

Abstract
Most orthopteran insects are phytophagous and some are important pests in agriculture and forests. Many intestinal microflora of Orthoptera insects have been reported, primarily from Acridoidea and there have been few reports of other taxa. In this study, we collected 15 individuals representing five species (Ruspolialineosa, Tetrixjaponica, Erianthusversicolor, Gryllotalpaorientalis and Teleogryllusemma) belonging to five orthopteran superfamilies (Tettigonioidea, Tetrigoidea, Eumastacoidea, Gryllotalpoidea and Grylloidea) to characterise and compare the gut microbiota with greater taxonomic width by performing sequencing analysis of the 16S rRNA V4 region in gut material. A total of 606,053 high-quality sequences and 3,105 OTUs were acquired from 15 gut samples representing 24 phyla, 48 classes, 69 orders, 133 families and 219 genera. Firmicutes and bacteria were the most abundant phyla, followed by Bacteroidetes, Cyanobacteria, Actinobacteria and Acidobacteria. At the genus level, Serratia, Citrobacter, Wolbachia, Lactobacillus and Parabacteroides were the most predominant genera in R.lineosa, T.japonica, E.versicolor, G.orientalis and T.emma, respectively. Both Principal Coordinates Analysis (PCoA) and heatmap results revealed significant differences in bacterial community composition across species. Additionally, alpha diversity analysis indicated the bacterial richness was significantly different amongst the five species.
Keywords: gut microbiota, DNA metabarcoding, Orthoptera, biodiversity
Introduction
Large numbers of microorganisms colonise the insect gut and form complex symbiotic relationships with their host. Insect-gut symbiotic microorganisms play important roles in parasitifer mating preference (Sharon et al. 2010), resistance to harmful microbes (Scott et al. 2008), expand the range of diet (Anonymous 2008), longevity (Behar et al. 2008), the regulation of phenolic compound bioavailability (Selma et al. 2009) and pheromone aggregation (Dillon and Charnley 2002). In addition, symbiotic microorganisms in the insect gut influence parasitifer nutrition, digestion and the immune response. Recent work has indicated that insect symbionts mediate insecticide resistance. Studies investigating the mid-gut microbiota of the diamondback moth have suggested roles for Lactobacillales or other scarcer taxa in conferring diamondback moth insecticide resistance (Xia et al. 2013). Many factors influence insect gut communities. Changes in the gut ecological conditions impact the structure and diversity of bacterial populations; for example, variations in the physicochemical conditions in different gut compartments of Cubitermes spp. are reflected in the diversity of their respective intestinal microbial communities (Schmitt-Wagner et al. 2003). Furthermore, sampling site location primarily reflects microbiota composition rather than taxonomy or ecology (Hird et al. 2014). According to a recent report, gut bacterial diversity is significantly higher in omnivorous insects than in stenophagous insects (Yun et al. 2014) and higher bacterial diversity may be related to the types of food consumed (Anderson et al. 2013). Dillon and Charnley studied the numbers and types of intestinal microflora in Schistocercagregaria and demonstrated how different diets influenced gut microbe numbers and varieties (Dillon and Charnley 2002). Shi et al. studied the microbial community structures of gut symbionts in woodbore, silkworm, grasshopper and cutworm and observed significant differences in symbiotic community structure correlated with food adaptation (Shi et al. 2011). However, because traditional sequencing technology is low-throughput and time-consuming, the exploration of insect gut bacterial diversity has been limited.
DNA metabarcoding, a high-throughput DNA barcoding technique, is a fast and efficient method to assess biodiversity (Yu et al. 2012, Carew et al. 2013, Leray and Knowlton 2015, Dowle et al. 2016). This approach has aroused widespread interest amongst scientists and has been widely employed to investigate soil, water, intestines, air and other ecologies (Chen et al. 2014, Kraaijeveld et al. 2015, Xiong et al. 2015, Zhao et al. 2015, Yu et al. 2015). DNA metabarcoding technologies facilitate accurate, rapid and highly efficient identification on a large scale and, to a large extent, compensate for the defects of traditional identification methods. DNA metabarcoding has been widely employed to study the intestinal microflora of insects. For example, Minard et al. performed DNA metabarcoding sequencing to compare the intestinal microflora of four autochthonous Aedesalbopictus populations in Vietnam and three populations recently introduced to metropolitan France and found that French invasive Asian tiger mosquito populations harbour reduced bacterial microbiota and genetic diversity compared to Vietnamese autochthonous relatives (Minard et al. 2015). According to Gauthier et al., who analysed the diversity of bacterial communities associated with nine biotypes of the pea aphid complex via DNA metabarcode sequencing, the aphid microbiota is dominated by a few heritable symbionts and plant specialisation is an important structural factor for bacterial communities associated with the pea aphid complex (Gauthier et al. 2015). The widespread use of DNA metabarcoding technology has revolutionised the study of insect intestinal microflora.
Most orthopterans are phytophagous and some are important pests in agriculture and forests. Most reports of intestinal microflora in Orthoptera have primarily concentrated on Acridoidea (Dillon et al. 2008, Idowu et al. 2009, Ademolu and Idowu 2011) and there have been few reports of other taxa. In this study, we used DNA metabarcoding to investigate the gut microbial composition and diversity in five superfamilies (Tetrigoidea, Eumastacoidea, Tettigonioidea, Gryllotalpoidea and Grylloidea) of Orthoptera.
Material and methods
Insect sampling
A total of 15 orthopteran specimens across five species (Ruspolialineosa belonging to Tettigonoidea, Gryllotalpaorientalis belonging to Gryllotalpoidea, Teleogryllusemma belonging to Grylloidea, Erianthusversicolor belonging to Eumastacoidea and Tetrixjaponica belonging to Tetrigoidea) were collected, with three specimens per species collected in the same region (see Table 1 for details). Before dissection, all specimens were starved for 24 hours to clear food residue from their guts and reduce chloroplast contamination. Then, all guts were dissected under sterile conditions with flame-sterilised forceps in 1X phosphate-buffered saline. The guts of each specimen were stored and frozen at -80°C before DNA extraction.
