

ABSTRACT
Taniborbactam (TAN; VNRX-5133) is a novel bicyclic boronic acid β-lactamase inhibitor (BLI) being developed in combination with cefepime (FEP). TAN inhibits both serine and some metallo-β-lactamases. Previously, the substitution R228L in VIM-24 was shown to increase activity against oxyimino-cephalosporins like FEP and ceftazidime (CAZ). We hypothesized that substitutions at K224, the homologous position in NDM-1, could impact FEP/TAN resistance. To evaluate this, a library of codon-optimized NDM K224X clones for minimum inhibitory concentration (MIC) measurements was constructed; steady-state kinetics and molecular docking simulations were next performed. Surprisingly, our investigation revealed that the addition of TAN restored FEP susceptibility only for NDM-1, as the MICs for the other 19 K224X variants remained comparable to those of FEP alone. Moreover, compared to NDM-1, all K224X variants displayed significantly lower MICs for imipenem, tebipenem, and cefiderocol (32-, 133-, and 33-fold lower, respectively). In contrast, susceptibility to CAZ was mostly unaffected. Kinetic assays with the K224I variant, the only variant with hydrolytic activity to FEP comparable to NDM-1, confirmed that the inhibitory capacity of TAN was modestly compromised (IC50 0.01 µM vs 0.14 µM for NDM-1). Lastly, structural modeling and docking simulations of TAN in NDM-1 and in the K224I variant revealed that the hydrogen bond between TAN’s carboxylate with K224 is essential for the productive binding of TAN to the NDM-1 active site. In addition to the report of NDM-9 (E149K) as FEP/TAN resistant, this study demonstrates the fundamental role of single amino acid substitutions in the inhibition of NDM-1 by TAN.
KEYWORDS: NDM-1, taniborbactam, K224, metallo-β-lactamase, β-lactamase inhibitor
INTRODUCTION
β-Lactam antibiotics play a central role in antimicrobial therapy, and, among them, carbapenems are the most potent against life-threatening infections caused by highly drug-resistant bacteria. Therefore, resistance to carbapenems, especially in Gram-negative bacteria, is a significant problem worldwide (1, 2). Although several mechanisms are involved, the production of carbapenemases, enzymes capable of hydrolyzing carbapenems, is the most common and effective mechanism of resistance to carbapenems in Gram-negative bacteria (3). Consequently, several new β-lactamase inhibitors (BLIs) have recently been developed to provide more treatment options for infections caused by carbapenemase-producing Gram-negative bacteria (4). However, due to fundamental structural and mechanistic differences between β-lactamases, clinically available BLIs are only effective against serine β-lactamases (SBLs; classes A, C, and D β-lactamases). SBLs use an active-site serine nucleophile and a transient covalent reaction intermediate to catalyze the hydrolysis of β-lactams. In contrast, class B contains the metallo-β-lactamases (MBLs), which catalyze the hydrolysis of β-lactam drugs through a non-covalent mechanism in which one or two equivalents of bound zinc ions promote the formation of a nucleophilic hydroxide that substitutes for the Ser residue used by SBLs (5) (Fig. 1).
FIG 1.

General catalysis mechanism of B1-metallo-β-lactamases. The resting unliganded B1 MBL contains a dizinc-bridging hydroxide ion within the hydrogen bonding distance of the side chain of Asp120 (E). Substrates bind with their β-lactam carbonyl oxygen coordinating to Zn1 and the conserved carboxylate coordinates to Zn2 as well as a backbone amide nitrogen provided by a structurally conserved K/R residue (ES). Upon the nucleophilic attack performed by the zinc-bound hydroxide, a high-energy tetrahedral intermediate (HE-TI) is formed, which quickly transitions to form the anionic intermediate (AI) after the cleavage of the C-N bond in the β-lactam ring. The negative charge is stabilized by a strong interaction with Zn2 and is delocalized within the structure of the hydrolyzed antibiotic. In the last step, protonation of the N atom (the rate-limiting step) and the addition of a water molecule complete the product and restore the enzyme for another cycle of catalysis (EP).
Besides the different catalytic mechanisms, the active sites of SBLs and MBLs also present different sizes and topologies (6–9). SBLs have a narrow and deep catalytic site (10). In contrast, the active site in MBLs is in a shallow groove, with only a few contact points to bind the inhibitor or substrate (11). In B1 MBLs, a positively charged side chain (K224 in IMP-1 and NDM-1 or R228 in VIM-2) is located in the active-site loop 10 (ASL-10) approximately 6 Å from Zn2 and provides an anchoring position for the conserved carboxylate found in all β-lactam drugs (12). Ample crystallographic evidence of MBLs in complex with different β-lactam hydrolysis products supports the anchoring role of K224, which has also been exploited in inhibitor designs (13–15). Another structurally conserved residue, N233, also found in the ASL10, is shown to be within hydrogen (H)-bonding distance to the newly formed carboxylate in the ring-opened β-lactam products (16, 17). Lastly, a conserved extended β-hairpin loop, the ASL3, towers over one side of the active site, providing a broad hydrophobic surface for binding the core ring structures of the β-lactam substrates (18–21).
The first BLIs introduced in the clinic (clavulanic acid, tazobactam, and sulbactam) are mechanism-based inhibitors. These β-lactam compounds are hydrolyzed by the SBLs but remain bound to the active-site serine residue, thereby inactivating the enzyme. None of these compounds are significantly active against any carbapenemase, including MBLs. Newly developed BLIs such as the diazabicyclooctanones (DBOs; avibactam and relebactam), and the boronate compounds (vaborbactam), inactivate class A (e.g., KPC) and some class D (e.g., OXA-48) carbapenemases (1, 4, 22). These compounds act as reversible inhibitors of SBLs, but none of them inhibits MBLs (4).
Taniborbactam (TAN; formerly VNRX-5133) (Fig. 2) is a novel BLI belonging to the group of bicyclic boronate BLIs (23). TAN is unique in that it inhibits not only SBLs but also some MBLs (excluding IMP and L1 types). Co-crystal structures of CTX-M-15 (PDB ID 6SP6) and VIM-2 (PDB ID 6SP7) with TAN revealed the structural basis of this dual inhibition. The catalytic Ser70 of CTX-M-15 is covalently bound to the boron atom of TAN, with one hydroxyl group on the boron atom located in the oxyanion hole, mimicking the acylation tetrahedral intermediate. On the other hand, the structure of VIM-2:TAN showed that the tetrahedral boron of TAN was bound to the active-site zinc (Zn1) through an oxygen atom, thus mimicking the tetrahedral intermediate formed following the nucleophilic attack of the β-lactam carbonyl by the hydroxide anion (24). Thus, the positioning of the boron atom with respect to the attacking nucleophile (the Ser residue in SBLs and the Zn-bound hydroxide in MBLs) is essential for enzyme inhibition of boron-based inhibitors (23, 24). In addition, TAN demonstrates potent in vitro rescue of cefepime activity against carbapenem-resistant, clinical isolates of Enterobacterales and Pseudomonas aeruginosa (25). Therefore, TAN is a promising candidate for the treatment of infections caused by MBL-harboring Gram-negative bacteria. The combination cefepime (FEP)/TAN has just completed a Phase III clinical trial (NCT03840148) to evaluate its safety and efficacy in patients with complicated urinary tract infections.
