

Abstract
Aquaporins are a family of ubiquitously expressed transmembrane water channels implicated in a broad range of physiological functions. We have previously reported that aquaporin 4 (AQP4) is expressed on T cells and that treatment with a small molecule AQP4 inhibitor significantly delays T cell mediated heart allograft rejection. Using either genetic deletion or small molecule inhibitor, we show that AQP4 supports T cell receptor mediated activation of both mouse and human T cells. Intact AQP4 is required for optimal T cell receptor (TCR)-related signaling events, including nuclear translocation of transcription factors and phosphorylation of proximal TCR signaling molecules. AQP4 deficiency or inhibition impairs actin cytoskeleton rearrangements following TCR crosslinking, causing inferior TCR polarization and a loss of TCR signaling. Our findings reveal a novel function of AQP4 in T lymphocytes and identify AQP4 as a potential therapeutic target for preventing TCR-mediated T cell activation.
Keywords: T lymphocytes, T cell receptors/receptor complex, signaling cascade, signal transduction, cell activation, aquaporin 4
Graphical Abstract
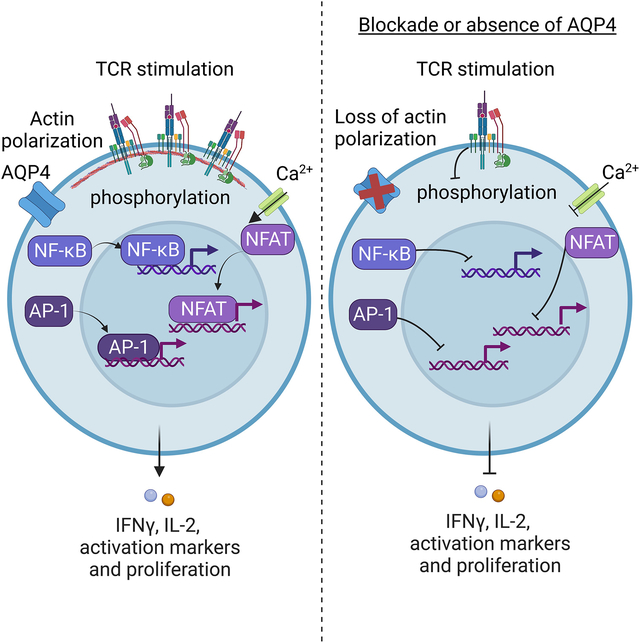
1. Introduction
Aquaporins (AQPs) are a family of plasma membrane channels that allow passive movement of small molecule ligands in and out of cells.1–3 There are 13 known mammalian AQPs (AQP0–12), which can be further subdivided into classical AQPs that allow passage of water and some gases such as AQP1 and AQP4, aquaglyceroporins that allow additional small molecules such as glycerol and hydrogen peroxide across the membrane such as AQP3 and AQP9, and the nonclassical AQP11.4 AQPs are broadly expressed within the body and mediate a range of physiological functions from water transport and fluid balance in the kidneys to neuroexcitation in the end feet of brain astrocytes.4,5
The role AQPs in the immune system has been largely unknown until the expression and functions of aquaglyceroporins were recently reported in different immune cell types. AQP3 and AQP7 expressed by immature dendritic cells facilitate macropinocytosis-mediated antigen uptake.6 AQP3 is also expressed in T cells and macrophages and is critical for the actin cytoskeleton remodeling required for chemokine-dependent cell migration.7,8 An aquaglyceroporin AQP9 is upregulated upon activation of CD8+ T cells and is essential for the development of T cell memory.9 Despite these important insights, the understanding of AQP involvement in immune cell biology and responses is far from complete.
AQP4 is expressed in renal collection ducts, renal epithelium, brain astrocytes, fast-twitch fibers of skeletal muscle,10 and cardiomyocytes11,12 and functions exclusively as a water channel.4 We recently demonstrated that AQP4 is expressed on CD4+ and CD8+ T cells and that blocking AQP4 with a small molecule inhibitor significantly delays T cell mediated rejection of mouse cardiac allografts.13 The extended graft survival was associated with decreased activation of donor-specific alloreactive T cells and in reduced infiltration into the graft.13 In the absence of antigen activation, AQP4 inhibition in resting T cells led to downregulation of S1PR1 and CCR7, resulting in impaired chemotaxis in vitro and altered lymphocyte trafficking in vivo.14 Remarkably, while resting T cells express several AQPs including AQP3 and AQP9, specific inhibition of AQP4 but not other AQPs was sufficient to protect T cells from lysis in a hypoosmotic shock swelling assay, indicating that AQP4 is an essential water channel in T lymphocytes.14 However, the mechanisms underlying the requirement for AQP4 in T cell homeostasis and functions remain to be determined.
The goal of this study was to investigate the role of AQP4 in TCR-mediated T cell activation. Using a small molecule inhibitor of AQP4 and AQP4-deficient mice, we demonstrated that TCR-mediated activation and proliferation of both murine and human T cells require functional AQP4. Following TCR crosslinking, AQP4 deficiency or inhibition results in diminished phosphorylation of key proximal signaling molecules, calcium flux, and nuclear translocation of transcription factors p65, NFAT, and AP-1. Targeting AQP4 altered actin remodeling kinetics and TCR capping, suggesting that AQP4 regulates actin cytoskeleton rearrangement required for optimal TCR signaling. These findings reveal a novel function of classical water channel AQP4 in adaptive immunity and identify AQP4 as a potential therapeutic target for modulating undesirable T cell activation.
2. Material and methods
2.1. Animals
Male and female C57BL/6J (H-2b) [B6] mice aged 6–8 weeks were purchased from The Jackson Laboratories (Bar Harbor, ME). AQP4 knockout (KO) mice on the C57BL/6 background were purchased from RIKEN Bioresource Center (Accession no. CDB0758K-1, Stock no. RBRC04364). All animals were maintained and bred in the pathogen-free facility at the Cleveland Clinic. All procedures involving animals were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic, and all experiments were performed in accordance with the relevant guidelines and regulations.
2.2. AQP inhibitors
A small molecule inhibitor of AQP4, AER-270 (Aeromics LLC, Cleveland, OH), was identified as previously described.11 During in vitro incubations AER-270 was added to the culture media at 0.25 μM or 1.0 μM.
2.3. Cell isolation and activation
Murine splenic T cells were enriched using negative selection mouse T cell isolation kit from STEMCELL technologies (Vancouver, Canada) to contain >96% of CD3+ cells. Purified cell aliquots were cultured in complete RPMI [RPMI (Gibco Life Technologies, Grand Island, NY) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA), 2 mM L-glutamine, 5 μM 2-beta-mercaptoethanol, 100 U/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate] ± AER-270 before spinning down and staining the cells for flow cytometry analysis.
For T cell activation studies, round-bottomed 96-well or flat-bottomed 6-well plates were coated with anti-CD3 mAb at 2 μg/ml in sterile PBS and stored overnight at 4 °C. Plates were then washed, and cells were plated at a density of 0.5 × 106/ml ± 4 μg/ml anti-CD28 mAb ± AER-270 and placed in an incubator at 5% CO2 and 37 °C for indicated time points (24 or 72 h). Cells were then harvested and washed before surface marker staining for flow cytometry analysis and subcellular fractionation for Western blot analysis. For phosphoflow analysis, phalloidin staining kinetics, and immunofluorescence, cells were prepared in aliquots of 5 × 106 T cells ± AER-270 in complete RPMI as described above. 5 μg/ml and 10 μg/ml of biotinylated anti-CD3 and anti-CD28 mAb, respectively, were added to the cells prior to incubation on ice for 30 min.
Human peripheral blood mononuclear cells (PBMCs) were isolated by standard methods using Ficoll-sp medium. In brief, peripheral blood was collected from healthy volunteers in tubes containing sodium heparin, diluted with phosphate buffered saline (PBS), and overlaid on Isoprep (Alere Technologies AS, Oslo, Norway), prior to centrifugation for 30 min at 400 × g. PBMCs obtained were then washed 3 times in PBS. Isolated PBMCs were counted and labeled with CFSE as previously published.13 5 × 104 to 1 × 105 CFSE-labeled cells were stimulated with plate-bound anti-CD3 (LEAF Purified antihuman CD3 antibody; Biolegend; 10 μg/ml) and anti-CD28 (LEAF Purified antihuman CD28 antibody; Biolegend; 1 μg/ml) in 200 μl of complete RPMI ± 0.25–1.0 μM AER-270 in flat-bottomed 96-well plastic plates at 37 °C. Cells were harvested after 72 h, and CFSE dilution was assessed by flow cytometry. Human T cell activation was quantified as normalized proliferative index relative to CFSE MFI in anti-CD3/anti-CD28 stimulated wells with no AQP4 inhibitor present.
