

Abstract
Recently, several mass spectrometry methods have utilized protein structural stability for the quantitative study of protein-ligand engagement. These protein-denaturation approaches, which include thermal proteome profiling (TPP) and stability of proteins from rates of oxidation (SPROX), evaluate ligand-induced denaturation susceptibility changes with a MS-based readout. The different techniques of bottom-up protein-denaturation methods each have their own advantages and challenges. Here, we report the combination of protein-denaturation principles with quantitative cross-linking mass spectrometry using isobaric quantitative protein interaction reporter technologies. This method enables the evaluation of ligand-induced protein engagement through analysis of cross-link relative ratios across chemical denaturation. As a proof-of-concept, we found ligand-stabilized cross-linked lysine pairs in well-studied bovine serum albumin and ligand bilirubin. These links map to known binding sites Sudlow Site I and subdomain IB. We propose that protein-denaturation and qXL-MS can be combined with similar peptide-level quantification approaches, like SPROX, to increase the coverage information profiled for facilitating protein-ligand engagement efforts.
INTRODUCTION
For many years, protein target drug discovery efforts have relied on mass spectrometry (MS)-based affinity capture techniques. While purification strategies have been useful for identifying protein targets, the required covalent modification of the drug demands expert medicinal chemistry and potentially complicates binding drug properties.1 More recently, a series of approaches exploiting the link between protein folding, thermodynamic stability, and ligand-induced changes have been developed. These methods, such as thermal proteome profiling (TPP), stability of proteins from rates of oxidation (SPROX), and pulse proteolysis (PP), rely on ligand-induced changes on the biophysical properties of proteins when exposed to increasing denaturation.2–4 To date, TPP has been the most popular approach for detecting protein-ligand interactions. Building from cellular thermal shift assay (CETSA), the TPP workflow exposes the lysates from cells or ligand-treated cells to a thermal gradient.5 Proteins will unfold, aggregate, and eventually precipitate during this heat treatment based on their inherent stability and interaction with other partners. By subjecting the soluble fraction to isobaric labeling through tandem mass tags, the MS-based output allows for an unbiased search for ligand targets by observed shifts in calculated melting curves. Despite the body of target discoveries made by TPP, its protein-level readout cannot provide information on ligand binding at the regional or domain level. The peptide-level readout of SPROX and PP therefore compliment the characterization of protein-ligand interactions. However, SPROX and PP are limited in detecting methionine-containing and semi-tryptic peptides, respectively, and expanding the readout of peptide-level bottom-up approaches would further our ability to catalogue protein-target interactions.
Chemical cross-linking mass spectrometry (XL-MS) complements current peptide-level readout methods by capturing residue-level interactions on a large-scale. The reaction of a small molecule with two reactive groups, separated by a spacer with a defined length, forms covalent bonds between residues that are in close spatial proximity. The identified cross-links serve as “molecular rulers” that impose distance restraints and serve as a basis for subsequent computational modeling of protein three-dimensional structure. XL-MS has been applied to protein complexes and living cells to probe protein topologies and protein-ligand conformational changes.6,7 However, the use of XL-MS to quantify ligand-induced structural stability shifts, to our knowledge, has not been documented.
In this work we describe protein-denaturation with quantitative cross-linking mass spectrometry, a general strategy that combines protein-denaturation with quantitative XL-MS (qXL-MS) workflows for the quantitation of cross-link ratios generated during protein unfolding. Quantitative XL-MS with isobaric quantitative Protein Interaction Reporter (iqPIR) has previously been performed to demonstrate conformational changes upon protein-drug binding.12 Wang et al.’s XL-MS in the presence of chaotropic agents characterized protein unfolding by MS1-based measurements.13 Here, we expand application of MS2-based qXL-MS with protein-denaturation to detect ligand-induced structural stability shifts. When protein-denaturation and qXL-MS is applied to bovine serum (BSA) and ligand bilirubin, we accurately identify ligand-induced stabilization at the known subdomain binding sites.
MATERIALS AND METHODS
All reagents were obtained through Fisher (Waltham, MA) unless otherwise noted. Bovine serum albumin (heat shock fraction, pH 7, ≥98%) was purchased from Sigma-Aldrich (Saint Louis, MO) and used without further purification. Isobaric cross-linker was synthesized in house as described by Chavez et al.14 Sequencing grade-trypsin was purchased from Promega (Madison, WI). C18 Sep-Pak cartridges for peptide desalting were purchased from Waters (Milford, MA).
BSA denaturation
The experimental workflow used in this study is shown in Figure 1. BSA (15 μM) was prepared in 170 mM disodium phosphate (Na2HPO4) buffer pH 8 and incubated with increasing concentrations of urea at room temperature for 10 minutes. For ligand-incubated samples, bilirubin stocks were prepared at 1 mM in dimethyl sulfoxide. The BSA-bilirubin solutions were mixed at a 1:1 molar ratio (30 μM). Mixtures were then incubated for 30 minutes at room temperature before the addition of urea and concentration adjustment in sodium phosphate buffer. The final BSA concentration in all samples was 15 μM for all experiments.
Figure 1.
