in Figure 3. Mice were randomly assigned to treatment (i.e., CB1R KO [Cre; rgFlp x fDIO-Cre] or control [mCherry/GFP]).
Generation of Cnr1flx/flx mice
See.16
METHOD DETAILS
Surgeries
Mice were anesthetized with isoflurane and then transferred to the stereotax (Kopf Instruments, Tujunga, CA) and kept under 3% isoflurane anesthesia. Mice were injected with 10 mg/kg ketoprofen (AlliVet, St. Hialeah, FL) as an analgesic. The hair over the incision site was shaved and the skin was prepped with alcohol and iodine. A midline sagittal incision was made to expose the skull; a hole was drilled bilaterally above the vHPC (AP, −3.6; ML, ±3.0; DV, −4.0) or BLA (AP: −1.25, ML: ±3.30, DV: −5.00) and virus was delivered using a motorized digital software (NeuroStar, Stoelting CO., Wood Dale, IL), a 10μL microinjection syringe (Hamilton CO., Reno, NV), and a Micropump Controller (World Precision Instruments, Sarasota, FL) at 0.1μL per minute. A local, topical anesthetic, benzocaine (Medline Industries, Brentwood, TN) was applied to the incision area. After surgery, post-operative treatment with ketoprofen was administered for at least 48 h, up to 72 h.
Ex vivo electrophysiology
Male and female wild-type (Figure 1) or CB1Rfl/fl (Figure 3) mice were briefly anesthetized with isoflurane and transcardially perfused with ice-cold oxygenated (95% v/v O2, 5% v/v CO2) N-methyl-D-glucamine (NMDG) based ACSF46 comprised (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na-ascorbate, 3 Na-pyruvate, 5 N-acetylcyctine, 0.5 CaCl2$4H2O and 10 MgSO4$7H2O. The brain was quickly removed and 250 μm coronal slices containing the BLA or NAc were cut using a vibratome (Leica Biosystems, model # VT1000S) in the NMDG solution. Slices were incubated for 13–20 min at 32°C in oxygenated NMDG-ACSF then stored at 24°C until recordings were performed in HEPES-based ACSF containing (in mM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 ascorbate, 3 Na-pyruvate, 5 N-acetylcyctine, 2 CaCl2$4H2O and 2 MgSO4$7H2O.
Recordings were performed in a submerged recording chamber during continuous perfusion of oxygenated ACSF containing (in mM): 113 NaCl, 2.5 KCl, 1.2 MgSO4$7H2O, 2.5 CaCl2$2H2O, 1 NaH2PO4, 26 NaHCO3, 1 ascorbate, 3 Na-pyruvate and 20 glucose; at a flow rate of 2.5–3 mL/min. Slices were visualized using a Nikon microscope (Eclipse FN1, Nikon Instruments Inc., Melville, NY) equipped with differential interference contrast microscopy. Whole-cell current clamp recordings were obtained under visual control using a 40× objective. 2–6 MΩ borosilicate glass pipettes were filled with (Figures 3 and S3), a Cs+ based internal solution (in mM): 120 CsOH, 120 D-gluconic acid, 2.8 NaCl, 20 HEPES, 5 TEA-Cl, 2.5 Mg-ATP, 0.25 Na-GTP; or (Figure 1), a K+-gluconate internal solution (in mM): 125 K+ -gluconate, 4 NaCl, 10 HEPES, 4 MgATP, 0.3 Na-GTP, and 10 Na-phosphocreatine.
Ex vivo optogenetics, “oDSE”
For electrophysiological recordings in Figure 1, to confirm that BLA stimulation-induced 2-AG production can activate endogenous CB1R at vHPC-BLA synapses, wild-type mice were unilaterally injected with AAV5-CaMKIIaChR2(H134R)-eYFP (300nL) into the BLA and AAV5-Syn-Chrimson-TdT (450nL) into the vHPC. At least 4 weeks after virus expression, mice were sacrificed for electrophysiological recordings in the BLA. Importantly, we showed that blue light (465nm) selectively activated somatic BLA ChR2 (Figure 1N), while red light (615nm) activated Chrimson at vHPC terminals to induce optically-evoked EPSCs (oEPSCs). Neurons were held at −70mV; ChR2 was pulsed for 30Hz, at 20s to mimic our in vivo BLA stimulation paradigm (Figure 1). A baseline of 10 oEPSC (615nm; 1ms exposure time) were taken prior to stimulation and data is plotted as an oEPSC amplitude normalized to the baseline period. To confirm that “oDSE” was 2-AG-dependent, slices were incubated in the DAGL inhibitor, DO34 (2.5μM) for at least 40min prior to oDSE recordings. DO34 was dissolved in DMSO.
