

Abstract
The stereoselective total synthesis of structure 1 assigned to the macrolide natural product neaumycin B is reported in a 2.3% overall yield on 90 mg scale. The synthesis features a gram-scale nickel-catalyzed reductive cross-coupling/spiroketalization tactic to construct the spiroketal core of neaumycin B. The stereostructures of the C3–C6, C8–C14, and C20–C41 segments of synthetic neaumycin B were unambiguously verified by X-ray crystallography.
The first congener of neaumycin was isolated in 2012 by Shen et al. from the soil actinomycete Streptomyces sp. NEAU-x21.1 The structure of neaumycin was then substantially revised in 2015, with the isolation of neaumycin A and the congener neaumycin B, albeit without stereochemistry.2 In 2018, Jenson and Fenical et al. isolated a substance from a marine microbial Micromonospora sp. (strain CNY-010) from the surface of the tropical brown alga Stypopodium zonale collected in the Bahamas Islands.3 A combination of genomic data and 2D NMR studies led to the assignment of neaumycin B to be 1. Preliminary in vitro study of neaumycin B against several cancer cell lines displayed significant potency (LD50: 5.6 × 10−5 μg/mL), in particular with selectivity toward U87 human glioblastoma, which is among the most malignant types of gliomas.4 The bioactivity of neaumycin B holds promise as a lead structure for drug design. The development of a total synthesis of neaumycin B (1) would thus be of significance and, as such, has drawn considerable interest.5a–c Herein we report the first total synthesis of the reported structure of neaumycin B (1).
From the retrosynthetic perspective (Scheme 1), we envisioned that neaumycin B (1) could be dissected into a linear southern (C1–C17) hemisphere (2) and a spirocyclic northern hemisphere (C18–C41) (3), which in turn could be united via Stille coupling6 and macrolactonization, to complete, upon deprotection, neaumycin B (1).
Scheme 1.
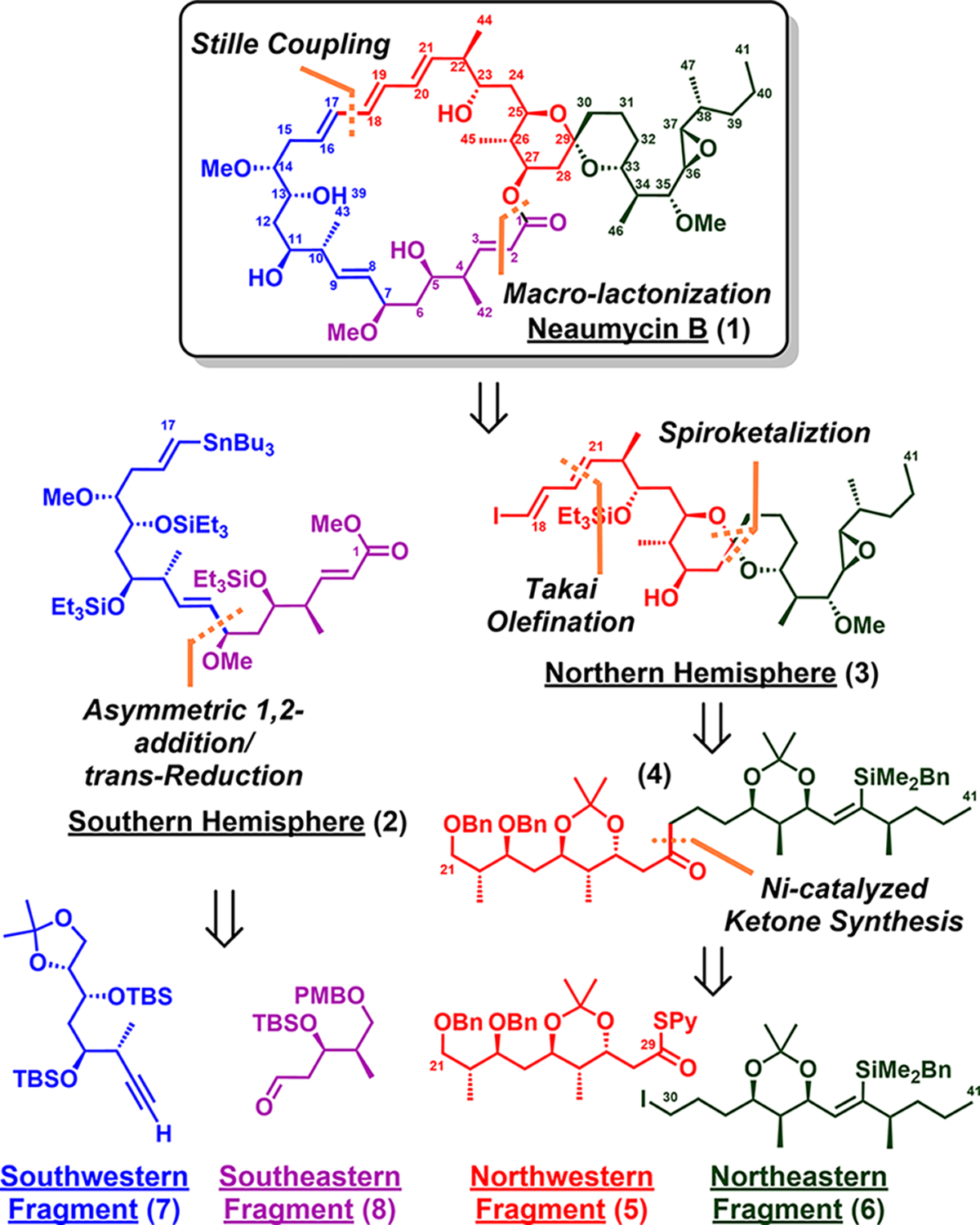
Structure and Retrosynthetic Analysis of Neaumycin B (1)
The linear southern hemisphere 2 (Scheme 1) was envisioned to arise from an asymmetric 1,2-addition of southwestern (7) and southeastern (8) fragments, while the spiroketal core of the northern hemisphere (3) could be constructed from the linear ketone precursor 4 (Scheme 1), which in turn could be dissected into the northwestern (5) and northeastern (6) fragments.
Our synthesis began with the northwestern fragment 5 (Scheme 2), the C21–C29 segment of neaumycin B. Union of epoxide (+)-9 (prepared in two steps from a known compound; see SI for details) and dithiane 107b proceeded smoothly via a Brook rearrangement/epoxide ring opening sequence7a–c to yield adduct 11, which underwent benzyl protection, hydrolysis of trimethylsilyl ether, and Cu/TEMPO-catalyzed aerobic oxidation8 to aldehyde 12 (see X-ray), gratifyingly with no oxidation of sulfur or epimerization at the α position of the carbonyl. A Felkin–Anh selective aldol reaction of the enolate derived from methyl acetate with 12 then delivered the desired syn adduct 13 with good diastereoselectivity (15:1). Dithioacetal hydrolysis9 and Evans–Saksena reduction10 followed by 1,3-diol protection led to compound 15, with the desired anti configuration. Saponification of the methyl ester and subsequent thioesterification completed synthesis of northwestern fragment 5 on a decagram scale.
Scheme 2.