FIG 2.

Structures of β-lactams used in this study and taniborbactam (VNRX-5133).
Notwithstanding its recent discovery, resistance mechanisms to FEP/TAN have already been described. Most cases of FEP/TAN resistance include co-production of carbapenemase and other β-lactamases such as CTX-M-type ESBLs, PBP2, and three mutations, nonfunctional OmpK35 due to an early stop codon in Enterobacterales, and mutations in PBP3 and OprD MexR/Z in Pseudomonas aeruginosa (26–32). Most recently, Le Terrier et al. reported that NDM-9, which differs from NDM-1 only by a single amino acid substitution (E149K), confers resistance to FEP/TAN (33). Crystallographic evidence of the binding mode of TAN to target enzymes, like NDM-1, could provide further clues regarding possible mechanisms of resistance. The crystal structure of the NDM-1:TAN complex obtained by Krajnc et al. revealed that K224 acts as the anchoring residue by forming two H-bonds with TAN (24). One boron (B)-bonded oxygen bridges the two zinc ions in the active site. However, it is positioned closer to Zn1 (1.9 Å) than Zn2 (3 Å). In addition, the aryl carboxylate interacts with Zn2 and with K224 via an H-bond in both bicyclic and tricyclic forms. Surprisingly, the ASL3, which is involved in substrate or inhibitor binding (20), does not seem to play an important role in TAN binding (24).
The homologous residue of K224 in VIM-2 is R228. Previously, it was shown that the R228L substitution in VIM-24 improves the hydrolytic activity of the enzyme toward oxyimino-cephalosporins, ceftazidime (CAZ), and FEP (34). Therefore, we hypothesized that substitution at position 224 in NDM-1 could also increase the tendency toward resistance to FEP and potentially, to FEP/TAN. To test this, a library of engineered variants of NDM-1 at position 224 was generated, and constructs were expressed in Escherichia coli DH10B. We then investigated their susceptibilities to several β-lactams, including the FEP/TAN combination. In addition, we conducted biochemical analysis and molecular dynamics for two NDM-1 K224X variants of interest and compared them with NDM-1. Our analyses revealed the important role of K224 in NDM-1 inhibition by TAN and elucidated important structure-activity relationships heretofore unexpected.
RESULTS
Antibiotic susceptibility assays
To determine the effect of substitutions at position 224 in NDM-1, we generated all 19 variants at this position and assessed their antibiotic susceptibility profile against a variety of β-lactams (Fig. 2). In general, while all the engineered K224X substitutions resulted in reduced minimum inhibitory concentrations (MICs) to FEP, imipenem (IMI), tebipenem (TBPM), and cefiderocol (FDC), 14/19 (74%) substitutions conferred elevated MIC values for FEP/TAN and CAZ (Fig. 3).
FIG 3.
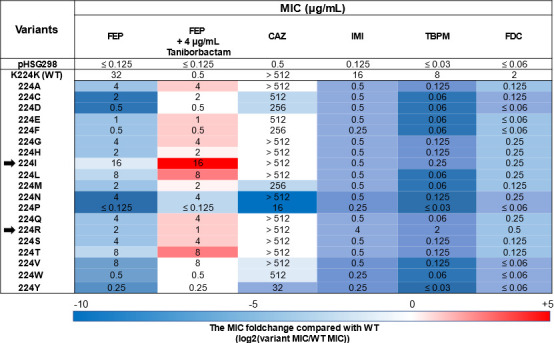
Minimum inhibitory concentrations (MICs) of different β-lactams against E. coli DH10B strains harboring the pHSG298 blaNDM-1(K224X) gene. The heat map represents the doubling dilution changes in the MIC values of the K224X variants compared with that of the NDM-1 wild type. The results shown are the mode of three biological replicates. FEP, cefepime; CAZ, ceftazidime; IMI, imipenem; TBPM, tebipenem; FDC, cefiderocol; WT, wild type.
Compared to NDM-1, the MICs of FEP against E. coli expressing the K224X variants were 2 to ≥8 doubling dilution lower. However, the K224I variant conferred a comparable level of resistance to FEP (16 µg/mL) as NDM-1 wild type (32 µg/mL). The expression of three variants (K224V, K224L, and K224T) yielded MIC values of 8 µg/mL, whereas the expression of five variants (K224G, K224A, K224S, K224N, and K224Q) conferred MIC values of 4 µg/mL. Finally, the expression of the K224P variant produced the lowest MIC value within the variants (MIC ≤0.125 µg/mL).
The MIC values of FEP were also measured in combination with TAN (at a fixed concentration of 4 µg/mL). Notably, while the MICs of the E. coli expressing NDM-1 were reduced from 32 μg/mL to 0.5 μg/mL upon the addition of TAN, a reduction of the MIC value for the FEP/TAN combination was not observed for any of the E. coli expressing the K224X variants. Therefore, the MIC values obtained by 12/19 (63%) of the K224X variants for the FEP/TAN combination were 2 to 5 doubling dilution higher than the MIC of the NDM-1 wild type. Importantly, the MIC values of FEP and FEP/TAN yielded by the expression of the K224I variant were equally high (16 µg/mL). Only the expression of the K224P variant displayed a MIC 2 doubling dilution lower than that in the NDM-1 wild type.
Contrary to FEP, decreased CAZ MIC for ≥2 doubling dilutions was observed only in 5/19 (26%) of the K224X variants. Some substitutions, including K224I and K224R, conferred the same level of resistance to CAZ as NDM-1 wild type. On the other hand, except for the K224R variant, the MIC values of IMI obtained by all the K224X variants were 2 to 6 doubling dilution lower than the MIC values of the wild type. Likewise, the MIC values of TBPM against all the K224X variants, but K224R, were 2 to 8 doubling dilution lower than the MIC of the wild type. Lastly, the MIC values of FDC against all K224X variants were 2 to ≥5 doubling dilution lower than the MIC values of the wild type. Of note, an MIC reduction was also observed for the K224I variant, for which significant MIC reductions for FEP and CAZ (both antibiotics are the two structural components of FDC) were not observed.
The effect of 50-µM EDTA addition on the MICs of FEP, CAZ, or IMI was also measured (Fig. 4). As expected, the MIC values obtained on media supplemented with EDTA were lower than those obtained under normal conditions, suggesting that most of the substitutions at this position 224 affect the Zn binding capabilities. Based on the doubling dilution difference between the MICs of antibiotics with and without EDTA, most of the K224X variants behaved as the NDM-1 WT. However, the effect of EDTA supplementation on the IMI MICs could not be fully explored, as the basal MIC value of the E. coli DH10B (0.25 µg/mL) was the same or just one dilution lower than the MIC value for the vast majority of the K224X variants.
FIG 4.
