

Abstract
Patients with pre-existing immunity to adeno-associated virus (AAV) are currently unable to receive systemic gene transfer therapies. In this nonhuman primate study, we investigated the impact of immunosuppression strategies on gene transfer therapy safety and efficacy and analyzed plasmapheresis as a potential pretreatment for circumvention of pre-existing immunity or redosing. In part 1, animals received delandistrogene moxeparvovec (SRP-9001), an AAVrh74-based gene transfer therapy for Duchenne muscular dystrophy. Cohort 1 (control, n = 2) received no immunosuppression; cohorts 2–4 (n = 3 per cohort) received prednisone at different time points; and cohort 5 (n = 3) received rituximab, sirolimus, and prednisone before and after dosing. In part 2, cohorts 2–4 underwent plasmapheresis before redosing; cohort 5 was redosed without plasmapheresis. We analyzed safety, immune response (humoral and cell-mediated responses and complement activation), and vector genome distribution. After 2 or 3 plasmapheresis exchanges, circulating anti-AAVrh74 antibodies were reduced, and animals were redosed. Plasmapheresis was well tolerated, with no abnormal clinical or immunological observations. Cohort 5 (redosed with high anti-AAVrh74 antibody titers) had hypersensitivity reactions, which were controlled with treatment. These findings suggest that plasmapheresis is a safe and effective method to reduce anti-AAV antibody levels in nonhuman primates prior to gene transfer therapy. The results may inform human studies involving redosing or circumvention of pre-existing immunity.
Keywords: antibodies, Duchenne muscular dystrophy, gene therapy, plasmapheresis, redosing, safety, pre-existing antibodies, adeno-associated virus, delandistrogene moxeparvovec, immunosuppression
Graphical abstract
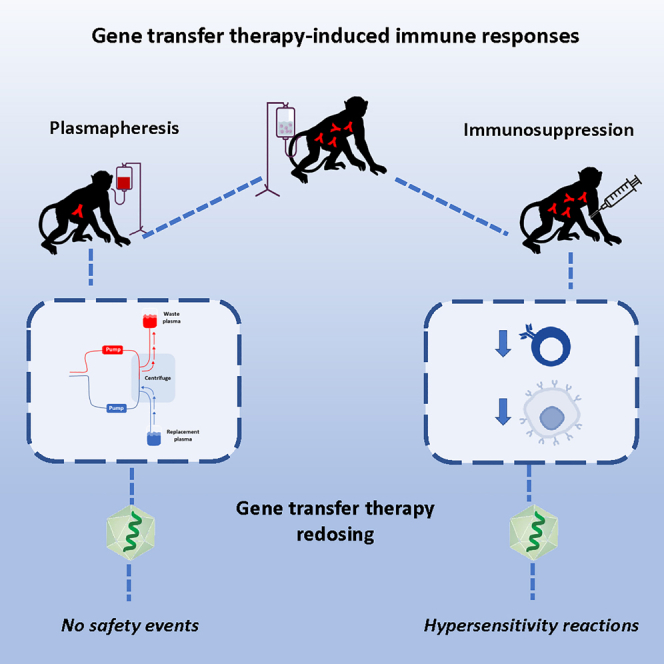
Potter and colleagues examine the impact of immunosuppression on gene transfer therapy safety and efficacy in nonhuman primates along with plasmapheresis for either circumventing pre-existing immunity to AAV or permitting redosing. The findings suggest that plasmapheresis is a safe, effective method to reduce anti-AAV antibody levels, which may inform human studies.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular disease caused by mutations in the DMD gene, resulting in the absence of functional dystrophin protein. Without this essential structural protein within muscle fibers, affected individuals develop muscle weakness in the first years of life and experience increasing difficulty walking, progressing to loss of ambulation (LoA) in the pre-teenage or early teenage years. Children with DMD ultimately succumb to respiratory or cardiac failure in their mid-20s to 30s.1,2,3 Delandistrogene moxeparvovec (SRP-9001) is a gene transfer therapy (GTT) that restores a functional SRP-9001 dystrophin to the skeletal and cardiac muscle of boys with DMD, directly addressing the root cause of disease pathology.4,5
GTTs are designed to express a therapeutic protein, commonly to compensate for the absence or dysfunction of an endogenous protein because of a genetic mutation. Adeno-associated viral (AAV) vectors have become the leading platform for GTT, as they have been shown to be safe and efficacious in humans. Several AAV-based GTTs have been approved by the US Food and Drug Administration (FDA).6 However, there are still multiple challenges that GTTs must overcome to ensure that they are available to all patients and are durable over a lifetime. One of the biggest challenges is overcoming the immune response to GTT administration,7 including both pre-existing immunity due to natural exposure to wild-type AAVs, which may prevent GTT dosing, and immunity resulting from initial GTT dosing, which may preclude subsequent redosing.
Pre-existing immunity to specific AAV serotypes is a common exclusion criterion across the GTT field and is evaluated using two types of tests: (1) binding assays that quantify total antibodies (TAbs), detecting both neutralizing antibodies (NAbs) and non-neutralizing antibodies (non-NAbs) that bind to the capsid, and (2) neutralizing assays that detect NAbs only. NAbs may directly inhibit transduction, reducing the efficacy of GTTs by blocking recombinant AAV (rAAV) attachment to receptors on target cells or interfering with uncoating of the vector. Although non-NAbs do not directly block transduction, any antibody that binds to the rAAV serotype used may affect GTT efficacy (e.g., by reducing transduction through vector opsonization, resulting in phagocytosis and elimination of the circulating rAAV by macrophages and neutrophils). In addition, both NAbs and non-NAbs that bind the vector may stimulate complement activation. Both opsonization and complement activation may lead to inflammation with safety implications.8,9,10 Furthermore, administration of GTT results in development of high-titer antibodies against AAV, which would likely prevent the use of AAV-based GTTs in those individuals in the future, should it become necessary.7,11 Therefore, it is important to evaluate immune modulation strategies to circumvent either pre-existing immunity or suppress post-dosing immune responses to AAV.
