

Abstract
Buruli ulcer, a chronic subcutaneous infection caused by Mycobacterium ulcerans, is increasing in prevalence in southeastern Australia. Possums are a local wildlife reservoir for M. ulcerans and, although mosquitoes have been implicated in transmission, it remains unclear how humans acquire infection. We conducted extensive field survey analyses of M. ulcerans prevalence among mosquitoes in the Mornington Peninsula region of southeastern Australia. PCR screening of trapped mosquitoes revealed a significant association between M. ulcerans and Aedes notoscriptus. Spatial scanning statistics revealed overlap between clusters of M. ulcerans-positive Ae. notoscriptus, M. ulcerans-positive possum excreta and Buruli ulcer cases, and metabarcoding analyses showed individual mosquitoes had fed on humans and possums. Bacterial genomic analysis confirmed shared single-nucleotide-polymorphism profiles for M. ulcerans detected in mosquitoes, possum excreta and humans. These findings indicate Ae. notoscriptus probably transmit M. ulcerans in southeastern Australia and highlight mosquito control as a Buruli ulcer prevention measure.
Subject terms: Bacteriology, Bacterial infection
Genomic evidence reveals that Mycobacterium ulcerans recovered from Aedes notoscriptus mosquitoes are genetically identical to bacteria from possums and humans, implicating mosquitoes as a vector for Buruli’s ulcer transmission in southeastern Australia.
Main
Mycobacterium ulcerans is the causative agent of a neglected tropical skin disease called Buruli ulcer, a necrotizing infection of skin and subcutaneous tissue1. Buruli ulcer is rarely a fatal condition but it can cause severe tissue destruction if not diagnosed and managed effectively2. Buruli ulcer has been described in more than 32 countries worldwide3 and is an ongoing public health issue in West and Central Africa2. Buruli ulcer has also been unexpectedly surging in temperate southeastern Australia (Extended Data Fig. 1) and encroaching on the major metropolitan centres of Melbourne (population 5.1 million) and Geelong (population 274,000), with more than 250 cases routinely notified each year since 2017 to the Victorian state government Department of Health4.
Extended Data Fig. 1. Location of the Mornington Peninsula and the Bellarine Peninsula of Victoria, Australia.

Population density is represented on the map from low to high (white to red) based on the human population per square kilometre. The study area is shown within an elipse (inset). Map boundaries in a from Australian Bureau of Statistics48 under a Creative Commons license CC BY 4.0.
How humans contract Buruli ulcer is a central question that has confounded public health control efforts and intrigued scientists since the discovery of M. ulcerans from patients in the Bairnsdale region of Australia in the 1930s and across Africa shortly thereafter1,5,6. Buruli ulcer epidemiology can be unpredictable, with a 4–5 month median incubation period and outbreaks emerging in specific geographical areas and then disappearing over a number of years. It is also very challenging to isolate the bacterium in pure culture from the environment, presumably due to its very slow growth, although it can be isolated from human skin lesions. These factors combined have made it incredibly challenging to establish how M. ulcerans is spread to humans, despite global research efforts over more than 80 years7.
The discovery that Buruli ulcer is a zoonosis and that Australian native possums are a major wildlife source of M. ulcerans that is intimately linked with disease transmission has addressed one key component of the transmission enigma8–13. The first indications that mosquitoes might be vectors of M. ulcerans from possums to humans in Australia came from a series of entomological field surveys in the southeast of the country in response to an increase in Buruli ulcer cases in the seaside township of Point Lonsdale, located on the Bellarine Peninsula14. Among the 12 species identified from a trapping effort that collected 11,500 mosquitoes, 5 different species were IS2404 PCR positive for M. ulcerans, including Aedes camptorhynchus, Aedes notoscriptus, Coquillettidia linealis, Culex australicus and Anopheles annulipes (maximum likelihood estimate (MLE) was 4.11 per 1,000 mosquitoes)14. IS2404 is a M. ulcerans-specific insertion sequence and molecular target for the gold-standard diagnostic PCR for Buruli ulcer15.
A concurrent case–control study performed in the same geographical area identified only two factors associated with the odds of being diagnosed with Buruli ulcer: insect repellent use reduced risk (odds ratio (OR), 0.37; 95% confidence interval (CI), 0.19–0.69) and being bitten by mosquitoes on the lower legs increased risk (OR, 2.60; 95% CI, 1.22–5.53). A variety of outdoor activities was also surveyed but was not independently predictive, suggesting that mosquito exposure specifically rather than environmental exposure generally might be the main mode of M. ulcerans transmission to humans16. In Africa, two case–control studies conducted in Cameroon found use of bed nets as a protective factor against Buruli ulcer (OR, 0.4; 95% CI, 0.2–0.9; P = 0.04 (ref. 17) and OR, 0.1; 95% CI, 0.03–0.3; P < 0.001 (ref. 18)).
However, case–control studies and entomological surveys alone are insufficient to implicate biological agents as vectors of pathogens. There are formal frameworks used in biomedicine, such as the Barnett criteria3, that build hierarchies of evidence to implicate a candidate disease vector. Here we build on this aforementioned research to formally address the Barnett criteria and to test the hypothesis that mosquitoes vector M. ulcerans to humans14,19,20. A summary of our findings is shown in Extended Data Table 1, including the new data shown in this study that specifically address criteria 1–3.
Extended Data Table 1.
Summary of evidence to indict mosquitoes as vectors of M. ulcerans

The research shown in this study is based on a substantial field survey of more than 65,000 mosquitoes undertaken over 4 months in 2019 and 2020, and four smaller ad hoc surveys conducted between 2016 and 2021. The study area was the Mornington Peninsula, an area of 350 km2, located 90 km south of Melbourne, the capital city of Victoria9 (Extended Data Fig. 1). The area was originally covered in low-lying coastal vegetation9, receives an average annual rainfall of 740 mm and sits at an average elevation of 60 m above sea level21. The Mornington Peninsula region continues to maintain among the highest incidences of Buruli ulcer in the world22, with a conservative local incidence estimate of 55 human cases per 100,000 population in 20224,23. Here we used mosquito trapping, bacterial enrichment genome sequencing, mosquito blood-meal metabarcoding and spatial clustering analyses to build a hierarchy of evidence that supports mosquitoes as vectors of M. ulcerans from local wildlife reservoirs to humans.
Results
Mosquito surveys of the Mornington Peninsula
A primary goal of this research was to test the hypothesis that mosquitoes are associated with M. ulcerans transmission, as reflected by IS2404 PCR positivity at a certain frequency in areas of the Mornington Peninsula with human Buruli ulcer cases. A total of 73,580 mosquitoes were collected, consisting of 72,263 females (Table 1) and 1,317 males (Supplementary Table 1). The majority (90%) of these insects were collected in the large survey of 2019–2020 (Supplementary dataset 1). Across all 5 surveys, 26 different mosquito species were collected covering 6 genera (Table 1). The most dominant species identified during the 2019–2020 survey was Culex molestus, accounting for 42% of mosquitoes, followed by Ae. notoscriptus (35%) and Culex australicus (8%). Twenty-three other species comprised the remaining 15% of mosquitoes. The distribution of the two dominant species across the survey area is shown in Fig. 1. This mapping revealed an asymmetric distribution of each species, with Ae. notoscriptus dominant to the eastern end and Culex molestus dominant towards the western end of the peninsula (Fig. 1).
Table 1.
Female mosquitoes trapped on the Mornington Peninsula and screened by IS2404 PCR
| Species | Number of mosquitoes/number of mono-species pools screened for M. ulcerans (number of pools positive for M. ulcerans, IS2404a) | |||||
|---|---|---|---|---|---|---|
| December 2016 to April 2017 | November 2017 to May 2018 | December 2018 to May 2019 | November 2019 to March 2020 | February 2021 to March 2021 | Total | |
| Aedes alboannulatus | 9/9 | 4/4 | 3/3 | 183/96 | 21/21 | 220/133 |
| Aedes australis | 0 | 0 | 0 | 1/0 | 0 | 1/0 |
| Aedes bancroftianus | 0 | 0 | 0 | 3/2 | 0 | 3/2 |
| Aedes camptorhynchus | 258/258 | 5/5 | 0 | 121/64 (1) | 8/8 | 392/335 (1) |
| Aedes clelandi | 0 | 0 | 0 | 41/32 | 0 | 41/32 |
| Aedes flavifrons | 1/1 | 0 | 0 | 31/26 | 13/13 | 45/40 |
| Aedes imperfectus | 0 | 0 | 0 | 266/226 | 0 | 226/226 |
| Ae. notoscriptus | 174/174 (2) | 367/367 (8) | 1,793/1,779 (15) | 23,360/4,330 (14) | 1,247/1,247 (7) | 26,941/7,897 (46) |
| Aedes rubrithorax | 9/9 | 27/27 | 230/230 | 1,402/335 | 45/45 | 1,713/646 |
| Aedes sagax | 0 | 0 | 0 | 2/2 | 0 | 2/2 |
| Aedes theobaldi | 0 | 0 | 0 | 1/1 | 0 | 1/1 |
| Aedes vigilax | 0 | 0 | 0 | 2/0 | 0 | 2/0 |
| Aedes vittiger | 0 | 0 | 0 | 2/0 | 0 | 2/0 |
| Anopheles annulipes | 1/1 | 24/24 | 4/4 | 751/376 | 1/1 | 781/406 |
| Anopheles atratipes | 0 | 0 | 0 | 2/0 | 0 | 2/0 |
| Coquillettidia linealis | 89/89 | 142/142 | 2/2 | 1,254/258 | 11/11 | 1,498/502 |
| Culiseta inconspicua | 0 | 0 | 0 | 2/2 | 3/3 | 5/5 |
| Culex annulirostris | 0 | 3/3 | 0 | 42/1 | 0 | 45/4 |
| Culex australicus | 3/3 | 198/198 | 17/17 | 4,910/534 | 33/33 | 5,161/785 |
| Culex cylindricus | 0 | 0 | 2/2 | 160/81 | 0 | 162/83 |
| Culex globocoxitus | 0 | 24/24 | 0 | 3,207/854 | 9/9 | 3,240/887 |
| Culex molestus | 61/61 | 29/29 | 384/377 | 28,107/3,308 | 1,465/1,465 | 30,046/5,240 |
| Culex orbostiensis | 0 | 0 | 0 | 131/69 | 0 | 131/69 |
| Culex quinquefasciatus | 3/3 | 257/257 | 37/37 | 1,098/401 | 163/163 | 1,558/861 |
| Tripteroides atripes | 0 | 0 | 0 | 18/0 | 12/12 | 30/12 |
| Tripteroides tasmaniensis | 0 | 0 | 0 | 15/13 | 0 | 15/13 |
| Total | 608/608 (2) | 1,080/1,080 (8) | 2,512/2,451 (15) | 65,112/11,011 (15) | 3,031/3,031 (7) | 72,263/18,181 (47) |
aPool size ranged from 1 individual up to 15 individuals.