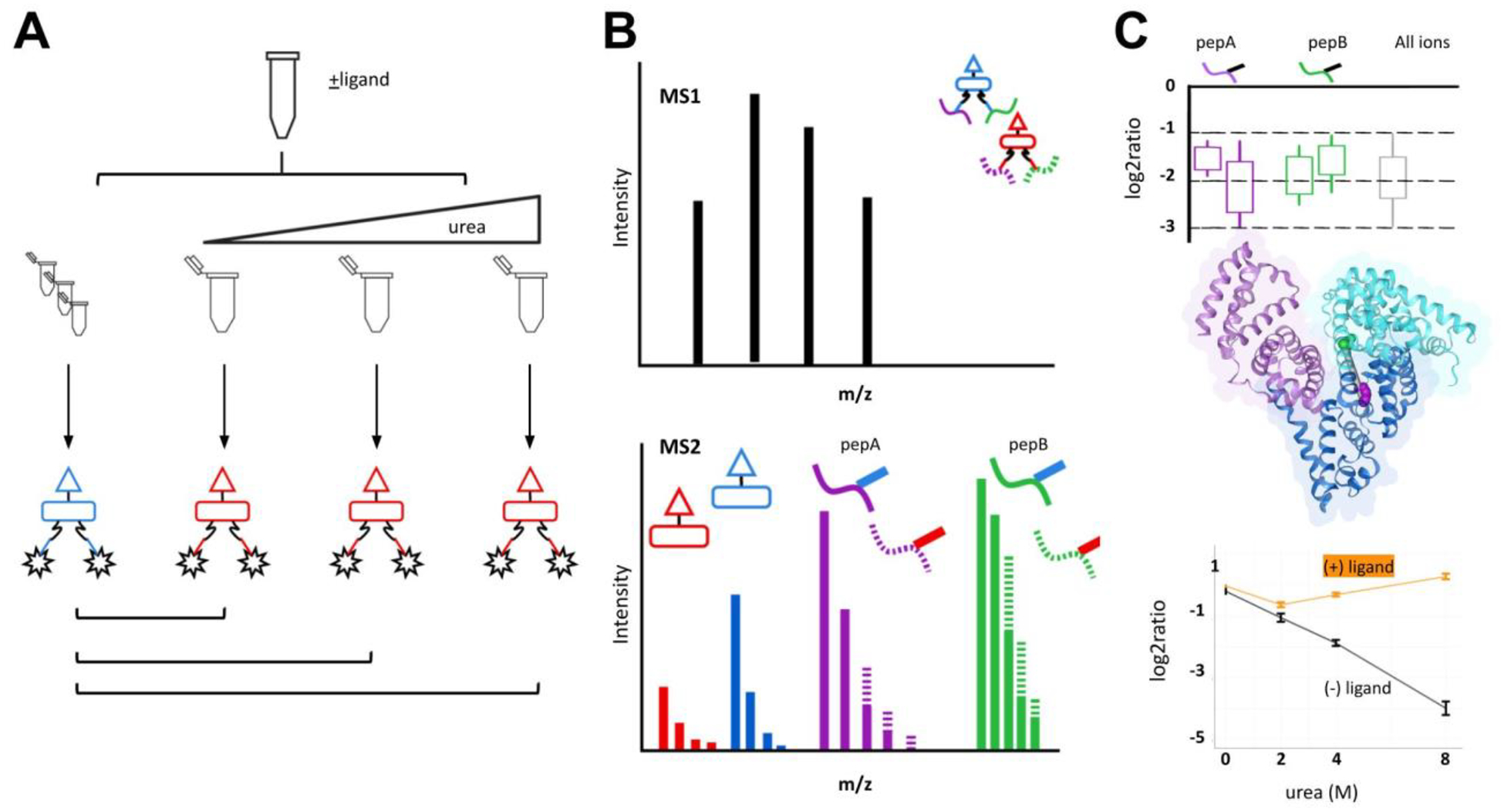
Protein denaturation and quantitative XL-MS workflow.
A) Chemical denaturation of BSA. A BSA stock solution is divided and diluted with increasing urea. Solutions are crosslinked with iqPIR reagent (reporter 808 or 812 m/z). The same crosslinking design was performed for BSA incubated prior with bilirubin. After crosslinking, samples are mixed with a non-denaturing control at a 1:1 ratio. Mixtures are reduced, alkylated, and digested prior to data acquisition. (B) LC-MS. Cross-linked peptide mixtures were prepared for LC-MS/MS using Mango. The data was searched using Comet with crosslink validation using XLinkProphet. (C) Crosslinking data was uploaded to XLinkDB for crosslink peptide pair quantitation viewing and structure mapping. Example peptide K211-K245 (EK211VLTSSAR-LSQK245FPK) was mapped to BSA structure (PDB: 4F5S).
BSA intrinsic fluorescence
Fluorescence measurements were acquired using a Cytation5 microplate reader (Biotek). All spectra were measured at 25 ± 0.1 °C. For measurements involving bilirubin, intrinsic fluorescence was measured by exciting the protein solution at 488 nm and recording the emission at 530 nm. For all other measurements, excitation and emission were performed at 285 and 340 nm, respectively.15 The fluorescence intensity data are averages of four independent measurements.
Cross-linking and sample preparation
BSA solutions were subjected to chemical cross-linking with 1 mM of iqPIR by adding either iqPIR reagent (reporter 808 or 812 m/z) from a concentrated stock in dimethyl sulfoxide. The crosslinking reaction was performed at room temperature for 45 minutes with constant shaking, as previously described.14 The reaction was quenched upon addition of ammonium bicarbonate for 10 minutes. Cross-linked BSA solutions were then mixed 1:1 (iqPIR-808:iqPIR-812) (mol:mol) to the native, non-denaturing cross-linked condition to generate 100 μg total mixtures.
