

Abstract
The oxetane functional group offers a variety of potential advantages when incorporated within appropriate therapeutic agents as a ketone surrogate. OXi8006, a 2-aryl-3-aroyl-indole analogue, functions as a small-molecule inhibitor of tubulin polymerization that has a dual mechanism of action as both an antiproliferative agent and a tumor-selective vascular disrupting agent. Replacement of the bridging ketone moiety in OXi8006 with an oxetane functional group has expanded structure activity relationship (SAR) knowledge and provided insights regarding oxetane incorporation within this class of molecules. A new synthetic method using an oxetane-containing tertiary alcohol subjected to Lewis acid catalyzed conditions led to successful Friedel–Crafts alkylation and yielded fourteen new oxetane-containing indole-based molecules. This synthetic approach represents the first method to successfully install an oxetane ring at the 3-position of a 2-aryl-indole system. Several analogues showed potent cytotoxicity (micromolar GI50 values) against human breast cancer cell lines (MCF-7 and MDA-MB-231) and a pancreatic cancer cell line (PANC-1), although they proved to be ineffective as inhibitors of tubulin polymerization. Molecular docking studies comparing colchicine with the OXi8006-oxetane analogue 5m provided a rationale for the differential interaction of these molecules with the colchicine site on the tubulin heterodimer.
Keywords: Inhibition of Tubulin Polymerization, Oxetane, Friedel-Crafts Alkylation
1. Background
Decades of fundamental science coupled with translational advancement have resulted in a wide variety of FDA approved therapeutic modalities for the treatment of cancer and led to the identification of many promising biological targets associated with cancer initiation, progression, and metastasis. Despite these important discoveries, there remains a continuing quest focused on the discovery of new therapeutic agents and methods to more effectively and selectively treat cancer and to further our understanding of the associated biological mechanisms of action.
Inspired by the natural products colchicine,1 combretastatin A-42 and combretastatin A-13 (Figure 1), our efforts to extend the known structure-activity relationship correlations associated with small-molecule inhibitors of tubulin polymerization led to the discovery of promising new molecules including dihydronaphthalenes,4,5 benzosuberenes,4,6–11 benzo[b]thiophenes,12 benzo[b]furans,13 and indoles14–18 (Figure 1). These molecules function as potent inhibitors of tubulin polymerization and show strong antiproliferative activity against human cancer cell lines. In addition, these molecules function as tumor-selective vascular disrupting agents (VDAs)17,19–23 that cause effective vascular damage to tumor-associated microvessels.
Figure 1.

Natural Products Colchicine,1 CA-42, CA-13 and Selected Synthetic Small-Molecule Analogues Designed and Synthesized by the Pinney Group4,6–11,14–18 as Inhibitors of Tubulin Polymerization.
The promising biological activity associated with the indole-based analogue OXi800615−17 has inspired continued synthetic efforts towards new analogues to enhance pharmacophore kinetics and bioactivity. These efforts included the identification of alternative functional groups to serve as surrogates for the carbonyl moiety. One promising candidate was the oxetane functional group, which is a four-membered ring containing three carbon atoms and one oxygen atom. The oxetane moiety emerged as a “hot topic” in medicinal chemistry, due to the physicochemical and biological properties associated with this functional group.24,25 The oxetane functionality offers several advantages as a good replacement group for gem-dimethyl26,27 and carbonyl groups.28 In particular, the use of oxetanes as replacements for carbonyl groups has drawn attention due to similar dipoles and H-bonding properties.28 Given the intriguing features of oxetane as a surrogate of ketone, this functionality was incorporated into our indole-based analogue OXi800615,17 (Figure 2) along with related analogues. Common strategies to prepare oxetanes include intramolecular cyclization,29–31 Paternò−Büchi [2+2] photocycloaddition,32,33 and derivatization from oxetane containing building blocks.26,27,34 Previously, the Bull group reported a method to a 3,3-diaryloxetane ring by Friedel–Crafts alkylation on oxetanols with a lithium catalyst.35 Installation of the oxetane moiety into the highly sterically congested quaternary center still poses a synthetic challenge and was further investigated.
Figure 2.

Molecular Design Paradigm: Oxetane Moiety as Ketone Surrogate in OXi8006
2. Results and Discussion
2.1. Synthesis
The oxetane containing tertiary alcohol 335 was synthesized by performing lithium-halogen exchange on aryl bromide 1 and subsequent addition to ketone 2, which delivered the oxetane containing building block 3 in 77% yield (Scheme 1). Our methodology drew inspiration from work accomplished by the Bull Group,35 using lithium-catalyzed Friedel–Crafts to access 3,3-diaryloxetanes and acylation of indoles under conditions developed by the Aquino Group.36 Through utilization of this method, SnCl4 was used as a Lewis acid and CH2Cl2:CH3NO2 was employed as a co-solvent under room temperature (overall mild conditions) to install the 3,4,5-trimethoxylphenyloxetane moiety within structurally diverse 2-arylated indole analogues 4. Various conditions utilizing different Lewis acids or Brønsted acids were also investigated in order to enhance the yield during Friedel-Crafts acylation, but SnCl4 as a Lewis acid showed the highest efficiency. This approach represents the first method that facilitates successful installation of the oxetane moiety within 2-arylated indoles.
Scheme 1.

Synthesis of Indole-Based Oxetane-Containing Analogues
Through this Friedel–Crafts alkylation strategy, the key compound (5m), oxetane-incorporated OXi8006 (Scheme 2), along with a variety of indole-based analogues (Table 1), were synthesized with functional group variation on the indole core as well as in the pendant aryl ring. Analogues selected for synthesis drew structure and functional group inspiration from OXi800615−18 (Pinney Group) along with related molecules from the Silvestri Group.37
Scheme 2.

Synthesis of Oxetane-Substituted OXi8006
Table 1.