Fig. 1. Dominant mosquito species distribution across the Mornington Peninsula.

Map showing the proportional distribution of the two dominant mosquito species trapped during 2019 and 2020. The pie charts are an aggregation of the 180 different trap sites. Trap groups containing mosquitoes that were PCR positive for M. ulcerans are also indicated. The mesh-block statistical areas are also shown, with those in red containing at least one human Buruli ulcer case diagnosed in 2019–2020. Map boundaries from Australian Bureau of Statistics69 under a Creative Commons license CC BY 4.0.
Ae. notoscriptus not Culex molestus harbour M. ulcerans
The IS2404 quantitative-PCR (qPCR) assay was used to infer the presence of M. ulcerans in association with mosquitoes15. Of the 73,580 mosquitoes trapped across all years, 18,610 (25%) were screened by IS2404 qPCR for the presence of M. ulcerans (Table 1). M. ulcerans qPCR positives were observed in 53 pools or individuals of Ae. notoscriptus, with detections occurring in each year across all survey events (Fig. 1 and Table 1). Only one other mosquito species tested IS2404 positive, which was a two-insect pool of Aedes camptorhynchus (Table 1). The positive association between M. ulcerans and Ae. notoscriptus compared with other mosquitoes, in particular the other abundant mosquito species, Culex molestus, was significant (P < 0.0001, Fisher’s exact test).
Subsets of Ae. notoscriptus were screened individually to better estimate the prevalence of M. ulcerans-positive mosquitoes in this species. Individually tested mosquitoes included all of the 2017–2018 collection comprising 367 of 367 screened individuals with 8 positives (2.2%), 448 of 4,330 screened individuals of the 2019–2020 collection with 3 positives (0.67%), and all 1,247 individuals of the 2021 collection with 7 positives (0.56%) (Supplementary Table 2). Thus, on the basis of screening individual insects, 18 of 2,062 (0.87%) Ae. notoscriptus were IS2404 PCR positive. All other Ae. notoscriptus and other mosquito species in this study were screened in pools (up to 15 insects per pool) with 24 of 747 Ae. notoscriptus pools (3%) being PCR positive (Supplementary Table 3).
Of the 46 Ae. notoscriptus that tested positive for IS2404, 26 (56%) were confirmed positive for IS2606 and 31 (67%) for KR, with 24 (52%) pools positive by all three qPCR assays (Supplementary Table 2). Of the 46 IS2404-positive Ae. notoscriptus, 8 (17%) pools were not tested using the IS2606 assay and 1 (2%) pool was not tested using the KR assay due to limited template DNA available. The average cycle threshold (Ct) value for IS2404-positive Ae. notoscriptus was 36.00 (range, 29.74–39.65). The average Ct value for IS2606 was 37.73 (range, 32.49–45.00) and the average Ct value for KR was 34.19 (range, 28.52–39.26). With reference to an IS2404 qPCR standard curve (Supplementary Fig. 2), we estimated the M. ulcerans burden per mosquito. The mean bacterial genome equivalents (GE) per insect was 294 GE (range, 11–4,200; Supplementary Table 3). The MLE of estimated infection rate for all Ae. notoscriptus was 5.88 (95% CI, 4.37–7.76) based on the M. ulcerans IS2404-qPCR-positive mosquitoes per 1,000 tested for all Ae. notoscriptus tested over the years and 2.96 (95% CI, 0.17–14.19) for Aedes camptorhynchus (Table 2).
Table 2.
M. ulcerans-positive pools tested by IS2404 qPCR
| Species | Number of mosquitoes | Number positive/number pools tested | MLEa | 95% CI |
|---|---|---|---|---|
| Aedes alboannulatus | 133 | 0/97 | 0 | 0–27.71 |
| Aedes bancroftianus | 2 | 0/2 | 0 | 0–657.62 |
| Aedes camptorhynchus | 335 | 1/85 | 2.96 | 0.17–14.19 |
| Aedes clelandi | 32 | 0/3 | 0 | 0–67.97 |
| Aedes flavifrons | 40 | 0/16 | 0 | 0–78.10 |
| Aedes imperfectus | 226 | 0/27 | 0 | 0–14.96 |
| Ae. notoscriptus | 7,897 | 46/2,937 | 5.88 | 4.37–7.76 |
| Aedes rubrithorax | 646 | 0/220 | 0 | 0–5.84 |
| Aedes sagax | 2 | 0/1 | 0 | 0–545.52 |
| Aedes theobaldi | 1 | 0/1 | 0 | 0–793.45 |
| Anopheles annulipes | 406 | 0/148 | 0 | 0–9.16 |
| Coquillettidia linealis | 502 | 0/139 | 0 | 0–7.43 |
| Culiseta inconspicua | 5 | 0/4 | 0 | 0–408.11 |
| Culex annulirostris | 4 | 0/3 | 0 | 0–450.75 |
| Culex australicus | 785 | 0/150 | 0 | 0–4.76 |
| Culex cylindricus | 83 | 0/38 | 0 | 0–41.60 |
| Culex globocoxitus | 887 | 0/150 | 0 | 0–4.22 |
| Culex molestus | 5,240 | 0/609 | 0 | 0–0.73 |
| Culex orbostiensis | 69 | 0/9 | 0 | 0–42.95 |
| Culex quinquefasciatus | 861 | 0/237 | 0 | 0–4.39 |
| Tripteroides atripes | 12 | 0/9 | 0 | 0–230.35 |
| Tripteroides tasmaniensis | 13 | 0/10 | 0 | 0–215.26 |
aMLE of M. ulcerans per 1,000 mosquitoes trapped in the Mornington Peninsula. MLE bias was corrected when one or more pool was positive.
M. ulcerans genomes from mosquitoes, possums and people match
Genomic epidemiological studies have shown there are characteristic single-nucleotide-polymorphism (SNP) signatures associated with M. ulcerans clinical isolates from specific endemic areas in southeastern Australia24. To test if the M. ulcerans genotypes present in mosquitoes from our study area matched those found in possum excreta and human Buruli ulcer cases in the same region, we conducted genome sequence enrichment. As reported in other sequence enrichment studies, we observed decreasing genome sequence recovery with decreasing pathogen load, as indicated by increasing IS2404 Ct values (Fig. 2a)25. Nevertheless, DNA sequence reads were obtained from five IS2404-positive mosquitoes and two IS2404-positive possum excreta specimens, the latter specimens collected as part of a large field survey of M. ulcerans in Australian native possum excreta in the region26. Although DNA sequence reads were obtained across the length of the 5.6 Mbp M. ulcerans reference chromosome, for all 5 mosquitoes and the 2 possum excreta specimens, only the excreta specimens (IS2404 Ct values <23) and 3 of the 5 mosquito DNA extracts (identifier (ID) 5675, Ct 32.62; ID 226, Ct 33.47; ID 819, Ct 31.20) yielded sufficient M. ulcerans reads for SNP calling (Fig. 2b).
Fig. 2. M. ulcerans genome from human Buruli ulcer cases compared with sequences recovered from possum excreta and mosquitoes on the Mornington Peninsula.

a, Summary of IS2404 qPCR screening of primary samples and the sequence-capture libraries pre- and post-enrichment. Note that possum excreta were enriched as barcoded sequence library pools so they share the same pre- and post-enrichment Ct values. b, Artemis coverage plots depicting sequence-capture reads mapped to the M. ulcerans JKD8409 chromosome from possum excreta samples and three qPCR-positive mosquitoes (labels are given as numbers of mapped reads; percentage of total chromosome bases mapped; number of core variable nucleotide positions (VNPs) covered). The grey horizontal bar above the chromosome map shows the sites of 117 core SNPs (black inverted triangles). c, Maximum likelihood phylogeny inferred from an alignment of 117 core genome SNPs using reads mapped to the JKD8049 reference chromosome, and with tips aligned with environmental sample origin or patient origin. The dataset includes reads from the 5 environmental samples listed in a with >21 VNPs, and a reference collection of 36 M. ulcerans genomes representing the genomic diversity of the M. ulcerans population in southeastern Australia24. The shortest vertical branch length represents a single SNP difference, as per the scale bar. Map boundaries in c from Google Maps under a Creative Commons license CC BY 4.0.
To assess the relatedness of M. ulcerans genomes from mosquito and excreta to human M. ulcerans isolates, we inferred a phylogeny using a 5.6 Mbp local M. ulcerans reference chromosome and sequence reads from 36 published clinical M. ulcerans genomes from southeastern Australia24, based on a core genome alignment with 117 SNP positions. These 36 genomes were selected because they spanned our previously reported population structure of M. ulcerans in this locale (Supplementary Data 2)24. Alignment of the five mosquito and two excreta sequence-capture datasets revealed reads mapping across the length of the M. ulcerans chromosome—confirming the presence of the pathogen on (or in) Ae. notoscriptus and in the possum faecal material (Fig. 2b).