Current GTT trials for patients with DMD that use the AAVrh74 vector have used prednisone daily during the first few months after GTT dosing and have found that adverse events (AEs), specifically hepatotoxicity, have been transient and manageable. Although prednisone is the current standard of care, other immunosuppressive drugs have shown promise in modulating immune response. Rituximab is a monoclonal antibody that targets CD20 on B cells, induces apoptosis, and decreases B cell activation.12 Sirolimus, also known as rapamycin, is a macrolide that targets mammalian target of rapamycin (mTOR) and inhibits activation of T cells and B cells, reducing the adaptive immune response.13,14 Both rituximab and sirolimus have been used to inhibit the immune response in many contexts, including AAV treatment.15,16
Plasmapheresis is an extracorporeal technique that removes large-molecular-weight substances, such as antibodies, immune complexes, cryoglobulins, and cholesterol-containing lipoproteins, from plasma. Because approximately 75% of immunoglobulin M (IgM) is intravascular with a short half-life, several consecutive rounds of plasmapheresis are typically sufficient to rapidly reduce their levels.17 In comparison, only 45% of IgG is intravascular, with the rest deposited in tissues, and has a longer half-life; plasma IgG levels return to 40% of the pre-apheresis level within 48 h post-plasmapheresis as IgG moves from tissues back into circulation. Serum IgG level is also characterized by a “rebound” phenomenon, with transient plasmapheresis effects and a return to pre-plasmapheresis, or even higher, values within a week or two, especially if the patient is not on immunosuppressive therapy.18 As a result, a more rigorous regimen, involving more frequent plasmapheresis sessions and concomitant immunosuppressive therapy, may be required to efficiently reduce IgG levels.17 Importantly, plasmapheresis is generally a safe medical procedure, performed worldwide, in both adults and children. In the United States, most plasmapheresis procedures are performed for neurologic, immunologic, or hematologic diseases. Of note, procedures similar to plasmapheresis are also performed each year on a large number of volunteer donors for plasma and platelet collection.
There is currently no established immunosuppression (IS) regimen or antibody removal protocol to allow patients with pre-existing immunity to receive AAV-based GTTs or to permit redosing. Therefore, it is important to evaluate the different durations of existing IS regimens to understand safety and efficacy outcomes and utility for dosing patients with either pre-existing immunity or immunity due to GTT dosing. Plasmapheresis has been shown to safely lower pre-existing AAVrh74 antibody levels following the administration of AAV-based GTTs via isolated limb perfusion in rhesus macaques.19 This study builds upon these findings and addresses TAb suppression following systemic administration of AAV GTT. The present study had two objectives: (1) investigate the impact of various IS strategies on the safety and efficacy of GTT, and (2) analyze the safety and efficacy of plasmapheresis as a tool to reduce the antibody response after GTT dosing and unlock the ability to redose GTT in the future, in addition to enabling dosing of individuals with natural pre-existing immunity. The data presented here suggest that the use of plasmapheresis before redosing with GTT will reduce anti-AAVrh74 antibody titers, allowing safer administration in seropositive individuals. These data also indicate that the use of prednisone is an effective immunosuppressive regimen to lower the risk for hepatotoxicity.
Results
In-life observations
The safety of IS, plasmapheresis, and redosing was examined in five cohorts of nonhuman primates (NHPs) (Figure 1; Tables S1 and S2). In part 1, cohort 1 (control, n = 2) received no IS, and cohorts 2–4 (n = 3 per cohort) received prednisone at different time points. Cohort 5 (n = 3) received rituximab, sirolimus, and prednisone before and after dosing. In part 2, cohorts 2–4 underwent plasmapheresis before redosing; cohort 5 was redosed without plasmapheresis, and cohort 1 remained as the control cohort that received only one dose of GTT. Detailed daily observations of each animal were performed and documented by the Animal Resource Core at Nationwide Children’s Hospital (Columbus, OH). There were no significant treatment-related clinical observations recorded for cohorts 1–4 throughout the duration of the study. In part 1, petechial-like rash was observed in the groin, torso, and lower limbs of all three animals in cohort 5 at 3 weeks after the initial GTT dosing, likely because of the triple IS regimen. All NHPs in cohort 5 received 25 mg diphenhydramine orally for 5–9 days until the rash resolved. Aspartate aminotransferase (AST) elevation was also observed at this time in all three cohort 5 animals. In part 2, animals in cohort 5 experienced sporadic nose bleeds, weight loss, and loss of appetite prior to redosing. Upon redosing with elevated anti-AAVrh74 TAb titers in cohort 5 (≥51,200), animals experienced acute and serious side effects consistent with a hypersensitivity reaction, including elevated respiratory rate, elevated heart rate, lethargy, and rash. They were treated with diphenhydramine, dexamethasone, oxygen, and fluids. All animals made a complete recovery within 40 min. No other treatment-related clinical observations occurred in cohorts 2–4 throughout the duration of the study. At the conclusion of part 2, week 12, all NHPs underwent necropsy according to American Animal Hospital Association (AAHA) guidelines. This time point at the conclusion of part 2 was the study endpoint.
Figure 1.

Study design
∗Supercoiled standard qPCR titer method. †Biopsy collected from gastrocnemius muscle. ‡One nonhuman primate (NHP) did not undergo plasmapheresis, because of a lack of antibody response to AAVrh74 (adeno-associated virus rhesus isolate serotype 74). §One NHP did not undergo plasmapheresis, because of poor vascular access. ||Cohort 5 did not undergo plasmapheresis, because of incompatibility with previous treatment with rituximab. ¶All NHPs received prednisone (2 mg/kg/day) from 1 day pre-redosing to 30 days post-redosing with delandistrogene moxeparvovec. ∗∗End-of-life (EOL) necropsy collected from gastrocnemius, heart, and diaphragm. ††Immediately post-plasmapheresis, the NHPs were disconnected from the apheresis unit and systemically redosed with delandistrogene moxeparvovec. ‡‡Sirolimus was started 3 days prior to the first GTT dose and was continued until three weeks after the second dose. NHPs received one dose of rituximab 14 and 7 days prior to the first GTT dose, as well as one dose on the day of GTT dosing and one dose prior to redosing. GT, gene transfer; PGT, post-gene therapy.
There were no treatment-related histopathological findings due to IS or systemic gene transfer in examined skeletal muscles from animals in cohorts 1–5. Minimal and moderate bile duct hyperplasia was noted in one animal in cohort 2 and one animal in cohort 5, respectively, but was undetermined to test article delivery. In addition, following apheresis, no treatment-related increases were seen in the blood chemistry values tested, including liver enzymes, gamma-glutamyl transpeptidase (GGT), and platelets (Tables S3‒S6).
