

Summary
Somatic genetic heterogeneity resulting from post-zygotic DNA mutations is widespread in human tissues and can cause diseases, however few studies have investigated its role in neurodegenerative processes such as Alzheimer’s Disease (AD). Here we report the selective enrichment of microglia clones carrying pathogenic variants, that are not present in neuronal, glia/stromal cells, or blood, from patients with AD in comparison to age-matched controls. Notably, microglia-specific AD-associated variants preferentially target the MAPK pathway, including recurrent CBL ring-domain mutations. These variants activate ERK and drive a microglia transcriptional program characterized by a strong neuro-inflammatory response, both in vitro and in patients. Although the natural history of AD-associated microglial clones is difficult to establish in human, microglial expression of a MAPK pathway activating variant was previously shown to cause neurodegeneration in mice, suggesting that AD-associated neuroinflammatory microglial clones may contribute to the neurodegenerative process in patients.
One-Sentence Summary:
A subset of Alzheimer Disease patients carry mutant microglia somatic clones which promote neuro-inflammation.
Introduction
Neurodegenerative diseases are a frequent cause of progressive dementia. Alzheimer’s disease (AD) is diagnosed in ~90% of cases, with an estimated prevalence of ~10% in the population over 65 years of age 1,2. The role of germline genetic variation in neurodegenerative diseases and AD has been studied intensely. Although autosomal dominant forms of AD due to rare germline variants with high penetrance account only for an estimated ~1% of cases 3–8, a number of common variants were also shown to contribute to disease risk. Carriers of one germline copy of the epsilon4 (E4) allele of the apolipoprotein E gene (APOE4), present in ~15 to 20% of population, have a three-fold higher risk of AD, while two copies (~2 to 3 % of population) increase the risk by ~10-fold 9–12. Genome-wide association studies (GWAS) have identified an additional ~50 common germline variants that more moderately increase the risk of AD, including TREM2, CD33, and MS4A6A variants 13–15. Interestingly, the APOE4 allele is responsible for an increased inflammatory and neurotoxic response of microglia and astrocytes in the brain of carriers 16–18, and it was noted that the majority of the other germline AD-risk variants are located within or near genes expressed in microglia 15 and in particular at microglia-specific enhancers 19. These data, together with transcriptional studies 20–22 support the hypothesis that genetic variation in microglia may contribute to the pathogenesis of neurodegeneration and AD.
Somatic genetic heterogeneity (mosaicism), resulting from post-zygotic DNA mutations, is widespread in human tissues, and a cause of tumoral, developmental, and immune diseases 23–26. Aditionally, a role of somatic variants in neuropsychiatric disorders is also suspected 27. Mosaicism has been documented in the brain tissue of AD patients in several deep-sequencing studies 28–30, showing that the enrichment of putative pathogenic somatic mutations in the PI3K-AKT, MAPK, and AMPK pathway do occur in the brain of patients in comparison to controls 30. However these studies performed in whole brain tissue lacked cellular resolution and mechanistic insights, and the role of somatic mutants in neurodegenerative diseases remains poorly understood 23. Somatic variants that activate the PI3K-AKT-mTOR or MAPK pathways in neural progenitors are a cause of cortical dysplasia and epilepsy 31–34 and developmental brain malformations 35, while somatic variants that activate the MAPK pathway in brain endothelial cells are associated with arteriovenous malformations 36. Interestingly, we reported that expression of a somatic variant activating the MAPK pathway in microglia causes neurodegeneration in mice 37, but the presence and contribution of microglial somatic clones in neurodegenerative diseases and AD remains unknown.
Here, we investigated the presence and nature of somatic variants in brain cells from control and AD patients. In an attempt to examine all brain cells at the same resolution, nuclei from neurons, glia cells and microglia, which only represent ~5% of brain cells, were pre-sorted. Human microglia are reported to develop in embryo and renew by local proliferation within the brain 38–40. However, bone marrow-derived myeloid cells can enter the brain, in particular during pathological processes, and may not be distinguishable from resident microglia by transcriptomics alone 41. In order to distinguish somatic variants carried by resident microglia from the one carried by myeloid cells of peripheral origin, we analyzed matched peripheral blood from control and patients, to ‘barcode’ somatic mutants shared between microglia and blood. Finally, in order to achieve high sensitivity in the detection of variants that confer a proliferative or activation advantage (pathogenic mutations) and support the emergence or pathogenicity of mosaic clones 42, and/or that have been previously associated with neurological diseases, we initially performed a targeted deep-sequencing of a panel of 716 genes covering somatic variants reported in clonal proliferative disorders and genes associated with neurodegenerative diseases diseases.
We found that microglia from AD patients were enriched for pathogenic variants in comparison to age-matched controls. Furthemore, we found that these microglia-specific AD-associated variants preferentially target the MAPK pathway, including recurrent CBL ring-domain mutations. In addition, we showed that these variants drive a microglia transcriptional program characterized by a strong neuro-inflammatory response previously associated with neurotoxicity, including the production of IL1 and TNF, both in in vitro microglia models and in patients. The natural history of the AD-associated microglia clonal inflammatory disorder we describe here is difficult to establish. Specifically, we do not know whether it contributes to the onset of the neuro-inflammatory process at an early stage of the disease, or if microglia carrying pathogenic mutations preferentially expand later during the course of the disease in response to tissue inflammation. Under both hypotheses however, the presence of neuro-inflammatory microglial clones may contribute to the neurodegenerative process in a subset of AD patients. This report reveals a previously unrecognized presence of AD-associated microglia harboring pathogenic somatic variants in humans and provides mechanistic insight for neurodegenerative diseases by delineating cell-type specific variant recurrence.
Results
Clonal diversity among brain cells and blood from controls and AD patients.
We examined post-mortem frozen brain samples and matching blood from 45 patients with intermediate-onset sporadic AD and 44 control individuals who died of other causes, including 27 donors age and sex-matched donors with the AD cohort (Fig. 1A; Supplementary Fig. 1A and Supplementary Table 1). APOE risk allele frequency for patients and controls was comparable to published series 10–12 (Supplementary Fig. 1A), and analysis of germline mutations did not identify deleterious variants in the 140 genes associated with neurological diseases. Myeloid/microglia, neurons, and glia/stromal cells were purified by flow cytometry using antibody against PU.1 and NeuN 43 (Fig. 1B, Supplementary Fig. 1B and 1C). Single nuclei (sn)RNAseq was performed on PU.1+ nuclei from one control and 3 AD patients to evaluate microglia enrichment following PU.1+ purification, and a cell-type annotation analysis indicated that ~94% of PU.1+ nuclei correspond to microglia (Fig. 1C and Supplementary Fig. 1D-1H). Cortex samples were obtained from all donors but hippocampus samples were mostly obtained from AD patients (Fig. 1A; Supplementary Table 1). A total of 744 DNA samples from blood, PU.1+ nuclei, NeuN+ nuclei, and Double Negative nuclei (glia/stromal cells) from patients and controls (Fig. 1A) were submitted to targeted hybridization/capture and deep-DNA targeted sequencing (TDS, Fig. 1D, see Methods), at mean coverage of ~1,100x (Supplementary Fig. 1I), for a panel of 716 genes (3.43 Mb, referred to below as BRAIN-PACT) which included genes reported to carry somatic variants in clonal proliferative disorders (n=576 genes)44,45 or that have been reported to be associated with neurodegenerativediseases (n=140) 46–54 (Supplementary Table 2, see Methods).
Fig. 1-. Detection of mutations in brain cell types and blood.