Despite chromosome-wide read mapping for the seven sequence-capture datasets, the relatively low and incomplete coverage meant that these datasets did not have reads spanning all 117 variable loci (range, 22–112 sites; Fig. 2b). Thus, to enable the inclusion of the sequence-capture datasets into a phylogenomic analysis of 117 core SNPs, a multivariate statistical approach using an imputation model was used (Extended Data Fig. 2). The model was trained on the 117 SNPs from the core genome alignment of all 36 clinical isolates to predict with high confidence (impute) the missing SNP alleles of the sequence-capture datasets (Methods). To validate the model, we initially masked 95 random SNP sites as missing data for a random set of 5 of the 36 clinical isolates, leaving 22 SNPs. Using the multivariate imputation model trained on the full 117 sites of the remaining 31 clinical isolate genomes, we predicted the missing alleles of the 5 masked clinical isolates (Extended Data Fig. 2). This approach yielded a mean accuracy of 97% in correctly predicting missing alleles, given the 22 available SNPs. To ensure the model was robust, we randomly selected five M. ulcerans clinical isolate genomes and then varied the number of masked chromosome SNP sites to simulate missing data and replicated this process 100 times. This analysis showed the high performance of the imputation model, with a mean accuracy of 94% when 100 of 117 SNP sites were randomly masked and then imputed (Extended Data Fig. 2). Using the validated imputation model to predict the missing alleles for the five sequence-capture datasets and create a sequence alignment (Supplementary Data 3), a maximum likelihood tree was inferred using all 117 variable sites with location of tree tips aligned with geographical origin. This phylogeny showed possum excreta and mosquito M. ulcerans genotypes co-clustered with each other and with human M. ulcerans isolates from the Mornington Peninsula, with 0–5 SNP differences between any pairwise comparison (Fig. 2c). This pattern is consistent with a shared transmission cycle between possums, mosquitoes and humans. Individual phylogenies inferred with and without SNP allele imputation showed that imputation did not create artefactual tree topologies (Extended Data Fig. 3).
Extended Data Fig. 2. Validation of allele imputation approach to assess robustness to level of missing data and genome selection.
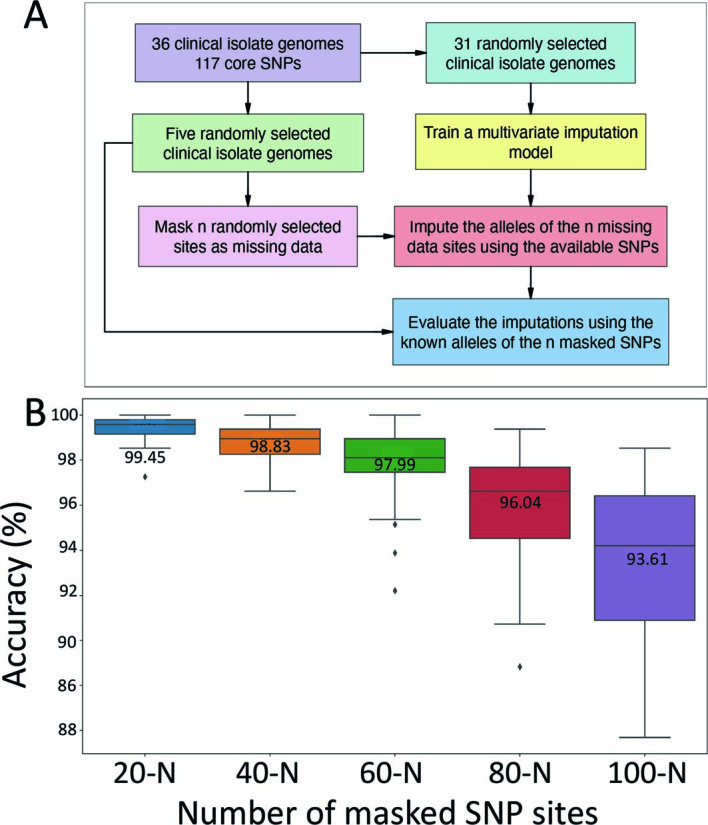
(a) Flow diagram of imputation method. (b) Boxplots showing the distribution of imputation accuracy across 100 replicates for varying numbers of M. ulcerans chromosome SNP sites masked as missing data for five randomly selected M. ulcerans clinical isolate genomes. The line inside each box represents the median accuracy and the annotated numbers above each box indicate the mean accuracy in percentage. The whiskers extend to 1.5× the interquartile range.
Extended Data Fig. 3. Tanglegrams analysis showing impact of imputation on phylogenetic inference.
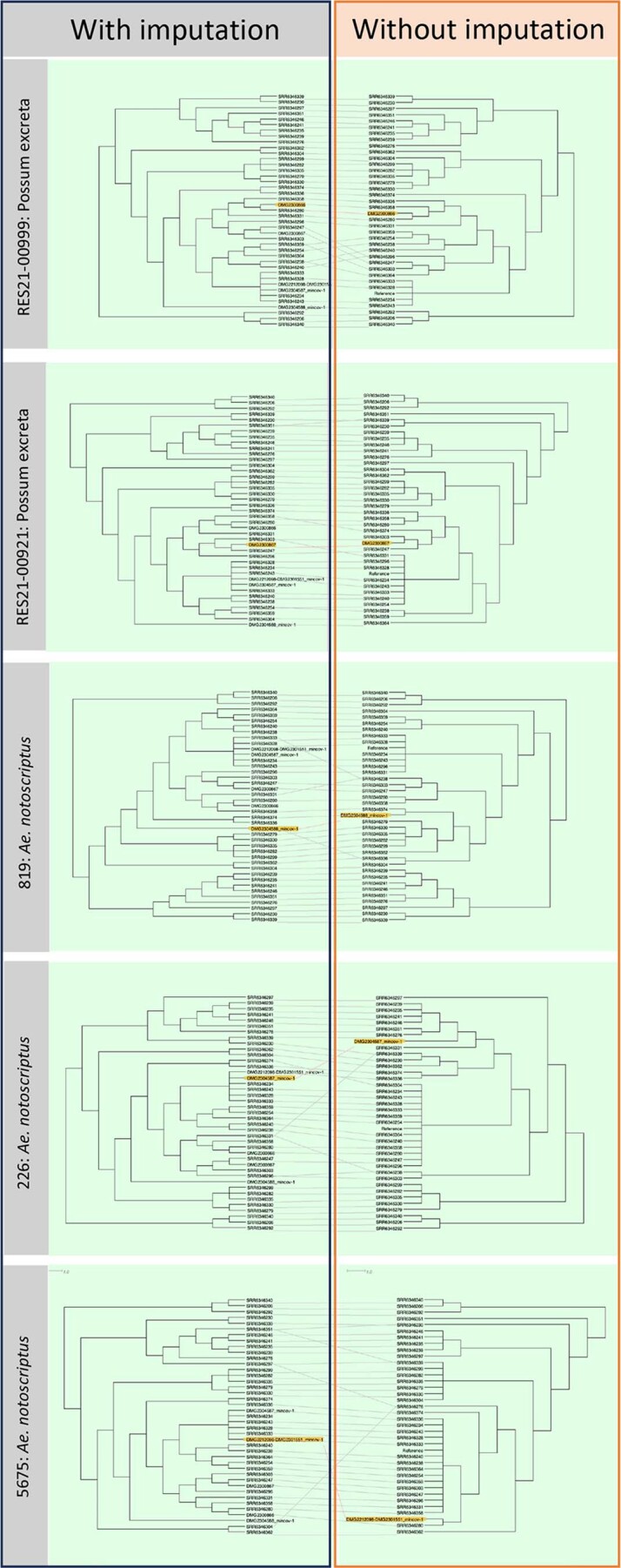
Depicted are tanglegrams (Dendroscope) showing phylogenies inferred with imputed SNP alignments containing each of the five, individual sequence enrichment and 36 clinical isolate M. ulcerans genome sequences compared to phylogenies of the individual sequence enrichment genomes and 36 clinical isolates without imputation. Yellow highlighted tips show the sequence enrichment genomes. Branch lengths transformed and presented as cladograms.
Genetic characterization of Ae. notoscriptus
To assess if M. ulcerans presence was associated with a particular clade of Ae. notoscriptus on the Mornington Peninsula, we compared cytochrome c oxidase subunit I (COI) gene sequences for 18 M. ulcerans-positive (confirmed by all IS2404, IS2606 and KR qPCR assays) Ae. notoscriptus and 19 M. ulcerans-negative Ae. notoscriptus. On the basis of the COI phylogenetic tree, Ae. notoscriptus from the Mornington Peninsula spanned the three previously identified clades and there was no association between M. ulcerans and a particular mosquito lineage (Supplementary Fig. 1)27.
IS2404 PCR screen of arthropods other than mosquitoes
We also investigated the association of M. ulcerans with arthropods other than mosquitoes in the region using yellow sticky traps (YST) and sticky ovitraps (SO). A total of 21,000 specimens were collected and sorted from 278 YST and 33 SO. We were able to classify 2,696 specimens as insects that may bite or pierce human skin (Extended Data Table 2). Flies were the largest group collected on the sticky traps. YST collected more insects than the SO, but this was proportional to the number of traps set (Extended Data Table 2). Of the 2,696 insects screened by PCR, only 2 flies tested positive for M. ulcerans (Supplementary Table 2). Both flies had high Ct values for IS2404 (Ct 35.92 and 37.54) and each sample was only confirmed by either the IS2606 or the KR assay (Supplementary Table 2), but not with all three assays. Both insects were blow flies (Calliphora hilli Patton) based on morphology and sequencing of the COI region, with sequences having >99.86 nt identity to Calliphora hilli.
Extended Data Table 2.
