

Abstract
Obesity-associated inflammation is a systemic process that affects all metabolic organs. Prominent among these is adipose tissue, where cells of the innate and adaptive immune system are markedly changed in obesity, implicating these cells in a range of processes linking immune memory to metabolic regulation. Furthermore, weight loss and weight cycling have unexpected effects on adipose tissue immune populations. Here, we review the current literature on the roles of various immune cells in lean and obese adipose tissue. Within this context, we discuss pharmacological and nonpharmacological approaches to obesity treatment and their impact on systemic inflammation.
INTRODUCTION
Obesity [defined by body mass index (BMI) > 30 kg/m2] is highly prevalent worldwide, with more than 42% of the US population considered obese (1), and obesity rates have more than doubled in over 70 countries since 1980 (2). Obesity is a major risk factor for a constellation of noncommunicable diseases including cardiovascular disease, type 2 diabetes mellitus, and cancer (3). Although obesity is known to be associated with a heightened inflammatory milieu in all metabolic organs, a unifying mechanistic link between immune responses to obesity and cardiometabolic complications has been challenging to delineate, limiting the potential for therapeutic advancements. However, evidence continues to suggest that nutrient excess elicits a chronic low-grade inflammatory response that may underlie cardiometabolic complications, which has expanded into the study of “metaflammation” (4). Here, we present an overview of the links between obesity and inflammation, discuss the immunologic memory triggered by obesity, and review the metabolic response to nutrient oversupply in immune cells (immune cell bioenergetics). Last, we identify clinical approaches targeting obesity and their impact on inflammation.
OVERVIEW OF OBESITY-ASSOCIATED INFLAMMATION
Obesity is accompanied by chronic low-grade inflammation in metabolically impactful tissues such as adipose tissue (AT), liver, skeletal muscle, pancreatic islets, and the brain (Fig. 1). Inflammatory cytokine signaling can interfere with insulin signaling pathways leading to impaired glucose uptake and uncontrolled lipolysis, ultimately resulting in ectopic lipid storage and propagating insulin resistance in a vicious cycle. Although this Review will focus on AT, immune contributions to other organs are summarized in these excellent articles (4–8). AT immune cells serve a wide range of homeostatic functions that are modified upon obesity. In addition to the loss of their regulatory functions, heightened inflammatory phenotypes of immune cells in AT are typically met with systemic complications such as insulin resistance, hyperglycemia, and dyslipidemia—contributing to greater cardiometabolic disease risk. In addition, excessive pro-inflammatory cytokine release into the circulation is often present in obesity and metabolic syndrome, suggesting the induction of inflammatory processes as a clinical biomarker for metabolic disease risk (9). Thus, localized and systemic inflammatory processes provide potential targets for immune-mediated therapies to treat metabolic disease.
Fig. 1. Organ-specific inflammatory responses in obesity.
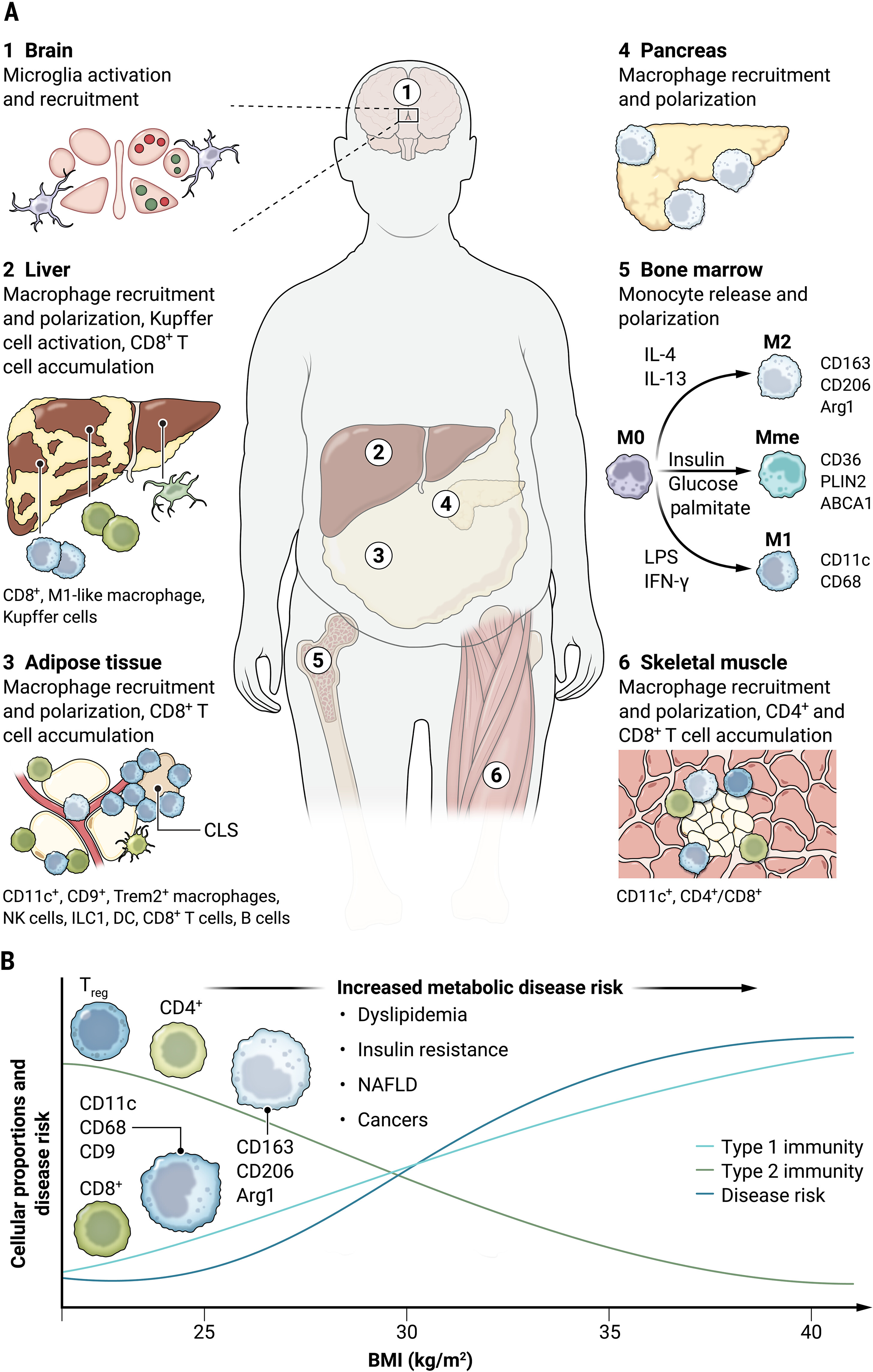
(A) Inflammation is a coordinated immune response aimed to alleviate stresses induced by obesity and occurs in tissues such as liver, pancreas, adipose tissue, skeletal muscle, and the brain. Ectopic lipid accumulation in these tissues can initiate monocyte recruitment, macrophage polarization toward pro-inflammatory states, CD8+ T cell accumulation, and excess accumulation of pro-inflammatory mediators. (B) Immune response to positive energy balance within the stromal compartments of these tissues increases type 1 immunity (light blue line) including M1-like innate immune cells and CD8+ T cells. Conversely, obesity attenuates type 2 immunity (green line) phenotype, resulting in reduced M2-like innate immune cells as well as Treg and CD4+ adaptive immune cells. This reciprocal relationship between type 1 and type 2 immunity in obesity is proposed to underlie tissue homeostasis and insulin resistance (dark blue line). CLS, crown-like structure; NAFLD, nonalcoholic fatty liver disease.
In mice, most studies have analyzed epididymal white AT from male animals. However, other visceral adipose depots (perirenal, mesenteric, and omental AT), as well as inguinal subcutaneous white AT and brown AT depots, may also be relevant to systemic dysfunction associated with AT inflammation. Furthermore, tissue-specific adipose depots such as perivascular and epicardial AT, as well as fat around skin and lymph nodes, are important for tissue homeostasis and may be altered in obesity, although their discussion is beyond the scope of this article. In humans, most studies have analyzed subcutaneous and omental white AT depots. Although the inflammatory response in AT is more extreme in mice, ample human data suggest that similar changes in the immune landscape occur in humans. For example, the number of macrophages in visceral AT is elevated in obese compared with lean individuals, associated with insulin resistance (10), and predictive of hemoglobin A1c (HbA1c) concentrations (11).
ADIPOSE TISSUE MACROPHAGES
Macrophage polarization
In a simplified and dichotomous paradigm, macrophages have been described as either classically activated M1-like or alternatively activated M2-like. These extreme states are based on in vitro polarization with cytokines in an effort to generate macrophage populations specialized to respond to environmental cues from stimuli within the tissues. M1-like macrophages defend against intracellular pathogens and are characterized by the production of pro-inflammatory cytokines [for example, interleukin-6 (IL-6), IL-1β, and tumor necrosis factor–α (TNF-α)] and expression of inducible nitric oxide synthase (iNOS). M2-like macrophages are induced by IL-4 and IL-13 and present a tissue-repair phenotype characterized by the release of anti-inflammatory cytokines [for example, IL-10, IL-12, and transforming growth factor–β (TGF-β)]. In contrast to in vitro stimulation of macrophage phenotypes, macrophages in vivo exist along a spectrum of polarization states, and individual cells can simultaneously express genes/proteins of both M1/M2 phenotypes (12, 13).
Macrophage polarization in AT has been an area of interest in the immunometabolism field. In AT of mice and humans, M1- and M2-like macrophages express immune cell surface markers F4/80, CD11b, and CD64 but also contain unique identifiers specific to their population. M1-like macrophages express CD11c in mice and humans (10, 14, 15) and major histocompatibility complex class II (MHCII) in mice (16), whereas M2-like signatures include CD206, CD163, and TIM4 in mice (17) and CD206 in humans (10, 11). Studies identifying macrophage subpopulations in obesity have also revealed dissimilarities between mice and humans (18). For example, subcutaneous AT CD206+ CD11c+ cells are positively associated with insulin resistance in humans, but this double-positive population may be less prevalent in mice (10, 19). As in other tissues, the simple M1/M2 dichotomy is not sufficient to explain the full range of macrophage phenotypes in AT. Fortunately, technological and analytical advances in single-cell RNA sequencing, mass cytometry, and spatial transcriptomics are emerging in the immunometabolism field (11, 19–22), allowing for the profiling of functionally diverse immune cell populations.
Additional insights into AT macrophage (ATM) polarization can be made when considering the local AT milieu, which is rich in lipids, glucose, and insulin. Thus, a metabolically activated (MMe) macrophage phenotype in obesity provides a more accurate description of the ATM polarization state than the “M1” nomenclature (12, 13). In vivo cell surface abundance of CD36, PLIN2, and ABCA1 in ATMs from humans with obesity is a signature of the lipid-handling functions of MMe cells (13). In vitro MMe macrophages can be generated by metabolic stimuli (palmitic acid, glucose, and insulin). A detailed description of the function of these lipid-handling macrophages in AT is provided below.
Specialized resident macrophage function in AT homeostasis
Macrophages are the most abundant immune cell population in AT in both genetic and diet-induced obesity (23, 24). In the lean state, macrophages represent about 10% of all AT immune cells and can expand to represent more than 40% of all immune cells in obesity (15, 23). Macrophages present a high degree of plasticity and readily adapt to environmental cues to preserve tissue homeostasis. The diversity of tissue-resident macrophage populations within tissues is indicative of their homeostatic functions ranging from tissue repair, clearance of cellular debris, and metabolic regulation. Specifically in AT, their functions include buffering lipids and other critical molecules, antigen presentation, and coordinating both intercellular and interorgan cross-talk.