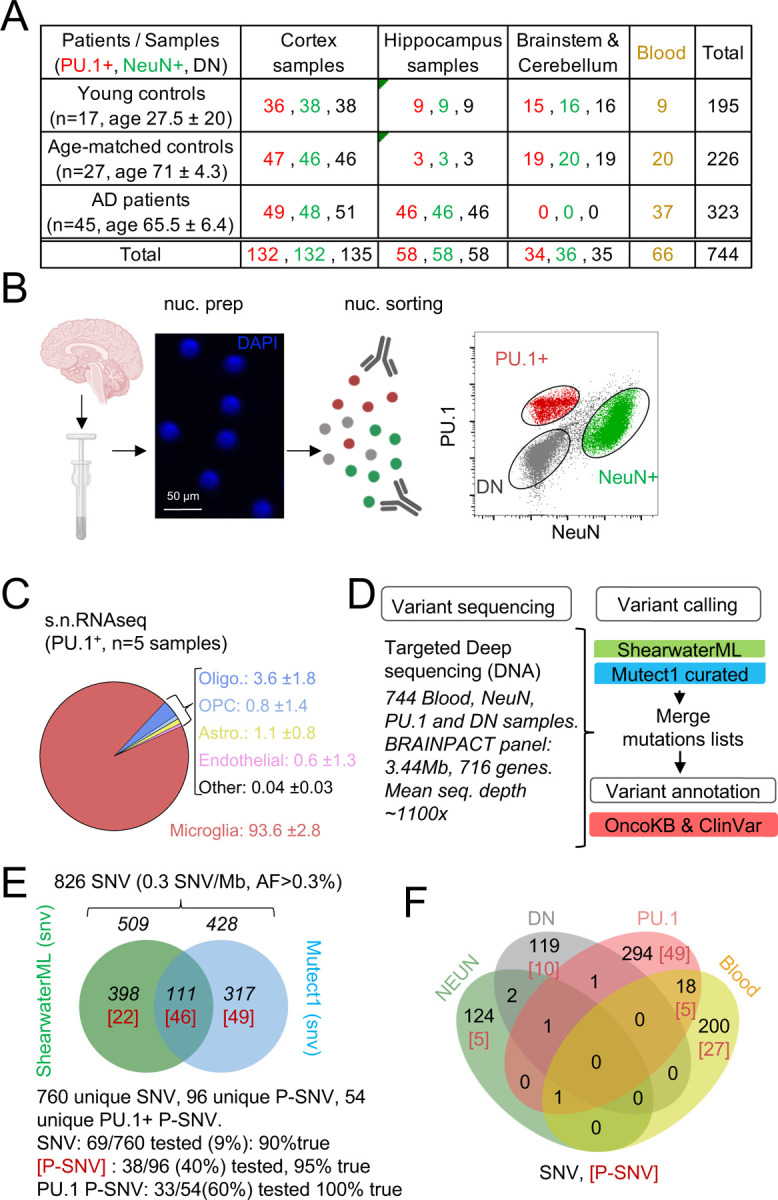
(A) Table with patient and sample information. (B) Schematic represents the isolation and labelling of nuclei from post-mortem frozen brain samples from controls and Alzheimer’s disease patients with DAPI and antibodies against PU.1+ (myeloid/microglia) and NeuN+ (neurons).Representative flow cytometry dot-plot of nuclei separation. Double negative nuclei are labeled ‘DN’. (C) Percentage of cell types obtained in sorted PU.1+ nuclei determined by single-nuclei RNAseq in 5 brain samples from 4 individuals. (D) Schematic represents the sequencing strategy. Two algorithms (ShearwaterML and Mutect1) were used for variant calling. After annotation, pathogeneity was determined using OncoKb and ClinVar. (E) Venn diagram represents the number of variants and overlap between the ShearwaterML and Mutect1. Numbers in red indicate pathogenic variants (P-SNV). Validation of variants was performed by droplet digital (dd)PCR on pre-amplified DNA when available. (F) Venn diagrams represent the repartition per cell type of the 826 single-nucleotide variations (SNVs) identified in NeuN+: Neurons, PU.1+: microglia, DN: glia, and matching blood. [Numbers] in red indicate pathogenic variants P-SNV
After QC and filtering of germline variants, variant calling using ShearwaterML and a curated Mutect1 analysis identified 826 somatic synonymous and non-synonymous single-nucleotide-variations (SNVs), at an allelic frequency > 0.3% (mean 1.3%) in the 744 samples, corresponding to an overall variant burden of 0.3 mut/Mb (Fig. 1E). Sixty-six/826 SNV were present in more than one sample (Supplementary Table 3). Droplet digital-PCR performed on pre-amplification DNA for ~10% of the 760 unique SNV was positive in 90% of cases (Fig.1E; Supplementary Table 3). After annotation using the OncoKB 55 and ClinVar 56 databases for disease- associated or causative variants (Fig. 2D,F; Supplementary Table 3), 96 unique SNV were classified as Pathogenic (P)-SNV. 40% of these P-SNV were tested by droplet digital-PCR and confirmed in 95% of cases (Fig. 1E and Supplementary Table 3). Positive and negative results in matching brain samples from individual donors were confirmed in 100% of samples at a mean depth of ~5000x (range 648–23.000x) (Supplementary Table 3). A venn-diagram analysis of SNVs detected in PU.1+, NeuN+, DN, and blood samples indicated that most (>90%) SNV and P-SNV were cell-type or tissue specific, with ~ 5% of SNV and ~ 8% of P-SNV shared between the blood and brain of individual donors (Fig. 1F; Supplementary Table 3). These data indicate that targeted deep-sequencing of purified nuclei allows to detect clonal mosaic variants with high sensitivity and specificity. In addition, ‘bar-coding’ of clonal variants across tissues suggests that infiltrating myeloid cells of peripheral origin account for ~5% of microglia somatic diversity, and therefore that blood clones have a detectable but minor contribution to microglia, consistent with its local maintainance and proliferation 38,39.
Fig. 2-. Pathogenic variants are enriched in microglia from AD patients.

(A) Correlation plot represents the mean number of variants per cell type and donor (n=89) (Y axis), as a function of age (X axis). Each dot represents mean value for a donor. Statistics: fitted lines, the correlation coefficients (rs) and associated p values were obtained by linear regression (Spearman’s correlation). (B) Number of SNV per Mb and cell types per donor, of age-matched controls (n=27) and AD patients (n=45). Each dot represents mean value for a donor. Statistics: p-values are calculated with unpaired two-tailed Mann-Whitney U. Note: non-parametric tests were used when data did not follow a normal distribution (D’Agostino-Pearson normality test). (C) Number of SNV per Mb in PU.1+ samples across brain regions, of age-matched controls (n=27) and AD patients (n=45). Each dot represents a sample. Statistics: p-values are calculated with Kruskal–Wallis, multiple comparisons. Note: non-parametric tests were used when data did not follow a normal distribution (D’Agostino-Pearson normality test). (D) Correlation plot represents the mean number of pathogenic variants (P-SNV) as determined by ClinVar and/or OncoKB, per cell type and donor (n=89) (Y axis), as a function of age (X axis). Each dot represents mean value for a donor. Statistics: fitted lines, the correlation coefficients (rs) and associated p values were obtained by linear regression (Spearman’s correlation). (E) Number of P-SNV per Mb and cell types per sample, of age-matched controls (n=27) and AD patients (n=45). Each dot represents a sample. Statistics: p-values are calculated with unpaired two-tailed Mann-Whitney U. Note: non-parametric tests were used when data did not follow a normal distribution (D’Agostino-Pearson normality test). (F) Number of P-SNV per Mb in PU.1 samples across brain regions, of age-matched controls (n=27) and AD patients (n=45). Each dot represents a sample. Statistics: p-values for comparison within each group (controls and patients) are calculated with Kruskal–Wallis test and Dunn’s test for multiple comparisons. p-values for the comparison of P-SNV between the cortex of C and the cortex of AD (0.01) was calculated with unpaired two-tailed Mann-Whitney U. Note: non-parametric tests were used when data did not follow a normal distribution (D’Agostino-Pearson normality test). (G) Number of P-SNV per Mb and and cell types per donor for age-matched controls (n=27) and AD patients (n=45). Each dot represents mean value for a donor. Statistics: p-values are calculated with unpaired two-tailed Mann-Whitney U test. Odds ratio (95% CI, 2.049 to 29.02) and p values for the association between AD and the presence of pathogenic variants are calculated by multivariate logistic regression, with age and sex as covariates. (H) P-SNV burden as a function of age and disease status (age-matched controls and AD). Linear lines represent trend lines from mixed-effects linear regression that incorporates individual donor as a random effect (blue, control: P=0.0025, R^2=0.13; red, NDD: P=9.1x10^-16, R^2=0.50 by Pearson’s correlation). The model’s total explanatory power is substantial (conditional R^2=0.48). Both age and AD are associated with a significant increase in SNV burden in this model (P<1x10^-4 and P=1x10^-4, respectively, by likelihood ratio test). Anatomical regions of the brain specimen and originating brain banks were not incorporated because the models incorporating those parameters did not significantly improve the overall model fitting by likelihood ratio test (see Methods). Graph depicts SNV burden corrected by the mixed-effects model (See Fig S2D for observed P-SNV burden)
Somatic clonal diversity of the different cell types, as evaluated by the SNV/megabase burden was higher in blood (1 mut/Mb) and PU.1+ nuclei (0.5 mut/Mb) than for DN and neurons (0.18 mut/Mb) (Supplementary Fig. 2A). The SNV/mb burden of blood and PU.1+ nuclei increased as a function of age (Fig. 2A and Supplementary Table 3) as previously reported for proliferating cells 24,57–59. Interestingly, the SNV/mb burden of blood cells from age-matched controls was higher than for AD patients (Fig. 2B). In contrast, there was no difference in SNV/mb burden between PU.1+, NEUN+ and DN samples from AD patients and age-matched controls (Fig. 2B), and between PU.1+ nuclei from the cortex, hippocampus, and brainstem/cerebellum samples (Fig. 2C). These data altogether indicate that the clonal diversity of microglia and blood both increase with age, and that the clonal diversity of blood cells is lower in AD than in age-matched controls who died of other causes including cancer and cardiovascular diseases (see Methods). This is consistent with recent studies showing that clonal hematopoiesis is associated with a higher risk of several diseases related to ageing such as cardiovascular diseases, but inversely associated with the risk of AD 60,61.
Microglia clones carrying pathogenic variants are enriched in AD patients.
In contrast to the global SNV burden, increased P-SNV burden was correlated not only with age (Fig. 2D), but also with the disease status (AD) (Fig. 2E-H). Within the control group, the SNV and P-SNV burden was higher in the blood of controls treated for cancer (Supplementary Fig. 2B). The P-SNV burden per Mb was selectively and highly enriched in PU.1+ samples from AD patients in comparison to age-matched controls (p=0.0003, Fig. 2E). Analysis of PU1+ P-SNV/Mb burden per brain region indicated that the P-SNV/Mb burden was similar between brain regions within each group (Fig. 2F and Supplementary Fig. 2C), and therefore attributable to AD status rather than sampling bias. Analysis of mutational load per donor confirmed that microglial clones carrying P-SNV were enriched in the brain of AD patients in comparison to age-matched controls (Fig. 2G). Despite the relatively modest cohort size, a logistic regression analysis confirmed the association between the presence of P-SNVs in PU.1+ nuclei and AD after adjusting for sex and age (OR= 7; p=0.0035, Fig. 2G and Supplementary Fig. 2D). A mixed-effects linear regression model analysis also showed an excess of P-SNVs in AD independently of the effect of age (p = 0.0215) (Fig. 2H and Supplementary Fig. 2E).