Immune response to delandistrogene moxeparvovec
ELISA was used to monitor anti-AAVrh74 antibody levels every 2 weeks throughout the study (Figures 2A and 2B). With the exception of one NHP (RA2481 in cohort 2), all treated animals ultimately showed similar anti-AAVrh74 antibody responses over the course of the study (Figure 2A), suggesting that the immune modulating treatments did not significantly affect the humoral immune response to delandistrogene moxeparvovec. In cohort 5, the triple IS regimen substantially dampened the anti-AAVrh74 TAb response in only one of the three animals in the cohort, and this suppression waned after 6 weeks, with this animal eventually achieving TAb titers equivalent to animals in the other cohorts. The remaining two animals in cohort 5 did not show TAb suppression.
Figure 2.

Humoral immune response to delandistrogene moxeparvovec
Anti-AAVrh74 antibody levels at various time points following initial dose (left of dashed line) and redosing (right of dashed line) of delandistrogene moxeparvovec for cohorts 2–4 (A) and cohort 5 (B). Animals in cohort 5 did not receive plasmapheresis. Vertical dotted line delineates part 1 from part 2. Horizontal dashed line delineates the threshold for 1:400, which indicates positive antibody titer response. (C) IgM response at 2 weeks after initial dose of GTT (part 1) and at 1 and 2 weeks post-redosing of GTT (part 2). Error bars are SEM and represents three animal samples per cohort run.
All animals in cohorts 1–4 that underwent plasmapheresis showed a decrease anti-AAVrh74 TAbs following the procedure (Table S7). Anti-AAVrh74 TAb levels returned to elevated levels after redosing. As expected, IgM titers had a similar profile in all animals, characterized by a increase after AAV vector redosing (Figure 2C).
Cohort 5 did not receive plasmapheresis; however, the animals continued their IS regime throughout the redosing phase. Figure 2B shows that triple IS had no effect on the already elevated humoral immune response exhibited by cohort 5 animals. However, animals experienced acute and serious side effects consistent with a hypersensitivity reaction upon redosing AAV vector, including elevated respiratory rate, elevated heart rate, lethargy, and rash.
Enzyme-linked immunosorbent spot (ELISpot) analysis was used to evaluate T cell response to both AAVrh74 and micro-dystrophin peptides. Transient T cell increases toward AAVrh74 peptides, SRP-9001 dystrophin peptides, or both following treatment were noted (Figure 3). However, these transient responses were not correlated with any clinical observations.
Figure 3.

T cell immune response to delandistrogene moxeparvovec
(A and B) T cell response to AAVrh74 and dystrophin peptides in cohorts 1–4 (A) and cohort 5 (B) across the study. Time points before redosing are denoted with “A,” and time points post-redosing are marked with “B.” Lines indicate mean ± SD. Each point represents a value from an individual animal. Immune response is considered positive if the number of spots is greater than 50, which is indicated with the dashed line. DMSO was used as a negative control.
Complement activation was assessed by examining C3 concentration in serum in a subset of animals (n = 6 at each time point because of sample availability). A minimal decrease in C3 was seen immediately post-plasmapheresis (Figure 4), but no complement activation was noted 24 h post-redosing. These data support the use of plasmapheresis to lower anti-AAV74 TAbs and enable redosing without antibody-mediated safety events.
Figure 4.

Effect of plasmapheresis on complement activation
Complement activation in animals before, immediately after, and 1 day post-plasmapheresis (n = 6 at each time point). C3 concentration in serum was measured in a subset of animals at the indicated time points.
Biodistribution of delandistrogene moxeparvovec
At 12 weeks after the initial dosing, there was no significant difference in vector genome (vg) copies in the biopsied muscle (gastrocnemius) between cohorts (Figure 5), suggesting that the various immune modulating regimens did not affect the biodistribution of delandistrogene moxeparvovec. Following redosing in part 2, vg copies were present in all tissues tested, including skeletal muscle, heart, diaphragm, and liver from postmortem tissue (Figure 5); however, although there was no statistically significant difference in vector copies levels between cohorts in any of the analyzed tissues, there was a nominal increase in cohorts that received a second dose of GTT compared with the single-dose cohort across target tissues. It is important to note that the assay did not differentiate vector copies derived from the first dose from those that may have resulted from the second dose.
Figure 5.

Vector genome biodistribution
Vector genome copies/nucleus at the end of part 1 (12 weeks after initial GTT dosing in the gastrocnemius biopsy) and at the end of part 2 (endpoint of study) in the tibialis anterior, gastrocnemius, heart, diaphragm, and liver in NHPs from cohort 1 (blue), cohorts 2–4 (red), and cohort 5 (green). Bars represent the mean ± SEM. The endpoint was 12 weeks post-dosing GTT in part 2.
Expression of SRP-9001 dystrophin protein
Western blot was used to examine the protein expression of SRP-9001 dystrophin in skeletal muscle of NHPs after plasmapheresis and redosing of rAAVrh74.MHCK7.micro-dystrophin with varying IS regimens. SRP-9001 dystrophin expression was seen across all cohorts at varying levels following redosing in skeletal muscle, heart, and diaphragm tissues, with increased level of expression in animals that received two doses of GTT (Figure 6). Importantly, there was no expression of SRP-9001 dystrophin in any off-target tissues, including the liver, in any animals (Figure S1). Additionally, mRNA was evaluated across skeletal muscle, diaphragm, and cardiac tissue and demonstrated nominal increases in animals that received two doses of GTT (Figure S2).
Figure 6.

Representative western blots across heart, diaphragm, and skeletal muscle (part 2)
Representative western blot images of heart, diaphragm, and tibialis anterior (TA) samples taken after a single dose of delandistrogene moxeparvovec or after redosing across all animals in cohorts 2–5. (B) Alpha-actinin was used as a loading control.