For reduction and alkylation, mixtures were incubated with 5 mM Tris-(2-carboxyethyl)phosphine from a 500 mM stock for 30 minutes followed by 10 mM iodoacetamide from a 500 mM stock for 45 minutes. Protein mixtures were digested overnight with a 1:200 ratio of trypsin to protein at 37°C. The resulting peptides were desalted by solid phase extraction using C18 SepPak cartridges. Dried cross-linked peptide material were reconstituted in 300 μl of 0.1% formic acid, 4% acetonitrile in water.
LC-MS analysis
Cross-linked peptide samples were analyzed by LC-MS/MS in technical triplicate using a nanoAcquity UPLC system (Waters) coupled to an QE Exactive Plus mass spectrometer (Thermo Scientific). Aliquots of 3 μl were loaded onto a 3 cm × 100 μM inner diameter fused silica trap column packed with a stationary phase consisting of 5 μM Reprosil C8 particles with 120 Å pores (Dr Maish GmbH) at a flow rate of 2 μL/min for 10 min. Peptides were then separated by reverse-phase chromatography analytical column (60 cm × 75 mM) by applying a linear gradient from 82% solvent A (water containing 0.1% formic acid) and 18% solvent B (acetonitrile containing 0.1% formic acid) to 60% solvent A and 40% solvent B over 1 hour at a flow rate of 300 nL/min. The column temperature was set at 55°C.
The QE+ mass spectrometer was operated using a top five data-dependent acquisition method of ions with a charge state from +4 to +8 with a resolving power setting of 70,000 for MS1 and MS2 scans. Additional settings include a 1e6 AGC value and 100 millisecond maximum ion time for MS1 scans and 5e4 AGC value and 300 millisecond maximum ion time for the MS2 scans. Ions selected for MS2 were dynamically excluded from further selection for 30 seconds.
For identification, raw files were converted to mzXML format and searched for PIR mass relationships with Mango.16 The resulting MS2 files were searched using Comet against a sequence database containing forward and reverse sequences.17 The database consisted of BSA and natural variant BSA-A214T along with a background of 4389 known false positive proteins from Escherichia coli.18 The same database was used to search the mzXML for cross-linked looplink peptides. Resulting pepXML files were analyzed with XLinkProphet and filtered to an estimated false discovery rate of 1% at a nonredundant peptide pair level.19
For quantification, analysis was performed as described by Chavez et al.14 Cross-links with a log2ratio having 95% confidence less or equal to 1 were considered quantified with high confidence. All analyses of cross-link log2ratios were calculated using R 4.0.2. Ratios are compared by Wilcoxon signed-rank test unless otherwise noted. K-means clustering of log2ratios was accomplished using the R-package kml.20 Protein bar diagrams were generated in xiNet.21
Cross-linking identification and quantification are available on XLinkDB as dataset inUrea_BSA_wBR_Bruce.22 The LC-MS data reported are available on PRIDE (ProteomeXchange Identifier PXD036649).
RESULTS AND DISCUSSION
We developed an experimental approach with quantitative XL-MS to measure protein unfolding across increasing chaotrope concentration in the presence and absence of ligand to reveal how protein-ligand interactions affect protein stability. To investigate this, we applied this experimental workflow to BSA, a well-studied protein system with documented chemical denaturant induced unfolding data (Figure 1).15,23,24 BSA shares structural homology with human serum albumin and is characterized as “heart shaped” with three homologous yet structurally distinct domains (I, II, III) each composed of two subdomains (A, B) with helical folding patterns connected by flexible loops.22–24
Aliquots of protein or protein-ligand stock solutions were added to a series of cross-linking buffer samples containing increasing amounts of urea. Then, samples were cross-linked using a constant amount of iqPIR reagent. After the cross-linking reaction, no protein aggregates were observed after high-speed centrifugation. Each cross-linked sample is mixed with a 0M urea, non-denatured sample prior at a 1:1 ratio (iqPIR-808:812, mol:mol) prior to tryptic digestion. As such, experimental replicates were performed with reciprocal iqPIR labeling schemes. Thus, a total of 24 cross-linked protein and protein-ligand samples were generated.
Isobaric PIR enables quantitative, reproducible measurements of protein denaturation
Analysis of triplicate LC-MS/MS injections of label-swapped samples identified 531 non-redundant cross-link peptide pairs at an estimated FDR of 1%, corresponding to 368 lysine site pairs. From the total network, 31% of crosslinks mapped to inter-domain links. Over 90% were quantified in at least one condition. To validate the qXL-MS for protein unfolding strategy, we compared the quantitation of cross-links quantified in both label-swapped experimental replicates. Analysis of cross-link quantitation was observed to be highly reproducible with R2 values of 0.47 to 0.95 (Figure 2). Representative MS2 results for cross-link quantifications are shown in Figure S1. Quantitative ion information from all experimental and technical replicates were consolidated to obtain a single log2ratio for each crosslink at a single urea concentration.
Figure 2.