Oxetane-Containing Indole-based Analogues
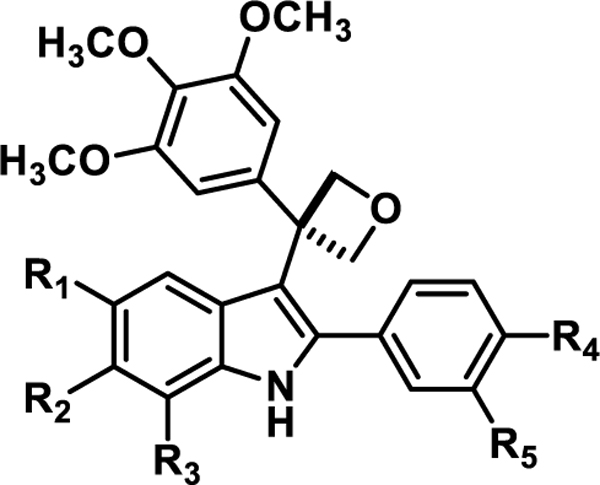
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | Yield |
| 5a | H | H | H | H | H | 23% |
| 5b | H | Cl | H | CH3 | H | 31% |
| 5c | H | Br | H | H | H | 14% |
| 5d | OCH3 | H | H | CH3 | H | 5% |
| 5e | H | Cl | H | OCH3 | H | 17% |
| 5f | H | Br | H | CH3 | H | 34% |
| 5g | H | H | H | H | OCH3 | 30% |
| 5h | H | H | OCH3 | H | H | 13% |
| 5i | H | H | H | OCH3 | H | 10% |
| 5j | H | H | H | CH3 | H | 27% |
| 5k | H | Cl | H | H | H | 11% |
| 5l | OCH3 | H | H | H | H | 22% |
| 5m | H | OCH3 | H | OCH3 | OH | 12% |
| 5n | Br | H | H | H | H | 14% |
2.2. Biological Evaluation:
The new oxetane-containing analogues were evaluated for their bioactivity against selected human cancer cell lines and their ability to inhibit tubulin polymerization. While none of the synthesized oxetane-containing analogues demonstrated significant activity as inhibitors of tubulin polymerization (IC50 > 20 μM), ten of the molecules displayed μM cytotoxicity as low as 0.47 ± 0.02 μM against MCF-7 cells (Table 2). The most potent analogues (5c, 5h, 5k) were further evaluated against additional cancer cell lines (MDA-MB-231 and PANC-1), and potent activity against these cell lines was also observed (Table 3 and SI Figs. S1–S6). It is instructive to note that OXi8006,15 the ketone counterpart of oxetane 5m, and the ketone analogues37 of compounds 5c, 5h, 5k, and 5l function as inhibitors of tubulin polymerization while all five corresponding oxetane analogues (5m, 5c, 5h, 5k, and 5l) were at most weakly active as inhibitors of tubulin polymerization. Some limited preliminary structure activity relationship (SAR) patterns can be drawn from these initial antiproliferative cell-based studies. The three most active compounds (5c, 5h, 5k) each incorporate a functional group at either the R2 position (Br and Cl, respectively for 5c and 5k) or the R3 position (OCH3 for 5h) and include an unfunctionalized pendant phenyl ring at the 2-position. The lack of any functionalization (5a) and functionalization at just R4 (5i and 5j) led to significantly decreased antiproliferative activity. Interestingly, antiproliferative activity was restored (albeit at a lower level) with substitution at R4 on the pendant ring when accompanied by substitution on the indole ring at R2 or R3 (5b, 5e, 5f), and in one case with additional functionalization at R5 (5m), but not at R1 (5d). Since these molecules are not highly active as inhibitors of tubulin polymerization, their cytotoxicity could result from an alternative biological target, which will be the subject of future investigation.
Table 2.
Cytotoxicity against the MCF-7 Human Breast Cancer Cell Line
| Compound | IC50 (μM) SRB assay MCF-7a |
|---|---|
| OXi8006 b | 0.048 ± 0.01 |
| 5a | >5 |
| 5b | 2.7 ± 0.2 |
| 5c | 0.47 ± 0.02 |
| 5d | >5 |
| 5e | 3.7 ± 0.07 |
| 5f | 1.7 ± 0.2 |
| 5g | 4.8 ± 0.3 |
| 5h | 0.78 ± 0.2 |
| 5i | >5 |
| 5j | >5 |
| 5k | 0.84 ± 0.09 |
| 5l | 3.2 ± 1.0 |
| 5m | 1.6 ± 0.4 |
| 5n | 2.2 ± 0.5 |
Average of n ≥ 3 independent determinations
See ref16 for additional OXi8006 data
Table 3.
Cytotoxicity Evaluation Against the MDA-MB-231 Human Breast Cancer and Pancreatic Cancer PANC-1 Cell Lines in Comparison with the MCF-7 Data
| Compound | IC50 (μM) SRB assay MCF-7a | IC50 (μM) SRB assay MDA-MB-231a | IC50 (μM) SRB assay PANC-1a |
|---|---|---|---|
| OXi8006 | 0.048 ± 0.01 | 0.044 ± 0.008 | NDb |
| 5c | 0.47 ± 0.02 | 1.9 ± 0.1 | 0.43 ± 0.1 |
| 5h | 0.78 ± 0.2 | 3.0 ± 1.0 | 0.44 ± 0.01 |
| 5k | 0.84 ± 0.09 | 2.2 ± 0.2 | 1.1 ± 0.5 |
Average of n ≥ 3 independent determinations
ND: not determined
2.2.1. Wound Healing Assay
In the multistep process that leads to cancer metastasis, cell migration is an essential element. Both compounds 5c and 5h inhibited PANC-1 cell migration in wound healing assays. At a higher concentration than the respective IC50 values, 0.5 μM, cells treated with compound 5c (Fig. 3B) or 5h (Fig. 4B) slowly moved into the gap introduced by the scratch as compared to controls (Figs. 3A, 4A). Upon treatment with 1 μM 5c (Fig. 3C) or 5h (Fig. 4C), the gaps were largely unclosed at 40 h while the control scratches were confluent (SI-2 Figs. S7, S8). Inhibition of cell migration is often associated with disruption of the microtubule cytoskeleton, which is required for cell body translocation. However, cell migration is a complex process that involves many proteins in addition to the actin and microtubule cytoskeleton. For example, anticancer agents homoharringtonine, an inhibitor of protein translation, and doxorubicin, an inhibitor of topoisomerase II, both inhibit human osteosarcoma U2OS cell migration by other mechanisms.38
Figure 3: Scratch Assay for PANC-1 Cells Treated with Compound 5c.

Representative images of PANC-1 cells at 1, 24, and 40 h after making scratches. Cells were incubated at 37 °C with 5% CO2. A(1) A(2) and A(3): control wells; scratch gap closed after 40 h; B(1), B(2) and B(3): 0.5 μM 5c treated cells; cells slowly move towards the center of the gap, but gap is not completely closed after 40 h; C(1), C(2) and C(3): 1 μM 5c treated cells; gap remains open after 40 h.
Figure 4: Scratch Assay for PANC-1 Cells Treated with Compound 5h.

Representative images of PANC-1 cells at time 1, 24, and 48 h after making scratches. Cells were incubated at 37 °C with 5% CO2. A(1) A(2) and A(3): control wells; scratch gap closed after 48 h; B(1), B(2) and B(3): 0.5 μM 5h treated cells; cells slowly move towards the center of the gap, but gap is not closed after 48 h; C(1), C(2) and C(3): 1 μM 5h treated cells; gap remains open after 48 h.
2.3. Docking Study
Molecular docking was implemented to predict binding affinity and docking poses of the oxetane indole analogues utilizing Discovery Studio Client 4.5 (Accelrys). Compound 5m was used as a model compound for the oxetane-containing analogues for the docking study. A previously characterized X-ray structure of N-deacetyl-N-(2-mercaptoacetyl)-colchicine (DAMA-colchicine) co-crystallized with tubulin (1SA0) served as the model structure.39 The protein was prepared, and DAMA-colchicine was removed and re-docked (CDocker) to validate docking parameters. Agreement between X-ray structure and docked ligand bound to tubulin demonstrated a suitable protein structure and parameters to dock compound 5m.