2.4. Flow cytometry
Fluorochrome-conjugated and unconjugated antibodies—anti-CD4, anti-CD8, anti-CD19, anti-CD44, anti-CCR7, donkey antirat IgG-Biotin, rat antimouse CD16/CD32 (Mouse BD FcBlock, clone 2.4G2), and APC-conjugated streptavidin were purchased from BD Biosciences (San Diego, CA), eBioscience (San Diego, CA), R&D (Minneapolis, MN), and Jackson ImmunoResearch (West Grove, PA) as listed in Table 1.
Table 1.
Antibody and reagent list.
| Vendor | Cat no. | Reagent | Notes |
|---|---|---|---|
| BD Biosciences | 553142 | BD Pharmigen: Rat antimouse CD16/CD32 | Clone: 2.4G2 |
| BD Biosciences | 567316 | BD Pharmigen: APC Rat antimouse CD25 | Clone: BC96 |
| BD Biosciences | 552772 | Bd Pharmigen: PE-Cy7 Rat antimouse B220 | Clone: RA3-6B2 |
| BD Biosciences | 553133 | BD Pharmigen: FITC antimouse CD44 | Clone: IM7 |
| BD Biosciences | 558448 | BD Phosflow: PE mouse anti-CD247 (pY142) | Clone: K25-40769 |
| BD Biosciences | 558577 | BD Phosflow: Alexafluor 647 mouse anti-Lck (pY505) | Clone: 4/LCK-Y505 |
| Biolegend | 933102 | PE mouse anti-Lck (pY394) | Clone: A18002D |
| BD Biosciences | 557818 | BD Phosflow: Alexafluor 488 mouse anti-ZAP70 (pY319)/Syk (pY352) | Clone: 17A/P-ZAP70 |
| BD Biosciences | 558519 | BD Phosflow: Alexafluor 488 mouse anti-LAT (pY171) | Clone: I58-1169 |
| BD Biosciences | 558438 | BD Phosflow: Alexafluor 647 mouse anti-SLP76 (pY128) | Clone: J141-668.36.58 |
| Cell Signaling Technology | 14461S | PE rabbit anti-PLCγ1 (pY83) | D6M9S |
| BD Biosciences | 558275 | BD Phosflow: PE anti-Akt (pT308) | J1-223.371 |
| BD Biosciences | 561670 | BD Phosflow: Alexafluor 647 mouse anti-Akt (pS473) | Clone: M89-61 |
| Biolegend | 300314 | LEAF Purified antihuman CD3 | Clone: HIT3a |
| Biolegend | 302914 | LEAF Purified antihuman CD28 | Clone: CD28.2 |
| Biolegend | 102104 | Biotin-antimouse CD28 | Clone: 37.51 |
| Biolegend | 100244 | Biotin-antimouse CD3 | Clone: 17A2 |
| Biolegend | 405151 | Purified Steptavidin | — |
| Cell Signaling Technology | 5861S | Rabbit anti-NFAT1 XP | Clone: D43B1 |
| Cell Signaling Technology | 8242S | Rabbit anti-NF-kappa B p65 XP | Clone: D14E12 |
| Cell Signaling Technology | 2250S | Rabbit anti-cFos | Clone: 9F6 |
| eBioscience | 17-7311-81 | APC antimouse IFNγ | Clone: XMG1.1 |
| Invitrogen | 16-0031-86 | Antimouse CD3e | Clone: 145-2C11 |
| Invitrogen | 16-0281-86 | Antimouse CD28 | Clone: 37.51 |
| Invitrogen | 31115 | Goat anti-hamster IgG (H + L) | |
| Invitrogen | A11011 | Alexafluor 568 goat antirabbit IgG (H + L) | |
| Invitrogen | A21451 | Alexafluor 647 goat anti-hamster IgG (H + L) | |
| Invitrogen | P36935 | Prolong Gold antifade reagent with DAPI | |
| Invitrogen | A12379 | Alexa Fluor 488 phalloidin |
Cell viability was determined using Sytox staining. Sytox Orange (S34861) was purchased from Thermofisher (San Diego, CA). Sytox was added to PBS at a dilution of 1:15000, and 300 μl was added to samples prestained for other surface markers and incubated at room temperature for 20 min protected from the light prior to acquisition. Unless specified elsewhere, cells were stained as described previously,15,16 and 50,000 events per sample were collected on a BD Biosciences LSRII flow cytometer. For cell size analysis after stimulation, the sample collection was performed using a Thermofisher Attune Ctypix flow cytometer. The data were analyzed using FlowJo software (Tree Star, Ashland, OR).
For phosphoflow staining, T cell stimulation was stopped by adding ice-cold 100% methanol dropwise to simultaneously fix and permeabilize the cells. Cells were then washed, resuspended in 100 μl of FcBlock—1:100 in FACS medium, and incubated for 10 min at room temperature. Phosphoflow antibodies against pCD3ζ-pY142 (BD Biosciences #558448), pLck-pY505 (BD Biosciences #558577), pLck-pY394 (Biolegend #933102), pZAP70-pY319/Syk-pY352 (BD Biosciences #557818), pLAT-pY171 (BD Biosciences #558519), pSLP-76-pY128 (BD Biosciences #558438), pPLC-γ1-pY783 (Cell Signaling Technology #14461S), pAkt-pT308 (BD Biosciences #558275), and pAkt-pS473 (BD Biosciences #561670) were diluted 1:100 in FACS medium and added to cells for 60 min at room temperature. Phosphoflow experiments were performed on a Thermofisher Attune NXT.
For phalloidin staining, T cells were activated, divided into aliquots of 5 × 105 cells, and fixed with 4% paraformaldehyde for 1 h at RT prior to permeabilization with 0.1% Triton X-100 in PBS solution. Cells were then blocked with a 1% BSA 0.1% Triton X-100 for 45 min at room temperature, stained with Alexafluor488 conjugated phalloidin (Invitrogen; A12379) at 300 units/assay for 20 min at room temperature, and washed with Triton X-100 based permeabilization buffer and analyzed by flow cytometry.
2.5. IL-2 ELISA
The supernatants following 72 h of stimulation of murine splenic T cells, as outlined in 2.3, were retained and stored at −20 °C. Supernatants were thawed, and IL-2 was detected with the R&D Systems Mouse IL-2 Quantikine ELISA kit (R&D Systems, #M2000) in accordance with the manufacturer instructions. Optical density of the assay was determined using the Bio-Rad iMark Microplate Reader (Bio-rad Laboratories, Hercules, CA).
2.6. Calcium flux assay
Calcium mobilization was measured using a FlexStation III (Molecular Devices, San Jose, CA). In brief, isolated spleen T cells were incubated with αCD3(4 μg/ml) and αCD28(8 μg/ml) in RPMI or RPMI only for 1 h on ice. Cells were washed with PBS, and aliquots of 107 cells were loaded with 5 μM of calcium indicator dye Indo-1AM (Thermofisher, #I1223) and 1 mM probenecid in 2 ml of RMPI for 30 min at 37 °C and 5% CO2. Cells were then washed twice with PBS, and 106 cells were added to a flat-bottomed black-walled 96-well plate in 100 μl of RPMI. The temperature in the FlexStation III was set to 37 °C for 5 min before the experiment was started, and both cells and stimulants were left to equilibrate to 37 °C for 10 min within the FlexStation III. Changes in the fluorescence ratio (violet/blue) were recorded every 5 s for a total of 360 s. The cells were left unstimulated for the first 60 s to record basal levels of fluorescence intensities, and this was followed by the addition of 25 μl of either RPMI, 5× streptavidin (50 μg/ml, Biolegend #405151), 5× streptavidin + 5× AER-270 (1.25 μM), 5× phorbol 12-myristate 13-acetate (50 ng/ml), (PMA, Sigma #P1585), ionomycin (2500 ng/ml), (Iono, Sigma #I3909) (PMAI) 5× PMAI + 5× AER-270. Data were imported into GraphPad Prism software for analysis and graph production.
2.7. Transcription factor luciferase reporter
Jukat cells, clone E6-1 (ATCC TIB-152), or Jurkat cells engineered to express luciferase under transcriptional control of either NF-κB (Jurkat-NF-κB Luc) or NFAT (Jurkat-NFAT-CD28 Luc) (both, Invivogen) were seeded in a flat-bottomed 96-well plate (3 × 106/200 μl, IMDM + 10% FBS, 25 mM HEPES, 2 mM L-glutamine, 100 U/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate). Cells were incubated at 37 °C for 24 or 6 h, respectively, in media ± αCD3(0.5 μg/ml) and αCD28 mAbs (0.5 μg/ml) and ±0.25 μM AER-270. Following stimulation, 20 ml of supernatant from each well was recovered and transferred to a black-walled clear-bottomed 96-well plate and 50 ml of Quanti-Luc (Invivogen), and luminosity was measured immediately using a Flexstation III.