Table 1.
Information of studied samples.
| Superfamily | Species | SampleID | Location | Date | |
| Tetrigoidea | Tetrixjaponica | Z | Z1 | Shaanxi, Xi’an | 21/08/2016 |
| Z2 | Shaanxi, Xi’an | 21/08/2016 | |||
| Z3 | Shaanxi, Xi’an | 21/08/2016 | |||
| Tettigoniidae | Ruspolialineosa | ZS | ZS1 | Shaanxi, Xi’an | 22/08/2016 |
| ZS2 | Shaanxi, Xi’an | 22/08/2016 | |||
| ZS3 | Shaanxi, Xi’an | 22/08/2016 | |||
| Eumastacoidea | Erianthusversicolor | M | M1 | Guangdong, Ruyuan | 15/09/2016 |
| M2 | Guangdong, Ruyuan | 15/09/2016 | |||
| M3 | Guangdong, Ruyuan | 15/09/2016 | |||
| Gryllotalpidae | Gryllotalpaorientalis | LG | LG1 | Henan, Nanyang | 29/08/2016 |
| LG2 | Henan, Nanyang | 29/08/2016 | |||
| LG3 | Henan, Nanyang | 29/08/2016 | |||
| Gryllidae | Teleogryllusemma | HLYHL | HLYHL1 | Shaanxi, Xi’an | 21/08/2016 |
| HLYHL2 | Shaanxi, Xi’an | 21/08/2016 | |||
| HLYHL3 | Shaanxi, Xi’an | 21/08/2016 | |||
DNA extraction and PCR amplification of the V4 region of 16S rRNA
Microbial genomic DNA was extracted from the gut samples using the phenol-chloroform method as previously described (Yang et al. 2008). Then, 0.8% agarose gel electrophoresis was performed to determine the molecular size of the extracted DNA and quantification was performed with a UV spectrophotometer. PCR amplification of the V4 region of the 16S rRNA gene was performed using the following primers: 520F (5’-barcode+GCACCTAAYTGGGYDTAAAGNG-3’) and 802R (5’-TACNVGGGTATCTAATCC-3’). The barcode in the forward primer (520F) is a seven-base oligonucleotide sequence used to distinguish between samples in the same library. A 25-μl reaction system was used for PCR amplification, containing 0.25 μl of NEB Q5 DNA high-fidelity polymerase, 0.5 μl of dNTPs (10 mM), 5 μl of 5× PCR reaction buffer, 5 μl of 5× high GC buffer, 1 μl of DNA template, 1 μl of forward primer (10 μM), 1 μl of reverse primer (10 μM) and 11.25 μl of sterile ultrapure water. The following PCR conditions were used: initial denaturation at 98°C for 30 sec, followed by 25-27 cycles of denaturation at 98°C for 30 sec, annealing at 50°C for 30 sec and extension at 72°C for 30 sec, with a final extension step of 5 min at 72°C. PCR products were detected by performing 2% agarose gel electrophoresis and target fragments were extracted and recovered using an Axygen Axy Prep DNA Gel Extraction Kit (AXYGEN Inc., Union City, CA USA, cat#AP-GX-500). V4 amplicons were pooled and 2 × 300 paired-end sequences were analysed by Illumina MiSeq at Personal Biotech Co., Ltd. (Shanghai, China).
Sequence analysis
To integrate raw paired-end sequences, we quality-screened for paired-end sequences in FASTQ format using Trimmomatic (v.0.36, http://www.usadellab.org/cms/index.php?page=trimmomatic) (Bolger et al. 2014). Ambiguous bases were not allowed and sequence lengths were longer than 150 bp. In addition, reads were removed if barcode errors or primer mismatches were found. We merged these reads using Flash software (v.1.2.7, http://ccb.jhu.edu/software/FLASH/) (Magoč and Salzberg 2011) and discarded unassembled reads. Chimeras were identified and removed using USEARCH (v.5.2.236, http://www.drive5.com/usearch/) in Qiime (v.1.8.0, http://qiime.org/) (Caporaso et al. 2010).
Operational taxonomic units (OTUs) were generated with sequence similarity greater than 97% using the uclust function (Edgar 2010) in Qiime. The sequence with the highest abundance for each OTU was selected as the representative sequence. Taxonomic information for each OTU was obtained by annotating the OTU representative sequence, based on the Greengenes database (Release 13.8, http://greengenes.secondgenome.com/) (DeSantis et al. 2006). A Venn diagram and the Dendrogram and Heatmap were generated using the Venn Diagram software package and ggtree software package in R. Unweighted clustering was performed using PCoA of UniFrac distance matrices.
Chao1, ACE, Shannon and Simpson indices for each sample were calculated using the summary.single command in the MOTHUR software package (http://www.mothur.org/) (Schloss et al. 2009). The relationship between the selected taxonomy group (abundant phyla and genera) and the bacterial community index (Chao1, ACE, Shannon and Simpson) was calculated using SPSS 20.0 software. Multiple-group analysis was carried out using ANOVA followed by the Tukey’s honestly significant difference test. P < 0.05 was considered as statistically significant.
Results
Barcoded 16S sequencing and OTUs composition
We utilised the V4 region of the 16S rRNA amplicon to assess the gut microbiota composition of five orthopterans using Illumina MiSeq DNA metabarcode sequencing. A total of 778,780 paired-end reads were acquired from all intestinal samples, with an average read length of 450 bp. After quality control, 606,053 high-quality reads were acquired. Based on 97% species similarity and chloroplast and mitochondrial sequences and OTUs with < 0.001% abundance in all samples being removed, a total of 3,105 OTUs were obtained from all intestinal samples. The fifteen insect samples were divided into five groups, each with three samples. The number of OTUs in each group (ZS, M, HLYHL, Z and LG) was 978, 818, 951, 952 and 1,417, respectively. Amongst these, 43 OTUs present in all groups were defined as core OTUs and 94, 81, 648, 90 and 1,039 OTUs were uniquely identified in ZS, M, HLYHL, Z and LG, respectively (Fig. 1).