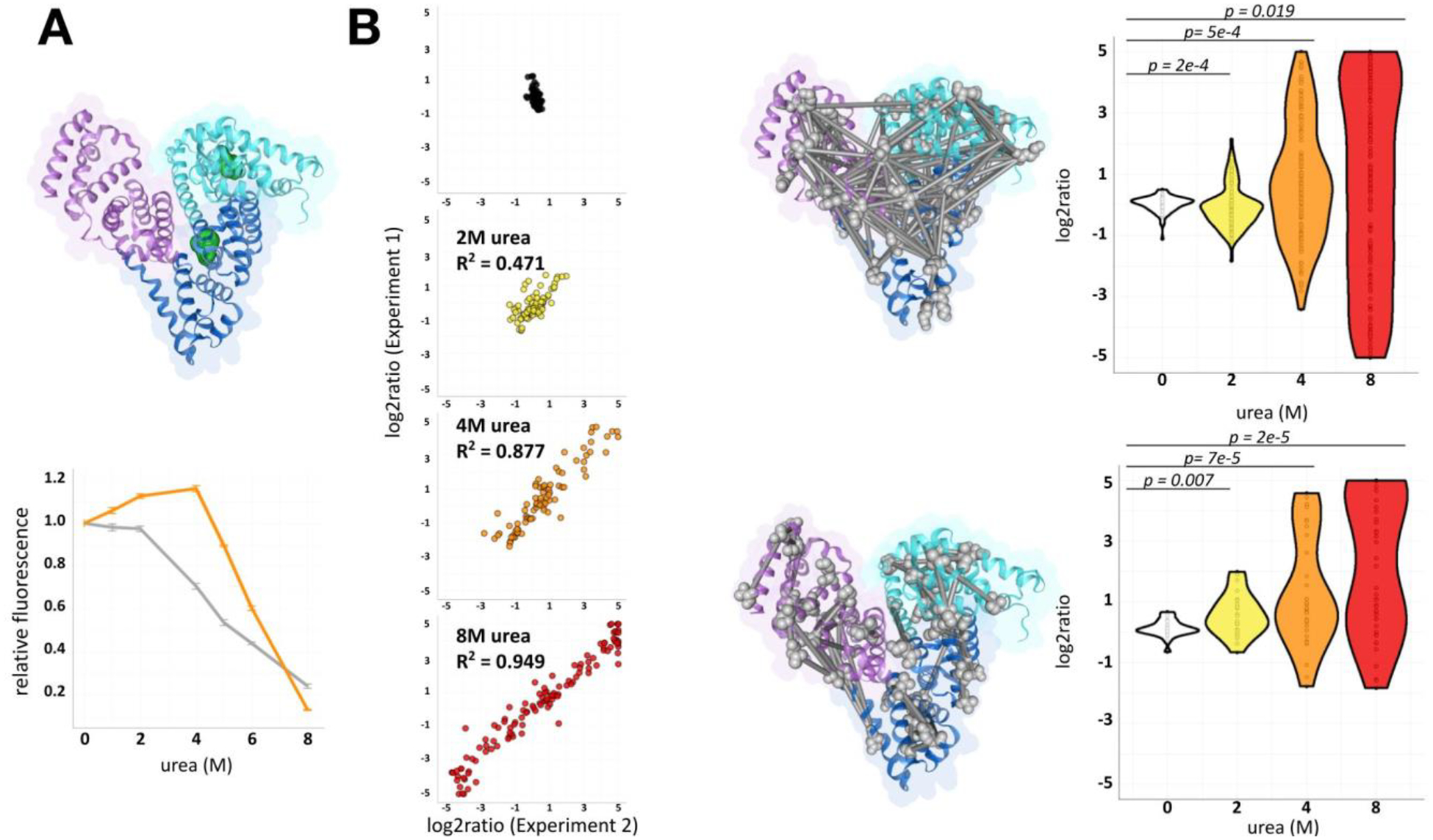
Evaluation of denaturation quantitative cross-linking mass spectrometry.
(A) Structure of BSA (PDB: 4F5S). Domains 1, 2, and 3 are indicated with the following color code: cyan, blue, and purple. Intrinsic fluorescent residues Trp-134 and Trp-212 are represented as green spheres. Relative intrinsic fluorescence of BSA with and without bilirubin (BSA, gray; BSA-bilirubin, orange). Upon urea exposure, loss of signal reflects changing Trp environments.15,26 (B) Correlation of label-swap experimental qXL-MS for protein unfolding replicates. Identified mapped cross-linked peptide pairs (top), looplinks (bottom) displayed as grey bars. Distribution of quantified cross-linked ratios in BSA.