Based on in silico results, the energetically favored orientation of compound 5m is dramatically different than that of known colchicine site agents. Displacement of the trimethoxy ring of 5m in comparison to DAMA-colchicine affects a multitude of hydrophobic interactions as well as hydrogen bonding (Figure 5). Relocation of the trimethoxy ring facing the alpha-tubulin subunit indicates hydrogen bonding with αASN101, but this produces an unfavorable acceptor-acceptor clash involving the 4’-methoxy group and αTHR179. Additionally, hydrophobic interactions within the binding pocket associated with β-tubulin are lacking, including those with LEU248 and LEU242 (Figure 6).40,41 These results suggest the importance of the bridging ketone between the aroyl and indole scaffold for maintaining potent activity as an inhibitor of tubulin polymerization.
Figure 5: Compound 5m Trimethoxy Ring (blue) Interaction with the α-Subunit Residues (Blue).

The DAMA-colchicine (co-crystalized structure with tubulin) trimethoxy ring indicated in yellow interaction with β-tubulin residues (green).
Figure 6:

Molecular Docking of Compound 5m, α-Subunit Residues (Blue); All Other Residues are Located on the β-Subunit (Green).
3. Conclusions
Incorporation of the oxetane functional group (as a surrogate for the corresponding ketone moiety) was evaluated as a design paradigm in a small but focused series of indole-based analogues. Several synthetic strategies were evaluated to circumvent difficulties associated with oxetane incorporation, which led to a general synthetic route capable of accessing several new analogues. Biological studies evaluated inhibition of tubulin polymerization and cytotoxicity against human cancer cell lines for these oxetane-bearing analogues. In comparison to their corresponding ketone counterparts, the oxetane-bearing analogues 5m, 5c, 5h, 5k, and 5l showed no significant tubulin assembly inhibition activity, however several oxetane analogues in this series demonstrated micromolar antiproliferative activity, expanding knowledge regarding SAR for tubulin binding at the colchicine site.
4. Experimental Section
4.1. Chemistry
4.1.1. General Materials and Methods
Tetrahydrofuran (THF) was used as anhydrous. Thin-layer chromatography (TLC) plates (pre-coated glass plates with silica gel 60 F254, 0.25 mm thickness) were used to monitor reactions. Purification of intermediates and products was carried out with a Biotage Isolera flash purification system using silica gel (200−400 mesh, 60 Å) or RP18 prepacked columns. Intermediates and products synthesized were characterized based on their 1H NMR (600 MHz) and 13C NMR (151 MHz) spectroscopic data using a Bruker DPX 600 MHz instrument. Spectra were recorded in CDCl3. All chemicals’ shifts are expressed in ppm (δ), and peak patterns are reported as singlet (s), doublet (d), triplet (t), double doublet (dd), double double doublet (ddd), and multiplet (m). The purity of the final compounds was further analyzed at 25 °C using an Agilent 1200 HPLC system with a diode-array detector (λ = 190−400 nm), a Zorbax XDB-C18 HPLC column (150 mm, 5 μm), and a Zorbax reliance cartridge guard-column; solvent A H2O, solvent B CH3CN; flow rate 1.0 mL/min; injection volume 20 μL; monitored at wavelengths of 210, 230, 240, 254, 280, 300 and 320 nm. Method A: gradient, 90% A/10% B for 5 min, 90% A/10% B to 0% A/100% B over 20 min, 0% A/100% B for 5 min; post-time 10 min; Method B: gradient, 70% A/30% B to 10% A/90% B over 25 min, 10% A/90% B for 5 min; post-time 10 min; Method C: gradient, 50% A/50% B to 10% A/90% B over 25 min; 10% A/90% B for 5 min; post-time 10 min. Mass spectrometry was carried out under either positive or negative ESI (electrospray ionization) or positive or negative APCI/APPI (atmospheric pressure chemical ionization/ atmospheric pressure photoionization) using a Thermo Scientific LTQ Orbitrap Discovery instrument. 4a was purchased from Ambeed Inc. Known commercially-available indoles 4b-4l, 4n were prepared following the reported procedure.42 Compound 4m was prepared using the procedure published previously by our group.16
4.1.2. Synthesis of Oxetane Analogues
4.1.2.1. 3-(3,4,5-Trimethoxyphenyl)oxetan-3-ol35 (3)
To an oven-dried flask, THF (30 mL) and 3,4,5-trimethoxyphenyl bromide (3.0 g, 12 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (5.3 mL, 13 mmol) was added dropwise to the reaction mixture, which was then stirred at −78 °C for 1 h. 3-Oxetanone (0.92 mL, 13 mmol) was then added slowly to the flask, and the reaction mixture was stirred while gradually warming from −78 °C to room temperature over 12 h. The reaction was quenched with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and the solvent removed by evaporation under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 100% ethyl acetate in hexanes) to afford alcohol 3 (2.2 g, 9.3 mmol, 77%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 6.80 (s, 2H), 4.87 (d, J = 1.3 Hz, 4H), 3.85 (s, 6H), 3.84 (s, 1H), 3.82 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 153.21, 138.44, 137.20, 101.70, 85.85, 75.46, 60.81, 56.13. HRMS (ESI+) calculated for C12H16O5 [M+Na] 263.0890, actual 263.0889.
4.1.2.2. 2-Phenyl-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5a)
To a well-stirred solution of commercially-available indole 4a (0.10 g, 0.50 mmol) in CH2Cl2 (5.0 mL) under N2 at 0 °C, SnCl4 (0.12 mL, 0.80 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.15 g, 0.60 mmol) was added in small portions to the suspension, followed by nitromethane (5.0 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 50 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5a (50 mg, 0.12 mmol, 23%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.28 (s, 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.41 – 7.36 (m, 3H), 7.30 (dd, J = 8.1, 2.0 Hz, 2H), 7.26 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.11 – 7.02 (m, 2H), 6.96 (s, 2H), 5.17 (d, J = 5.7 Hz, 2H), 4.93 (d, J = 5.7 Hz, 2H), 3.92 (s, 3H), 3.75 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 153.30, 142.62, 136.92, 136.00, 134.51, 132.96, 128.99, 128.35, 127.98, 127.34, 122.63, 120.11, 119.82, 115.17, 111.16, 103.69, 85.35, 60.88, 56.22, 47.16; HRMS (ESI+) calculated for C26H25NO4 [M+Na] 438.1676, actual 438.1678. HPLC (Method B): retention time 15.3 min.
