

Abstract
Background:
Age is the most common risk factor for Alzheimer’s disease (AD), a neurodegenerative disorder characterized by the hallmarks of toxic amyloid-β (Aβ) plaques and hyperphosphorylated tau tangles. Moreover, sub-physiological brain insulin levels have emerged as a pathological manifestation of AD.
Objective:
Identify age-related changes in the plasma disposition and blood-brain barrier (BBB) trafficking of Aβ peptides and insulin in mice.
Methods:
Upon systemic injection of 125I-Aβ40, 125I-Aβ42, or 125I-insulin, the plasma pharmacokinetics and brain influx were assessed in wild-type (WT) or AD transgenic (APP/PS1) mice at various ages. Additionally, publicly available single-cell RNA-Seq data [GSE129788] was employed to investigate pathways regulating BBB transport in WT mice at different ages.
Results:
The brain influx of 125I-Aβ40, estimated as the permeability-surface area product, decreased with age, accompanied by an increase in plasma AUC. In contrast, the brain influx of 125I-Aβ42 increased with age, accompanied by a decrease in plasma AUC. The age-dependent changes observed in WT mice were accelerated in APP/PS1 mice. As seen with 125I-Aβ40, the brain influx of 125I-insulin decreased with age in WT mice, accompanied by an increase in plasma AUC. This finding was further supported by dynamic single-photon emission computed tomography (SPECT/CT) imaging studies. RAGE and PI3K/AKT signaling pathways at the BBB, which are implicated in Aβ and insulin transcytosis, respectively, were upregulated with age in WT mice, indicating BBB insulin resistance.
Conclusion:
Aging differentially affects the plasma pharmacokinetics and brain influx of Aβ isoforms and insulin in a manner that could potentially augment AD risk.
Keywords: Aging, amyloid-β, blood-brain barrier, insulin, pharmacokinetics
INTRODUCTION
The blood-brain barrier (BBB) is responsible for maintaining the dynamic equilibrium between endogenous solute levels in the blood and the brain. The BBB endothelium serves dual functions, acting as both a gatekeeper to restrict the uptake of toxic substances circulating in the blood, and also as a trafficking portal to mediate the highly-selective delivery of essential nutrients to the brain. Additionally, the BBB plays a crucial role in clearing metabolic waste products from the brain. Disruptions to these spatially coordinated trafficking events have been implicated in the etiology of several neurodegenerative disorders, including sporadic Alzheimer’s disease (AD). Progression of physiological age is widely recognized as the greatest risk factor for AD [1]. While normal aging is associated with gradual loss of BBB function [2] and appearance of neuropathological changes, these are accelerated in AD brain and trigger catastrophic neurocognitive changes [3].
One major consequence of age-related BBB dysfunction in AD brain is the increased accumulation of amyloid-β (Aβ) peptides in the cerebral vasculature as amyloid deposits and in the brain parenchyma as senile plaques [4]. Of the two major Aβ isoforms that predominate in AD brain, Aβ40 is the major isoform present in cerebrovascular amyloid deposits, whereas Aβ42 is the major isoform present in parenchymal plaques. Notably, Aβ42 is considered to be more neurotoxic and amyloidogenic than Aβ40 [5]. Interestingly, Aβ40 is also suggested to be protective against Aβ42-induced neurotoxicity [6, 7]. However, changes in how the BBB handles each Aβ isoform during aging and AD remain unclear. Brain Aβ deposition precedes cognitive decline and continues to increase substantially over the course of AD progression. It has been estimated that 20–30% of all cognitively-normal older adults display significant Aβ deposition in the brain [4]. It is well established that decreased Aβ clearance from the brain promotes Aβ accumulation and plaque formation during aging and AD [8]. In contrast, very little is known about how alterations in Aβ handling by the periphery contribute to brain Aβ deposition in AD. Although the Aβ concentrations in plasma are typically ~ 6-fold lower than the concentrations in the brain interstitial fluid, the absolute amount of Aβ in plasma is estimated to be ~ 10-fold greater than the amount of soluble Aβ in the brain [9]. In aged nonhuman primates, it was shown that systemically injected Aβ efficiently crossed the BBB and became incorporated into existing parenchymal plaques [10]. Further, increased brain Aβ deposition in aged rats was associated with increased BBB expression of the receptor for advanced glycation end products (RAGE), which handles Aβ influx to the brain [11]. Elevated serum Aβ levels are also known to increase the risk of AD in elderly patients [12]. Therefore, it is likely that systemic Aβ can directly, by trafficking Aβ into the brain, or indirectly, by reducing the ability of BBB to clear brain Aβ, contribute to the brain Aβ load.
BBB dysfunction in aging and AD is associated with decreased influx of essential nutrients to the brain [13]. In particular, glucose and insulin are produced almost exclusively in peripheral tissues and must be delivered to the brain across the BBB. Decreased insulin levels in the cerebrospinal fluid are associated with progression of cognitive decline in early-stage AD patients [14]. In another study, insulin concentrations and insulin receptor (IR) density in postmortem brain decreased with age in subjects with or without AD [15]. We and others have demonstrated that Aβ peptide exposure interferes with the neurobiological functions mediated by insulin and IR [16–19]. Further, we reported that insulin modulates Aβ peptide transport at the BBB in mice, as well as the membrane expression of Aβ receptors (RAGE, LRP-1) in a BBB cell culture model [20]. Thus, the effects of insulin and Aβ on the BBB are strongly interconnected, and disruptions in this relationship are expected to promote BBB dysfunction in AD.
To investigate the contributions of aging to BBB dysfunction, we examined the plasma distribution and brain influx kinetics of 125I radiolabeled Aβ40, Aβ42, and insulin in female wild-type (WT) and/or AD transgenic (APP/PS1) mice at various ages. Using single-cell RNA-Seq data previously published by Ximerakis et al. [21], we further assessed the activity of various transport/signaling pathways in brain endothelial cells obtained from WT mice at different ages. Our findings reveal an intricate switch in the brain influx of Aβ isoforms and insulin with age, accompanied by disruptions to transport/signaling pathways at the BBB. These changes are expected to contribute to pathological alterations in Aβ and insulin levels in the brain that are intricately linked with neuropathological changes observed in AD patients.
MATERIALS AND METHODS
Preparation of Aβ peptides
Aβ40 and Aβ42 peptides were custom synthesized on a fluorenylmethyloxycarbonyl (FMOC) column by the Mayo Clinic proteomics core facility (Rochester, MN). Solutions of soluble Aβ were prepared using the procedure developed by Klein et al. [22]. Briefly, solid Aβ was dissolved in ice-coldhexafluoro-2-propanol (HFIP) (MP Biomedicals, Santa Ana, CA) and then incubated at room temperature for 1h. The solvent was evaporated overnight, then further dried under vacuum, and the resultant Aβ films were stored at −20°C prior to use. Before labeling with 125I, the Aβ films were dissolved in DMSO, diluted in Ham’s F-12 media (Mediatech, Manassas, VA), and centrifuged at 18,000 rpm to remove any insoluble Aβ aggregates.
Radioiodination of Aβ peptides and insulin
Carrier-free Na125I and Na131I radionuclides were obtained from PerkinElmer Life and Analytical Sciences(Boston, MA). The following peptides/proteins were labeled with 125I or 131I using the chloramine-T procedure as described previously [23, 24]: Aβ40, Aβ42, bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO), and human insulin (Sigma-Aldrich). Free radioactive iodine was separated from the radiolabeled peptide/protein by dialysis against 0.01M phosphate-buffered saline (PBS) at pH 7.4 (Sigma-Aldrich). Purity of the radiolabeled peptides/proteins was assessed by trichloroacetic acid (TCA) precipitation. The preparation was deemed acceptable if the precipitable radioactivity counts were greater than 95% of the total counts.
Animals
Animal studies were carried out in compliance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocols were approved by the Institutional Animal Care and Use Committee at Mayo Clinic, Rochester, MN (Mayo IACUC #A00006176–21). The studies were documented according to the ARRIVE reporting guidelines. Wild-type (C57BL/6J) were purchased from The Jackson Laboratory (Bar Harbor, ME). The APP/PS1 mice were generated by mating hemizygous transgenic mice (mouse strain C57B6/SJL; i.d. no. Tg2576) expressing mutant human amyloid precursor protein (APP695) with a second strain of hemizygous transgenic mice (mouse strain Swiss-Webster/B6D2; i.d. no. M146L6.2) expressing mutant human presenilin 1 (PS1). APP/PS1 mice exhibit Aβ overexpression, accelerated brain Aβ deposition, and cognitive decline. Mice were housed in a virus-free barrier facility with 12-h light and dark cycles and were provided with pellet food and purified water ad libitum. All studies were performed using female mice. Prior to the experiments, the mice were confirmed by visual inspection to be in the diestrus phase of the estrous cycle [25], during which hormonal fluctuations are reported to be minimal [26].