Fiber photometry
For somatic recordings of BLA activity, AAVrg-hSyn-GCaMP7f (300–400nL) was delivered unilaterally to the BLA. For assessment of eCB production and release, AAV9-hSyn-GRABeCB2.0 (650nL) or the control, AAV9-hSyn-GRABeCB-mut (650nL) was delivered unilaterally to the vHPC. At least 4 weeks following virus expression, a 400μM mono fiberoptic cannula (Doric) was implanted above the BLA (DV: −4.96). Mice were given at least one week to recover from fiber optic implantation before undergoing behavioral testing. A 465nm LED (Lx465; Tucker Davis Technologies [TDT]) was used to detect GFP-dependent changes in fluorescence; a 415nm LED (Lx415; TDT) was used as the isosbestic point. Light was emitted from the LEDs and passed through a minicube (Doric), that connected to the fiber implant via a 0.57 NA fiber optic patch cord (Doric). GFP-dependent changes in fluorescence were recorded by a real-time processor (RZ10x; TDT).
To confirm fiber placement and virus expression, mice were perfused, and brains were sectioned for histological verification of injection sites and fiber optic placement with a fluorescent microscope (Axio Imager M2 epifluorescent microscope). To confirm functional eCB2.0 sensor activity, mice were injected i.p. with the CB1R agonist, CP-55,940 (1 mg/kg; i.p.; dissolved in DMSO and injected at 1 μL/g bodyweight, or dissolved in a saline:kollifor:ethanol solution prepared in a dilution of 18:1:1 and injected 10 μL/g bodyweight); around 45min following CP-55,940 injection, mice were administered the CB1R inverse agonist, rimonabant (10 mg/kg; i.p.; dissolved in DMSO and injected at 1 μL/g bodyweight). Mice were excluded if there was no change in fluorescence following injection, because this suggests that eCB2.0 sensor was not functional, if the fiber optic was not above the BLA, or if there was no AAV-mediated fluorescence. A total of N = 5 mice were excluded based on these criteria.
BLA activity-dependent sensor activity
To assess activity-dependent, eCB production, a virus expressing Chrimson (AAV5-hSyn-Chrimson-TdT) was delivered to the BLA, and the eCB2.0 was delivered to the vHPC (Figure 1). A fiber optic was implanted above the BLA. Chrimson was activated at various stimulation frequencies (1–30Hz) for 20s. To confirm activity-dependent 2-AG production, mice were pretreated with DO34 (50 mg/kg; i.p.; dissolved in 18:1:1 saline:kollifor:ethanol and injected at 10 μL/g bodyweight), or JZL184 (20 mg/kg; i.p.; dissolved in DMSO and injected at 1 μL/g bodyweight) 2 h before testing (Figure 1).
To confirm BLA-dependent 2-AG production, a separate cohort of mice were injected with a virus that expresses the excitatory Gq-DREADD (Designer Receptor Exclusively Activated by Designer Drugs) (AAV5-CaMKII-hM3Dq-mCherry) (Figure S1) or inhibitory Gi-DREADD (AAV5-hSyn-hM4Di-mCherry) (Figure S2) in the BLA (300–400nL), and the eCB2.0 sensor was delivered to the vHPC. As before, a fiber optic was implanted above the BLA. Chemogenetic activation/inhibition of the BLA was induced by CNO injection (5–10 mg/kg; i.p.; dissolved in saline and injected at 10 μL/g bodyweight). Mice were tested 30min post CNO-injection.
Acute stress
Mice injected with eCB2.0/eCB-mut in the vHPC or GCaMP7f in the BLA were exposed to an acute footshock stress. This consisted of 2–6 unpredictable 0.7mA shocks (depending on specific experiment) using a MED Associated fear-conditioning chamber (St. Albans, VT, USA). Onset of shock was synchronized to fear conditioning software (FreezeFrame, acimetrics) via a TTL pulse or coded by hand. For restraint stress, mice were restrained in custom made (Vanderbilt Machine Shop, Nashville, TN) restraint tubes for 5–10 min.47 Active struggle bouts were coded posthoc, by hand, by a blinded experimenter (i.e., does not know what virus was injected). 1–5 struggle bouts were flagged per mouse per session.