2.8. Immunofluorescence staining
Aliquots of 5 × 106 isolated murine T cells from WT or AQP4−/− mice were incubated with 2 μg/ml antimouse CD3 and 4 μg/ml antimouse CD28 ± 0.25–1.0 μM AER-270 on ice for 30 min. Cells were resuspended in complete RPMI containing 10 μg/ml Alexafluor647 conjugated anti-Armenian hamster antibody and incubated either on ice for 30 min or at 37 °C for 30–60 min to allow binding and or crosslinking of TCR, respectively. Cessation of the reaction occurred following the addition of Fix/Perm buffer from eBioscience Foxp3/Transcription factor staining kit (eBioscience; 00-5523-00) and incubated at RT for 1 h. For experiments visualizing transcription factor translocation, cells were then washed 3 times with permeabilization buffer prior to overnight incubation with the following primary antibodies; rabbit antimouse p65 (abcam-ab32536), goat antimouse NFAT1 (Cell Signaling-5861S), and goat antimouse cFOS (abcam-ab214672). Cells were then washed in permeabilization buffer and stained with Alexafluor568 goat antirabbit IgG (H + L) (Invitrogen; A11011) for 30 min at RT. For experiments visualizing phalloidin polarization, cells were stained following activation as described above for flow cytometry experiments. Cells were resuspended and applied to poly-L-lysine-coated coverslips (Electron Microscopy Sciences; 7229-02) and allowed to rest at RT for 1 h, protected from light. Excess moisture was wicked away, and coverslips were applied to slides with Prolong Gold antifade reagent containing DAPI (Invitrogen; P36935) and cured overnight at RT, before sealing with nail polish. Cells were visualized using a Leica SP-8 confocal microscope, and images were analyzed using the Fiji suite of ImageJ.
2.9. Statistical analysis
Results were analyzed by using a 2-tailed Student’s t-test. The difference between groups was considered significant if the P value was <0.05. Unless noted otherwise, the data are presented as mean ± SD values. Total numbers of animals in each experimental group are indicated in respective figure legends.
3. Results and discussion
3.1. AQP4 is required for t cell activation, proliferation, and differentiation after tcr crosslinking
We previously reported that AQP4 is expressed by both CD4 and CD8 T lymphocytes11 and is essential for water movement in T cells.12 Our data also indicated that pharmacological inhibition of AQP4 diminished T cell alloresponses following heart transplantation and in vitro T cell proliferation.11 To this end, we used a recently identified small molecule inhibitor of AQP4, AER-270, that inhibits human AQP4a and AQP4b (60.5 ± 1.4% and 55.2 ± 1.6% inhibition, respectively) and mouse AQP4-M23 (52.0 ± 2.0%) water permeability, with only limited inhibition of AQP1 and AQP5.13 Importantly, we previously reported that AER-270 protected AQP4+/+ but not AQP4−/− T lymphocytes from lysis in a hypoosmotic shock swelling assay, indicating the specificity of AQP4 inhibition.14
In the current study, we used AER-270 as well as AQP4−/− T cells to test the role of AQP4 in TCR-mediated T cell activation. Stimulation with plate-bound αCD3/αCD28 mAbs for 24 h increased surface expression of early activation marker CD25 in B6.WT. The addition of AQP4 inhibitor AER-270, results in a modest decrease in CD25 expression. Interestingly, stimulation of B6.AQP4−/− T cells did induce CD25 expression but to a lesser extent than WT T cells (Fig. 1A). However, 72 h of stimulation with plate-bound αCD3/αCD28 mAbs resulted in a more pronounced induction of CD25 expression in WT controls (Fig. 1B and C). In contrast, the blockade or absence of AQP4 significantly impairs CD25 expression by T cells following 72 h of stimulation (Fig. 1 B and C). We then assessed the importance of T cell AQP4 expression on induction of late activation marker CD44 after 72 h of TCR stimulation. WT T cells stimulated in the presence of 0.25 μM AER-270 demonstrated a significant decrease in CD44 upregulation compared to untreated WT controls (Fig. 1D–E). Furthermore, the absence or inhibition of AQP4 prevented the increase in T cell size associated with activation and blasting17 (Fig. 1E). IL-2 production is a canonical marker of activated T cells. Upon testing the supernatants from 72 h stimulated T cells, we found that stimulation of B6.WT T cells in the presence of 0.25 μM AER-270 or B6.AQP4−/− T cells resulted in diminished IL-2 production compared to B6.WT controls (Fig. 1G). Annexin V/7-AAD staining showed that the viability of T cells was not significantly influenced by the addition of 0.25 μM AER-270 (13 and not shown). To test the effects of AQP4 deficiency or inhibition on T cell effector functions, T cells were stimulated with αCD3/αCD28 mAbs in the presence of rIL-12 and αIL-4 mAbs to promote Th1 differentiation. Under these conditions, up to 53.8% of untreated B6.WT T cells produced IFNγ (Fig. 1H and I). The addition of AER-270 or absence of AQP4 resulted in reduced frequencies of IFNγ-producing T cells (Fig. 1H and I). In conjunction with our previous findings, these data demonstrate that AQP4 is required for mouse T cell activation and effector cytokines production.
Fig. 1.
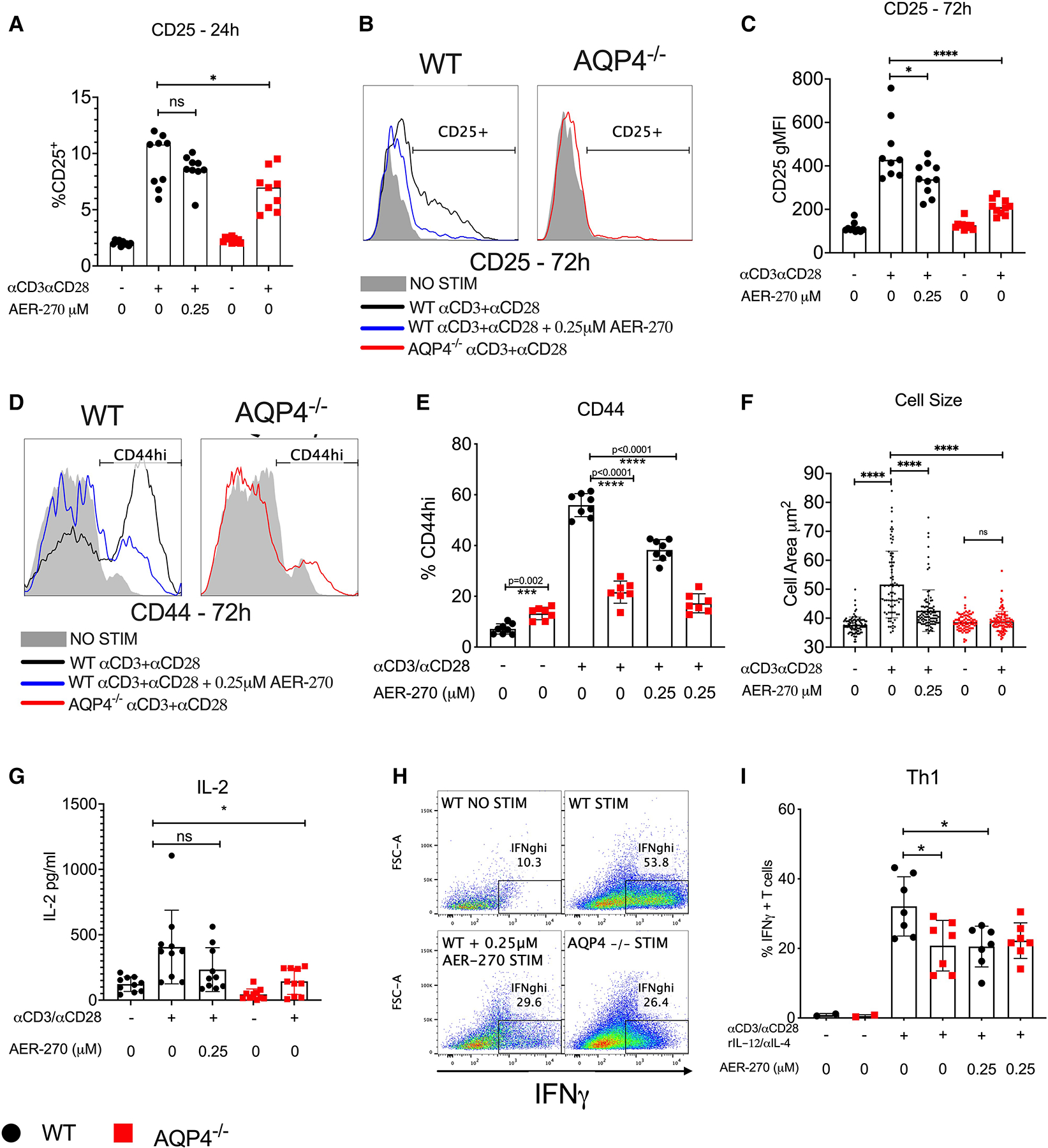
Intact AQP4 is required for optimal mouse T cell activation and cytokine production. Mouse WT (●) or AQP4−/− ( ) splenic T cells were stimulated with αCD3/αCD28 antibodies in the presence or absence of 0.25 μM AER-270. (A–C) Expression of early activation marker CD25 was evaluated after 24 h (A) and 72 h (B and C) of stimulation by flow cytometry. (D–G) After 72 h of stimulation, surface activation marker CD44 (D and E) as well as cell size (F) were measured by flow cytometry, and IL-2 was detected from culture supernatants by ELISA from the same stimulation conditions (G). (H–I) WT or AQP4−/− splenic T cells were stimulated for 72 h with αCD3/αCD28 antibodies with the addition of rIL-12/αIL-4, in the presence or absence of 0.25 μM AER-270 and Th1 polarization was evaluated by intracellular IFNγ detection (H&I) via flowcytometry. Data shown are representative of 3 experiments where n = 2–7. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns—P > 0.05 via a 1-tailed Student’s t-test.