Lipid uptake and partitioning
MMe polarization is characterized by the expression of lipid-handling genes and proteins (13). Similar lipid-handling ATM populations have also been identified by expression of CD9 (called CD9+ATMs) (25) and Trem2 [called lipid-associated macrophages (LAMs)] (26). The association of lipid-laden ATMs with CD9 and Trem2 markers has also been identified in humans (11). MMe, CD9+, and LAMs in AT are proposed to aid in the uptake and storage of lipids in an effort to limit ectopic lipid accumulation. In addition to their transcriptional profile linked to lipid handling, they are also localized to crown-like structures to aid with lipid scavenging around dead adipocytes. Other studies of lipid-handling ATMs report transcriptional characteristics of lysosomal biogenesis and lysosomal-dependent lipid metabolism, suggesting that an autophagy-dependent mechanism is up-regulated to aid in dead adipocyte degradation and fatty acid recycling (27). ATM localization to crown-like structures around dead adipocytes, coupled with a transcriptional pattern for lipid uptake and catabolism, offers a unique role for lipid uptake and storage in ATMs. In addition, adipocytes have been shown to release lipid-filled exosomes that are sensed by immune cells and directly contribute to bone marrow–derived monocyte differentiation into an MMe-like phenotype (28). Although not intuitive that macrophages would be required to buffer lipids in AT when adipocytes themselves can store large amounts of triglyceride, the reports described here support this function for ATMs.
Iron handling
Iron is required for normal homeostatic functions but is detrimental when in excess. We have described a subset of iron-rich macrophages—coined MFehi—that protect adipocytes from iron overload through an intrinsic capacity for iron uptake and storage (29, 30). Obesity results in fewer MFehi cells and lower iron content within the MFehi cells with concurrent adipocyte overload, suggesting that iron handling by MFehi cells is important for adipocyte iron homeostasis (29). Furthermore, iron transfer between macrophages and adipocytes in coculture was accelerated when the macrophages were MMe-polarized, with a net increased iron accumulation in the adipocytes (31). Adipocyte iron concentrations are relevant to systemic metabolic health because adipocyte-specific overload via ferroportin deficiency results in glucose intolerance (32), whereas reducing adipocyte iron concentrations via transferrin receptor deficiency results in protection from obesity-related metabolic dysfunction (33). In addition, increased iron localization within the mitochondrial matrix of ATMs promotes an inflammatory phenotype in ATMs and directly elevates mitochondrial iron content in adipocytes. In contrast, low macrophage iron content is associated with an M2-like phenotype and improved glucose tolerance (34). Thus, published data strongly support the role of ATMs in AT iron homeostasis, which may be particularly important for limiting the oxidative stress and insulin resistance common in obesity.
Catecholamine uptake and catabolism
Sympathetic innervation by norepinephrine signaling robustly promotes lipolysis. Nerve-associated macrophages in AT have been termed sympathetic neuron-associated macrophages (SAMs) (35). SAMs have been reported to increase with obesity (35) and to decrease with aging (36). SAMs are localized to nerve bundles, and their role is to coordinate sympathetic tone by regulating norepinephrine uptake and catabolism. In the context of obesity, increased activation and recruitment of SAMs to AT aim to protect from norepinephrine spillover. Ablation of the ability of SAMs to endocytose and degrade norepinephrine results in the browning of white fat and weight loss (35). Furthermore, this cell population appears to be conserved in humans (35), making it a potential target for weight loss. The decline of SAMs with aging also provides a partial explanation for why aging humans have a defect in white AT lipolysis (36). Given the link between catecholamine and lipid metabolism, SAMs appear to be an important immune cell population poised to limit lipolysis from AT by catecholamines and likely preserve further glucose intolerance in obesity and aging.
Antigen presentation
Another intriguing function of ATMs is antigen presentation, which should not come as a surprise given the existence of memory CD4+ and CD8+ T cells in AT (described further below). Murine ATMs express MHCII and human ATMs express HLA-D, indicative of antigen presentation capability (11). Interestingly, the antigen presentation markers CD1A-E are elevated in lipid-rich CD11c+ ATMs. In mouse models, a dynamic interaction between ATMs and CD4+ T cells was demonstrated, and deficiency of MHCII resulted in fewer CD4+ T cells in the AT (37). Furthermore, the transfer of activated antigen-presenting cells into lean mice results in AT inflammation and impaired insulin action systemically (38). Antigen presentation may also be relevant to CD8+ T cell activation, because in obesity, CD8+ T cells increase in number and in clonality (39).
Intercellular mitochondria transport
Recent studies have suggested that macrophages are able to coordinate both intercellular and interorgan mitochondrial transfer through the uptake of extracellular vesicles or free mitochondria (40, 41). In the context of obesity, mitochondrial transfer between adipocytes and ATMs can be attenuated by saturated fatty acids, resulting in mitochondrial release into the circulation either as free mitochondria or within extracellular vesicles for peripheral tissue uptake (40). Furthermore, genetic disruption of mitochondrial uptake by macrophages results in systemic metabolic dysregulation (41). When considering the iron-handling phenotype of ATMs, it is possible that some of the iron transfer between adipocytes and ATMs could be attributed to this mitochondrial transfer. Whether all ATMs can participate in mitochondrial transfer or whether a unique subset is responsible for this function is unknown.
Together, these data demonstrate the pleiotropic homeostatic functions of resident ATMs. Therefore, therapeutic strategies targeting ATMs need to be considered carefully because some populations serve a conserved and beneficial impact relative to their anatomic location and role in metabolism.
Macrophage recruitment and differentiation
Tissue macrophages originate from either resident yolk-sac progenitors that proliferate within the tissue environment or the recruitment of bone marrow–derived monocytes into regions of tissue inflammation. Specifically, bone marrow–derived monocytes expressing Ly6C migrate from their peripheral origins to inflammatory sites signaled by chemokine ligands/receptors such as CCL2/CCR2 (42). For example, CCR2+ Ly6C+ monocytes are thought to differentiate into M1-like macrophages after recruitment to AT, and deletion of CCR2 on macrophages results in decreased monocyte influx into AT (15, 42).
After monocyte recruitment to the site of tissue inflammation, environmental cues from the tissue niche become drivers for macrophage differentiation and polarization. Unlike adaptive immune cells that have high antigen specificity, innate immune cells do not express specific antigen recognition receptors (43). Therefore, macrophage polarization is activated by cell surface receptor response to ligands in the tissue microenvironment. Specifically, pattern recognition receptors (PRRs) are key components of the host immune system, initiating pathogen recognition and transcriptional activation to respond to specific pathogens of interest. Within the PRR family, Toll-like receptor (TLR) signaling has been demonstrated in early activation of the innate immune response, in which TLRs are proposed to “sense” nutrient excess by stimulation of their ligands [from saturated fatty acids and lipopolysaccharide (LPS)] to initiate signaling pathways aimed to meet the cellular and bioenergetic needs of the cell (44). The canonical model for TLR activation has been important for understanding signals responsible for inducing a pro-inflammatory environment in obesity and AT insulin resistance (45). For example, TLR activation by ligands activates nuclear factor κB (NF-κB) signaling to produce inflammatory cytokines as well as the Nod like receptor (NLR) family of innate immune sensors, specifically the NLRP3 inflammasome, which in turn regulates the activation of caspase-1 to cleave cytokines such as pro-IL-1β to their active form (46, 47). Therefore, IL-1β represents another inflammatory mediator that may impair insulin signaling and induce lipolysis. In summary, monocyte activation through the innate immune response aims to alleviate tissue stress caused by obesity but may follow with metabolic consequences.
Macrophage bioenergetics in obesity
Macrophage bioenergetics and polarization
Over the past 15 years, immunologists have developed a greater appreciation for the role of intrinsic immunometabolism, or bioenergetic pathways, in immune cell function (48). Preferential use of glycolysis or oxidative phosphorylation (OX PHOS) for adenosine 50-triphosphate (ATP) production allows metabolites to be generated for cellular functions such as proliferation, redox balance, inflammatory signaling, hypoxia-inducible factor–1α (HIF1α) activation, and epigenetic modifications. Interestingly, the relationship between fuel utilization and cell polarization is reciprocal because activating certain metabolic pathways can influence cell differentiation and polarization, and cell polarization can affect the use of bioenergetic pathways (Fig. 2). For example, fatty acid oxidation of oleic acid can induce M2 polarization (49), and using IL-4 for M2 polarization drives OX PHOS (50).
Fig. 2. AT macrophages have distinct metabolic phenotypes.
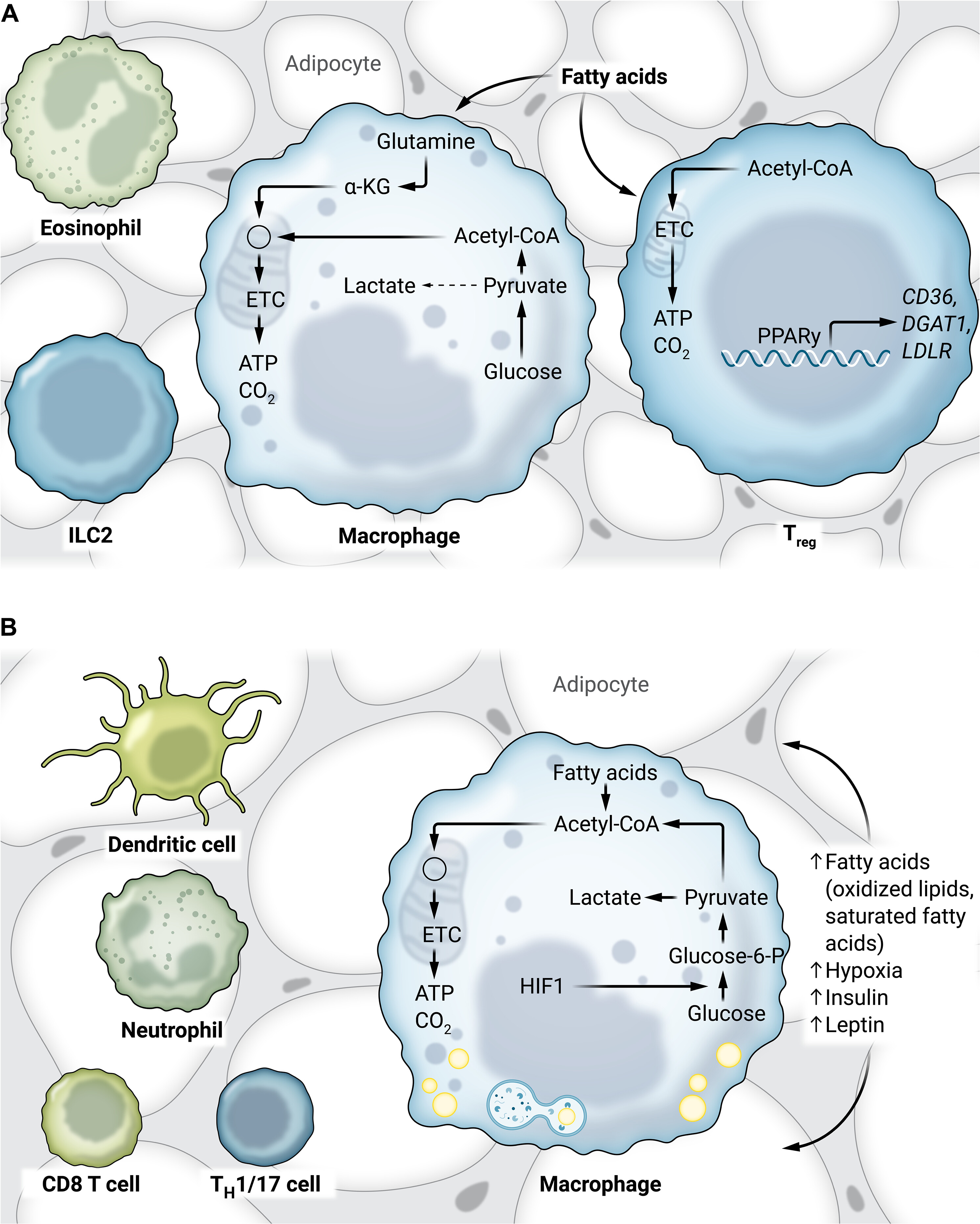
(A) In lean AT, macrophages have a low metabolic demand. Tregs use oxidative phosphorylation, and PPARγ drives gene expression of lipid handling–related genes. The metabolism of other adipose immune cells is unknown. (B) In obese adipose tissue, macrophages have heightened glycolysis and oxidative phosphorylation, likely driven by increases in more inflammatory lipids, hypoxia, insulin, and leptin signaling. Other cell types including TH1/17, T lymphocytes, neutrophils, and dendritic cells are also speculated to modify their metabolic phenotype in obesity.