INTRSECT behavior
Littermate-matched homozygous male and female CB1Rfl/fl mice were bilaterally injected with 450 nL AAV5-CMV-fDIO-Cre-mNeonGreen or AAV5-CMV-PI-eGFP-WPRE-bGH into the vHPC and 300 nL rAAV2-EF1a-mCherry-IRES-Flp into the BLA (Coh1); or 450 nL AAV5-CMV-fDIO-Cre-mNeonGreen into the vHPC and 300 nL rAAV2-EF1a-mCherry-IRES-Flp or mCherry control virus (rAAV2-CAG-tdTomato) into the BLA (Coh2). These virus combinations were used to confirm that rgFlp had no effect on its own, and to confirm that fDIO-Cre had no effect in the absence of rgFlp. There were no differences between control mice of both cohorts, so they were pooled together. Mice were allowed at least 5 weeks of recovery following virus surgery. Mice were singly housed for all experiments below, except locomotor testing.
Struggle behavior during restraint stress
Mice were restrained for 4min, and whole-body struggle behavior was quantified post-hoc, by 2 blinded experimenters. Struggle behavior was scored if the struggle lasted at least 1s, and separate bouts of struggle behavior was scored if there was at least 1s of immobility between bouts. The average between the two experimenters was used as final quantification.
Tail suspension test
Mice were hung by the tail with tape to a force meter. Mice were suspended for 6min, during which struggling behavior was measured through the force meter. Immobility time was gathered and processed by Med Associates Inc. tail suspension software, or by hand.
Forced swim test
Mice were placed in a 2L glass beaker filled with ~1.5L cold water. Mice were placed in the beaker and immobility was assessed for 5min.
Sucrose preference test
SPT was performed similarly to previously described testing.48 Mice were habituated to two identical 50mL canonical tubes that were filled with drinking water, for 24 h. One canonical tube was then replaced with 1.5% sucrose solution, made fresh. Bottle locations were flipped after 12hrs to prevent preferential drinking based on the side. Following a total of 24hrs with sucrose in the cage, the total consumption of water and sucrose solution was measured by weighing the tubes.
Mice were then exposed to an acute stress exposure, where mice were exposed to six 0.7mA foot shocks delivered 1 min apart using a MED Associates fear-conditioning chamber (St. Albans, VT). Each shock coincided with the last 2 s of a 30 s auditory tone. After footshock, mice were returned to their homecage with two identical 50mL canonical tubes (one containing water, and one containing 1.5% sucrose). Bottle locations were flipped after 12hrs. 24hrs after footshock, total consumption of water and sucrose solutions was measured by weighing the tubes. Importantly, two empty cages were also given two canonical tubes and the “drip” was measured after both 24hrs and 48hrs after the start of the initial test. This was averaged and subtracted from the final weight. The sucrose preference was calculated as (Δweight sucrose)/(Δweight sucrose + Δweight water) × 100%.
Locomotion
Mice were tested for deficits in locomotion in an open-field arena (27.9 × 27.9 × 20.3 cm; MED-OFA-510; MED Associates, St. Albans, Vermont), contained within a sound-attenuating chamber. Locomotion was monitored for 10min. Walls of the open-field arena were made of clear plexiglass. Beam breaks from 16 infrared beams were recorded by Activity Monitor v5.10 (MED Associates) to monitor activity.
Repeated novelty-induced hypophagia
rNIH was performed as previously described.19 Briefly, mice were habituated to liquid vanilla Ensure (Abobott Laboratries, Abbott Park, IL) in their home cages for 30 min per day for 4 days (“Training”). On test day, mice were transferred to a novel, empty cage in a well-lit room and given access to liquid vanilla Ensure for 30 min (“NIH Test”). The next week, mice underwent home-cage “Training” for 2 consecutive days. After the 2nd home-cage training, mice were exposed to foot shock stress as previously described.19 This foot shock stress is a 7.5-min session which consists of six 0.7mA foot shocks delivered 1 min apart using a MED Associates fear-conditioning chamber (St. Albans, VT). Each shock coincided with the last 2 s of a 30 s auditory tone. After stress exposure, mice were returned to their home cages. Approximately 24 h after foot shock, mice underwent novel-cage NIH testing (“1 FS NIH”). Mice were exposed to five days of consecutive foot shock stress and returned back to their home-cages after each foot shock session. 24 h after the 5th foot shock, mice underwent NIH testing (“5 FS NIH”). The stress-induced latency was calculated as the stress latency after 5 days of FS (“5 FS NIH”) – latency on basal NIH Test (“Basal NIH Test”); if the stress-induced change in latency was <120s, mice were classified as resilient, but if the stress-induced change in latency was >120s, mice were classified as susceptible.19
Ex vivo optogenetic Validation
To confirm that the CB1R was deleted selectively from vHPC-BLA synapses, CB1Rfl/fl mice were bilaterally injected with 450 nL AAV5-CMV-fDIO-Cre-mNeonGreen + AAV5-EF1a-DIO-ChR2(H134R)-eYFP-WPRE into the vHPC (1:1 ratio), and 300 nL rAAV2-EF1a-mCherry-IRES-Flp into the BLA (KO). Control mice were injected with 450 nL AAV-GFP + ChR2 into vHPC, and 300 nL rAAV2-EF1a-mCherry-IRES-Flp into the BLA. Following at least 5 weeks of virus expression, mice were sacrificed and slices of the BLA were obtained. vHPC-mediated oEPSCs were stimulated for a baseline period of 3–5min; the CB1R agonist, CP-55,940 (10μM) was washed on, and results are presented as oEPSC amplitude normalized to the baseline period.