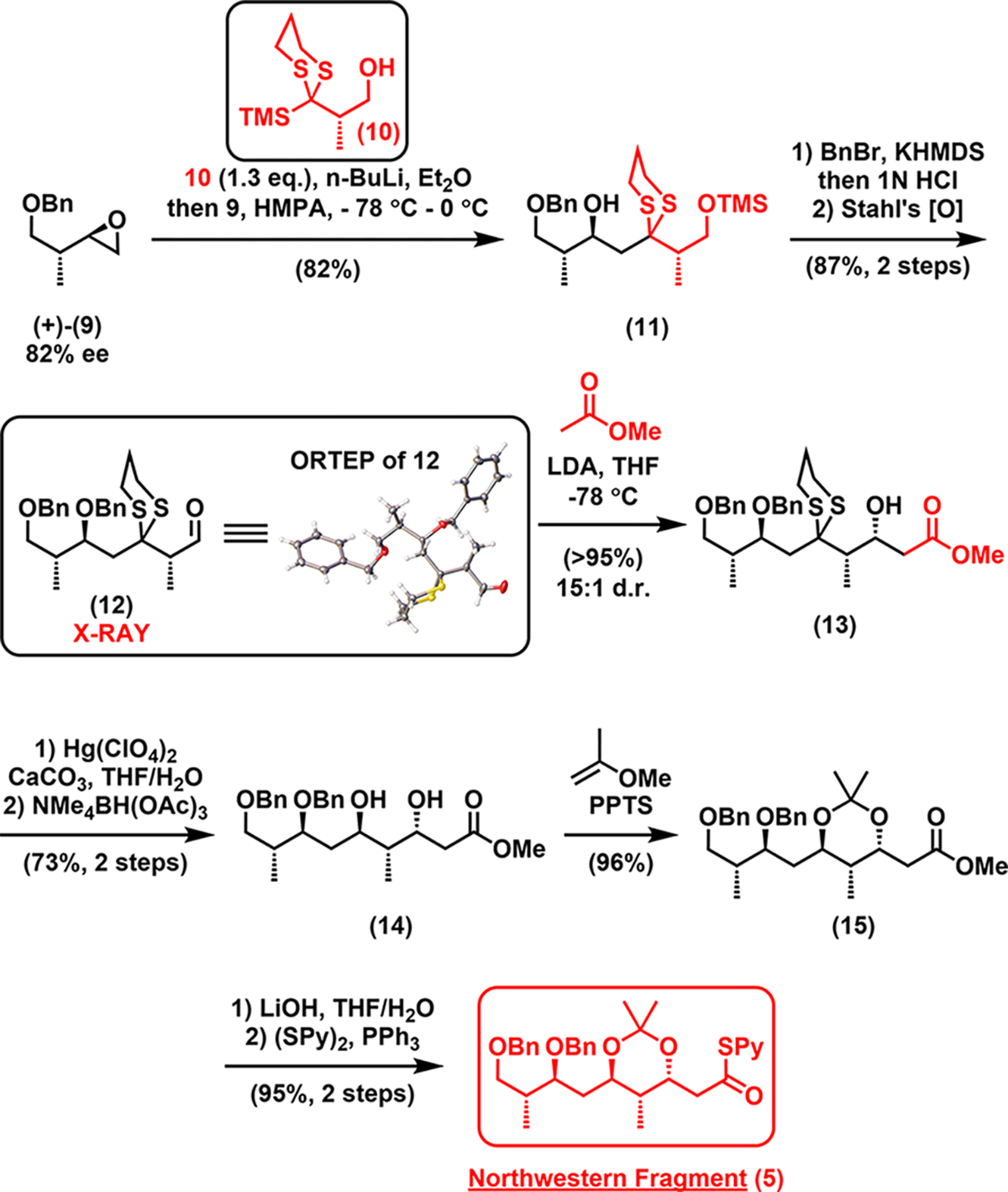
Synthesis of the Northwestern Fragment (5)
We next turned to northeastern (C30–C41) fragment 6 (Scheme 3). Regioselective opening of epoxy alcohol (−)-16 (91% ee, readily prepared from (E)-2-hexene-1-ol via Sharpless epoxidation11) with trimethylaluminum12 led almost exclusively to 1,2-diol 17 (>20:1 1,2-diol/1,3-diol) as indicated by the crude 1H NMR (see Figure S27), which upon biphasic periodate cleavage and dibromo-olefination13 furnished dibromo-alkene 18 on a 38 g scale. Exposure of 18 to n-BuLi and capture of the lithium alkynylide with formaldehyde delivered propargylic alcohol 19. Trans-hydrosilylation directed by the hydroxyl group employing ruthenium catalysis14a–d furnished alcohol 20, with both good regio-, Z/E selectivity and yield. Oxidation of the resulting allylic hydroxyl with MnO2 then afforded quantitatively aldehyde 21, which underwent an Evans aldol15 reaction with 22 to form 23; subsequent transamidation yielded Weinreb amide 24, with the desired syn configuration (82%, 2 steps). Monoaddition of allylmagnesium bromide to amide 24 then yielded the β-hydroxyl ketone, which was followed by Narasaka–Prasad reduction16a–c (Et2BOMe, NaBH4) and acetal protection to access 25. Hydroboration/oxidation, followed by an Appel reaction,17 completed the northeastern fragment 6.
Scheme 3.
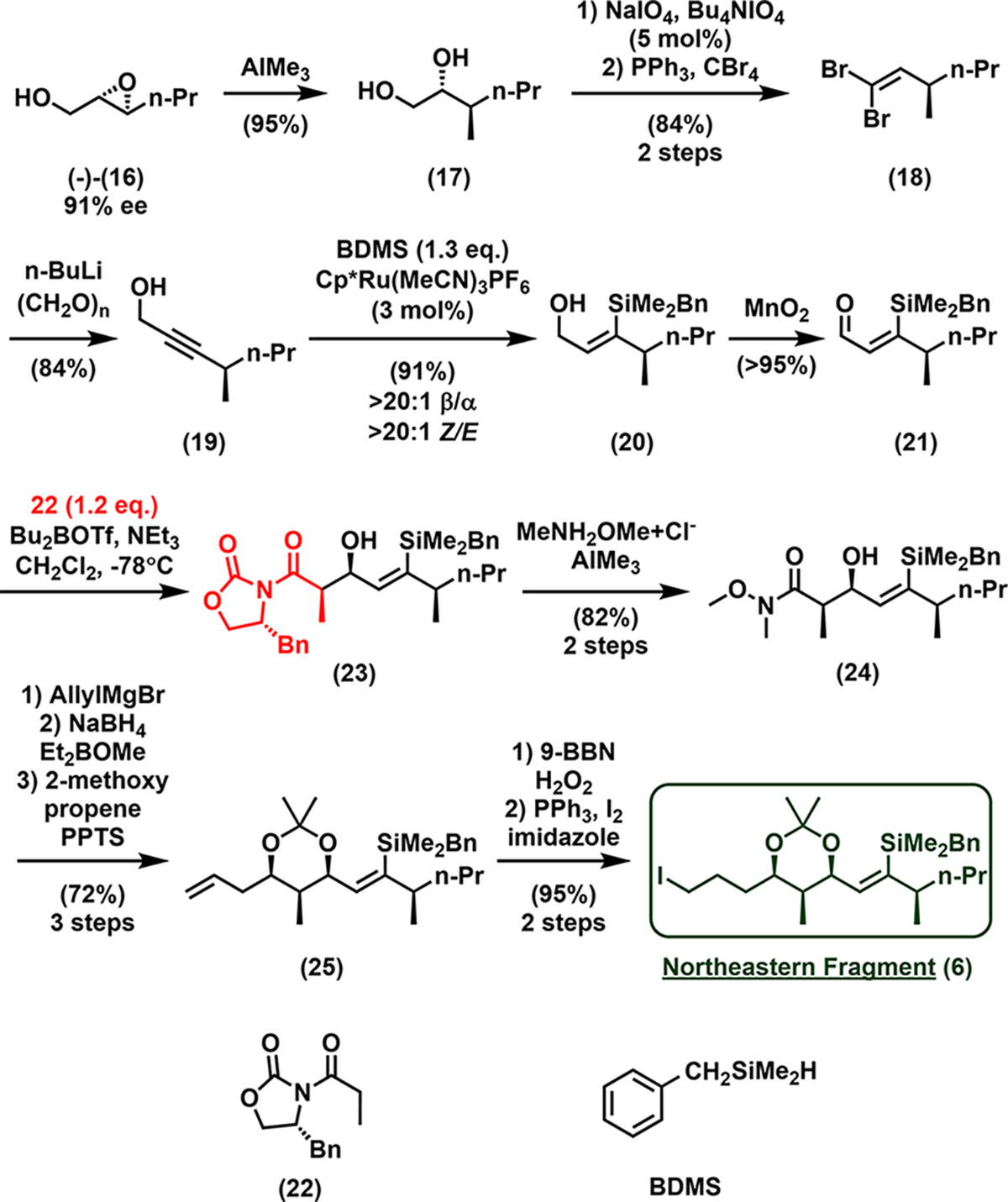
Synthesis of the Northeastern Fragment (6)
With northwestern and northeastern fragments 5 and 6 in hand, we set out to examine suitable methods for their union (Scheme 4). Although neither organolithium nor Grignard chemistry successfully delivered the desired coupled ketone in satisfactory yield, fragment coupling was achieved via the recently developed nickel-catalyzed reductive cross-coupling protocol18 to afford ketone 4 in good yield (76%) on a gramscale. Ketone 4 was then exposed to p-toluenesulfonic acid in methanol to achieve deprotection/spiroketalization and to furnish 26 and the epimer 26′, as a 1:0.8 mixture, which were separable by chromatography. Pleasingly, exposure of the pure undesired epimer 26′ to the same acidic condition reestablished the equilibrium, thereby permitting harvest of 26 upon each chromatography separation/re-equilibration to achieve 26 in 67% overall yield. A key NOE correlation between H-33 and H-28 (Scheme 4, see Figure S53) verified the stereogenicity of 26.