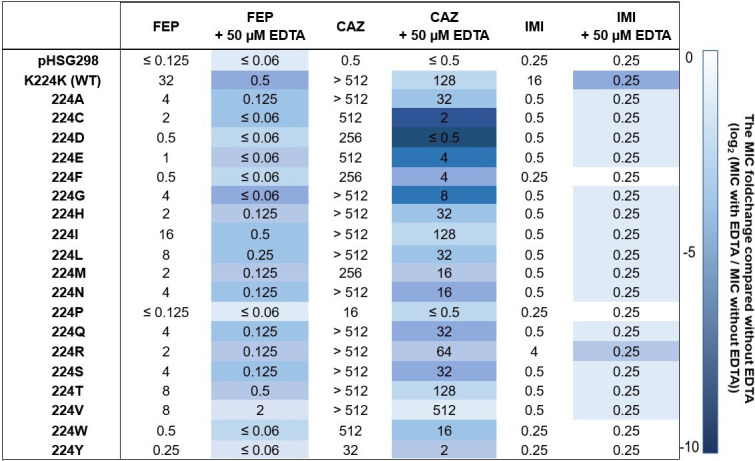
Minimum inhibitory concentrations (MICs) of different β-lactams with or without EDTA against E. coli DH10B strains harboring the pHSG298 blaNDM-1(K224X) gene. The heat map represents the doubling dilution changes in the MIC values with EDTA compared with those without EDTA. MIC fold change calculations were conducted by converting “>” to the next higher dilution (e.g., >512 µg/mL was converted to 1,024 µg/mL), and “≤” was assumed to be the same value (e.g., ≤0.125 µg/mL was converted to 0.125 µg/mL). The results shown are the mode of three biological replicates. Abbreviations: FEP, cefepime; CAZ, ceftazidime; IMI, imipenem; WT, wild type.
Immunoblotting
The expression level of the K224X variants is another factor that can possibly alter the antibiotic resistance profiles (35). Therefore, the expression of the K224X variants expressed from pHSG298 in E. coli DH10B cells by immunoblotting was analyzed. As shown in Fig. 5, most variants were produced at a lower level than the NDM-1 wild type; only the K224A and K224Y variants were produced as much as the NDM-1 wild type. Then, to examine the relationship between the expression level and antibiotic resistance, Spearman’s rank correlation test was conducted. However, a significant correlation was not observed between the MIC values and the expression levels (FEP/TAN: |r| = 0.20, P = 0.40).
FIG 5.

Expression of the blaNDM K224X library in E. coli DH10B cells. The steady-state expression of the K224X library in E. coli DH10B was analyzed by immunoblotting, as described in Material and Methods. * NDM-1 K224I, ** NDM-1 K224R.
Steady-state kinetics
Based on the MIC results, we examined two distinct variants: K224I, for which TAN did not have any inhibitory effect but displayed lower IMI MICs compared to the NDM-1 wild type, and K224R, which showed the smallest impact on the IMI and FDC MICs. Thus, the steady-state kinetics parameters of FEP and IMI hydrolysis by NDM-1, K224I, and K224R variants were determined.
As shown in Table 1, the FEP MIC values for the wild type and K224I- and K224R-producing E. coli DH10B strains were 32, 16, and 2 µg/mL, respectively. The kcat/KM values were 1.02, 0.63, and 0.08 µM−1 s−1, respectively (Table 1). The differences observed in kcat/KM were mainly due to the large changes in KM values, as kcat values were similar. Likewise, the kcat/KM values obtained for the hydrolysis of IMI by the NDM-1 wild type, and the K224I and K224R variants (5.85, 0.11, and 2.43 µM−1 s−1, respectively) followed the same trend as their respective MICs (16, 0.5, and 4 µg/mL). The extremely high KM value of K224I (KM = 1644 µM) contributed to its lowest catalytic efficiency compared to the other two enzymes.
TABLE 1.
Kinetics values of the hydrolysis of imipenem and cefepime by NDM-1 wild type and the K224R and K224I variants
| Imipenem | Cefepime | Taniborbactam | |||||
|---|---|---|---|---|---|---|---|
| k cat | K M | kcat/KM | k cat | K M | kcat/KM | IC50 | |
| (s−1) | (µM) | (µM−1 s−1) | (s−1) | (µM) | (µM−1 s−1) | (µM) | |
| NDM-1 (WT) | 164 ± 5 | 28 ± 4 | 5.85 ± 0.85 | 82 ± 8 | 80 ± 5 | 1.02 ± 0.12 | 0.010 ± 0.001 |
| NDM-1 (K224I) | 184 ± 19 | 1644 ± 170 | 0.11 ± 0.01 | 90 ± 9 | 144 ± 15 | 0.63 ± 0.09 | 0.140 ± 0.014 |
| NDM-1 (K224R) | 73 ± 2 | 30 ± 4 | 2.43 ± 0.32 | 61 ± 6 | 750 ± 80 | 0.08 ± 0.01 | NDa |
ND, not determined.
IC50
We then determined the half maximal inhibitory concentrations (IC50s) of TAN against the K224I variant compared to NDM-1. TAN effectively inhibited NDM-1 wild type, as predicted by the decreased FEP/TAN MIC values. Conversely, as expected by the unchanged FEP and FEP/TAN MICs, the TAN IC50 value determined for the K224I variant was 14 times higher than that of the NDM-1 wild type (0.14 vs 0.01 µM; Table 1).
Molecular modeling studies
Conventional docking simulations with AutoDock 4 software were next performed using the crystal structure of NDM-1 PDB ID 3SPU. One hundred different docking runs being clustered by an RMSD 2-Å cutoff were conducted for each docking calculation. In the best docking pose, which had the highest number of individual docking results as the identical ligand pose, of TAN in the active site of NDM-1 wild type shown in Figure 6A, TAN is anchored by two H-bonds from K224 and E149. With this H-bond anchoring, the boron atom is in an appropriate position for nucleophilic attack by NDM-1 (Boron–OH- distance is 2.71 Å). Conversely, we obtained two poses of similar energy in docking studies of the K224I variant with TAN (Fig. 6B and C). As shown, the K224I substitution also causes TAN to lose the H-bonding interaction with E149, and consequently, the boron atom is inappropriately positioned to undergo a nucleophilic attack.
Fig 6.