To validate the CB1R-agonism still induced depression at other vHPC outputs, CB1Rfl/fl mice were bilaterally injected with 450 nL AAV5-CMV-fDIO-Cre-mNeonGreen + ChR2 into the vHPC (1:1 ratio), and 300 nL rAAV2-EF1a-mCherry-IRES-Flp (KO) or rg-GFP (GFP) into the BLA. Following virus expression, slices of the NAc were obtained and vHPC-mediated oEPSCs were stimulated for a baseline period of 3–5min. The CB1R agonist, CP-55,940 (10μM) was washed on, and results are presented as oEPSC amplitude normalized to the baseline period. In both experiments, CP-55,940 was dissolved in DMSO and included in ACSF and holding/incubation chambers also containing 0.1–0.5 mg/mL bovine serum albumin.
vHPCCB1R global KO behavior
Littermate-matched homozygous CB1Rfl/fl mice were bilaterally injected with 650 nL AAV8-Ef1a-mCherry-IRES-Cre (AAV-Cre) or AAV5-CMV-PI-eGFP-WPRRE-bGH (AAV-GFP) into the vHPC. Mice were given at least 5 weeks recovery post-virus injection, before behavioral testing. Mice were singly housed for NIH, but not locomotor activity.
Locomotion
Mice were tested for deficits in locomotion as described above for INTRSECT cohorts of mice.
Ex vivo optogenetic validation
To confirm that Cre-injection induced deletion of vHPCCB1R, CB1Rfl/fl mice were bilaterally injected with 450 nL AAV5-Cre + AAV5-EF1a-DIO-ChR2(H134R)-eYFP-WPRE into the vHPC (1:1 ratio). Following virus expression, slices of the NAc were obtained and vHPC-mediated oEPSCs were stimulated for a baseline period of 3–5min. The CB1R agonist, CP-55,940 (10μM) was washed on, and results are presented as oEPSC amplitude normalized to the baseline period. In both experiments, CP-55,940 was dissolved in DMSO and included in ACSF containing 0.1–0.5 mg/mL bovine serum albumin.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests are reported in the figure legends. For analysis of 2 groups, an unpaired or paired student’s t-test was performed. For analysis of two or more groups across two or more treatments or time points, a two-way ANOVA with Holm-Sidak post hoc correction was used. For all datasets, significance was defined by a p value of <0.05. The following is used: *, p < 0.05; **, p < 0.01; ***,p < 0.001. The ROUT outlier test was run on each dataset individually; when multiple measures were taken from an identified outlier, measures were treated independently, and the corresponding data point was removed from only from that dataset.
Behavior
Behavior was analyzed as reported under individual method sections. For stress susceptibility in Figures 3O–3Q, a one-sided Chi squared test was performed, because we have previously determined that dysfunctional 2-AG/CB1R signaling at vHPC-BLA synapses is linked to enhanced susceptibility.19 Mice were excluded from behavioral experiments if the viral injections were misplaced or if there was no viral expression. Furthermore, mice were excluded if there was a technical issue during the behavioral experiment (e.g., mouse fell out the EZM/EPM or there was a malfunction on the behavioral hardware or software). All data presented as mean ± SEM.