To validate conformational changes induced by urea exposure, BSA solutions were examined by fluorescence spectroscopy (Figure 2A). BSA has two tryptophan residues that possess intrinsic fluorescence and are sensitive to their local environment. Trp-134 is located on the hydrophilic surface of subdomain IB. Trp-212 is located within the hydrophobic binding pocket of IIA, which possesses principle ligand-binding site Sudlow’s Site I.9 The intensity of their fluorescent emission band decreases markedly with increasing urea. Khan et al. and others ascribed this observance to unfolding induced conformational changes on the two tryptophan’s local environments (Figure 2A).15,25,26
The distribution of cross-linked log2ratios was observed to increase with urea exposure. A negative control of non-denaturing 0M urea BSA mixed with itself did not alter the distribution of cross-link log2ratios. The absolute fold change increased from two-fold to sixty-four-fold towards 8M urea exposure (Figure 2B). Cross-link and looplink ratios compared to the negative control showed significant changes during denaturing conditions (p-value = 0.05) (Figure 2B). To exclude the possibility that this was an artifact of increased variance, we asked whether the 95% confidence interval increased relative to denaturant. Quantitation confidence intervals were significant only at 8M urea exposure (Figure S2, mean ΔCI = +0.12, p-value = 1e-4, paired Wilcoxon test). The increased error represented 1% of the full range of log2ratios calculated at 8M urea exposure (Figure 2B), supporting crosslink quantitation changes are the result of large-scale conformational changes during denaturation. These observed changes occurred when crosslinking before reduction and alkylation, with intact repeating structural disulfide bonds. Links between helices connected by disulfide bonds that were quantified by protein-denaturation qXL-MS demonstrated no log2ratio changes greater than two-fold at the majority of assayed points of protein denaturation (Figure S3B).
BSA denaturation with qXL-MS demonstrates local domain and subdomain specific unfolding information
The cross-link peptide relative ratios generated by qXL-MS for protein unfolding report on the biophysical properties to which the constraints map. Previous XL-MS studies on denatured BSA demonstrated loss of inter-domain cross-links and increase of near-sequence cross-links by k-means clustering of MS1-based spectral counts.13 Similar analysis to our MS2-based qXL-MS for protein unfolding data also shows sharp losses of inter-domain cross-links (Figure 3). However, significant ratio differences were only observed in intra-domain increases across all quantified cross-links (Figure 3A, mean Δlog2ratio = +1.09, p-value = 1e-4).
Figure 3.
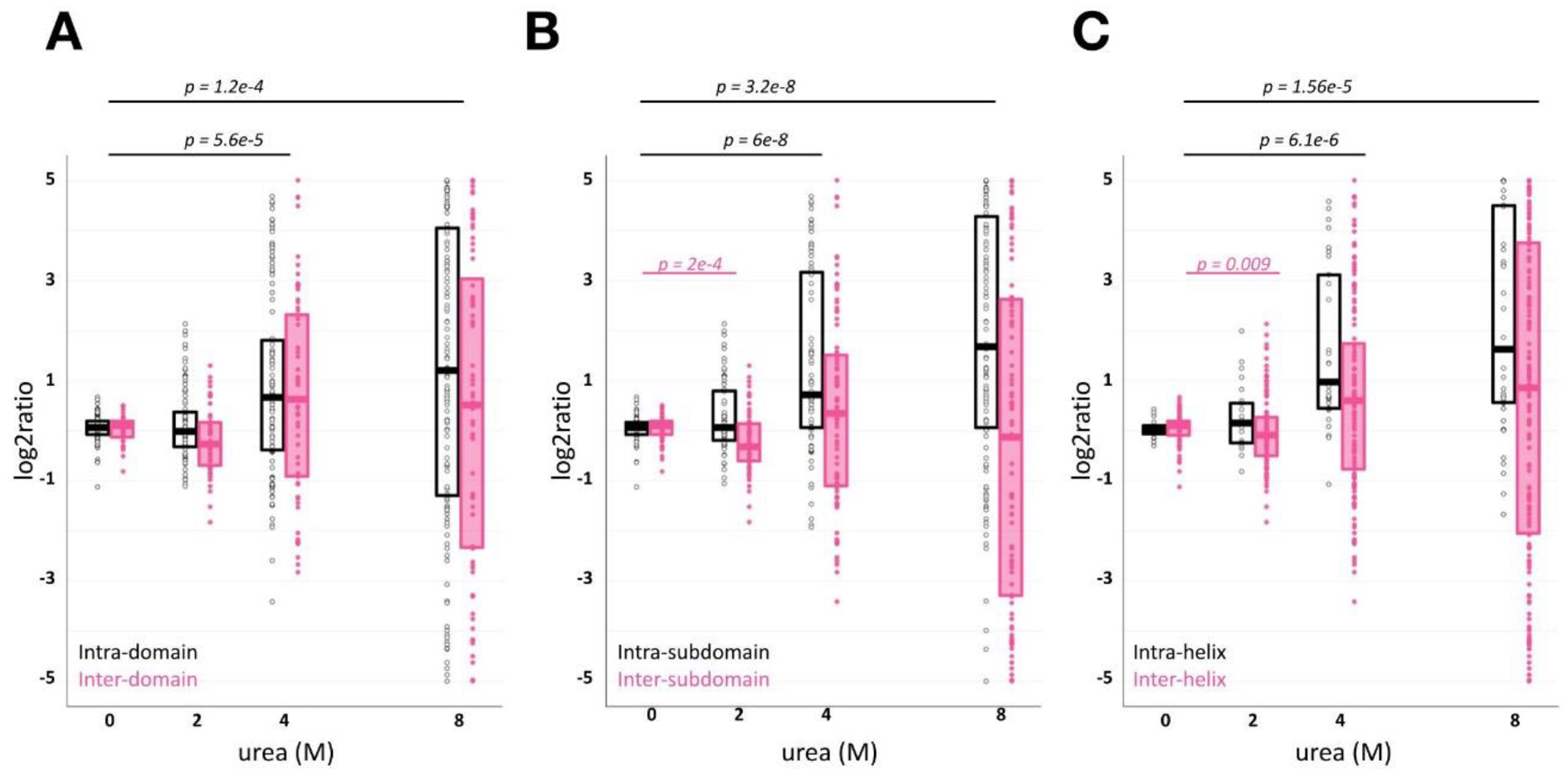
Characterizing BSA denaturation by qXL-MS for protein unfolding
Each dot represents an individual crosslink or looplink relative ratio at a given urea exposure. Relative ratios are separated on the intra- (black) and inter- (pink) structure mapping on the domain (A), subdomain (B), or helix (C) level.