Discussion
At present, patients with pre-existing antibodies to GTT vectors are unable to receive treatment as NAbs and TAbs can inhibit the therapeutic utility by preventing successful gene transfer and pose safety concerns.20,21 Although the prevalence of pre-existing immunity to AAV vectors derived from nonhuman species, including AAVrh74, has been shown to be relatively lower, 14%–17% of the target population (i.e., muscular dystrophy population) is seropositive for pre-existing antibodies to AAVrh74.22,23 Furthermore, elevated anti-AAV antibody levels resulting from their first dose precludes patients from redosing, if necessary. It should be noted that AAV GTT to muscle has potential to be long lasting. Currently available data from delandistrogene moxeparvovec clinical trials provide evidence for durability of the treatment out to at least 4 years.24,25 Moreover, as scientific advancements are made, one can envision the development of other AAV-based therapies that may complement dystrophin restoration. Therefore, methods to suppress or circumvent the immune response to GTTs are currently being evaluated. In part 1 of the study, we showed that the humoral response to AAVrh74 was similar irrespective of the immunosuppressive regimen with the exception of one animal that did not develop anti-AAVrh74 antibodies. AAV titers in cohort 5 were not suppressed for the duration of the triple IS regimen, thus demonstrating that the triple IS regimen was not effective in durable suppression of the AAV-mediated immune response and was also accompanied by adverse clinical observations. In contrast, animals receiving prednisone had no safety events attributed to the IS. Part 2 of the study showed that plasmapheresis was well tolerated with no abnormal clinical or immunologic observations, and levels of circulating antibodies to AAVrh74 were reduced after 2 or 3 consecutive rounds. Furthermore, this technique successfully reduced levels of circulating antibodies to AAVrh74, permitting safe redosing with delandistrogene moxeparvovec. Overall, none of the immunosuppressive regimens affected transduction, SRP-9001 dystrophin protein expression, or immune response (humoral or cell mediated).
Safety analyses indicated that initial treatment with delandistrogene moxeparvovec, plasmapheresis, and redosing were well tolerated with no abnormal clinical or immunological observations. Some NHPs from cohorts 1–4 experienced transient liver enzyme elevations, an expected AE with GTT treatment, and levels returned to normal within a few days. NHPs that received any of the evaluated prednisone regimens had generally no or lower elevations in liver enzymes, indicating that prednisone may decrease the risk for hepatotoxicity after AAV GTT. Most AEs observed in part 1 were related to the triple IS regimen administered to cohort 5. In part 2, NHPs in cohort 5 redosed, at high antibody titer, experienced increased heart rate and ventilation rate, vomiting, rash near the delivery site, paleness of the skin, and shallow breathing, suggesting that they were related to high anti-AAVrh74 titers (>1:51,200) and that these AEs were avoided by plasmapheresis prior to redosing.
Two animals had a blunted humoral immune response to AAVrh74. Specifically, RA2481 (cohort 2) did not mount an immune response during part 1 of the study for undetermined reasons. Although this animal did show increased anti-AAVrh74 antibodies following redosing, titers remained lower than the other animals at the redosing stage. One NHP in cohort 5, 13-156, also exhibited a blunted TAb response for the initial 6 weeks post-GTT, which may have been due to the triple IS regimen administered to animals in this cohort. However, after 6 weeks, antibody levels rose to be comparable (1:6,400–1:204,800) with the rest of the animals in cohort 5 for the remainder of the study.
Seven NHPs (cohorts 2–4) transitioned to part 2, underwent plasmapheresis, and were immediately redosed. One animal (RA2480 from cohort 3 redosed at a TAb titer of 1:25,600) did not receive plasmapheresis because of poor vascular access and was redosed without adverse clinical observations. Additionally, one of the animals, RA2481 (cohort 2), that did not have an initial antibody response to AAVrh74, was redosed safely and efficaciously without plasmapheresis because of poor vascular access. Three NHPs from cohort 5 were also transitioned to part 2 of the study; two were redosed without plasmapheresis at a high titer (≥51,200) of anti-AAVrh74 antibodies. These animals with highly elevated anti-AAVrh74 antibody titers exhibited adverse clinical observations consistent with a hypersensitivity reaction upon redosing, demonstrating the need to lower antibody titers from highly elevated levels before AAV administration.
The animals in cohort 5 with elevated antibodies did not show evidence of complement-mediated AEs on the basis of evaluation of C3. This is notable because of the observation of severe complement-mediated AEs in trials of AAV9-based GTTs for DMD, with the sponsors of these trials hypothesizing these resulted from antibody-mediated activation of complement.26,27 The reasons complement-mediated events were not apparent in this study are unclear but may be related to specific characteristics of the AAVrh74 vector used here given that complement activation can also occur via direct contact with the viral capsid.28 None of the clinical trials using AAVrh74 vector (NCT03375164, NCT03769116, NCT04626674, NCT05096221, NCT01976091, NCT03652259, and NCT02710500) have reported complement-mediated AEs, further suggesting that propensity for complement-mediated AEs may be related to AAV serotype.
Robust SRP-9001 dystrophin expression was observed across skeletal muscle, diaphragm, and heart with no abnormal histopathology upon redosing in NHPs that express endogenous dystrophin. Expression of GFP in human cardiomyocytes derived from induced pluripotent stem cells (IPSCs) treated with AAVrh74.MHCK7.GFP indicates that the MHCK7 promoter is able to effectively transduce and express protein in human heart cells (unpublished data). These data suggest that protein expression driven by the MHCK7 promoter in NHP heart would likely be translatable to protein expression in human heart.
The present study is limited by the plasmapheresis procedure used in NHPs, which differs from that used in humans. For example, there is a lack of donor NHP blood, and human albumin is used as a replacement fluid. In addition, plasmapheresis was performed in only 1 day, with a maximum of three cycles. Another limitation was that complement activation was not analyzed earlier in the study. However, the present study was designed and initiated prior to the reported complement activation safety finding observed in patients treated with another GTT designed for DMD, SGT-001.29
In conclusion, anti-AAVrh74 TAb response was similar among all NHP cohorts irrespective of IS regimen and with no evidence of abnormal immunological responses. The data also highlight the utility of plasmapheresis for circumventing pre-existing antibodies to AAV, whether those antibodies arose from environmental AAV exposure or previous AAV gene therapy. NHPs redosed with highly elevated anti-AAVrh74 antibody titers experienced significant safety issues, but when those titers were reduced with plasmapheresis, minimal safety issues were observed, highlighting the importance of reducing antibodies before dosing with AAV. Plasmapheresis has been demonstrated as a safe and effective tool to reduce pre-existing antibodies to AAV vectors in previous literature,18 and this study expands on establishing the safety of this procedure to enable redosing of GTT.