Electrophysiology
Electrophysiological data was initially analyzed using ClampFit 10.5 software (Molecular Devices, San Jose, California). Datasets were organized in Microsoft Excel and then transferred to GraphPad Prism 6.0 for generation of graphs and statistical analyses. Mice were excluded from physiological experiments if there was improper virus targeting or if there was no viral expression. Neurons were excluded from physiological experiments for five reasons. 1: if the holding current dropped below −300 pA at any time during the recording. 2: if the access resistance was >30 MΩ. 3: if the access resistance fluctuated by more than 20% throughout the recording. 4: The optogenetically-evoked response was <100pA during baseline. 5: if the baseline responses were not stable.
Fiber photometry
Fiber photometry was analyzed using code modified from TDT (https://www.tdt.com/docs/sdk/offline-data-analysis/offline-data-matlab/fiber-photometry-epoch-averaging-example/) and thus does not report original code. Traces are presented as ΔF/F, which was calculated via algorithm sourced from Tom Davidson’s Github (https://github.com/tjd2002/tjd-shared-code/blob/master/matlab/photometry/FP_normalize.m). This involved fitting the 415nm channel onto the 465nm channel to detrend signal bleaching via the polyfit function. The Z score is calculated as the change in ΔF/F, using the seconds −3 to −1 before stimulus onset (0s) as the baseline (Figures 1 and 2). The Z score was used to account for between-subject variability in signal magnitude.
For long-term recordings (Figure S1), we identified significant photobleaching of the eCB2.0 sensor; as such, the fluorescent signal of each mouse was baseline corrected, individually, using an exponential decay model. The first 20min of each recording were used to obtain the k (rate) and y0 (for each recording) using the nonlin fit function in PRISM. Exponential decay models were fitted over each recording to ensure proper plateau values and confirm that the model predicted the photobleaching decay. Data that underwent baseline correction is presented as ΔF/F*.
The change in the area under the curve (AUC) was calculated using the time before stimulus onset (0) as baseline. The resulting AUC was calculated using the entirety of the recording post-baseline (Figures 1 and 2) or during the time course depicted by the shaded region (Figure S1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AAV5-CMV-fDIO-Cre-mNeonGreen-wPRE | Marcus et al.16 | VB180530–1030aad |
| rAAV2-EF1a-mCherry-IRES-Flpo | Fenno et al.49 | RRID: Addgene_55634 |
| AAV5-CaMKII-ChR2(H134R)-eYFP-wPRE | Lee et al.50 | RRID: Addgene_26969 |
| AAV5-EF1a-DIO-ChR2(H134R)-eYFP-wPRE | Gift from Karl Deisseroth | Addgene Catalog #: 20298-AAV5 |
| AAV8-Ef1a-mCherry-IRES-Cre | Fenno et al.49 | Addgene Catalog #: 55632-AAV8 |
| AAV5-CMV-PI-eGFP-WPRE-bGH | Gift from James M. Wilson | RRID: Addgene_105530 |
| AAV9-hSyn-GRABeCB2.0 | Gift from Yulong Li Dong et al.31 | |
| AAV9-hSyn-GRABeCB2.0-mut | WZ Biosciences, Inc | |
| rAAV2-CAG-tdTomato | Chan et al.51 | RRID: Addgene_59462 |
| AAV5-Syn-Chrimson-TdT | Klapoetke et al.52 | Addgene Catalog #: 59171-AAV5 |
| rgAAV-syn-jGCaMP7f-WPRE | Dana et al.53 | Addgene #: 104488-AAVrg |
| AAV5-hSyn-hM4Di-mCherry | Gift from Brryan Roth Krashes et al.54 | Addgene #: 44362-AAV5 |
| AAV5-CaMKII-hM3Dq-mCherry | Gift from Bryan Roth | RRID:Addgene_50476 |
| Chemicals, peptides, and recombinant proteins | ||
| Rimonabant | Cayman Chemicals | 9000484 |
| DO34 | Glixx Laboraties | GLXC-09757 |
| JZL184 | Cayman Chemicals | 13158 |
| CP-55,940 | Cayman Chemicals | 13608 |
| CNO-HCl | Cayman Chemical | 25780 |
| Experimental models: Organisms/strains | ||
| CB1Rfl/fl mice | Dr. Eric Delpire | N/A |
| C57 WT mice | Jackson Laboratory | IMSR_JAX000664 |
| Software and algorithms | ||
| Prism | Graphpad | https://www.graphpad.com/ |
| pClamp10.5 | Molecular Devices | https://www.moleculardevices.com/ |
| MATLAB | Mathworks | https://www.mathworks.com/ |
| ANY-Maze | Stoelting Co | https://stoelting.com/ |
| Other | ||
| Fear Conditioning Chamber | Med Associates | MED-VFC-SCT-M |
| TDT Fiber Photometry System | TDT | |
Highlights.