Figure 1.
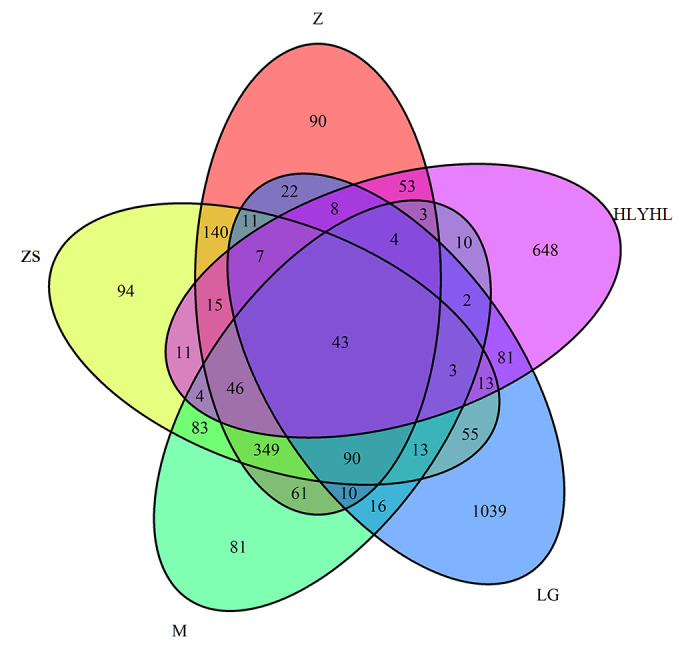
Venn diagram depicting the number of shared and exclusive bacterial OTUs in the bacterial community of five groups. Z: Tetrixjaponica; ZS: Ruspolialineosa; M: Erianthusversicolor; LG: Gryllotalpaorientalis; HLYHL: Teleogryllusemma.
The Dendrogram and heatmap revealed the differences of the top 100 OTUs amongst the 15 samples (Fig. 2). The most abundant and prevalent OTUs belonged to the families Ruminococcaceae (belonging to Firmicutes) and Enterobacteriaceae (belonging to bacteria). Ruminococcaceae was very abundant across the samples of LG and HLYHL, but virtually absent from Z, ZS and M. On the contrary, Enterobacteriaceae was very abundant across Z, but ZS, M, LG and HLYHL were relatively absence (Fig. 2). From the 100 most prevalent OTUs, 47 belonged to Firmicutes, 24 belonged to bacteria, 21 belonged to Bacteroidetes and there were a few Acidobacteria, Actinobacteria, Cyanobacteria and Planctomycetes. Within the Firmicutes, all OTUs belonged to Clostridiales and Lactobacillales order, except for two Bacillales OTUs. Within the Bacteroidetes, all OTUs, except for one [Saprospirales] OTU, belonged to Bacteroidales order (Fig. 2). The Principal Coordinates Analysis (PCoA), based on an unweighted UniFrac distance matrix, revealed differences in microbiota composition for different groups; the bacterial composition of each group were distinctly different, except for Z and ZS (Fig. 3). The ANOSIM and Adonis analysis (P = 0.001 and P = 0.001, respectively) also indicated different groups differed significantly.
Figure 2.

Dendrogram and heatmap of bacterial distributions of the top 100 abundant OTUs present in the microbial community of the fifteen samples. The numbers indicate the actual reads number of the OTU. The heatmap plot depicted the relative abundance of each sample and the relative values for OTUs are indicated by colour intensity. Z: Tetrixjaponica; ZS: Ruspolialineosa; M: Erianthusversicolor; LG: Gryllotalpaorientalis; HLYHL: Teleogryllusemma.
Figure 3.

PCoA plot based on an unweighted UniFrac distance matrix depicting differences in the composition of the gut microbiota of the five groups. In the unweighted UniFrac analysis of the gut samples, the first principal coordinate, explained 40.11% of sample variation and separated groups of LG and HLYHL from others. The third principal coordinate (7.49% of sample variation) separated groups (M) from others. Z: Tetrixjaponica; ZS: Ruspolialineosa; M: Erianthusversicolor; LG: Gryllotalpaorientalis; HLYHL: Teleogryllusemma.
Analysis of alpha diversity
Gut microbiota alpha diversity was estimated using alpha diversity curves (rarefaction curves and Shannon–Wiener curves) and alpha diversity indices (Chao1, ACE, Simpson and Shannon indices). The rarefaction curves (Amato et al. 2013) and Shannon–Wiener curves (Wang et al. 2012) for each sample are shown in Suppl. material 1: Figure S1. The rarefaction curves reached a saturation phase at 20,000 reads, indicating sufficient recovery of the OTUs present in the datasets. The Shannon-Wiener curves also reached saturation, indicating the addition of more sequences did not alter the saturation of microbial diversity.
The diversity indices for each sample are shown in Table 2. The Chao1 and ACE indices reflected microbial community richness and the Simpson and Shannon indices reflected microbial community diversity. ANOVA indicated significant differences for Chao1 (P = 0.001), ACE (P = 0.002) and Shannon (P = 0.027) and Simpson (P = 0.100) showed no difference (Table 2). According to the Chao1 and ACE indices, the bacterial richness of LG was significantly higher than ZS, HLYHL, Z and M (P < 0.05) (Suppl. material 1: Figure S2A). According to the Shannon Index, the bacterial diversity of LG was significantly higher than Z (P < 0.05), but the Simpson Index showed no difference amongst the five groups (Suppl. material 1: Figure S2B, C).
Table 2.