Scheme 4.
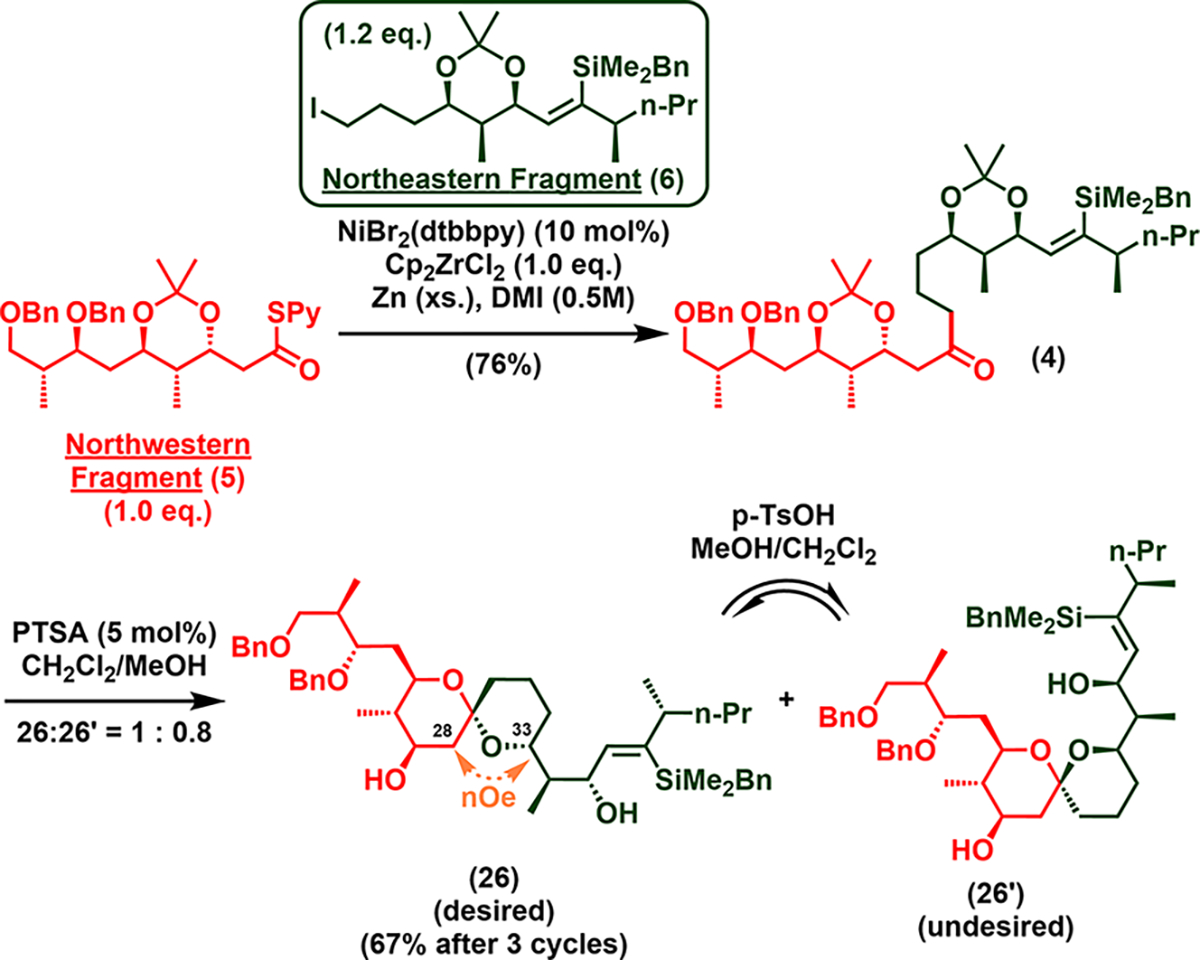
Fragment Union/Spiroketalization
To achieve 3,4-dimethoxybenzyl protection at the C27-OH (Scheme 5), 26 was treated first with catalytic tetra-n-butylammonium fluoride (TBAF)19 to affect closure of the five-membered siloxane ring across C35–C37 and leave the C27-OH exposed for selective DMB protection (see Supporting Information for details). Subsequent exposure of the siloxane to methyllithium then unmasked the C35-OH to give 27, which now serves as the directing group for the future stereoselective epoxidation.
Scheme 5.
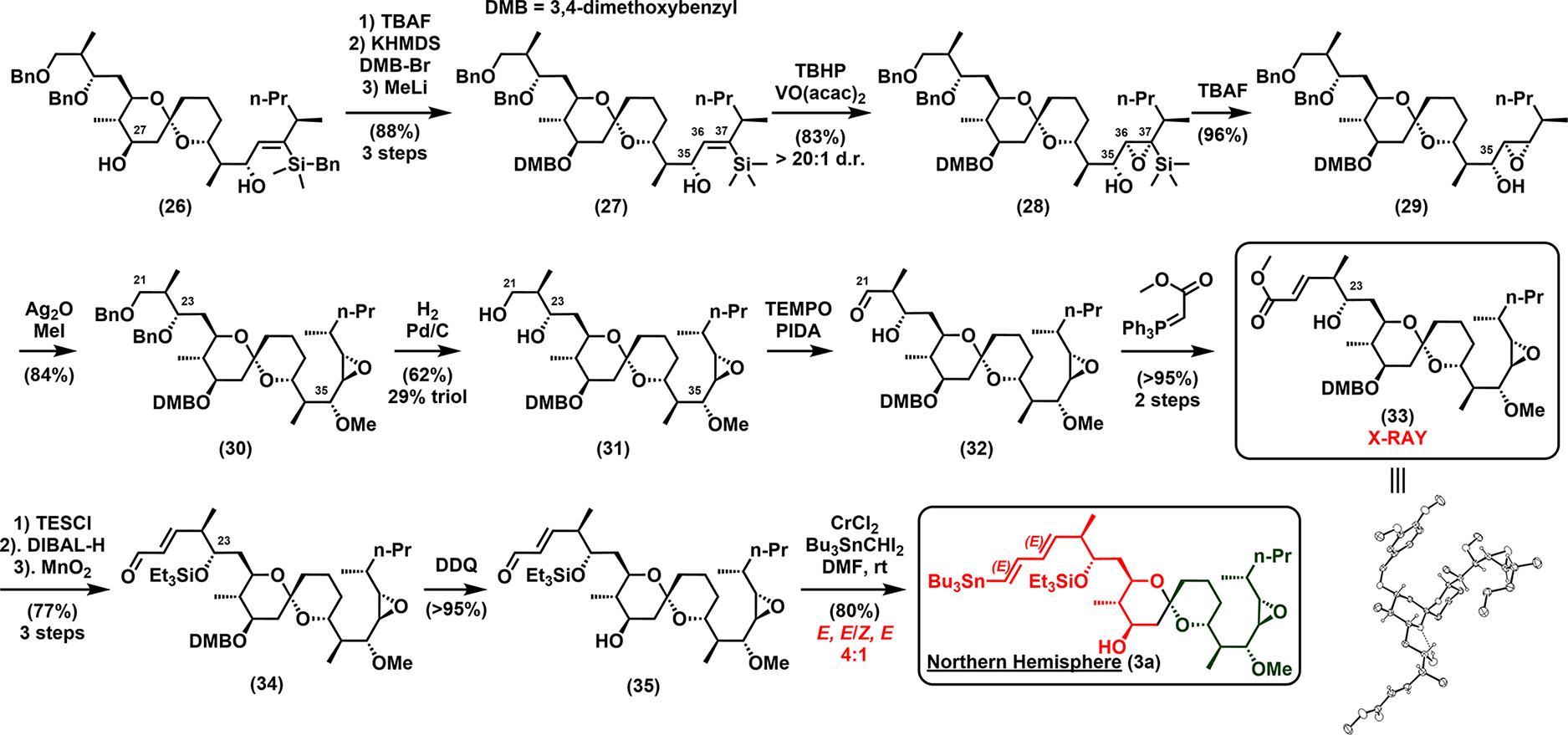
Synthesis of the Northern Hemisphere (3a)
With the thus locked conformation at the C35–C36 bond in 27, due to A1,3 strain invoked by the preinstalled C37 silyl group,14 epoxidation of the C36–C37 olefin proceeded with exclusive syn-selectivity with vanadyl catalysis20 to furnish 28. Desilylation with TBAF21 and methylation of C35-OH then gave 30, followed by hydrogenolysis of the C21 and C23 benzyl ethers to give diol 31. Chemoselective oxidation of the C21 primary alcohol next delivered β-hydroxy aldehyde 32, which upon treatment with the Wittig phosphoranylidene reagent,22a,b yielded olefin 33 with excellent E selectivity. Pleasingly, compound 33 was crystalline, thus enabling unambiguous verification of the stereostructure spanning the C20–C41 segment by X-ray crystallography (see Scheme 5).