In addition, genes targeted by P-SNV were all expressed in microglia (Supplementary Fig. 2F and Supplementary Table 3) and the analysis of P-SNV/Mb mutational load restricted to genes that are not expressed in microglia did not show an enrichment of candidate pathogenic variants in AD patients (Supplementary Fig. 2G and Supplementary Table 3). Altogether, these results show an association between microglia clones carrying P-SNV and AD in this series.
AD patients carry microglial clones with MAP-Kinase pathway variants including recurrent CBL variants.
Pathways analysis of genes carrying P-SNV in microglia from AD patients, against the background of the 716 genes sequenced, showed that the most significant pathways enriched were the receptor tyrosine kinase /MAP-Kinase pathways (Reactome, GO, and canonical pathways, Fig. 3A and Supplementary Table 4), corresponding to pathogenic/oncogenic variants in 6 of the 15 genes of the classical MAPK pathway 62 (CBL, BRAF, RIT1, NF1, PTPN11, KRAS), TEK, and the KEGG Chronic Myeloid Leukemia (CML) pathway, which includes the former plus SMAD5 and TP53 (Fig. 3B, 3C and Supplementary Fig. 3). Mutational load for MAPK genes was significantly higher in AD patients in comparison to age-matched control (Fig. 3C). Other enriched pathways, albeit less significant, included genes involved in DNA repair and chromatin binding/ methyltransferase activity (Fig. 3B; Supplementary Table 4). No pathway was enriched in age matched controls. Of note we did not observe microglia P-SNVs within genes reported to be associated with neurological disorders (Supplementary Table 2) in patients (Supplementary Table 3). P-SNV targeting genes of the classical RTK/ MAPK pathway (Fig. 3C) were detected in the PU.1+ samples from ~25% of the AD patients tested (p=0.0145 vs age-matched controls, Fig. 3D and Supplementary Fig. 3). Strikingly, half of these patients (6 patients, 13% of AD patients in this series) carried reccurent P-SNV in the RING domain of CBL 63–73 (Fig. 3B–3E). Two additional patients presented with P-SNV in the Switch II domain of RIT1 74 (Fig. 3B–3F). Microglia from the 3 other patients carried activating KRAS (p.A59G), PTPN11 (p.T73I) and TEK (p.R1099*) oncogenic variants previously described in cancer and sporadic venous malformations 75–77 (Fig. 3B and 3D). In addition, a 12th patient carried a gain of function (GOF) U2AF1 (p.S34F) variant 78, which is not a ‘classical MAPK gene’ but activates the MAPK pathway in myeloid malignancies 79 (Fig. 3B and Supplementary Fig. 3). Two patients carried 2 different MAPK activating variants: microglia from 1 patient carried an activating BRAF (p.L505H) variant 80 in addition to loss of function (LOF) variant CBL (p.C416S), and another patient carried the NF1 (p.L2442*) LOF variant 81,82 in addition to the activating RIT1 (p.M90I) variant (Fig. 3D and Supplementary Fig. 3). Five patients also carried additional P-SNV targeting genes involved in DNA repair with tumor suppressor function 83,84, including the loss of function variants in ATR (c.6318A>G) 85 and SMC1A (p.X285_splice) (Fig. 3D and Supplementary Fig. 3), and in DNA/histone methylation including TET2 (p.Q1627*) 63,86, IDH2 (p.R140Q) 87, and PBRM1 (c.996–7T>A) 88) (Fig. 3D and Supplementary Fig. 3). Finally, two patients carried oncogenic variants in genes from the KEGG Chronic Myeloid Leukemia (CML) pathway, SMAD3 (p.R373C) 89 and TP53 (pX261_splice) 90 (Fig. 3D and Supplementary Fig. 3). The detection of multiple oncogenic variants in the same patients is reminiscent of the features observed in myeloproliferative disorders described outside the brain 73,86.
Fig. 3-. Somatic microglial clones with multiple and recurrent CBL and MAP-Kinase pathway activating variants.

(A) Pathway enrichment analysis for the genes target of P-SNVs using the panel of 716 genes as background set. Graph shows the most enriched pathways by: Reactome Gene Sets, GO Molecular Functions, Canonical Pathways and KEGG Pathway (see complete list in Supplementary Table 4). (B) Bar plot indicates the genes carrying P-SNV (y-axis) and the % of AD patients carrying P-SNV for each gene (x-axis). Genes are color-coded by pathway. (C) Representation of the classical MAPK pathway, the 6 genes mutated in AD patients are labeled in red, TEK is labeled in blue, and larger font size indicate reccurence of variants in a given gene. Violin plot shows distribution of P-SNV in genes from classical MAPK pathway per Mb sequenced and per sample in patients and controls, p-value: unpaired two-tailed Mann-Whitney U test. (D) Summary Table showing patients carrying P-SNV in the classical RTK/MAPK pathway and CML associated genes (see Supplementary Table 3) and indicating the detection of variants in blood, and their association with other variants in microglia. (E) Recurrent variants in the ring-like domain of CBL are indicated in red on the diagram structure of gene, above representative Western blot from cell lysates from HEK293T cells expressing WT, positive control (Y371H), or CBL variants alleles found in patients, and stimulated with EGF or control, probed with antibodies against Phospho-p44/42 MAPK (Erk 1/2, Thr202/Tyr204), total p44/42 MAPK (Erk1/2), HA-tag and tubulin (BOTTOM). Histogram (RIGHT) represents quantification of the increase of the Phospho-ERK1/2 /total ERK1/2 ratio in western blots in n=5 independent experiments, statistics: unpaired one-tailed t-test. (F) RIT1 M90I and F82L are represented on the 3D structure of the gene (pdb code: 4klz, F82 is within a segment whose structure was not resolved) and representative western blot from HEK293T cells expressing Flag-RIT1 (WT and mutants) and treated -/+ 20% FBS before harvesting. Lysates were probed with antibodies against Phospho-p44/42 MAPK (Erk 1/2, Thr202/Tyr204), total p44/42 MAPK (Erk1/2, (MAPK)), Flag, and tubulin. Histogram (RIGHT) represents quantification of the increase of the Phospho-ERK1/2 /total ERK1/2 ratio in western blots in n=4 independent experiments, statistics: unpaired one-tailed t-test. (G) Variant allelic frequency (VAF, %) for the BRAFV600E allele in PU.1+ nuclei from brain samples from histiocytosis patients (each dot represents a sample) and for P-SNVs in in PU.1+ nuclei from brain of AD patients (each dot represent a variant). Note: non-parametric tests were used when data did not follow a normal distribution (D’Agostino-Pearson normality test). (H) Percentage of P-SNVs detected by targeted deep sequencing (TDS) which were also detected by Whole-Exome-Sequencing (WES).
Recurrent CBL and RIT1 variants activate the MAPK pathway.
CBL is an E3 ubiquitin-protein ligase that negatively regulates RTK signaling via MAPK 91. CBL somatic and germ-line LOF variants such as R420Q have been previously associated with tumoral diseases including clonal myeloproliferative disorders 63–73 and RASopathies 92 respectively. We confirmed that CBL RING-domain variants found in AD patients increased MAPK phosphorylation in response to EGF upon expression of HA-tagged WT or mutant alleles in HEK293T cells (Fig. 3E and Supplementary Fig. 4A). RIT1 is a RAS GTPase, and somatic or germ-line GOF variants such as RIT1 F82L and RIT1 M90I, also enhance MAPK signaling in malignancies 74 and RASopathies 93,94 respectively. As in the case of CBL variants, the 2 RIT1 variants found in AD patients increased MAPK phosphorylation in response to FBS in HEK293T cells expressing these mutant alleles (Fig. 3F, Supplementary Fig. 4B and 4C). These data altogether indicate that a subset of AD patients (12/45, ~ 27% of this series) present with microglial clones carrying one or several oncogenic variants that activate the RTK/MAPK pathway, and are characterized by recurrent oncogenic variants in CBL and RIT1.
Allelic frequency of the patients’ MAPK activating variants
The allelic frequencies at which MAPK activating variants are detected in brain samples from AD patients range from ~1–6% in microglia (Fig. 3G), which correspond to mutant clones representing 2 to 12% of all microglia in these samples, assuming heterozygosity. This range of allelic frequency is frequently observed for the MAPK activating BRAFV600E variant in microglia isolated from brain samples of 6 patients diagnosed with BRAFV600E+ histiocytosis, a rare clonal myeloid disorder associated with neurodegeneration 37,95–98 (Fig. 3G and Supplementary Table 5). These data suggested that the size of the mutant microglial clones in AD patients was compatible with a role in a neuro-inflammatory/neurodegeneration process.