Number of arthropods collected on Yellow Sticky Traps (YST) and Sticky Ovitraps (SO) on the Mornington Peninsula that might bite or pierce human skin

Mosquito blood-meal analysis
Of the mosquitoes collected, a proportion of individuals identified as being engorged (blood fed) were PCR screened and the resulting amplicon sequenced for cytochrome b (cytb) to identify host blood-meal sources. A total of 90 individual engorged mosquitoes were extracted, with 70 DNA preparations producing high-quality amplicons that were of sufficient concentration to permit Illumina amplicon sequencing. After quality filtering, 36 individuals were identified as having had a recent blood meal: 14 Culex molestus, 13 Ae. notoscriptus, 2 Aedes rubrithorax, 2 Culex globocoxitus, 2 Culex quinquefasciatus, 2 Coquillettidia linealis and 1 Culex australicus. Of the blood meals detected, the common ringtail possum was the most commonly identified with 20 detections across the 36 samples, followed by 17 blackbirds, 13 humans, 11 red wattle birds and 5 little wattle birds; a further 16 blood meals identified 10 minor host species (Fig. 3 and Extended Data Fig. 4). Dual blood meals were commonly identified, with 55% (20 of 36) of individuals having more than one blood meal identified. In addition, two mosquitoes had evidence of three different blood-meal sources within an individual insect. Three individuals (two Ae. notoscriptus and one Aedes rubrithorax) were also identified as having dual blood meals from ringtail possum and human origins (Fig. 3 and Extended Data Fig. 4).
Fig. 3. Mosquito blood-meal analysis.

Summary of cytb gene sequences from the 36 blood-fed mosquitoes to identify host blood-meal sources. A blue circle indicates positive for host blood source; the larger the circle, the more individual mosquitoes with an identical blood-meal profile. Red boxes indicate individual mosquitoes that have dual blood meals for both humans and ringtail possum sources.
Extended Data Fig. 4. Mosquito bloodmeal analysis of all detected bloodmeal sources.

A blue dot indicates positive for host blood source; the larger the dot, the more individual mosquitoes with an identical bloodmeal profile. Red boxes indicate individual mosquitoes which had dual bloodmeals from both humans and ringtail possum sources.
Clustering of mosquitoes, possums and human Buruli ulcer
In our previous research, we showed a spatial association between possum excreta containing M. ulcerans, as detected by structured surveys, and clusters of human Buruli ulcer cases26. To expand on this finding, we investigated whether further spatial clustering associations could be detected by analysing qualitative qPCR (IS2404) data of trapped mosquitoes using SaTScan. Here three separate analyses were conducted: (1) human Buruli ulcer case data from individuals reported to have acquired the disease in the study area during 2019–2020, (2) qPCR data from possum excreta collected in a previous investigation (2018–2019) and (3) qPCR data from trapped mosquitoes (2019–2020). These analyses identified a single mosquito cluster, four possum clusters and six human Buruli ulcer clusters (Fig. 4 and Extended Data Table 3). Notably, one human Buruli ulcer cluster and two possum excreta clusters had higher numbers of observed Buruli ulcer cases or M. ulcerans detections, respectively, than expected if uniformly distributed (P < 0.05; Extended Data Table 3). Importantly, the analyses revealed an instance of triple-cluster overlap (mosquito, possum excreta and human) in the Mornington Peninsula suburb of Rye, where all three SaTScan analyses had an overlapping cluster (Fig. 4). A permutation test to assess the probability of these three categories (mosquitoes, possums and humans) overlapping showed that a triple-cluster overlap occurred with randomly rearranged location labels at a low frequency of 8.9%. This analysis adds support to a causal relationship between the presence of M. ulcerans in possums and mosquitoes and with humans contracting Buruli ulcer in the Mornington Peninsula suburb of Rye.
Fig. 4. Spatial clustering of trapped mosquitoes, possum excreta and human Buruli ulcer cases on the Mornington Peninsula.

Map showing the clusters identified by the three separate SaTScan analyses: (1) trapped mosquitoes (177 traps screened for IS2404; collected November 2019 to March 2020; denoted M1 and M2), (2) M. ulcerans detected in possum excreta collected during the summer of 2019 (December 2018 to February 2019; P1–P4) using data from a previous study, and (3) notified human Buruli ulcer cases from the study area in the years 2019–2020 (H1–H6). The inset shows an instance where all three analyses had overlapping clusters in the suburb of Rye. Map boundaries from Australian Bureau of Statistics69 under a Creative Commons license CC BY 4.0.
Extended Data Table 3.
Summary of the SaTScan cluster analysis

Discussion
Using insect field surveys augmented with genomics, we have addressed Barnett criteria 1–3 (Extended Data Table 1) to add to a hierarchy of evidence implicating mosquitoes as the principal vectors in the spread of M. ulcerans from the environment to humans3,14,16,19,20. Our survey area was the Mornington Peninsula, where cases of human Buruli ulcer have increased progressively over the past 20 years11. Our key findings were that M. ulcerans was almost exclusively associated with one mosquito species in this region, Ae. notoscriptus, at a frequency of 0.87%. The estimated pathogen burden in each insect (Supplementary Table 2) was consistent with the reported low infectious dose for M. ulcerans20. Pathogen genomics provided support for a linked transmission chain between mosquitoes, native possums and humans. Metabarcoding mosquito blood-meal analysis revealed dual feeding by individual mosquitoes on both possums and humans, providing an example of a potential transmission pathway between infected possums and humans. Finally, spatial clustering analysis showed overlap between clusters of possums shedding M. ulcerans, mosquitoes harbouring M. ulcerans, and human Buruli ulcer cases, reinforcing the importance of possums and mosquitoes in the spread of M. ulcerans to humans.
A role for mosquitoes in the transmission of M. ulcerans was initially revealed in entomological surveys in the mid-2000s on the Bellarine Peninsula and then more recently in Far North Queensland14,19,28. Other observations that supported a role for mosquitoes in transmission included an analysis of the Buruli ulcer lesion location on the human body, which included more than 600 human Buruli ulcer cases, and showed a preponderance of lesions on ankles, elbows and backs of legs. These areas are frequently exposed and insects may preferentially blood-feed there29. Laboratory studies under one transmission scenario have also shown the competence of Ae. notoscriptus as mechanical vectors of M. ulcerans (Barnett criterion 4; Extended Data Table 1)20.
Of the ∼18,600 mosquitoes tested by IS2404 PCR from 5 different survey periods in this study, Ae. notoscriptus (the second-most-abundant species trapped) was consistently positively associated with M. ulcerans. Where and how M. ulcerans is contaminating (or infecting) these mosquitoes remains to be determined. However, we made the somewhat unexpected observation that the most abundant mosquito species on the Mornington Peninsula, Culex molestus, was consistently IS2404 PCR negative. This observation might be explained by a difference in the ecology of these two mosquito species, such as how (or where) they are encountering M. ulcerans. The apparent Ae. notoscriptus–M. ulcerans tropism might also indicate a specific biological interaction between this insect and the mycobacterium or, alternatively, a Culex molestus antagonism to mycobacterial carriage. Unfortunately, the ecology of mosquito subspecies in this region is generally poorly understood, including that of Ae. notoscriptus, which occurs as a series of genetically differentiated clades within Australia27.
To explore host sources for mosquitoes trapped in our study, we used metabarcoding sequencing of engorged mosquitoes30. Of the 90 blood-fed mosquitoes processed, 36 individual insects successfully had their host blood source identified. The 54 unidentified blood meals were probably due to degradation of the host DNA that occurs approximately 36 hours post-feeding31. Of the host blood sources identified, the common ringtail possum was the most commonly identified blood meal, followed by birds and humans, with dual blood meals observed in 55% of mosquitoes (Fig. 4 and Extended Data Fig. 3). Notably, three insects were determined to have blood meals from both common ringtail possums and humans. These insects consisted of two Ae. notoscriptus and one Aedes rubrithorax. The identification of these dual blood meals provides evidence that Ae. notoscriptus mosquitoes are blood-feeding between possums and humans within a relatively short timeframe. Dual blood meals may provide an opportunity for a mechanical transmission event of M. ulcerans from a possum with a Buruli ulcer teeming with M. ulcerans, from which a mosquito blood-feeds and then moves on to a nearby human. However, the observed positive association specifically between Ae. notoscriptus and M. ulcerans is not explained by our blood-meal analysis, which showed that several other mosquito species that were not M. ulcerans positive, such as Culex molestus, Aedes rubrithorax and Coquillettidia linealis, also fed on possums. An alternative source of M. ulcerans acquisition for Ae. notoscriptus might be from possum excreta contaminating the Ae. notoscriptus breeding sites, as this species breeds in small artificial containers32. If possum excreta, which can have a high concentration of M. ulcerans10, falls into these artificial breeding sites, then this may provide a means to contaminate the adult Ae. notoscriptus during eclosion and emergence on top of the water’s surface.
In common with other arthropod-borne bacterial pathogens, M. ulcerans also has the genomic hallmarks of niche adaptation33,34. For instance, the bacterial diseases vectored by blood-feeding arthropods—such as bubonic plague, spread to humans through the bite of fleas harbouring the bacterium Yersinia pestis35; the tick-borne infections such as Lyme disease, Rocky Mountain spotted fever and ehrlichiosis (among others) caused by Borrelia burgdorferi, Rickettsia rickettsii and Ehrlichia sp., respectively36; or tularaemia, caused by infection with Francisella tularensis spread through the bite of infected ticks (with some subspecies also spread by mosquitoes)37–39—are all caused by bacteria that have degenerating genomes. That is, as with M. ulcerans, their genomes bear the distinctive hallmarks of evolutionary bottlenecks and niche adaptation (plasmid acquisition, pseudogene accumulation and insertion sequence expansion), a genomic pattern thought indicative of a shared trajectory towards symbiosis with the arthropod host40. In addition, the absence of recombination and highly clonal population structure of M. ulcerans is aligned with the population structure of bacteria in ‘closed symbiosis’ with their host animal41.
Mosquitoes were not the only arthropods from which M. ulcerans were detected in the Mornington Peninsula. With over 20,500 arthropods collected using sticky traps (YST and SO), this study screened other insects that might broadly act as a mechanical vector, that is, an insect that is involved in the accidental transport of a pathogen20,42. M. ulcerans was detected on 2 of ∼1,800 flies tested, identified as blow flies (Calliphora hilli). Previous studies have screened limited numbers of other fly species, such as March flies (Tabanidae), with no positive detections28. Calliphora hilli is a native fly occurring along the Australian east coast and in South Australia43. It is a carrion breeder occurring year round throughout Victoria, with the female laying her eggs in decaying flesh from which the larvae emerge43. Possum carcasses are readily infested by Calliphora hilli44, which might explain how this fly species is becoming contaminated with M. ulcerans, as possums are wildlife reservoirs of M. ulcerans9. The likelihood of Calliphora hilli carrying M. ulcerans to humans is relatively low because this species rarely bites humans45.