We next analyzed ratios on the subdomain and helix level in order to understand deeper structural stability features with urea exposure. We expected increasing denaturant to decrease inter-subdomain cross-link ratios. Additionally, intact disulfide bridges during chemical cross-linking should introduce constraints for intra-subdomain cross-links. We found significant increases of intra-domain links (Figure 3B, mean Δlog2ratio = +1.788, p-value = 3e-8) as a consequence of urea exposure. However, inter-domain cross-link log2ratios decreases were interpreted as not statistically significant (Figure 3B, mean ΔCI = −0.0698). Similar results were observed for links mapped on the helix-level (Figure 3C). Taken together, qXL-MS for protein unfolding data captures the BSA denaturation landscape that subsists of increased intra-structure contacts and decrease of their inter-structure counterparts.
Protein denaturation with qXL-MS allows identification of bilirubin-stabilized cross-links in known ligand binding subdomains
Lastly, protein-denaturation and qXL-MS was used to investigate BSA conformational properties induced by the binding of known ligand bilirubin. Serum albumin binding to bilirubin, a catalytic degradation product of hemoglobin, has been extensively studied.15,25,27–29 Previous studies on the binding of BSA to bilirubin have suggested a 1:1 ligand:protein molar ratio generally occupies only the primary binding site.25,27,29 Reciprocal-labeled experimental replicates of qXL-MS BSA-bilirubin solutions showed similar quantitative reproducibility to BSA (Figure S5). Intrinsic fluorescent readings after 15 minutes of bilirubin incubation showed a marked increase at 4–5M urea exposure as previously observed (Figure 2A).15,24 Tayyab and colleagues proposed this observed spectroscopic shift indicates structural rearrangements by Trp-134 and Trp-212 without integrity losses.15 To identify a set of cross-links that exhibited ratio differences given ligand incubation, we focused on 4M urea exposure data given the fluorescent signal differences. Figure 4A shows the calculated 2-way ANOVA for each cross-link given bilirubin incubation. Applying a 2-fold cutoff filter and Bonferonni corrected p-value less than 0.05, we measured significant stability changes for eleven cross-links. Lysine residues of IB and IIA represented the entirety of cross-links whose ratios increased in the presence of bilirubin (Figure 4B). The observed ligand sensitivity at IIA are consistent with known binding site Sudlow Site I. While fluorescent and circular dichroism studies have placed the primary bilirubin binding site at IIA, X-ray crystallography of bilirubin with human serum albumin places the high affinity site at IB. While the primary binding site has not yet been fully clarified, both sites are supported by the qXL-MS for protein unfolding data. Nonetheless, observed ligand induced structural changes can occur beyond the specific binding site.
Figure 4.
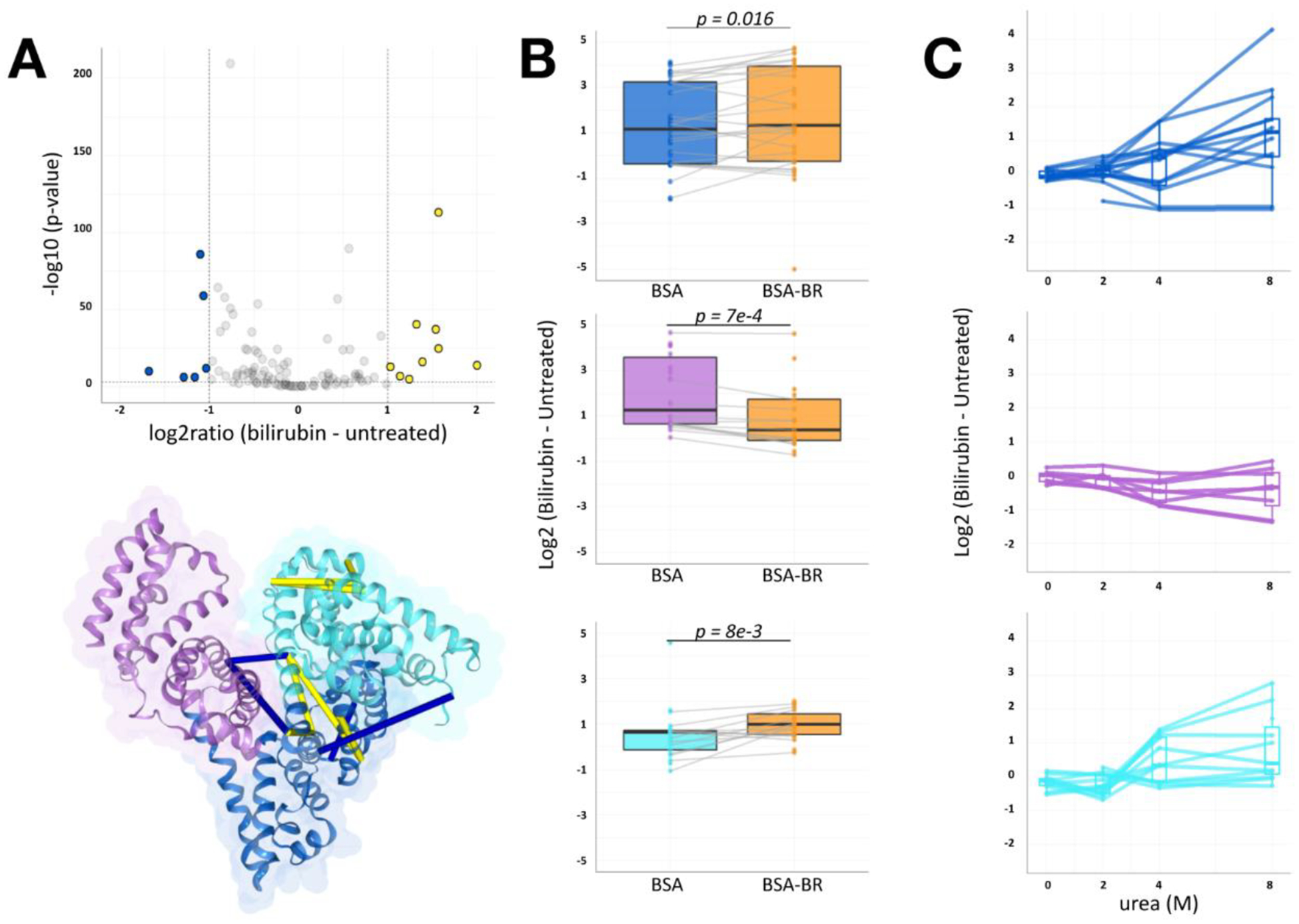
BSA-bilirubin experiments of qXL-MS for protein unfolding highlight IB, IIA denaturation stabilization