4.1.2.3. 6-Chloro-2-(p-tolyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5b)
To a well-stirred solution of indole 4b (0.30 g, 1.2 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.23 mL, 1.9 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.33 g, 1.4 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5b (0.18 g, 0.38 mmol, 31%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), 7.43 (d, J = 1.8 Hz, 1H), 7.21 – 7.12 (m, 4H), 7.03 (dd, J = 8.4, 1.9 Hz, 1H), 6.96 – 6.87 (m, 3H), 5.13 (d, J = 5.8 Hz, 2H), 4.92 (d, J = 5.9 Hz, 2H), 3.92 (s, 3H), 3.75 (s, 6H), 2.38 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 153.36, 142.49, 138.53, 136.90, 136.38, 135.60, 129.69, 129.63, 128.09, 127.17, 126.53, 120.60, 120.34, 114.42, 111.23, 103.65, 85.24, 60.89, 56.21, 47.03, 21.28; HRMS (ESI+) calculated for C27H26ClNO4 [M+Na] 486.1443, actual 486.1443. HPLC (Method A): retention time 22.8 min.
4.1.2.4. 6-Bromo-2-phenyl-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5c)
To a well-stirred solution of indole 4c (0.4 g, 1.5 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.32 mL, 2.2 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.39 g, 1.6 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5c (98 mg, 0.21 mmol, 14%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 7.62 (d, J = 1.7 Hz, 1H), 7.42 – 7.32 (m, 3H), 7.32 – 7.23 (m, 2H), 7.18 (dd, J = 8.5, 1.7 Hz, 1H), 6.97 – 6.87 (m, 3H), 5.12 (d, J = 5.8 Hz, 2H), 4.91 (d, J = 5.9 Hz, 2H), 3.92 (s, 3H), 3.75 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 153.37, 142.30, 137.00, 136.78, 135.26, 132.44, 129.05, 128.63, 127.31, 126.83, 123.40, 120.91, 116.05, 115.14, 114.21, 103.61, 85.20, 60.88, 56.24, 46.96.; HRMS (ESI+) calculated for C26H24BrNO4 [M+Na] 516.0781, actual 516.0794. HPLC (Method A): retention time 22.3 min.
4.1.2.5. 5-Methoxy-2-(p-tolyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5d)
To a well-stirred solution of indole 4d (0.50 g, 2.1 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.42 mL, 3.2 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.56 g, 2.3 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 60% ethyl acetate in hexanes) to afford oxetane analogue 5d (49 mg, 0.11 mmol, 5%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.14 (s, 1H), 7.39 – 7.32 (m, 1H), 7.21 – 7.13 (m, 4H), 6.97 (s, 2H), 6.90 (dd, J = 8.8, 2.4 Hz, 1H), 6.43 (d, J = 2.5 Hz, 1H), 5.17 (d, J = 5.7 Hz, 2H), 4.91 (d, J = 5.6 Hz, 2H), 3.92 (s, 3H), 3.78 (s, 3H), 3.76 (s, 6H), 2.38 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 154.12, 153.31, 142.63, 138.18, 136.85, 135.64, 131.23, 130.16, 129.62, 128.49, 127.12, 114.25, 111.96, 111.93, 103.70, 101.80, 85.29, 60.88, 56.21, 56.00, 47.13, 21.25; HRMS (ESI+) calculated for C28H29NO5 [M+Na] 482.1938, actual 482.1935. HPLC (Method C): retention time 6.6 min.
4.1.2.6. 6-Chloro-2-(4-methoxyphenyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5e)
To a well-stirred solution of indole 4e (0.50 g, 2.0 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.41 mL, 3.0 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.51 g, 2.1 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5e (0.16 g, 0.34 mmol, 17%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.38 (s, 1H), 7.39 (d, J = 1.8 Hz, 1H), 7.21 – 7.06 (m, 2H), 7.00 (dd, J = 8.4, 1.8 Hz, 1H), 6.94 – 6.81 (m, 5H), 5.11 (d, J = 5.8 Hz, 2H), 4.88 (d, J = 5.8 Hz, 2H), 3.89 (s, 3H), 3.80 (s, 3H), 3.72 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 159.80, 153.33, 142.49, 136.88, 136.28, 135.41, 128.58, 127.92, 126.54, 124.90, 120.55, 120.21, 114.41, 113.94, 111.13, 103.61, 85.17, 60.85, 56.20, 55.31, 46.97; HRMS (ESI+) calculated for C27H26ClNO5 [M+Na] 502.1392, actual 502.1395. HPLC (Method A): retention time 21.9 min.
4.1.2.7. 6-Bromo-2-(p-tolyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5f)
To a well-stirred solution of indole 4f (0.25 g, 0.90 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.22 mL, 1.3 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.23 g, 1.0 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5f (0.15 g, 0.30 mmol, 34%) as a yellow solid: 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.60 (d, J = 1.7 Hz, 1H), 7.23 – 7.10 (m, 5H), 7.03 – 6.81 (m, 3H), 5.12 (d, J = 5.8 Hz, 2H), 4.91 (d, J = 5.9 Hz, 2H), 3.91 (s, 3H), 3.75 (s, 6H), 2.38 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 153.35, 142.35, 138.70, 136.97, 136.67, 135.40, 129.77, 129.50, 127.16, 126.90, 123.33, 120.81, 115.86, 114.72, 114.09, 103.59, 85.19, 60.88, 56.23, 46.98, 21.29; HRMS (ESI+) calculated for C27H26BrNO4 [M+Na] 530.0937, actual 530.0941. HPLC (Method A): retention time 23.1 min.
4.1.2.8. 2-(3-Methoxyphenyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5g)
To a well-stirred solution of indole 4g (0.50 g, 2.2 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.42 mL, 3.3 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.59 g, 2.4 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 60% ethyl acetate in hexanes) to afford oxetane analogue 5g ( 0.31 g, 0.67 mmol, 30%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.33 – 7.20 (m, 2H), 7.14 – 7.01 (m, 2H), 6.97 (s, 2H), 6.92 – 6.87 (m, 2H), 6.85 – 6.81 (m, 1H), 5.20 (d, J = 5.8 Hz, 2H), 4.95 (d, J = 5.9 Hz, 2H), 3.90 (s, 3H), 3.75 (s, 6H), 3.65 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 159.82, 153.30, 142.65, 136.93, 135.88, 134.39, 134.18, 130.07, 127.95, 122.66, 120.15, 119.78, 119.57, 115.33, 114.35, 112.44, 111.18, 103.68, 85.34, 60.86, 56.24, 55.01, 47.17; HRMS (ESI+) calculated for C27H27NO5 [M+Na] 468.1781, actual 468.1788. HPLC (Method A): retention time 20.6 min.
4.1.2.9. 7-Methoxy-2-phenyl-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5h)
To a well-stirred solution of indole 4h (0.50 g, 2.2 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.40 mL, 3.3 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.59 g, 2.4 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 60% ethyl acetate in hexanes) to afford oxetane analogue 5h (0.12g, 0.30 mmol, 13%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.53 – 8.42 (m, 1H), 7.47 – 7.18 (m, 5H), 7.08 – 6.90 (m, 3H), 6.71 (d, J = 7.7 Hz, 1H), 6.66 (d, J = 8.0 Hz, 1H), 5.16 (d, J = 5.9, 2H), 4.92 (d, J = 6.0, 2H), 4.04 (s, 3H), 3.91 (s, 3H), 3.76 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 153.28, 146.12, 142.68, 136.87, 134.13, 132.99, 129.22, 128.93, 128.24, 127.34, 126.47, 120.52, 115.55, 112.48, 103.68, 102.40, 85.37, 60.87, 56.23, 55.37, 47.21; HRMS (ESI+) calculated for C27H27NO5 [M+Na] 468.1781, actual 468.1781. HPLC (Method A): retention time 20.7 min.