) splenic T cells were stimulated with αCD3/αCD28 antibodies in the presence or absence of 0.25 μM AER-270. (A–C) Expression of early activation marker CD25 was evaluated after 24 h (A) and 72 h (B and C) of stimulation by flow cytometry. (D–G) After 72 h of stimulation, surface activation marker CD44 (D and E) as well as cell size (F) were measured by flow cytometry, and IL-2 was detected from culture supernatants by ELISA from the same stimulation conditions (G). (H–I) WT or AQP4−/− splenic T cells were stimulated for 72 h with αCD3/αCD28 antibodies with the addition of rIL-12/αIL-4, in the presence or absence of 0.25 μM AER-270 and Th1 polarization was evaluated by intracellular IFNγ detection (H&I) via flowcytometry. Data shown are representative of 3 experiments where n = 2–7. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns—P > 0.05 via a 1-tailed Student’s t-test.
We next tested whether AQP4 plays an analogous role in human T cells. PBMCs were isolated from healthy donors, labeled with CFSE, and stimulated with plate-bound αCD3/αCD28 mAbs for 72 h. The addition of a high dose (1.0 μM) of AER-270 significantly impaired T cell viability (Fig. 2C) and completely abrogated cell proliferation (Fig. 2A and B). An intermediate AER-270 dose (0.25 μM) had minimal impact on cell viability (Fig. 2C) while still demonstrating a significant inhibition of T cell proliferation (Fig. 2A and B). These data reflect our observations in murine T cells exposed to the same AER-270 concentrations13 and suggest similar roles for AQP4 in human and murine adaptive immune responses.
Fig. 2.
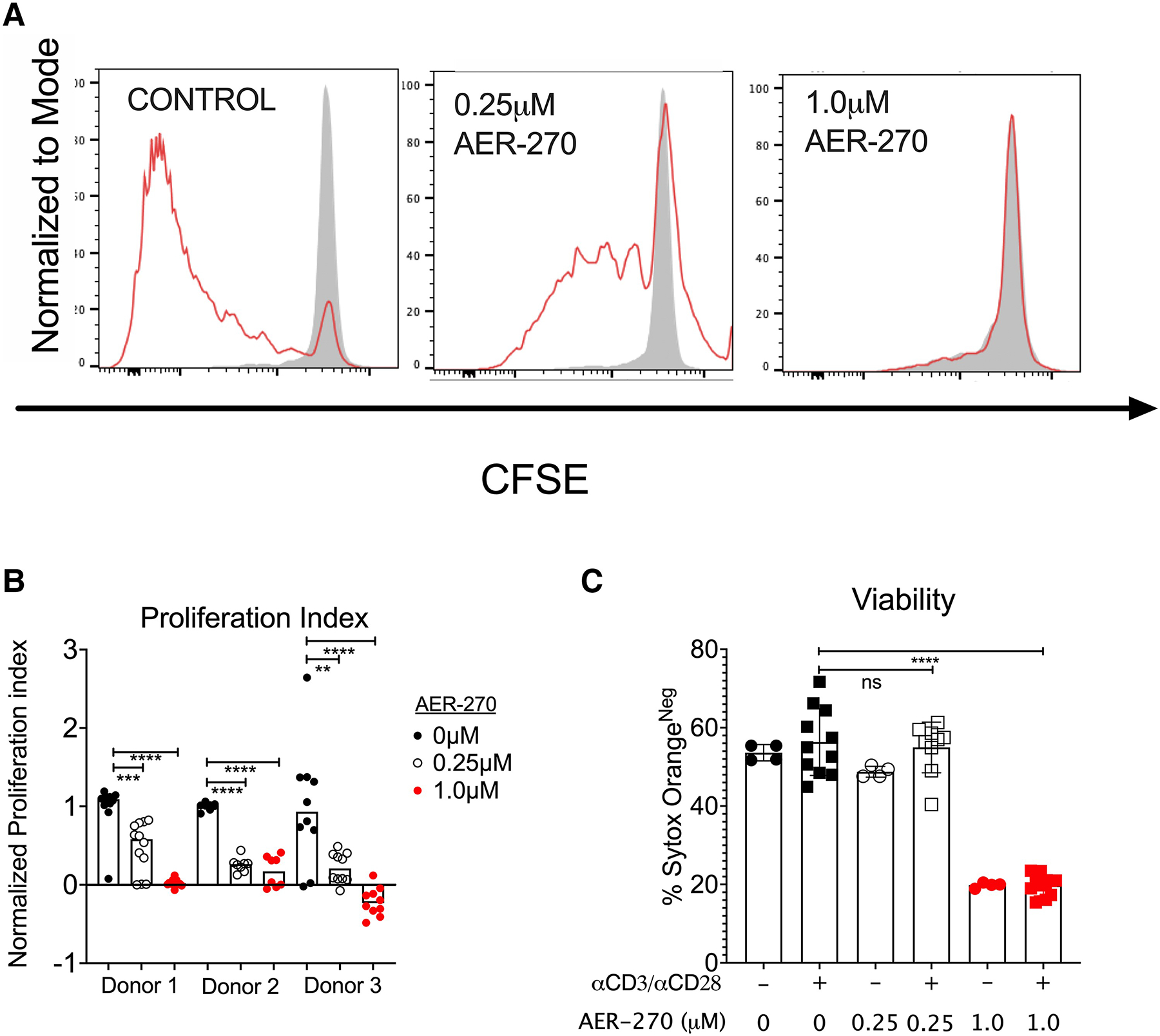
Aqp4 blockade inhibits human T cell proliferation. Human PBMCs isolated from healthy volunteers (n = 3) were labeled with CFSE and stimulated with αCD3/αCD28 antibodies for 72 h ± 0.25μM AER-270. (A) Representative flow cytometry histograms (Donor 1) gated on live CD3+ cells. Shaded histogram represents CFSE levels in unstimulated T cells. (B) Normalized proliferative index of T cells from all 3 donors with 0 mM AER-270 (•), 0.25 mM AER-270 ( ), or 1.0 mM AER-270 (
), or 1.0 mM AER-270 ( ). (C) Quantification of cell viability (Donor 1) by Sytox staining. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns—P > 0.05 via a 1-tailed Student’s t-test.
). (C) Quantification of cell viability (Donor 1) by Sytox staining. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns—P > 0.05 via a 1-tailed Student’s t-test.
3.2. AQP4 supports the TCR signaling cascade
Activation of canonical transcription factors AP-1, NF-κB, and NFAT is a hallmark feature of T cell stimulation via TCR.18–21 We next tested the effect of AQP4 deficiency or AER-270 treatment on nuclear translocation of cFOS-AP-1, NFAT1, and p65-NF-κB following TCR activation (Fig. 3A–C and Supplementary Fig. S1). Immunofluorescence staining and confocal microscopy imaging performed 1 h after TCR crosslinking revealed that the nuclear translocation of all analyzed key T cell transcription factors was markedly reduced in WT T cells treated with AQP4 inhibitor compared to untreated controls (Fig. 3D–F). Similar defects in AP-1, NFAT1, and p65 translocation were observed in AQP4−/− T cells (Fig. 3A–F). We confirmed the impaired transcription factor activity using Jurkat reporter where NFκB or NFAT activity drives luciferase activity. Inhibition of AQP4 significantly diminished both NFκB (Fig. 3G) and NFAT (Fig. 3H) after stimulation with anti-CD3 and anti-CD28 antibodies.
Fig. 3.