(A) Volcano plot of 111 quantified BSA cross-link ratios at 4M urea with and without bilirubin (log2 [bilirubin - apo]) versus statistical significance. Significance threshold set to 1e−2 and converted to −log10(p-value). The vertical and horizontal dotted lines show cut-off for fold-change and p-value, respectively. Blue and yellow points indicate cross-linked peptide ratios with decreasing and increasing quantitation, respectively. These cross-linked lysine pairs are mapped onto the BSA structure (PDB: 4F5S). (B) Boxplot showing log2ratio changes of subdomain intra-links at 4M urea with and without bilirubin (orange) pretreatment. Median log2ratios across quantified links are shown. The total number of quantified paired sites per group was: IB n=14, IIA n=24, IIIA n=12 and p-values for group comparisons were computed using paired Wilcoxon signed rank test. (C) Log2 difference denaturation curve obtained by qXL-MS for protein unfolding. Each quantified cross-link represents 1 line. Highlighted crosslinks map to intra-links within subdomain IB (cyan), IIA (blue), or IIIA (purple).
We next analyzed whether bilirubin incubation affected log2ratios for links involving known ligand binding site Sudlow Site II (IIIA), Sudlow Site I, and subdomain IB. We quantified changes in intra-subdomain links mapped to these regions that reached statistical significance (Figure 4, paired Wilcoxon signed rank test). On average, at 4M urea exposure, prior bilirubin incubation had stabilizing effects on IB and IIA cross-link relative abundance (mean Δlog2ratio = +0.42 and +0.45, p-value = 0.034 and 0.027, respectively). Additionally, we observed a destabilizing effect in subdomain IIIA (mean Δlog2ratio = −0.87, p-value = 1.5e-05). The direction of these bilirubin incubation effects persisted at 8M urea exposure (Figure 4C). Taken together, these results show how patterns of MS/MS-quantified cross-links reveal regions relevant to structural stability changes upon protein-complex ligand binding.
CONCLUDING REMARKS
A new approach, protein-denaturation and qXL-MS, combines protein-denaturation with quantitative XL-MS Protein Interaction Reporter sample workflows for the quantitative assessment of protein-ligand engagement studies. First, we demonstrate protein-denaturation and qXL-MS enables reproducible quantification of cross-link changes that occur during urea induced protein unfolding. Analyses that explore BSA denaturation patterns by quantified cross-link peptide pair data correlate with previously published structural data and the loss of inter-domain cross-links.13 Second, the application of protein-denaturation and qXL-MS to the BSA-bilirubin complex identifies ligand-stabilization of Sudlow Site IB and IIA during chemical denaturation. Although these characterizations have been identified in previous studies, our findings present, to our knowledge, the first use of quantitative cross-linked peptide-pair readout for ligand-induced changes in protein chemical denaturation properties. The integration of protein-denaturation principles with lysine-reactive qXL-MS provides a protein-ligand assay. The strategy presented has the potential to complement other protein-denaturation MS-based approaches in studying drug-target interactions on the cellular scale.
Supplementary Material
ACKNOLWEDGEMENTS
The authors thank all members of the Bruce Lab for helpful insights and suggestions in the course of this work. We also thank Drs. Shao-En Ong and Devin Schweppe for their helpful comments that improved the manuscript. This project was supported by the following grants from the National Institutes of Health 5R01GM086688, 5R35GM136255 and 5R01HL144778.