Next, triethyl silyl protection of the C23-OH in 33, reduction of ester, and MnO2 oxidation furnished enal 34, which was treated with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to affect deprotection of the C27-OH to give 35. Finally, Takai–Utimoto olefination23a,b of aldehyde 35 employing diiodo(tributylstannyl)methane led to 3a in good yield, but as an unacceptable 4:1 mixture of (E,E)/(Z,E) isomers.
We therefore revised our synthetic route to a second-generation northern hemisphere (3b, see Scheme 6). β-Hydroxy aldehyde 32 was first protected with triethylsilyl, and a Takai–Utimoto reaction23a,b was carried out. The steric bulk at the α-carbon next to the carbonyl permitted Takai olefination with CrCl2/Bu3SnCHI2 to proceed with excellent E-selectivity (>20:1), leading to the corresponding vinylstannane.23a,b Iodination then afforded E-iodide 37 in 78% yield over two steps. DDQ removal of the 3,4-dimethoxybenzyl group then gave 38. Next Stille cross-coupling24c between alkenyl iodide 38 and germylstannane 3924a–c afforded dienyl germane 40, with no isomerization of the diene. Iodination of germane 40 with N-iodosuccinimide then provided access to the second-generation northern hemisphere (3b) with excellent stereospecificity.
Scheme 6.
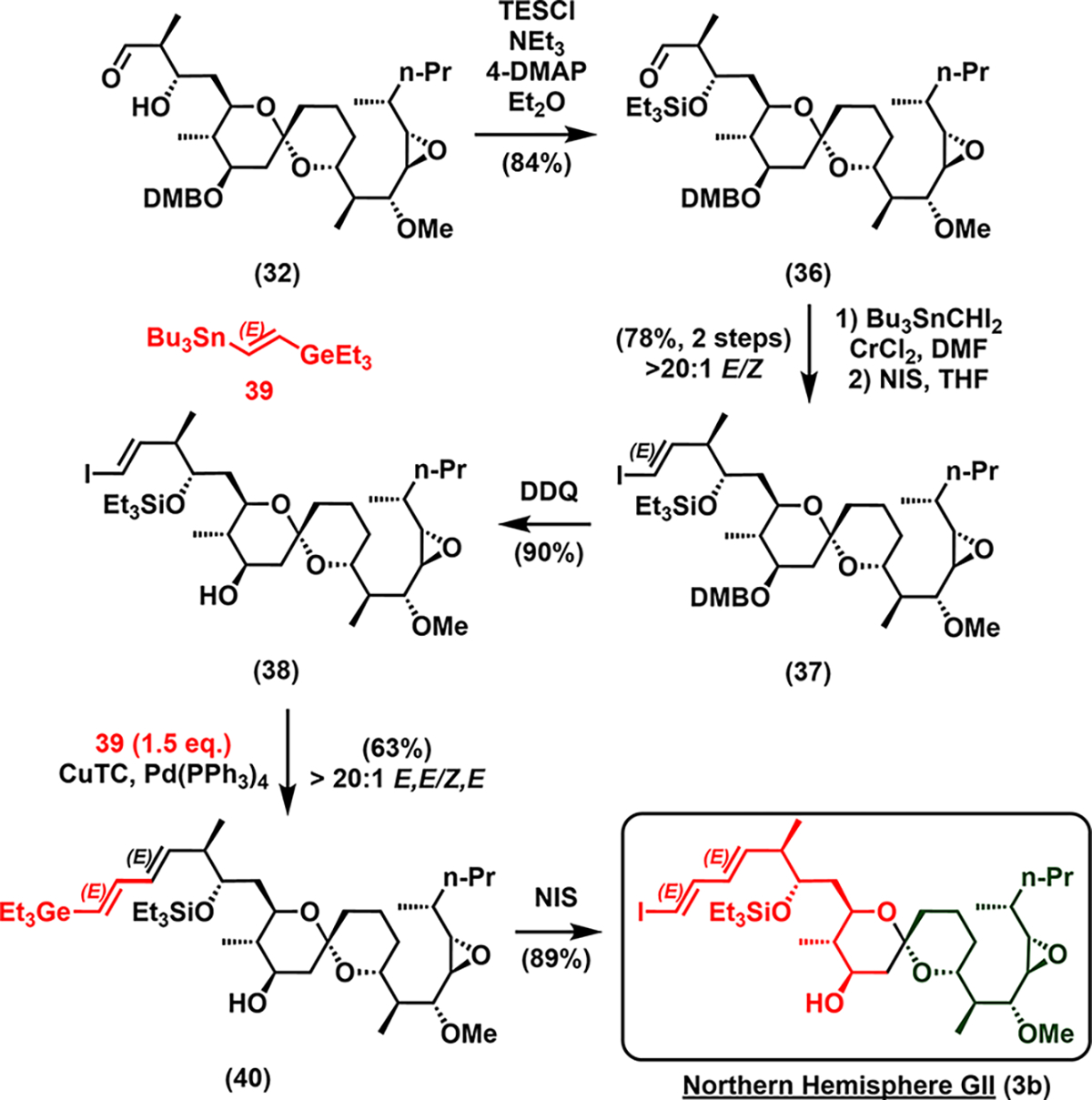
Synthesis of the Second-Generation Northern Hemisphere (3b)
Turning to the linear southwestern and southeastern fragments (Scheme 7a and b), synthesis of alkyne 7 began from known aldehyde 41 (prepared from d-xylose in five steps25a,b). Marshall asymmetric propargylation26 delivered the anti-configured homopropargylic alcohol 42 in good yield, but with modest 4:1 diastereoselectivity (Scheme 7). Adduct 42 was then treated with TBAF to afford diol 43 with excellent purity after chromatography. To gain proof of the stereostructure of 7, PMP acetalization of diol 43 furnished crystalline 44, which upon X-ray crystallography analysis verified the structure of the southwestern segment (C8–C14). Silyl ether protection then completed the southwestern fragment (7).
Scheme 7.