Plasma pharmacokinetics and brain permeability of 125I-Aβ42 and 125I-Aβ40
These experiments were carried out as described in our earlier publications [24, 27]. The age groups were selected based on our previous findings which demonstrated that in APP/PS1 mice, brain Aβ plaques were undetectable at 12 weeks of age, with initiation of plaque formation at 24 weeks, and full-scale plaque burden at 52 weeks [28]. Briefly, wild-type (WT) or APP/PS1 transgenic mice at various ages (8, 24, or 52 weeks) weighing 25–35g were catheterized at the femoral vein and femoral artery while under general anesthesia (1.5% isoflurane; 4L/min oxygen). A single dose (100μCi) of 125I-Aβ40 or 125I-Aβ42 in PBS (100μL) was bolus injected into the femoral vein. Blood was sampled serially (20μL) from the femoral artery at 0.25-,1-,3-,5-,10-, and 15-min post-injection. Immediately after the 15-min sampling event, 131I-BSA (100μCi) was bolus injected into the femoral vein to serve as a measure of the residual plasma volume (Vp). One minute after the 131I-BSA injection, a final blood sample was collected, and the animal was euthanized. Blood samples were diluted in PBS and centrifuged to separate the plasma. The plasma was subjected to TCA precipitation and then centrifuged. The total 125Iand 131I activity counts in the pellet, corresponding to intact radiolabeled peptide/protein, were assayed using a two-channel gamma counter (Cobra II; PerkinElmer Life and Analytical Sciences, Boston, MA).
The 125I-Aβ40 and 125I-Aβ42 plasma concentration versus time data was fitted with the following bi-exponential equation using Phoenix WinNonlin® 6.4 (Certara, St. Louis, MO):
| (1) |
where is the concentration (μCi/mL) in plasma at time (min), and are the intercepts of the distribution and elimination phases, respectively, and and are the distribution and elimination macro-rate constants (min−1), respectively. The secondary plasma pharmacokinetic parameters are reported in Table 1.
Table 1.
Plasma pharmacokinetics of 125I-Aβ40 and 125I-Aβ42 in WT and APP/PS1 transgenic mice at 8, 24, and 52 weeks
| Wild-type | ||||||
|---|---|---|---|---|---|---|
| Age (weeks) |
125I-Aβ40 |
125I-Aβ42 |
||||
| Cmax (μCi/mL) | AUC (μCi.min/mL) | Clearance (mL/min) | Cmax (μCi/mL) | AUC (μCi.min/mL) | Clearance (mL/min) | |
|
| ||||||
| 8 | 23.2 ± 0.6 | 63.5 ± 6.5 | 1.6 ± 0.2 | 11.3 ± 0.5### | 78.9 ± 11.8 | 1.3 ± 0.2 |
| 24 | 16.4 ± 0.4 | 43.5 ± 3.1 | 2.3 ± 0.2 | 5.2 ± 0.3### | 40.1 ± 9.4 | 2.5 ± 0.3* |
| 52 | 12.8 ± 0.1 | 373 ± 63*** | 0.26 ± 0.04** | 6.0 ± 0.3### | 31.2 ± 3.5#### | 3.2 ± 0.3*** |
|
| ||||||
| APP/PS1 | ||||||
|
| ||||||
| 8 | 17.7 ± 1.8 | 49.7 ± 12.4 | 2.1 ± 0.5 | 3.4 ± 0.3### | 27.6 ± 9.4 | 3.6 ± 1.2 |
| 24 | 12.5 ± 0.2 | 103.0 ± 34.5 | 1.0 ± 0.3 | 7.1 ± 0.2## | 72.2 ± 19.7 | 1.4 ± 0.3 |
| 52 | 13.7 ± 0.2 | 86.8 ± 22.8 | 1.2 ± 0.3 | 5.1 ± 1.0### | 30.9 ± 7.8 | 3.2 ± 0.8 |
Data represent mean±SE, n = 3–5. Significance was assessed over age (*p<0.05, **p<0.01, and ***p<0.001) and 125I-Aβ40 versus 125I-Aβ42 (##p<0.01, ###p<0.001, and ###p<0.0001; two-way ANOVA with Bonferroni post-tests).
At the end of the experiment, the brain was removed from the cranial cavity and dissected into anatomical regions (cortex, caudate putamen, hippocampus, thalamus, brain stem, and cerebellum). Individual brain regions were assayed for 125I and 131I activity using a two-channel gamma counter. The residual plasma volume at each brain region (mL per gram tissue) was estimated using the equation:
| (2) |
where is the total 131I-BSA activity in the brain region (μCi), is the 131I-BSA concentration in plasma (μCi/mL), and is the weight of the brain region (g). Given the total 125I-Aβ40 or 125I-Aβ42 activity in the brain region , the amount that permeated into the brain extravascular space ( per gram tissue) was estimated using the equation:
| (3) |
where is the final 125I-Aβ concentration in plasma (μCi/mL). The permeability-surface area (PS) product (mL/s per gram tissue) for 125I-Aβ40 or 125I-Aβ42 at each brain region was estimated using the equation:
| (4) |
where is the area under the observed plasma concentration versus time profile (AUC) from 0–15 min (min∗μCi/mL) obtained using the linear-trapezoidal method. The PS product for the whole brain was estimated based on the sum of in all six dissected brain regions. The % of the injected dose that permeated into the whole brain per gram tissue (% ID/g) was estimated using the equation:
| (5) |
Plasma pharmacokinetics and brain permeability of 125I-insulin
These experiments were performed as described above for 125I-Aβ. Briefly, WT mice at various ages (8, 24, or 52 weeks) were bolus injected with a single dose (100μCi) of 125I-insulin in PBS (100μL) into the femoral vein. Blood was sampled serially (20μL) from the femoral artery at 0.25-, 1-, 3-, 5-, 10-, and 14-min post-injection. Immediately after the 14-min sampling event, 131I-BSA (100μCi) was bolus injected into the femoral vein to serve as a measure of . One minute after the 131I-BSAinjection,a final blood sample was collected, and the animal was euthanized. The plasma pharmacokinetics and brain uptake of 125I-insulin were assessed as described above for 125I-Aβ. The secondary plasma pharmacokinetic parameters for 125I-insulin are reported in Table 2.
Table 2.
Plasma pharmacokinetics of 125I-insulin in WT mice at 8, 24, and 52 weeks
|
125I-insulin | |||
|---|---|---|---|
| Age (weeks) | Cmax (μCi/mL) | AUC (μCi.min/mL) | Clearance (mL/min) |
|
| |||
| 8 | 19.6 ± 0.2 | 83.7 ± 7.9 | 1.19 ± 0.11 |
| 24 | 23.5 ± 0.5**** | 170 ± 15 | 0.61 ± 0.06*** |
| 52 | 28.0 ± 0.4**** | 357 ± 68** | 0.28 ± 0.05**** |
Data represent mean±SE, n = 3–5. Significance was assessed over age (**p <0.01, ***p <0.001, ****p <0.0001; one-way ANOVA with Bonferroni post-tests).
Dynamic SPECT/CT imaging of 125I-insulin influx to the brain
WT mice at 12 or 104 weeks of age weighing 25–35g were catheterized at the femoral vein and femoral artery while under general anesthesia (1.5% isoflurane; 4L/min oxygen). A single dose (500μCi) of 125I-Insulin diluted in PBS was bolus injected into the femoral vein. Immediately following which, the entire animal was placed inside the single-photon emission computed tomography (SPECT/CT) scanner (Gamma Medica, Northridge, CA), and the biodistribution of radioactive signal was imaged at 1-min intervals over the next 40 min, followed by a 5-min CT scan to locate regions of interest (ROIs). The brain influx of 125I-insulin was assessed by Gjedde-Patlak graphical analysis [23], which involved plotting:
| (6) |
where is the measured amount (μCi) of radioactive signal in the brain ROI at time (min), is the plasma concentration (μCi/mL) at time , and is the plasma AUC (min∗μCi/mL) from . Both and were predicted based on the primary plasma pharmacokinetic parameters obtained in a separate experiment. The slope obtained after linear regression corresponds to the brain influx clearance, (mL/min) of 125I-insulin.
Transcriptomic analysis of brain endothelial cells from WT mice
A comprehensive, transcriptomic analysis of gene expression profiles of brain endothelial cells obtained from WT mice at 12 and 104 weeks of age was recently published by Ximerakis et al. [21]. In these studies, the transcriptomes of 50,212 single cells obtained from the brains of young or aged mice were assessed by single-cell RNA-Seq. Following which, an established clustering method was used to group transcriptionally similar cells based on the expression of multiple cell-type-specific marker genes, such as those specific for brain endothelial cells (CD31, tight junction proteins, etc.).
Using the single-cell RNA-Seq data publicly available at http://shiny.baderlab.org/AgingMouseBrain/, we applied a non-parametric Gene Set Variation Analysis (GSVA) method [29] to assess the pathway-level activity in young and aged cohorts at an individual sample level. Curated gene sets defining specific pathways were obtained from MSigDB. The GSVA method condenses a gene-level expression matrix into pathway enrichment scores. The directionality of pathway scores was determined from the mean difference between the cohorts. The significance was assessed by unpaired, two-tailed t-tests. The analysis was performed using the GSVA package in R studio (R software; Boston, MA).
We also performed Gene Set Enrichment Analysis (GSEA) on the brain endothelial cell expression data published by Ximerakis et al. [21]. Similar to GSVA, the GSEA method can also be used to assess differences in pathway-level activity when comparing cohorts [3]. The same pathways (gene sets) assessed by GSVA were assessed by GSEA. Pathway enrichment scores were calculated based on the list of genes ranked according to their differential expression. The significance was assessed by a permutation test, which determines the probability of obtaining an enrichment score that is as strong as or stronger than that observed under the permutation-generated null distribution. The analysis was performed using the fgsea package in R studio.