Other variants found in microglia from AD patients
Pathogenic variants that did not involve the MAPK pathway included LOF variants in the DNA repair gene CHEK2 including CHEK2 c.319+1G>A 99 and CHEK2 R346H (Supplementary Fig. 3 and 4D), Mediator Complex gene MED12 100, Histone methyltransferases SETD2 101 and KMT2C/MLL3, the DNA methyltransferase DNMT3A 86,102, DNA demethylating enzymes TET2 and the Polycomb proteins ASXL1 86. Of note, TET2, DNMT3 and KMT2C variants when present, were frequently detectable in the patients’ matching blood at low allelic frequency (Supplementary Fig. 3). TET2, DNMT3 and KMT2C are frequently mutated in clonal hematopoiesis 58,59, suggesting that in contrast to other variants, the presence of TET2, DNMT3 and KMT2C/MLL3 in the brain of patients may reflect the entry of blood clones in the brain.
In half of the AD patients, no microglia pathogenic variants were identified. Targeted deep sequencing (TDS) cannot identify variants located outside of the BRAIN-PACT panel, such as other potential additional variants that would activate the MAPK pathway. Therefore, we performed whole exome sequencing (WES) of PU.1+ nuclei at an average depth ~400x, in selected samples from 48 donors, including samples from most of the patients negative for pathogenic variants by TDS (n=17 out of 22), a selection of patients with variants identified by TDS (n=16 out of 23), and 15 controls, followed by a curated Mutect analysis. Only 6/15 (40%) of the pathogenic SNVs previously identified by TDS and confirmed by ddPCR were detectable by WES in these samples (Fig. 3H), indicating a lower sensitivity of WES. Nevertheless, after annotation by 4 modeling predictors (Polyphen, SIFT, CADD/MSC and FATHMM-XF 103–108 additional SNVs predicted to be deleterious with high confidence were identified in 8/22 patients without pathogenic variants identified by TDS (Supplementary Fig. 3 and Supplementary Table 6). Interestingly, 4 of the predicted deleterious variants identified by WES targeted genes that regulate the MAPK pathway (ARHGAP9, ARHGEF26, CHD8, and DIXDC1 (Supplementary Fig. 3 and SupplementaryTable 6).
The patients’ MAPK activating variants increases ERK phosphorylation, proliferation, inflammatory and mTOR pathways in murine microglia and macrophages.
CBL variants increased ERK phosphorylation upon lentiviral transduction in BV2 murine microglial cells 109,110 (Supplementary Fig. 4E). However as this line was immortalized by v-Raf, which might interfere with the study of the MAPK pathway, we also stably expressed WT and variant CBL, RIT1, KRAS, PTPN11 alleles in SV-U19–5 transformed mouse ‘MAC’ lines 111,112 (see Methods and Supplementary Fig. 5A,B). MAC lines expressing CBL, RIT1, KRAS and PTPN11 variants presented with increased ERK phosphorylation and/or increased proliferation in comparison to their WT controls, as measured by Western immunoblotting and EdU incorporation (Fig. 4A and Supplementary Fig. 5A,B). In addition, Hallmark and KEGG pathway analysis of RNAseq data from control and mutant lines showed increased RAS, TNF, IL6 and JAK STAT signaling, complement, inflammatory responses, and mTOR pathway activation signatures in mutants (Fig. 4B and Supplementary Table 7). These data indicated that microglia variants from patient’s activate murine microglial cells and growth factor-dependent macrophages with proliferative and inflammatory responses in vitro. However, overexpression of mutant alleles in mouse cell lines does not necessarily recapitulate or predict the effects of a heterozygous genetic variant in physiological conditions. Thus, we investigated the role of CBLC404Y allele in heterozygous human primary microglia-like cells.
Fig. 4-. MAPK pathway activating variants in mouse macrophages and human iPSC-derived microglia-like cells.

(A) Representative western-blot analysis (Top panels) and quantification (Middle panels) of phospho- and total-ERK in lysates from a murine CSF-1 dependent macrophage cell line expressing CBLWT, CBLI383M, CBLC384Y, CBLC404Y, CBLC416S (n=3–6), and RIT1WT, RIT1F82L and RIT1M90I (n=3), KRASWT and KRASA59G (n=3). Bottom panels depicts flow cytometry analysis of EdU incorporation in the same lines. Statistics, Unpaired t-test. (B) HALLMARK and KEGG pathways (FDR / adj.p value <0.25, selected from Supplementary Table 7) enriched in gene set enrichment analysis (GSEA) of RNAseq from mutant CSF-1 dependent macrophages lines CBLI383M, CBLC384Y, CBLC404Y, CBLC416S, CBLR420Q, RIT1F82L RIT1M90I, KRASA59G, and PTPN11T73I (n=3–6) in comparison with their wt controls. NES: normalized enrichment score. (C) Sanger sequencing of 2 independent hiPSC clones (#93 and #91) of CBL404C/Y heterozygous mutant carrying the c.1211G/A transition on one allele and 2 independent isogenic control CBL404C/C clones (#71 and #89) all obtained by prime editing. (D) Photomicrographs in CBL404C/C and CBL404C/Y iPSC-derived microglia-like cells.(E) Quantification of leading edge and lateral lamellipodia in CBL404C/C and CBL404C/Y iPSC-derived microglia-like cells. n=3–7, statistics: p-value are obtained by nested one-way ANOVA. (F) Flow cytometry analysis of cell size for the same lines (n>3) statistics: p-value are obtained with nested one-way ANOVA,). (G) Flow cytometry analysis of EdU incorporation in CBL404C/C and CBL404C/Y microglia-like cells after a 2 hours EdU pulse. n=3, unpaired t-test). (H) Western-blot analysis (left) and quantification (right) of phospho- and total-ERK proteins in lysates from CBL404C/C and CBL404C/Y microglia-like cells starved of CSF-1 for 4 h and stimulated with CSF-1 (5 min, 100 ng/mL) (n=4), statistics: pvalue are obtained with two-way ANOVA.
Heterozygosity for a CBL variant allele activates human microglia-like cells
We used prime editing 113 of human induced pluripotent stem cells (hiPSCs, see Methods) to generate isogenic hiPSCs clones heterozygous for the patients’ variants (Fig. 3C and Supplementary Fig. 5). We focused our analysis on CBL404C/Y mutant lines because CBL was mutated in 6 patients and 2 of them carried the same CBL c.1211G>A p.C404Y variant (Fig. 3D). Microglia-like cells were differentiated from two independent hiPSC-derived CBL404C/Y lines and their isogenic CBL404C/C controls (Fig. 4C and Supplementary Fig. 5C). CBL404C/Y and isogenic CBL404C/C microglia-like cells expressed similar amount of CBL total mRNA and protein, and CBL404C/Y cells expressed wt and mutant mRNA in similar amounts, as expected assuming bi-allelic expression of CBL (Supplementary Fig. 5D-5F). CBL404C/Y cells presented with a phenotype comparable to isogenic CBL404C/C microglia-like cells for expression of IBA1, CSF1R, NGFR, EGFR, CD11b, MRC1, CD36, CD11c, Tim4, CD45, and MHC Class II (Supplementary Fig. 5G). Their viability was also comparable to control (Supplementary Fig. 5H). However, CBL404C/Y cells were larger and presented with more lamellipodia, resulting in an amoeboid morphology less frequently observed in isogenic controls (Fig. 4D,4E), and their proliferation rate was slightly increased, as measured by EdU incorporation (Fig. 4F). Moreover CBL404C/Y cells cultured in CSF1-supplemented medium also presented with a higher basal pERK level than control when restimulated with CSF1 (Supplementary Fig. 5I), and ERK phosphorylation after stimulation of starved microglia-like cells with CSF-1 was increased by ~2 fold in comparison to isogenic WT (Fig. 4G). Altogether, these results showed that heterozygosity for a CBLC404Y allele is sufficient to activate human microglia-like cells increasing their proliferation and ERK activation.
Heterozygosity for a CBLC404Y allele drives a microglial neuroinflammatory /AD associated signature
Gene Set Enrichment Analyses (GSEA) of RNAseq comparing CBL404C/Y and isogenic CBL404C/C microglia-like cells showed upregulation of Glycolysis, Oxidative Phosphorylation, and mTORC1 signatures, indicating increased metabolism and energy consumption by the mutant cells (Fig. 5A and Supplementary Table 8). In addition, as observed in MAC lines, CBL404C/Y cells upregulated complement, TNF, and JAK STAT signaling and inflammatory signatures (Fig. 5A; Supplementary Table 8) 114. Increased production of TNF, IL-6, IFN-γ, IL-1β, C3 and complement Factor H (CFH) by CBL404C/Y cells was confirmed by ELISA (Fig. 5B). In addition, CBL404C/Y microglia-like cells also presented with signatures from the KEGG database associated with neurodegenerative disorders (Fig. 5A; Supplementary Table 8), and for the recently published human microglia AD scRNAseq signature, obtained by analysis of 24 sporadic AD patients and 24 controls 21 (Fig. 5C). These data indicated that heterozygosity for the CBLC404Y allele is sufficient to drive expression of a neuroinflammatory/AD signature in a human microglia-like cell type, characterized by increased metabolism and the production of neurotoxic cytokines known to interfere with normal brain homeostasis.
Fig. 5-. CBL404C/Y microglia signature.