In the AT, intrinsic immunometabolism has been primarily studied in macrophages (51). In a resting state, lean ATMs have relatively low metabolic activity (52, 53). After adipose expansion, ATMs increase OX PHOS, typically associated with M2 polarization, and glycolysis, typically associated with M1 polarization. This unique bioenergetic state supports the data suggesting that ATMs are not M1 or M2 but have a unique “metabolically activated” polarization (13).
Whereas cytokine production in lean ATMs is supported by fatty acid, glucose, and glutamine utilization, cytokine production in obese ATMs is supported by glycolysis (52). Glycolytic macrophages often express high levels of the glucose transporter Glut1, and thus, Glut1 myeloid deficiency was hypothesized to improve fatty acid uptake and oxidation in ATMs (54). However, there were no differences in body weight, adiposity, or fasting blood glucose in myeloid Glut1-deficient mice compared with controls after high-fat diet (HFD) feeding. There were, however, unstable atherosclerotic lesions and defective phagocytosis in Ldlr−/− mice lacking myeloid Glut1, suggesting several metabolic functions of Glut1 in macrophages to maintain tissue homeostasis.
Together, the current body of literature suggests that signals in the AT environment (lipids, glucose, and hormones) contribute not only to MMe polarization but also to the unique bioenergetics of ATMs in obesity. However, there is still much to understand about how we can target ATM metabolism in obesity. Microarray analysis of ATMs has highlighted changes in gene expression across many metabolic pathways, including one-carbon metabolism, amino acid metabolism, amino/nucleotide sugar metabolism, fructose/mannose metabolism, glycerolipid metabolism, and sphingolipid metabolism (52). Moreover, obese AT is an environment with elevated concentrations of lactate and iron, which likely drive their own metabolic programs in adipose immune cells (55). Thus, more research is warranted to understand the contribution of each metabolic pathway to macrophage function and to determine whether one or more of the metabolic pathways can be targeted to improve ATM function in obesity.
Other innate immune cells in AT
Other populations of innate immune cells also reside in AT. These include eosinophils, mast cells, neutrophils, dendritic cells (DCs), and innate lymphoid cells (ILCs). Whether eosinophils and mast cells contribute to AT homeostasis or dysfunction remains controversial (56–58) (Fig. 3). Thus, we focus our attention here on DCs and ILCs.
Fig. 3. Innate and adaptive immune cell response to weight cycling.
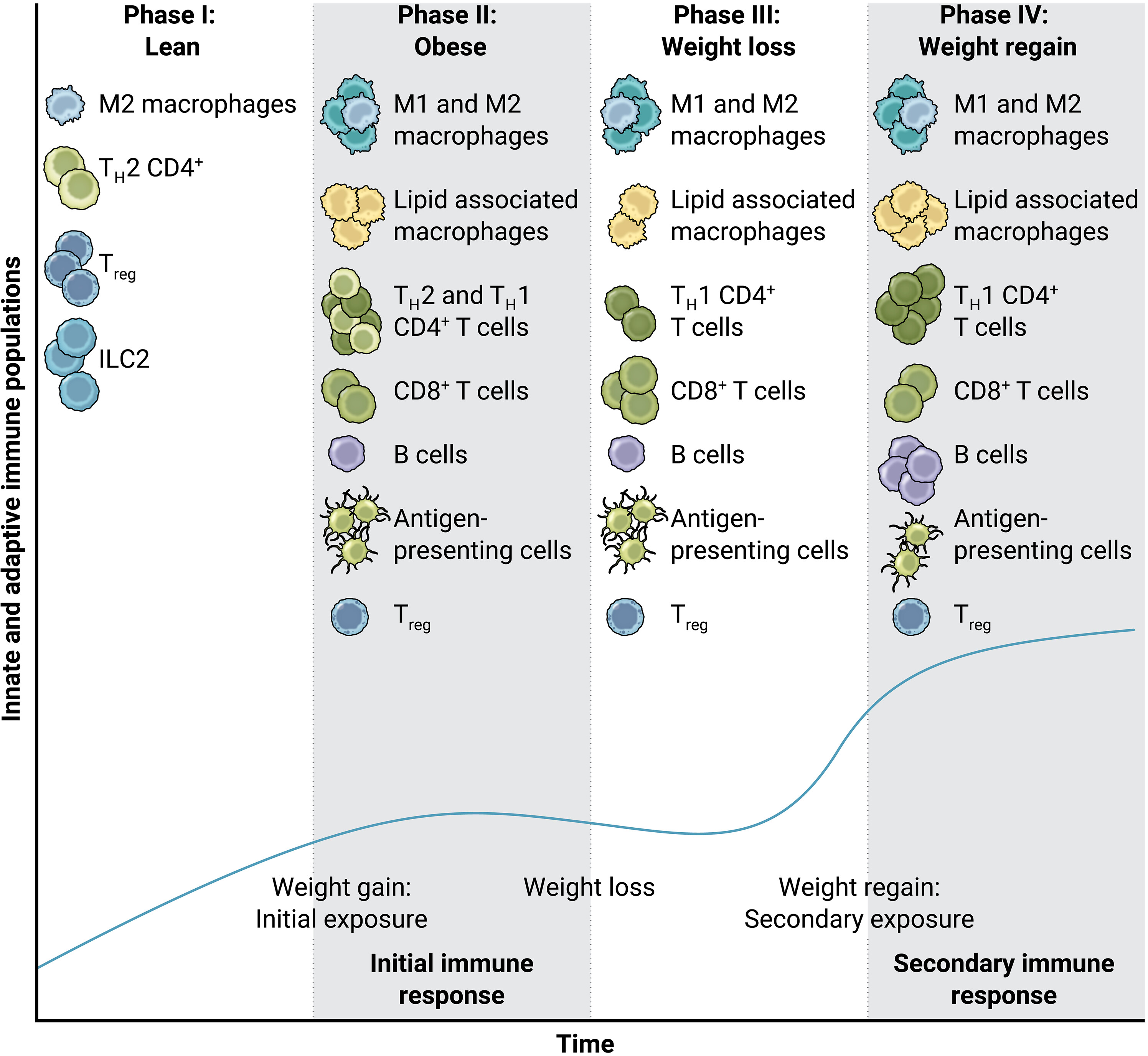
Lean AT is often composed of type 2 immune cells (for example, M2-like macrophages, TH2 CD4+ T cells, and Tregs). In obesity, both innate and adaptive immune cell recruitment result in greater type 1 immune cell populations, resulting in the influx of M1-like macrophages, TH1 CD4+ T cells, and CD8+ T cells. Conversely, the Treg population is diminished in obesity. In the event of weight loss, inflammatory macrophages and memory T cells remain intact, whereas Treg populations do not increase back to their lean proportion. Therefore, in the event of weight regain (as in weight cycling), effector T cell populations interact with antigen-presenting cells that had accumulated from the previous obesity cycle, resulting in even greater adaptive immune cell populations (including CD8+ T cells, CD4+ T cells, and B cells). The dark blue curve on the graph indicates the innate and adaptive immune cell populations shown per bout of weight gain, loss, and regain.
Dendritic cells
DCs in AT have been consistently grouped within macrophage clusters because of overlapping CD11c+ expression despite their functional diversity (59). Specifically, DCs have been identified in humans as CD11c+ CD1c+ (60) and in mice as CD11c+ CD64− (59) or CD11chigh F4/80low (60). Furthermore, DC subpopulations can be classified into conventional DC (cDC) subsets, comprising cDC1s and cDC2s. Previous reports have demonstrated that DC recruitment into AT is mediated by CCR2 and CCR7 and is increased in HFD-fed mice (59) and positively associated with BMI in humans (60). Specific functions for DCs are still incompletely understood, but they are proposed to serve as antigen-presenting cells to signal CD4+ T cell activation. In addition, DCs induce T helper 17 (TH17) differentiation in mice and associate with TH17 T cells in humans (60).
Innate lymphoid cells
ILCs are proposed to be regulators and responders to the inflammatory response to obesity. ILCs can be clustered by group 1 (ILC1), group 2 (ILC2), and group 3 (ILC3) based on cell surface markers and cytokine release (61). Consistent with innate cells, ILCs respond to cytokines produced by local macrophages and DCs and are absent of antigen receptors (62, 63). ILC1 activation is induced by IL-12 cytokines, by which IL-12R/STAT4 signaling mediates ILC1 activation and stimulates interferon-γ (IFN-γ) release (62, 63). In mice, ILC1s are largely tissue-resident whereas natural killer (NK) cells are recruited from bone marrow precursors and are present in circulation (64). Activation of ILC1 and NK cells by local cytokines serves as a feed-forward response to increase a pro-inflammatory environment necessary to induce immune responses to obesity. ILC2s are an innate immune cell population associated with type 2 immunity and maintained by IL-33 (65). ILC2s are attenuated in HFD-fed mice and humans with obesity (65). Interestingly, ILC-2s promote adipocyte precursor differentiation toward a beiging phenotype in mice, suggesting a potential role for type 2 ILCs to regulate tissue homeostasis by environmental cues triggered by IL-33 (65, 66).
Summary and future directions of innate AT immunity
Together, extensive literature now shows that macrophages and other innate immune cells play a key role in the pathogenesis of obesity and inflammation. Environmental cues that trigger the recruitment and differentiation of innate immune cells are exacerbated by obesity, and the polarization of macrophages into distinct phenotypes serves to modify AT function. Understanding how obesity alters the molecular and functional characteristics of macrophage subpopulations will continue to be critical for developing therapeutic strategies targeting inflammation.
ADAPTIVE IMMUNE CELL POPULATIONS IN OBESITY
T lymphocytes
T cells comprise the largest proportion of adaptive immune cells in AT, with CD4+ and CD8+ T cells predominating. They each play a unique role in AT homeostasis in the lean condition and in AT inflammation with obesity.
CD4+ T cells
CD4+ T cells can be divided into two broad categories: pro-inflammatory (TH1 and TH17) and anti-inflammatory [TH2 and Treg (regulatory T cells)]. In lean AT, the CD4+ to CD8+ ratio is 3:1; however, obesity increases the CD8+ T cell population (67). This in turn increases the ratio of inflammatory TH1 to anti-inflammatory TH2 CD4+ T cells. In addition, the Treg population, important for maintaining homeostasis within the AT, is reduced by up to 70% in HFD-fed mice (68). Forced reduction in the Treg population markedly increases the macrophage and effector T cell population within AT (68). This shift toward a more pro-inflammatory population is most prevalent in visceral AT and contributes to obesity-associated AT inflammation. In models of diet-induced obesity, Tregs are reduced and concentrated in the crown-like structures surrounding adipocytes, indicating their role in the maintenance of systemic metabolism (78). Tregs are abundant in lean AT and express genes involved in lipid metabolism (CD36, DGAT1, and LDLR) driven by peroxisome proliferator–activated receptor γ (PPARγ) (79). Deletion of PPARγ specifically in Tregs increases the infiltration of pro-inflammatory macrophages and monocytes after HFD feeding and abolishes the insulin-sensitizing effect of pioglitazone, a US Food and Drug Administration (FDA)–approved medication for type 2 diabetes.
CD8+ T cells
Diet-induced obesity evokes an early increase in CD8+ effector T cells and subsequent macrophage infiltration into visceral AT depots in mice (67) and subcutaneous AT in humans (69). CD8+ T cells interact with local macrophages to elicit macrophage differentiation, activation, and migration, inciting inflammation that contributes to insulin resistance. CD8+ T cell infiltration precedes the reduction in the Treg population. CD8 depletion studies resulted in an inhibition of inflammatory macrophage infiltration and the downstream inflammatory cascade typically observed in obese AT (69). An expanding area of research is related to the observation that T cell exhaustion, most commonly associated with the tumor microenvironment and viral infections, also increases within obese AT in mice and humans (20, 70, 71). Specifically, these exhausted CD8+ T cells are effector memory T cells, which express PD1 and TIGIT at the surface protein level and Pdcd1, Tox, Entpd1, Tigit, and Lag3 at the gene level. The relevance of T cell exhaustion to AT inflammation and function in obesity is not fully known.