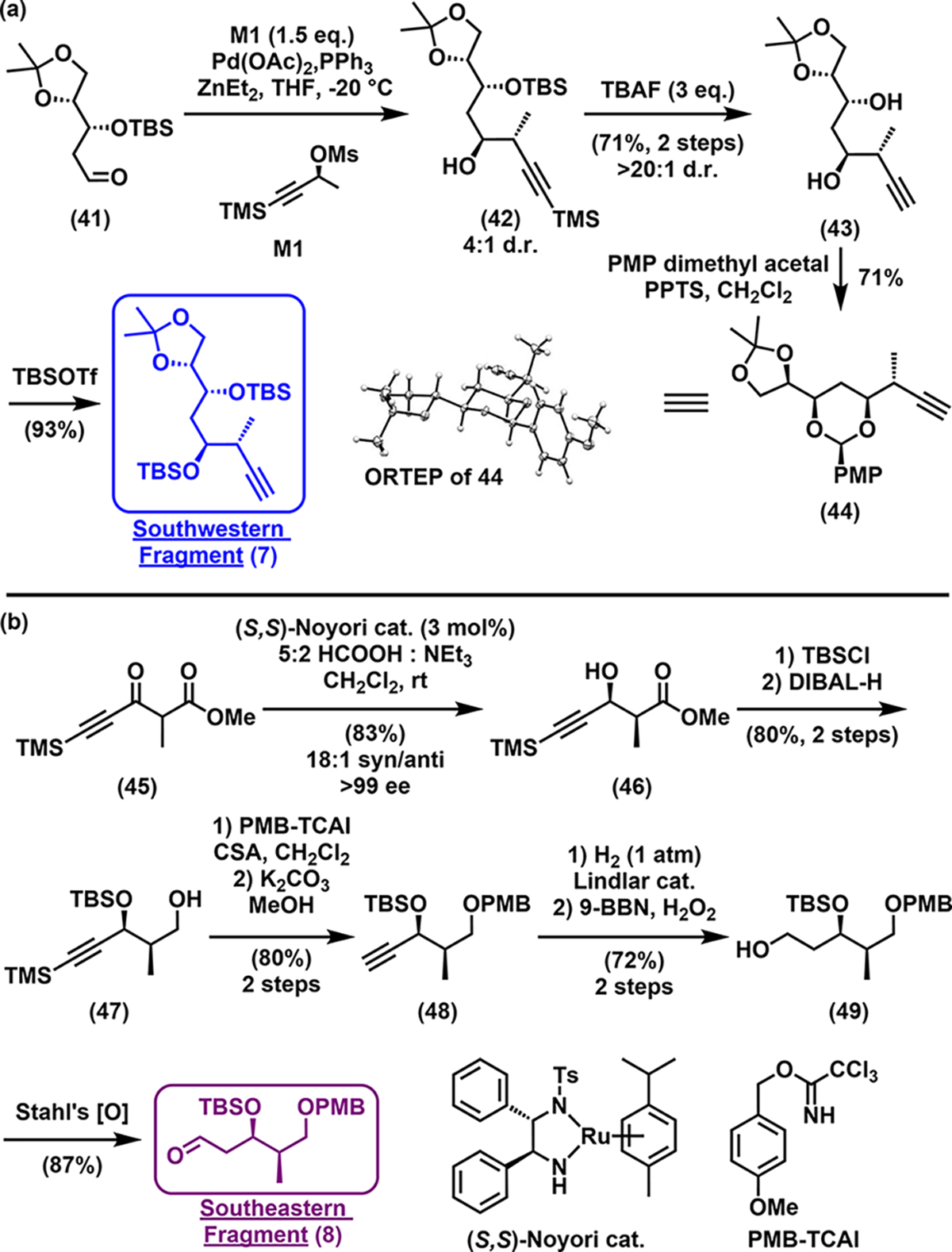
Synthesis of Southwestern (7) and Southeastern (8) Fragments
Synthesis of the southeastern fragment (8, Scheme 7b) began with β-keto ester 45. Catalytic asymmetric dynamic kinetic resolution27 successfully delivered alcohol 46, in good yield and excellent enantio- and diastereoselectivity. Tert-Butyldimethylsilyl (TBS) protection and diisobutylaluminum (DIBAL) reduction of the ester then afforded compound 47, which upon 4-methoxybenzyl (PMB) protection of the primary alcohol followed by removal of the TMS group under mild basic conditions then revealed terminal alkyne 48. Semihydrogenation, hydroboration/oxidation, and Cu/(2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)-catalyzed aerobic oxidation8 completed the Southeastern fragment 8.
Having prepared alkyne 7 and aldehyde 8 (Scheme 8) on a multigram scale, Carreira asymmetric alkynylation28 cleanly afforded adduct 51 with excellent yield and diastereoselectivity. Subsequent trans-reduction of the triple bond with conventional aluminum hydride reagents (LiAlH4, Red-Al), however, failed to give the desired allylic alcohol in appreciable yield. We therefore turned to a Ru-catalyzed hydrostannation/destannation tactic,29a,b which gratifyingly led to the desired trans-reduction in good yield (72%). Stereochemical assessment of the C7-hydroxyl of 53 was achieved via Mosher ester analysis30 (see Table S27). Methylation then led to compound 54, which, after exposure to 50% aqueous trifluoroacetic acid (TFA), cleanly furnished triol 55 as the major product. Selective tosylation and epoxide ring closure, followed by reinstallation of the silyl protecting group, then gave 56 in high overall yield (92%, three steps), which was subjected to BF3-assisted nucleophilic ring opening with lithium trimethylsilyl (TMS) acetylide to furnish alcohol 57 in near quantitative yield. Methylation employing Meerwein’s reagent gave 58, which was followed by global silyl group removal with TBAF and the hydroxyl groups reprotected with triethylsilyl, which at the end of our synthesis proved much easier to remove. The PMB protecting group in 59 was next removed, and in turn Dess–Martin oxidation31 of the resulting alcohol followed by Horner–Wadsworth–Emmons (HWE) olefination32 led to 61. Finally, bromination of the terminal alkyne and hydrostannation33 permitted regio- and stereoselective formation of the vinylstannane, completing the synthesis of the linear southern hemisphere 2 on a 1.26 g scale.
Scheme 8.