Statistical analysis
Statistical tests were performed using GraphPad Prism 8.4 (GraphPad software; La Jolla, CA). A p-value of less than 0.05 was regarded as statistically significant. Two-way ANOVA followed by Bonferroni post-hoc tests were used to compare the plasma pharmacokinetic parameters or PS products of 125I-Aβ40 and 125I-Aβ42 at 8, 24, and 52 weeks of age in WT mice, as well as in APP/PS1 mice. One-way ANOVA followed by Bonferroni post-hoc tests were used to compare the plasma pharmacokinetic parameters or PS products of 125I-insulin in WT mice at 8, 24, and 52 weeks of age. Unpaired, two-tailed t-tests were used to compare the slopes obtained from Gjedde-Patlak plots for 125I-insulin in WT mice at 12 and 104 weeks of age.
RESULTS
Age-dependent changes in plasma pharmacokinetics of 125I-Aβ40 and 125I-Aβ42 in WT and APP/PS1 mice (Fig. 1, Table 1)
Fig. 1.
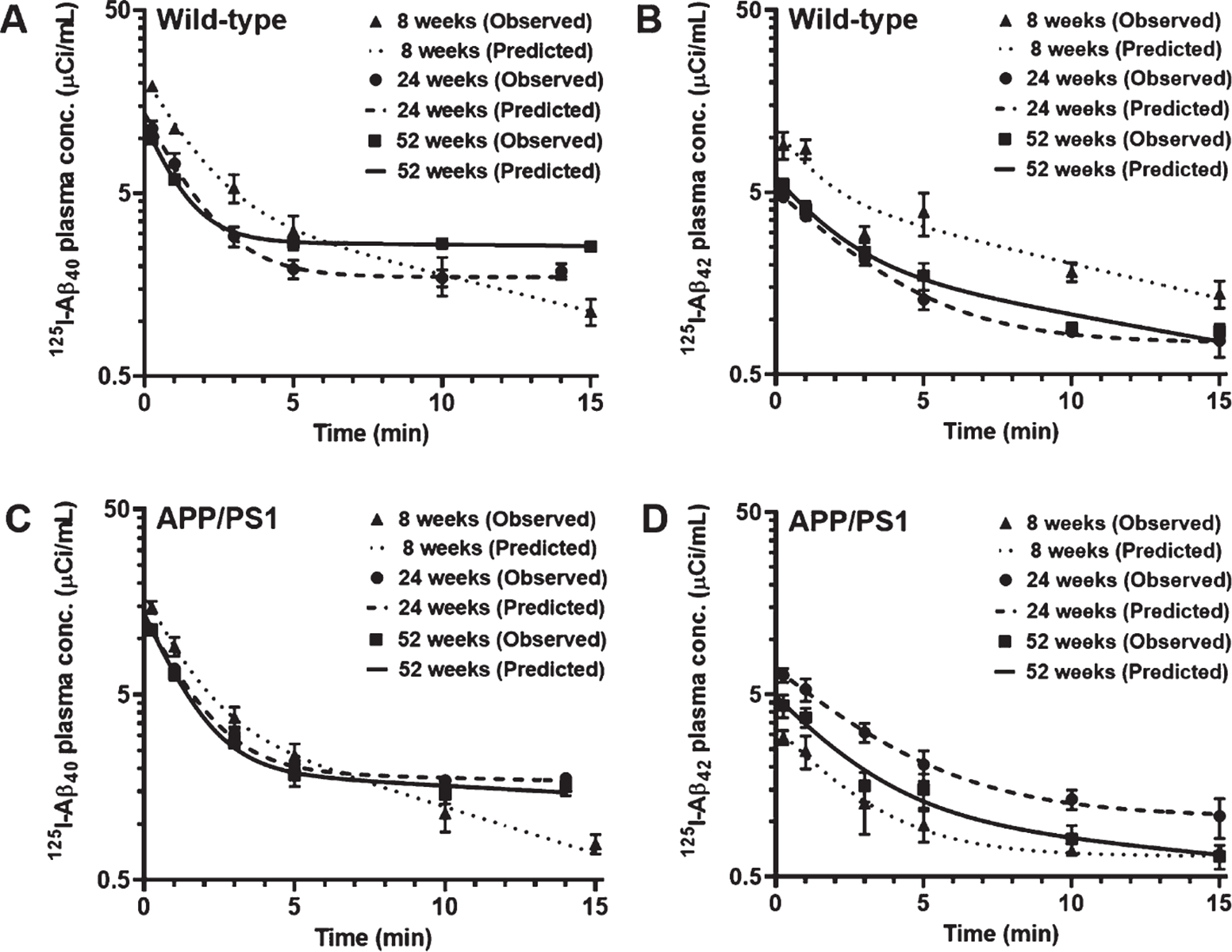
Plasma pharmacokinetics of 125I-Aβ40 and 125I-Aβ42 in WT and APP/PS1 mice at 8, 24, and 52 weeks. WT or APP/PS1 transgenic mice were bolus injected with 100Ci of 125I-Aβ40 (A, C) or 125I-Aβ42 (B, D) into the femoral vein. Plasma was sampled periodically between 0–15min, and radioactivity in the intact 125I-Aβ fraction was measured. The plasma concentration versus time data was fit to a bi-exponential equation. Shown are the observed values (mean±SD, n=3–5) overlaid on the predicted curves.
The plasma pharmacokinetics of 125I-Aβ40 and 125I-Aβ42 were assessed in WT and APP/PS1 transgenic mice at 8, 24, and 52 weeks of age. Female mice were used for the studies, given that AD disproportionately affects women, and that female APP/PS1 transgenic mice display higher brain Aβ levels and greater occurrence of histopathological hallmarks compared to their male littermates [18, 30]. Following a bolus injection into the femoral vein, both peptides exhibited bi-exponential decline in their plasma concentrations with time (Fig. 1), which is consistent with our earlier publications [20, 24]. The plasma exposure to 125I-Aβ40 in WT mice, estimated by the area under the concentration versus time curve (AUC), was unaltered between 8 and 24 weeks, but increased by ~ 7-fold at 52 weeks; a concomitant decrease in plasma clearance was observed (Fig. 1A; Table 1). In contrast, the plasma AUC of 125I-Aβ42 reduced by ~ 2-fold in WT mice around 24 or 52 weeks of age compared to 8 weeks; a concomitant increase in plasma clearance was observed (Fig. 1B; Table 1). Further, at 52 weeks, the plasma AUC of 125I-Aβ40 in WT mice was ~ 12-fold higher than that of 125I-Aβ42, whereas no significant differences were observed between the two peptides at 8 or 24 weeks (Table 1).
In APP/PS1 mice, the plasma AUC of 125I-Aβ40 was not significantly altered across all three age groups, although an increasing trend was observed at 24 and 52 weeks compared to 8 weeks (Fig. 1C; Table 1). The plasma AUC of 125I-Aβ42 was also not significantly different across all three age groups of APP/PS1 mice, but an increasing trend was observed at 24 weeks compared to 8 or 52 weeks (Fig. 1D; Table 1). In APP/PS1 mice, no significant differences were observed between the plasma AUC of 125I-Aβ40 and 125I-Aβ42 at any of the three age groups, although an increasing trend was observed for 125I-Aβ40 compared to 125I-Aβ42 at 52 weeks (Table 1). Interestingly, in both WT and APP/PS1 mice, the maximum observed plasma concentration (Cmax) was higher for 125I-Aβ40 compared to 125I-Aβ42 at all three age groups.
Brain uptake of 125I-Aβ40 decreases and that of 125I-Aβ42 increases with age in WT mice, which is disrupted in APP/PS1 mice (Fig. 2)
Fig. 2.