In this study, we also used targeted enrichment genome capture to show beyond reasonable doubt the presence of M. ulcerans in association with mosquitoes25. Although the method lost sensitivity for IS2404 qPCR Ct values >32, we generated sufficient genome coverage from three IS2404-PCR-positive mosquitoes and from two possum excreta specimens to show that the M. ulcerans genotypes of the captured genomes were identical to those associated with human Buruli ulcer cases in the study area, rather than Buruli ulcer cases linked to other regions (Fig. 2). We are now exploring the use of genome sequence enrichment to dissect the genomic epidemiology from the perspective of environmental sources, in particular to track the spread of Buruli ulcer in Australian native possums across the region26.
The triple-cluster overlap observed in our SaTScan analyses (Fig. 4) suggests that the spatial distribution of these three factors is likely to be closely linked and might be contributing to the persistence of Buruli ulcer in the Mornington Peninsula. Our findings highlight the importance of ongoing surveillance of possum and mosquito populations, which may provide critical insights into the epidemiology of Buruli ulcer in the region and inform targeted public health interventions to control the disease.
Our research has some limitations. Although we found a spatial association between the presence of M. ulcerans in possums and mosquitoes and with human Buruli ulcer cases, we cannot absolutely conclude causality or directionality. In addition, the very specific set of circumstances that have led to the rise of Buruli ulcer in temperate southeastern Australia restricts the generalizability of our results. One must be cautious to draw parallels with African Buruli ulcer endemic countries, for instance, where a highly susceptible mammalian reservoir equivalent to the Australian native possum is yet to be identified and evidence for mosquitoes as possible vectors is lacking46. Further investigations are needed to better understand the underlying mechanisms that drive the association between the presence of M. ulcerans in these different species and the development of human Buruli ulcer cases.
Despite these caveats, our collective research over more than 15 years makes it very clear that mosquitoes are probable vectors and native possums are major wildlife reservoirs of M. ulcerans in southeastern Australia. Mosquito surveillance with M. ulcerans screening coupled with mosquito control and public health messaging to avoid mosquito bites are practical interventions that would be expected to reduce the incidence of human Buruli ulcer.
Methods
Ethics
Ethical approval for the use in this study of de-identified human Buruli ulcer case location, aggregated at the mesh-block level, was obtained from the Victorian government Department of Health Human Ethics Committee under HREC/54166/DHHS-2019-179235(v3), ‘Spatial risk map of Buruli ulcer infection in Victoria’.
Study site
Insects were collected from the Mornington Peninsula suburbs of Rye (population 8,416), Blairgowrie (population 2,313), Tootgarook (population 2,869) and Capel Sound (population 4,930)47 (Extended Data Fig. 1)48.
Arthropod collection and identification
Mosquito-trapping campaigns used Biogent Sentinal (BGS) traps (Biogent) that were baited with dry-ice pellets to provide a source of CO2 over an intended 12 hour period. BGS traps were set out at dusk, and collected at dawn, in shaded locations on the grassed edges of the roadways in the study area. GPS locations for all traps were recorded, with data collection managed using Atlas of Medical Entomology (v.3.4.4; Gaia Resources). Trapped mosquitoes were knocked down with CO2 by placing the catch bag in dry ice before being transported back to the laboratory and kept at −20 °C until processing. Mosquito species were morphologically identified using a stereo dissecting microscope (SMZ800N; Nikon) and reference to taxonomic keys49–51. Data collection was managed using Microsoft Excel (v.16.73).
To assess the potential presence of M. ulcerans beyond mosquitoes, other arthropods were collected using YST and SO. Two YST and SO were placed in residential properties where householders had previously noted insect activity to the researchers8. The SO were placed on the ground and had hay grass infusion (3 Jack Rabbit (clover or lucerne) pellets (Laucke Mills) in 500 ml water) added to them, whereas the YST (Bugs for Bugs) were placed on the ground with a 14 cm plant tag plastic T-support (Garden City Plastics). Within 3–4 days of being set, residents were asked to pack up the YST and SO by covering the sticky card with a plastic film and return the sealed traps to the laboratory, where they were stored at −20 °C. Non-mosquito arthropods were morphologically identified to family level and, if PCR positive for M. ulcerans, were DNA barcoded for species confirmation by targeting the COI gene52.
DNA extraction from mosquitoes
Mosquitoes were sorted by species per trap and by sex and then pooled in 2 ml O-ring tubes, with a maximum of 15 individuals in each pool. A subset of Ae. notoscriptus mosquitoes were also screened individually. Mosquitoes were homogenized with 10 mm × 1.0 mm zirconia/silica beads (BioSpec Products), with 597 µl of Buffer RLT and 2.8 µl of carrier RNA. Homogenization was performed using a TissueLyser II (Qiagen) at 30 oscillations per second for 100 s, repeated twice. Tubes were then centrifuged at 16,000g for 3 min. A 550 µl volume of supernatant was transferred into a 96-well deep-well plate, with extraction performed according to the protocol for the BioSprint 96 One-For-All Vet Kit (Qiagen). Every 11th of 12 wells in a 96-well plate was a blank DNA extraction control (7 in total) and a synthetic IS2404-positive control was spiked into 1 of these 7 wells to act as a positive extraction control. Extraction was performed on a KingFisher Flex Magnetic Particle Processor (Thermo Scientific).
DNA extraction from arthropods
Arthropods other than mosquitoes collected on YST or SO were removed from the sticky cards and placed in 1.5 ml microtubes (Eppendorf). Insects were separated by family and trap location, and were pooled with a maximum of 10 individuals from each family per 1.5 ml tube. Samples were extracted non-destructively to allow species confirmation if positive detections occurred. DNA was extracted using the ISOLATE II Genomic DNA Kit (Bioline). Briefly, 25 µl of Proteinase K and 180 µl of Lysis Buffer GL was added to each tube with samples incubated overnight at 56 °C. Following incubation, the insects were removed and stored to allow for further morphological identification if required, with the DNA extraction completed on the incubation solution as per manufacturer instructions.
Synthetic PCR positive control
A synthetic PCR positive control DNA molecule was designed to discriminate false positives due to contamination with positive control DNA versus the authentic IS2404 amplicon. The synthetic positive control was designed to have an amplicon size of 120 bp to easily differentiate it from a true IS2404 PCR positive (59 bp)15. The synthetic positive was added at the DNA extraction stage on all 96-well plates as a positive control for this step and for the subsequent qPCR. The additional DNA sequence used to construct the synthetic positive control was randomly selected from a DNA sequence unlikely to be in the laboratory, in this case, Irrawaddy dolphin (MK032252).
The synthetic positive control had the sequence 5′-TCCTAAAGCACCACGCAGCATCTATCGCGAGCTTAATCACCATGCCGCGTCCAACGCGATCCCCGCTCGGCAGGGATCCCTCTTCTCGCACCGGGCCACAATCCACTGGGGTCGCTATGA-3′ and was synthesized as a single-stranded DNA oligonucleotide (Sigma-Aldrich). The underlined regions indicate the forward primer, probe and reverse primer sequences, respectively. The synthesized IS2404 synthetic positive was resuspended in nuclease-free water and diluted to 0.001 pM, with 2 µl being used for extraction and positive controls. To confirm the presence of a true positive as opposed to contamination, 5 µl of the qPCR product was added to 1 µl of DNA Gel Loading Dye 6X (Thermo Scientific) and run on a 2% agarose gel (TopVision Agarose Tablets; Thermo Scientific), with 1% SYBR Safe DNA Gel Stain (Invitrogen). The size of any positive IS2404 detection was assessed against 2 µl of 100 bp DNA Ladder (Promega) and run at 50 V for 1.5 h before being visualized with an EZ Gel Documentation System (Bio-Rad). Before the screening of insects began, the synthetic positive control for IS2404 was successfully designed and tested. By running the amplified PCR products on an agarose gel with the synthetic positive control and a real positive control, visual differentiation could be determined between a synthetic positive occurring at 120 bp and a true positive at 59 bp (Supplementary Fig. 2).
Screening insects by qPCR for M. ulcerans
The qPCR screening was performed using three independent assays IS2404, IS2606 and KR15. All samples were first screened with the IS2404 qPCR; if a positive was detected, additional confirmation was attempted with IS2606 and KR qPCR assays. Reactions were performed using 7.5 µl TaqMan Fast Universal PCR Master Mix (2X), no AmpErase UNG (Applied Biosystems), 1 µl of the primer–probe mix, 2 µl DNA and 4.5 µl nuclease-free water. A final primer–probe concentration for the IS2404 assay was as follows: 250:650:450 nM for the forward primer, reverse primer, and probe; and 800:800:220 nM for the forward primer, reverse primer, and probe for the IS2606 and KR assay. A 2 µl volume of the synthetic positive control was added for the IS2404 reactions, whereas 2 µl of M. ulcerans DNA was used for IS2606 and KR. All reactions included 6 no-template extraction controls and were run in a 96-well-plate format. Cycling conditions were as follows: denaturation at 95 °C for 2 min, followed by 45 cycles at 95 °C for 10 s and 60 °C for 30 s, with qPCR performed on a QuantStudio 5 Real-Time PCR System (Applied Biosystems). Positives were classified as reactions that produced a cycle quantification value less than 40. Data were analysed using the QuantStudio Design and Analysis Software v.1.4.3 with the ΔRn threshold set at 0.04 for IS2404 and IS2606, and a ΔRn threshold of 0.1 for KR. The MLE per 1,000 mosquitoes tested (bias-corrected MLE for point estimation of infection rate and a skew-corrected score CI) was calculated from the pooled samples53. Fisher’s exact test for assessing the significance of differences in IS2404 PCR positivity between mosquito species was calculated in R 4.0.2 (ref. 54).