Materials and methods
Animal model
Chinese rhesus macaques (more than 2 years of age; cohorts 1–4, females; cohort 5, males) were obtained from Covance (Madison, WI). Animals were housed in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited vivarium facility in stainless steel cages, which conformed to the requirements set forth in the 2011 Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC). Animals were group housed on the basis of positive or negative anti-AAVrh74 antibody titer and compatibility with other NHPs. NHPs were separated if necessary because of distress or fighting. Primates were fed either high- or low-fat standard biscuits as their main source of nutrition. Daily enrichment consisted of fruits or vegetables and additional treats to allow oral drug consumption. Body weights were recorded for animals starting 2 weeks post-dosing (day 1), and weekly thereafter. Environmental controls were set to maintain a temperature of 18°C–26°C (64°F–79°F) with a relative humidity of 30%–70%. A 12:12 h light/dark cycle was maintained. Primates were provided enrichment by Animal Resource Core technical staff members, including foraging enrichment, television, music, chew and play toys, at least three times per week, following American Association for Laboratory Animal Science guidelines. All procedures were approved by the Research Institute at Nationwide Children’s Hospital Institutional Animal Care and Use Committee (IACUC; protocol AR06-00016). Necropsies were done 12 weeks after gene therapy in part 2 and followed AAHA guidelines. Skeletal muscle, heart, diaphragm, and liver were processed for histopathology.
AAV vector production
For clinical translation, we placed the SRP-9001 dystrophin transgene under the control of an MHCK7 promoter, which drives high-level transgene expression in skeletal muscles, including the diaphragm, and includes an enhancer for expression in the heart, with minimal off-target tissue expression.30 A cassette containing the MHCK7 promoter and SRP-9001 dystrophin transgene were packaged into AAVrh74 capsids using the standard triple transfection protocol, as described elsewhere.4,31,32,33,34 A quantitative polymerase chain reaction (qPCR)-based titration method based on a supercoiled plasmid standard was used to determine an encapsulated vg titer using a Prism 7500 Fast TaqMan detector system (PE Applied Biosystems, Foster City, CA).33
In vivo gene delivery with IS (part 1)
This study was divided into two parts (Figure 1). In part 1, animals were divided into five cohorts (cohort 1 [n = 2] and cohorts 2–5 [n = 3]). Each cohort received different IS regimens during the study (Table S1). All animals received delandistrogene moxeparvovec at a dose of 2.00 × 1014 vg/kg (on the basis of the supercoiled standard qPCR titer method and equivalent to 1.33 × 1014 vg/kg on the basis of the linear standard qPCR titer method). Cohort 1 received delandistrogene moxeparvovec only. Cohort 2 received prednisone 1 day prior to dosing and continued prednisone for 30 days post-dosing. Cohort 3 received prednisone 1 day prior to dosing and continued prednisone for 60 days post-dosing. Cohort 4 received prednisone 14 days prior to dosing and continued prednisone for 60 days post-dosing. For all groups, prednisone was administered orally at a dose of 2 mg/kg. Cohort 5 was treated with a triple IS regime that included rituximab, sirolimus, and prednisone. Rituximab (750 mg/m2) was dosed via intravascular infusion for three dose sessions 14 and 10 days before dosing and on the same day as GTT administration (immediately prior to GTT dosing). Sirolimus (4 mg/m2/day) was dosed orally 3 days before dosing and continued daily for the remainder of part 1. Prednisone (2 mg/kg) was dosed orally 1 day prior to GTT dosing and 30 days post-dosing. At 12 weeks after initial administration of delandistrogene moxeparvovec, all animals transitioned to part 2 of the study.
Total plasma exchange and redosing (part 2)
In part 2 of the study, cohort 1 did not receive any additional dose or treatment regimen (Figure 1; Table S2). In addition, one animal did not undergo any further treatment (cohort 5). Seven NHPs from cohorts 2–4 underwent 2 or 3 rounds of plasmapheresis before being redosed with delandistrogene moxeparvovec at a dose of 2.00 × 1014 vg/kg. At 28 and 14 days prior to plasmapheresis, a maximum blood collection (10% of the circulating blood) was performed. Collected whole blood was preserved and stored for no more than 30 days in anticoagulant acid-citrate-dextrose (ACDA) solution at 4°C. Additionally, NHPs were provided extra iron-rich supplements and enrichment. On the day of apheresis, animals were sedated intramuscularly with tiletamine (3–6 mg/kg), intubated, and secured to a heated procedure table. Anesthetic maintenance was achieved with isoflurane in oxygen 1%–4%. Angiocatheters were placed in both legs (saphenous vein), with one access port to withdraw whole blood and a second in the opposite leg to re-deliver red blood cells and replacement fluid. Additional catheters were placed in the arms (cephalic vein) for support fluid and blood draws throughout the procedure. After vascular access was obtained, animals were dosed with heparin (50–100 U/kg) to maintain adequate blood flow and prevent clotting during plasmapheresis. One plasmapheresis exchange equates to the entire amount of circulating blood being removed and replaced one time, and 2 or 3 plasma exchanges were performed in animals in cohorts 2–4 to achieve an estimated antibody removal of 70%. Blood was collected after each completed exchange for blood chemistry analysis and anti-AAVrh74 TAb testing. Immediately after the plasma exchanges, the animals were disconnected from the apheresis unit and systemically redosed with delandistrogene moxeparvovec. Animals in cohorts 2–4 also received prednisone (2 mg/kg) once per day, 1 day prior to and up to 30 days post-plasmapheresis and after gene transfer. Two animals were redosed without plasmapheresis: venous access was lost in one animal in cohort 3, so plasmapheresis was not feasible; the other animal (cohort 2) did not mount an antibody response to the initial dose of delandistrogene moxeparvovec, so plasmapheresis was not warranted. However, these animals were redosed and continued prednisone (2 mg/kg) once per day at 1 day prior to and up to 30 days after gene transfer.