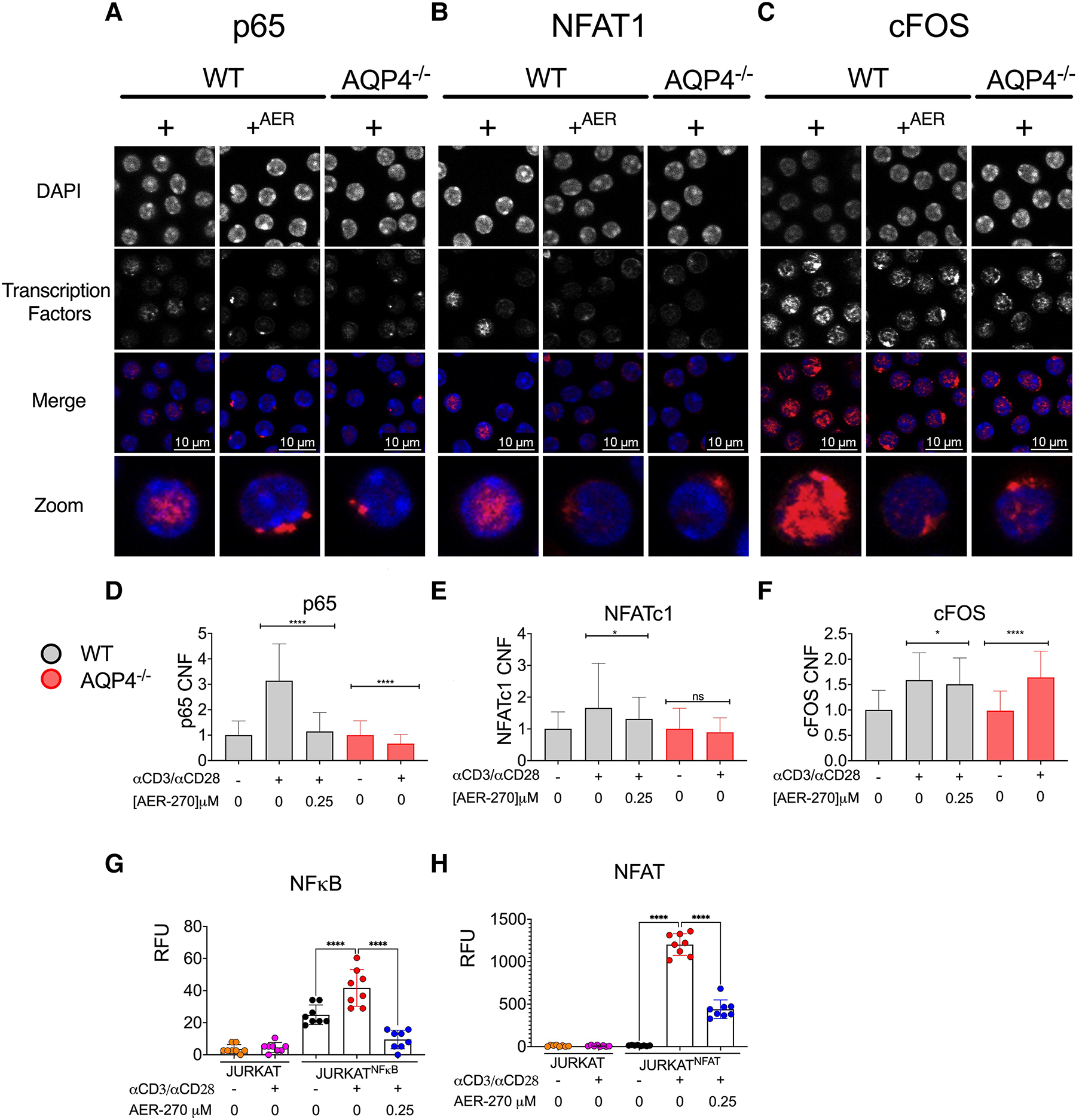
AQP4 is required for optimal activation of key T cell transcription factors. WT and AQP4−/− T cells were stimulated with αCD3/αCD28 mAb (+) ± 0.25 μM AER-270 (+AER). (A–C) Immunofluorescence of nuclear translocation of p65 (A), NFAT1 (B), and AP1 (C) after 30 min of stimulation. Cells were stained with DAPI (blue), in addition to transcription factors (red) anti-p65, anti-NFAT1, anti-c-FOS. Representative cells shown. (D–F). Quantification of corrected nuclear fluorescence (CNF) from full-field images from transcription factors p65 (A), NFAT1 (B), and cFOS (C), following stimulation as described above. (G–H) Jurkat cell lines were incubated for 6 h with αCD3/αCD28 mAb ± 0.25 μM AER-270 (n = 8). Luciferase activity regulated by NF-kB (Jurkat-Duo) (G) or NFAT (Jurkat-NFAT-CD28) (H) was measured to determine transcription factor activity. Jurkat E6-1 were included as a negative control for reporter activity (orange and pink). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns—P > 0.05 via a 1-tailed Student’s t-test.
As AP-1, NFAT, and NF-κB are activated via distinct pathways, the observed reduction in their nuclear translocation suggested the defects in signaling more proximal to TCR. Because of the importance of water movement in balancing ionic exchange, we next tested the impact of AQP4 on Ca2+ flux, a key component of the TCR signaling cascade. While Ca2+ flux following TCR-crosslinking was diminished in T cells treated with AER-270 (Fig. 4A), AQP4 inhibition had a minimal effect on Ca2+ flux induced by treatment with the PKC activator PMA and calcium ionophore ionomycin that bypass proximal TCR signaling (Fig. 4B). These results implied that AQP4 inhibition impairs events upstream of calcium flux in the TCR signaling cascade. Consistent with this scenario, phosphoflow cytometry demonstrated that inhibition or absence of AQP4 results in diminished phosphorylation of Lck Y394, ZAP70 Y319, LAT Y171, SLP7 Y128, PLC-γ1 Y783, Akt T308, and Akt S473 (Fig. 4C and Supplementary Fig. S2) for up to 10 min following TCR crosslinking. Taken together, these data suggested that AQP4 regulates the initial steps of TCR signaling, as early as Lck phosphorylation.
Fig. 4.
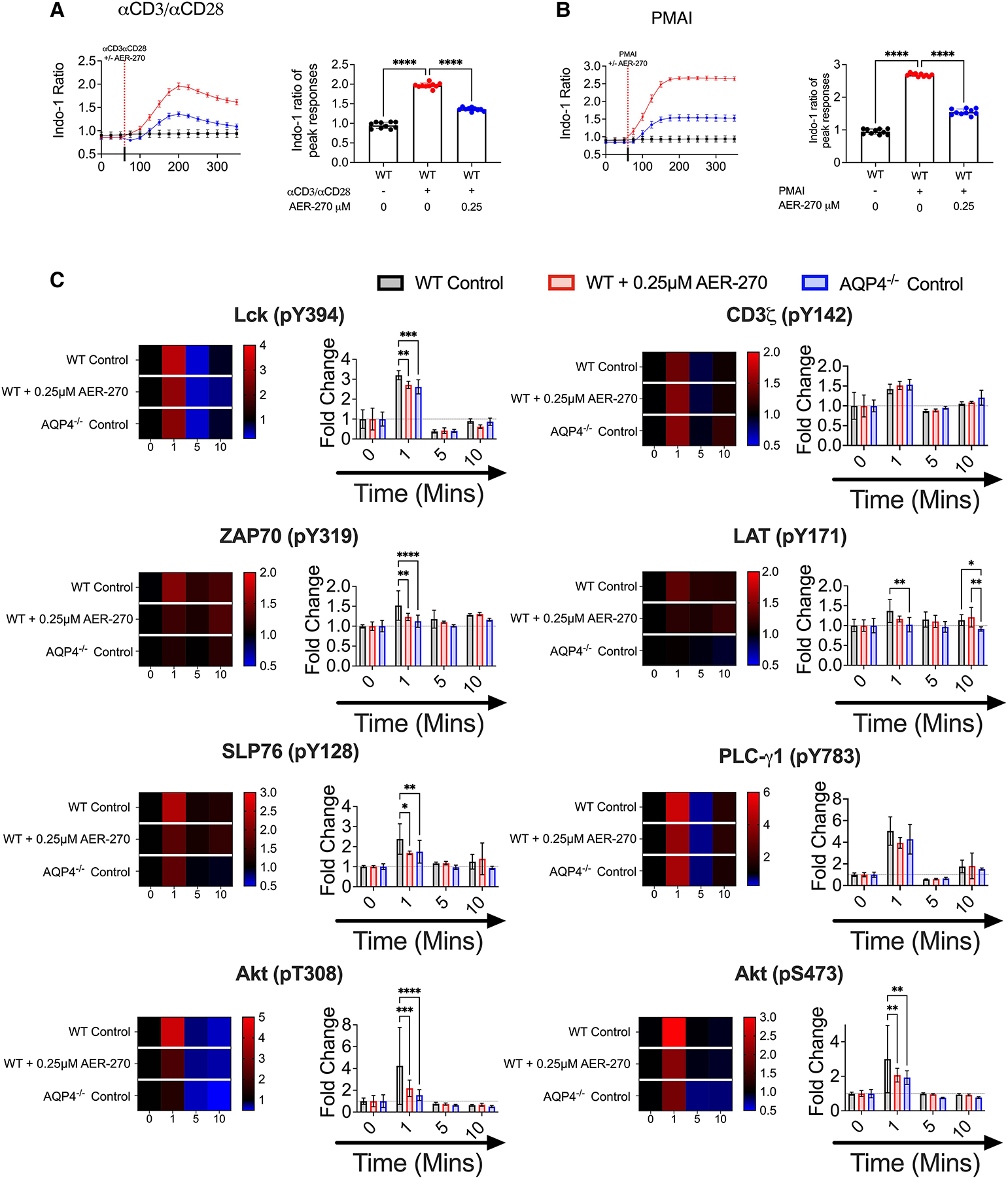
AQP4 is required for optimal TCR-proximal signaling events. (A–B) WT T cells were stimulated with ± 0.25 μM AER-270, and Ca2+ flux following T cell activation was measured as described in the Materials and Methods. Cells were loaded with the calcium sensitive dye Indo-1 AM and a baseline reading was recorded for 60 s (n = 10). After 60 s, cells were stimulated with αCD3/αCD28 mAb (A) or PMA-Ionomycin (B) ± 0.25 μM AER-270 (indicated by dashed line), and the changes in relative calcium-sensitive fluorescence ratios over time were recorded for a further 300 s. Plots show the mean of the maximal calcium peak upon stimulation ± 0.25 μM AER-270. (C) WT T cells were stimulated with ± 0.25 μM AER-270, and phosphorylation of critical signaling events was measured by flow cytometry. T cells from WT and AQP4−/− mice were stimulated with αCD3/αCD28 for 0–10 min ± AER270 and stained for flow cytometric analysis with antibodies against Lck (pY394), CD3ζ (pY142), ZAP70 (pY319), LAT (pY171), SLP76 (pY128), PLC-γ1 (pY783), Akt (pT308), and Akt (pS473) (n = 5–8). gMFI was normalized to 0 s of stimulation for each condition to generate fold increase data visualized as heatmaps and bar graphs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns—P > 0.05 via a 1-tailed Student’s t-test.