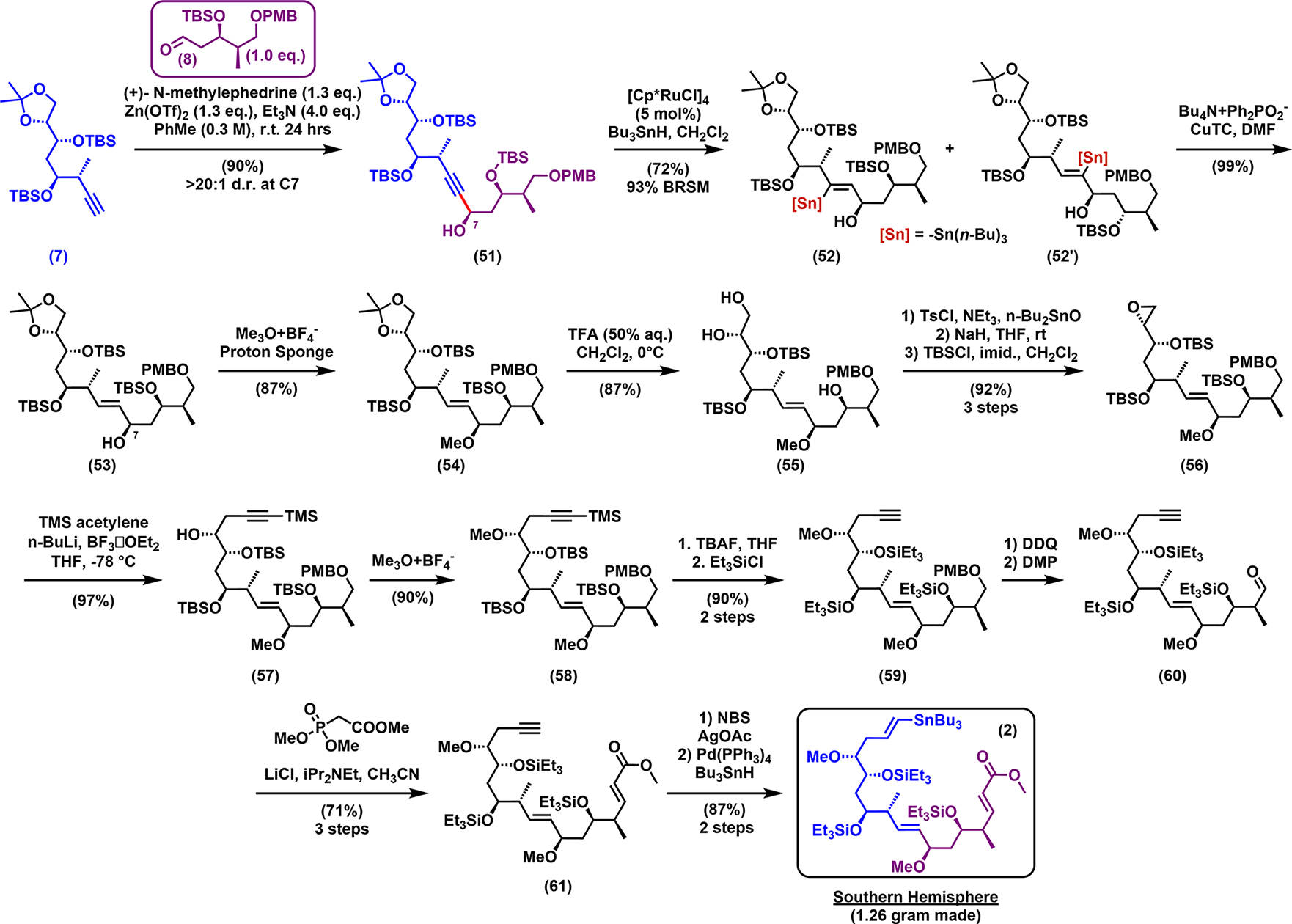
Synthesis of the Southern Hemisphere (2)
With both the northern (3b) and southern (2) hemispheres successfully prepared (Scheme 9), a Stille union reaction6 united 2 and 3. To our delight, the desired (E,E,E)-1,3,5-triene 62 was formed in excellent yield on an appreciable 200 mg scale with no isomerization of double bonds! The methyl ester was next hydrolyzed to afford seco-acid 63 via transesterification with trimethyltin hydroxide.34 Macrolactonization, employing Mukaiyama’s conditions,35 afforded the silylprotected macrocycle 64 in good yield.36 Finally, global removal of triethylsilyl groups gratifyingly proceeded cleanly under mild conditions (TBAF, HOAc, and 0 °C) to complete the synthesis of the reported structure of neaumycin B (1) isolated as a white powder. Notably, 1 was prepared on a 90 mg scale in a single batch, with a 2.3% overall yield.
Scheme 9.
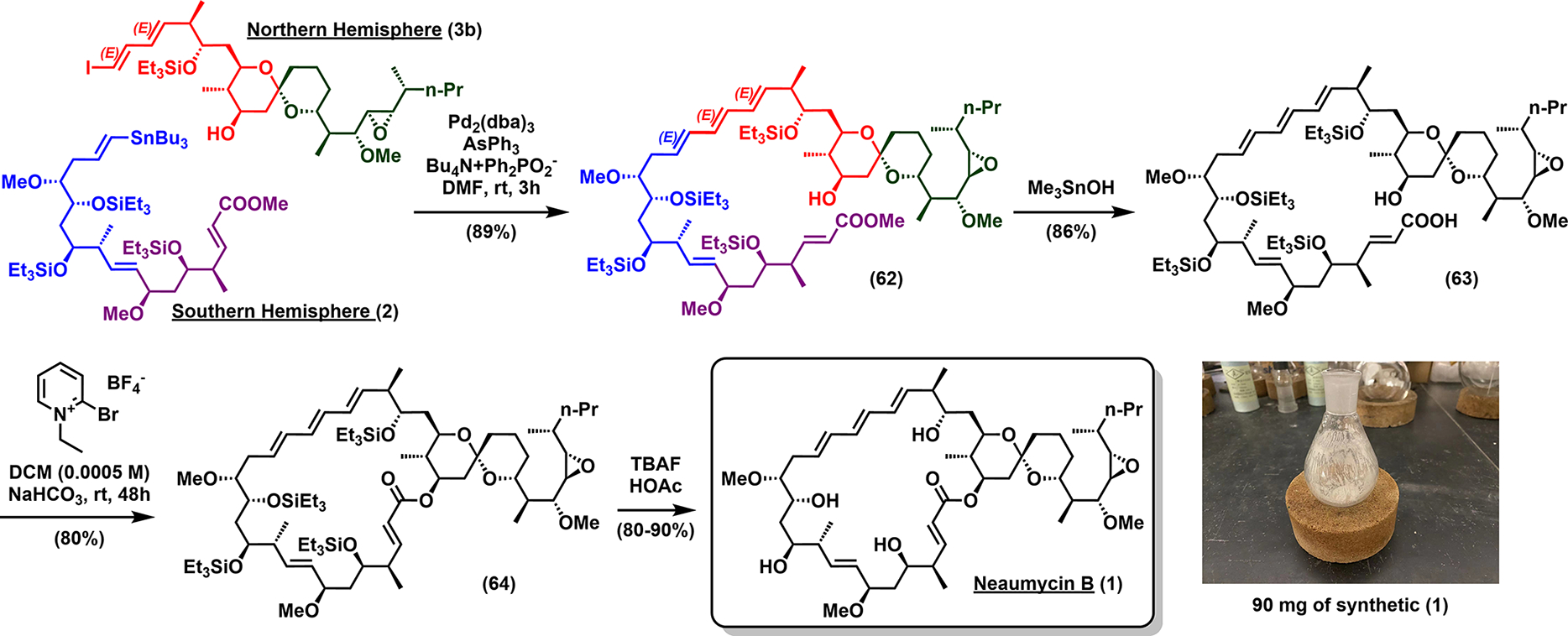
End Game for the Proposed Structure of Neaumycin B (1)
Unfortunately, the 1H NMR spectra of synthetic 1 displayed significant deviations from that of the spectra reported by Fenical et al.3 In addition, 1 displayed no activity against glioblastoma cells (see Figure S5). Importantly, the stereo-chemical assignments of synthetic 1 were derived from X-ray crystallography analysis of each fragment (Figure 1). That is, crystal structures of compounds 33, 44, and 50 (see SI for details) confirmed the stereochemistry that spans C20–C41, C8–C14, and C3–C6 of synthetic neaumycin B (see Figure 1), respectively. The stereogenicity at C7 was also confirmed by Mosher ester analysis (see Table S27 for details). Based on the evidence reported here, we are confident that the synthetic neaumycin B (1) prepared here matches the structure reported by Fenical et al.3
Figure 1.