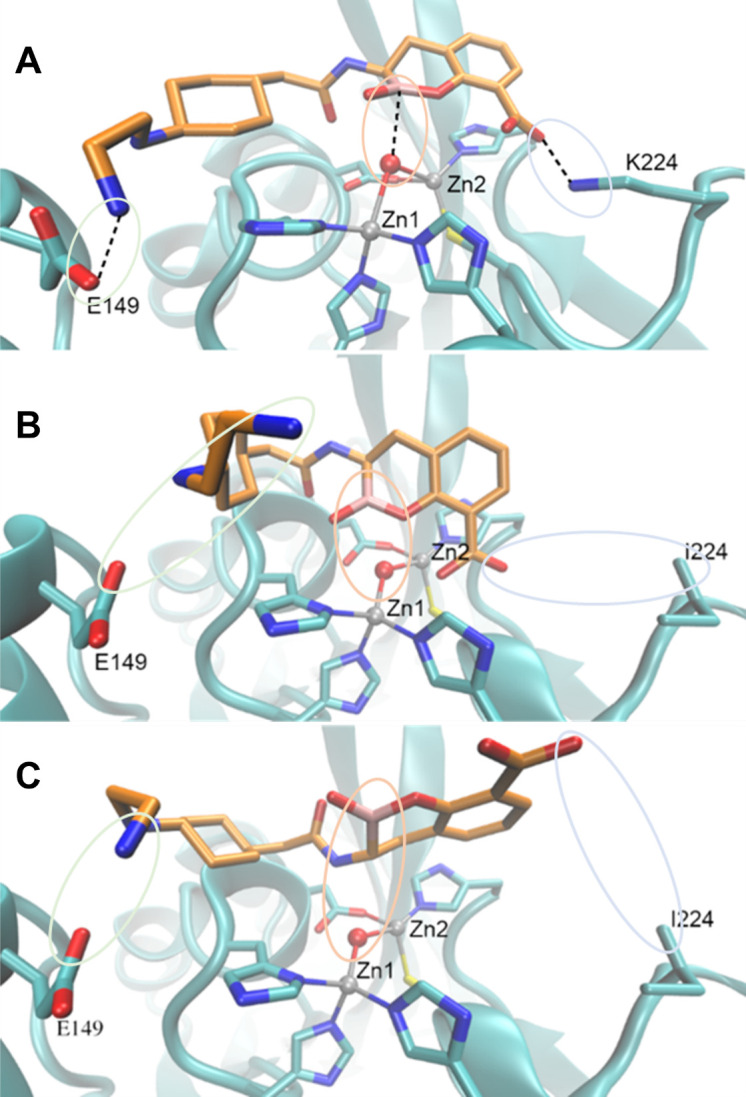
Productive binding of taniborbactam to NDM-1 is mediated by K224.Energy-minimized models of NDM-1 wild-type (A) and NDM K224I (B and C) in complex with taniborbactam. Notice that in (A) taniborbactam is anchored by hydrogen-bonds from(K224 and E149, and the boron (B) atom is properly positioned for nucleophilic attack. In contrast, in (B and C) taniborbactam loses the interaction with K224 and E149, resulting in a non-productive binding that impairs inhibition. C atoms of the protein are shown in cyan, C atoms of taniborbactam in orange, O atoms in red, N atoms in blue, S atoms in yellow and B atom in pink. Zn(II) ions are shown as grey spheres. Taniborbactam-protein interactions are depicted in dash lines.
DISCUSSION
Although several new BLIs are being developed and made clinically available, BLIs against MBL producers are not available at the time of this writing. TAN is a novel BLI belonging to the cyclic boronic acid BLI group and is characterized by its ability to inhibit both SBL and some MBL (23). Although the FEP/TAN combination has not been approved for clinical use, several resistance mechanisms have already been reported.
The R228L amino acid mutation in VIM-24, a VIM-2 clinical variant conferring enhanced hydrolytic activity toward CAZ and FEP, was previously reported (34). Since the homologous position of R228 in NDM-1 is K224, we sought to investigate if substitutions at this position could also affect FEP hydrolysis, thus leading to a decrease in FEP/TAN susceptibility. Contrary to our expectations, 15/19 of the substitutions at position 224 decreased the catalytic activity of the enzyme toward most of the substrates tested, as reflected by the drastic decrease in the MIC values for FEP, IMI, TBPM, and FDC. However, this was not the case for CAZ, as 17/19 of the variants continued displaying MIC values comparable to that of the NDM-1 wild type. This finding is consistent with the previous report by Sun et al. in their work exploring the active-site sequence requirements for β-lactam hydrolysis by NDM-1 (36). This investigation used ampicillin (AMP), cefotaxime (CTX), or IMI to select functional mutants of randomized codon libraries. Analysis of the deep sequencing data from those libraries allowed them to classify the residues into three categories: essential, substrate-specific, or nonessential. Position 224 in NDM-1 was classified as a substrate-specific residue group, as the distribution of residue types observed was dependent on the antibiotic used for selection. Briefly, a broad distribution of amino acid types was observed among the sequences from AMP-selected libraries, whereas a narrower range of amino acid types was observed among sequences from IMI-selected libraries. This study also found that in general, substitutions at these residues decrease the kcat/KM value for IMI hydrolysis more than those for AMP and cefotaxime (CTX) hydrolysis, suggesting that stringent amino acid residues are required for the hydrolysis of carbapenems. In a similar view, Feng et al. provided crystallographic evidence supporting that NDM-1 utilizes a distinct catalytic mechanism for carbapenem hydrolysis compared to penicillin or cephalosporin (37). In addition, carbapenems contact at fewer points with the active site of NDM-1 than penicillins and cephalosporins. The structural analysis of hydrolyzed IMI with NDM-1 revealed that the R2 group contacts minimally with the enzyme and the R1 hydroxyethyl group contacts only W87 as an interaction with the hydrophobic area in the active site (36). In contrast, AMP and cefuroxime display more hydrophobic interactions than IMI. For example, AMP exhibits interactions with L59, M61, and W87, aside from a H-bond with Q119. However, only a handful of the NDM-1:β-lactam interactions have been elucidated, and differences among the same β-lactam class are anticipated. We propose that this might explain why the K224 substitutions did not affect CAZ hydrolysis to the same extent it affected FEP hydrolysis.
Among the variants of the K224X library, K224I and K224R were chosen for further studies. The K224I variant displayed the highest FEP MICs and, consequently, the highest FEP/TAN MIC values among the K224X variants. Conversely, the K224R variant was the variant with the best performance against the carbapenems tested (IMI and TBPM) and FDC. Table 1 shows the changes in the steady-state kinetic constants for the FEP and IMI hydrolysis by these variants, compared to the NDM-1 wild type. In the case of FEP, the kinetic values obtained by NDM-1 and its K224I variant are comparable. In contrast, the dramatic increase of the KM of the K224R variant yields a catalytic efficiency that is one order of magnitude lower than of the wild type. Similar changes explained the different hydrolytic efficiencies toward IMI. This time, however, the K224I variant displays a catalytic efficiency one order of magnitude lower than the wild type as a result of the increased KM. Of note, our kinetic data are consistent with previously published data for NDM-1 and, importantly, the K224R variant. The K224R variant was characterized by Sun et al., as it was consistently among the most common variants selected under AMP, CTX, and IMI selective pressure (36).
Previously, we showed that preservation of the hydrolytic activity under Zn scarcity is the main driver of NDM clinical evolution (38–40). Moreover, enhanced Zn binding capabilities in vivo translate into an increased resistance phenotype (41). On this premise, we tested the susceptibility of the K224X library to FEP, CAZ, and IMI on media supplemented with EDTA. Overall, the MIC values obtained with EDTA are lower than the MIC values obtained under normal conditions, suggesting that most of the substitutions at this position affect the Zn affinity or the stability of the apoprotein (Fig. 4). Interestingly, the effect of EDTA on the MIC values obtained by K224I, K224R, and the NDM-1 WT against FEP and CAZ was comparable. Moreover, the K224V variant was the variant least affected by the Zn depriving environment, as the MIC values obtained against FEP + EDTA and CAZ + EDTA were only 2 and 1 doubling dilutions lower, respectively, compared with the agents alone. More biophysical experiments are needed to establish the effect of these substitutions on the Zn binding affinities or the stability of the apo variants. Lastly, although a statistically significant association between the production level and the MIC values was not demonstrated, the K224P variant had the lowest MIC values for each antimicrobial and condition tested and the lowest expression level. These results resemble the behavior of the VIM R228P variant previously studied (34). Collectively, these results suggest substantial stability issues of this protein probably derived from the major structural perturbation that the substitution by a proline residue causes in Loop 10.