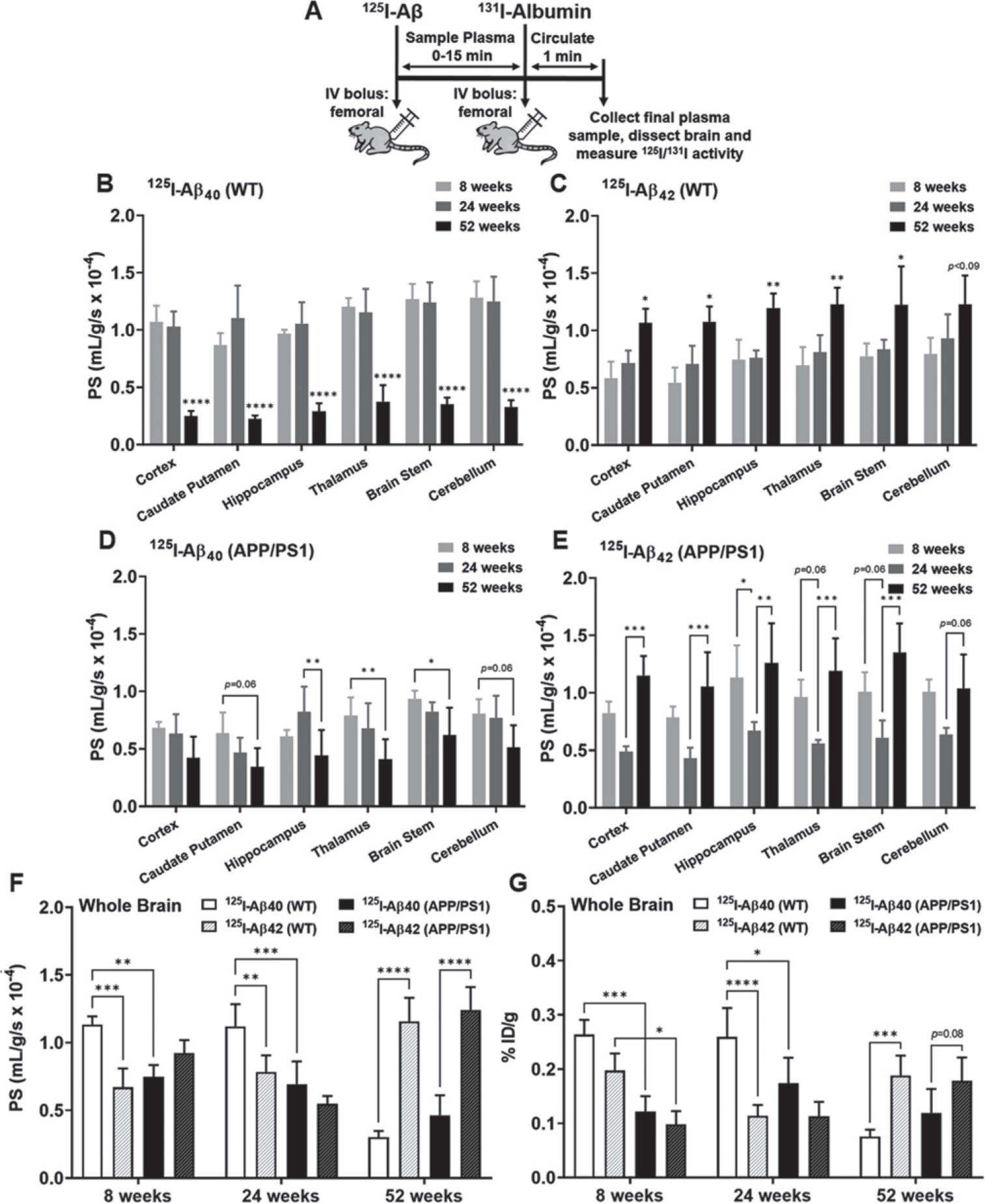
Brain uptake of 125I-Aβ40 and 125I-Aβ42 in WT and APP/PS1 mice at 8, 24, and 52 weeks. A) Experiment scheme. WT or APP/PS1 transgenic mice were bolus injected with 100Ci of 125I-Aβ40 or 125I-Aβ42 into the femoral vein. At the end of the experiment, 100μCi of 131I-albumin was injected to serve as a marker of Vp. The brain regions were dissected and assayed for radioactivity. Shown are the PS value estimates for 125I-Aβ40 (B, D) and 125I-Aβ42 (C, E) uptake at various brain regions. F) The PS values are shown for the whole brain. G) The overall brain accumulation was assessed as % ID/g. Data represent mean±SD, n = 3–5. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; two-way ANOVA with Bonferroni post-tests).
Following a bolus injection of 125I-Aβ40 or 125I-Aβ42 into the femoral vein of WT or APP/PS1 transgenic mice at 8, 24, and 52 weeks of age, the brain uptake was assessed as the permeability-surface area (PS) product. In WT mice, the PS values of 125I-Aβ40 in various brain regions were unaltered at 8 and 24 weeks but decreased by ~ 4-fold at 52 weeks (Fig. 2B). In contrast, the PS values of 125I-Aβ42 were unaltered at 8 and 24 weeks but increased by ~ 1.5-fold at 52 weeks in the WT mice (Fig. 2C). In APP/PS1mice,thePSvaluesof 125I-Aβ40 were unaltered at 8 and 24 weeks but decreased by ~ 1.5-fold at 52 weeks (Fig. 2D). Interestingly, the PS values of 125I-Aβ42 at various brain regions in APP/PS1 mice were unaltered at 8 and 52 weeks but decreased by ~ 1.5-fold at 24 weeks (Fig. 2E).
When comparing age-matched WT and APP/PS1 mice, the PS products of 125I-Aβ40 in the whole brain were ~ 1.5-fold lower in APP/PS1 mice compared to WT mice, at both 8 and 24 weeks. However, by 52 weeks, at which point the PS products of 125I-Aβ40 had decreased substantially in WT mice, no differences were observed between WT and APP/PS1 mice (Fig. 2F). Interestingly, for 125I-Aβ42, no significant differences between WT and APP/PS1 mice were observed at any of the age groups (Fig. 2F). When comparing Aβ40 versus Aβ42 in WT mice, the PS products in the whole brain were ~ 1.5-fold higher for 125I-Aβ40 compared to 125I-Aβ42 at both 8 and 24 weeks; however, by 52 weeks, a dramatic shift was observed in the PS product of 125I-Aβ42, which increased by ~ 4-fold compared to 125I-Aβ40 (Fig. 2F). Conversely, in APP/PS1 mice, the PS products in the whole brain were similar for 125I-Aβ40 compared to 125I-Aβ42 at both 8 and 24 weeks; however, by 52 weeks, again a shift was observed in the PS product of 125I-Aβ42,whichincreasedby~3-fold compared to 125I-Aβ40 (Fig. 2F). Similar trends were observed in the overall brain accumulation, which was expressed as the % of the injected dose (ID) accumulated in the brain per gram of tissue (% ID/g) (Fig. 2G).
Age-dependent changes in plasma pharmacokinetics of 125I-insulin in WT mice (Fig. 3, Table 2)
Fig. 3.
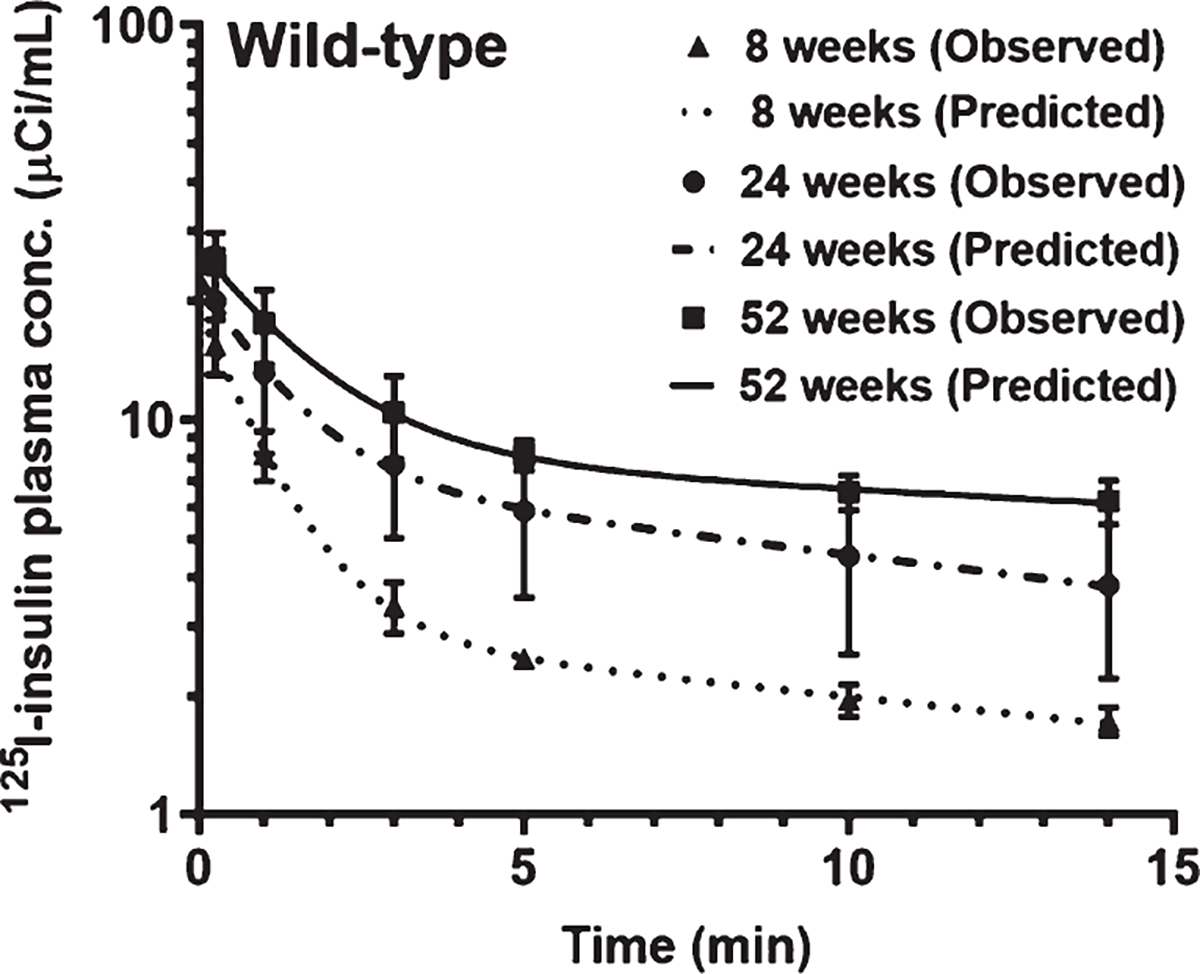
Plasma pharmacokinetics of 125I-insulin in WT mice at 8, 24, and 52 weeks. Mice were bolus injected with 100μCi of 125I-insulin into the femoral vein. Plasma was sampled periodically between 0–14 min, and radioactivity in the intact 125I-insulin fraction was measured. The plasma concentration versus time data was fit to a bi-exponential equation. Shown are the observed values (mean±SD, n = 3–5) overlaid on the predicted curves.