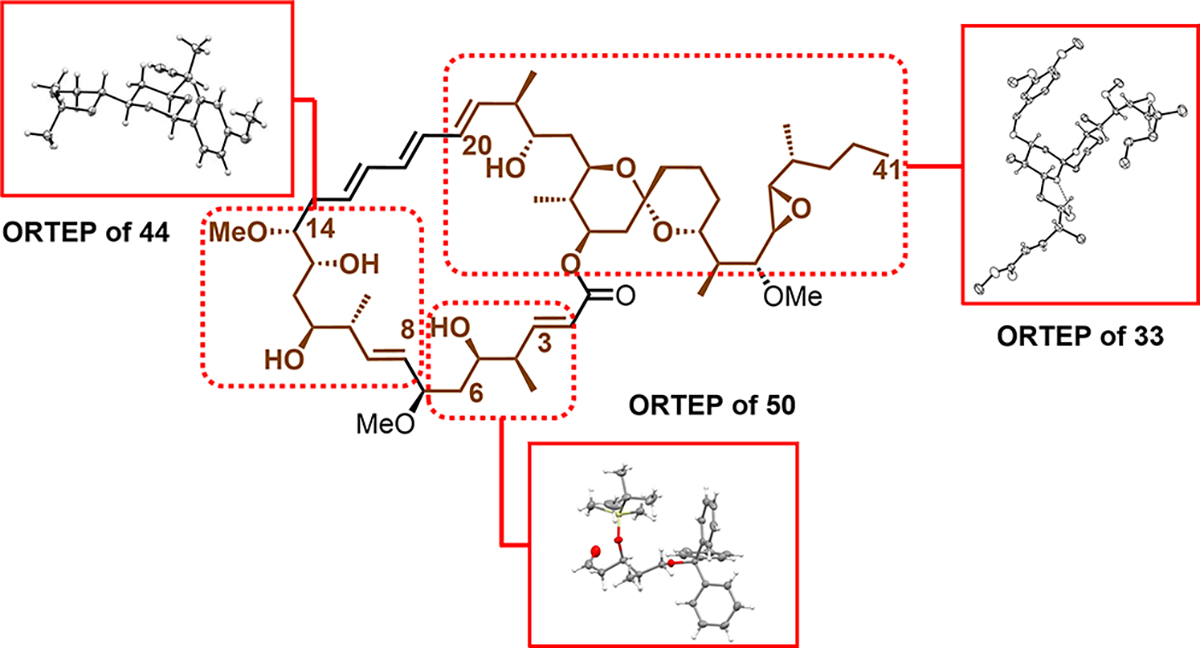
Crystal Structures of Each Fragment.
Supplementary Material
ACKNOWLEDGMENTS
We thank NIH grant no. CA-19033 for financial support. We thank Drs. Patrick J. Carroll, Michael Gau, and C. Ross, III, at the University of Pennsylvania for assistance in X-ray structure and HRMS. We thank Dr. Jun Gu and Dr. Yike Zou for their generous help on NMR analysis. We also thank Dr. David C. Schultz and the staff of the University of Pennsylvania High-throughput Screening Core (RRID: SCR_022379) for providing the in vitro toxicity testing in U87-MG cells.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c06573.
Experimental procedures and analytical data for all new compounds (PDF)
Accession Codes
CCDC 2271141-2271144 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Contributor Information
Jiaming Ding, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Amos B. Smith, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
REFERENCES
- (1).Huang SX; Wang XJ; Yan YJ; Wang JD; Zhang J; Liu CX; Xiang WS; Shen B Neaumycin: A New Macrolide from Streptomyces sp. NEAU-x211. Org. Lett. 2012, 14 (5), 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Simone M; Maffioli SI; Tocchetti A; Tretter S; Cattaneo M; Biunno I; Gaspari E; Donadio S Additional congeners of the macrolide neaumycin: structure revision and biological activity. J. Antibiot. 2015, 68 (6), 406–408. [DOI] [PubMed] [Google Scholar]
- (3).Kim MC; Machado H; Jang KH; Trzoss L; Jensen PR; Fenical W Integration of Genomic Data with NMR Analysis Enables Assignment of the Full Stereostructure of Neaumycin B, a Potent Inhibitor of Glioblastoma from a Marine-Derived Micromonospora. J. Am. Chem. Soc. 2018, 140 (34), 10775–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ostrom QT; Bauchet L; Davis FG; Deltour I; Fisher JL; Langer CE; Pekmezci M; Schwartzbaum JA; Turner MC; Walsh KM The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncology 2014, 16 (7), 896–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Takeshita H; Sugai T; Fuwa H Stereoselective Synthesis of the Southern Hemisphere Acyclic Domain of Neaumycin B. J. Org. Chem. 2021, 86 (9), 6787–6799. [DOI] [PubMed] [Google Scholar]; (b) Pyun YM; Cho SI; Lee SJ; Lee DH Synthesis of the C1-C18 fragment of Neaumycin B. Bull. Korean Chem. Soc. 2022, 43 (12), 1364–1366. [Google Scholar]; (c) Liang XT; Yoo M; Schempp T; Maejima S; Krische MJ Ruthenium-Catalyzed Butadiene-Mediated Crotylation and Oxazaborolidine-Catalyzed Vinylogous Mukaiyama Aldol Reaction for The Synthesis of C1–C19 and C23–C35 of Neaumycin B. Angew. Chem., Int. Ed. 2022, 61, e202214786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stille JK The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles [New Synthetic Methods (58)]. Angew. Chem., Int. Ed. Engl. 1986, 25 (6), 508–523. [Google Scholar]
- (7).(a) Smith AB; Adams CM Evolution of dithiane-based strategies for the construction of architecturally complex natural products. Acc. Chem. Res. 2004, 37 (6), 365–377. [DOI] [PubMed] [Google Scholar]; (b) Melillo B; Chen MZ; Forestieri R; Smith AB An Effective Bifunctional Aldehyde Linchpin for Type II Anion Relay Chemistry: Development and Application to the Synthesis of a C16–C29 Fragment of Rhizopodin. Org. Lett. 2015, 17 (24), 6242–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deng YF; Smith AB Evolution of anion relay chemistry: construction of architecturally complex natural products. Acc. Chem. Res. 2020, 53 (4), 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ryland BL; Stahl SS Practical aerobic oxidations of alcohols and amines with homogeneous copper/TEMPO and related catalyst systems. Angew. Chem., Int. Ed. 2014, 53 (34), 8824–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fujita E; Nagao Y; Kaneko K Useful dethioacetalization with soft acid metal salts: Thallium trinitrate and mercuric perchlorate. Chem. Pharm. Bull. 1978, 26 (12), 3743–3751. [Google Scholar]
- (10).(a) Evans DA; Chapman KT; Carreira EM Directed reduction of β-hydroxy ketones employing tetramethylammonium triacetoxyborohydride. J. Am. Chem. Soc. 1988, 110 (11), 3560–3578. [Google Scholar]; (b) Saksena AK; Mangiaracina P Recent studies on veratrum alkaloids: a new reaction of sodium triacetoxyborohydride [NaBH-(OAc)3]. Tetrahedron Lett. 1983, 24 (3), 273–276. [Google Scholar]
- (11).Katsuki T; Sharpless KB The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102 (18), 5974–5976. [Google Scholar]
- (12).Suzuki I; Saimoto H; Tomioka H; Oshima K; Nozaki H Regio- and stereoselective ring opening of epoxy alcohols with organoaluminium compounds leading to 1, 2-diols. Tetrahedron Lett. 1982, 23, 3597–3600. [Google Scholar]
- (13).Corey EJ; Fuchs PL A synthetic method for formyl→ ethynyl conversion (RCHO→ RC≡CH or RC≡CR′). Tetrahedron Lett. 1972, 13, 3769–3772. [Google Scholar]
- (14).(a) Tomioka H; Suzuki T; Oshima K; Nozaki H The role of trimethylsilyl group in highly stereoselective eposidation of allylic alcohols. Tetrahedron Lett. 1982, 23 (33), 3387–3390. [Google Scholar]; (b) Trost BM; Ball ZT; Laemmerhold KM An alkyne hydrosilylationoxidation strategy for the selective installation of oxygen functionality. J. Am. Chem. Soc. 2005, 127 (28), 10028–10038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) .Trost BM; Sieber JD; Qian W; Dhawan R; Ball ZT. Asymmetric Total Synthesis of Soraphen A: A Flexible Alkyne Strategy. Angew. Chem., Int. Ed. 2009, 48 (30), 5478–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]; For review article on applications of related chemistry to natural product synthesis, see: [Google Scholar]; (d) Frihed TG; Fuürstner A. Progress in the trans-Reduction and trans-Hydrometalation of Internal Alkynes. Applications to Natural Product Synthesis. Bull. Chem. Soc. Jpn. 2016, 89 (2), 135–160. [Google Scholar]
- (15).Evans DA; Bartroli J; Shih TL Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. J. Am. Chem. Soc. 1981, 103 (8), 2127–2129. [Google Scholar]