Gamma-delta (γδ) T cells
γδ T cells are a class of tissue-resident lymphocytes present in both mice and humans with primary roles in defending against pathogens in epithelial tissues such as the skin, gut, and lungs (72). γδ T cells are also present in AT, comprising ~4 to 11% of CD3+ T cells in mice (73). γδ T cells are unique in that they respond to antigen presentation similar to conventional T cells but also respond to cytokines, bridging a gap between innate and adaptive immunity. In mice, γδ T cells in AT have been shown to increase in obesity and are noted to secrete cytokines IL-17 (73, 74) and TNF-α (75). IL-17 released by γδ T cells may limit adipogenesis (73), increase M1-like (CD11c+ CD206−) macrophages (75), and limit glucose tolerance (73, 75). In humans, circulating γδ T cells are decreased with obesity (76), yet omental AT γδ T cells are increased (77). Future studies profiling γδ T cells in obesity should identify innate signals responsible for γδ T cell activation and downstream T cell activation by γδ T cells.
T cell clonality in obesity
One hallmark of adaptive immunity is the clonal expansion of specific T and B cell populations after exposure to specific antigens. Remarkably, restricted T cell receptor (TCR) sequences have been identified on CD4+ and CD8+ T cells isolated from AT in mice (37, 69). What is even more interesting is that with obesity, Treg clonal expansion decreases (68), whereas CD8+ clonal expansion increases. Tregs isolated from lean visceral AT have a restricted TCR sequence, suggesting that they are responding to a select population of local antigens. Regarding CD8+ T cells in obese AT, little is known about potential antigens. In our study, the amino acid sequences corresponding to the complementarity determining region 3 of clonal TCRs were characterized as positively charged and nonpolar (80). Isolevuglandins are negatively charged and detected in high amounts within ATMs, suggesting that they may be one component of the AT microenvironment capable of stimulating CD8+ T cell proliferation (80). However, the identification of specific antigens driving CD8+ T cell clonal expansion is still needed. Furthermore, the mechanisms regulating CD8+ T cell expansion in human obesity remain unclear.
B lymphocytes
In contrast to regulatory B-1 cells, B-2 cells in AT create an inflamed environment, secreting more pro-inflammatory cytokines, such as IL-6 and IFN-γ, at the expense of anti-inflammatory cytokines, specifically IL-10 (81). Excess adiposity results in the accumulation of B-2 cells in visceral AT, worsening inflammation of the tissue. In pan B cell-null mice, adipocyte hypertrophy was mitigated, macrophage infiltration was substantially decreased, and glucose tolerance was improved (81). The hypertrophic obesity and worsened glucose tolerance seen in B cell null animals are influenced by the ability of the B cells to regulate T cells in the AT. In T cell activation studies, cells from B cell–deficient mice had reduced pro-inflammatory cytokine production. The role of B cell and T cell interactions in AT inflammation was further confirmed when RAG1−/− mice were reconstituted with B cells from 16 weeks in HFD-fed mice. Despite the reconstitution, fasting glucose, insulin, and glucose tolerance were not worsened, suggesting that B cells require the presence of T cells for their impact on metabolic function (82).
Summary and future directions of adaptive AT immunity
The contribution of adaptive immune cells to AT homeostasis and dysfunction remains an intriguing area of research. Outstanding questions include the following: Why do T cells change in degree of clonality with obesity (with decreased Tregs and increased CD4+ and CD8+ cells)? Are there specific antigens that can be targeted to reduce AT inflammation? And how are T cell exhaustion and B cell antibody production related to AT function? Furthermore, whereas the bioenergetics of T cells in the context of cancer is well documented, whether the AT milieu changes intrinsic T cell metabolism is not known. For example, although Tregs are well primed to metabolize lipids for their regulatory functions in homeostasis, there is still much to learn about the metabolism of other adipose immune cell types (T cells and B cells, and their subpopulations) in lean and obese AT.
IMMUNE RESPONSE TO WEIGHT LOSS AND REGAIN
Weight cycling is loosely defined as the repeated gain and loss of body mass. Most of the literature on weight cycling was published several decades ago and sought to determine whether fluctuations in body weight have a negative impact on food efficiency and energy requirements. More recent studies have revealed an association between weight cycling and increased risk of insulin resistance and cardiovascular disease (83, 84). One of the first studies addressing this issue reported a 10% increase in the 25-year risk of coronary death in men who weight-cycled compared with those who maintained a stable weight (83). In addition, multiple recent publications suggest that weight regain after substantial weight loss has negative consequences on metabolic health in humans (84, 85). Emerging studies provide evidence that AT immune memory may be involved in the metabolic consequences of weight cycling.
Adaptive immune memory to metabolic signals
To interrogate the cell types and molecular contributors to weight cycling, researchers have turned to mouse models that can recapitulate many of the adverse metabolic sequelae of weight cycling. In mice, prior obesity results in weight regain at a faster rate, greater fat accumulation, and an adaptive immune response characterized by type I immune cells (86–88). CD4+ T cells have been thoroughly studied in mouse models of weight cycling (82). Rag1−/− (lacking T and B cells), TCRβ−/− (lacking CD4 and CD8 T cells), and H2A−/− (lacking CD4 T cells) mice were protected from accelerated weight regain (87). Interestingly, when reconstituted with CD4+ T cells from wild-type mice with a history of obesity, both Rag1−/− and TCRβ−/− mice gained weight more quickly than those that received CD4+ T cells from lean controls (87). Work from our laboratory showed an increase in CD8+ effector memory T cells in AT of weight-cycled mice (20, 88). Unanswered questions remain regarding how extreme the weight loss and regain must be, whether the numberof weight cycles matters, whether T cell clonality is different in weight cycling versus maintained obesity, and the degree to which the immune landscape in human AT is affected by weight cycling. These findings suggest that adaptive immune cell populations within AT are highly plastic and that further work should be conducted to better characterize these populations in AT and within metabolically active tissues at each stage of weight cycling.
The reason for the immune changes induced by weight cycling can be garnered by assessing the AT immune landscape after weight loss. Surprisingly, AT of animals that have lost weight does not fully revert to a lean anti-inflammatory phenotype (Fig. 3) (20, 89, 90). In addition, recent work indicates that the highest proportion of T and B cells occurs during the weight-loss phase of weight cycling (20). These data suggest that although weight loss improves systemic metabolic processes, the AT immune system may “remember” previous obesity and become overly activated upon weight regain.
Innate immune memory to metabolic signals
Although immunological memory is historically considered characteristic of only adaptive immune cells, recent work suggests that innate immune cells can also develop nonspecific memory (91). Activation of PRRs by β-glucan, the Mycobacterium tuberculosis vaccine (BCG), and even oxidized low-density lipoprotein (LDL) can drive metabolic activation and persistent epigenetic remodeling to prime cells for a more robust response after second stimuli. Specifically, innate stimuli can drive glycolytic ATP production so that metabolic intermediates like acetyl-CoA can help acetylate histones (92). These histone modifications can hold open regions of chromatin at glycolytic and inflammatory genes for faster transcription upon subsequent activation.
Although this response may be evolutionarily protective in responses to infection, it appears detrimental in chronic disease. We have recently shown that bone marrow–derived macrophages can develop enhanced secondary activation after priming with AT-conditioned media or palmitate, which is dependent on TLR4 and suppressed with metformin or pan methyltransferase inhibition. In addition, ATMs from previously obese mice secrete more inflammatory cytokines after ex vivo activation with LPS and are more inflammatory after weight regain (93). Similarly, others have shown that a ketogenic diet and palmitate injections worsen systemic LPS-induced inflammation via ceramide production (94) and stearic acid (via TLR4) and that previous obesity induces chromatin remodeling in ATMs, which predisposes mice to worsened macular degeneration (95). Other innate immune stimuli have shown a role in mevalonate and changes in genes related to fatty acid metabolism and OX PHOS (92). Thus, ATMs appear uniquely situated to develop innate immune memory in response to activation and uptake of lipids, and future research should aim to understand how ATM memory affects both metabolic and immunological diseases.
Together, these data suggest that multiple arms of the immune system remember previous obesity, and clinical approaches to weight loss should strive for long-term efficacy. Moreover, further understanding of the signals that drive and maintain immunological memory to obesity may provide additional therapeutic targets to mitigate diseases associated with weight regain.
CLINICAL APPROACHES TARGETING OBESITY AND METAFLAMMATION IN HUMANS
Trials with anti-inflammatory therapies
Although chronic inflammation has been strongly linked to metabolic diseases such as atherosclerotic cardiovascular disease and type 2 diabetes, compared with lipid- and glucose-lowering drugs, anti-inflammatory therapies are nascent with regard to clinical application. One of the first studies focused on salsalate and glycemic control in patients with type 2 diabetes (96). At three different doses, salsalate treatment resulted in significant decreases in HbA1c concentrations and other markers of glycemic control. More recently, the CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) trial, with ~10,000 patients, provided proof of concept that targeting an inflammatory cytokine (in this case, IL-1β) could reduce the incidence of a composite primary end-point of myocardial infarction, stroke, and cardiovascular death (97). Interestingly, cancer mortality was also decreased in the canakinumab-treated groups. However, the incidence of serious infection was seen in one out of every 750 patients. This does not diminish enthusiasm for the idea of using anti-inflammatory agents to treat cardiometabolic disease; however, other approaches, such as improving pro-resolving processes to limit the acute inflammatory response, are now considered to be a better approach (98).
Lifestyle interventions for obesity
Weight loss, either via lifestyle modification, medication, or surgery, is the primary recommended intervention for obesity and its associated cardiometabolic complications. Moderate amounts of weight loss (~5% body weight) result in improved metabolic outcomes in humans (99). Clinical trials have found that moderate weight loss does not affect AT inflammation in adults with obesity despite improved insulin sensitivity. Rather, more progressive weight loss (~11 to 16%) sufficiently reduced both systemic and AT inflammation (99). Therefore, we speculate that the immune response to weight loss in humans is a biphasic response, where macrophage subpopulations are increased in AT to undergo tissue remodeling and buffer lipid accumulation derived by lipolysis (100) but may be reduced with sustained weight loss. However, given the strong data showing sustained inflammatory immune cells in AT of mice that have lost weight, more extensive studies are needed in humans. Here, we highlight pharmacological and surgical approaches for weight loss and discuss their impact on AT and systemic inflammation.
Dietary modifications
The effects of negative energy balance on pro-inflammatory markers have been studied clinically in individuals with obesity. In the clinical weight management setting, patients are advised to focus on unprocessed foods with an emphasis on whole grains, lean protein, fresh fruits and vegetables, and limited refined sugars and flour. One study suggested that a reduction in high-sensitivity C reactive protein (CRP) and IL-6 occurs when energy homeostasis is achieved after a decline in fat mass (101). However, antioxidant-rich foods containing carotenoids, mixed tocopherols, vitamin C, or selenium have not consistently been shown to reduce inflammatory markers (102). Oftentimes, patients choose a dairy-free diet as a method to induce weight loss despite a lack of evidence to suggest increases in weight, fat, visceral AT, and subcutaneous ATM number, subcutaneous AT inflammatory gene expression, and circulating cytokines from dairy foods (103). The Mediterranean diet on the other hand, characterized by a high consumption of legumes, nuts, seeds, and olive oil, and a moderate consumption of protein, has been shown to reduce inflammation (104).
Physical activity
The American Heart Association recommends at least 150 min/week of exercise to promote and maintain health. In the clinical weight management setting, patients are counseled on physical activity, both structured and unstructured, consistent with standard of care guidelines. In one study, routine exercise over 6 and 12 months resulted in decreased levels of leptin, adiponectin, resistin, homocysteine, and IL-6, particularly in patients who were overweight (BMI, ≥25 and <30 kg/m2) (105). Intensive exercise focused on structure and consistency may be necessary to produce a hypo-inflammatory state, especially in persons with obesity (106).