REFERENCES
- 1.Budayeva HG, Cristea IM. A mass spectrometry view of stable and transient protein interactions. Adv Exp Med Biol 2014;806:263–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.West GM, Tang L, Fitzgerald MC. Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Anal Chem 2008;80(11):4175–85. [DOI] [PubMed] [Google Scholar]
- 3.Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods 2005;2(3):207–12. [DOI] [PubMed] [Google Scholar]
- 4.Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S and others. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014;346(6205):1255784. [DOI] [PubMed] [Google Scholar]
- 5.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013;341(6141):84–7. [DOI] [PubMed] [Google Scholar]
- 6.Chavez JD, Schweppe DK, Eng JK, Bruce JE. In Vivo Conformational Dynamics of Hsp90 and Its Interactors. Cell Chem Biol 2016;23(6):716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavez JD, Liu NL, Bruce JE. Quantification of protein-protein interactions with chemical cross-linking and mass spectrometry. J Proteome Res 2011;10(4):1528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sand KM, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT. Unraveling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front Immunol 2014;5:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sudlow G, Birkett DJ, Wade DN. The characterization of two specific drug binding sites on human serum albumin. Mol Pharmacol 1975;11(6):824–32. [PubMed] [Google Scholar]
- 10.Kragh-Hansen U Evidence for a large and flexible region of human serum albumin possessing high affinity binding sites for salicylate, warfarin, and other ligands. Mol Pharmacol 1988;34(2):160–71. [PubMed] [Google Scholar]
- 11.He XM, Carter DC. Atomic structure and chemistry of human serum albumin. Nature 1992;358(6383):209–15. [DOI] [PubMed] [Google Scholar]
- 12.Wippel HH, Chavez JD, Keller AD, Bruce JE. Multiplexed Isobaric Quantitative Cross-Linking Reveals Drug-Induced Interactome Changes in Breast Cancer Cells. Anal Chem 2022;94(6):2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JH, Tang YL, Gong Z, Jain R, Xiao F, Zhou Y, Tan D, Li Q, Huang N, Liu SQ and others. Characterization of protein unfolding by fast cross-linking mass spectrometry using di-ortho-phthalaldehyde cross-linkers. Nat Commun 2022;13(1):1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chavez JD, Keller A, Mohr JP, Bruce JE. Isobaric Quantitative Protein Interaction Reporter Technology for Comparative Interactome Studies. Anal Chem 2020;92(20):14094–14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tayyab S, Sharma N, Mushahid Khan M. Use of domain specific ligands to study urea-induced unfolding of bovine serum albumin. Biochem Biophys Res Commun 2000;277(1):83–8. [DOI] [PubMed] [Google Scholar]
- 16.Mohr JP, Perumalla P, Chavez JD, Eng JK, Bruce JE. Mango: A General Tool for Collision Induced Dissociation-Cleavable Cross-Linked Peptide Identification. Anal Chem 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics 2013;13(1):22–4. [DOI] [PubMed] [Google Scholar]
- 18.Leptos KC, Sarracino DA, Jaffe JD, Krastins B, Church GM. MapQuant: open-source software for large-scale protein quantification. Proteomics 2006;6(6):1770–82. [DOI] [PubMed] [Google Scholar]
- 19.Keller A, Chavez JD, Bruce JE. Increased sensitivity with automated validation of XL-MS cleavable peptide crosslinks. Bioinformatics 2019;35(5):895–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genolini C, Falissard B. KmL: a package to cluster longitudinal data. Comput Methods Programs Biomed 2011;104(3):e112–21. [DOI] [PubMed] [Google Scholar]
- 21.Combe CW, Fischer L, Rappsilber J. xiNET: cross-link network maps with residue resolution. Mol Cell Proteomics 2015;14(4):1137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller A, Chavez JD, Eng JK, Thornton Z, Bruce JE. Tools for 3D Interactome Visualization. J Proteome Res 2019;18(2):753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.M a B, Tie Z, Z ou D, L i J, W ang W. Urea- and thermal-induced unfolding of bovine serum albumin. Modern Physics Letters B 2006;20(29):1909–1916. [Google Scholar]
- 24.Katz S, Denis J. Structural transformations in serum albumin as demonstrated by urea-perturbation technique. Biochem Biophys Res Commun 1967;28(5):711–7. [DOI] [PubMed] [Google Scholar]
- 25.Ni Y, Su S, Kokot S. Spectrofluorimetric studies on the binding of salicylic acid to bovine serum albumin using warfarin and ibuprofen as site markers with the aid of parallel factor analysis. Anal Chim Acta 2006;580(2):206–15. [DOI] [PubMed] [Google Scholar]
- 26.Khan MY, Agarwal SK, Hangloo S. Urea-induced structural transformations in bovine serum albumin. J Biochem 1987;102(2):313–7. [DOI] [PubMed] [Google Scholar]
- 27.Goncharova I, Orlov S, Urbanová M. The location of the high- and low-affinity bilirubin-binding sites on serum albumin: ligand-competition analysis investigated by circular dichroism. Biophys Chem 2013;180–181:55-65 . [DOI] [PubMed] [Google Scholar]
- 28.Zunszain PA, Ghuman J, McDonagh AF, Curry S. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J Mol Biol 2008;381(2):394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tatlidil D, Ucuncu M, Akdogan Y. Physiological concentrations of albumin favor drug binding. Phys Chem Chem Phys 2015;17(35):22678–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.