- (16).(a) Narasaka K; Pai FC Stereoselective reduction of β-hydroxyketones to 1,3-diols highly selective 1,3-asymmetric induction via boron chelates. Tetrahedron 1984, 40 (12), 2233–2238. [Google Scholar]; (b) Narasaka K; Pai HC Stereoselective Synthesis of Meso(or Erythro) 1,3-Diols from Β-Hydroxyketones. Chem. Lett. 1980, 9 (11), 1415–1418. [Google Scholar]; (c) Chen KM; Hardtmann GE; Prasad K; Repic O; Shapiro MJ 1, 3-syn diastereoselective reduction of β-hydroxyketones utilizing alkoxydialkylboranes. Tetrahedron Lett. 1987, 28 (2), 155–158. [Google Scholar]
- (17).Appel R Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angew. Chem., Int. Ed. Engl. 1975, 14 (12), 801–811. [Google Scholar]
- (18).Ai YR; Ye N; Wang QY; Yahata K; Kishi Y Zirconium/nickel-mediated one-pot ketone synthesis. Angew. Chem., Int. Ed. 2017, 56 (36), 10791–10795. [DOI] [PubMed] [Google Scholar]
- (19).Gudmundsson HG; Kuper CJ; Cornut D; Urbitsch F; Elbert BL; Anderson EA Synthesis of Cyclic Alkenyl Dimethylsiloxanes from Alkynyl Benzyldimethylsilanes and Application in Polyene Synthesis. J. Org. Chem. 2019, 84 (22), 14868–14882. [DOI] [PubMed] [Google Scholar]
- (20).Itoh T; Jitsukawa K; Kaneda K; Teranishi S Vanadium-catalyzed epoxidation of cyclic allylic alcohols. Stereoselectivity and stereocontrol mechanism. J. Am. Chem. Soc. 1979, 101 (1), 159–169. [Google Scholar]
- (21).Chan TH; Lau PWK; Li MP Cleavage of the siliconcarbon bond by fluoride ion in triorganosilyloxiranes. The stereochemistry of substitution at oxiranyl carbon. Tetrahedron Lett. 1976, 17 (31), 2667–2670. [Google Scholar]
- (22).(a) Wittig G; Haag W Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien (I. Mitteil. Chem. Ber. 1955, 88 (11), 1654–1666. [Google Scholar]; (b) Isler O; Gutmann H; Montavon M; Ruegg R; Ryser G; Zeller P Synthesen in der carotinoid-reihe. 10. Mitteilung. Anwendung der Wittig-reaktion zur synthese von estern des bixins und crocetins. Helv. Chim. Acta 1957, 40 (5), 1242–1249. [Google Scholar]
- (23).(a) Hodgson DM Chromium (II)-mediated synthesis of E-alkenylstannanes from aldehydes and Bu3SnCHBr2. Tetrahedron Lett. 1992, 33 (38), 5603–5604. [Google Scholar]; (b) Hodgson DM; Foley AM; Lovell PJ Improved Cr (II)-mediated synthesis of E-alkenyl-stannanes from aldehydes using Bu3SnCHI2 in DMF. Tetrahedron Lett. 1998, 39 (35), 6419–6420. [Google Scholar]
- (24).(a) David-Quillot F; Thibonnet J; Marsacq D; Abarbri M; Duchene A Cross-coupling reaction: stereoselective synthesis of (E)-aryl or heteroarylvinylgermanes. Tetrahedron Lett. 2000, 41 (51), 9981–9984. [Google Scholar]; (b) David-Quillot F; Marsacq D; Balland A; Thibonnet J; Abarbri M; Duchene A Efficient synthesis of (E)-vinylgermanes via Stille cross-coupling reaction from 1-tributylstannyl-2-trialkyl (triphenyl) germylethylenes. Synthesis-Stuttgart 2003, 2003 (3), 448–454. [Google Scholar]; (c) Lee SJ; Anderson TM; Burke MD A Simple and General Platform for Generating Stereochemically Complex Polyene Frameworks by Iterative Cross-Coupling. Angew. Chem., Int. Ed. 2010, 49 (47), 8860–8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Wong MYH; Gray GR 2-Deoxypentoses. Stereoselective reduction of ketene dithioacetals. J. Am. Chem. Soc. 1978, 100 (11), 3548–3553. [Google Scholar]; (b) Wang B; Hansen TM; Wang T; Wu DM; Weyer L; Ying L; Engler MM; Sanville M; Leitheiser C; Christmann M; Lu M; Chen J; Zunker N; Cink RD; Ahmed F; Lee Chi-Sing; Forsyth CJ. Total Synthesis of Phorboxazole A via de Novo Oxazole Formation: Strategy and Component Assembly. J. Am. Chem. Soc. 2011, 133 (5), 1484–1505. [DOI] [PubMed] [Google Scholar]
- (26).Marshall JA; Adams ND Addition of Allenylzinc Reagents, Prepared in Situ from Nonracemic Propargylic Mesylates, to Aldehydes. A New Synthesis of Highly Enantioenriched Homopropargylic Alcohols. J. Org. Chem. 1999, 64 (14), 5201–5204. [DOI] [PubMed] [Google Scholar]
- (27).Fang ZJ; Wills M Asymmetric transfer hydrogenation of functionalized acetylenic ketones. J. Org. Chem. 2013, 78 (17), 8594–8605. [DOI] [PubMed] [Google Scholar]
- (28).Frantz DE; Fassler R; Carreira EM Facile enantioselective synthesis of propargylic alcohols by direct addition of terminal alkynes to aldehydes. J. Am. Chem. Soc. 2000, 122 (8), 1806–1807. [Google Scholar]
- (29).(a) Fuürstner A. trans-Hydrogenation, gem-Hydrogenation, and trans-Hydrometalation of Alkynes: An Interim Report on an Unorthodox Reactivity Paradigm. J. Am. Chem. Soc. 2019, 141 (1), 11–24. [DOI] [PubMed] [Google Scholar]; (b) Mo XB; Letort A; Rosca DA; Higashida K; Fuürstner A. Site-Selective trans-Hydrostannation of 1,3- and 1,n-Diynes: Application to the Total Synthesis of Typhonosides E and F, and a Fluorinated Cerebroside Analogue. Chem.—Eur. J. 2018, 24 (38), 9667–9674. [DOI] [PubMed] [Google Scholar]
- (30).Hoye TR; Jeffrey CS; Shao F Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2 (10), 2451–2458. [DOI] [PubMed] [Google Scholar]
- (31).Dess DB; Martin JC Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48 (22), 4155–4156. [Google Scholar]
- (32).Blanchette MA; Choy W; Davis JT; Essenfeld AP; Masamune S; Roush WR; Sakai T Horner-Wadsworth-Emmons reaction: Use of lithium chloride and an amine for base-sensitive compounds. Tetrahedron Lett. 1984, 25 (21), 2183–2186. [Google Scholar]
- (33).Boden CDJ; Pattenden G; Ye T Palladium-catalysed hydrostannylations of 1-bromoalkynes. A practical synthesis of (E)-1-stannylalk-1-enes. J. Chem. Soc. Perkin Trans. 1 1996, No. 20, 2417–2419. [Google Scholar]
- (34).Nicolaou KC; Estrada AA; Zak M; Lee SH; Safina BS A mild and selective method for the hydrolysis of esters with trimethyltin hydroxide. Angew. Chem., Int. Ed. 2005, 44 (9), 1378–1382. [DOI] [PubMed] [Google Scholar]
- (35).Narasaka K; Maruyama K; Mukaiyama T A useful method for the synthesis of macrocyclic lactone. Chem. Lett. 1978, 7, 885–888. [Google Scholar]
- (36).The first conditions employed for the macrocyclization were the Yamaguchi conditions (ref 37). Although these conditions proved successful (60–70%), the Mukaiyama macrocyclization (ref 35) is simpler and milder and provided a cleaner conversion (80%). [Google Scholar]
- (37).Nanaga J; Hirata K; Saeki H; Katsuki T; Yamaguchi M A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.