Diversity index of each sample.
| SampleID | Chao1 | ACE | Simpson | Shannon |
| Z1 | 371 | 478.18 | 0.78 | 3.36 |
| Z2 | 522 | 648.34 | 0.89 | 4.82 |
| Z3 | 446 | 577.26 | 0.83 | 4.11 |
| ZS1 | 498 | 582.61 | 0.86 | 5.11 |
| ZS2 | 665 | 788.12 | 0.97 | 6.43 |
| ZS3 | 594 | 694.67 | 0.97 | 6.43 |
| M1 | 339 | 395.71 | 0.89 | 4.83 |
| M2 | 306 | 396.69 | 0.77 | 3.56 |
| M3 | 579 | 629.24 | 0.98 | 7.19 |
| LG1 | 865 | 865.00 | 0.99 | 7.87 |
| LG2 | 898 | 969.45 | 0.92 | 6.36 |
| LG3 | 932 | 971.13 | 0.98 | 7.62 |
| HLYHL1 | 436 | 468.90 | 0.96 | 6.18 |
| HLYHL2 | 582 | 621.68 | 0.97 | 6.76 |
| HLYHL3 | 602 | 621.76 | 0.98 | 6.87 |
| p-value | 0.001 | 0.002 | 0.100 | 0.027 |
Microbial composition and intestinal sample abundance
Amongst the identified sequences, a total of 219, 133, 69, 48 and 24 microbes at the genus, family, order, class and phylum taxonomic levels, respectively, were identified across all samples. Table 3 shows the gut microbial composition at different taxonomic levels. In this study, we primarily compared and analysed microbial composition and abundance at the genus and phylum taxonomic levels.
Table 3.
The gut microbial composition at different taxonomic levels.
| SampleID | Phylum | Class | Order | Family | Genus | OTUs |
| Z1 | 13 | 25 | 37 | 84 | 111 | 581 |
| Z2 | 18 | 33 | 41 | 93 | 131 | 694 |
| Z3 | 16 | 29 | 41 | 92 | 125 | 616 |
| ZS1 | 15 | 26 | 36 | 83 | 114 | 624 |
| ZS2 | 16 | 28 | 42 | 95 | 136 | 811 |
| ZS3 | 16 | 31 | 43 | 91 | 124 | 726 |
| M1 | 15 | 29 | 40 | 90 | 113 | 515 |
| M2 | 12 | 25 | 35 | 83 | 105 | 455 |
| M3 | 16 | 31 | 38 | 95 | 151 | 656 |
| LG1 | 16 | 28 | 39 | 63 | 83 | 1049 |
| LG2 | 14 | 27 | 39 | 66 | 78 | 1104 |
| LG3 | 15 | 25 | 35 | 55 | 65 | 1080 |
| HLYHL1 | 5 | 13 | 20 | 40 | 41 | 512 |
| HLYHL2 | 7 | 15 | 25 | 42 | 50 | 725 |
| HLYHL3 | 7 | 14 | 24 | 34 | 36 | 680 |
| Z | 18 | 35 | 48 | 105 | 160 | 955 |
| ZS | 18 | 36 | 53 | 109 | 155 | 980 |
| M | 17 | 35 | 46 | 107 | 165 | 827 |
| LG | 18 | 34 | 45 | 75 | 101 | 1417 |
| HLYHL | 8 | 18 | 29 | 51 | 65 | 951 |
| Total | 24 | 48 | 69 | 133 | 219 | 3105 |
Amongst 24 phyla, Firmicutes (45.0%), bacteria (31.4%), Bacteroidetes (17.8%), Actinobacteria (2.1%) and Acidobacteria (2.0%) were present in all samples and abundant in the majority of samples, representing more than 98% of total sequences (Fig. 4A). The bacterial composition and abundance of distinct phyla differed amongst the five groups (Suppl. material 1: Figure S3A). Firmicutes was the most predominant phylum in ZS, LG and HLYHL, accounting for 42.0%, 57.6% and 62.2% of sequences, respectively. bacteria was the most predominant phylum in Z and M, accounting for 59.6% and 36.9% of sequences, respectively. Composition and abundance at the phylum taxonomic level were investigated for each gut microbiota sample (Suppl. material 1: Figure S3B). For Z1, Z2 and Z3, bacteria was the most predominant phylum, accounting for 63.4%, 60.0% and 55.5% of sequences, respectively. For LG1, LG2 and LG3, Firmicutes was the most predominant phylum, accounting for 43.4%, 76.0% and 49.0% of sequences, respectively. For HLYHL1 and HLYHL2, Firmicutes was the most predominant phylum, accounting for 85.9% and 51.6% of sequences, respectively; however, Bacteroidetes was the most predominant phylum for HLYHL3, accounting for 48.8% of sequences. In M1, M2, and ZS2, ZS3, the most predominant phylum was Firmicutes (accounting for 38.5%, 43.9%, 49.3% and 44.4% of sequences, respectively); however, bacteria predominated in M3 and ZS1 (accounting for 41.6% and 49.9% of sequences, respectively).
Figure 4.

Distribution of the gut microbiota composition. A Five groups at phylum level; B Five groups at genus level.
Amongst 219 genera, Lactococcus (9.95%), Lactobacillus (9.00%), Citrobacter (7.87%), Parabacteroides (7.67%), Sediminibacterium (6.77%), Serratia (6.65%), Bacteroides (5.18%), Streptococcus (4.37%), Wolbachia (4.27%), Geobacillus (3.14%), Bacillus (2.72%), Rhodanobacter (1.89%), Pseudomonas (1.69%), Ralstonia (1.63%), Ochrobactrum (1.58%), Burkholderia (1.49%), Ruminococcus (1.48%), Sphingomonas (1.42%), Rhodococcus (1.41%) and Oscillospira (1.07%) were the most abundant genera, accounting for more than 81% of total sequences (Fig. 4B). Amongst these, Bacteroides, Parabacteroides, Bacillus, Lactococcus, Oscillospira, Ruminococcus, Ochrobactrum and Citrobacter were present in all samples. Microbial composition and abundance varied significantly across groups (Suppl. material 1: Figure S4A). Citrobacter was the most predominant genus in Z (accounting for 39.8% of sequences), but its abundance was very low in ZS, M, LG and HLYHL. Serratia was the most predominant genus in ZS (accounting for 18.3% of sequences), but was not found in LG and HLYHL. Wolbachia, Lactobacillus and Parabacteroides were the most predominant genera in M (accounting for 17.1% of sequences), LG (accounting for 50.2% of sequences) and HLYHL (accounting for 49.0% of sequences), respectively. Microbial composition and abundance in different samples within the same groups also varied significantly (Suppl. material 1: Figure S4B). Serratia was the most predominant genus in ZS1, but Lactococcus was the most predominant genus in ZS2 and ZS3. Lactobacillus was the most predominant genus in LG2, but demonstrated very low abundance in LG1 and LG3.