(A) HALLMARK and KEGG pathways (FDR /adj.p value <0.25, selected from Supplementary Table 8) enriched in gene set enrichment analysis (GSEA) of RNAseq from from CBL404C/Y iPSC-derived macrophages and isogenic controls NES, normalized enrichment score. (B) ELISA for pro-inflammatory cytokines (n=3) and complement proteins (n=2) in the supernatant from CBL404C/Y iPSC-derived microglial like cells and isogenic controls. Statistics: p-value are obtained by nonparametric Mann-Whitney U test,* 0.05, ** 0.01, *** 0.001, **** 0.0001. (C) GSEA analysis for enrichment of the human AD-microglia snRNAseq signature (MIC1) 21 in differentially expressed genes between CBL404Y/C microglial like cells and isogenic controls. (D) Dot plot represents the GSEA analysis of HALLMARK and KEGG pathways enriched in snRNAseq microglia clusters (samples from all donors). Genes are pre-ranked per cluster using differential expression analysis with SCANPY and the Wilcoxon rank-sum method. Statistical analyses were performed using the fgseaMultilevel function in fgsea R package for HALLMARK and KEGG pathways. Selected gene-sets with p-value < 0.05 and adjusted p-value < 0.25 are visualized using ggpubr and ggplot2 R package (gene sets/pathways are selected from Supplementary Fig. 6B, Supplementary Table 9). (E) Dot plot represents the GSEA analysis (as in (E)) of HALLMARK and KEGG pathways enriched in cluster 2 / 2B and deconvoluted by donor samples (selected from Supplementary Fig. 6A).
The MAPK variant neuroinflammatory microglial signature is detectable in patients.
Analysis of the snRNAseq data from 5 samples of purified microglia nuclei from 4 donors (control, AD without and with pathogenic variants (Fig.1C, Supplementary Fig. 1D-H and Supplementary Table 9) using unsupervised Louvain clustering and GSEA showed that microglia samples from patients carrying variants were enriched for the signatures observed in the MAC lines and CBL404C/Y cells (Fig. 5D,E and Supplementary Fig. 6). In particular microglia cluster 2 and 2B, were most enriched for the inflammatory, TNF, mTOR and oxidative phosphorylation and glycolysis signatures in patients carrying variants (AD52, AD53) but not the controls (C11, AD34) (Fig. 5D,E and Supplementary Fig. 6). Despite the small size of the mutant clones and the low sensitivity of scRNAseq to detect rare allelic variants, KRAS A59G variant reads were detected in cluster 2/2B from patient AD52.
Altogether, the above results support the hypothesis that patients’ microglial clones carrying pathogenic mutations are associated with a metabolic and neuroinflammatory signature that includes the production of neurotoxic cytokines in vitro and in vivo.
Discussion
We report here that microglia from a cohort of 45 AD patients with intermediate-onset sporadic AD (mean age 65 y.o) is enriched for clones carrying pathogenic/oncogenic variants in genes associated with clonal proliferative disorders (Supplementary Table 2) in comparison to 44 controls. Of note we did not observed microglia P-SNVs within genes reported to be associated with neurological disorders in the patients.
These pathogenic variants are absent from blood, glia or neurons in most cases. They are found predominantly in the MAPK pathway and include reccurent variants (CBL RING domain variants in 6 patients), which promote microglial proliferation, activation, and expression of a neuroinflammatory/neurodegereration-associated transcriptional programme in vitro and in vivo, and the production of neurotoxic cytokines IL1b, TNF, and IFNg 115–118. Heterozygous expression of pathogenic CBL variant in human microglia-like cells was sufficient to drive a transcriptional program that associates with increased metabolic activity and a neurotoxic inflammatory response, also observed in microglia from patients with MAPK-activating variants.
The association between AD and MAPK pathway variants is consistent with a previous study where WES performed on unseparated brain tissue from AD patients showed that putative pathogenic somatic variants were enriched for the MAPK pathway, despite the lower sensitivity of the approach and the lack of cellular specificity 30. The pathogenic role of the somatic pathogenic variants in the MAPK pathway associated with the microglia of AD patients is supported by several lines of evidence. We show here that they promote a neuroinflammatory/neurodegereration-associated transcriptional programme in microglia like cells. In addition, somatic variants that activate the MAPK pathway in tissue macrophages cause a clonal proliferative and inflammatory disease called Histiocytosis, strongly associated with neurodegeneration 37,95–97, and introduction in mouse microglia of the variant allele most frequently associated with histiocytosis (BRAFV600E) causes neurodegeneration in mice 37. The allelic frequencies of pathogenic variants found in AD patients is lower than values classically observed in solid tumors or leukemia, but within the range of the clonal frequency of pathogenic T cells observed in auto-immune diseases 119, and we found that they were in the range of the allelic frequencies observed for the BRAFV600E variant in microglia in the brain of Histiocytosis patients. Moreover, the RAS/MAPK signaling pathway is involved in microglia proliferation, activation and inflammatory response 120–122, neuronal death, neurodegeneration, and AD pathogenesis 15,19,37,123, and its activation has been proposed to be an early event in the pathophysiology of AD in human 124. Neuroinflammation is an early event in AD pathogenesis, increasingly considered as critical in pathogenesis initiation and progression 16,125,126. This is underscored by the observation that the main known genetic risk factor for sporadic AD is the APOE4 allele, responsible for an increased inflammatory response in the brain of APOE4 carriers 16. In this regard, the contributing role of MAPK activating variants could be comparable to that of the APOE4 allele, and we noted that the allelic frequency of APOE4 allele is lower in patients with pathogenic variants (16/46 alleles, 34%) than patients without detected variant (23/44 alleles, 53%) although the difference did not reach significance in this series.
Variants targeting the DNA-repair and DNA/histone methylation pathways are also enriched among AD patients, sometimes associated in the same patients, albeit their functional significance was not investigated here. Of note however, germline variants of the DNA-repair transcription factor TP53, and DNA damage sensors ATR and CHEK2 were shown to promote accelerated neurodegeneration in human 83,84.
Microglia variants are frequently absent from blood, and our DNA sequencing barcoding approach does not support a model where blood cells massively infiltrate the brain or replace the microglia pool in patients from our series, but instead consistent with the local maintenance and proliferation of microglia 38,39. In addition, our results are consistent with a recent study showing that clonal hematopoiesis was inversely associated with the risk of AD 61.
The association of microglia clones carrying pathogenic variants with AD in a subset of patients is not a consequence of an overall increase in microglia mutational load (SNV) in AD. Together with evidence that pathogenic variants drive neuroinflammation, these data suggest that these clones could contribute to AD pathogenesis, together with other genetic and environmental factors. Lewy bodies, amyloid angiopathy, tauopathy, or alpha synucleinopathy, were equally distributed among AD patients with or without microglia clones carrying MAPK activating variants. The natural history of the microglial clones is difficult to study in human. It is possible that microglial clones with proliferative and activation advantage and a neuroinflammatory and neurotoxic profile may be present at the onset and contribute to the early stages of the disease. Alternatively it is also possible that the microglial clones carrying the pathogenic mutations appear or are selected later during the course of the disease in the inflammatory milieu of the AD brain. In the latter case, pathogenic microglial clones may contribute to disease progression, i.e. neuroinflammation and neurodegeneration.
Methods
Tissue samples.
The study was conducted according to the Declaration of Helsinki. Human tissues were obtained with patient-informed consent and used under approval by the Institutional Review Boards from Memorial Sloan Kettering Cancer Center (IRB protocols #X19–027). Snap-frozen human brain and matched blood were provided by the Netherlands Brain Bank (NBB), the Human Brain Collection Core (HBCC, NIH), Hospital Sant Joan de Déu and the Rapid Autopsy Program (MSKCC, IRB #15–021). Samples were neuropathologically evaluated and classified by the collaborating institutions as Alzheimer’s disease (AD) 1-5 or non-dementia controls. The mean age of AD patients is 65 years old (55.5% female, 44.5% male). The mean age of all controls is 54 years old (60% female, 40% male), and the mean age of AD age-matched controls was 70 years old (60% female, 40% male). The overall mean of the post-mortem delay interval was 9.8 hours. Patients did not present with germline pathogenic PSEN1/2/3 or APP AD’s associated variants. For additional information on donor’s brain regions, sex, age, cause of death, Apoe status, Braak status see Supplementary Table S1. To avoid possible contamination of sequencing data with mutations associated with donor’s tumoral disease in the group of non-dementia controls, we refrained from selecting cases with blood malignancies or with brain tumors. Samples from histiocytosis patients were collected under GENE HISTIO study (approved by CNIL and CPP Ile-de France) from Pitié-Salpêtrière Hospital and Hospital Trousseau and from Memorial Sloan Kettering Cancer Center.
Nuclei isolation from frozen brain samples, FACS-sorting and DNA extraction.