All qPCR screening was performed blind with mixed-species 96-well plates. The synthetic positive control was added at the DNA extraction phase to one well of each plate and to qPCR plates to check that both extraction and qPCR detections were successful. All no-template controls (extraction and qPCR stage) were checked to ensure they remained negative, and that synthetic positive controls were detected for both the DNA extraction and qPCR stage in each run. Positive samples were run on agarose gels to confirm they were true positives and not contamination from the synthetic positive control.
An IS2404 qPCR standard curve was prepared using 10-fold serial dilutions of M. ulcerans genomic DNA, with quadruplicate testing of each dilution. The DNA was extracted from M. ulcerans JKD8049 and quantified using fluorimetry (Qubit dsDNA HS; Thermo Fisher Scientific)20. A limit of detection was defined as the lowest dilution that returned a positive signal for all four replicates. GE were calculated based on the estimated mass of the M. ulcerans genome of 5.7 fg (ref. 20). IS2404 Ct values were converted to GE to estimate bacterial load within a sample by reference to a standard curve (r2 = 0.9956, y = [−3.829ln(x) + 37.17]Z, where r2 = correlation coefficient y = Ct and x = amount of DNA (in fg) and Z = the dilution factor; Supplementary Fig. 2). An IS2404 qPCR standard curve was fitted using nonlinear regression in GraphPad Prism (v.9.5.1) (Supplementary Fig. 3).
M. ulcerans genome sequencing
Whole-genome sequencing was performed directly on DNA extracted from selected PCR-positive mosquito samples and possum excreta specimens using a hybridization capture approach, based on 120 nt RNA baits spanning the 5.8 Mbp chromosome of the M. ulcerans JKD8049 reference genome (BioProject ID PRJNA771185) (SureSelect Target Enrichment System; Agilent; Supplementary dataset 4) and the Illumina Nextera Flex for Enrichment with RNA Probes protocol55. Resulting sequence reads were submitted to National Centre for Biotechnology Information (NCBI) GenBank and are available under BioProject PRJNA943595 (Supplementary Table 2).
M. ulcerans SNP calling, SNP imputation and phylogenetic analysis
To compare genomic variations between M. ulcerans clinical isolate genome sequences and sequence-capture enrichment datasets, we mapped the sequence reads and called nucleotide variations using Snippy (v.4.4.5) against a finished M. ulcerans reference chromosome, reconstructed from a Victorian clinical isolate (JKD8049; GenBank accession NZ_CP085200.1; https://github.com/tseemann/snippy). Although standard parameters, including a minimum coverage of 10×, were used for the clinical isolates and two possum sequence-capture datasets, the mosquito sequence-capture datasets had lower read coverage, necessitating the adjustment of parameters. Thus, the minimum coverage threshold was lowered to 1× to facilitate SNP calling for the mosquito sequence-capture datasets. The resulting SNPs were combined with 117 SNPs obtained from a reference set of 36 M. ulcerans genomes that represented the previously defined population structure of the pathogen in Victoria24. Due to the low read coverage however, the number of core variable nucleotide positions mapped among the 5 sequence-capture datasets was variable (range, 22–112 variable nucleotide positions). To enable inclusion of the sequence-capture datasets that had missing SNP sites, we used a multivariate imputation approach, using the IterativeImputer function from scikit-learn56. The combined alignment of 117 core genome SNPs from the sequence-capture datasets and clinical isolate genomes served as the foundation for inferring a maximum likelihood phylogeny. This phylogeny was established using the GTR model of nucleotide substitution and executed with FastTree (v.2.1.10)57. The incorporation of R packages phytools (v.1.0-1)58 and mapdata (v.2.3.1)59 allowed for the alignment of tree tips against a base map, facilitating the visualization of geographical origins of the samples. Further details, including the code used for missing SNP imputation and phylogeographical analysis, can be found in our GitHub repository60.
Ae. notoscriptus typing and species confirmation sequencing
Mosquito genotyping was performed by sequence comparisons of a partial fragment of the COI gene52. DNA was extracted using the above protocols. PCR was performed using 5 µl of 5× MyFi Reaction Buffer, 1 µl MyFi DNA polymerase, 5 µl DNA and primer concentrations27, with the reaction made up to 25 µl with nuclease-free water. Reaction conditions were as follows for COI: initial denaturation at 95 °C for 1 min, followed by 35 cycles at 95 °C for 20 s, 46 °C for 20 s and 72 °C for 60 s, before a final extension at 72 °C for 5 min. A 5 µl volume of the amplified PCR product was added to 1 µl of DNA Gel Loading Dye 6X (Thermo Scientific) and run on a 1% agarose gel (TopVision Agarose Tablets; Thermo Scientific), with 1% SYBR Safe DNA Gel Stain (Invitrogen). A 2 µl volume of 100 bp DNA Ladder (Promega) was added to confirm amplicon size and run at 100 V for 45 min. PCR products that produced bands of the correct size were purified using the ISOLATE II PCR and Gel Kit (Bioline), as per the manufacturer’s protocol, and submitted for sequencing using an Applied Biosystems 3730xl capillary analyser (Macrogen), with sequencing occurring on both strands. Sequences were analysed in Geneious Prime (v.2019.2.1) and trimmed to high-quality bases, aligned using ClustalW v.2.1 and trimmed to a consensus region, Ae. notoscriptus COI (874 bp) and for species identification COI (816–882 bp). Sequences were analysed using blastn against the NCBI database. COI sequences generated for species identification are available under accession numbers OQ600123–OQ6001234, and COI sequences for Ae. notoscriptus phylogenetics are under accession numbers OQ588831–OQ588867 (Supplementary Fig. 1).
Mosquito blood-meal analysis
Ninety blood-fed mosquitoes were identified as having an engorged abdomen and still having a red pigment (Sella score 2–3), indicating a fresh blood meal, and were dissected with a sterile scalpel blade. Blood from the dissected abdomen was absorbed onto a 3 mm × 20 mm piece of a Whatman FTA card (Merck) and placed in a 2 ml tube. DNA was extracted from the FTA card using an ISOLATE II Genomic DNA Kit (Bioline) with a pre-lysis in 180 µl of Lysis Buffer GL and 25 µl Proteinase K for 2 hours before completing the extraction as per the manufacturer’s protocol. Extracted DNA was amplified for cytb using primers previously described61, with MyTaq HS Red Mix (Bioline), and the thermocycling conditions used were as follows: 95 °C for 1 min, 30 cycles of 95 °C for 15 s, 50 °C for 20 s, 72 °C for 20 s and a final extension at 72 °C for 2 min. Negative extraction and negative PCR controls were included with each PCR reaction. A volume of 5 µl of the amplified products was run on a 1% agarose gel containing 0.1% SYBR Safe DNA gel stain (Invitrogen) and visualized with a G:BOX Syngene blue-light visualization instrument. If a band was visualized at ∼480 bp, the PCR product was purified using an ISOLATE II PCR and Gel Kit (Bioline).
PCR products were quantified using a Qubit (Invitrogen) fluorometer with an HS dsDNA kit. Sequencing libraries were prepared using 10 ng of purified PCR product with a NEXTFLEX Rapid XP DNA-Seq Kit (PerkinElmer) barcoded using NEXTFLEX UDI Barcodes (PerkinElmer). As a result, 70 blood-meal libraries were sequenced, along with 6 PCR negative controls and 3 extraction negatives, on a NovaSeq 6000 (Illumina), with 2 Gb requested per sample.
Sequence data were analysed by identifying poor-quality reads using Rcorrector62 and removed with TrimGalore v.0.6.5. De novo assembly was performed on the remaining sequence reads using Trinity v.2.8.663. The resulting contigs were filtered to be between 400 bp and 480 bp and analysed using BLASTN against the nucleotide database publicly available on the NCBI website. The resulting hits were filtered to exclude those sequences that had <97% sequence identity to the database. Contigs identified as a host blood meal were confirmed by mapping raw reads using BWA-MEM v.0.7.1764. Sequence reads were submitted to GenBank (BioProject ID PRJNA943595).
Mosquito phylogenetic analysis
Mosquito genotyping was performed with COI because this genetic marker has previously been used to identify potential cryptic species within Ae. notoscriptus and can provide better resolution than other markers such as ND5, CAD or EPIC (exon-primed intron crossing)27. Phylogenetic analysis was performed on trimmed consensus regions of Ae. notoscriptus COI. The substitution model was selected using jModelTest2 v.2.1.10, with the topology taking the best of nearest-neighbour interchange, subtree pruning and regrafting65. The most appropriate substitution model was selected based on the Akaike information criterion. Maximum likelihood trees were constructed in PhyML v.3.3.2 with 1,000 bootstrap replicates; the gamma distribution parameter was used to estimate rate variation across sites66. The Hasegawa–Kishino–Yano (HKY) substitution model was selected for the COI tree.
Geographical data acquisition and spatial cluster analysis
The population map was created using QGIS geographical information system software (v.3.16.7)67, using a 1 km2 population grid68 with 2011 Victorian mesh-block data. Since 2004, Buruli ulcer has been a notifiable condition in Victoria, requiring health department reporting by doctors and laboratories. De-identified case notification data of patients with Buruli ulcer who had laboratory-confirmed M. ulcerans infection and who lived on the Mornington Peninsula during the years 2019–2020 were provided by the Victorian Department of Health. The cases were defined as patients with a clinical lesion that was diagnosed using IS2404 qPCR and culture15. To conduct high-resolution spatial analyses, the data were aggregated at the mesh-block level, the smallest geographical census units which typically contain 30–60 dwellings. The 2011 Victorian mesh-block boundaries and the Victorian mesh-block census population counts datasets were obtained from the Australian Bureau of Statistics website69. The datasets were joined using the unique mesh-block IDs using QGIS (v.3.16.7)67. The latitude and longitude (projected in GDA94) were derived from the centroids of the mesh-block polygon. The dataset was then downsampled to include only the Mornington Peninsula study area, specifically the Point Nepean and Rosebud–McCrae Australian Bureau of Statistics level 2 Statistical Area.