Analysis of differences amongst groups
At the phylum level, we analysed the differences in Firmicutes, bacteria, Bacteroidetes, Actinobacteria and Acidobacteria in different groups. Amongst these, Acidobacteria (P < 0.01) and bacteria (P < 0.001) demonstrated significant differences and Actinobacteria, Bacteroidetes and Firmicutes showed no differences. We further calculated multiple comparisons to show differences between each two groups of Acidobacteria and bacteria, the relative abundance of the phylum bacteria in Z was mostly significantly higher than others and the relative abundance of the phylum Acidobacteria in ZS and M were significantly higher than LG and HLYHL (Fig. 5).
Figure 5.

The relative abundance (% of individual taxonomic group) of Acidobacteria and bacteria present in the microbial community of the different groups. Differences were analysed by employing ANOVA analysis and Tukey Post Hoc HSD Significance Test (* P < 0.05, ** P < 0.01, *** P < 0.001). Z: Tetrixjaponica; ZS: Ruspolialineosa; M: Erianthusversicolor; LG: Gryllotalpaorientalis; HLYHL: Teleogryllusemma.
Amongst the 20 most abundant genera, ANOVA indicated significant differences for Lactococcus (P < 0.05), Citrobacter (P < 0.001), Parabacteroides (P < 0.01), Sediminibacterium (P < 0.01), Wolbachia (P < 0.001), Geobacillus (P < 0.01), Bacillus (P < 0.05), Rhodanobacter (P < 0.05), Pseudomonas (P < 0.05), Ralstonia (P < 0.01), Ochrobactrum (P < 0.05), Burkholderia (P < 0.01) and Rhodococcus (P < 0.01) (Suppl. material 1: Figure S5).
Discussion
Based on the results obtained for 15 samples across five orthopteran species using DNA metabarcoding, the predominant phyla in the insect gut were Firmicutes and bacteria, representing 70.1% of total sequences. This result is quite similar to those obtained in previous studies. Yun et al. studied gut samples from 305 individuals belonging to 218 species in 21 taxonomic orders and found the predominant phyla to be Firmicutes and bacteria, representing 82.8% of total sequences (Yun et al. 2014). Additionally, Colman et al. studied 62 insect gut samples and found Firmicutes and bacteria to be the predominant phyla, comprising 79.1% of total sequences (Colman et al. 2012). Bacteroidetes, the third most predominant phylum, generally produces butyrate, a chemical thought to have antineoplastic properties, in the mammalian gut (Kim and Milner 2007).
According to our study, the predominant genera in the gut were Lactococcus and Lactobacillus, belonging to the order Lactobacillales and the class Bacilli. Bacilli species reportedly exert beneficial effects in terms of preventing intestinal disorders and reducing inflammation (Hong et al. 2005); they are also the microbial communities responsible for biogas production (Schlüter et al. 2008). Lactobacillales are known for their beneficial effects in insects, such as their ability to mediate insecticide resistance (Xia et al. 2013), modulate the microflora composition to protect the host against infections (Ouwehand et al. 2002), promote intestinal peptidase expression, increase intestinal proteolytic activity (Erkosar et al. 2015) and enhance the systemic production of host ecdysone and insulin-like peptides (Storelli et al. 2011). As described in several reports, Wolbachia induce male-killing, regulate host reproduction (Jiggins et al. 2000, Hiroki et al. 2002, Sebastien et al. 2012) and defend some insects against natural enemies (Hedges et al. 2008, Teixeira et al. 2008). Wolbachia (14.1%) was the most prevalent genera in a study of 305 individuals belonging to 21 taxonomic orders (Yun et al. 2014). However, in our study, Wolbachia abundance only reached 4.27% and ANOVA results indicated Wolbachia differed significantly amongst the five groups. The abundance of Wolbachia was highest in M and there were no Wolbachia bacteria in ZS, LG and HLYHL. Jeyaprakash and Hoy (2000) and Russell et al. (2012) observed Wolbachia strains in Orthoptera and Yun et al. showed Wolbachia to be the dominant species in Orthoptera (Yun et al. 2014). We compared Wolbachia in five species of Orthoptera and found this genus in E.versicolor and T.japonica, but not R.lineosa, G.orientalis or T.emma.
When comparing gut bacteria amongst samples, we identified differences in diversity and abundance. Stanley et al. analysed samples from 207 chicken caecal microbiota across three similar trials and demonstrated the ability of host genes and environmental factors to alter the composition of the intestinal microflora (Stanley et al. 2013). A previous study investigating Mormon crickets suggested gut bacteria are either acquired from the environment in each generation or are not restricted over appreciable periods of evolutionary time (Smith et al. 2017). Dynamic variations in the gut microbiota are attributable to ecological conditions in the gut, including pH levels, redox conditions, oxygen levels and biologically active compounds (Dillon and Dillon 2004, Engel and Moran 2013). Variations are also attributable to ecological relationships between gut microorganisms. Positive interactions may promote the symbiosis of intestinal microbiota, while negative interactions inhibit symbiosis, resulting in changes in the gut microflora composition amongst individual hosts (Coyne et al. 2005, Donohoe et al. 2011, Rosenthal et al. 2011).