The plasma pharmacokinetics of 125I-Insulin were assessed in WT mice at 8, 24, and 52 weeks of age. Following a bolus injection into the femoral vein, a bi-exponential decline in 125I-insulin plasma concentrations was observed with time (Fig. 3), which is consistent with earlier publications [31]. The plasma AUC of 125I-insulin increased by ~ 3-fold at 52 weeks compared to 8 or 24 weeks; a concomitant decrease in clearance was observed. The Cmax was also increased at 52 weeks compared to 8 or 24 weeks (Table 2).
Brain uptake of 125I-insulin decreases with age in WT mice (Figs. 4 and 5)
Fig. 4.
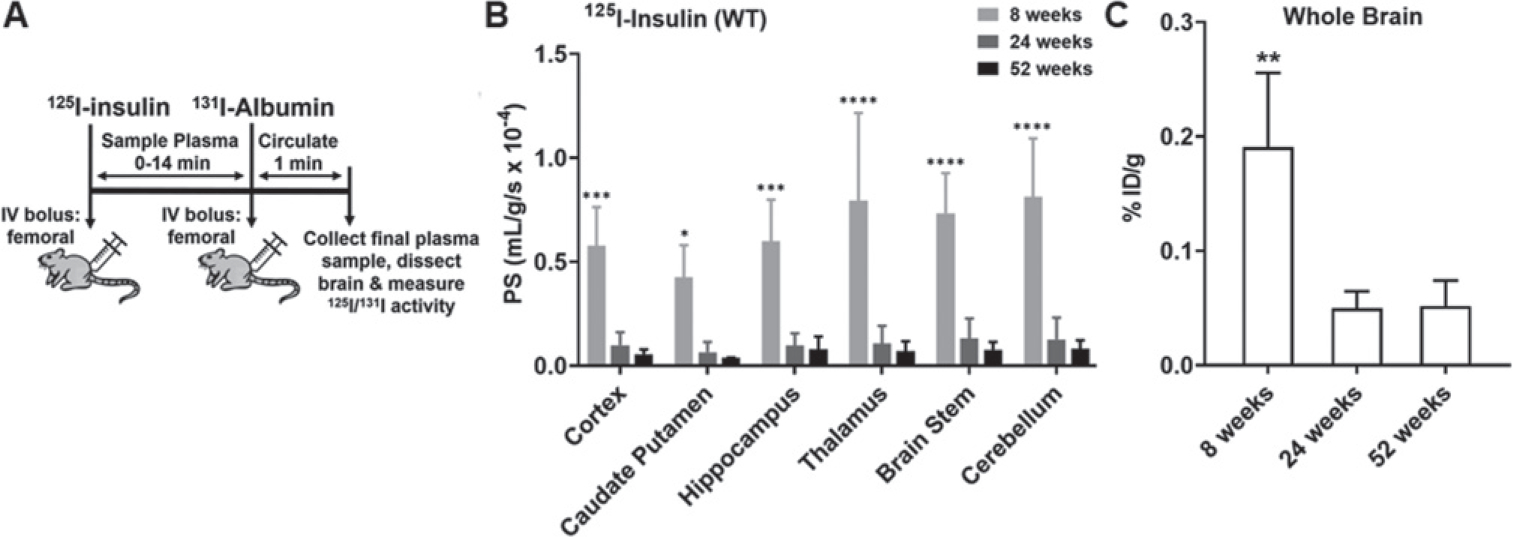
Brain uptake of 125I-insulin in WT mice at 8, 24, and 52 weeks. A) Experimental scheme. WT mice were bolus injected with 100μCi of 125I-insulin into the femoral vein. At the end of the experiment, 100μCi of 131I-albumin was injected to serve as a marker of Vp. The brain regions were dissected and assayed for radioactivity. B) The PS value estimates for 125I-insulin uptake in various brain regions are shown. Data represent mean±SD, n = 3–5. *p<0.05, ***p<0.001, ****p<0.0001; two-way ANOVA with Bonferroni post-tests. C) The overall brain accumulation was assessed as % ID/g. **p<0.01; one-way ANOVA with Bonferroni post-tests.
Fig. 5.
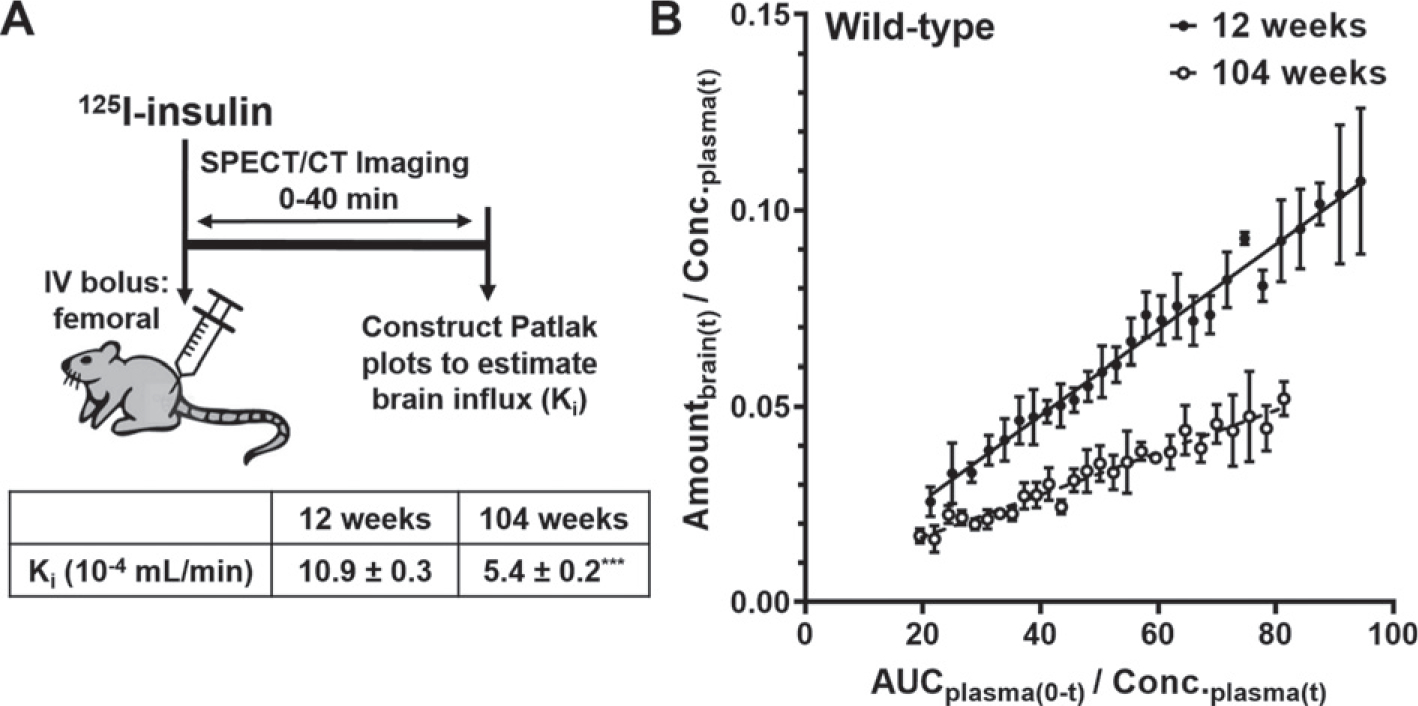
Dynamic SPECT/CT imaging of 125I-insulin uptake to the brain. A) Experimental scheme. WT mice were bolus injected with 500μCi of 125I-insulin into the femoral vein and the accumulation of 125I-insulin in the brain was monitored between 0–40 minutes post-injection by dynamic SPECT/CT imaging. B) The brain influx clearance (Ki) of 125I-insulin was estimated by the slope obtained from Gjedde-Patlak graphical analysis. In the graph, Amountbrain(t) is the measured radioactivity in the brain ROI (Ci) at each time point (data are mean±SD, n=4), whereas AUCplasma(0-t) (min.μCi/mL) and Conc.plasma(t) (μCi/mL) were predicted using the plasma pharmacokinetic parameters. The Ki predictions presented in the table are mean±SE, n=4. ***p<0.001; unpaired two-tailed t-test.