As mentioned previously, K224 is a conserved residue that facilitates β-lactam binding to the active site via an electrostatic interaction with the negatively charged carboxylate group common to this class of antibiotic (12, 39). Therefore, characteristics of the substituted amino acid residue, such as size and polarity can affect substrate binding. As a positively charged residue, arginine should be able to replace lysine in this functional role. However, our results and the crystal structure of the K224R/G232A/N233Q triple mutant obtained by Sun et al. (36) suggest that although the side chain of R224 is positioned to interact with the substrate, it cannot efficiently accommodate the substrates for catalysis. This notion is consistent with the fact that the extent of the changes of KM values was greater than that of kcat (Table 1).
Regarding TAN, the results showed that the inhibitory activity of this compound is dependent upon the precise positioning of the boron atom with respect to the nucleophile hydroxyl/water molecule bridged between the two Zn ions in the active site. As shown in our docking studies, such positioning in NDM-1 is achieved by two anchoring H-bonds with K224 and E149 (Fig. 6A). Our results showed that all the substitutions at position 224 yielded resistance to TAN inhibition, not only because of the loss of the H-bond between the residue at position 224 with the aryl carboxylate but also probably due to the loss of the H-bond between with the ethylamino side chain with E149 (Fig. 6B and C). Consequently, the boron atom is positioned far away from the OH- ion unable to elicit a nucleophilic attack. This also explains the increase in the IC50 value of TAN for the K224I variant. The difference in the mode of TAN interaction with the wild type and the K224I variant highlights the importance of K224 in the productive binding of TAN to the active site.
Surprisingly, the interaction of the TAN ethylamino side chain with E149 was not observed in the crystal structure of the complex NDM-1:TAN previously solved (24). However, the importance of this interaction for TAN-mediated inhibition of NDM-1 and VIM-2 was recently implied (33). In this work, NDM-9, which only differs from NDM-1 by the E149K substitution, was reported to confer FEP/TAN resistance. This study also demonstrated that the homologous substitution in VIM-2 confers the same resistant phenotype (33). The homologous residue of E149 in IMP-1 is D149. As aspartic acid has a lateral chain one carbon shorter than glutamic acid, it is possible that it might not be at the H-bond distance of the ethylamino motif of TAN. This could explain why TAN inhibits NDM-1 and VIM-2, but not IMP-1, even though they are all categorized in the same MBL B1 subgroup. Furthermore, the trade-off between resistance to TAN inhibition and decreased β-lactam hydrolysis is intriguing and may be a factor to consider in future studies of β-lactam/TAN combinations other than FEP/TAN.
Lastly, although this study was conducted only with laboratory variants using site-directed mutagenesis methods, the major findings from this study, the essential role of K224 for TAN-mediated inhibition of NDM-1, and that substitutions at this position impair efficient hydrolysis to some β-lactams (especially carbapenems) are important results that enrich our structure-activity relationship (SAR) knowledge of NDM-1.
In summary, the interaction of TAN with K224 is central to the inhibition of NDM-1. Surprisingly, most of the K224X substitutions in NDM-1 did not influence the ability of NDM to hydrolyze CAZ, but it impaired the hydrolysis of all other substrates, especially carbapenems. Besides the occurrence of NDM-9, this study revealed the fundamental role of K224 in TAN-mediated inhibition of NDM-1. Further studies are needed to elucidate more SAR of MBL to guide the design of the next generation of MBL inhibitors.
MATERIALS AND METHODS
Cloning of blaNDM variants for cell viability assays
The blaNDM-1 gene was originally cloned from Klebsiella pneumoniae 246-61A, a clinical isolate from New Delhi, in 2007 as previously described (40). Based on this construct, E. coli codon-optimized NDM-1 clones containing all 20 amino acid variants at position 224 were synthesized (Genscript Biotech Corp., Piscataway, NJ, USA) into a uniform vector (pHSG298) and codon-optimized for E. coli DH10B for use in antibiotic susceptibility assays.
MIC measurements
MIC measurements were performed in triplicate using the Mueller-Hinton (MH) agar dilution method, according to the Clinical Laboratory and Standards Institute (CLSI) protocol (42). Ceftazidime (CAZ) and cefepime (FEP) were obtained from Sigma-Aldrich (Burlington, MA, USA). TAN, imipenem (IMI), tebipenem (TBPM), and cefiderocol (FDC) were obtained from MedChemExpress (Monmouth Junction, NJ, USA). To assess the resistance profile when zinc availability is limited, the MICs of FEP, CAZ, and IMI were also determined with the addition of 50µM EDTA.
Cloning, expression, and purification of NDM variants
For protein purification, a blaNDM-1 gene construct in the pET-28 (+) plasmid lacking the first 38 amino acids was previously described (40). Based on this construct, E. coli codon-optimized NDM-1 clones containing the K224I and K224R variations were synthesized (Genscript Biotech Corp., Piscataway, NJ, USA). Constructs were transformed into BL21(DE3) E. coli (Thermo Fisher Scientific, Waltham, MA ).
Expression and purification of blaNDM were performed based on a previously described method (14). A bacterial culture was grown at 37°C in 500-mL SOB media until OD600 = 0.6. blaNDM production was induced by the addition of 0.25-mM isopropyl β-D-1-thiogalactopyranoside (IPTG). This culture was also supplemented with 1-mM ZnSO4, and the cells were incubated overnight at 18°C. The cells were harvested and resuspended in 50-mM Tris-Cl (pH 8.0) and 200-mM NaCl and supplemented with 10-mM β-mercaptoethanol. Cells were disrupted by sonication (five cycles of 30 s at 40% amplitude), and the insoluble material was removed by centrifugation for 60 min at 15,000 rpm. The crude extract was loaded onto a 1-mL Ni-Sepharose column equilibrated with buffer A (50-mM Tris-Cl, pH 8.0, 200-mM NaCl, 10-mM β-mercaptoethanol), the column was washed with 10-mL of buffer A + 20-mM imidazole, and His6x-blaNDM was eluted with buffer B (50-mM Tris-Cl, pH 8.0, 200-mM NaCl, 10-mM β-mercaptoethanol, 500-mM imidazole). His6x-blaNDM was mixed with the TEV protease (1:20 TEV:His6x-blaNDM), and the mixture was incubated for 16 h at 4°C during dialysis against 100 volumes of 50-mM Tris-Cl (pH 8.0), 200-mM NaCl, and 10-mM β-mercaptoethanol. NDM was then loaded onto a Ni-Sepharose column to separate it from the His6x tag, the uncleaved fusion protein, and the His6x-tagged TEV protease. NDM was collected in the flow-through of the column. β-Mercaptoethanol was removed from the protein sample by one 12-h dialysis step of NDM against 100 volumes of 10-mM HEPES, pH 7.5, 200-mM NaCl, and 200-µM ZnSO4, followed by three 4-h dialysis steps against 100 volumes of 10-mM HEPES (pH 7.5) and 200-mM NaCl. Protein concentrations were determined from the absorbance at 280 nm using a molar absorption coefficient (ε280) of 27,960 M−1 cm−1.