To evaluate the relationships between the gut microbiota and host in five species, we collected 15 samples and classified them into five groups. Amongst the six most abundant phyla, ANOVA analysis revealed that Acidobacteria and bacteria differed significantly. bacteria abundance was highest in Z, followed by ZS, M, LG and HLYHL. Acidobacteria abundance was highest in ZS, followed by M and Z and low abundance in LG and HLYHL. Bacteroidetes, Cyanobacteria and Firmicutes did not differ significantly. Amongst the 20 most abundant genera, 13 to 20 were significantly different. Of these, all were low in LG and HLYHL with the exception of Parabacteroides. According to our PCoA and heatmap analysis, different individuals in the same group had relatively close relationships and, thus, bacterial community composition similarity was higher in same-group individuals than in different-group individuals. Alpha diversity analysis showed significant differences for Chao1, ACE and Shannon, illustrating higher bacterial community richness and diversity in the different groups.
In summary, our study revealed the composition and diversity of the gut microbiota of 15 individuals belonging to five orthopteran species using DNA metabarcode sequencing. The results revealed a bacterial community composition comprising 24 phyla and 219 genera. The most abundant phyla were Firmicutes and bacteria and the most abundant genera were Lactococcus and Lactobacillus. We also compared differences in bacterial composition of distinct species at the phylum and genus levels. The results suggested the gut bacteria composition differed significantly across the five species.
Data resources
The raw data are available at the National Center for Biotechnology Information (NCBI) SRA (https://www.ncbi.nlm.nih.gov/sra/): SRR20722952 - SRR20722966.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The gut microbiota diversity of five Orthoptera insects determined by DNA metabarcoding
Yantong Liu1, Lina Zhao2, Zhongying Qiu1, Hao Yuan1*
Data type
images
File: oo_767535.docx
Acknowledgements
This study was supported by the Key Research and Development Project of Shaanxi Province (2019SF-172).
References
- Ademolu Kehinde O, Idowu Adewunmi B. Occurrence and distribution of microflora in the gut regions of the variegated grasshopper Zonocerusvariegatus (Orthoptera: Pyrgomorphidae) during development. Zoological Studies. 2011;50(4):409–415. [Google Scholar]
- Amato Katherine R, Yeoman Carl J, Kent Angela, Righini Nicoletta, Carbonero Franck, Estrada Alejandro, Rex Gaskins H, Stumpf Rebecca M, Yildirim Suleyman, Torralba Manolito. Habitat degradation impacts black howler monkey (Alouattapigra) gastrointestinal microbiomes. The ISME Journal. 2013;7(7):1344–1353. doi: 10.1038/ismej.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson Kirk E, Sheehan Timothy H, Mott Brendon M, Maes Patrick, Snyder Lucy, Schwan M R, Walton Alexander, Jones B M, Corby-Harris Vanessa. Microbial ecology of the hive and pollination landscape: bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apismellifera) PLOS One. 2013;8(12) doi: 10.1371/journal.pone.008312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar A, Yuval B, Jurkevitch E. Gut bacterial communities in the Mediterranean fruit fly (Ceratitiscapitata) and their impact on host longevity. Journal of Insect Physiology. 2008;54(9):1377–1383. doi: 10.1016/j.jinsphys.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Bolger Anthony M, Lohse Marc, Usadel Bjoern. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J Gregory, Kuczynski Justin, Stombaugh Jesse, Bittinger Kyle, Bushman Frederic D, Costello Elizabeth K, Fierer Noah, Peña Antonio Gonzalez, Goodrich Julia K, Gordon Jeffrey I. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew Melissa E, Pettigrove Vincent J, Metzeling Leon, Hoffmann Ary A. Environmental monitoring using next generation sequencing: rapid identification of macroinvertebrate bioindicator species. Frontiers in Zoology. 2013;10(1):1–15. doi: 10.1186/1742-9994-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Dawei, Yang Zhenquan, Chen Xia, Huang Yujun, Yin Boxing, Guo Feixiang, Zhao Haiqing, Zhao Tangyan, Qu Henxian, Huang Jiadi. The effect of Lactobacillusrhamnosus hsryfm 1301 on the intestinal microbiota of a hyperlipidemic rat model. BMC Complementary and Alternative Medicine. 2014;14(1):1–9. doi: 10.1186/1472-6882-14-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman DR, Toolson EC, Takacs‐Vesbach CD. Do diet and taxonomy influence insect gut bacterial communities? Molecular Ecology. 2012;21(20):5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x. [DOI] [PubMed] [Google Scholar]
- Coyne Michael J, Reinap Barbara, Lee Martin M, Comstock Laurie E. Human symbionts use a host-like pathway for surface fucosylation. Science. 2005;307(5716):1778–1781. doi: 10.1126/science.1106469. [DOI] [PubMed] [Google Scholar]
- DeSantis Todd Z, Hugenholtz Philip, Larsen Neils, Rojas Mark, Brodie Eoin L, Keller Keith, Huber Thomas, Dalevi Daniel, Hu Ping, Andersen Gary L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon Rod, Charnley Keith. Mutualism between the desert locust Schistocercagregaria and its gut microbiota. Research in Microbiology. 2002;153(8):503–509. doi: 10.1016/S0923-2508(02)01361-X. [DOI] [PubMed] [Google Scholar]
- Dillon RJ, Webster Gordon, Weightman Andrew John, Dillon VM, Blanford S, Charnley AK. Composition of Acridid gut bacterial communities as revealed by 16S rRNA gene analysis. Journal of Invertebrate Pathology. 2008;97(3):265–272. doi: 10.1016/j.jip.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Dillon Rod J, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annual Reviews in Entomology. 2004;49(1):71–92. doi: 10.1146/annurev.ento.49.061802.123416. [DOI] [PubMed] [Google Scholar]