All samples were handled and processed under Air Clean PCR Workstation. An average of 400 mg of frozen brain tissues were homogenized with a sterile Dounce tissue grinder using a sterile non-ionic surfactant-based buffer to isolate cell nuclei (‘homogenization buffer’: 250 mM Sucrose, 25 mM KCL, 5 mM MgCl2, 10 mM Tris buffer pH 8.0, 0.1% (v/v) Triton X-100, 3 μM DAPI, Nuclease Free Water). Homogenate was filtered in a 40-μm cell strainer and centrifuged 800g 8 min 4°C. To clean-up the homogenate, we performed a iodixanol density gradient centrifugation as follow: pellet was gently mixed 1:1 with iodixanol medium at 50% (50% Iodixanol, 250 mM Sucrose, 150 mM KCL, 30 mM MgCl2, 60 mM Tris buffer pH 8.0, Nuclease Free Water) and homogenization buffer. This solution layered to a new tube containing equal volume of iodixanol medium at 29% and centrifuged 13.500g for 20 min at 4°C. Nuclei pellet was gently resuspended in 200 μl of FACS buffer (0.5% BSA, 2mM EDTA) and incubated on ice for 10 min. After centrifugation 800g 5 min 4°C, sample was incubated with anti-NeuN (neuronal marker, 1:500, Anti-NeuN-PE, clone A60 Milli-Mark™) for 40 min. After centrifugation 800g 5 min 4°C, sample was washed with 1X Permeabilization buffer (Foxp3 / Transcription Factor Staining Buffer Set, eBioscience™) and centrifuged 1300g for 5 min, without breaks to improve nuclei recovery. Staining with anti-Pu.1 antibody in 1X Permeabilization buffer (myeloid marker 1:50, Pu.1-AlexaFluor 647, 9G7 Cell Signaling) was performed for 40 min. After a wash with FACS buffer samples were prepared for FACS. Nuclei were FACS-sorted in a BD FACS Aria with a 100-μm nozzle and a sheath pressure 20 psi, operating at ~1000 events per second. Nuclei were sorted into 1.5 ml certified RNAse, DNAse DNA, ATP and Endotoxins tubes containing 100μl of sterile PBS. For detailes on sorted samples see Supplementary Table S1. Sorting purity was >95%. Sorting strategy is depicted in Supplementary Fig 1. Of note, the Double-negative gate is restricted to prevent cross-contamination between cell types. Nuclei suspensions were centrifuged 20 min at 6000g and processed immediately for gDNA extraction with QIAamp DNA Micro Kit (Qiagen) following manufacture instructions. DNA from whole-blood samples was extracted with QIAamp DNA Micro Kit (Qiagen) following manufacture instructions. Flow cytometry data was collected using DiVa 8.0.1 Software. Subsequent analysis was performed with FlowJo_10.6.2. For sorting strategy, see Supplementary Fig 1.
DNA library preparation and sequencing.
DNA samples were submitted to the Integrated Genomics Operation (IGO) at MSKCC for quality and quantity analysis, library preparation and sequencing. DNA quality mas measured with Tapestation 2200. All samples had a DNA Integrity Number (DIN) >6. After PicoGreen quantification, ~200ng of genomic DNA were used for library construction using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with 8 cycles of PCR. After sample barcoding, 2.5ng-1µg of each library were pooled and captured by hybridization with baits specific to either the HEME-PACT (Integrated Mutation Profiling of Actionable Cancer Targets related to Hematological Malignancies) assay, designed to capture all protein-coding exons and select introns of 576 (2.88Mb) commonly implicated oncogenes, tumor suppressor genes 6 and/or HEME/BRAIN-PACT (716 genes, 3.44 Mb, Supplementary Table S2) an expanded panel that included additional custom targets related to neurological diseases including, Alzheimer’s Disease, Parkinson’s Disease, Amyotrophic Lateral Sclerosis (ALS) and others (tableS1) 7-15. To simplify, in the manuscript the combined panel is referred to as ‘BRAIN-PACT’. In Supplementary Table S3, ‘Heme-only’ or ‘Brain-only’ is indicated in the cases for which only one or the other panels were used. Capture pools were sequenced on the HiSeq 4000, using the HiSeq 3000/4000 SBS Kit (Illumina) for PE100 reads. Samples were sequenced to a mean depth of coverage of 1106x (Control samples: 1071x, AD samples 1100x). For detailed information on the sample quality control checks used to avoid potential sample and/or barcode mix-ups and contamination from external DNA, see 6.
Mutation data analysis.
The data processing pipeline for detecting variants in Illumina HiSeq data is as follows. First the FASTQ files are processed to remove any adapter sequences at the end of the reads using cutadapt (v1.6). The files are then mapped using the BWA mapper (bwa mem v0.7.12). After mapping the SAM files are sorted and read group tags are added using the PICARD tools. After sorting in coordinate order the BAM’s are processed with PICARD MarkDuplicates. The marked BAM files are then processed using the GATK toolkit (v 3.2) according to best practices for tumor normal pairs. They are first realigned using ABRA (v 0.92) and then the base quality values are recalibrated with the BaseQRecalibrator. Somatic variants are then called in the processed BAMs using MuTect (v1.1.7) for SNV and ShearwaterML16-18.
muTect (v1.1.7): to identify somatic variants and eliminate germline variants, we run the pipeline as follow: PU.1, DN and Blood samples against matching-NeuN samples, and NeuN samples against matching-PU.1. In addition, we ran all samples against a Frozen-Pool of 10 random genomes. We selected Single Nucleotide Variations (SNVs) [Missense, Nonsense, Splice Site, Splice Regions] that were supported by at least 4 or more mutant reads and with coverage of 50x or more. Fill-out file for each project (~27 samples per sequencing pool), were used to exclude by manual curation, variants with high background noise. This resulted in 428 variants (Missense, Nonsense, Splice_site, Splice_Region).
ShearwaterML, was used to look for low allelic frequency somatic mutations as it has been shown to efficiently call variants present in a small fraction of cells with true positives being ~90%. Briefly, the basis of this algorithm is that is uses a collection of deep-sequenced samples to learn for each site a base-specific error model, by fitting a beta-binomial distribution to each site combining the error rates across all normal samples both the mean error rate at the site and the variation across samples, and comparing the observed variant rate in the sample of interest against this background model using a likelihood-ratio test. For detailed description of this algorithm please refer to 16,17. In our data set, for each cell type (NeuN, DN, PU.1) we used as “normal” a combination of the other cell types, i.e PU.1 vs NeuN+DN, DN vs NeuN+PU.1, NEUN vs PU.1+DN, Blood vs NeuN+DN. Since all samples were processed and sequenced using the same protocol, we expect the background error to be even across samples. More than 400 samples were used as background leading to an average background coverage >400.000x. Resulting variants for each cell type were filtered out as germline if they were present in more than 20% of all reads across samples. Additionally, variants with coverage of less than 50x and more than 35% variant allelic frequency (VAF) were removed from downstream analysis. P-values were corrected for multiple testing using Benjamini & Hochberg’s False Discovery Rate (FDR) 19 and a q-value of cutoff of 0.01 was used to call somatic variants. Variants were required to have a least one supporting read in each strand. Somatic variants within 10bp of an indel were filtered out as they typically reflect mapping errors. We selected Single Nucleotide Variations (SNVs) [Intronic, Intergenic, Missense, Nonsense, Splice Site, Splice Regions] that were supported by at least 4 or more mutant reads and annotated them using VEP. Finally, to reduce the risk of SNP contamination, we excluded variants with a MAF (minor allelic frequency) cutoff of 0.01 using the gnomeAD database. This resulted in 509 SNVs.
We compared the final mutant calls from Muetct1 and ShearwaterML and found that 30% of the events (111 variants) that were called by MuTect1 were also called by ShearwaterML. Overall a total of 826 variants (Supplementary Table S3) were found, with a mean coverage at the mutant site of 668.3X (10% percentile: 276X, 90% percentile: 1181X) and a mean of 29.1 mutant reads (10% percentile: 4, 90% percentile: 52), with 84% of mutated supported by at least 5 mutant reads (Supplementary Table S3). The median allelic frequency was ~1.34% (Supplementary Table S3). Negative results for matching brain negative samples were confirmed in 100% of samples at a mean depth of ~5000x (range 648–23.000x) (Supplementary Table S3), confirming nuclei sorting purity of >95% for PU.1+, DN, and NEUN+ populations.
Validation of variants by droplet-digital-PCR (ddPCR).
We performed validation of ~11% of unique variants (69/760) by droplet-digital PCR (ddPCR) on pre-amplified DNA or on libraries (in the cases where DNA was not sufficient). Around 15% (15/69) of the variants analyzed by ddPCR were called by ShearwaterML, ~44% (34/69) were called by Mutect1 and 40% (24/69) by both ShearwaterML+ Mutect1. Altogether we confirmed 62/69 of variants tested (~90%). In addition, 61 assays (from variants detected in PU.1+ nuclei) were tested in paired cell types isolated from the same brain region). Assays were also run in matching blood when available. The mean depth of ddPCR was ~5000x and mutant counts of 3 or more were considered positive. VAF obtained by ddPCR correlated with original VAF by sequencing (R2 0.93, p<0.0001). For KRAS_G12D: Bio-Rad validated assay (Unique Assay ID: dHsaMDV2510596) and MTOR_Arg1616His_c.4847G>A: Bio-Rad validated assay (Unique Assay ID: dHsaMDV2510596) were used. The remaining assays were designed and ordered through Bio-Rad. For setting-up the right conditions for newly designed assays, cycling conditions were tested to ensure optimal annealing/extension temperature as well as optimal separation of positive from empty droplets. All reactions were performed on a QX200 ddPCR system (Bio-Rad catalog # 1864001). When possible, each sample was evaluated in technical duplicates or quartets. Reactions contained 10ng gDNA, primers and probes, and digital PCR Supermix for probes (no dUTP). Reactions were partitioned into a median of ~31,000 droplets per well using the QX200 droplet generator. Emulsified PCRs were run on a 96-well thermal cycler using cycling conditions identified during the optimization step (95°C 10’; 40–50 cycles of 94°C 30’ and 52–56°C 1’; 98°C 10’; 4°C hold). Plates were read and analyzed with the QuantaSoft sotware to assess the number of droplets positive for mutant DNA, wild-type DNA, both, or neither. ddPCR results are listed in Supplementary Table S3.