Pharmacological intervention
Pharmacologic interventions directly aimed at reducing inflammatory cytokines have demonstrated success as measured by reductions in CRP and IL-6 in the circulation and a resultant decrease in cardiovascular events (97), yet none is standard of care in obesity at this time. Pharmacological interventions aimed to mitigate the metabolic effects of obesity, specifically type 2 diabetes, have focused on pathways involved in insulin action. Serendipitously, these have sometimes resulted in the added benefit of weight loss. These pathways include incretins and sodium-glucose co-transporter 2 (SGLT2) inhibitors, which we will highlight below.
Glucagon-like peptide 1 receptor agonists
The incretin effect describes the phenomenon that oral glucose increases pancreatic insulin secretion to a greater degree than intravenous glucose. This is due to the secretion of incretin hormones glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1) from the gut. GIPand GLP-1 act directly on β cells to promote insulin secretion. GLP-1 is lower in individuals with obesity and increases after bariatric surgery (107). GLP-1 receptor agonists (GLP-1RAs) decrease blood glucose, cause weight loss, and decrease composite death from cardiovascular causes, nonfatal stroke, and myocardial infarction in patients with diabetes (108, 109).
In animal models, GLP-1RAs have been shown to decrease inflammation. In obese animals, treatment with GLP-1RAs decreases AT inflammatory cytokines and causes a switch from a pro-inflammatory to an anti-inflammatory macrophage phenotype (110, 111). GLP-1RA treatment of nonobese diabetic mice with new-onset diabetes increases Tregs (112). Furthermore, GLP-1RA treatment reduces atherosclerosis in ApoE and LDLr knockout animals by inhibiting inflammatory pathways in the vasculature (113). Despite the therapeutic potential for GLP-1RAs to modify inflammation in animals and limit cardiovascular outcomes in humans, their effects on AT immune profile in humans are still unconfirmed.
Human studies have provided contradictory results. In two studies of adults with obesity and type 2 diabetes, GLP-1RAs increased circulating adiponectin concentration and decreased circulating concentrations of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6; soluble CD163; and M1-like ATMs (114, 115). However, a different group found that GLP-1RAs increased circulating MCP-1, expression of TNF-α, MCP-1, and markers of extracellular matrix deposition in AT (116). Despite the promising effects of GLP-1RAs to improve cardiometabolic outcomes and glucose tolerance, variations in the length of these clinical trials range from 8 weeks to 1 year and are typically in patients with type 2 diabetes (Table 1). Therefore, the effect of GLP-1RAs on obesity independent of metabolic disease onset is unknown. In addition, more studies rigorously profiling the impact of GLP1-RAs on AT immune cell phenotype are warranted. Last, controlled trials comparing the effect of GLP-1RAs independent of weight loss or in combination with lifestyle interventions could identify additional therapeutic potential.
Table 1. Anti-inflammatory effects of GLP-1RAs and SGLT2is in human clinical studies.
Direction of statistically significant effects shown for the drug of interest (GLP-1RA or SGLT2i). CRP, c-reactive protein; HbA1c, hemoglobin A1c; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; ND, not done; RCT, randomized controlled trial; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TNF, tumor necrosis factor.
| Population characteristics | Study design | n | CRP | Cytokines | Adipokines | Notes | |
|---|---|---|---|---|---|---|---|
| GLP-1 receptor agonists | |||||||
| Bunck et al. (140) | T2DM on metformin | RCT GLP-1RA exenatide versus insulin for 1 year | 36 exenatide | ↓ | ↔IL-6 and MCP-1 | ↑Adiponectin | Change in weight correlated with CRP and leptin |
| 33 insulin | ↓Leptin | ||||||
| Derosa et al. (141) | Uncontrolled T2DM on metformin | RCT GLP-1RA exenatide versus sulfonylurea for up to 12 months | 63 exenatide | ↓ | ND | ND | Similar degree of HbA1c lowering between treatments |
| 65 sulfonylurea | |||||||
| Chaudhuri et al. (115) | Obesity and T2DM | RCT of GLP-1RA exenatide versus placebo for up to 12 weeks | 12 exenatide | ND | ↓TNFA, IL1B, TLR2, and TLR4 expression in PBMCs | ND | Decrease in SAA |
| 12 placebo | ↓Circulating MCP-1 and IL-6 | ||||||
| Hogan et al. (114) | T2DM | Observational study of add-on GLP-1RA therapy for 8 weeks | 10 | ND | ↓sCD163, TNF-α, IL-1β, and IL-6 | ↑Adiponectin | Inflammatory changes independent of weight loss or change in glycemia |
| Mazidi et al. (142) | Variable | Meta-analysis of RCTs for effects of GLP-1RA on CRP | 7 trials | ↓ | ND | ND | |
| Pastel et al. (116) | T2DM | RCT GLP-1RA liraglutide versus diet for 4 months | 22 liraglutide | ND | ↑TNFA, MCP-1, and TGFB expression in adipose | ND | |
| 8 diet | ↑MCP-1 | ||||||
| Anholm et al. (143) | Newly diagnosed T2DM with coronary artery disease on stable statin therapy | RCT crossover of GLP-1RA liraglutide + metformin versus placebo + metformin for 12 weeks | 39 | ↓ | ↔TNF-α | ND | |
| Savchenko et al. (144) | T2DM on metformin and glimepiride | Observational study of add-on GLP-1RA liraglutide for 6 weeks | 15 | ND | ↓TNFA, TLR2, and TLR4 expression in PBMCs | ND | |
| ↔IL-2 and NOD-1 | |||||||
| ↑SIRT1 expression | |||||||
| SGLT2 inhibitors | |||||||
| Matsumura et al. (128) | T2DM on insulin | Observational study of add-on SGLT2i canagliflozin for 3 days | 15 | ND | ↔TNF-α | ↑Adiponectin | ↓Markers of oxidative stress |
| Garvey et al. (127) | T2DM inadequately controlled on metformin | RCT of SGLT2i canagliflozin versus sulfonylurea for 52 weeks | 100 canagliflozin | ↔ | ↓IL-6 | ↓Leptin | |
| 100 sulfonylurea | ↑TNF-α | ↑Adiponectin | |||||
| Hattori (129) | T2DM | RCT SGLT2i empagliflozin versus placebo for 1 year | 51 empagliflozin | ↓ | ND | ND | |
| 51 placebo | |||||||
| Sato et al. (145) | T2DM with coronary artery disease | RCT SGLT2i dapagliflozin versus conventional treatment for 6 months | 20 dapagliflozin | ND | ↓TNF-α | ND | |
| 20 conventional treatment | |||||||
| Heerspink et al. (146) | T2DM and albuminuria | RCT of SGLT2i canagliflozin versus sulfonylurea for up to 2 years | 207 canagliflozin | ND | ↓TNFR1 and IL-6 | ND | ↓MMP-7 and fibronectin 1 |
| 89 sulfonylurea | ↔CCL2, CCL5 | ||||||
| Iannantuoni et al. (147) | T2DM | Observational study of SGLT2i empagliflozin for 24 weeks | 15 | ↓ | ↑IL-10 | ND | Reduction in leukocyte superoxide production |
| Latva-Rasku et al. (148) | T2DM | RCT SGLT2i dapagliflozin versus placebo for 8 weeks | 15 dapagliflozin | ND | ↓IL-6 | ND | Decreased visceral adipose tissue |
| 16 placebo | ↔TNF-α and MCP-1 | ||||||
| Sezai et al. (149) | T2DM and heart failure | SGLT2i canagliflozin for 12 months | 35 | ↓ | ND | ND | |
| Bray et al. (119) | T2DM ± secondary characteristics | Systematic review of effects of SGLT2 inhibition on biomarkers of inflammation | 23 studies | ↓ | ↓IL-6 and TNF-α | ↑Adiponectin | ↓markers of oxidative stress |
| Kim et al. (125) | T2DM and high cardiovascular risk | RCT of SGLT2i empagliflozin versus sulfonylurea effects on blood-derived macrophages ex vivo | 29 empagliflozin | ND | ↓IL-1β and TNF-α from macrophages ex vivo | ND | |
| 32 sulfonylurea | |||||||
| Nishimiya et al. (150) | T2DM and nonalcoholic fatty liver disease | SGLT2i canagliflozin for 6 months | 9 | ↓ | ↔TNF-α | ↔Adiponectin and leptin | |
| Sposito et al. (151) | T2DM and increased carotid intima-media thickness | RCT of SGLT2i dapagliflozin versus sulfonylurea for 12 weeks | 48 dapagliflozin | ↔ | ↔IL-2, IL-6, and TNF-α | ND | |
| 49 sulfonylurea | |||||||
Sodium-glucose cotransporter 2 inhibitors
Sodium-glucose cotransporter 2 (SGLT2) is responsible for the reabsorption of 90 to 97% of the filtered glucose in the kidneys. SGLT2 inhibitors cause glucosuria, reduce HbA1c, and induce weight loss of 2 to 3 kg (117). Large cardiovascular outcome trials have also demonstrated that SGLT2 inhibitors significantly decrease major adverse cardiovascular events in patients with type 2 diabetes and with established cardiovascular disease and are effective at primary and secondary prevention of heart failure and chronic kidney disease (118).
Similar to GLP-1RAs, SGLT2 inhibitors also decrease inflammation in animals and may decrease inflammation in humans, as reviewed recently (119). SGLT2 inhibitors decrease the production of pro-inflammatory cytokines (IL-6 and TNF-α) in vitro in mouse and human cell lines and in vivo after LPS stimulation (120). In diabetic, atherosclerosis-prone ApoE knockout mice, SGLT2 inhibitors decrease aortic pro-inflammatory gene expression independently of weight loss (121) and decrease NLRP3 inflammasome activation (122, 123). Last, SGLT2 inhibitors skew ATMs from pro-inflammatory to anti-inflammatory phenotypes in a mouse model of diet-induced obesity (124). In humans, SGLT2 inhibitors decrease NLRP3 inflammasome activation in blood-derived macrophages after 30 days of in vivo treatment (125) and in human aortic smooth muscle cells after in vitro IL-17A stimulation (126). In addition, SGLT2 inhibitors increase circulating adiponectin in multiple studies (119, 127, 128). Add-on SGLT2 inhibitors in people with type 2 diabetes decrease CRP, which correlates with improvements in insulin resistance (129). Controlled clinical studies identifying the effect of SGLT2 inhibitors on tissue-specific immune cell populations (such as those in AT) are still lacking, as well as the effect of therapies in combination with or independent of weight loss. Despite the promising impact of SGLT2 inhibitors in a diabetic population, the effect in nonclinical populations with obesity is as of yet unknown.
Bariatric surgery
Adipocyte-derived inflammatory factors are elevated in individuals with obesity and contribute to the metabolic and vascular derangements common in obesity. Given the well-established link between adiposity and a pro-inflammatory state, it is plausible that marked weight loss seen after bariatric surgery would yield a reduction in inflammation. Surprisingly, studies show variable changes in inflammatory markers postoperatively [reviewed in (130)].
Diabetes remission occurs in more than 75% of patients 2 years after Roux-en-Y gastric bypass (RYGB), and oftentimes, the improvement in glycemic control is seen immediately postoperatively, before significant weight loss occurs (131). There is debate about the exact mechanism underlying this phenomenon. We know that glycemic improvement is partially attributed to increased incretin secretion, but one other theory is that inflammatory cytokines are markedly reduced after surgery. This could also account for the reduction in cardiovascular complications of obesity after bariatric surgery as well (132).