Following a bolus injection of 125I-insulin into the femoral vein of WT mice at 8, 24, and 52 weeks of age, the brain influx was assessed as the PS product (Fig. 4A). The PS products of 125I-insulin at various brain regions decreased by ~ 5-fold at 24 weeks compared to 8 weeks but did not further decrease at 52 weeks (Fig. 4B). Similarly, the % ID/g of 125I-insulin in the whole brain decreased by ~ 3-fold at 24 and 52 weeks compared to 8 weeks. To study the effects of advanced aging, the brain influx of 125I-insulin was further assessed in WT mice at 12 and 104 weeks of age by dynamic SPECT/CT imaging. After femoral injection, the accumulation of 125I-insulin in the brain was imaged from 0–40min (Fig. 5A). Gjedde-Patlak plots were constructed to estimate the brain influx clearance [32], which corresponds to the slope (Ki; mL/min) obtained after linear regression (Fig. 5B). The brain influx clearance of 125I-insulin decreased by ~ 2-fold at 104 weeks compared to 12 weeks (Fig. 5 inset).
Age-dependent changes in transporter/signaling pathways at the BBB in WT mice (Fig. 6)
Using the publicly available single-cell RNA-Seq data published by Ximerakis et al. [21], we performed Gene Set Variation Analysis (GSVA) and also Gene Set Enrichment Analysis (GSEA) to assess the activity of various cellular pathways that are implicated in Aβ or insulin trafficking at the BBB. Pathways relating to RAGE signaling, insulin/Akt signaling, and T2DM pathology were upregulated in brain endothelial cells obtained from WT mice at 104 weeks compared to 12 weeks (Table 3). In contrast, ABC transporter and tight junction networks were downregulated in the brain endothelial cells obtained at 104 weeks compared to 12 weeks (Table 3).
Table 3.
Enrichment of various transporter/signaling pathways in brain endothelial cells from WT mice at 12 and 104 weeks
| Pathway | GSVA |
GSEA |
||
|---|---|---|---|---|
| Directionality | p | Directionality | p | |
|
| ||||
| KEGG AGE RAGE Signaling Pathway | Up | 0.088 | Up | 0.25 |
| PID_Insulin Pathway | 0.035 | 0.11 | ||
| PID.PI3KCI AKT Pathway | 1.5E-08 | 0.0043 | ||
| KEGG_Type II Diabetes Mellitus | 0.012 | 0.12 | ||
| KEGG.ABC Transporters | Down | 0.0058 | Down | 0.086 |
| REACTOME_Tight Junction Interactions | 2.9E-05 | 0.13 | ||
The GSVA and GSEA methods were used to compare the enrichment of various transporter/signaling pathways in brain endothelial cells obtained from WT mice at different ages, using the single-cell RNA-Seq data published by Ximerakis et al. [21]. The directionality indicates upregulation or downregulation of the pathway in the aged compared to young mice.
DISCUSSION
BBB dysfunction is observed in the aging brain [3] and is thought to be among the earliest changes driving AD pathogenesis [33]. The BBB dysfunction is customarily studied from the perspective of loss of tight junction integrity [34] and reduced cerebral blood flow [35]. However, age-related changes in cellular and molecular mechanisms that drive these changes in BBB functions are only partially understood. Even less understood are age-related changes in diverse BBB functions that coordinate the delivery of essential nutrients like glucose and insulin to the brain [13], as well as the clearance of toxic metabolites like Aβ peptides from the brain [36].
Our findings demonstrate that plasma pharmacokinetics of Aβ isoforms and insulin are differentially affected by aging in WT mice. The liver and kidneys are the major organs responsible for clearing Aβ from the periphery [37], but it remains poorly understood how systemic Aβ clearance is affected during normal aging and in AD. We showed that in WT mice, the plasma AUC of 125I-Aβ40 increased with age, but that of 125I-Aβ42 decreased. This demonstrates an age-dependent switch in the plasma pharmacokinetics of the two major Aβ isoforms. Consistent with these findings, the plasma AUC of Aβ40 was reported to increase with age in squirrel monkeys [38]. The observed age-dependent changes in Aβ40 plasma pharmacokinetics in WT mice were evident at an even earlier age in APP/PS1 transgenic mice, likely due to the accelerated AD pathology in these animals. Similarly, it was reported that Aβ40 plasma concentrations increased with age in healthy patients, and substantially elevated in AD patients [39]. This is expected to promote a decrease in the Aβ42/Aβ40 ratio in plasma with age, which has been strongly linked to amyloid plaque deposition and AD risk in patients [40, 41]. The increase in 125I-Aβ40 plasma AUC with age predicts increased exposure of this isoform to the luminal surface of the BBB. Increased plasma Aβ40 levels are associated with cerebrovascular disease, which is prevalent in approximately 90% of all AD patients [42].
The current study demonstrates that plasma-to-brain influx of Aβ isoforms and insulin are progressively and differentially affected by aging in WT mice. Using the publicly available single-cell RNA-Seq data of BBB endothelial cells in WT mice [21], we identified age-dependent changes to various trafficking/signaling pathways implicated in Aβ and insulin delivery to the brain. Changes in Aβ trafficking are evident in AD transgenic mice at a much younger age, suggesting that age-related pathophysiological changes are accelerated in AD. The brain influx of 125I-Aβ40 is lower in APP/PS1 mice than in WT mice at both 8 and 24 weeks of age; but no significant differences in 125I-Aβ42 influx were observed between WT and APP/PS1 mice in these age groups. At 52 weeks, the differences between WT and APP/PS1 mice were entirely lost, with Aβ42 influx dominating over Aβ40 influx. Previously, we reported that in APP/PS1 mice, initiation of Aβ plaque formation occurs at 24 weeks of age, with full-scale plaque burden observed by 52 weeks [28]. It was also reported that cognitive deficits first appear in APP/PS1 mice at 24 weeks, and become more severe by 52 weeks [43]. Thus, we speculate that the shift in the preferential brain influx of Aβ42 versus Aβ40 observed at 52 weeks is associated with enhanced amyloid deposition and cognitive decline in the APP/PS1 mice.
Normal aging, AD, and T2DM have all been associated with hyperinsulinemia and insulin resistance, which manifests as increased insulin plasma levels and impaired insulin sensitivity in the brain [44, 45]. We showed that the plasma AUC of 125I-insulin increased from 8 to 24 weeks, and then further increased at 52 weeks in WT mice. This could be due to impaired renal clearance of insulin, which was claimed to be a major driver of hyperinsulinemia [46], and/or decreased distribution from plasma to peripheral tissues and the brain. In the AD brain, increased levels of toxic Aβ42 peptides are paralleled by decreased levels of the essential growth factor insulin [14]. The PS products of 125I-insulin in WT mice declined steeply from 8 to 24 weeks, then remained unchanged from 24 to 52 weeks. In contrast, the PS products of 125I-Aβ40 and 125I-Aβ42 remained unchanged from 8 to 24 weeks, then decreased and increased, respectively, at 52 weeks. It was also shown that WT mice demonstrate learning/memory deficits at 52 weeks compared to 24 weeks [47]. Taken together, these findings suggest that age-related pathological changes in insulin transport at the BBB precede the changes in Aβ transport at the BBB and the onset of cognitive decline in WT mice.
The brain uptake of insulin is extremely rapid relative to other serum proteins, indicating a highly specialized and efficient trafficking system for delivering insulin across the BBB [23]. To capture the initial rate of insulin uptake, dynamic SPECT/CT imaging studies were performed to measure 125I-insulin uptake to the brain after systemic injection in WT mice at 12 and 104 weeks. These age groups were selected based on a previous study that established age-dependent changes in insulin sensitivity in WT mice [48]. Based on Gjedde-Patlak graphical analysis, the 125I-insulin brain influx clearance decreased at 104 weeks compared to 12 weeks. Thus, we showed that compared to young WT mice, insulin uptake to the brain was decreased in both middle-aged (52 weeks) and advanced-aged (104 weeks) mice. Furthermore, we recently showed that 125I-insulin uptake to the brain is decreased in APP/PS1 mice compared to age-matched WT mice [19]. Together, these findings suggest that reduced insulin transport at the BBB during aging in WT mice may decrease further in APP/PS1 mice and reduce brain insulin levels.
Both insulin and Aβ peptides are known to exhibit saturable, receptor-mediated transcytosis at the BBB [49, 50]. The brain uptake of Aβ peptides is thought to be primarily mediated by RAGE, expressed on the luminal surface of the BBB [9]. Membrane RAGE is reported to localize in caveolae microdomains [51], while IR is reported to localize in clathrin-coated pits [52]. Although the role of IR at the BBB in insulin delivery to the brain is controversial [49, 53], insulin transcytosis across microvascular endothelial cells was reported to be clathrin-dependent [54]. We previously showed that Aβ42 uptake in BBB endothelial cells is caveolae-dependent, whereas Aβ40 uptake is clathrin-dependent [64]. This suggests that in addition to RAGE, Aβ40 uptake at the BBB may be handled by other receptor(s) that involve clathrin-dependent endocytosis. In a recent study, aging in WT mice was shown to be associated with increased expression of caveolin-1 and decreased expression of clathrin heavy chain in the brain microvessels [55]. Therefore, our current findings that aging in WT mice is associated with increased brain uptake of Aβ42 and decreased uptake of Aβ40 and insulin could be resulting from the age-dependent switch from clathrin to caveolae-mediated endocytosis at the BBB.