SaTScan v.10.1.0 (ref. 70) was used to identify spatial clusters among trapped mosquitoes positive for M. ulcerans, possum excreta positive for M. ulcerans and human Buruli ulcer cases. The software searches for instances where the observed number of spatial incidences exceeds the expected number within a circular window of varying size across a defined study area. A log-likelihood ratio statistic is calculated for each window by comparing the number of observed and expected cases inside and outside the circle against the assumption of randomly distributed cases. In addition to the most likely cluster, there are usually secondary clusters with almost as high likelihood that substantially overlap with the primary clusters. These secondary clusters can be indicative of subclusters within the primary cluster or potentially distinct clusters that are spatially adjacent to the primary cluster. The Mornington Peninsula surveillance data used in these analyses consisted of trapped mosquitoes (177 traps screened for IS2404 collected 12 November 2019 to 20 March 2020), M. ulcerans detected in possum excreta collected during the summer (December to February) of 2019 using data from a previous study26 and notified human Buruli ulcer cases from the study area in the years 2019–2020. The use of possum excreta collected during the summer was appropriate as Buruli ulcer transmission is most likely to occur during that time of year71,72. For each of the three data sources (trapped mosquitoes, possum excreta and human cases), the null hypothesis assumes that M. ulcerans detections or Buruli ulcer cases are uniformly distributed across the study area, where the alternative hypothesis suggests that there may be certain locations with higher rates than expected if the risk was evenly distributed. Primary and secondary clusters were accepted only if the secondary clusters did not overlap with previously reported clusters with a higher likelihood. Given that the trapped mosquito and possum excreta IS2404 PCR results were binary (positive or negative), a Bernoulli model was used to scan for spatial clusters, with the maximum cluster size set to 50% of the population size. The human Buruli ulcer case data aggregated at the mesh-block level varied in number, with some mesh blocks having zero cases and others having one or more. We applied the Poisson probability model to the notified Buruli ulcer case counts, using a background population at risk that was derived from the 2011 population census. The maximum cluster size was limited to 14,481 individuals, 10% of the total population at risk. To determine the likelihood of a triple-cluster overlap between the three SaTScan analyses occurring by chance, we conducted a permutation test73. In each of 10,000 iterations, the geographical coordinates for each variable were randomly shuffled. The number of SaTScan clusters with triple overlap was determined using the sf package74 in the R statistical programming language54.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Supplementary Figs. 1–3 and Tables 1–3.
Mosquito trapping details. Accession details for M. ulcerans DNA sequence reads used in this study. M. ulcerans SNP alleles used for phylogenomic inference. Oligonucleotide sequences of RNA baits used for M. ulcerans genome-enrichment sequencing.
Source data
Statistical source data.
Acknowledgements
This research was funded by the National Health and Medical Research Council of Australia (GNT1152807 and GNT1196396) to E.L.T., S.R.C., P.D.R.J., A.A.H., K.B.G., T.P.S. and S.E.L., Investigator Award (GNT1194325) to T.P.S and Investigator Award (MRF1193727) to K.B.G. The Millersville University Faculty Grant and Sabbatical Leave Grant Program supported J.R.W. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We thank the residents of the Mornington Peninsula for assisting in collecting and sending sticky cards to our laboratory as part of our surveillance and to M. Tachedjian, M. Dunn, S. Clayton, J. Gaburro and V. Boyd for assistance during field work.
Extended data
Author contributions
P.T.M., J.O., R.F., S.R.C., P.D.R.J, J.R.W., A.A.H., K.B.G., T.P.S. and S.E.L. conceptualized the study. P.T.M., K.B. and J.O. were responsible for data curation. P.T.M., A.H.B. and T.P.S. were responsible for formal analysis. S.R.C., P.D.R.J., K.B.G., J.R.W., A.A.H., K.B.G., T.P.S. and S.E.L. were responsible for funding acquisition. P.T.M., A.H.B., J.O., J.L.P., E.C.H., J.C.C., L.M.J., M.O.M., G.T., N.P., D.A.W., K.R.B., E.L.T., J.R.W., D.J.P., T.P.S. and S.E.L. were responsible for methodology. P.T.M., A.H.B., S.E.L. and T.P.S. wrote the original draft. All authors reviewed and edited the article.
Peer review
Peer review information
Nature Microbiology thanks Anthony Ablordey, Sebastien Gagneux and Thomas Walker for their contribution to the peer review of this work.
Data availability
DNA sequencing data generated in this project are available under the following NCBI GenBank accession numbers: PRJNA943595, PRJNA943595, NZ_CP085200.1, OQ600123, OQ6001234, OQ588831–OQ58883167. Source data are provided with this paper.
Code availability
The code is available via Github at https://github.com/abuultjens/BU-3-way-SatScan for SatScan analysis and https://github.com/abuultjens/Mosquito_possum_human_genomic_analysis.for sequence enrichment data analysis.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Peter T. Mee, Andrew H. Buultjens, Jane Oliver.
These authors jointly supervised this work: Ary A. Hoffmann, Katherine B. Gibney, Timothy P. Stinear, Stacey E. Lynch.
Change history
4/15/2024
A Correction to this paper has been published: 10.1038/s41564-024-01693-y
Contributor Information
Peter T. Mee, Email: peter.mee@agriculture.vic.gov.au
Timothy P. Stinear, Email: tstinear@unimelb.edu.au
Extended data
is available for this paper at 10.1038/s41564-023-01553-1.
Supplementary information
The online version contains supplementary material available at 10.1038/s41564-023-01553-1.
References
- 1.MacCullum P, Tolhurst JC, Buckle G, Sissons HA. A new mycobacterial infection in man. J. Pathol. Bacteriol. 1948;60:93–102. doi: 10.1002/path.1700600111. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Buruli ulcer (Mycobacterium ulcerans infection) https://www.who.int/news-room/fact-sheets/detail/buruli-ulcer-(mycobacterium-ulcerans-infection) (2022).
- 3.Merritt RW, et al. Ecology and transmission of Buruli ulcer disease: a systematic review. PLoS Negl. Trop. Dis. 2010;4:e911–e911. doi: 10.1371/journal.pntd.0000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Department of Health and Human Services (Victoria). Interactive infectious disease surveillance reports https://www2.health.vic.gov.au/public-health/infectious-diseases/infectious-diseases-surveillance/interactive-infectious-disease-reports (2021).
- 5.Janssens PG, Quertinmont MJ, Sieniawski J, Gatti F. Necrotic tropical ulcers and mycobacterial causative agents. Trop. Geogr. Med. 1959;11:293–312. [PubMed] [Google Scholar]
- 6.Clancey JK, Dodge OG, Lunn HF, Oduori ML. Mycobacterial skin ulcers in Uganda. Lancet. 1961;2:951–954. doi: 10.1016/S0140-6736(61)90793-0. [DOI] [PubMed] [Google Scholar]
- 7.Muleta AJ, Lappan R, Stinear TP, Greening C. Understanding the transmission of Mycobacterium ulcerans: a step towards controlling Buruli ulcer. PLoS Negl. Trop. Dis. 2021;15:e0009678. doi: 10.1371/journal.pntd.0009678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blasdell KR, et al. Environmental risk factors associated with the presence of Mycobacterium ulcerans in Victoria, Australia. PLoS ONE. 2022;17:e0274627. doi: 10.1371/journal.pone.0274627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carson C, et al. Potential wildlife sentinels for monitoring the endemic spread of human buruli ulcer in south-east Australia. PLoS Negl. Trop. Dis. 2014;8:e2668. doi: 10.1371/journal.pntd.0002668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fyfe JAM, et al. A major role for mammals in the ecology of Mycobacterium ulcerans. PLoS Negl. Trop. Dis. 2010;4:e791. doi: 10.1371/journal.pntd.0000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson, P. D. R. in Buruli Ulcer: Mycobacterium ulcerans Disease (eds Pluschke, G. & Roltgen, K.) 61–76 (Springer, 2019).
- 12.O’Brien CR, et al. Clinical, microbiological and pathological findings of Mycobacterium ulcerans infection in three Australian possum species. PLoS Negl. Trop. Dis. 2014;8:e2666. doi: 10.1371/journal.pntd.0002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu RW, Stinear TP, Johnson PD, O’Brien DP. Possum bites man: case of Buruli ulcer following possum bite. Med J. Aust. 2022;216:452–453. doi: 10.5694/mja2.51505. [DOI] [PubMed] [Google Scholar]
- 14.Johnson PDR, et al. Mycobacterium ulcerans in mosquitoes captured during outbreak of Buruli ulcer, southeastern Australia. Emerg. Infect. Dis. 2007;13:1653–1660. doi: 10.3201/eid1311.061369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fyfe JA, et al. Development and application of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl. Environ. Microbiol. 2007;73:4733–4740. doi: 10.1128/AEM.02971-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quek TY, et al. Risk factors for Mycobacterium ulcerans infection, southeastern Australia. Emerg. Infect. Dis. 2007;13:1661–1666. doi: 10.3201/eid1311.061206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landier J, et al. Adequate wound care and use of bed nets as protective factors against Buruli ulcer: results from a case control study in Cameroon. PLoS Negl. Trop. Dis. 2011;5:e1392. doi: 10.1371/journal.pntd.0001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pouillot R, et al. Risk factors for Buruli ulcer: a case control study in Cameroon. PLoS Negl. Trop. Dis. 2007;1:e101. doi: 10.1371/journal.pntd.0000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavender CJ, et al. Risk of Buruli ulcer and detection of Mycobacterium ulcerans in mosquitoes in southeastern Australia. PLoS Negl. Trop. Dis. 2011;5:e1305. doi: 10.1371/journal.pntd.0001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallace JR, et al. Mycobacterium ulcerans low infectious dose and mechanical transmission support insect bites and puncturing injuries in the spread of Buruli ulcer. PLoS Negl. Trop. Dis. 2017;11:e0005553. doi: 10.1371/journal.pntd.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Australian Bureau of Meteorology. Summary statistics Mornington http://www.bom.gov.au/climate/averages/tables/cw_086079.shtml (2022).
- 22.World Health Organization. Number of new reported cases of Buruli ulcer https://apps.who.int/gho/data/node.main.A1631 (2023).
- 23.Mornington Peninsula Shire. Mornington Peninsula Shire population forecasts https://forecast.id.com.au/mornington-peninsula (2023).