4.1.2.10. 2-(4-Methoxyphenyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5i)
To a well stirred solution of indole 4i (0.50 g, 2.2 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.41 mL, 3.3 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.58 g, 2.4 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 60 % ethyl acetate in hexanes) to afford oxetane analogue 5i (96 mg, 0.22 mmol, 10%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.17 (s, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.26 – 7.19 (m, 3H), 7.10 – 7.00 (m, 2H), 6.95 (s, 2H), 6.93 – 6.89 (m, 2H), 5.18 (d, J = 5.8 Hz, 2H), 4.92 (d, J = 5.9 Hz, 2H), 3.91 (s, 3H), 3.84 (s, 3H), 3.75 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 159.72, 153.28, 142.74, 136.88, 135.84, 134.49, 128.61, 128.06, 125.37, 122.30, 120.01, 119.60, 114.43, 114.27, 111.00, 103.67, 85.29, 60.87, 56.22, 55.35, 47.16; HRMS (ESI+) calculated for C27H27NO5 [M+Na] 468.1781, actual 468.1783. HPLC (Method B): retention time 15.6 min.
4.1.2.11. 2-(p-Tolyl)-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5j)
To a well-stirred solution of indole 4j (0.50 g, 2.4 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.40 mL, 3.6 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.64 g, 2.7 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5j (0.21 g, 0.65 mmol, 27%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.25 (s, 1H), 7.46 (d, J = 8.2, 1H), 7.24 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.18 (s, 4H), 7.07 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 7.03 (dd, J = 7.9, 1.1 Hz, 1H), 6.96 (s, 2H), 5.18 (d, J = 5.9 Hz, 2H), 4.93 (d, J = 6.0 Hz, 2H), 3.91 (s, 3H), 3.75 (s, 6H), 2.39 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 153.28, 142.70, 138.34, 136.87, 135.91, 134.66, 130.04, 129.68, 128.03, 127.19, 122.42, 120.01, 119.70, 114.69, 111.08, 103.68, 85.33, 60.87, 56.21, 47.18, 21.28; HRMS (ESI+) calculated for C27H27NO4 [M+Na] 452.1832, actual 452.1837. HPLC (Method B): retention time 17.1 min.
4.1.2.12. 6-Chloro-2-phenyl-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5k)
To a well-stirred solution of indole 4k (0.50 g, 2.2 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.41 mL, 3.3 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.58 g, 2.4 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 50% ethyl acetate in hexanes) to afford oxetane analogue 5k (0.11 g, 0.24 mmol, 11%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.39 – 8.20 (m, 1H), 7.48 – 7.45 (m, 1H), 7.41 – 7.35 (m, 3H), 7.30 – 7.26 (m, 2H), 7.05 (dd, J = 8.4, 2.0 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.92 (s, 2H), 5.12 (d, J = 5.8 Hz, 2H), 4.91 (d, J = 6.0 Hz, 2H), 3.92 (s, 3H), 3.76 (s, 6H).; 13C NMR (151 MHz, CDCl3) δ 153.37, 142.28, 137.01, 136.33, 135.29, 132.46, 129.07, 128.63, 128.47, 127.29, 126.55, 120.90, 120.60, 115.19, 111.18, 103.59, 85.20, 60.89, 56.23, 46.97; HRMS (ESI+) calculated for C26H24ClNO4 [M+Na] 472.1286, actual 472.1288. HPLC (Method B): retention time 5.9 min.
4.1.2.13. 5-Methoxy-2-phenyl-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5l)
To a well-stirred solution of indole 4l (0.80 g, 3.6 mmol) in CH2Cl2 (10 mL) under N2 at 0 °C, SnCl4 (0.60 mL, 5.4 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.95 g, 4.0 mmol) was added in small portions to the suspension, followed by nitromethane (10 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (30 mL), the organic material was extracted with CH2Cl2 (3×30 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column (0 to 60% ethyl acetate in hexanes) to afford oxetane analogue 5l (0.35 g, 0.80 mmol, 22%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 8.17 (s, 1H), 7.41 – 7.32 (m, 4H), 7.32 – 7.23 (m, 2H), 6.97 (s, 2H), 6.92 (dd, J = 8.8, 2.4 Hz, 1H), 6.44 (d, J = 2.4 Hz, 1H), 5.16 (d, J = 5.8 Hz, 2H), 4.92 (d, J = 5.9 Hz, 2H), 3.92 (s, 3H), 3.78 (s, 3H), 3.76 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 154.24, 153.32, 142.49, 136.92, 135.40, 133.04, 131.23, 128.96, 128.51, 128.27, 127.24, 114.92, 112.29, 111.91, 103.67, 101.92, 85.27, 60.88, 56.23, 55.99, 47.10; HRMS (ESI+) calculated for C27H27NO5 [M+Na] 468.1781, actual 468.1795. HPLC (Method A): retention time 19.8 min.
4.1.2.14. 2-Methoxy-5-(6-methoxy-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indol-2-yl)phenol (5m)
To a well-stirred solution of indole 4m (0.10 g, 0.26 mmol) in CH2Cl2 (5 mL) under N2 at 0 °C, SnCl4 (0.05 mL, 0.4 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (69 mg, 0.29 mmol) was added in small portions to the suspension, followed by nitromethane (5 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (10 mL), the organic material was extracted with CH2Cl2 (3×10 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent. The crude reaction product was added to THF (5 mL) and TBAF (5 mL), and the reaction solution was stirred at room temperature for 30 min. After the reaction was quenched with ice and water (5 mL), the organic material was extracted with CH2Cl2 (3×5 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product purified by flash chromatography using a prepacked 25 g silica column (0 to 60 % ethyl acetate in hexanes) to afford oxetane analogue 5m (15 mg, 0.03 mmol, 12%) as a colorless solid: 1H NMR (600 MHz, CDCl3) δ 7.95 (s, 1H), 6.85 (m, 3H), 6.83 (d, J = 2.1 Hz, 1H), 6.80 (d, J = 8.7 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 6.63 (dd, J = 8.6, 2.2 Hz, 1H), 6.56 (dd, J = 8.3, 2.1 Hz, 1H), 5.60 (s, 1H), 5.07 (d, J = 5.6 Hz, 2H), 4.83 (d, J = 5.6 Hz, 2H), 3.82 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H), 3.66 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 156.65, 153.26, 146.52, 145.88, 142.65, 136.85, 136.65, 133.19, 126.49, 122.40, 120.28, 119.37, 114.37, 113.08, 110.87, 109.76, 103.65, 94.62, 85.25, 60.86, 56.21, 55.99, 55.66, 47.14. HRMS (ESI+) calculated for C28H29NO7 [M+Na] 514.1836, actual 514.1833. HPLC (Method B): retention time 11.5 min.