Two primates from cohort 5 transitioned to part 2 of the study; the remaining primate from cohort 5 was not redosed because of adverse reactions to the IS regimen. Cohort 5 animals did not receive plasmapheresis, because of the receipt of a triple IS regimen. Also, rituximab caused poor venous access in these animals, resulting in incompatibility with plasmapheresis. Cohort 5 animals received two doses of rituximab (750 mg/m2/day) 10–14 days prior to second gene transfer and an additional dose on the day of gene transfer. The second gene transfer was delivered systemically at 2.00 × 1014 vg/kg.
Blood samples for analysis of immunology, complete blood count, blood chemistries, and sirolimus levels for cohort 5 were drawn biweekly through the entirety of the study. Blood samples were also used for ELISA and interferon-gamma (IFN-γ) ELISpot assays.
Biopsy collection
Biopsies were collected from the tibialis anterior and gastrocnemius at baseline, prior to study initiation, and at 12 weeks after initial GTT administration. Biopsies were performed using the Vacora Biopsy System (Bard Peripheral Vascular, Tempe, AZ) as previously described.35 Histopathology review was performed on tissue samples by an independent, external veterinary pathologist.
ELISA
Anti-AAVrh74 antibody levels were assessed by ELISA. Briefly, ELISA plates were coated with AAVrh74 capsid at 2 × 1010 vg/mL in carbonate buffer (pH 9.4) alongside wells that contained only coating buffer. After an incubation at 4°C overnight, the wells were then blocked with 5% milk and 1% goat serum in PBS (blocking buffer) for 1 h at 37°C. The sera samples for the specific time points were diluted in blocking solution (1:100) then serially diluted (2-fold) from 1:25 to 1:1:26,214,400 in the blocking solution and incubated for 1 h at room temperature. The plate was washed five times with 200 μL Dulbecco’s 1× PBS with 0.1% Tween 20 detergent (DPBS-T; Thermo Fisher Scientific, Pittsburgh, PA) and incubated for 30 ± 5 min with either rabbit anti-monkey IgG (for anti-AAVrh74) or goat anti-monkey IgM μ-chain (for IgM detection) labeled with horseradish peroxidase (HRP; 1:10,000 in blocking buffer) (Sigma-Aldrich, St. Louis, MO). After washing the plate five times with DPBS-T, the plate was incubated with 100 μL 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Thermo Fisher Scientific, Waltham, MA) for 2–3 min at room temperature followed by the addition of 100 μL sulfuric acid 1N to stop the reaction. Signal intensity was quantitated by reading the absorbance at 450nm using a microplate reader (Bio Tek, Winooski, VT). The optical density (OD) values of the capsid-coated and coating buffer-only wells were used to determine the absorbance ratio. The endpoint titer was determined by identifying the last serum dilution (highest) that yielded a ratio of ≥2.00.
ELISpot analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood through density gradient centrifugation as previously described.36 PBMCs were tested in an ELISpot assay for reactivity to AAVrh74 and micro-dystrophin using a monkey IFN-γ ELISpot kit (U-CyTech, Utrecht, the Netherlands). Briefly, multiscreen-IP filter plates were treated with 100 μL diluted coating antibody and incubated for 1 h at 37°C. Wells were washed five times with 200 μL 1× PBST. After washing, plates were blocked with 200 μL 1× Blocking Buffer, and 2 × 105 cells were plated in each well except for the concavalin A wells, which received 2.5 × 104 cells in 200 μL of AIM-V medium per well. Plated cells were stimulated with AAVrh74 peptides, micro-dystrophin peptides, concavalin A (positive control), DMSO (vehicle control), and eGFP (negative control). Plates were incubated at 37°C, 5% CO2, and 95% humidity for 36–48 h. Following incubation, plates were washed five times with 200 μL 1× PBST. Wells were then treated with biotinylated detection antibody diluted in 1× dilution buffer B and incubated for 1 h at 37°C. Plates were next washed five times with 200 μL 1× PBST, and 50 μL GABA substrate diluted in 1× dilution buffer B was added and incubated for 1 h at 37°C. Following the wash, activator solutions I and II were combined in a 1:1 ratio in dark, and 30 μL of the combined reagents were added to each well. Plates were incubated with activator solution in the dark at room temperature for at least 10 min for spot development. The activating reaction was then stopped by rinsing all plate surfaces with demineralized water for a minimum of 3 min. Plates were dried and then imaged and analyzed using a CTL ImmunoSpot Analyzer (CTL, Shaker Heights, OH).
Complement analysis
Complement activation was assessed in all NHPs that underwent plasmapheresis using a C3 complement detection kit (Novus Biologicals LLC, Centennial, CO). C3-pre-coated microtiter plates were blocked using reagent diluent and were incubated for 1 h at room temperature. Serum samples were prepared and added to the plates and incubated for 2 h at room temperature. Biotinylated detection antibody was then added to each plate, and after a 1 h incubation at room temperature, plates were washed, and the HRP conjugate was added and incubated for 30 min at room temperature. After a series of washes, the plates were developed by adding the substrate reagent and reading the absorbance (OD) at 450 nm. Results were expressed as micrograms per milliliter.
qPCR
qPCR was used to quantify the number of vg copies in collected tissue samples as described previously.37,38 DNA isolation of tissue samples was performed in Airclean PCR Workstations using the QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, Germantown, MD). DNA concentrations were determined using a Qubit Flex with a high-sensitivity double-stranded DNA dye. MHCK7 primers (forward: 5′-CCAACACCTGCTGCCTCTAAA-3′; reverse: 5′-GTCCCCCACAGCCTTGTTC-3′) and probe (/56-FAM/TGGATCCCC/ZEN/TGCATGCGAAGATC/3IABkFQ) were added to the TaqMan Fast Advanced Master Mix (Applied Biosystems, San Francisco, CA), and a 6-point standard curve of linearized plasmid containing the MHCK7 promoter sequence was used with 10-fold decreases from 1 × 106 to 1 × 101 copies/μL. A QuantStudio 6 machine (Thermo Fisher Scientific) was used to analyze each plate. Plates were accepted if R2 was ≥0.980, the standard curve slope was between −3.1 and −3.7, and gDNA and no template controls were below the lower limit of quantification (BLLOQ; defined as CT values greater than the final standard curve point). All test samples BLLOQ were considered negative for MHCK7.