3.3. Intact AQP4 functions are required for actin cytoskeleton rearrangement following TCR stimulation
Optimal TCR signaling requires clustering and polarization of the receptors with other signaling components such as costimulatory and adhesion molecules.22–24 To address whether AQP4 is involved in TCR aggregation, we stimulated WT and AQP4−/− T cells ± AER-270 for 15 min and analyzed the formation of polarized TCR caps by immunofluorescence. Compared to control untreated WT T cells, inhibition or absence of AQP4 resulted in decreased TCR capping (Fig. 5A and B).
Fig. 5.
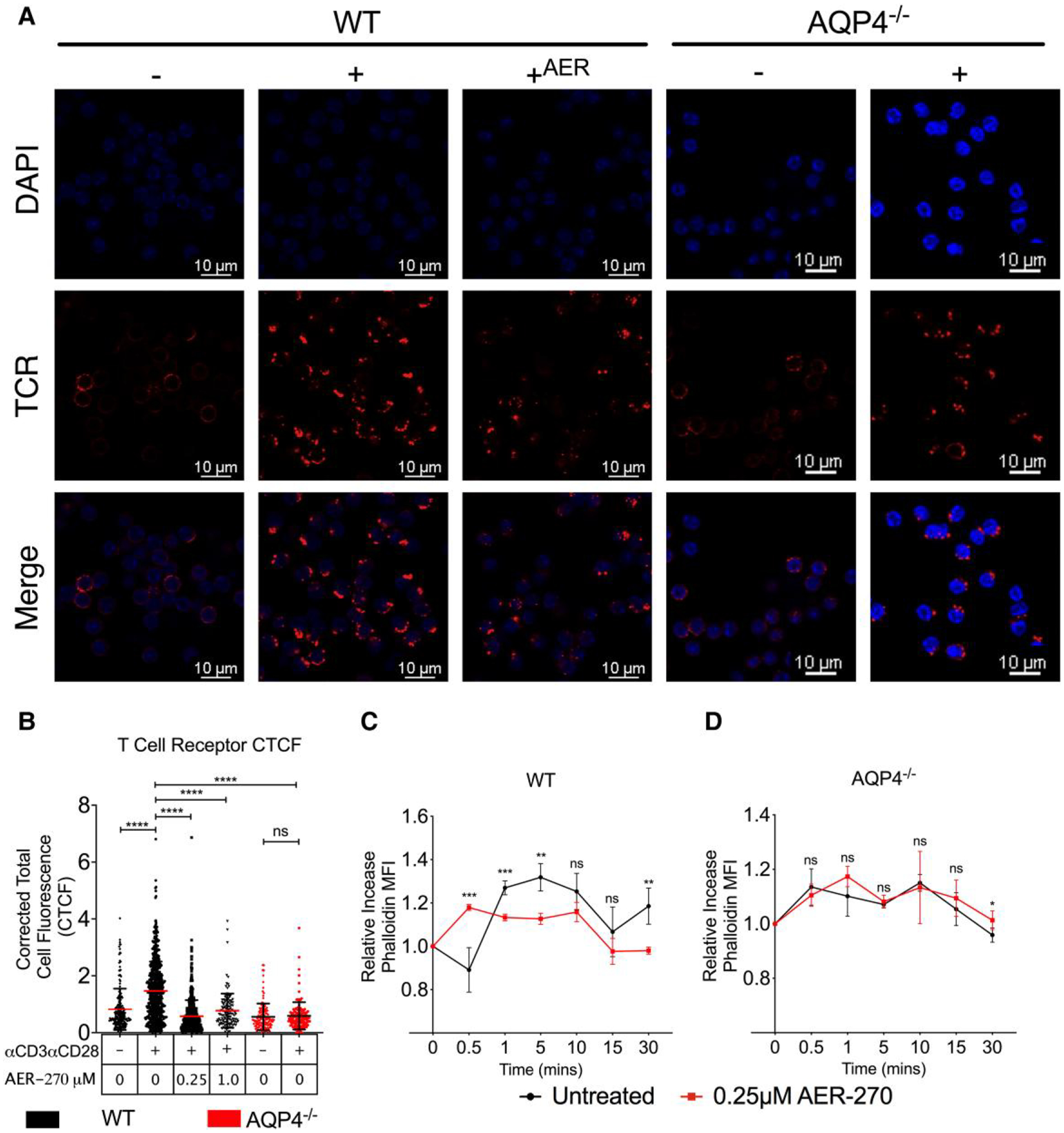
AQP4 is required for actin cytoskeletal remodeling and T cell receptor polarization. A–C. Immunofluorescence analysis of TCR polarization. WT or AQP4−/− T cells were stimulated with αCD3/αCD28 ± 0.25μM AER-270 for 15 min. Cells were stained with DAPI (blue) and TCR (red) and analyzed by confocal microscopy. (A) Representative cell staining. (B) Quantification of TCR corrected total cell fluorescence (CTCF) from full-field images (raw full-field images available in the Supplementary Materials). (C–D) Kinetics of actin polymerization following TCR crosslinking in WT (C) and AQP4−/−. (D) T cells measured by flow cytometry after intracellular phalloidin staining. Data shown are representative of multiple experiments where *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns—P.0.05 via a 1-tailed Student’s t-test.
As the process of TCR capping is critically dependent on actin cytoskeleton,25–28 we investigated how inhibition or lack of AQP4 affect actin remodeling dynamics in T cells. Consistent with previous reports,25,29,30 TCR crosslinking in WT T cells led to a slight initial decline in the amount of polymerized F-actin (0.5 min) followed by rapid repolymerization starting at 1 min (Fig. 5C). In contrast, T cells stimulated in the presence of AQP4 inhibitor had significantly altered actin remodeling kinetics with marked absence of characteristic changes within 5 min after TCR ligation (Fig. 5C). Similar defects in actin cytoskeletal remodeling were observed in AQP4−/− T cells regardless of AER-270 treatment (Fig. 5D).
Our data indicate that intact AQP4 is essential for cytoskeletal rearrangement underlying TCR aggregation and polarization on the cell surface and formation of the intracellular signaling complex. Given this finding, we would anticipate that this has an effect on thymic selection in AQP4-deficient mice. Previous studies found no difference in TCR Vβ utilization in AQP4−/− mice compared to WT controls.31 To determine the impact of AQP4 absence on the thymus, we assessed thymocyte (Supplementary Fig. S3) and found no major change or shift in the numbers of thymocyte subsets. Although our data from AQP4−/− mice clearly show a defect in TCR signaling in peripheral T cells, the thymic selection and TCR repertoire under these conditions requires further investigation.