- 24.Buultjens, A. H. et al. Comparative genomics shows that mycobacterium ulcerans migration and expansion preceded the rise of Buruli ulcer in Southeastern Australia. Appl. Environ. Microbiol.10.1128/AEM.02612-17 (2018). [DOI] [PMC free article] [PubMed]
- 25.Taouk ML, et al. Characterisation of Treponema pallidum lineages within the contemporary syphilis outbreak in Australia: a genomic epidemiological analysis. Lancet Microbe. 2022;3:e417–e426. doi: 10.1016/S2666-5247(22)00035-0. [DOI] [PubMed] [Google Scholar]
- 26.Vandelannoote, K. et al. Statistical modelling based on structured surveys of Australian native possum excreta harbouring Mycobacterium ulcerans predicts Buruli ulcer occurrence in humans. eLife10.7554/eLife.84983 (2023). [DOI] [PMC free article] [PubMed]
- 27.Endersby NM, et al. Evidence of cryptic genetic lineages within Aedes notoscriptus (Skuse) Infect. Genet. Evol. 2013;18:191–201. doi: 10.1016/j.meegid.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 28.Singh A, McBride WJH, Govan B, Pearson M, Ritchie SA. A survey on Mycobacterium ulcerans in mosquitoes and march flies captured from endemic areas of northern Queensland, Australia. PLoS Negl. Trop. Dis. 2019;13:e0006745. doi: 10.1371/journal.pntd.0006745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yerramilli A, et al. The location of Australian Buruli ulcer lesions—implications for unravelling disease transmission. PLoS Negl. Trop. Dis. 2017;11:e0005800. doi: 10.1371/journal.pntd.0005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flies EJ, Flies AS, Fricker SR, Weinstein P, Williams CR. Regional comparison of mosquito bloodmeals in south Australia: implications for Ross River virus ecology. J. Med. Entomol. 2016;53:902–910, 909. doi: 10.1093/jme/tjw035. [DOI] [PubMed] [Google Scholar]
- 31.Oshaghi MA, et al. Effects of post-ingestion and physical conditions on PCR amplification of host blood meal DNA in mosquitoes. Exp. Parasitol. 2006;112:232–236. doi: 10.1016/j.exppara.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Kay BH, Boyd AM, Ryan PA, Hall RA. Mosquito feeding patterns and natural infection of vertebrates with Ross River and Barmah Forest viruses in Brisbane, Australia. Am. J. Trop. Med. Hyg. 2007;76:417–423. doi: 10.4269/ajtmh.2007.76.417. [DOI] [PubMed] [Google Scholar]
- 33.Doig KD, et al. On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genomics. 2012;13:258. doi: 10.1186/1471-2164-13-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stinear TP, et al. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. 2007;17:192–200. doi: 10.1101/gr.5942807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eisen RJ, Gage KL. Transmission of flea-borne zoonotic agents. Annu. Rev. Entomol. 2012;57:61–82. doi: 10.1146/annurev-ento-120710-100717. [DOI] [PubMed] [Google Scholar]
- 36.Kernif, T., Leulmi, H., Raoult, D. & Parola, P. Emerging tick-borne bacterial pathogens. Microbiol. Spectr.10.1128/microbiolspec.EI10-0012-2016 (2016). [DOI] [PubMed]
- 37.Ryden P, et al. Outbreaks of tularemia in a boreal forest region depends on mosquito prevalence. J. Infect. Dis. 2012;205:297–304. doi: 10.1093/infdis/jir732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thelaus J, et al. Francisella tularensis subspecies holarctica occurs in Swedish mosquitoes, persists through the developmental stages of laboratory-infected mosquitoes and is transmissible during blood feeding. Microb. Ecol. 2014;67:96–107. doi: 10.1007/s00248-013-0285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdellahoum, Z., Maurin, M. & Bitam, I. Tularemia as a mosquito-borne disease. Microorganisms10.3390/microorganisms9010026 (2020). [DOI] [PMC free article] [PubMed]
- 40.Hendry, T. A. et al. Ongoing transposon-mediated genome reduction in the luminous bacterial symbionts of deep-sea ceratioid Anglerfishes. mBio10.1128/mBio.01033-18 (2018). [DOI] [PMC free article] [PubMed]
- 41.Perreau J, Moran NA. Genetic innovations in animal-microbe symbioses. Nat. Rev. Genet. 2022;23:23–39. doi: 10.1038/s41576-021-00395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drucker M, Then C. Transmission activation in non-circulative virus transmission: a general concept? Curr. Opin. Virol. 2015;15:63–68. doi: 10.1016/j.coviro.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Archer, M. S. The Ecology of Invertebrate Associations with Vertebrate Carrion in Victoria, with Reference to Forensic Entomology. PhD thesis, Univ. of Melbourne (2002).
- 44.Lang MD, Allen GR, Horton BJ. Blowfly succession from possum (Trichosurus vulpecula) carrion in a sheep-farming zone. Med. Vet. Entomol. 2006;20:445–452. doi: 10.1111/j.1365-2915.2006.00654.x. [DOI] [PubMed] [Google Scholar]
- 45.Skopyk AD, Forbes SL, LeBlanc HN. Recognizing the inherent variability in Dipteran colonization and decomposition rates of human donors in Sydney. Aust. J. Clin. Health Sci. 2021;6:102–119. doi: 10.24191/jchs.v6i1(Special).13993. [DOI] [Google Scholar]
- 46.Djouaka R, et al. Evidences of the low implication of mosquitoes in the transmission of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Can. J. Infect. Dis. Med. Microbiol. 2017;2017:1324310. doi: 10.1155/2017/1324310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Australian Bureau of Statistics. 2016 Census QuickStats https://quickstats.censusdata.abs.gov.au/census_services/getproduct/census/2016/quickstat/SSC22187?opendocument (2021).
- 48.Australian Bureau of Statistics. (Australian Bureau of Statistics, 2014).
- 49.Russell, R. C. A Colour Photo Atlas of Mosquitoes of Southeastern Australia (Univ. of Sydney Printing Service, 1996).
- 50.Dobrotworsky, N. V. The Mosquitoes of Victoria (Diptera, Culicidae) (Melbourne Univ. Press, 1965).
- 51.Liehne, P. F. S. An Atlas of the Mosquitoes of Western Australia (Health Department of Western Australia, 1991).
- 52.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- 53.PooledInfRate v.4.0: a Microsoft Office Excel add-in to compute prevalence estimates from pooled samples (Fort Collins, 2009).
- 54.R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2021).
- 55.Illumina. Nextera Flex for enrichment with RNA probes https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_nextera/nextera-flex-enrichment/nextera-flex-for-enrichment-rna-probes-demonstrated-protocol-1000000070581-01.pdf (2023).
- 56.van Buuren S, Groothuis-Oudshoorn K. mice: multivariate imputation by chained equations in R. J. Stat. Softw. 2011;45:1–67. doi: 10.18637/jss.v045.i03. [DOI] [Google Scholar]
- 57.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Revell L. phytools: An R package for phylogenetic comparative biology (and other things) Methods Ecol. Evol. 2012;3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 59.Becker, R. A. & Wilks, A. R. Constructing a Geographical Database (1995).
- 60.Buultjens, A. H. Genomic analysis of mosquito, possum and human derived Mycobacterium ulcerans genomes https://github.com/abuultjens/Mosquito_possum_human_genomic_analysis (2023).
- 61.Townzen JS, Brower AVZ, Judd DD. Identification of mosquito bloodmeals using mitochondrial cytochrome oxidase subunit I and cytochrome b gene sequences. Med. Vet. Entomol. 2008;22:386–93. doi: 10.1111/j.1365-2915.2008.00760.x. [DOI] [PubMed] [Google Scholar]
- 62.Song L, Florea L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience. 2015;4:48. doi: 10.1186/s13742-015-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grabherr MG, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 67.QGIS Geographic Information System v.3.16.7 (Open Source Geospatial Foundation Project, 2022).
- 68.Australian Bureau of Statistics. Victoria mesh blocks ASGS edition 2011 digital boundaries in ESRI shapefile https://www.abs.gov.au/AUSSTATS/abs@.nsf/DetailsPage/1270.0.55.001July%202011 (2011).
- 69.Australian Bureau of Statistics. https://www.abs.gov.au (2023).
- 70.Kulldorff M. A spatial scan statistic. Commun. Stat. Theory Methods. 1997;26:1481–1496. doi: 10.1080/03610929708831995. [DOI] [Google Scholar]
- 71.Loftus MJ, et al. The incubation period of Buruli ulcer (Mycobacterium ulcerans infection) in Victoria, Australia—remains similar despite changing geographic distribution of disease. PLoS Negl. Trop. Dis. 2018;12:e0006323. doi: 10.1371/journal.pntd.0006323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trubiano JA, Lavender CJ, Fyfe JA, Bittmann S, Johnson PD. The incubation period of Buruli ulcer (Mycobacterium ulcerans infection) PLoS Negl. Trop. Dis. 2013;7:e2463. doi: 10.1371/journal.pntd.0002463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buultjens, A. H. BU-3-way-SatScan https://github.com/abuultjens/BU-3-way-SatScan (2023).
- 74.Pebesma E. Simple features for R: standardized support for spatial vector data. R J. 2018;10:439–446. doi: 10.32614/RJ-2018-009. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–3 and Tables 1–3.
Mosquito trapping details. Accession details for M. ulcerans DNA sequence reads used in this study. M. ulcerans SNP alleles used for phylogenomic inference. Oligonucleotide sequences of RNA baits used for M. ulcerans genome-enrichment sequencing.
Statistical source data.
Data Availability Statement
DNA sequencing data generated in this project are available under the following NCBI GenBank accession numbers: PRJNA943595, PRJNA943595, NZ_CP085200.1, OQ600123, OQ6001234, OQ588831–OQ58883167. Source data are provided with this paper.
The code is available via Github at https://github.com/abuultjens/BU-3-way-SatScan for SatScan analysis and https://github.com/abuultjens/Mosquito_possum_human_genomic_analysis.for sequence enrichment data analysis.