Western blot analysis
Western blotting was performed as previously described, with additional modifications for the type of tissue and antibodies used for this study.39 Homogenized protein samples were prepared at a concentration of 30 μg total protein per sample. After 7.5 μL 4× loading dye and NuPAGE reducing agent (NuPAGE, Invitrogen, Carlsbad, CA) was added, each sample was brought to 20 μL with homogenization buffer. Samples were heated at 105°C for 10 min. Running buffer (1× NuPAGE Tris-acetate running buffer) was added to the outside chamber of the electrophoresis unit, and interior running buffer (1× NuPAGE Tris-acetate running buffer, 0.25% NuPAGE reducing agent) was added to the interior chamber between the gels. Samples were run on 3%–8% Tris-acetate gels using the NuPAGE electrophoresis system at 150 V for 1 h 5 min. Using the NuPAGE transfer electrophoresis system and buffers, protein was transferred to a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Invitrolon, Invitrogen). PVDF membranes were prepared by soaking in methanol for 1 min followed by soaking in transfer buffer (1× NuPAGE transfer buffer, 10% methanol, and 0.1% NuPAGE reducing agent) for 1 min. Transfer was conducted in transfer buffer at 35 V for 1 h 30 min. Primary antibody staining was performed using an antibody specific for the N terminus of human dystrophin (DYS3 1:20 dilution; Leica Biosystems, Deer Park, IL) diluted in 5% nonfat dry milk and incubating overnight at 4°C. Membranes were next incubated for 1 h at room temperature with secondary antibody that was diluted in 5% nonfat dry milk (1:1,000; anti-mouse HRP). Blots were imaged using an Alliance Q9 Advanced Imaging System (UVITEC Ltd., Cambridge, UK).
Data and code availability
Qualified researchers may request access to the data that support the findings of this study from Sarepta Therapeutics, Inc., by contacting medinfo@sarepta.com.
Acknowledgments
This study and medical writing support were funded by Sarepta Therapeutics. Medical writing and editorial support were provided by Marjet Heitzer, PhD, of 360 Medical Writing and funded by Sarepta Therapeutics. The immunological assays were supported by Sohrab Khan, Shannon Miller, and Zach Purney of Sarepta Therapeutics.
Author contributions
Conceptualization, R.A.P., E.L.P., D.G., and L.R.R.-K.; investigation, R.A.P., E.L.P., D.G., G.C.O., S.L., and K.C.; writing – original draft, R.A.P. and G.C.O.; writing – review & editing, R.A.P., E.L.P., D.G., G.C.O., S.L., K.C., J.R.M., and L.R.R.-K.; supervision, L.R.R.-K and J.R.M
Declaration of interests
R.A.P., E.L.P., D.G., G.C.O., S.L., K.C., and L.R.R.-K. are employees of Sarepta Therapeutics, Inc. J.R.M. has received grants from Parent Project Muscular Dystrophy and personal fees from Sarepta Therapeutics Inc. and Nationwide Children’s Hospital outside the submitted work and holds a pending patent for a micro-dystrophin cassette for gene therapy and an issued patent for rAAV.SGCA delivery isolated limb infusion.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101195.
Supplemental information
References
- 1.Flanigan K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014;32:671–688. doi: 10.1016/j.ncl.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Eagle M., Baudouin S.V., Chandler C., Giddings D.R., Bullock R., Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002;12:926–929. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 3.Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L., Kaul A., Kinnett K., McDonald C., Pandya S., et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 4.Mendell J.R., Sahenk Z., Lehman K., Nease C., Lowes L.P., Miller N.F., Iammarino M.A., Alfano L.N., Nicholl A., Al-Zaidy S., et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020;77:1122–1131. doi: 10.1001/jamaneurol.2020.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potter R.A., Griffin D.A., Heller K.N., Peterson E.L., Clark E.K., Mendell J.R., Rodino-Klapac L.R. Dose-Escalation Study of Systemically Delivered rAAVrh74.MHCK7.micro-dystrophin in the mdx Mouse Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. 2021;32:375–389. doi: 10.1089/hum.2019.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He X., Urip B.A., Zhang Z., Ngan C.C., Feng B. Evolving AAV-delivered therapeutics towards ultimate cures. J. Mol. Med. 2021;99:593–617. doi: 10.1007/s00109-020-02034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calcedo R., Wilson J.M. Humoral Immune Response to AAV. Front. Immunol. 2013;4:341. doi: 10.3389/fimmu.2013.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 9.Wang L., Calcedo R., Bell P., Lin J., Grant R.L., Siegel D.L., Wilson J.M. Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum. Gene Ther. 2011;22:1389–1401. doi: 10.1089/hum.2011.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendell J.R., Connolly A.M., Lehman K.J., Griffin D.A., Khan S.Z., Dharia S.D., Quintana-Gallardo L., Rodino-Klapac L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: rationale, lessons and future considerations. Mol. Ther. Methods Clin. Dev. 2022;25:74–83. doi: 10.1016/j.omtm.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mays L.E., Vandenberghe L.H., Xiao R., Bell P., Nam H.J., Agbandje-McKenna M., Wilson J.M. Adeno-associated virus capsid structure drives CD4-dependent CD8+ T cell response to vector encoded proteins. J. Immunol. 2009;182:6051–6060. doi: 10.4049/jimmunol.0803965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith M.R. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359–7368. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 13.Limon J.J., So L., Jellbauer S., Chiu H., Corado J., Sykes S.M., Raffatellu M., Fruman D.A. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc. Natl. Acad. Sci. USA. 2014;111:E5076–E5085. doi: 10.1073/pnas.1407104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgoffe G.M., Kole T.P., Zheng Y., Zarek P.E., Matthews K.L., Xiao B., Worley P.F., Kozma S.C., Powell J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corti M., Cleaver B., Clément N., Conlon T.J., Faris K.J., Wang G., Benson J., Tarantal A.F., Fuller D., Herzog R.W., Byrne B.J. Evaluation of Readministration of a Recombinant Adeno-Associated Virus Vector Expressing Acid Alpha-Glucosidase in Pompe Disease: Preclinical to Clinical Planning. Hum. Gene Ther. Clin. Dev. 2015;26:185–193. doi: 10.1089/humc.2015.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller C., Berry J.D., McKenna-Yasek D.M., Gernoux G., Owegi M.A., Pothier L.M., Douthwright C.L., Gelevski D., Luppino S.D., Blackwood M., et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020;383:151–158. doi: 10.1056/NEJMoa2005056. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan A.A. Therapeutic plasma exchange: a technical and operational review. J. Clin. Apher. 2013;28:3–10. doi: 10.1002/jca.21257. [DOI] [PubMed] [Google Scholar]