The role of water channels in cytoskeletal regulation and cell motility has been previously demonstrated for several aquaporins and different cell types, including endothelial cells, astroglia, and cancer cells.32–34 In particular, AQP3 deficiency leads to impaired chemotaxis in T lymphocytes and macrophages.7,35 Several potential explanations were proposed for these findings, including a role for H2O2 transport and rapid changes in cytoplasm osmolality within the leading edge. However, the exact mechanisms by which aquaporins regulate cytoskeleton dynamics are still not well understood and may not be limited to aquaporin functions as membrane channels. Notably, previous studies demonstrated a molecular scaffold function of AQP4 and its direct association with the actin cytoskeleton. The sequence and structural analysis of AQP4 from different species show a conserved motif Ser-Ser-Val (SSV) at the intracellular C-terminus that can potentially serve as a binding site for PDZ domain–containing proteins.36,37 PDZ domain–containing proteins mediate a broad range of physiological functions through interactions with various binding partners.38,39 Studies of astrocytes and skeletal muscle cells demonstrated a SSV motif–dependent association between AQP4 and α-syntrophin (αSyn), a component of the dystrophin-glycoprotein complex (DGC) mediating interactions between plasma membrane and cytoskeleton.36 This interaction allows for the polarization at the end feet of astrocytes, a requirement for optimal signal transduction across the synapse. In T cells, previous studies revealed a network of PDZ-containing proteins such as Scribble, Crumbs3, Par3, and ZO-2 that regulate T cell polarity and morphology during migration and formation of the immune synapse.40,41 Therefore, it is possible that inhibition or absence of AQP4 prevents optimal recruitment of PDZ domain–containing proteins to the plasma membrane, resulting in impaired actin cytoskeleton rearrangement and TCR aggregation.
An alternate and PDZ independent mechanism may also contribute to our findings. Tybulewicz and colleagues have demonstrated that WNK1, a kinase that regulates ion flux into cells via OXSR1 and STK39 kinases, is critical in both activation both T and B cells.42,43 WNK1-mediated ion flux was followed by water flux into the cell, in their studies via AQP3. Furthermore, post-stimulus water flux induces membrane swelling at the leading edge of the cell generating space into which actin filaments can polymerize and facilitate cell migration.44 This is in agreement with our previous studies where we demonstrated that AQP4 blockade resulted in altered T cell trafficking and defective T cell chemotaxis.14 It should be noted that those studies focused on AQP3, not AQP4, and AQP3 lacks the PDZ-binding domain, so the link to actin is less direct than in AQP4 but still does not preclude a role for AQP4 in WNK1-mediated control of T cell activation and migration. Furthermore, the contribution of H2O2 flux in those models is unclear but has been shown by others to be necessary for T cell migration.7 Ongoing studies in the lab are focused on identifying potential PDZ domain–containing protein candidates binding to AQP4 in resting and activated T lymphocytyes and testing the links between the WNK1 pathway and AQP4 in our models.
If AQP4 regulates cytoskeletal changes in T lymphocytes, its functions may not be limited to supporting TCR signaling. Consistent with this scenario, we previously demonstrated that AQP4 inhibition results in severely diminished T cell chemokine-driven chemotaxis both in vitro and in vivo.14 Short-term treatment with an AQP4 inhibitor significantly altered T cell trafficking patterns and anatomical distribution in naïve mice, and it prevented T cell infiltration into heart allografts.13,14 Furthermore, AQP4 inhibition markedly decreased the expression of S1PR1 and CCR7. In the light of the current findings, it remains to be determined whether AQP4 impacts these processes via influencing chemokine receptor recycling, signaling, or subsequent actin cytoskeleton rearrangements leading to cell polarization and migration.
The use of a small molecule inhibitor targeting AQP4 raises a concern of potential off-target effects and necessitates demonstration of specificity. It was previously reported that AER-270, under the moniker of IMD-0354, inhibits IKKβ,45–48 a key signaling molecule in the canonical-NFκB pathway.49 Whereas AER-270/IMD-0354 treatment led to diminished NFκB nuclear translocation in cardiomyocytes, the study neither demonstrated direct IKKβ inhibition nor ruled out the possibility of an indirect effect. Nevertheless, it was recently suggested that the ability of AER-270 to mediate water transport on cells might not be AQP4 dependent as NFκB can mediate this process.50 We have previously performed screening of a panel of 456 kinases including IKKβ and observed no significant inhibitory activity by AER-270.51 Our current results indicate that AER-270 modulates proximal TCR signaling upstream of NFκB activation and has no effect on these events in AQP4−/− T cells (Figs. 3 and 4). In addition, the inhibitory effects of AER-270 on T cell water transport or activation are critically dependent on the presence of AQP4 (14 and Fig. 1) and cannot be explained entirely by the direct inhibition of IKKβ.
The functions of several AQPs were previously reported in T lymphocytes. However, the existing findings are limited to aquaglyceroporins (AQP3, 7, and 9) and attributed to the transport of glycerol or H2O2.7,9 Although the classical water transporter AQP4 has been extensively studied in nonimmune cells, this is the first demonstration that AQP4 is required for optimal TCR signaling and T cell activation. Taken together, our findings provide novel insights into mechanisms of T cell activation and identify AQP4 as an important therapeutic target for modulating undesirable T cell responses.
Supplementary Material
Funding
This work was supported by NIH R56 AI152368, RO1-AI113142-01A1, and NIH PO1-AI087586 (AV).
Footnotes
Competing interests
We would like to disclose that George Farr, Paul McGuirk, and Marc Pelletier are all holders of stock or stock options in Aeromics, Inc.—provider of AER-270/271, the small molecule AQP4 inhibitor used in the study. All authors concur with the submission.
Supplementary material
Supplementary materials are available at Journal of Leukocyte Biology online.
References
- 1.Borgnia M, Nielsen S, Engel A, Agre P. Cellular and molecular biology of the aquaporin water channels. Annu Rev Biochem. 1999:68(1):425–458. 10.1146/annurev.biochem.68.1.425 [DOI] [PubMed] [Google Scholar]
- 2.Ishibashi K, Hara S, Kondo S. Aquaporin water channels in mammals. Clin Exp Nephrol. 2009:13(2):107–117. 10.1007/s10157-008-0118-6 [DOI] [PubMed] [Google Scholar]
- 3.Meli R, Pirozzi C, Pelagalli A. New perspectives on the potential role of aquaporins (AQPs) in the physiology of inflammation. Front Physiol. 2018:9:101. 10.3389/fphys.2018.00101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkman AS, Anderson MO, Papadopoulos MC. Aquaporins: important but elusive drug targets. Nature Reviews Drug Discovery. 2014:13(4):259–277. 10.1038/nrd4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Day RE, Kitchen P, Owen DS, Bland C, Marshall L, Conner AC, Bill RM, Conner MT. Human aquaporins: regulators of transcellular water flow. Biochim Biophys Acta. 2014:1840(5):1492–1506. 10.1016/j.bbagen.2013.09.033 [DOI] [PubMed] [Google Scholar]
- 6.de Baey A, Lanzavecchia A. The role of aquaporins in dendritic cell macropinocytosis. J Exp Med. 2000:191(4):743–748. 10.1084/jem.191.4.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hara-Chikuma M, Chikuma S, Sugiyama Y, Kabashima K, Verkman AS, Inoue S, Miyachi Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J Exp Med. 2012:209(10):1743–1752. 10.1084/jem.20112398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hara-Chikuma M, Satooka H, Watanabe S, Honda T, Miyachi Y, Watanabe T, Verkman AS. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-kappaB signalling in keratinocytes and development of psoriasis. Nat Commun. 2015:6(1):7454. 10.1038/ncomms8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, Kaech SM. IL-7-Induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell. 2015:161(4):750–761. 10.1016/j.cell.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frigeri A, Nicchia GP, Verbavatz JM, Valenti G, Svelto M. Expression of aquaporin-4 in fast-twitch fibers of mammalian skeletal muscle. J Clin Invest. 1998:102(4):695–703. 10.1172/JCI2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutkovskiy A, Stenslokken KO, Mariero LH, Skrbic B, Amiry-Moghaddam M, Hillestad V, Valen G, Perreault MC, Ottersen OP, Gullestad L, et al. Aquaporin-4 in the heart: expression, regulation and functional role in ischemia. Basic Res Cardiol. 2012:107(5):280. 10.1007/s00395-012-0280-6 [DOI] [PubMed] [Google Scholar]