- Donohoe Dallas R, Garge Nikhil, Zhang Xinxin, Sun Wei, O'Connell Thomas M, Bunger Maureen K, Bultman Scott J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metabolism. 2011;13(5):517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowle Eddy J, Pochon Xavier, Jonathan C. Banks,, Shearer Karen, Wood Susanna A. Targeted gene enrichment and high‐throughput sequencing for environmental biomonitoring: A case study using freshwater macroinvertebrates. Molecular Ecology Resources. 2016;16(5):1240–1254. doi: 10.1111/1755-0998.12488. [DOI] [PubMed] [Google Scholar]
- Edgar Robert C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Engel Philipp, Moran Nancy A. The gut microbiota of insects–diversity in structure and function. FEMS Microbiology Reviews. 2013;37(5):699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- Erkosar Berra, Storelli Gilles, Mitchell Mélanie, Bozonnet Loan, Bozonnet Noémie, Leulier François. Pathogen virulence impedes mutualist-mediated enhancement of host juvenile growth via inhibition of protein digestion. Cell Host & Microbe. 2015;18(4):445–455. doi: 10.1016/j.chom.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier Jean-Pierre, Outreman Yannick, Mieuzet Lucie, Simon Jean-Christophe. Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLOS One. 2015;10(3) doi: 10.1371/journal.pone.0120664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges Lauren M, Brownlie Jeremy C, O'Neill Scott L, Johnson Karyn N. Wolbachia and virus protection in insects. Science. 2008;322(5902):702–702. doi: 10.1126/science.1162418. [DOI] [PubMed] [Google Scholar]
- Hird Sarah M, Carstens Bryan C, Cardiff Steven W, Dittmann Donna L, Brumfield Robb T. Sampling locality is more detectable than taxonomy or ecology in the gut microbiota of the brood-parasitic Brown-headed Cowbird (Molothrusater) PeerJ. 2014;2 doi: 10.7717/peerj.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroki Masato, Kato Yoshiomi, Kamito Takehiko, Miura Kazuki. Feminization of genetic males by a symbiotic bacterium in a butterfly, Euremahecabe (Lepidoptera: Pieridae) Naturwissenschaften. 2002;89(4):167–170. doi: 10.1007/s00114-002-0303-5. [DOI] [PubMed] [Google Scholar]
- Hong Huynh A, Duc Le Hong, Cutting Simon M. The use of bacterial spore formers as probiotics. FEMS Microbiology Reviews. 2005;29(4):813–835. doi: 10.1016/j.femsre.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Idowu AB, Edema MO, Oyedepo MT. Extracellular enzyme production by microflora from the gut region of the variegated grasshopper Zonocerusvariegatus (Orthoptera: Pyrgomorphidae) International Journal of Tropical Insect Science. 2009;29(4):229–235. doi: 10.1017/S1742758409990312. [DOI] [Google Scholar]
- Jeyaprakash Ayyamperumal, Hoy Marjorie A. Long PCR improves Wolbachia DNA amplification: wsp sequences found in 76% of sixty‐three arthropod species. Insect Molecular Biology. 2000;9(4):393–405. doi: 10.1046/j.1365-2583.2000.00203.x. [DOI] [PubMed] [Google Scholar]
- Jiggins FM, Hurst GDD, Jiggins CD, vd Schulenburg JHG, Majerus MEN. The butterfly Danauschrysippus is infected by a male-killing Spiroplasma bacterium. Parasitology. 2000;120(5):439–446. doi: 10.1017/S0031182099005867. [DOI] [PubMed] [Google Scholar]
- Kim Young S, Milner John A. Dietary modulation of colon cancer risk. The Journal of Nutrition. 2007;137(11) doi: 10.1093/jn/137.11.2576S. [DOI] [PubMed] [Google Scholar]
- Kraaijeveld Ken, De Weger Letty A, Ventayol García Marina, Buermans Henk, Frank Jeroen, Hiemstra Pieter S, Den Dunnen Johan T. Efficient and sensitive identification and quantification of airborne pollen using next‐generation DNA sequencing. Molecular Ecology Resources. 2015;15(1):8–16. doi: 10.1111/1755-0998.12288. [DOI] [PubMed] [Google Scholar]
- Leray Matthieu, Knowlton Nancy. DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proceedings of the National Academy of Sciences. 2015;112(7):2076–2081. doi: 10.1073/pnas.1424997112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč Tanja, Salzberg Steven L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minard Guillaume, Tran Florence Hélène, Van Van Tran, Goubert Clement, Bellet Christophe, Lambert Guillaume, Kim Khanh Ly Huynh, Thuy Trang Huynh Thi, Mavingui Patrick, Valiente Moro Claire. French invasive Asian tiger mosquito populations harbor reduced bacterial microbiota and genetic diversity compared to Vietnamese autochthonous relatives. Frontiers in Microbiology. 2015;6:970. doi: 10.3389/fmicb.2015.00970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouwehand Arthur C, Salminen Seppo, Isolauri Erika. Probiotics: an overview of beneficial effects. Lactic Acid Bacteria: Genetics, Metabolism and Applications. 2002;82:279–289. [PubMed] [Google Scholar]
- Rosenthal Adam Z, Matson Eric G, Eldar Avigdor, Leadbetter Jared R. RNA-seq reveals cooperative metabolic interactions between two termite-gut spirochete species in co-culture. The ISME Journal. 2011;5(7):1133–1142. doi: 10.1038/ismej.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell Jacob A, Funaro Colin F, Giraldo Ysabel M, Goldman-Huertas Benjamin, Suh David, Kronauer Daniel JC, Moreau Corrie S, Pierce Naomi E. A veritable menagerie of heritable bacteria from ants, butterflies, and beyond: broad molecular surveys and a systematic review. PLOS One. 2012;7(12):e51027. doi: 10.1371/journal.pone.0051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss Patrick D, Westcott Sarah L, Ryabin Thomas, Hall Justine R, Hartmann Martin, Hollister Emily B, Lesniewski Ryan A, Oakley Brian B, Parks Donovan H, Robinson Courtney J. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter Andreas, Bekel Thomas, Diaz Naryttza N, Dondrup Michael, Eichenlaub Rudolf, Gartemann Karl-Heinz, Krahn Irene, Krause Lutz, Krömeke Holger, Kruse Olaf. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. Journal of Biotechnology. 2008;136(1-2):77–90. doi: 10.1016/j.jbiotec.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Schmitt-Wagner Dirk, Friedrich Michael W, Wagner Bianca, Brune Andreas. Axial dynamics, stability, and interspecies similarity of bacterial community structure in the highly compartmentalized gut of soil-feeding termites (Cubitermes spp.) Applied and Environmental Microbiology. 2003;69(10):6018–6024. doi: 10.1128/AEM.69.10.6018-6024.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott Jarrod J, Oh Dong-Chan, Yuceer M Cetin, Klepzig Kier D, Clardy Jon, Currie Cameron R. Bacterial protection of beetle-fungus mutualism. Science. 2008;322(5898):63–63. doi: 10.1126/science.1160423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastien A, Gruber MAM, Lester PJ. Prevalence and genetic diversity of three bacterial endosymbionts (Wolbachia, Arsenophonus, and Rhizobiales) associated with the invasive yellow crazy ant (Anoplolepisgracilipes) Insectes Sociaux. 2012;59(1):33–40. doi: 10.1007/s00040-011-0184-8. [DOI] [Google Scholar]
- Selma Maria V, Espin Juan C, Tomas-Barberan Francisco A. Interaction between phenolics and gut microbiota: role in human health. Journal of Agricultural and Food Chemistry. 2009;57(15):6485–6501. doi: 10.1021/jf902107d. [DOI] [PubMed] [Google Scholar]
- Sharon Gil, Segal Daniel, Ringo John M, Hefetz Abraham, Zilber-Rosenberg Ilana, Rosenberg Eugene. Commensal bacteria play a role in mating preference of Drosophilamelanogaster. Proceedings of the National Academy of Sciences. 2010;107(46):20051–20056. doi: 10.1073/pnas.1009906107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi WB, Uzuner U, Jesudhasan PR, Pillai SD, Yuan JY. Comparative analysis of insect gut symbiotic composition and diversity as adaptation to different food type. Biofuels. 2011;2:529–544. [Google Scholar]
- Smith Chad C, Srygley Robert B, Healy Frank, Swaminath Karthikeyan, Mueller Ulrich G. Spatial structure of the mormon cricket gut microbiome and its predicted contribution to nutrition and immune function. Frontiers in Microbiology. 2017;8:801. doi: 10.3389/fmicb.2017.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley Dragana, Geier Mark S, Hughes Robert J, Denman Stuart E, Moore Robert J. Highly variable microbiota development in the chicken gastrointestinal tract. PLOS One. 2013;8(12):e84290. doi: 10.1371/journal.pone.0084290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storelli Gilles, Defaye Arnaud, Erkosar Berra, Hols Pascal, Royet Julien, Leulier François. Lactobacillusplantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metabolism. 2011;14(3):403–414. doi: 10.1016/j.cmet.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Teixeira Luís, Ferreira Álvaro, Ashburner Michael. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophilamelanogaster. PLOS Biology. 2008;6(12) doi: 10.1371/journal.pbio.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida Tsutomu, Koga Ryuichi, Fukatsu Takema. In: Insect Symbiosis, Volume 3. 1st. , editor. Vol. 3. CRC Press; 2008. Endosymbiont that broadens food plant range of host insect19. [Google Scholar]
- Wang Yu, Sheng Hua-Fang, He Yan, Wu Jin-Ya, Jiang Yun-Xia, Tam Nora Fung-Yee, Zhou Hong-Wei. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Applied and Environmental Microbiology. 2012;78(23):8264–8271. doi: 10.1128/AEM.01821-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Xiaofeng, Zheng Dandan, Zhong Huanzi, Qin Bingcai, Gurr Geoff M, Vasseur Liette, Lin Hailan, Bai Jianlin, He Weiyi, You Minsheng. DNA sequencing reveals the midgut microbiota of diamondback moth, Plutellaxylostella (L.) and a possible relationship with insecticide resistance. PLOS One. 2013;8(7):e68852. doi: 10.1371/journal.pone.0068852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Wu, Li Zhigang, Liu Hongjun, Xue Chao, Zhang Ruifu, Wu Huasong, Li Rong, Shen Qirong. The effect of long-term continuous cropping of black pepper on soil bacterial communities as determined by 454 pyrosequencing. PLOS One. 2015;10(8):e0136946. doi: 10.1371/journal.pone.0136946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Jin-Long, Wang Ming-Shu, Cheng An-Chun, Pan Kang-Cheng, Li Chuan-Feng, Deng Shu-Xuan. A simple and rapid method for extracting bacterial DNA from intestinal microflora for ERIC-PCR detection. World Journal of Gastroenterology: WJG. 2008;14(18):2872. doi: 10.3748/wjg.14.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Douglas W, Ji Yinqiu, Emerson Brent C, Wang Xiaoyang, Ye Chengxi, Yang Chunyan, Ding Zhaoli. Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods in Ecology and Evolution. 2012;3(4):613–623. doi: 10.1111/j.2041-210X.2012.00198.x. [DOI] [Google Scholar]
- Yun Ji-Hyun, Roh Seong Woon, Whon Tae Woong, Jung Mi-Ja, Kim Min-Soo, Park Doo-Sang, Yoon Changmann, Nam Young-Do, Kim Yun-Ji, Choi Jung-Hye. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Applied and Environmental Microbiology. 2014;80(17):5254–5264. doi: 10.1128/AEM.01226-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Zheng, Yang Jun, Liu Lemian, Zhang Wenjing, Amalfitano Stefano. Bacterioplankton community shifts associated with epipelagic and mesopelagic waters in the Southern Ocean. Scientific Reports. 2015;5(1):1–10. doi: 10.1038/srep12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Wenjing, Wang Yapeng, Liu Shuyun, Huang Jiaojiao, Zhai Zhengxiao, He Chuan, Ding Jinmei, Wang Jun, Wang Huijuan, Fan Weibing. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLOS One. 2015;10(2):e0117441. doi: 10.1371/journal.pone.0117441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The gut microbiota diversity of five Orthoptera insects determined by DNA metabarcoding
Yantong Liu1, Lina Zhao2, Zhongying Qiu1, Hao Yuan1*
Data type
images
File: oo_767535.docx