- 18.Reverberi R., Reverberi L. Removal kinetics of therapeutic apheresis. Blood Transfus. 2007;5:164–174. doi: 10.2450/2007.0032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chicoine L.G., Montgomery C.L., Bremer W.G., Shontz K.M., Griffin D.A., Heller K.N., Lewis S., Malik V., Grose W.E., Shilling C.J., et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol. Ther. 2014;22:338–347. doi: 10.1038/mt.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chicoine L.G., Rodino-Klapac L.R., Shao G., Xu R., Bremer W.G., Camboni M., Golden B., Montgomery C.L., Shontz K., Heller K.N., et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin alpha2 surrogates. Mol. Ther. 2014;22:713–724. doi: 10.1038/mt.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mingozzi F., High K.A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goedeker N.L., Griffin D., Dharia S., Santra S., Coy J., Yocum N., Zaidman C.M. Muscular Dystrophy Association Clinical & Scientific Conference. 2022. Evaluation of Total Binding Antibodies Against rAAVrh74 in Patients with Duchenne Muscular Dystrophy. [Google Scholar]
- 23.Griffin D.A., Potter R.A., Pozsgai E.R., Peterson E.L., Rodino-Klapac L.R. Adeno-associated virus serotype rh74 prevalence in muscular dystrophy population. Mol. Ther. 2019;27:342. [Google Scholar]
- 24.Asher D.R., Thapa K., Dharia S.D., Khan N., Potter R.A., Rodino-Klapac L.R., Mendell J.R. Clinical development on the frontier: gene therapy for duchenne muscular dystrophy. Expet Opin. Biol. Ther. 2020;20:263–274. doi: 10.1080/14712598.2020.1725469. [DOI] [PubMed] [Google Scholar]
- 25.Mendell J.R., Sahenk Z., Lehman K., Lowes L.P., Reash N.F., Iammarino M., Alfano L., Lewis S., Church K., Shell R., et al. Presented at the 17th International Congress on Neuromuscular Diseases. 2022. Phase 1/2a trial of delandistrogene moxeparvovec in patients with DMD: 4-year update. [Google Scholar]
- 26.Pfizer press release Pfizer Presents Initial Clinical Data on Phase 1b Gene Therapy Study for Duchenne Muscular Dystrophy (DMD) 2023. https://www.pfizer.com/news/press-release/press-release-detail/pfizer_presents_initial_clinical_data_on_phase_1b_gene_therapy_study_for_duchenne_muscular_dystrophy_dmd
- 27.Solid Biosciences press release Solid Biosciences Provides SGT-001 Program Update. 2023. https://www.solidbio.com/about/media/press-releases/solid-biosciences-provides-sgt-001-program-update
- 28.Agrawal P., Nawadkar R., Ojha H., Kumar J., Sahu A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017;8:1117. doi: 10.3389/fmicb.2017.01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris C., Dreghici R.D., Redican S. Presented at the American Society of Gene and Cell Therapy Annual Meeting 2022. 2022. IGNITE DMD Study of SGT-001 Microdystrophin Gene Therapy for DMD: Long-Term Outcomes and Biomarker Update. [Google Scholar]
- 30.Salva M.Z., Himeda C.L., Tai P.W., Nishiuchi E., Gregorevic P., Allen J.M., Finn E.E., Nguyen Q.G., Blankinship M.J., Meuse L., et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 31.Mendell J.R., Rodino-Klapac L.R., Rosales X.Q., Coley B.D., Galloway G., Lewis S., Malik V., Shilling C., Byrne B.J., Conlon T., et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodino-Klapac L.R., Janssen P.M.L., Montgomery C.L., Coley B.D., Chicoine L.G., Clark K.R., Mendell J.R. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J. Transl. Med. 2007;5:45. doi: 10.1186/1479-5876-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnepp B.C., Jensen R.L., Chen C.L., Johnson P.R., Clark K.R. Characterization of adeno-associated virus genomes isolated from human tissues. J. Virol. 2005;79:14793–14803. doi: 10.1128/JVI.79.23.14793-14803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sondergaard P.C., Griffin D.A., Pozsgai E.R., Johnson R.W., Grose W.E., Heller K.N., Shontz K.M., Montgomery C.L., Liu J., Clark K.R., et al. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015;2:256–270. doi: 10.1002/acn3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarnopolsky M.A., Pearce E., Smith K., Lach B. Suction-modified Bergstrom muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve. 2011;43:717–725. doi: 10.1002/mus.21945. [DOI] [PubMed] [Google Scholar]
- 36.Labikova J., Vcelakova J., Ulmannova T., Petruzelkova L., Kolouskova S., Stechova K. The cytokine production of peripheral blood mononuclear cells reflects the autoantibody profile of patients suffering from type 1 diabetes. Cytokine. 2014;69:189–195. doi: 10.1016/j.cyto.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Clark K.R., Liu X., McGrath J.P., Johnson P.R. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum. Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- 38.Pozsgai E.R., Griffin D.A., Heller K.N., Mendell J.R., Rodino-Klapac L.R. beta-Sarcoglycan gene transfer decreases fibrosis and restores force in LGMD2E mice. Gene Ther. 2016;23:57–66. doi: 10.1038/gt.2015.80. [DOI] [PubMed] [Google Scholar]
- 39.Pozsgai E.R., Griffin D.A., Heller K.N., Mendell J.R., Rodino-Klapac L.R. Systemic AAV-Mediated beta-Sarcoglycan Delivery Targeting Cardiac and Skeletal Muscle Ameliorates Histological and Functional Deficits in LGMD2E Mice. Mol. Ther. 2017;25:855–869. doi: 10.1016/j.ymthe.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Qualified researchers may request access to the data that support the findings of this study from Sarepta Therapeutics, Inc., by contacting medinfo@sarepta.com.