BLA activity promotes eCB release at vHPC-BLA synapses
Active stress coping recruits vHPC-BLA eCB signaling
vHPC-BLA CB1 receptor deletion exacerbates stress-induced avoidance/anhedonia
ACKNOWLEDGMENTS
These studies were supported by NIH grants F31MH126I460 (V.K.), MH107435 (S.P.), and MH119817 (S.P.). The Cnr1 floxed mouse generation was supported by the Integrative Neuroscience Initiative on Alcoholism (INIA stress) grant AA9013514 (E.D.). Behavior experiments were performed in part through the use of the Murine Neurobehavioral Core lab at the Vanderbilt University Medical Center. Figure 3O was created with BioRender.com.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113027.
DECLARATION OF INTERESTS
S.P. is a scientific consultant for Psy Therapeutics, Janssen Pharmaceuticals, and Jazz Pharmaceuticals, unrelated to the present work.
REFERENCES
- 1.Arnsten AFT (2015). Stress weakens prefrontal networks: molecular insults to higher cognition. Nat. Neurosci 18, 1376–1385. 10.1038/nn.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McEwen BS, Eiland L, Hunter RG, and Miller MM (2012). Stress and anxiety: structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology 62, 3–12. 10.1016/j.neuropharm.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma S, Powers A, Bradley B, and Ressler KJ (2016). Gene x Environment Determinants of Stress- and Anxiety-Related Disorders. Annu. Rev. Psychol 67, 239–261. 10.1146/annurev-psych-122414-033408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondev V, Winters N, and Patel S (2021). Cannabis use and posttraumatic stress disorder comorbidity: Epidemiology, biology and the potential for novel treatment approaches. Int. Rev. Neurobiol 157, 143–193. 10.1016/bs.irn.2020.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Bedse G, Hill MN, and Patel S (2020). 2-Arachidonoylglycerol Modulation of Anxiety and Stress Adaptation: From Grass Roots to Novel Therapeutics. Biol. Psychiatr 88, 520–530. 10.1016/j.biopsych.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill MN, Campolongo P, Yehuda R, and Patel S (2018). Integrating Endocannabinoid Signaling and Cannabinoids into the Biology and Treatment of Posttraumatic Stress Disorder. Neuropsychopharmacology 43, 80–102. 10.1038/npp.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bedse G, Bluett RJ, Patrick TA, Romness NK, Gaulden AD, Kingsley PJ, Plath N, Marnett LJ, and Patel S (2018). Therapeutic endocannabinoid augmentation for mood and anxiety disorders: comparative profiling of FAAH, MAGL and dual inhibitors. Transl. Psychiatry 8, 92. 10.1038/s41398-018-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedse G, Hartley ND, Neale E, Gaulden AD, Patrick TA, Kingsley PJ, Uddin MJ, Plath N, Marnett LJ, and Patel S (2017). Functional Redundancy Between Canonical Endocannabinoid Signaling Systems in the Modulation of Anxiety. Biol. Psychiatr 82, 488–499. 10.1016/j.biopsych.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill MN, Patel S, Campolongo P, Tasker JG, Wotjak CT, and Bains JS (2010). Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. J. Neurosci 30, 14980–14986. 10.1523/JNEUROSCI.4283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel SA, DeMichele A, Cheer JF, Wotjak CT, and Holmes A (2017). The endocannabinoid system as a target for novel anxiolytic drugs. Curr. Oncol. Rep 19, 56–66. 10.1016/j.neubiorev.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowe DJE, Sasiadek JD, Coles AS, and George TP (2019). Cannabis and mental illness: a review. Eur. Arch. Psychiatr. Clin. Neurosci 269, 107–120. 10.1007/s00406-018-0970-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morena M, Patel S, Bains JS, and Hill MN (2016). Neurobiological Interactions Between Stress and the Endocannabinoid System. Neuropsychopharmacology 41, 80–102. 10.1038/npp.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorzalka BB, Hill MN, and Hillard CJ (2008). Regulation of endocannabinoid signaling by stress: implications for stress-related affective disorders. Neurosci. Biobehav. Rev 32, 1152–1160. 10.1016/j.neubiorev.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Hill MN, and McEwen BS (2010). Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 34, 791–797. 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondev V, Morgan A, Najeed M, Winters ND, Kingsley PJ, Marnett L, and Patel S (2022). The Endocannabinoid 2-Arachidonoylglycerol bidirectionally modulates acute and protracted effects of predator odor exposure. Biol. Psychiatr 92, 739–749. 