Although inflammation may be difficult to quantify, there are key inflammatory markers that have been studied in the post-RYGB population, with the most cited being IL-6, TNF-α, CRP, and serum amyloid A (SAA). IL-6 is a cytokine that is implicated in obesity-related metabolic decline. Most RYGB studies agree that this cytokine can increase in the acute postoperative period but show a long-term downtrend over 1 year postoperatively (130). One study showed a different triphasic pattern of interleukin response: a sharp decrease at 3 months, then a steady rise to presurgical values at 6 months, and at 12 months a downtrend that depended on BMI and metabolic status change (133). Smaller studies looking at changes in TNF-α post-RYGB do not show consistent trends, where TNF-α has been shown to increase 3 months after surgery and return to presurgical baseline (134). However, a recent meta-analysis of 116 studies evaluating TNF-α response 12 months after surgery showed a significant decrease compared with baseline (135). CRP is an acute-phase liver protein strongly associated with inflammatory states and cardiovascular disease. Studies show a consistent reduction in CRP after various types of bariatric surgical procedures, including RYGB, vertical sleeve gastrectomy, and laparoscopic adjustable gastric banding. CRP was reduced by 82% at 1 year postoperative in a study of 66 participants with obesity who underwent RYGB, and the reduction was higher in those without insulin resistance (136). Furthermore, a longitudinal analysis using high-sensitivity CRP testing showed a progressive decline in this marker at 3, 6, and 12 months after RYGB, and this correlated with BMI, insulin, and HOMA-IR (134). SAA is another acute-phase reactant produced in the liver in response to inflammation and has shown positive correlations with atherosclerosis. The aforementioned studies show a consistent reduction in SAA as well across various bariatric surgical procedures, although a decrease in SAA is not as pronounced as a reduction in CRP (136).
In summary, the evidence confirms a reduction in the acute-phase reactants CRP and SAA after bariatric surgery, but changes in inflammatory cytokines are inconsistent. This may be partially due to drastic changes in calorie intake postoperatively and perhaps macronutrient composition as well. Additional studies are needed to assess the long-term impact of bariatric surgery on inflammatory markers.
Emerging therapies
Whereas older-generation anti-obesity pharmacotherapies can achieve approximately 3 to 7% weight loss from baseline, more recent therapeutics have shown more than 15% weight loss. These include incretin therapies such as semaglutide and tirzepatide, both of which have proven safety and tolerability. Semaglutide (2.4 mg) is now FDA-approved for chronic weight management, and tirzepatide is expected to be FDA-approved for obesity in light of the recent SURMOUNT-1 study showing 22.5% weight loss (137). Although 3 to 5% weight loss signifies clinically meaningful weight loss, >10 to 15% weight loss can revert or improve more serious inflammatory diseases such as nonalcoholic fatty liver and cardiovascular disease. In addition, the cardiovascular risk profile of semaglutide in patients with type 2 diabetes has been shown to be noninferior to placebo (138, 139). Clinically, for patients with metabolic derangements and worsening inflammatory processes, these newer therapies are highly preferred over the older-generation compounds. Presently, studies are ongoing, with SELECT (Semaglutide Effects on Cardiovascular Outcomes in People with Overweight or Obesity) being the first cardiovascular clinical trial to measure the superiority of major adverse cardiovascular events reduction for an anti-obesity medication, semaglutide. As more novel obesity agents gain FDA approval, insights into their mechanisms of action and effect on inflammation can enhance our clinical application of these drugs to target not only severe obesity but also weight-related complications.
CONCLUSION
Understanding the direct and indirect effects of obesity-induced metaflammation will be important to further understand how obesity accelerates cardiometabolic disease. Despite tremendous efforts to identify cellular mediators contributing to metabolic dysfunction in obesity, pharmacological approaches targeting a single mechanism remain difficult because of the diversity of innate and adaptive immune cells altered in obesity. Technological advancements and availability of multiomics platforms will continue to improve our understanding of immune populations that are either associated with or directly causative of metabolic dysfunction and to propose new therapeutic targets. Researchers should continue to address whether obesity-induced modifications in immune cell populations are causal to poor metabolic health outcomes. Similarly, an important question for pharmacological research in obesity is to identify whether therapies directly targeting weight loss additionally mitigate inflammation driven by obesity or whether the weight-loss effects of obesity drive the attenuated inflammatory phenotype independent of pharmacological intervention.
Acknowledgments
Funding:
Authors on this work are funded by the Department of Veterans Affairs Research Career Scientist IK6 BX005649 (to A.H.H.), the National Institutes of Diabetes and Digestive and Kidney Diseases R01DK121520 (to A.H.H.), the NIH Molecular Endocrinology Training Program T32DK007563 (to J.N.G. and M.W.S.), the American Heart Association Postdoctoral Fellowship 20POST35120547 (to H.L.C.), and the National Center for Advancing Translational Sciences KL2TR002245 (to M.M.).
Footnotes
Competing interests: G.S. reports advisory fees from Rhythm Pharmaceuticals, Novo Nordisk, and Eli Lilly outside the submitted work. The other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Hales CM, Carroll MD, Fryar CD, Ogden CL, Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief, 1–8 (2020). [PubMed] [Google Scholar]
- 2.The GBD 2015 Obesity Collaborators, Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heymsfield SB, Wadden TA, Mechanisms, pathophysiology, and management of obesity. N. Engl. J. Med. 376, 254–266 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Lumeng CN, Saltiel AR, Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 121, 2111–2117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu H, Ballantyne CM, Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Invest. 127, 43–54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ, Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat. Rev. Endocrinol. 17, 455–467 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jais A, Brüning JC, Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Invest. 127, 24–32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koyama Y, Brenner DA, Liver inflammation and fibrosis. J. Clin. Invest. 127, 55–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park HS, Park JY, Yu R, Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-α and IL-6. Diabetes Res. Clin. Pract. 69, 29–35 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE, Harrison LC, Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59, 1648–1656 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muir LA, Cho KW, Geletka LM, Baker NA, Flesher CG, Ehlers AP, Kaciroti N, Lindsly S, Ronquist S, Rajapakse I, O’Rourke RW, Lumeng CN, Human CD206+ macrophages associate with diabetes and adipose tissue lymphoid clusters. JCI Insight 7, e146563 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, Peris E, Cui C, Hoffman A, Zhou G, Fernandez S, Zhai L, Hall BA, Haka AS, Shah AM, Reardon CA, Brady MJ, Rhodes CJ, Maxfield FR, Becker L, Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep. 20, 3149–3161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L, Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 20, 614–625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li P, Lu M, Nguyen MTA, Bae EJ, Chapman J, Feng D, Hawkins M, Pessin JE, Sears DD, Nguyen AK, Amidi A, Watkins SM, Nguyen U, Olefsky JM, Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J. Biol. Chem. 285, 15333–15345 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lumeng CN, Bodzin JL, Saltiel AR, Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua W-J, Hansen TH, Turley SJ, Merad M, Randolph GJ; Immunological Genome Consortium, Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 13, 1118–1128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Félix I, Jokela H, Karhula J, Kotaja N, Savontaus E, Salmi M, Rantakari P, Single-cell proteomics reveals the defined heterogeneity of resident macrophages in white adipose tissue. Front. Immunol. 12, 719979 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emont MP, Jacobs C, Essene AL, Pant D, Tenen D, Colleluori G, Di Vincenzo A, Jørgensen AM, Dashti H, Stefek A, McGonagle E, Strobel S, Laber S, Agrawal S, Westcott GP, Kar A, Veregge ML, Gulko A, Srinivasan H, Kramer Z, De Filippis E, Merkel E, Ducie J, Boyd CG, Gourash W, Courcoulas A, Lin SJ, Lee BT, Morris D, Tobias A, Khera AV, Claussnitzer M, Pers TH, Giordano A, Ashenberg O, Regev A, Tsai LT, Rosen ED, A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nance SA, Muir L, Lumeng C, Adipose tissue macrophages: Regulators of adipose tissue immunometabolism during obesity. Mol. Metab. 66, 101642 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cottam MA, Caslin HL, Winn NC, Hasty AH, Multiomics reveals persistence of obesity-associated immune cell phenotypes in adipose tissue during weight loss and weight regain in mice. Nat. Commun. 13, 2950 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildreth AD, Ma F, Wong YY, Sun R, Pellegrini M, O’Sullivan TE, Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity. Nat. Immunol. 22, 639–653 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bäckdahl J, Franzén L, Massier L, Li Q, Jalkanen J, Gao H, Andersson A, Bhalla N, Thorell A, Rydén M, Ståhl PL, Mejhert N, Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. 33, 1869–1882.e6 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr., Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H, Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill DA, Lim H-W, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HCB, Chegireddy K, Kim J, Habertheuer A, Vallabhajosyula P, Kambayashi T, Won K-J, Lazar MA, Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 115, E5096–E5105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, Keren-Shaul H, David E, Zmora N, Eldar SM, Lubezky N, Shibolet O, Hill DA, Lazar MA, Colonna M, Ginhoux F, Shapiro H, Elinav E, Amit I, Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr., Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 18, 816–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flaherty III SE, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr., A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 363, 989–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orr JS, Kennedy A, Anderson-Baucum EK, Webb CD, Fordahl SC, Erikson KM, Zhang Y, Etzerodt A, Moestrup SK, Hasty AH, Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes 63, 421–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubler MJ, Erikson KM, Kennedy AJ, Hasty AH, MFehi adipose tissue macrophages compensate for tissue iron perturbations in mice. Am. J. Physiol. Cell Physiol. 315, C319–C329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ameka MK, Beavers WN, Shaver CM, Ware LB, Kerchberger VE, Schoenfelt KQ, Sun L, Koyama T, Skaar EP, Becker L, Hasty AH, An iron refractory phenotype in obese adipose tissue macrophages leads to adipocyte iron overload. Int. J. Mol. Sci. 23, 7417 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabrielsen JS, Gao Y, Simcox JA, Huang J, Thorup D, Jones D, Cooksey RC, Gabrielsen D, Adams TD, Hunt SC, Hopkins PN, Cefalu WT, McClain DA, Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Invest. 122, 3529–3540 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z, Funcke JB, Zi Z, Zhao S, Straub LG, Zhu Y, Zhu Q, Crewe C, An YA, Chen S, Li N, Wang MY, Ghaben AL, Lee C, Gautron L, Engelking LJ, Raj P, Deng Y, Gordillo R, Kusminski CM, Scherer PE, Adipocyte iron levels impinge on a fat-gut crosstalk to regulate intestinal lipid absorption and mediate protection from obesity. Cell Metab. 33, 1624–1639 e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joffin N, Gliniak CM, Funcke J-B, Paschoal VA, Crewe C, Chen S, Gordillo R, Kusminski CM, Oh DY, Geldenhuys WJ, Scherer PE, Adipose tissue macrophages exert systemic metabolic control by manipulating local iron concentrations. Nat. Metab. 4, 1474–1494 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, Mendes R, Gres V, Kubasova N, Morris I, Arús BA, Larabee CM, Vasques M, Tortosa F, Sousa AL, Anandan S, Tranfield E, Hahn MK, Iannacone M, Spann NJ, Glass CK, Domingos AI, Sympathetic neuron–associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 23, 1309–1318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm Y-H, Brown CW, Elsworth J, Rodeheffer MS, Schultze JL, Dixit VD, Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho KW, Morris DL, DelProposto JL, Geletka L, Zamarron B, Martinez-Santibanez G, Meyer KA, Singer K, O’Rourke RW, Lumeng CN, An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep. 9, 605–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB, RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 19, 512–526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, Stephens JM, Mynatt RL, Dixit VD, Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: Implications for systemic inflammation and insulin resistance. J. Immunol. 