In addition to serving as an endocytosis receptor, RAGE also functions as a signaling receptor to trigger the activation of pro-inflammatory pathways, which have been implicated in Aβ uptake at the BBB [56]. It was reported that RAGE expression at the BBB increased with age in rats [11]. By performing pathway-level analyses (GSVA and GSEA) on publicly available single-cell RNA-Seq data [21], we found that the RAGE signaling pathway was upregulated in brain endothelial cells obtained from WT miceat104weekscomparedto12weeks.Incontrast, ABC transporter proteins, which were claimed to mediate Aβ efflux from the brain [57] as well as tight junction proteins, were downregulated at 104 weeks. Thus, increased RAGE expression and signaling at the BBB likely contributed to the increased 125I-Aβ42 uptake observed in the aged mice. However, these trends cannot explain the concomitant decrease in 125I-Aβ40 uptake. We speculate these differences are engendered by other unidentified luminal receptors reliant on clathrin-dependent endocytosis that demonstrate higher selectivity for Aβ40 over Aβ42. During normal aging and AD progression, downregulation of such receptors could promote a decrease in the brain influx of Aβ40. The differential handling of Aβ40 versus Aβ42 by cell-surface receptors and intracellular mechanisms governing their disposition require further investigation.
Insulin signaling networks in the brain are disrupted during normal aging and AD, which triggers brain insulin resistance and contributes to cognitive decline [58,59].ThePI3K-AKTsignalingpathwayis downstream of IR and has canonical functions in glucose metabolism. By performing GSVA and GSEA on the single-cell RNA-Seq data [21], we found that insulin signaling and PI3K-AKT pathways were upregulated at the BBB in WT mice at 104 weeks compared to 12 weeks. Previously, it was reported that sustained over-activation of PI3K-AKT signaling in the aging brain is associated with brain insulin resistance and Aβ pathology [60, 61]. Consistent with these findings, we observed upregulation of a set of proteins implicated in insulin resistance/T2DM in the 104-week-old mice. These age groups match with those used in the 125I-insulin SPECT/CT imaging studies. Based on these reports and our current findings, we speculate that in aged WT mice, insulin signaling disruptions at the BBB triggered by hyperinsulinemia and peripheral insulin resistance inhibit insulin transcytosis at the BBB, resulting in decreased insulin uptake to the brain.
Insulin signaling networks have also been implicated in the regulation of Aβ trafficking receptors at the BBB [62]. We previously reported that stimulation with insulin modulates the plasma membrane expression of LRP-1 in hCMEC/D3 cell monolayers (BBB cell model) [20], while others demonstrated similar findings in hepatocytes [63]. Insulin is also suggested to modulate expression of other important Aβ receptors, specifically RAGE and P-glycoprotein, at the BBB [20, 64, 65]. Moreover, we have previously shown that insulin differentially regulates the trafficking of Aβ isoforms at the BBB. Specifically, systemic insulin injection in WT mice decreased the brain influx of 125I-Aβ42, but increased the brain influx of 125I-Aβ40, with concomitant changes in their plasma pharmacokinetics [20]. Therefore, we speculate that impaired insulin signaling in the BBB endothelium contributes to the Aβ trafficking perturbations observed in aged mice.
In summary, we demonstrated age-dependent changes in the plasma disposition and brain influx of Aβ isoforms and insulin in female WT mice. The brain influx of both 125I-insulin and 125I-Aβ40 decreased and 125I-Aβ42 influx increased in WT mice with age. The increase in plasma AUC of 125I-insulin with age is indicative of hyperinsulinemia, whereas the increase in 125I-Aβ40 plasma AUC and decrease in 125I-Aβ42 plasma AUC could reduce the Aβ42/Aβ40 ratio in plasma. Both of these changes are heavily implicated in AD risk. Thus, pathological changes during normal aging alter systemic clearance and brain influx of both insulin and Aβ peptides in a manner that could augment AD risk. Additionally, aging was associated with various transporter/signaling deficits in BBB endothelial cells, which is indicative of BBB dysfunction and insulin resistance. Further studies are needed to clarify the molecular mechanisms by which insulin and Aβ trafficking in the brain and periphery are impacted during normal aging and in AD.
ACKNOWLEDGMENTS
This work was supported by the Minnesota Partnership for Biotechnology and Medical Genomics [Grant 00056030], the Dr. Paul B. Myrdal Memorial Pre-Doctoral Fellowship in Pharmaceutics, the Ronald J. Sawchuk Fellowship in Pharmacokinetics, and the Theodore H. Rowell Graduate Fellowship.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-5128r1).
REFERENCES
- [1].Gao S, Hendrie HC, Hall KS, Hui S (1998) The relationships between age, sex, and the incidence of dementia and Alzheimer disease: A meta-analysis. Arch Gen Psychiatry 55, 809–815. [DOI] [PubMed] [Google Scholar]
- [2].Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Popescu BO, Toescu EC, Popescu LM, Bajenaru O, Muresanu DF, Schultzberg M, Bogdanovic N (2009) Blood-brain barrier alterations in ageing and dementia. J Neurol Sci 283, 99–106. [DOI] [PubMed] [Google Scholar]
- [4].Rodrigue KM, Kennedy KM, Park DC (2009)Beta-amyloid deposition and the aging brain. Neuropsychol Rev 19, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Qiu T, Liu Q, Chen YX, Zhao YF, Li YM (2015) Abeta42 and Abeta40: Similarities and differences. J Pept Sci 21, 522–529. [DOI] [PubMed] [Google Scholar]
- [6].Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E (2007) Abeta40 inhibits amyloid deposition in vivo. J Neurosci 27, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Murray MM, Bernstein SL, Nyugen V, Condron MM, Teplow DB, Bowers MT (2009) Amyloid beta protein: Abeta40 inhibits Abeta42 oligomerization. J Am Chem Soc 131, 6316–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yoon SS, Jo SA (2012) Mechanisms of amyloid-β peptide clearance: Potential therapeutic targets for Alzheimer’s disease. Biomol Ther (Seoul) 20, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deane R, Wu Z, Zlokovic BV (2004) RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35, 2628–2631. [DOI] [PubMed] [Google Scholar]
- [10].Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV (2002) Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer’s disease lesions. Vascul Pharmacol 38, 303–313. [DOI] [PubMed] [Google Scholar]
- [11].Silverberg GD, Miller MC, Messier AA, Majmudar S, Machan JT, Donahue JE, Stopa EG, Johanson CE (2010) Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J Neuropathol Exp Neurol 69, 98–108. [DOI] [PubMed] [Google Scholar]
- [12].Okereke OI, Xia W, Selkoe DJ, Grodstein F (2009) Ten-year change in plasma amyloid beta levels and late-life cognitive decline. Arch Neurol 66, 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sartorius T, Peter A, Heni M, Maetzler W, Fritsche A, Haring HU, Hennige AM (2015) The brain response to peripheral insulin declines with age: A contribution of the blood-brain barrier? PLoS One 10, e0126804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gil-Bea FJ, Solas M, Solomon A, Mugueta C, Winblad B, Kivipelto M, Ramirez MJ, Cedazo-Minguez A (2010) Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer’s disease. J Alzheimers Dis 22, 405–413. [DOI] [PubMed] [Google Scholar]
- [15].Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna) 105, 423–438. [DOI] [PubMed] [Google Scholar]
- [16].Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R (2002) Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci 22, Rc221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL (2008) Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 22, 246–260. [DOI] [PubMed] [Google Scholar]
- [18].Gali CC, Fanaee-Danesh E, Zandl-Lang M, Albrecher NM, Tam-Amersdorfer C, Stracke A, Sachdev V, Reichmann F, Sun Y, Avdili A, Reiter M, Kratky D, Holzer P, Lass A, Kandimalla KK, Panzenboeck U (2019) Amyloid-beta impairs insulin signaling by accelerating autophagy-lysosomal degradation of LRP-1 and IR-β in blood-brain barrier endothelial cells in vitro and in 3XTg-AD mice. Mol Cell Neurosci 99, 103390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Swaminathan SK, Min H-K, Sarma VV, Ahlschwede KM, Bruinsma TJ, Curran GL, Decklever T, Lowe VJ, Kandimalla KK (2018) P1–197: Amyloid beta effects on insulin permeabilityfromplasmatobrainmeasuredbyI-125insulin SPECT in APP/PS1 mice. Alzheimers Dement 14, P354–P354. [Google Scholar]
- [20].Swaminathan SK, Ahlschwede KM, Sarma V, Curran GL, Omtri RS, Decklever T, Lowe VJ, Poduslo JF, Kandimalla KK (2018) Insulin differentially affects the distribution kinetics of amyloid beta 40 and 42 in plasma and brain. J Cereb Blood Flow Metab 38, 904–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, Mayweather BA, Nguyen L, Niziolek Z, Ozek C, Butty VL, Isserlin R, Buchanan SM, Levine SS, Regev A, Bader GD, Levin JZ, Rubin LL (2019) Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 22, 1696–1708. [DOI] [PubMed] [Google Scholar]
- [22].Klein WL, Stine WB Jr., Teplow DB (2004) Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging 25, 569–580. [DOI] [PubMed] [Google Scholar]