10.1016/j.biopsych.2022.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcus DJ, Bedse G, Gaulden AD, Ryan JD, Kondev V, Winters ND, Rosas-Vidal LE, Altemus M, Mackie K, Lee FS, et al. (2020). Endocannabinoid Signaling Collapse Mediates Stress-Induced Amygdalo-Cortical Strengthening. Neuron 105, 1062–1076.e6. 10.1016/j.neuron.2019.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan A, Kondev V, Bedse G, Baldi R, Marcus D, and Patel S (2019). Cyclooxygenase-2 inhibition reduces anxiety-like behavior and normalizes enhanced amygdala glutamatergic transmission following chronic oral corticosterone treatment. Neurobiol. Stress 11, 100190. 10.1016/j.ynstr.2019.100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel S, Roelke CT, Rademacher DJ, and Hillard CJ (2005). Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur. J. Neurosci 21, 1057–1069. 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- 19.Bluett RJ, Báldi R, Haymer A, Gaulden AD, Hartley ND, Parrish WP, Baechle J, Marcus DJ, Mardam-Bey R, Shonesy BC, et al. (2017). Endocannabinoid signalling modulates susceptibility to traumatic stress exposure. Nat. Commun 8, 14782. 10.1038/ncomms14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diamond DM, and Zoladz PR (2016). Dysfunctional or hyperfunctional? The amygdala in posttraumatic stress disorder is the bull in the evolutionary China shop. J. Neurosci. Res 94, 437–444. 10.1002/jnr.23684. [DOI] [PubMed] [Google Scholar]
- 21.Hughes KC, and Shin LM (2011). Functional neuroimaging studies of post-traumatic stress disorder. Expert Rev. Neurother 11, 275–285. 10.1586/ern.10.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheynin J, and Liberzon I (2017). Circuit dysregulation and circuit-based treatments in posttraumatic stress disorder. Neurosci. Lett 649, 133–138. 10.1016/j.neulet.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin LM, Rauch SL, and Pitman RK (2006). Amygdala, medial pre-frontal cortex, and hippocampal function in PTSD. Ann. N. Y. Acad. Sci 1071, 67–79. 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Huang Z, Ren L, Liu J, Wang X, Yu T, Hu M, Wang X, Du J, Ni D, et al. (2020). Amygdalar and hippocampal beta rhythm synchrony during human fear memory retrieval. Acta Neurochir 162, 2499–2507. 10.1007/s00701-020-04276-y. [DOI] [PubMed] [Google Scholar]
- 25.Bienvenu TCM, Busti D, Magill PJ, Ferraguti F, and Capogna M (2012). Cell-type-specific recruitment of amygdala interneurons to hippocampal theta rhythm and noxious stimuli in vivo. Neuron 74, 1059–1074. 10.1016/j.neuron.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Likhtik E, and Gordon JA (2014). Circuits in sync: decoding theta communication in fear and safety. Neuropsychopharmacology 39, 235–236. 10.1038/npp.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill JL, and Martinowich K (2016). Activity-dependent signaling: influence on plasticity in circuits controlling fear-related behavior. Curr. Opin. Neurobiol 36, 59–65. 10.1016/j.conb.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh S, Laxmi TR, and Chattarji S (2013). Functional connectivity from the amygdala to the hippocampus grows stronger after stress. J. Neurosci 33, 7234–7244. 10.1523/JNEUROSCI.0638-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruces-Solis H, Babaev O, Ali H, Piletti Chatain C, Mykytiuk V, Balekoglu N, Wenger S, and Krueger-Burg D (2021). Altered theta and beta oscillatory synchrony in a genetic mouse model of pathological anxiety. Faseb. J 35, e21585. 10.1096/fj.202002028RR. [DOI] [PubMed] [Google Scholar]
- 30.Pitkanen A, Pikkarainen M, Nurminen N, and Ylinen A (2000). Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann N Y Acad Sci 911, 369–391. 10.1111/j.1749-6632.2000.tb06738.x. [DOI] [PubMed] [Google Scholar]
- 31.Dong A, He K, Dudok B, Farrell JS, Guan W, Liput DJ, Puhl HL, Cai R, Wang H, Duan J, et al. (2022). A fluorescent sensor for spatio-temporally resolved imaging of endocannabinoid dynamics in vivo. Nat. Biotechnol 40, 787–798. 10.1038/s41587-021-01074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, and Watanabe M (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev 89, 309–380. 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 33.Stella N, Schweitzer P, and Piomelli D (1997). A second endogenous cannabinoid that modulates long-term potentiation. Nature 388, 773–778. 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 34.Wilson RI, Kunos G, and Nicoll RA (2001). Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31, 453–462. 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 35.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, et al. (2009). Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol 5, 37–44. 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin LM, and Liberzon I (2010). The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology 35, 169–191. 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortiz S, Latsko MS, Fouty JL, Dutta S, Adkins JM, and Jasnow AM (2019). Anterior Cingulate Cortex and Ventral Hippocampal Inputs to the Basolateral Amygdala Selectively Control Generalized Fear. J. Neurosci 39, 6526–6539. 10.1523/JNEUROSCI.0810-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seidenbecher T, and Lesting J (2012). Amygdala-hippocampal Theta Synchrony in Learning, Memory, and Disease (Nova Science Publishers, Inc.). [Google Scholar]
- 39.Zhang Q, Gao SH, Shen ZS, Wang Y, Hu SW, Duan GB, Liu Y, Zhong DY, Liu J, Sun MH, et al. (2022). The Slack Channel Regulates Anxiety-Like Behaviors via Basolateral Amygdala Glutamatergic Projections to Ventral Hippocampus. J. Neurosci 42, 3049–3064. 10.1523/JNEUROSCI.2027-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Servonnet A, Hernandez G, El Hage C, Rompré PP, and Samaha AN (2020). Optogenetic Activation of the Basolateral Amygdala Promotes Both Appetitive Conditioning and the Instrumental Pursuit of Reward Cues. J. Neurosci 40, 1732–1743. 10.1523/JNEUROSCI.2196-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel S, and Hillard CJ (2008). Adaptations in endocannabinoid signaling in response to repeated homotypic stress: a novel mechanism for stress habituation. Eur. J. Neurosci 27, 2821–2829. 10.1111/j.1460-9568.2008.06266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TTY, Gray JM, Hillard CJ, Gorzalka BB, and Viau V (2010). Endogenous cannabinoid signaling is essential for stress adaptation. Proc. Natl. Acad. Sci. USA 107, 9406–9411. 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terburg D, Scheggia D, Triana Del Rio R, Klumpers F, Ciobanu AC, Morgan B, Montoya ER, Bos PA, Giobellina G, van den Burg EH, et al. (2018). The Basolateral Amygdala Is Essential for Rapid Escape: A Human and Rodent Study. Cell 175, 723–735.e16. 10.1016/j.cell.2018.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim WB, and Cho JH (2020). Encoding of contextual fear memory in hippocampal-amygdala circuit. Nat. Commun 11, 1382. 10.1038/s41467-020-15121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amir A, Headley DB, Lee SC, Haufler D, and Paré D (2018). Vigilance-Associated Gamma Oscillations Coordinate the Ensemble Activity of Basolateral Amygdala Neurons. Neuron 97, 656–669.e7. 10.1016/j.neuron.2017.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luchsinger JR, Fetterly TL, Williford KM, Salimando GJ, Doyle MA, Maldonado J, Simerly RB, Winder DG, and Centanni SW (2021). Delineation of an insula-BNST circuit engaged by struggling behavior that regulates avoidance in mice. Nat. Commun 12, 3561. 10.1038/s41467-021-23674-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng J, Umschweif G, Leung J, Sagi Y, and Greengard P (2019). HCN2 Channels in Cholinergic Interneurons of Nucleus Accumbens Shell Regulate Depressive Behaviors. Neuron 101, 662–672.e5. 10.1016/j.neuron.2018.12.018. [DOI] [PubMed] [Google Scholar]
- 49.Fenno LE, Mattis J, Ramakrishnan C, Hyun M, Lee SY, He M, Tucciarone J, Selimbeyoglu A, Berndt A, Grosenick L, et al. (2014). Targeting cells with single vectors using multiple-feature Boolean logic. Nat. Methods 11, 763–772. 10.1038/nmeth.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim DS, Fenno LE, Ramakrishnan C, and Deisseroth K (2010). Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature 465, 788–792. 10.1038/nature09108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, and Gradinaru V (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci 20, 1172–1179. 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dana H, Sun Y, Mohar B, Hulse BK, Kerlin AM, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, et al. (2019). High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657. 10.1038/s41592-019-0435-6. [DOI] [PubMed] [Google Scholar]
- 54.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest 121, 1424–1428. 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request
This paper does not contain original code
Access to any additional information can be requested from the lead contact