4.1.2.15. 5-Bromo-2-phenyl-3-(3-(3,4,5-trimethoxyphenyl)oxetan-3-yl)-1H-indole (5n)
To a well-stirred solution of indole 4n (0.60 g, 2.2 mmol) in CH2Cl2 (5 mL) under N2 at 0 °C, SnCl4 (0.45 mL, 3.3 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then 3-(3,4,5-trimethoxyphenyl)oxetan-3-ol (0.60 g, 2.4 mmol) was added in small portions to the suspension, followed by nitromethane (5 mL). The mixture was stirred for 8 h at room temperature. After the reaction was quenched with ice and water (10 mL), the organic material was extracted with CH2Cl2 (3×10 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to remove the solvent, and the crude reaction product was purified by flash chromatography using a prepacked 50 g silica column (0 to 60 % ethyl acetate in hexanes) to afford oxetane analogue 5n (0.15 g, 0.31 mmol, 14%) as a yellow solid: 1H NMR (600 MHz, CDCl3) δ 8.49 (s, 1H), 7.39 – 7.36 (m, 3H), 7.34 – 7.32 (m, 2H), 7.30 – 7.25 (m, 2H), 7.16 – 7.13 (m, 1H), 6.92 (s, 2H), 5.12 (d, J = 5.7 Hz, 2H), 4.91 (d, J = 5.8 Hz, 2H), 3.93 (s, 3H), 3.76 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 153.39, 142.19, 137.07, 135.93, 134.64, 132.40, 129.57, 129.06, 128.71, 127.34, 125.52, 121.98, 114.62, 113.23, 112.75, 103.67, 85.21, 60.90, 56.26, 46.90; HRMS (ESI+) calculated for C26H24BrNO4 [M+Na] 516.0781, actual 516.0784. HPLC (Method B): retention time 5.7 min.
4.2. Biological Evaluation
4.2.1. Cell Lines and Sulforhodamine B (SRB) Assay.
The SRB assay was used to assess growth inhibition of human cancer cells, as previously described.43–45 The MDA-MB-231 and PANC-1 cancer cell lines (obtained from ATCC) were plated in triplicate at 12,000 cells/well into 96-well plates (Corning) and incubated for 24 h at 37 °C in a humidified incubator in a 5% CO2 atmosphere using high glucose DMEM supplemented with 10% fetal bovine serum (FBS) (Gibco) and 30 μg/mL gentamicin sulfate. Compounds to be tested were dissolved in DMSO to generate 10 mg/mL stock solutions. Serial dilutions were made in media and added to the plates. Paclitaxel (Tokyo Chemical) and CA4 were used as positive controls. After a 48 h drug treatment, the cells were fixed with 6% final concentration trichloroacetic acid (Acros), washed with deionized water, air-dried, stained with 100 μL 7.1 mM SRB dye, washed with 1% acetic acid to remove excess dye, and air-dried. SRB was solubilized with 10 mM Tris-base, and absorbances were measured at 530 nm using a Varioskan LUX Multimode Microplate Reader. A growth inhibition of 50% (GI50 or the drug concentration causing 50% reduction in net protein increase) relative to control was calculated from the absorbance data using GraphPad. A minimum of three independent experiments were performed. The MCF-7 breast cancer cells were obtained from the NCI Cell Screen, and the procedure used was that described by Monks et al.45 The GI50 values were obtained following 96 h of growth in varying drug concentrations, essentially as described above for the MDA-MB-231 and PANC-1 cells. CA4 was used as a positive control.
4.2.2. Wound Healing Assay
PANC-1 cells (ATCC CRL-1469) were cultured in T75 (Corning U420720U) flasks with Dulbecco’s modified eagle media (DMEM) supplemented with 10% heat inactivated FBS and 30 μg/mL gentamicin sulfate. The cells were grown in a humidified incubator supplemented with 5% CO2 at 37 °C. Cells were detached with Tryple, plated at a seeding density of approximately 1 × 105 cells in a 6-well plate (sterile tissue culture treated Corning 3516) and allowed to reach 70–80% confluency before treatment. A scratch was made with a sterile 10 μL pipette tip in each well before removing the media and any detached cells. Two wells of PANC-1 cells were treated with either 1 μM or 0.5 μM compound 5c or 5h, with two wells as controls with the final DMSO being no greater than 0.2% and the final media volume being 3 mL in each well. Wound status was monitored with an automated Biotek Lionheart ELx800 microscope with a 4X objective for 48 h. We utilized the supplemented Gen5 software to perform a cellular and statistical analysis on the wound closure data.
4.2.3. Inhibition of Tubulin Polymerization.46
Tubulin polymerization was evaluated in 0.25 mL reaction mixtures (final volume) containing 1 mg/mL (10 μM) purified bovine brain tubulin, 0.8 M monosodium glutamate (pH 6.6), 4% (v/v) dimethyl sulfoxide, 0.1 mM GTP, and different compound concentrations. All components except GTP were preincubated for 15 min at 30 °C in 0.24 mL. The assay mixtures were cooled to 0 °C, and 10 μL of 0.0025 M GTP was added to each sample. Reaction mixtures were transferred to cuvettes held at 0 °C in Beckman DU-7400 and DU-7500 spectrophotometers equipped with electronic temperature controllers. The temperature was increased to 30 °C, over about 30 s, and polymerization was followed turbidimetrically at 350 nm for 20 min. The IC50 was defined as the compound concentration inhibiting extent of polymerization by 50% after 20 min.
Supplementary Material
Acknowledgements
The authors are grateful for financial support of this project provided, in part, by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under award numbers R01CA244579 (to RPM, KGP, MLT) and R01CA238624 (to KGP, MLT). Additional valuable funding was provided by the Cancer Prevention and Research Institute of Texas (CPRIT, Grant RP200614 to KGP, MLT). This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supporting Information
1H NMR,13C NMR, HRMS, HPLC for final compounds and intermediates (1H NMR, 13C NMR, HRMS) associated with this paper and Molecular Docking details (SI-1); Selected traces for cytotoxicity and wounding healing assay (SI-2); Wound healing assay videos for 5c and 5h are uploaded as separated video files.
This study appears as a chapter in: Wen Ren, Ph.D. Dissertation, Baylor University, August 2023, Synthesis and Biological Evaluation of Structurally Diverse Indole-based Analogues as Inhibitors of Tubulin Polymerization and Synthesis of Drug-Linker Constructs Cleavable by Enzymes Present in the Tumor Microenvironment.