- 12.Warth A, Eckle T, Kohler D, Faigle M, Zug S, Klingel K, Eltzschig HK, Wolburg H. Upregulation of the water channel aquaporin-4 as a potential cause of postischemic cell swelling in a murine model of myocardial infarction. Cardiology. 2007:107(4):402–410. 10.1159/000099060 [DOI] [PubMed] [Google Scholar]
- 13.Ayasoufi K, Kohei N, Nicosia M, Fan R, Farr GW, McGuirk PR, Pelletier MF, Fairchild RL, Valujskikh A. Aquaporin 4 blockade improves survival of murine heart allografts subjected to prolonged cold ischemia. Am J Transplant. 2018:18(5):1238–1246. 10.1111/ajt.14624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicosia M, Miyairi S, Beavers A, Farr GW, McGuirk PR, Pelletier MF, Valujskikh A. Aquaporin 4 inhibition alters chemokine receptor expression and T cell trafficking. Sci Rep. 2019:9(1):7417. 10.1038/s41598-019-43884-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayasoufi K, Yu H, Fan R, Wang X, Williams J, Valujskikh A. Pretransplant antithymocyte globulin has increased efficacy in controlling donor-reactive memory T cells in mice. Am J Transplant. 2013:13(3):589–599. 10.1111/ajt.12068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rabant M, Gorbacheva V, Fan R, Yu H, Valujskikh A. CD40-independent help by memory CD4 T cells induces pathogenic alloantibody but does not lead to long-lasting humoral immunity. Am J Transplant. 2013:13(11):2831–2841. 10.1111/ajt.12432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obst R The timing of T cell priming and cycling. Front Immunol. 2015:6:563. 10.3389/fimmu.2015.00563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018:18(8):485–497. 10.1038/s41577-018-0020-8 [DOI] [PubMed] [Google Scholar]
- 19.Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. 2013:13(4): 257–269. 10.1038/nri3403 [DOI] [PubMed] [Google Scholar]
- 20.Trebak M, Kinet JP. Calcium signalling in T cells. Nat Rev Immunol. 2019:19(3):154–169. 10.1038/s41577-018-0110-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courtney AH, Lo WL, Weiss A. Tcr signaling: mechanisms of initiation and propagation. Trends Biochem Sci. 2018:43(2):108–123. 10.1016/j.tibs.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dustin ML. The immunological synapse. Cancer Immunol Res. 2014:2(11):1023–1033. 10.1158/2326-6066.CIR-14-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999:285(5425):221–227. 10.1126/science.285.5425.221 [DOI] [PubMed] [Google Scholar]
- 24.Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, Yagita H, Tokunaga M, Saito T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. 2010:33(3):326–339. 10.1016/j.immuni.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 25.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Ann Rev Immunol. 2008:26(1):233–259. 10.1146/annurev.immunol.26.021607.090347 [DOI] [PubMed] [Google Scholar]
- 26.Ueda H, Morphew MK, McIntosh JR, Davis MM. Cd4+ T-cell synapses involve multiple distinct stages. Proc Natl Acad Sci U S A. 2011:108(41):17099–17104. 10.1073/pnas.1113703108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryser JE, Rungger-Brandle E, Chaponnier C, Gabbiani G, Vassalli P. The area of attachment of cytotoxic T lymphocytes to their target cells shows high motility and polarization of actin, but not myosin. J Immunol. 1982:128(3):1159–1162. 10.4049/jimmunol.128.3.1159 [DOI] [PubMed] [Google Scholar]
- 28.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy: role of T cell actin cytoskeleton. J Exp Med. 1995:181(2):577–584. 10.1084/jem.181.2.577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta. 2014:1838(2):546–556. 10.1016/j.bbamem.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Y, Smoligovets AA, Groves JT. Modulation of T cell signaling by the actin cytoskeleton. J Cell Sci. 2013:126(5):1049–1058. 10.1242/jcs.098210 [DOI] [PubMed] [Google Scholar]
- 31.Sagan SA, Winger RC, Cruz-Herranz A, Nelson PA, Hagberg S, Miller CN, Spencer CM, Ho PP, Bennett JL, Levy M, et al. Tolerance checkpoint bypass permits emergence of pathogenic T cells to neuromyelitis optica autoantigen aquaporin-4. Proc Natl Acad Sci U S A. 2016:113(51):14781–14786. 10.1073/pnas.1617859114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch. 2008:456(4):693–700. 10.1007/s00424-007-0357-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005:434(7034):786–792. 10.1038/nature03460 [DOI] [PubMed] [Google Scholar]
- 34.Satooka H, Hara-Chikuma M. Aquaporin-3 controls breast cancer cell migration by regulating hydrogen peroxide transport and its downstream cell signaling. Mol Cell Biol. 2016:36(7): 1206–1218. 10.1128/MCB.00971-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu N, Feng X, He C, Gao H, Yang L, Ma Q, Guo L, Qiao Y, Yang H, Ma T. Defective macrophage function in aquaporin-3 deficiency. Faseb J. 2011:25(12):4233–4239. 10.1096/fj.11-182808 [DOI] [PubMed] [Google Scholar]
- 36.Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci U S A. 2001:98(24):14108–14113. 10.1073/pnas.241508198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995:269(5231): 1737–1740. 10.1126/science.7569905 [DOI] [PubMed] [Google Scholar]
- 38.Lee HJ, Zheng JJ. Pdz domains and their binding partners: structure, specificity, and modification. Cell Commun Signal. 2010:8(1): 8. 10.1186/1478-811X-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christensen NR, Calyseva J, Fernandes EFA, Luchow S, Clemmensen LS, Haugaard-Kedstrom LM, Stromgaard K. Pdz domains as drug targets. Adv Ther (Weinh). 2019:2:1800143. 10.1002/adtp.201800143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludford-Menting MJ, Oliaro J, Sacirbegovic F, Cheah ET, Pedersen N, Thomas SJ, Pasam A, Iazzolino R, Dow LE, Waterhouse NJ, et al. A network of PDZ-containing proteins regulates T cell polarity and morphology during migration and immunological synapse formation. Immunity. 2005:22(6):737–748. 10.1016/j.immuni.2005.04.009 [DOI] [PubMed] [Google Scholar]
- 41.Tello-Lafoz M, Martinez-Martinez G, Rodriguez-Rodriguez C, Albar JP, Huse M, Gharbi S, Merida I. Sorting nexin 27 interactome in T-lymphocytes identifies zona occludens-2 dynamic redistribution at the immune synapse. Traffic. 2017:18(8):491–504. 10.1111/tra.12492 [DOI] [PubMed] [Google Scholar]
- 42.Hayward D, Vanes L, Wissmann S, Sivapatham S, Hartweger H, O’May JB, Boer LD, Mitter R, Köchl R, Stein JV, et al. B cell-intrinsic requirement for WNK1 kinase in T cell-dependent antibody responses. bioRxiv. 2021:2021.09.09.459588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’May JB, Vanes L, Hartweger H, de Boer LL, Hayward D, Köchl R, Tybulewicz VLJ. Water influx is required for CD4+ T cell activation and T cell-dependent antibody responses. bioRxiv. 2022:2022.03.16.484637. [Google Scholar]
- 44.de Boer LL, Melgrati S, Vanes L, O’May JB, Hayward D, Köchl R, Tybulewicz VLJ. T cell migration requires ion and water influx to regulate actin polymerization. bioRxiv. 2022:2022.03.16.484584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, Muto S, Itai A, Isobe M. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004:63(1):51–59. 10.1016/j.cardiores.2004.03.002 [DOI] [PubMed] [Google Scholar]
- 46.Onai Y, Suzuki J, Maejima Y, Haraguchi G, Muto S, Itai A, Isobe M. Inhibition of NF-{kappa}B improves left ventricular remodeling and cardiac dysfunction after myocardial infarction. Am J Physiol Heart Circ Physiol. 2007:292(1):H530–H538. 10.1152/ajpheart.00549.2006 [DOI] [PubMed] [Google Scholar]
- 47.Inayama M, Nishioka Y, Azuma M, Muto S, Aono Y, Makino H, Tani K, Uehara H, Izumi K, Itai A, et al. A novel IkappaB kinase-beta inhibitor ameliorates bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2006:173(9):1016–1022. 10.1164/rccm.200506-947OC [DOI] [PubMed] [Google Scholar]
- 48.Tanaka A, Muto S, Jung K, Itai A, Matsuda H. Topical application with a new NF-kappaB inhibitor improves atopic dermatitis in NC/NgaTnd mice. J Invest Dermatol. 2007:127(4):855–863. 10.1038/sj.jid.5700603 [DOI] [PubMed] [Google Scholar]
- 49.Paul S, Schaefer BC. A new look at T cell receptor signaling to nuclear factor-kappaB. Trends Immunol. 2013:34(6):269–281. 10.1016/j.it.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitchen P, Salman MM, Halsey AM, Clarke-Bland C, MacDonald JA, Ishida H, Vogel HJ, Almutiri S, Logan A, Kreida S, et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell. 2020:181(4):784–799.e19. 10.1016/j.cell.2020.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farr GW, Hall CH, Farr SM, Wade R, Detzel JM, Adams AG, Buch JM, Beahm DL, Flask CA, Xu K, et al. Functionalized phenylbenzamides inhibit aquaporin-4 reducing cerebral edema and improving outcome in two models of CNS injury. Neuroscience. 2019;404: 484–498. 10.1016/j.neuroscience.2019.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.