Steady-state kinetic and inhibitor analysis
Steady-state kinetic and inhibitor parameters were determined by using an Agilent 8453 diode array spectrophotometer at room temperature (~25°C) as previously described (14). Each assay was performed in 10-mM HEPES (pH 7.5), 0.2-M NaCl, 50-µg/mL bovine serum albumin, and 50-µM ZnSO4 in a quartz cuvette with a 0.2-cm path length. Measurements were obtained using IMI and FEP. The kinetic parameters, Vmax (maximum velocity) and KM, were obtained with a nonlinear least-square fit of the data (Henri-Michaelis-Menten, equation 1) using Origin 2020b (OriginLab, Northampton, MA).
| (1) |
where [S] = substrate concentration and v = velocity. In these cases where Vmax was not achievable, a Lineweaver-Burk plot was generated by plotting 1/[S] vs 1 /v generating a line in which Vmax = 1/y-intercept and the x-intercept = −(1/KM). For IC50 determination, reaction mixtures were prepared in 10-mM HEPES (pH 7.5), 0.2-M NaCl, 50-µg/mL bovine serum albumin, and 50-µM ZnSO4 with various concentrations of TAN. The IC50 values were performed after a 5-min preincubation. Reactions were initiated by the addition of 50-µM nitrocefin as a reporter substrate. Initial velocities at λ = 482 nm were recorded. The IC50 is the concentration of inhibitor that reduces the velocity by 50%. Plots of inverse initial velocity vs inhibitor concentration were fitted to a linear equation, and the IC50 values for TAN were determined by dividing the slope by the y-intercept.
Immunoblotting
E. coli DH10B cells harboring the pHSG298 blaNDM-1(K224X) gene were grown in MH broth with 50-µg/mL kanamycin to mid-log phase (OD600 = 0.8). Immunoblotting was performed as previously reported (43), with the following two changes. First, 50 µL of the cells were spun down, and after the supernatant was removed, they were resuspended in 20 µL of loading buffer (19:1 mixture of coomassie blue and β-mercaptoethanol; whole cell extracts). Then, the whole cell extracts were stored at −20°C overnight. Next, after boiling for 10 min, the 20 µL of whole cell extracts were loaded into each lane of a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. Second, as a loading control, a 1 to 20,000 dilution of anti-DnaK rabbit monoclonal antibody was added (Enzo Life Sciences, Farmingdale, NY, USA). Images of the western blotting were quantified by the Fiji image processing package (44). To examine the relationship between the expression level of the K224 variants and antibiotic resistance profiles, Spearman’s rank correlation test was conducted to evaluate the relationship between the MICs and expression level of individual K224 variants. Statistical analysis was performed using Stata statistical software version 15 (Stata Corp, TX, USA). A P-value of <0.05 was considered statistically significant.
Structural modeling and docking simulations
The structure for NDM-1 was taken from PDB code 3SPU (45). The amino acid substitution for the K224I variant was made using LEAP software from the Amber22 package (46) and subjected to a molecular dynamic simulation protocol using the PMEMD software from the same package with the FF14SB forcefield (47). The protein was first immersed in a periodical octahedral box of TIP3P water molecules, and the structure was then optimized, taken from 0 to 300 K at constant volume for 100 ps, equilibrated at a constant pressure of 1 bar for 200 ps, and finally subjected to 200 ns of production simulation. Temperature control was performed using the Langevin thermostat (48), and pressure control was performed using the Monte Carlo barostat (49).
Docking experiments were carried out using AutoDock 4.2.6 (50). Charges for the Zn2+ and hydroxyl ions of the catalytic center of the MBLs were obtained from the literature (51). For the boron AutoDock parameters, the van der Waals radius for the desolvated atom and the associated van der Waals well-depth value were adapted from the literature, and the default AutoDock atomic solvation value for carbon was used (52, 53). The structure for TAN was generated from the SMILES representation using obabel (54), and its geometry was optimized using Gaussian09 (55). The grid maps were set to 70 × 70 × 70 points with a grid spacing of 0.375 Å centered on the oxygen atom of the hydroxyl ion in the catalytic site. For each docking calculation, 100 different docking runs were performed, which were clustered using an RMSD cutoff of 2 Å. The poses selected as correct were those that had the highest population (number of individual docking results that were clustered as the same ligand pose).
ACKNOWLEDGMENTS
This work was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, Award Number 1I01B × 001974 (R.A.B.), from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and the Geriatric Research Education and Clinical Center VISN 10. The content is solely the authors’ responsibility and does not necessarily represent the official views of the Department of Veterans Affairs.
Contributor Information
Maria F. Mojica, Email: mfm72@case.edu.
Robert A. Bonomo, Email: robert.bonomo@va.gov.
Alessandra Carattoli, Universita degli studi di roma La Sapienza, Rome, Italy.
REFERENCES
- 1. Mojica MF, Rossi M-A, Vila AJ, Bonomo RA. 2022. The urgent need for metallo-β-lactamase inhibitors: an unattended global threat. Lancet Infect Dis 22:e28–e34. doi: 10.1016/S1473-3099(20)30868-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. WHO . 2017. WHO publishes list of bacteria for which new antibiotics are urgently needed. Available from: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed. Retrieved 10 Oct 2023.