References:
- 1.Hastie SB Interactions of Colchicine with Tubulin. Pharmacol Ther 1991, 51, 377–401. [DOI] [PubMed] [Google Scholar]
- 2.Pettit G; Singh S; Hamel E; Lin C; Alberts D; Garcia-Kendal D. Antitumor Activity of Combretastatin-A4 Phosphate, a Natural Product Tubulin Inhibitor. Experientia 1989, 45, 209–211.2920809 [Google Scholar]
- 3.Pettit GR; Lippert JW 3rd. Antineoplastic Agents 429. Syntheses of the Combretastatin A-1 and Combretastatin B-1 Prodrugs. Anticancer Drug Des 2000, 15, 203–16. [PubMed] [Google Scholar]
- 4.Sriram M; Hall JJ; Grohmann NC; Strecker TE; Wootton T; Franken A; Trawick ML; Pinney KG Design, Synthesis and Biological Evaluation of Dihydronaphthalene and Benzosuberene Analogs of the Combretastatins as Inhibitors of Tubulin Polymerization in Cancer Chemotherapy. Bioorgan Med Chem 2008, 16, 8161–8171. [DOI] [PubMed] [Google Scholar]
- 5.Maguire CJ; Chen Z; Mocharla VP; Sriram M; Strecker TE; Hamel E; Zhou HL; Lopez R; Wang YF; Mason RP; Chaplin DJ; Trawick ML; Pinney KG Synthesis of Dihydronaphthalene Analogues Inspired by Combretastatin A-4 and Their Biological Evaluation as Anticancer Agents. Medchemcomm 2018, 9, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo YH; Wang HH; Gerberich JL; Odutola SO; Charlton-Sevcik AK; Li MP; Tanpure RP; Tidmore JK; Trawick ML; Pinney KG; Mason RP; Liu L. Imaging-Guided Evaluation of the Novel Small-Molecule Benzosuberene Tubulin-Binding Agent KGP265 as a Potential Therapeutic Agent for Cancer Treatment. Cancers 2021, 13, 4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niu HC; Strecker TE; Gerberich JL; Campbell JW; Saha D; Mondal D; Hamel E; Chaplin DJ; Mason RP; Trawick ML; Pinney KG Structure Guided Design, Synthesis, and Biological Evaluation of Novel Benzosuberene Analogues as Inhibitors of Tubulin Polymerization. J Med Chem 2019, 62, 5594–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mondal D; Niu H; Pinney KG Efficient Synthetic Methodology for the Construction of Dihydronaphthalene and Benzosuberene Molecular Frameworks. Tetrahedron Lett 2019, 60, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herdman CA; Devkota L; Lin CM; Niu H; Strecker TE; Lopez R; Liu L; George CS; Tanpure RP; Hamel E; Chaplin DJ; Mason RP; Trawick ML; Pinney KG Structural Interrogation of Benzosuberene-Based Inhibitors of Tubulin Polymerization. Bioorg Med Chem 2015, 23, 7497–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanpure RP; George CS; Strecker TE; Devkota L; Tidmore JK; Lin CM; Herdman CA; MacDonough MT; Sriram M; Chaplin DJ; Trawick ML; Pinney KG Synthesis of Structurally Diverse Benzosuberene Analogues and their Biological Evaluation as Anti-Cancer Agents. Bioorgan Med Chem 2013, 21, 8019–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanpure RP; George CS; Sriram M; Strecker TE; Tidmore JK; Hamel E; Charlton-Sevcik AK; Chaplin DJ; Trawick ML; Pinney KG An Amino-Benzosuberene Analogue that Inhibits Tubulin Assembly and Demonstrates Remarkable Cytotoxicity. Medchemcomm 2012, 3, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinney KG; Bounds AD; Dingeman KM; Mocharla VP; Pettit GR; Bai R; Hamel E. A New Anti-Tubulin Agent Containing the Benzo[b]thiophene Ring System. Bioorg Med Chem Lett 1999, 9, 1081–6. [DOI] [PubMed] [Google Scholar]
- 13.Pinney KG; Pettit GR; Mocharla VP; del Pilar Mejia M; Shirali A. Description Anti-Mitotic Agents which Inhibit Tubulin Polymerization. In U.S. Patent 6,350,777 B2, issued February 26, 2002. [Google Scholar]
- 14.Hadimani MB; Kessler RJ; Kautz JA; Ghatak A; Shirali AR; O’Dell H; Garner CM; Pinney KG 2-(3-Tert-Butyldimethylsiloxy-4-Methoxyphenyl)-6-Methoxy-3-(3,4,5-Trimethoxybenzoyl)Indole. Acta Crystallogr C 2002, 58, O330–O332. [DOI] [PubMed] [Google Scholar]
- 15.Hadimani MB; MacDonough MT; Ghatak A; Strecker TE; Lopez R; Sriram M; Nguyen BL; Hall JJ; Kessler RJ; Shirali AR; Liu L; Garner CM; Pettit GR; Hame E; Chaplin DJ; Mason RP; Trawick ML; Pinney KG Synthesis of a 2-Aryl-3-aroyl Indole Salt (OXi8007) Resembling Combretastatin A-4 with Application as a Vascular Disrupting Agent. J Nat Prod 2013, 76, 1668–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacDonough MT; Strecker TE; Hamel E; Hall JJ; Chaplin DJ; Trawick ML; Pinney KG Synthesis and Biological Evaluation of Indole-Based, Anti-Cancer Agents Inspired by the Vascular Disrupting Agent 2-(3 ′-Hydroxy-4 ′-Methoxyphenyl)-3-(3 ″,4 ″,5 ″-Trimethoxybenzoyl)-6-Methoxyindole (OXi8006). Bioorgan Med Chem 2013, 21, 6831–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strecker TE; Odutola SO; Lopez R; Cooper MS; Tidmore JK; Charlton-Sevcik AK; Li L; MacDonough MT; Hadimani MB; Ghatak A; Liu L; Chaplin DJ; Mason RP; Pinney KG; Trawick ML The Vascular Disrupting Activity of OXi8006 in Endothelial Cells and its Phosphate Prodrug OXi8007 in Breast Tumor Xenografts. Cancer Lett 2015, 369, 229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacDonough MT; Shi Z; Pinney KG Mechanistic Considerations in the Synthesis of 2-Aryl-Indole Analogues under Bischler-Mohlau Conditions. Tetrahedron Lett 2015, 56, 3624–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tozer GM; Kanthou C; Baguley BC Disrupting Tumour Blood Vessels. Nat Rev Cancer 2005, 5, 423–35. [DOI] [PubMed] [Google Scholar]
- 20.Smolarczyk R; Czapla J; Jarosz-Biej M; Czerwinski K; Cichon T. Vascular Disrupting Agents in Cancer Therapy. Eur J Pharmacol 2021, 891, 173692. [DOI] [PubMed] [Google Scholar]
- 21.Liu L; O’Kelly D; Schuetze R; Carlson G; Zhou H; Trawick ML; Pinney KG; Mason RP Non-Invasive Evaluation of Acute Effects of Tubulin Binding Agents: A Review of Imaging Vascular Disruption in Tumors. Molecules 2021, 26, 2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siemann DW; Horsman MR Small-Molecule Vascular Disrupting Agents in Cancer Therapy. Antiangiogenic Agents in Cancer Therapy 2008, 297–310. [Google Scholar]