185, 1836–1845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borcherding N, Jia W, Giwa R, Field RL, Moley JR, Kopecky BJ, Chan MM, Yang BQ, Sabio JM, Walker EC, Osorio O, Bredemeyer AL, Pietka T, Alexander-Brett J, Morley SC, Artyomov MN, Abumrad NA, Schilling J, Lavine K, Crewe C, Brestoff JR, Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte-derived mitochondria into the blood. Cell Metab. 34, 1499–1513 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brestoff JR, Wilen CB, Moley JR, Li Y, Zou W, Malvin NP, Rowen MN, Saunders BT, Ma H, Mack MR, Hykes BL Jr., Balce DR, Orvedahl, Williams JW, Rohatgi N, Wang X, McAllaster MR, Handley SA, Kim BS, Doench JG, Zinselmeyer BH, Diamond MS, Virgin HW, Gelman AE, Teitelbaum SL, Intercellular mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity. Cell Metab. 33, 270–282.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Jr AWF, CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest. 116, 115–124 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li D, Wu M, Pattern recognition receptors in health and diseases. Signal Transduct. Target Ther. 6, 291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM, A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 282, 35279–35292 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM, Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 10, 419–429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, Neale GA, Hooiveld GJ, Hijmans A, Vroegrijk I, van den Berg S, Romijn J, Rensen PC, Joosten LA, Netea MG, Kanneganti T-D, Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 15324–15329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD, The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Neill LA, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Camell C, Smith CW, Dietary oleic acid increases m2 macrophages in the mesenteric adipose tissue. PLOS ONE 8, e75147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van den Bossche J, Baardman J, de Winther MPJ, Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J. Vis. Exp, 53424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caslin HL, Bhanot M, Bolus WR, Hasty AH, Adipose tissue macrophages: Unique polarization and bioenergetics in obesity. Immunol. Rev. 295, 101–113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R, Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia 61, 942–953 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serbulea V, Upchurch CM, Schappe MS, Voigt P, DeWeese DE, Desai BN, Meher AK, Leitinger N, Macrophage phenotype and bioenergetics are controlled by oxidized phospholipids identified in lean and obese adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 115, E6254–E6263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, Johnson AR, Milner JJ, Lim MF, Galanko JA, Beck MA, Bear JE, Rotty JD, Bezavada L, Smallwood HS, Puchowicz MA, Liu J, Locasale JW, Lee DP, Bennett BJ, Abel ED, Rathmell JC, Makowski L, Myeloid Slc2a1-deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT1. J. Immunol. 202, 1265–1286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caslin HL, Abebayehu D, Pinette JA, Ryan JJ, Lactate is a metabolic mediator that shapes immune cell fate and function. Front. Physiol. 12, 688485 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM, Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS, Shi G-P, Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 15, 940–945 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutierrez DA, Muralidhar S, Feyerabend TB, Herzig S, Rodewald H-R, Hematopoietic kit deficiency, rather than lack of mast cells, protects mice from obesity and insulin resistance. Cell Metab. 21, 678–691 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, Geletka L, Meyer KA, O’Rourke RW, Lumeng CN, Adipose tissue dendritic cells are independent contributors to obesity-induced inflammation and insulin resistance. J. Immunol. 197, 3650–3661 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, Blin-Wakkach C, Anty R, Iannelli A, Gugenheim J, Tran A, Bouloumié A, Gual P, Wakkach A, Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 61, 2238–2247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Artis D, Spits H, The biology of innate lymphoid cells. Nature 517, 293–301 (2015). [DOI] [PubMed] [Google Scholar]
- 62.O’Sullivan TE, Rapp M, Fan X, Weizman O-E, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, Sun JC, Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. J. Immunity 45, 428–441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A, Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY, Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P, Artis D, Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, Yun K, Locksley RM, Chawla A, Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS, T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity (Silver Spring) 18, 1918–1925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D, Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R, CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 15, 914–920 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Porsche CE, Delproposto JB, Geletka L, O’Rourke R, Lumeng CN, Obesity results in adipose tissue T cell exhaustion. JCI Insight 6, e139793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Damouche A, Pourcher G, Pourcher V, Benoist S, Busson E, Lataillade JJ, Le Van M, Lazure T, Adam J, Favier B, Vaslin B, Muller-Trutwin M, Lambotte O, Bourgeois C, High proportion of PD-1-expressing CD4+ T cells in adipose tissue constitutes an immunomodulatory microenvironment that may support HIV persistence. Eur. J. Immunol. 47, 2113–2123 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Chien Y.-h, Meyer C, Bonneville M, γδ T cells: First line of defense and beyond. Ann. Rev. Immunol. 32, 121–155 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Zúñiga LA, Shen W-J, Joyce-Shaikh B, Pyatnova EA, Richards AG, Thom C, Andrade SM, Cua DJ, Kraemer FB, Butcher EC, IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J. Immunol. 185, 6947–6959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bruno MEC, Mukherjee S, Powell WL, Mori SF, Wallace FK, Balasuriya BK, Su LC, Stromberg AJ, Cohen DA, Starr ME, Accumulation of γδ T cells in visceral fat with aging promotes chronic inflammation. GeroScience 44, 1761–1778 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mehta P, Nuotio-Antar AM, Smith CW, γδ T cells promote inflammation and insulin resistance during high fat diet-induced obesity in mice. J. Leukoc. Biol. 97, 121–134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costanzo AE, Taylor KR, Dutt S, Han PP, Fujioka K, Jameson JM, Obesity impairs γδ T cell homeostasis and antiviral function in humans. PLOS ONE 10, e0120918 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kohlgruber AC, Gal-Oz ST, LaMarche NM, Shimazaki M, Duquette D, Koay HF, Nguyen HN, Mina AI, Paras T, Tavakkoli A, von Andrian U, Uldrich AP, Godfrey DI, Banks AS, Shay T, Brenner MB, Lynch L, γδ T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat. Immunol. 19, 464–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muñoz-Rojas AR, Mathis D, Tissue regulatory T cells: Regulatory chameleons. Nat. Rev. Immunol. 21, 597–611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D, PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McDonnell WJ, Koethe JR, Mallal SA, Pilkinton MA, Kirabo A, Ameka MK, Cottam MA, Hasty AH, Kennedy AJ, High CD8 T-cell receptor clonality and altered CDR3 properties are associated with elevated isolevuglandins in adipose tissue during diet-induced obesity. Diabetes 67, 2361–2376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, Markham D, Strissel KJ, Watkins AA, Zhu M, Allen J, Bouchard J, Toraldo G, Jasuja R, Obin MS, McDonnell ME, Apovian C, Denis GV, Nikolajczyk BS, B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. U.S.A. 110, 5133–5138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, Leong HX, Glassford A, Caimol M, Kenkel JA, Tedder TF, McLaughlin T, Miklos DB, Dosch HM, Engleman EG, B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 17, 610–617 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamm P, Shekelle RB, Stamler J, Large fluctuations in body weight during young adulthood and twenty-five-year risk of coronary death in men. Am. J. Epidemiol. 129, 312–318 (1989). [DOI] [PubMed] [Google Scholar]
- 84.Fothergill E, Guo J, Howard L, Kerns JC, Knuth ND, Brychta R, Chen KY, Skarulis MC, Walter M, Walter PJ, Hall KD, Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring) 24, 1612–1619 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bangalore S, Fayyad R, Laskey R, DeMicco DA, Messerli FH, Waters DD, Body-weight fluctuations and outcomes in coronary disease. N. Engl. J. Med. 376, 1332–1340 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Kyung DS, Sung HR, Kim YJ, Kim KD, Cho SY, Choi JH, Lee YH, Kim IY, Seong JK, Global transcriptome analysis identifies weight regain-induced activation of adaptive immune responses in white adipose tissue of mice. Int. J. Obes. (Lond) 42, 755–764 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zou J, Lai B, Zheng M, Chen Q, Jiang S, Song A, Huang Z, Shi P, Tu X, Wang D, Lu L, Lin Z, Gao X, CD4+ T cells memorize obesity and promote weight regain. Cell Mol. Immunol. 15, 630–639 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anderson EK, Gutierrez DA, Kennedy A, Hasty AH, Weight cycling increases T-cell accumulation in adipose tissue and impairs systemic glucose tolerance. Diabetes 62, 3180–3188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blaszczak AM, Bernier M, Wright VP, Gebhardt G, Anandani K, Liu J, Jalilvand A, Bergin S, Wysocki V, Somogyi A, Bradley D, Hsueh WA, Obesogenic memory maintains adipose tissue inflammation and insulin resistance. Immunometabolism 2, e200023 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zamarron BF, Mergian TA, Cho KW, Martinez-Santibanez G, Luan D, Singer K, DelProposto JL, Geletka LM, Muir LA, Lumeng CN, Macrophage proliferation sustains adipose tissue inflammation in formerly obese mice. Diabetes 66, 392–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, Riksen NP, Schlitzer A, Schultze JL, Stabell Benn C, Sun JC, Xavier RJ, Latz E, Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ferreira AV, Domiguez-Andres J, Netea MG, The role of cell metabolism in innate immune memory. J. Innate Immun. 14, 42–50 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caslin HL, Cottam MA, Pinon JM, Boney LY, Hasty AH, Weight cycling induces innate immune memory in adipose tissue macrophages. Front. Immunol. 13, 984859 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seufert AL, Hickman JW, Traxler SK, Peterson RM, Waugh TA, Lashley SJ, Shulzhenko N, Napier RJ, Napier BA, Enriched dietary saturated fatty acids induce trained immunity via ceramide production that enhances severity of endotoxemia and clearance of infection. eLife 11, e76744 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hata M, Andriessen EMMA, Hata M, Diaz-Marin R, Fournier F, Crespo-Garcia S, Blot G, Juneau R, Pilon F, Dejda A, Guber V, Heckel E, Daneault C, Calderon V, Des Rosiers C, Melichar HJ, Langmann T, Joyal J-S, Wilson AM, Sapieha P, Past history of obesity triggers persistent epigenetic changes in innate immunity and exacerbates neuroinflammation. Science 379, 45–62 (2023). [DOI] [PubMed] [Google Scholar]
- 96.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE; TINSAL-T2D Study Team, The effects of salsalate on glycemic control in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 152, 346–357 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group, Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Doran AC, Inflammation resolution: Implications for atherosclerosis. Circ. Res. 130, 130–148 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, de las Fuentes L, He S, Okunade AL, Patterson BW, Klein S, Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metabolism 23, 591–601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, Ferrante AW Jr., Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J. Clin. Invest. 120, 3466–3479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Belza A, Toubro S, Stender S, Astrup A, Effect of diet-induced energy deficit and body fat reduction on high-sensitive CRP and other inflammatory markers in obese subjects. Int. J. Obes. (Lond) 33, 456–464 (2009). [DOI] [PubMed] [Google Scholar]