- 3. Nordmann P, Poirel L. 2019. Epidemiology and diagnostics of carbapenem resistance in gram-negative bacteria. Clin Infect Dis 69:S521–S528. doi: 10.1093/cid/ciz824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bush K, Bradford PA. 2019. Author correction: interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol 17:459–460. doi: 10.1038/s41579-019-0221-6 [DOI] [PubMed] [Google Scholar]
- 5. Bahr G, González LJ, Vila AJ. 2021. Metallo-β-lactamases in the age of multidrug resistance: from structure and mechanism to evolution, dissemination, and inhibitor design. Chem Rev 121:7957–8094. doi: 10.1021/acs.chemrev.1c00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bush K. 2018. Past and present perspectives on β-lactamases. Antimicrob Agents Chemother 62:e01076-18. doi: 10.1128/AAC.01076-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lisa M-N, Palacios AR, Aitha M, González MM, Moreno DM, Crowder MW, Bonomo RA, Spencer J, Tierney DL, Llarrull LI, Vila AJ. 2017. A general reaction mechanism for carbapenem hydrolysis by mononuclear and binuclear metallo-β-lactamases. Nat Commun 8:538. doi: 10.1038/s41467-017-00601-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meini MR, Llarrull LI, Vila AJ. 2014. Evolution of metallo-β-lactamases: trends revealed by natural diversity and in vitro evolution. Antibiotics (Basel) 3:285–316. doi: 10.3390/antibiotics3030285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drawz SM, Taracila M, Caselli E, Prati F, Bonomo RA. 2011. Exploring sequence requirements for C₃/C₄ carboxylate recognition in the Pseudomonas aeruginosa cephalosporinase: insights into plasticity of the AmpC β-lactamase. Protein Sci 20:941–958. doi: 10.1002/pro.612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mehta SC, Furey IM, Pemberton OA, Boragine DM, Chen Y, Palzkill T. 2021. KPC-2 β-Lactamase enables carbapenem antibiotic resistance through fast deacylation of the covalent intermediate. J Biol Chem 296:100155. doi: 10.1074/jbc.RA120.015050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang H, Hao Q. 2011. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J 25:2574–2582. doi: 10.1096/fj.11-184036 [DOI] [PubMed] [Google Scholar]
- 12. Mojica MF, Bonomo RA, Fast W. 2016. B1-metallo-β-lactamases: where do we stand ? Curr Drug Targets 17:1029–1050. doi: 10.2174/1389450116666151001105622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palacios AR, Rossi M-A, Mahler GS, Vila AJ. 2020. Metallo-β-lactamase inhibitors inspired on snapshots from the catalytic mechanism. Biomolecules 10:854. doi: 10.3390/biom10060854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. González MM, Kosmopoulou M, Mojica MF, Castillo V, Hinchliffe P, Pettinati I, Brem J, Schofield CJ, Mahler G, Bonomo RA, Llarrull LI, Spencer J, Vila AJ. 2015. Bisthiazolidines: a substrate-mimicking scaffold as an inhibitor of the NDM-1 carbapenemase. ACS Infect Dis 1:544–554. doi: 10.1021/acsinfecdis.5b00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinchliffe P, González MM, Mojica MF, González JM, Castillo V, Saiz C, Kosmopoulou M, Tooke CL, Llarrull LI, Mahler G, Bonomo RA, Vila AJ, Spencer J. 2016. Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc Natl Acad Sci U S A 113:E3745–54. doi: 10.1073/pnas.1601368113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. King DT, Worrall LJ, Gruninger R, Strynadka NCJ. 2012. New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J Am Chem Soc 134:11362–11365. doi: 10.1021/ja303579d [DOI] [PubMed] [Google Scholar]
- 17. Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frère JM, Payne DJ, Bateson JH, Abdel-Meguid SS. 2000. Crystal structure of the IMP-1 metallo beta-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298. doi: 10.1021/bi992569m [DOI] [PubMed] [Google Scholar]
- 18. Rydzik AM, Brem J, van Berkel SS, Pfeffer I, Makena A, Claridge TDW, Schofield CJ. 2014. Monitoring conformational changes in the NDM-1 metallo-β-lactamase by 19F NMR spectroscopy. Angew Chem Int Ed Engl 53:3129–3133. doi: 10.1002/anie.201310866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomatis PE, Fabiane SM, Simona F, Carloni P, Sutton BJ, Vila AJ. 2008. Adaptive protein evolution grants organismal fitness by improving catalysis and flexibility. Proc Natl Acad Sci U S A 105:20605–20610. doi: 10.1073/pnas.0807989106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palacios AR, Mojica MF, Giannini E, Taracila MA, Bethel CR, Alzari PM, Otero LH, Klinke S, Llarrull LI, Bonomo RA, Vila AJ. 2019. The reaction mechanism of metallo-β-lactamases is tuned by the conformation of an active-site mobile loop. Antimicrob Agents Chemother 63:e01754-18. doi: 10.1128/AAC.01754-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moali C, Anne C, Lamotte-Brasseur J, Groslambert S, Devreese B, Van Beeumen J, Galleni M, Frère JM. 2003. Analysis of the importance of the metallo-beta-lactamase active site loop in substrate binding and catalysis. Chem Biol 10:319–329. doi: 10.1016/s1074-5521(03)00070-x [DOI] [PubMed] [Google Scholar]
- 22. Coleman K. 2011. Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr Opin Microbiol 14:550–555. doi: 10.1016/j.mib.2011.07.026 [DOI] [PubMed] [Google Scholar]
- 23. Liu B, Trout REL, Chu G-H, McGarry D, Jackson RW, Hamrick JC, Daigle DM, Cusick SM, Pozzi C, De Luca F, Benvenuti M, Mangani S, Docquier J-D, Weiss WJ, Pevear DC, Xerri L, Burns CJ. 2020. Discovery of taniborbactam (VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J Med Chem 63:2789–2801. doi: 10.1021/acs.jmedchem.9b01518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krajnc A, Brem J, Hinchliffe P, Calvopiña K, Panduwawala TD, Lang PA, Kamps JJAG, Tyrrell JM, Widlake E, Saward BG, Walsh TR, Spencer J, Schofield CJ. 2019. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J Med Chem 62:8544–8556. doi: 10.1021/acs.jmedchem.9b00911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamrick JC, Docquier J-D, Uehara T, Myers CL, Six DA, Chatwin CL, John KJ, Vernacchio SF, Cusick SM, Trout REL, Pozzi C, De Luca F, Benvenuti M, Mangani S, Liu B, Jackson RW, Moeck G, Xerri L, Burns CJ, Pevear DC, Daigle DM. 2020. VNRX-5133 (taniborbactam) a broad-spectrum inhibitor of serine- and metallo-β-lactamases, restores activity of cefepime in enterobacterales and Pseudomonas aeruginosa. Antimicrob Agents Chemother 64:e01963-19. doi: 10.1128/AAC.01963-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vázquez-Ucha JC, Lasarte-Monterrubio C, Guijarro-Sánchez P, Oviaño M, Álvarez-Fraga L, Alonso-García I, Arca-Suárez J, Bou G, Beceiro A, GEMARA-SEIMC/REIPI Enterobacterales Study Group . 