- 23.Siemann DW; Shi W. Efficacy of Combined Antiangiogenic and Vascular Disrupting Agents in Treatment of Solid Tumors. Int J Radiat Oncol Biol Phys 2004, 60, 1233–40. [DOI] [PubMed] [Google Scholar]
- 24.Pell A; Pilcher G. Measurements of Heats of Combustion by Flame Calorimetry. Part 3.—Ethylene Oxide, Trimethylene Oxide, Tetrahydrofuran and Tetrahydropy. Transactions of the Faraday Society 1965, 61, 71–77. [Google Scholar]
- 25.Eigenmann H; Golden D; Benson S. Revised Group Additivity Parameters for the Enthalpies of Formation of Oxygen-Containing Organic Compounds. The Journal of Physical Chemistry 1973, 77, 1687–1691. [Google Scholar]
- 26.Wuitschik G; Rogers-Evans M; Muller K; Fischer H; Wagner B; Schuler F; Polonchuk L; Carreira EM Oxetanes as Promising Modules in Drug Discovery. Angew Chem Int Edit 2006, 45, 7736–7739. [DOI] [PubMed] [Google Scholar]
- 27.Wuitschik G; Carreira EM; Wagner B; Fischer H; Parrilla I; Schuler F; Rogers-Evans M; Muller K. Oxetanes in Drug Discovery: Structural and Synthetic Insights. J Med Chem 2010, 53, 3227–46. [DOI] [PubMed] [Google Scholar]
- 28.Wuitschik G; Rogers-Evans M; Buckl A; Bernasconi M; Marki M; Godel T; Fischer H; Wagner B; Parrilla I; Schuler F; Schneider J; Alker A; Schweizer WB; Muller K; Carreira EM Spirocyclic Oxetanes: Synthesis and Properties. Angew Chem Int Ed Engl 2008, 47, 4512–5. [DOI] [PubMed] [Google Scholar]
- 29.Searles S Jr; Nickerson RG; Witsiepe WK Oxetanes. IX. Structural and Solvent Effects in the Reaction of γ-Bromoalcohols with Base1, 2. The Journal of Organic Chemistry 1959, 24, 1839–1844. [Google Scholar]
- 30.Searles S; Gortatowski M. Cleavage of 3-Bromo-2, 2-Dimethyl-1-Propanol by Base1. Journal of the American Chemical Society 1953, 75, 3030–3031. [Google Scholar]
- 31.Picard P; Leclercq D; Bats JP; Moulines J. An Efficient One-Pot Synthesis of Oxetanes from 1,3-Diols. Synthesis-Stuttgart 1981, 550–551. [Google Scholar]
- 32.Vogt F; Jodicke K; Schroder J; Bach T. Paterno-Buchi Reactions of Silyl Enol Ethers and Enamides. Synthesis-Stuttgart 2009, 2009, 4268–4273. [Google Scholar]
- 33.Bach T. The Paterno-Buchi Reaction of N-Acyl Enamines and Aldehydes - The Development of a New Synthetic Method and its Application to Total Synthesis and Molecular Recognition Studies. Synlett 2000, 2000, 1699–1707. [Google Scholar]
- 34.Wojtowicz JA 3-Substituted Oxetanes. J Org Chem 1973, 38, 2061–2066. [Google Scholar]
- 35.Croft RA; Mousseau JJ; Choi C; Bull JA Structurally Divergent Lithium Catalyzed Friedel-Crafts Reactions on Oxetan-3-ols: Synthesis of 3,3-Diaryloxetanes and 2,3-Dihydrobenzofurans. Chemistry 2016, 22, 16271–16276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ottoni O; Neder AV; Dias AK; Cruz RP; Aquino LB Acylation of Indole Under Friedel-Crafts Conditions-an Improved Method to Obtain 3-Acylindoles Regioselectively. Org Lett 2001, 3, 1005–7. [PubMed] [Google Scholar]
- 37.La Regina G; Bai RL; Coluccia A; Farniglini V; Pelliccia S; Passacantilli S; Mazzoccoli C; Ruggieri V; Verrico A; Miele A; Monti L; Nalli M; Alfonsi R; Di Marcotullio L; Gulino A; Ricci B; Soriani A; Santoni A; Caraglia M; Porto S; Da Pozzo E; Martini C; Brancale A; Marinelli L; Novellino E; Vultaggio S; Varasi M; Mercurio C; Bigogno C; Dondio G; Hamel E; Lavia P; Silvestri R. New Indole Tubulin Assembly Inhibitors Cause Stable Arrest of Mitotic Progression, Enhanced Stimulation of Natural Killer Cell Cytotoxic Activity, and Repression of Hedgehog-Dependent Cancer. J Med Chem 2015, 58, 5789–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X; Decker CC; Zechner L; Krstin S; Wink M. In vitro Wound Healing of Tumor Cells: Inhibition of Cell Migration by Selected Cytotoxic Alkaloids. BMC Pharmacol Toxicol 2019, 20, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravelli RBG; Gigant B; Curmi PA; Jourdain I; Lachkar S; Sobel A; Knossow M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-Like Domain. Nature 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z; Maderna A; Sukuru SC; Wagenaar M; O’Donnell CJ; Lam MH; Musto S; Loganzo F. New Cytotoxic Benzosuberene Analogs. Synthesis, Molecular Modeling and Biological Evaluation. Bioorg Med Chem Lett 2013, 23, 6688–94. [DOI] [PubMed] [Google Scholar]
- 41.Bane SL Molecular Features of the Interaction of Colchicine and Related Structures with Tubulin. The Role of Microtubules in Cell Biology, Neurobiology, and Oncology 2008, 259–279. [Google Scholar]
- 42.Yang SD; Sun CL; Fang Z; Li BJ; Li YZ; Shi ZJ Palladium-Catalyzed Direct Arylation of (Hetero)Arenes with Aryl Boronic Acids. Angew Chem Int Ed Engl 2008, 47, 1473–6. [DOI] [PubMed] [Google Scholar]
- 43.Siles R; Ackley JF; Hadimani MB; Hall JJ; Mugabe BE; Guddneppanavar R; Monk KA; Chapuis JC; Pettit GR; Chaplin DJ; Edvardsen K; Trawick ML; Garner CM; Pinney KG Combretastatin Dinitrogen-Substituted Stilbene Analogues as Tubulin-Binding and Vascular-Disrupting Agents. J Nat Prod 2008, 71, 313–320. [DOI] [PubMed] [Google Scholar]
- 44.Vichai V; Kirtikara K. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nat Protoc 2006, 1, 1112–6. [DOI] [PubMed] [Google Scholar]
- 45.Monks A; Scudiero D; Skehan P; Shoemaker R; Paull K; Vistica D; Hose C; Langley J; Cronise P; Vaigro-Wolff A; et al. Feasibility of a High-Flux Anticancer Drug Screen using a Diverse Panel of Cultured Human Tumor Cell Lines. J Natl Cancer Inst 1991, 83, 757–66. [DOI] [PubMed] [Google Scholar]
- 46.Hamel E. Evaluation of Antimitotic Agents by Quantitative Comparisons of their Effects on the Polymerization of purified tubulin. Cell Biochem Biophys 2003, 38, 1–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.