- 102.Dewell A, Tsao P, Rigdon J, Gardner CD, Antioxidants from diet or supplements do not alter inflammatory markers in adults with cardiovascular disease risk. A pilot randomized controlled trial. Nutr. Res. 50, 63–72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Van Loan MD, Keim NL, Adams SH, Souza E, Woodhouse LR, Thomas A, Witbracht M, Gertz ER, Piccolo B, Bremer AA, Spurlock M, Dairy foods in a moderate energy restricted diet do not enhance central fat, weight, and intra-abdominal adipose tissue losses nor reduce adipocyte size or inflammatory markers in overweight and obese adults: A controlled feeding study. J. Obes. 2011, 989657 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Esposito K, Ciotola M, Giugliano D, Mediterranean diet, endothelial function and vascular inflammatory markers. Public Health Nutr. 9, 1073–1076 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Gondim OS, de Camargo VTN, Gutierrez FA, de Martins PF, Passos MEP, Momesso CM, Santos VC, Gorjão R, Pithon-Curi TC, Cury-Boaventura MF, Benefits of regular exercise on inflammatory and cardiovascular risk markers in normal weight, overweight and obese adults. PLOS ONE 10, e0140596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Christiansen T, Paulsen SK, Bruun JM, Pedersen SB, Richelsen B, Exercise training versus diet-induced weight-loss on metabolic risk factors and inflammatory markers in obese subjects: A 12-week randomized intervention study. Am. J. Physiol. Endocrinol. Metab. 298, E824–E831 (2010). [DOI] [PubMed] [Google Scholar]
- 107.Izaguirre M, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Becerril S, Valenti V, Moncada R, Unamuno X, Silva C, de la Higuera M, Salvador J, Monreal I, Fruhbeck G, Catalan V, GLP-1 limits adipocyte inflammation and its low circulating pre-operative concentrations predict worse type 2 diabetes remission after bariatric surgery in obese patients. J. Clin. Med. 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cherney DZI, Udell JA, Drucker DJ, Cardiorenal mechanisms of action of glucagon-like-peptide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors. Med. 2, 1203–1230 (2021). [DOI] [PubMed] [Google Scholar]
- 109.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB, Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 375, 311–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee Y-S, Park M-S, Choung J-S, Kim S-S, Oh H-H, Choi C-S, Ha S-Y, Kang Y, Kim Y, Jun H-S, Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 55, 2456–2468 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Zhuge F, Ni Y, Nagashimada M, Nagata N, Xu L, Mukaida N, Kaneko S, Ota T, DPP-4 inhibition by linagliptin attenuates obesity-related inflammation and insulin resistance by regulating m1/m2 macrophage polarization. Diabetes 65, 2966–2979 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Xue S, Wasserfall CH, Parker M, Brusko TM, McGrail S, McGrail K, Moore M, Campbell-Thompson M, Schatz DA, Atkinson MA, Haller MJ, Exendin-4 therapy in NOD mice with new-onset diabetes increases regulatory T cell frequency. Ann. N. Y. Acad. Sci. 1150, 152–156 (2008). [DOI] [PubMed] [Google Scholar]
- 113.Rakipovski G, Rolin B, Nohr J, Klewe I, Frederiksen KS, Augustin R, Hecksher-Sorensen J, Ingvorsen C, Polex-Wolf J, Knudsen LB, The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE −/− and LDLr −/− mice by a mechanism that includes inflammatory pathways. JACC Basic Transl. Sci. 3, 844–857 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hogan AE, Gaoatswe G, Lynch L, Corrigan MA, Woods C, O’Connell J, O’Shea D, Glucagon-like peptide 1 analogue therapy directly modulates innate immune-mediated inflammation in individuals with type 2 diabetes mellitus. Diabetologia 57, 781–784 (2014). [DOI] [PubMed] [Google Scholar]
- 115.Chaudhuri A, Ghanim H, Vora M, Sia CL, Korzeniewski K, Dhindsa S, Makdissi A, Dandona P, Exenatide exerts a potent antiinflammatory effect. J. Clin. Endocrinol. Metab. 97, 198–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pastel E, McCulloch LJ, Ward R, Joshi S, Gooding KM, Shore AC, Kos K, GLP-1 analogue-induced weight loss does not improve obesity-induced AT dysfunction. Clin. Sci. (Lond) 131, 343–353 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Brown E, Wilding JPH, Barber TM, Alam U, Cuthbertson DJ, Weight loss variability with SGLT2 inhibitors and GLP-1 receptor agonists in type 2 diabetes mellitus and obesity: Mechanistic possibilities. Obes. Rev. 20, 816–828 (2019). [DOI] [PubMed] [Google Scholar]
- 118.Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RHM, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Sabatine MS, SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 393, 31–39 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Bray JJH, Foster-Davies H, Stephens JW, A systematic review examining the effects of sodium-glucose cotransporter-2 inhibitors (SGLT2is) on biomarkers of inflammation and oxidative stress. Diabetes Res. Clin. Pract. 168, 108368 (2020). [DOI] [PubMed] [Google Scholar]
- 120.Xu C, Wang W, Zhong J, Lei F, Xu N, Zhang Y, Xie W, Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 152, 45–59 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Nakatsu Y, Kokubo H, Bumdelger B, Yoshizumi M, Yamamotoya T, Matsunaga Y, Ueda K, Inoue Y, Inoue MK, Fujishiro M, Kushiyama A, Ono H, Sakoda H, Asano T, The SGLT2 inhibitor luseogliflozin rapidly normalizes aortic mRNA levels of inflammation-related but not lipid-metabolism-related genes and suppresses atherosclerosis in diabetic ApoE KO mice. Int. J. Mol. Sci. 18, 1704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, Levasseur JL, Jahng JWS, Vos D, Parajuli N, El-Kadi AOS, Braam B, Young ME, Verma S, Light PE, Sweeney G, Seubert JM, Dyck JRB, Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) inflammasome activation in heart failure. Circ. Heart Fail. 13, e006277 (2020). [DOI] [PubMed] [Google Scholar]
- 123.Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y, SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. further augmentation of the effects with saxagliptin, a DPP4 Inhibitor. Cardiovasc Drugs Ther 31, 119–132 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, Mayoux E, Kaneko S, Ota T, SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 20, 137–149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim SR, Lee S-G, Kim SH, Kim JH, Choi E, Cho W, Rim JH, Hwang I, Lee CJ, Lee M, Oh C-M, Jeon JY, Gee HY, Kim J-H, Lee B-W, Kang ES, Cha B-S, Lee M-S, Yu, Cho JW, Kim J-S, Lee Y-H, SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 11, 2127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sukhanov S, Higashi Y, Yoshida T, Mummidi S, Aroor AR, Jeffrey Russell J, Bender SB, DeMarco VG, Chandrasekar B, The SGLT2 inhibitor empagliflozin attenuates interleukin-17A-induced human aortic smooth muscle cell proliferation and migration by targeting TRAF3IP2/ROS/NLRP3/Caspase-1-dependent IL-1β and IL-18 secretion. Cell. Signal. 77, 109825 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Garvey WT, Van Gaal L, Leiter LA, Vijapurkar U, List J, Cuddihy R, Ren J, Davies MJ, Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 85, 32–37 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Matsumura M, Nakatani Y, Tanka S, Aoki C, Sagara M, Yanagi K, Suzuki K, Aso Y, Efficacy of additional canagliflozin administration to type 2 diabetes patients receiving insulin therapy: Examination of diurnal glycemic patterns using continuous glucose monitoring (CGM). Diabetes Ther. 8, 821–827 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hattori S, Anti-inflammatory effects of empagliflozin in patients with type 2 diabetes and insulin resistance. Diabetol. Metab. Syndr. 10, 93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hafida S, Mirshahi T, Nikolajczyk BS, The impact of bariatric surgery on inflammation: Quenching the fire of obesity? Curr. Opin. Endocrinol. Diabetes Obes. 23, 373–378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, Schoelles K, Bariatric surgery: A systematic review and meta-analysis. JAMA 292, 1724–1737 (2004). [DOI] [PubMed] [Google Scholar]
- 132.Biobaku F, Ghanim H, Monte SV, Caruana JA, Dandona P, Bariatric surgery: Remission of inflammation, cardiometabolic benefits, and common adverse effects. J. Endocr. Soc 4, bvaa049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dalmas E, Rouault C, Abdennour M, Rovere C, Rizkalla S, Bar-Hen A, Nahon JL, Bouillot JL, Guerre-Millo M, Clement K, Poitou C, Variations in circulating inflammatory factors are related to changes in calorie and carbohydrate intakes early in the course of surgery-induced weight reduction. Am. J. Clin. Nutr. 94, 450–458 (2011). [DOI] [PubMed] [Google Scholar]
- 134.Illan-Gomez F, Gonzalvez-Ortega M, Orea-Soler I, Alcaraz-Tafalla MS, Aragon-Alonso A, Pascual-Diaz M, Perez-Paredes M, Lozano-Almela ML, Obesity and inflammation: Change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes. Surg. 22, 950–955 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Askarpour M, Khani D, Sheikhi A, Ghaedi E, Alizadeh S, Effect of bariatric surgery on serum inflammatory factors of obese patients: A systematic review and meta-analysis. Obes. Surg. 29, 2631–2647 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Holdstock C, Lind L, Engstrom BE, Ohrvall M, Sundbom M, Larsson A, Karlsson FA, CRP reduction following gastric bypass surgery is most pronounced in insulin-sensitive subjects. Int. J. Obes. (Lond) 29, 1275–1280 (2005). [DOI] [PubMed] [Google Scholar]
- 137.Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, Kiyosue A, Zhang S, Liu B, Bunck MC, Stefanski A; SURMOUNT-1 Investigators, Tirzepatide once weekly for the treatment of obesity. N. Engl. J. Med. 387, 205–216 (2022). [DOI] [PubMed] [Google Scholar]
- 138.Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, Jeppesen OK, Lingvay I, Mosenzon O, Pedersen SD, Tack CJ, Thomsen M, Vilsbøll T, Warren ML, Bain SC; PIONEER 6 Investigators, Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 381, 841–851 (2019). [DOI] [PubMed] [Google Scholar]
- 139.Husain M, Bain SC, Jeppesen OK, Lingvay I, Sørrig R, Treppendahl MB, Vilsbøll T, Semaglutide (SUSTAIN and PIONEER) reduces cardiovascular events in type 2 diabetes across varying cardiovascular risk. Diabetes Obes. Metab. 22, 442–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bunck MC, Diamant M, Eliasson B, Corner A, Shaginian RM, Heine RJ, Taskinen MR, Yki-Jarvinen H, Smith U, Exenatide affects circulating cardiovascular risk biomarkers independently of changes in body composition. Diabetes Care 33, 1734–1737 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Derosa G, Maffioli P, Salvadeo SA, Ferrari I, Ragonesi PD, Querci F, Franzetti IG, Gadaleta G, Ciccarelli L, Piccinni MN, D’Angelo A, Cicero AF, Exenatide versus gliben-clamide in patients with diabetes. Diabetes Technol. Ther. 12, 233–240 (2010). [DOI] [PubMed] [Google Scholar]
- 142.Mazidi M, Karimi E, Rezaie P, Ferns GA, Treatment with GLP1 receptor agonists reduce serum CRP concentrations in patients with type 2 diabetes mellitus: A systematic review and meta-analysis of randomized controlled trials. J. Diabetes Complicat. 31, 1237–1242 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Anholm C, Kumarathurai P, Pedersen LR, Samkani A, Walzem RL, Nielsen OW, Kristiansen OP, Fenger M, Madsbad S, Sajadieh A, Haugaard SB, Liraglutide in combination with metformin may improve the atherogenic lipid profile and decrease C-reactive protein level in statin treated obese patients with coronary artery disease and newly diagnosed type 2 diabetes: A randomized trial. Atherosclerosis 288, 60–66 (2019). [DOI] [PubMed] [Google Scholar]
- 144.Savchenko LG, Digtiar NI, Selikhova LG, Kaidasheva EI, Shlykova OA, Vesnina LE, Kaidashev IP, Liraglutide exerts an anti-inflammatory action in obese patients with type 2 diabetes. Rom. J. Intern. Med. 57, 233–240 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Sato T, Aizawa Y, Yuasa S, Kishi S, Fuse K, Fujita S, Ikeda Y, Kitazawa H, Takahashi M, Sato M, Okabe M, The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 17, 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Heerspink HJL, Perco P, Mulder S, Leierer J, Hansen MK, Heinzel A, Mayer G, Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 62, 1154–1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Iannantuoni F, de Marañon AM, Diaz-Morales N, Falcon R, Bañuls C, Abad-Jimenez Z, Victor VM, Hernandez-Mijares A, Rovira-Llopis S, The SGLT2 inhibitor empagliflozin ameliorates the inflammatory profile in type 2 diabetic patients and promotes an anti-oxidant response in leukocytes. J. Clin. Med. 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Latva-Rasku A, Honka M-J, Kullberg J, Mononen N, Lehtimaki T, Saltevo J, Kirjavainen AK, Saunavaara V, Iozzo P, Johansson L, Oscarsson J, Hannukainen JC, Nuutila P, The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect tissue insulin sensitivity: A randomized, double-blind, placebo-controlled study with 8-week treatment in type 2 diabetes patients. Diabetes Care 42, 931–937 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Sezai A, Sekino H, Unosawa S, Taoka M, Osaka S, Tanaka M, Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc. Diabetol. 18, 76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nishimiya N, Tajima K, Imajo K, Kameda A, Yoshida E, Togashi Y, Aoki K, Inoue T, Nakajima A, Utsunomiya D, Terauchi Y, Effects of canagliflozin on hepatic steatosis, visceral fat and skeletal muscle among patients with type 2 diabetes and non-alcoholic fatty liver disease. Intern. Med. 60, 3391–3399 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sposito AC, Breder I, Soares AAS, Kimura-Medorima ST, Munhoz DB, Cintra RMR, Bonilha I, Oliveira DC, Breder JC, Cavalcante P, Moreira C, Moura FA, de Lima-Junior JC, do Carmo HRP, Barreto J, Nadruz W, Carvalho LSF, Quinaglia T, Dapagliflozin effect on endothelial dysfunction in diabetic patients with atherosclerotic disease: A randomized active-controlled trial. Cardiovasc. Diabetol. 20, 74 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]