- [23].Poduslo JF, Curran GL, Berg CT (1994) Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A 91, 5705–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kandimalla KK, Curran GL, Holasek SS, Gilles EJ, Wengenack TM, Poduslo JF (2005) Pharmacokinetic analysis of the blood-brain barrier transport of 125I-amyloid beta protein 40 in wild-type and Alzheimer’s disease transgenic mice (APP,PS1) and its implications for amyloid plaque formation. J Pharmacol Exp Ther 313, 1370–1378. [DOI] [PubMed] [Google Scholar]
- [25].Champlin AK, Dorr DL, Gates AH (1973) Determining the stage of the estrous cycle in the mouse by the appearance of the vagina. Biol Reprod 8, 491–494. [DOI] [PubMed] [Google Scholar]
- [26].McLean AC, Valenzuela N, Fai S, Bennett SA (2012) Performing vaginal lavage, crystal violet staining, and vaginal cytological evaluation for mouse estrous cycle staging identification. J Vis Exp, e4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Agyare EK, Leonard SR, Curran GL, Yu CC, Lowe VJ, Paravastu AK, Poduslo JF, Kandimalla KK (2013) Traffic jam at the blood-brain barrier promotes greater accumulation of Alzheimer’s disease amyloid-beta proteins in the cerebral vasculature. Mol Pharm 10, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jack CR Jr., Wengenack TM, Reyes DA, Garwood M, Curran GL, Borowski BJ, Lin J, Preboske GM, Holasek SS, Adriany G, Poduslo JF (2005) In vivo magnetic resonance microimaging of individual amyloid plaques in Alzheimer’s transgenic mice. J Neurosci 25, 10041–10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hänzelmann S, Castelo R, Guinney J (2013) GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang J, Tanila H, Puoliväli J, Kadish I, Groen Tv (2003) Gender differences in the amount and deposition of amyloid in APPswe and PS1 double transgenic mice. Neurobiol Dis 14, 318–327. [DOI] [PubMed] [Google Scholar]
- [31].Sato H, Tsuji A, Hirai K-I, Kang YS (1990) Application of HPLC in disposition study of A14-125I-labeled insulin in mice. Diabetes 39, 563–569. [DOI] [PubMed] [Google Scholar]
- [32].Patlak CS, Blasberg RG, Fenstermacher JD (1983) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 3, 1–7. [DOI] [PubMed] [Google Scholar]
- [33].Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV (2016) Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta 1862, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sweeney MD, Sagare AP, Zlokovic BV (2018) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tarumi T, Zhang R (2018) Cerebral blood flow in normal aging adults: Cardiovascular determinants, clinical implications, and aerobic fitness. J Neurochem 144, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, Donahue JE, Johanson CE (2010) Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J Neuropathol Exp Neurol 69, 1034–1043. [DOI] [PubMed] [Google Scholar]
- [37].Ghiso J, Shayo M, Calero M, Ng D, Tomidokoro Y, Gandy S, Rostagno A, Frangione B (2004) Systemic catabolism of Alzheimer’s Abeta40 and Abeta42. J Biol Chem 279, 45897–45908. [DOI] [PubMed] [Google Scholar]
- [38].Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV (1998) Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 70, 210–215. [DOI] [PubMed] [Google Scholar]
- [39].Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM (2000) Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol 57, 100–105. [DOI] [PubMed] [Google Scholar]
- [40].Lui JK, Laws SM, Li QX, Villemagne VL, Ames D, Brown B, Bush AI, De Ruyck K, Dromey J, Ellis KA, Faux NG, Foster J, Fowler C, Gupta V, Hudson P, Laughton K, Masters CL, Pertile K, Rembach A, Rimajova M, Rodrigues M, Rowe CC, Rumble R, Szoeke C, Taddei K, Taddei T, Trounson B, Ward V, Martins RN, AIBL Research Group (2010) Plasma amyloid-beta as a biomarker in Alzheimer’s disease: The AIBL study of aging. J Alzheimers Dis 20, 1233–1242. [DOI] [PubMed] [Google Scholar]
- [41].Rembach A, Faux NG, Watt AD, Pertile KK, Rumble RL, Trounson BO, Fowler CJ, Roberts BR, Perez KA, Li QX, Laws SM, Taddei K, Rainey-Smith S, Robertson JS, Vandijck M, Vanderstichele H, Barnham KJ, Ellis KA, Szoeke C, Macaulay L, Rowe CC, Villemagne VL, Ames D, Martins RN, Bush AI, Masters CL (2014) Changes in plasma amyloid beta in a longitudinal study of aging and Alzheimer’s disease. Alzheimers Dement 10, 53–61. [DOI] [PubMed] [Google Scholar]
- [42].Brenowitz WD, Nelson PT, Besser LM, Heller KB, Kukull WA (2015) Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol Aging 36, 2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Filali M, Lalonde R (2009) Age-related cognitive decline and nesting behavior in an APPswe/PS1 bigenic model of Alzheimer’s disease. Brain Res 1292, 93–99. [DOI] [PubMed] [Google Scholar]
- [44].Young SE, Mainous AG 3rd, Carnemolla M (2006) Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care 29, 2688–2693. [DOI] [PubMed] [Google Scholar]
- [45].Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Häring H-U (2016) Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. PhysiolRev 96, 1169–1209. [DOI] [PubMed] [Google Scholar]
- [46].Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, Yki-Jarvinen H (2008) Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 135, 122–130. [DOI] [PubMed] [Google Scholar]
- [47].Wong AA, Brown RE (2007) Age-related changes in visual acuity, learning and memory in C57BL/6J and DBA/2J mice. Neurobiol Aging 28, 1577–1593. [DOI] [PubMed] [Google Scholar]
- [48].Reynolds TH, Dalton A, Calzini L, Tuluca A, Hoyte D, Ives SJ (2019) The impact of age and sex on body composition and glucose sensitivity in C57BL/6J mice. Physiol Rep 7, e13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rhea EM, Rask-Madsen C, Banks WA (2018) Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol 596, 4753–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B (1993) Blood-brain barrier transport of circulating Alzheimer’s amyloid beta. Biochem Biophys Res Commun 197, 1034–1040. [DOI] [PubMed] [Google Scholar]
- [51].Stitt AW, Burke GA, Chen F, McMullen CBT, Vlassara H (2000) Advanced glycation end product receptor interactions on microvascular cells occur within caveolin-rich membrane domains. FASEB J 14, 2390–2392. [DOI] [PubMed] [Google Scholar]
- [52].Carpentier JL (1994) Insulin receptor internalization: Molecular mechanisms and physiopathological implications. Diabetologia 37(Suppl 2), S117–124. [DOI] [PubMed] [Google Scholar]
- [53].Gray SM, Aylor KW, Barrett EJ (2017) Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 60, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Azizi PM, Zyla RE, Guan S, Wang C, Liu J, Bolz S-S, Heit B, Klip A, Lee WL (2015) Clathrin-dependent entry and vesicle-mediated exocytosis define insulin transcytosis across microvascular endothelial cells. Mol Biol Cell 26, 740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yang AC, Stevens MY, Chen MB, Lee DP, Stähli D, Gate D, Contrepois K, Chen W, Iram T, Zhang L, Vest RT, Chaney A, Lehallier B, Olsson N, du Bois H, Hsieh R, Cropper HC, Berdnik D, Li L, Wang EY, Traber GM, Bertozzi CR, Luo J, Snyder MP, Elias JE, Quake SR, James ML, Wyss-Coray T (2020) Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 583, 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen F, Ghosh A, Hu M, Long Y, Sun H, Kong L, Hong H, Tang S (2018) RAGE-NF-κB-PPARγ Signaling is Involved in AGEs-Induced Upregulation of Amyloid-β Influx Transport in an In Vitro BBB Model. Neurotox Res 33, 284–299. [DOI] [PubMed] [Google Scholar]
- [57].Abuznait AH, Kaddoumi A (2012) Role of ABC transporters in the pathogenesis of Alzheimer’s disease. ACS Chem Neurosci 3, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Akintola AA, van Heemst D (2015) Insulin, aging, and the brain: Mechanisms and implications. Front Endocrinol (Lausanne) 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gabbouj S, Ryhänen S, Marttinen M, Wittrahm R, Takalo M, Kemppainen S, Martiskainen H, Tanila H, Haapasalo A, Hiltunen M, Natunen T (2019) Altered insulin signaling in Alzheimer’s disease brain - special emphasis on PI3K-Akt pathway. Front Neurosci 13, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].O’ Neill C (2013) PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48, 647–653. [DOI] [PubMed] [Google Scholar]
- [61].Chen YR, Li YH, Hsieh TC, Wang CM, Cheng KC, Wang L, Lin TY, Cheung CHA, Wu CL, Chiang H (2019) Aging-induced Akt activation involves in aging-related pathologies and A-induced toxicity. Aging Cell 18, e12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vandal M, Bourassa P, Calon F (2015) Can insulin signaling pathways be targeted to transport Aβ out of the brain? Front Aging Neurosci 7, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Laatsch A, Merkel M, Talmud PJ, Grewal T, Beisiegel U, Heeren J (2009) Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis 204, 105–111. [DOI] [PubMed] [Google Scholar]
- [64].Sun YN, Liu LB, Xue YX, Wang P (2015) Effects of insulin combined with idebenone on blood-brain barrier permeability in diabetic rats. J Neurosci Res 93, 666–677. [DOI] [PubMed] [Google Scholar]
- [65].Liu L, Liu X (2014) Alterations in function and expression of ABC transporters at blood-brain barrier under diabetes and the clinical significances. Front Pharmacol 5, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]