Classification of variants.
To classify somatic variants according to their pathogenicity we did as follow: Variants were classified as ‘pathogenic (P-SNV)’ if reported as ‘pathogenic/likely pathogenic’ by ClinVar20 and/or ‘oncogenic/predicted oncogenic/likely oncogenic’ by OncoKb21 (Supplementary Table S3). These two databases report pathogenicity in cancer and other diseases, based on supporting evidence from curated literature (see corresponding citations in Supplementary Table S3). We considered classical-MAPK-pathway genes those reported to be mutated in RASopathies: BRAF, CBL, KRAS, MAP2K1, NF1, PTPN11, SOS1, RIT1, SHOC2, NRAS, RAF1, RASA1, HRAS, MAP2K2,SPRED1 22,23 (Supplementary Table S3).
Quantification of mutational load and statistics.
We defined mutational load or mutational burden as the number of synonymous and non-synonymous somatic single-nucleotide-variant (SNV) per megabase of genome examined 24. Overall, a total of 826 single-nucleotide-variants (SNV) were detected resulting in 0.3 mutations/Mb sequenced. As detailed in the manuscript, the mutational load varies considerably across cell types and patients. To quantify mutational load we took into consideration the panel used for sequencing each sample: HEME-PACT (2.88 Mb) or the extended panel BRAIN-PACT (3.44 Mb) (see Supplementary Table S2). Therefore, the number of mutations was normalized by the number of Mb sequenced for that specific sample. In the cases where we calculated mutational load per patient, we averaged the mutational load of each sample from that patient for a given cell type [(i.e if for one patient, 2 PU.1 samples were sequenced, one from hippocampus and one from superior parietal cortex (with BRAIN-PACT) then the mutational load for PU.1 for that patient is the mean of the mutational load of the 2 PU.1 samples analyzed). For the quantification of ‘pathogenic’ variants, the same analysis is performed, quantifying only variants that are reported as pathogenic by ClinVar and/or OncoKb. Statistical significance was analyzed with GraphPad Prism (v9) and R (3.6.3). Non-parametric tests were used when data did not follow a normal distribution (Normality test: D’Agostino-Pearson and Shapiro-Wilk test). For normally distributed data, unpaired t-test was used to compare two groups and one-way, nested one-way or two-way analyses of variance (ANOVA) were used for comparing more than two groups, as indicated in the Figure legends. For data that did not have a normal distribution, the tests performed were unpaired two-tailed Mann-Whitney U test and Kruskal–Wallis test and Dunn’s test for multiple comparisons. Pearson and Spearman were used for correlation analysis. In Fig. 2G, we used multivariate logistic regression analysis to test if there was an association between Alzheimer’s disease and the presence of pathogenic variants in PU.1+ nuclei. We used Alzheimer’s disease as a dependent variable, and age, sex, and the presence of pathogenic variant/s (Yes/No) as co-variates. In all the statistical tests, significance was considered at P < 0.05. For Venn Diagram plots, we used 25.
Mixed-effects modelling of somatic P-SNV burden.
To evaluate the correlation between P-SNV burden and disease status (non-dementia controls and AD) after adjusting for other factors such as individual donor and age, we performed linear mixed-effects regression modeling using the nlme package in R. In the subsequent analysis, we also tested another framework implemented in the lme4 package and confirmed similar findings. We estimated the linear mixed model via maximum likelihood method with nlminb optimizer. The model included individual donor as a random effect. We also tested if inclusion of other co-variates (i.e., sex, anatomical location of the brain, and source biobank of brain samples) as random effects inproved the overall model fitting via likelihood ratio test. However, none of these co-variates improved the model fitting (P>0.99). Thus, we used a relatively simple model that incorporated disease status and age as fixed effects and donor as random effect. The total explanatory power of the final model is substantial (conditional R^2=0.48). To assess the significance of age or disease status in predicting P-SNV burden, we constructed another model that does not incorporate the variable as fixed effect, and compared the two models via likelihood ratio test. The variable was considered significantly associated with P-SNV burden when the P value was below 0.05 and the AIC increased after removing the variable.
Pathway enrichment analysis of genes target of variants was performed using Metascape 26 and the following ontology sources: KEGG Pathway 27,28, GO Molecular function 29,30, Reactome Gene Sets 29,30 and Canonical Pathways 31. The list of 716 genes from the targeted panel were used as the enrichment background. Terms with a p-value< 0.05, a minimum count of 3, and an enrichment factor > 1.5 (the enrichment factor is the ratio between the observed counts and the counts expected by chance) are shown. p-values are calculated based on the cumulative hypergeometric distribution 32.
Expression of target genes in microglia.
To evaluate the expression levels of the genes identified in this study as target of somatic variants, we consulted a publicly available database (https://www.proteinatlas.org/), and also plotted their expression as determined by RNAseq in 2 studies (Galatro et al. GSE99074 33, and Gosselin et al. 34) (Table S3 and Figure S2). For data from Galatro et al. (GSE99074) 33, normalized gene expression data and associated clinical information of isolated human microglia (N = 39) and whole brain (N = 16) from healthy controls were downloaded from GEO. For data from Gosselin et al. 34, raw gene expression data and associated clinical information of isolated microglia (N = 3) and whole brain (N = 1) from healthy controls were extracted from the original dataset. Raw counts were normalized using the DESeq2 package in R 35.”
Nuclei isolation from frozen brain samples for sn-RNAseq.
For sn-RNAseq studies we only selected samples with a RIN score in whole tissue of 6 or more. All samples were handled and processed under Air Clean PCR Workstation. About 250–400 mg of frozen brain tissues were homogenized with a sterile Dounce tissue grinder using a sterile homogenization buffer to isolate cell nuclei (250 mM Sucrose, 25 mM KCL, 5 mM MgCl2, 10 mM Tris buffer pH 8.0, 0.1% (v/v) Triton X-100, 3 μM DAPI, Nuclease Free Water and 20 U/ml of Superase-In RNase inhibitor and 40 U/ml RNasin ribonuclease inhibitor). Homogenate was filtered in a 40-μm cell strainer and centrifuged 800g 8 min 4°C. To clean-up the homogenate, we performed a iodixanol density gradient centrifugation as follow: pellet was gently mixed 1:1 with iodixanol medium at 50% (50% Iodixanol, 250 mM Sucrose, 150 mM KCL, 30 mM MgCl2, 60 mM Tris buffer pH 8.0, Nuclease Free Water) and homogenization buffer. This solution layered to a new tube containing equal volume of iodixanol medium at 29% and centrifuged 13.500g for 20 min at 4°C. Nuclei pellet was resuspended in FACS buffer with RNAse inhibitors (0.5% BSA, 2mM EDTA, Superase-In RNase inhibitor and 40 U/ml RNasin ribonuclease inhibitor) and centrifuged 800g 5 min, 4 °C. Nuclei pellet was fixed with 90% ice-cold methanol and incubated for 10 min on ice, followed by a centrifugation at 1300g (without brakes, which improves with nuclei recovery after fixation). The pellet was resuspended in permeabilization buffer (6% BSA, Superase-In RNase inhibitor 20 U/mL, RNasin ribonuclease inhibitor 40 U/mL and 0.05% Triton) followed by a centrifugation at 1300g. Sample was incubated with anti-Pu.1 antibody (microglia marker 1:50, Pu.1-AlexaFluor 647, 9G7 Cell Signaling) in permeabilization buffer. After a wash with FACS buffer sample were ready for sorting. Nuclei are FACS-sorted in a BD FACS Aria with a 100-μm nozzle and a sheath pressure 20 psi, operating at ~1000 events per second. Nuclei were sorted into 1.5 ml certified RNAse, DNAse DNA, ATP and Endotoxins tubes containing 100μl of sterile PBS. For each population we sorted >105 nuclei into FACS buffer.
Sn-RNAseq library preparation and sequencing.
The single-nuclei RNA-Seq of FACS-sorted nuclei suspensions was performed on Chromium instrument (10X genomics) following the user guide manual (Reagent Kit 3’ v3.1). Each sample, containing approximately 10,000 nuclei at a final dilution of ~1,000 cells/µl was loaded onto the cartridge following the manual. The individual transcriptomes of encapsulated cells were barcoded during RT step and resulting cDNA purified with DynaBeads followed by amplification per manual guidelines. Next, PCR-amplified product was fragmented, A-tailed, purified with 1.2X SPRI beads, ligated to the sequencing adapters and indexed by PCR. The indexed DNA libraries were double-size purified (0.6–0.8X) with SPRI beads and sequenced on Illumina NovaSeq S4 platform (R1 – 26 cycles, i7 – 8 cycles, R2 – 70 cycles or higher). Sequencing depth was ~200 million reads per sample on average. FASQ files were processed using SEQC pipeline 36 for quality control, mapping to GRCH38 reference genome, and log2 transformation of the data with the default SEQC parameters to obtain the gene-cell count matrix.
Sn-RNAseq analysis.