2022. Assessment of activity and resistance mechanisms to cefepime in combination with the novel β-lactamase inhibitors zidebactam taniborbactam, and enmetazobactam against a multicenter collection of carbapenemase-producing enterobacterales. Antimicrob Agents Chemother 66:e0167621. doi: 10.1128/AAC.01676-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Golden AR, Baxter MR, Karlowsky JA, Mataseje L, Mulvey MR, Walkty A, Bay D, Schweizer F, Lagace-Wiens PRS, Adam HJ, Zhanel GG. 2022. Activity of cefepime/taniborbactam and comparators against whole genome sequenced ertapenem-non-susceptible enterobacterales clinical isolates: CANWARD 2007-19. JAC Antimicrob Resist 4:dlab197. doi: 10.1093/jacamr/dlab197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Satapoomin N, Dulyayangkul P, Avison MB. 2022. Klebsiella pneumoniae mutants resistant to ceftazidime-avibactam plus aztreonam, imipenem-relebactam, meropenem-vaborbactam, and cefepime-taniborbactam. Antimicrob Agents Chemother 66:e0217921. doi: 10.1128/aac.02179-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Terrier C, Nordmann P, Sadek M, Poirel L. 2023. In vitro activity of cefepime/zidebactam and cefepime/taniborbactam against aztreonam/avibactam-resistant NDM-like-producing Escherichia coli clinical isolates. J Antimicrob Chemother 78:1191–1194. doi: 10.1093/jac/dkad061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lasarte-Monterrubio C, Fraile-Ribot PA, Vázquez-Ucha JC, Cabot G, Guijarro-Sánchez P, Alonso-García I, Rumbo-Feal S, Galán-Sánchez F, Beceiro A, Arca-Suárez J, Oliver A, Bou G. 2022. Activity of cefiderocol, imipenem/relebactam, cefepime/taniborbactam and cefepime/zidebactam against ceftolozane/tazobactam- and ceftazidime/avibactam-resistant Pseudomonas aeruginosa. J Antimicrob Chemother 77:2809–2815. doi: 10.1093/jac/dkac241 [DOI] [PubMed] [Google Scholar]
- 31. Hernández-García M, García-Castillo M, Ruiz-Garbajosa P, Bou G, Siller-Ruiz M, Pitart C, Gracia-Ahufinger I, Mulet X, Pascual Á, Tormo N, Cantón R. 2022. In vitro activity of cefepime-taniborbactam against carbapenemase-producing enterobacterales and Pseudomonas aeruginosa isolates recovered in Spain. Antimicrob Agents Chemother 66:e0216121. doi: 10.1128/aac.02161-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Piccirilli A, Segatore B, Brisdelli F, Amicosante G, Perilli M. 2021. Potent inhibitory activity of taniborbactam towards NDM-1 and NDM-1(Q119X) mutants, and in vitro activity of cefepime/taniborbactam against MBLS producing enterobacterales. Int J Antimicrob Agents 57:106228. doi: 10.1016/j.ijantimicag.2020.106228 [DOI] [PubMed] [Google Scholar]
- 33. Le Terrier C, Gruenig V, Fournier C, Nordmann P, Poirel L. 2023. NDM-9 resistance to taniborbactam. Lancet Infect Dis 23:401–402. doi: 10.1016/S1473-3099(23)00069-5 [DOI] [PubMed] [Google Scholar]
- 34. Mojica MF, Mahler SG, Bethel CR, Taracila MA, Kosmopoulou M, Papp-Wallace KM, Llarrull LI, Wilson BM, Marshall SH, Wallace CJ, Villegas MV, Harris ME, Vila AJ, Spencer J, Bonomo RA. 2015. Exploring the role of residue 228 in substrate and inhibitor recognition by VIM metallo-β-lactamases. Biochemistry 54:3183–3196. doi: 10.1021/acs.biochem.5b00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. López C, Delmonti J, Bonomo RA, Vila AJ. 2022. Deciphering the evolution of metallo-β-lactamases: a journey from the test tube to the bacterial periplasm. J Biol Chem 298:101665. doi: 10.1016/j.jbc.2022.101665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun Z, Hu L, Sankaran B, Prasad BVV, Palzkill T. 2018. Differential active site requirements for NDM-1 β-lactamase hydrolysis of carbapenem versus penicillin and cephalosporin antibiotics. Nat Commun 9:4524. doi: 10.1038/s41467-018-06839-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng H, Liu X, Wang S, Fleming J, Wang D-C, Liu W. 2017. The mechanism of NDM-1-catalyzed carbapenem hydrolysis is distinct from that of penicillin or cephalosporin hydrolysis. Nat Commun 8:2242. doi: 10.1038/s41467-017-02339-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng Z, Thomas PW, Ju L, Bergstrom A, Mason K, Clayton D, Miller C, Bethel CR, VanPelt J, Tierney DL, Page RC, Bonomo RA, Fast W, Crowder MW. 2018. Evolution of New Delhi metallo-β-lactamase (NDM) in the clinic: effects of NDM mutations on stability, zinc affinity, and mono-zinc activity. J Biol Chem 293:12606–12618. doi: 10.1074/jbc.RA118.003835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stewart AC, Bethel CR, VanPelt J, Bergstrom A, Cheng Z, Miller CG, Williams C, Poth R, Morris M, Lahey O, Nix JC, Tierney DL, Page RC, Crowder MW, Bonomo RA, Fast W. 2017. Clinical variants of new Delhi metallo-β-lactamase are evolving to overcome zinc scarcity. ACS Infect Dis 3:927–940. doi: 10.1021/acsinfecdis.7b00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bahr G, Vitor-Horen L, Bethel CR, Bonomo RA, González LJ, Vila AJ. 2018. Clinical evolution of New Delhi metallo-β-lactamase (NDM) optimizes resistance under Zn(II) deprivation. Antimicrob Agents Chemother 62:e01849-17. doi: 10.1128/AAC.01849-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. González LJ, Bahr G, González MM, Bonomo RA, Vila AJ. 2023. In-cell kinetic stability is an essential trait in metallo-β-lactamase evolution. Nat Chem Biol 19:1116–1126. doi: 10.1038/s41589-023-01319-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. CLSI . 2020. Performance standards for antimicrobial susceptibility testing. 30th ed. CLSI, Wayne PA. [Google Scholar]
- 43. Pegg KM, Liu EM, George AC, LaCuran AE, Bethel CR, Bonomo RA, Oelschlaeger P. 2014. Understanding the determinants of substrate specificity in IMP family metallo-β-lactamases: the importance of residue 262. Protein Sci 23:1451–1460. doi: 10.1002/pro.2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. King D, Strynadka N. 2011. Crystal structure of New Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Sci 20:1484–1491. doi: 10.1002/pro.697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. HMA DAC, Belfon K, Ben-Shalom IY, Berryman JT, Brozell SR, Cerutti DS, Cheatham, III GA, Cruzeiro VWD, Darden TA, Duke RE, et al. 2022. Amber 202. University of California SF. [Google Scholar]
- 47. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. 2015. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99Sb. J Chem Theory Comput 11:3696–3713. doi: 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiang T, Liu F, Grant DM. 1991. Generalized langevin equations for molecular dynamics in solution. J Chem Phys 94:4463–4471. doi: 10.1063/1.460602 [DOI] [Google Scholar]
- 49. Faller R, de Pablo JJ. 2002. Constant pressure hybrid molecular dynamics–monte carlo simulations. J Chem Phys 116:55–59. doi: 10.1063/1.1420460 [DOI] [Google Scholar]
- 50. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. doi: 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suárez D, Brothers EN, Merz KM. 2002. Insights into the structure and dynamics of the dinuclear zinc beta-lactamase site from bacteroides fragilis. Biochemistry 41:6615–6630. doi: 10.1021/bi0121860 [DOI] [PubMed] [Google Scholar]
- 52. Tiwari R, Mahasenan K, Pavlovicz R, Li C, Tjarks W. 2009. Carborane clusters in computational drug design: a comparative docking evaluation using AutoDock, FlexX, glide, and surflex. J Chem Inf Model 49:1581–1589. doi: 10.1021/ci900031y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Otkidach DS, Pletnev IV. 2001. Conformational analysis of boron-containing compounds using gillespie–kepert version of molecular mechanics. Theochem 536:65–72. doi: 10.1016/S0166-1280(00)00602-3 [DOI] [Google Scholar]
- 54. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. 2011. Open babel: an open chemical toolbox. J Cheminform 3:33. doi: 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frisch MJ, Trucks GW, Schlegel HB, Scuseria E, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. 2009. Gaussian 09. Gaussian, Inc., Wallingford CT [Google Scholar]