Seurat v4.0.3 with default parameters was used to perform sctransform (SCT) normalization, integration and Uniform Manifold Approximation and Projection (UMAP) for dimensionality reduction. The FindClusters function was used for cell clustering. To improve clustering, all samples were analyzed in an integrated analysis, based on canonical correlation analysis (CCA). Cell types were annotated using the top 500 DEGs of each cell type in a human cortex database. Data can be accessed at https://weillcornellmed.shinyapps.io/Human_brain/. The removal of doublets using DoubletFinder and cells with high mitochondrial content (>10% mitochondrial RNA) yielded between 6,437 and 9,241 nuclei per patient and sample. Microglia represented 94 ± 3% of total cells. Unique Molecular Identifiers (UMIs) per nucleus and gene count per nucleus were comparable between donors. Integrated_snn at resolution 0.2 outlined 16 microglia clusters. Except for cluster 13 consisting at 97% of cells from the healthy control Control 11_AG, all donors and samples were represented in every cluster. One cluster contained few cells (0.84% of total microglia, for an average of 6.20 ± 1.60% for other clusters) and was marked by a low number of cluster-enriched genes and was excluded from further analyses. For pathway enrichment analysis, genes were pre-ranked using differential expression analysis in SCANPY 37 with Wilcoxon rank-sum method. Statistical analysis were performed using the fgseaMultilevel function in fgsea R package 38 for HALLMARK and KEGG pathways. Gene sets with p-value < 0.05 and adjusted p-value < 0.25 were selected and visualized using ggpubr and ggplot2 39 R package. For the variant analysis of AD52_HIP harboring a KRASA59G (c.176C>G) clone, Integrative Genomics Viewer (IGV) software was used to display sequencing reads at KRAS c.176C (exon 3; GRCh38 chr12:25,227,348). Cells within each cluster were identified based on the 16-digit barcodes from SEQC-aligned reads. Barcodes were converted to the 10X Genomics format and used to sample reads from each cluster within the original BAM file. BAM subsets for each cluster were read with IGV and reads with identical UMIs were filtered out to account for amplification bias.
Whole-Exome-Sequencing and analysis.
Remaining libraries from a selected group of PU.1 and NEUN samples sequenced with BRAIN-PACT (see above) were sequenced by Whole-Exome-Sequencing (WES). Matching NEUN samples were sequenced to extract the germline variants. Around 100 ng of library were captured by hybridization using the xGen Exome Research Panel v2.0 (IDT) according to the manufacturer’s protocol. PCR amplification of the post-capture libraries was carried out for 12 cycles. Samples were run on a NovaSeq 6000 in a PE100 run, using the NovaSeq 6000 S4 Reagent Kit (200 Cycles) (Illumina). Samples were covered to an average of 419X. The data processing pipeline for detecting variants in Novaseq data is as follows. First the FASTQ files are processed to remove any adapter sequences at the end of the reads using cutadapt (v1.6). The files are then mapped using the BWA mapper (bwa mem v0.7.12 ). After mapping the SAM files are sorted and read group tags are added using the PICARD tools. After sorting in coordinate order the BAM’s are processed with PICARD MarkDuplicates. The marked BAM files are then processed using the GATK toolkit (v 3.2) according to the best practices for tumor normal pairs. They are first realigned using ABRA (v 0.92) and then the base quality values are recalibrated with the BaseQRecalibrator. Somatic variants are then called in the processed BAMs using muTect (v1.1.7) for SNV and the Haplotype caller from GATK with a custom post-processing script to call somatic indels. The full pipeline is available here https://github.com/soccin/BIC-variants_pipeline and the post processing code is at https://github.com/soccin/Variant-PostProcess. We selected Single Nucleotide Variants (SNVs) [Missense, Nonsense, Splice Site, Splice Regions] that were supported by at least 8 or more mutant reads, variant allelic frequency above 5% and with coverage of 50x. Annotation was performed using VEP. Finally, to reduce the risk of SNP contamination, we excluded variants with a MAF (minor allelic frequency) cutoff of 0.01 using the genomeAD database. Variants were classified as ‘candidate pathogenic’ when SNV is predicted to affect the protein as determined by PolyPhen-2 (possibly and probably damaging) and SIFT (deleterious) and CADD-MSC (high) and FATHMM-XF (Functional Analysis through Hidden Markov Models (pathogenic) 40 (Supplementary Table S5).
Cell lines
HEK293T cell culture and transfection.
HEK 293T cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (Mediatech, Inc.) supplemented with 10% fetal bovine serum (Sigma) and 1000 IU/ml penicillin, 1000 IU/ml streptomycin.
BV2 microglial cell line.
BV2 murine microglial cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) High Glucose medium (Gibco), Glutamax (Gibco), sodium pyruvate, 1% non-essential amino acids (Invitrogen) and 10% heat-inactivated fetal bovine serum (FBS, EMD Millipore). For MAPK activation experiments, cells were treated with M-CSF1 100 ng/ml for 5 min.
MAC cell lines.
Mouse primary CSF-1 dependent macrophages immortalized with the SV-U19–5 retrovirus 41 were a gift of Dr. E. R. Stanley (Albert Einstein College of Medicine, Bronx, NY). They were cultured in RPMI 1640 medium with Glutamax (Gibco), 10 % heat-inactivated fetal bovine serum (FBS, EMD Millipore) and 100 ng/mL recombinant CSF-1 (gift from Dr. E. R. Stanley). Growth medium was renewed every second day. When confluency reached 80%, cells were passaged by cell scraping and plated at 5 X 104 cells/ cm2 in tissue culture treated plates. For signaling pathway analyses, cell proliferation assays or collection for RNA sequencing, cells were plated one day prior at 5 X 104 cells/ cm2 in medium containing 10 ng/mL CSF-1 for lines expressing wild-type (WT) and mutant CBL, RIT1 and KRAS proteins and 100 ng/mL CSF-1 for lines expressing WT and mutant PTPN11 proteins. Cells were grown at 37°C and 5% CO2.
Human induced pluripotent stem cell (hiPSC) culture.
Human induced Pluripotent Stem Cell (hiPSC) lines were derived from peripheral blood mononuclear cells (PBMCs) of a healthy donor. Written informed consent was obtained according to the Helsinki convention. The study was approved by the Institutional Review Board of St Thomas’ Hospital; Guy’s hospital; the King’s College London University; the Memorial Sloan Kettering Cancer Center and by the Tri- institutional (MSKCC, Weill-Cornell, Rockefeller University) Embryonic Stem Cell Research Oversight (ESCRO) Committee. hiPSC were derived using Sendai viral vectors (ThermoFisher Scientific; A16517). Newly derived hiPSC clones were maintained in culture for 10 passages (2–3 months) to remove any traces of Sendai viral particles. Over 90% of hiPSCs in the derived lines expressed high levels of the pluripotency markers NANOG and OCT4 by flow cytometry. The C12 hiPSC WT line was engineered to carry a CBL p.C404Y, c.1211G>A heterozygous variant at the endogenous CBL locus. HiPSCs of passage 25–35 were cultured on confluent irradiated CF1 mouse embryo fibroblasts (MEFs, Gibco) in hiPSC medium consisting of knock-out DMEM (Invitrogen), 10% knock-out-Serum Replacement (Invitrogen), 2 mM L-glutamine (Gibco), 100 U/mL penicillin-streptomycin (Invitrogen), 1% non-essential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (R&D). hiPSC medium was supplement with 10 ng/mL bFGF (PeproTech) and changed every second day. Two days before culture with hiPSCs, MEFs were plated at 20,000 cells/cm2 in DMEM supplemented with 10 % heat-inactivated fetal bovine serum (FBS, EMD Millipore), 100 U/mL penicillin-streptomycin (Invitrogen), 1% non-essential amino acids (Invitrogen) and 0.1 mM β-mercaptoethanol (R&D Systems) on 150 mm tissue culture plates coated with 0.1% gelatin (Sigma). hiPSCs were passaged weekly with 250 U/mL collagenase type IV (ThermoFisher Scientific) at a 1:4 to 1:6 ratio onto MEF cells in hiPSC medium supplemented with 10 µM Rock inhibitor (Y-27632 dihydrochloride, Sigma). Cells were maintained at 37°C and 5% CO2 and they were routinely tested for mycoplasma and periodically assessed for genomic integrity by karyotyping. Microglia-like cells were obtained from hiPSCs using an embryoid body (EB)-based protocol as previously described 42. Briefly, hiPSC were loosened with 250 U/mL collagenase type IV (ThermoFisher Scientific) and lifted with cell scraping. For EBs formation, hiPSC colonies were transferred to suspension plates on an orbital shaker in hiPSC medium supplemented with 10 µM Rock inhibitor (Y-27632 dihydrochloride, Sigma). After 6 days, EBs were transferred to 6-wells tissue culture treated plates in STEMdiff APEL 2 medium (Stem Cell Technology) with 5% Protein Free Hybridoma Media (Gibco), 100 U/mL penicillin-streptomycin (Invitrogen), 25 ng/mL IL-3 (Peprotech) and 50 ng/mL CSF-1 (Peprotech). Microglia-like cells were harvested every week from the supernatant of EBs cultures. Collected microglia-like cells were used immediately for signaling pathway analyses or plated for 6–7 days in RPMI 1640 medium with Glutamax supplement (Gibco), 10 % heat-inactivated fetal bovine serum (FBS, EMD Millipore) and 100 ng/mL human recombinant CSF-1 (Peprotech) in tissue culture plates for cytology, flow cytometry, RNA sequencing and supernatant analyses of cytokines release. Microglia like cells differentiation was monitored by May-Grunwald Giemsa staining and flow cytometry analyses of myeloid markers.
Plasmids used in in-vitro studies (HEK293, BV2, and MAC lines).
The expression vectors for Flag-tagged CHK2 kinase and RIT1 were from Sino Biological and Origene, respectively. The vector encoding pcDNA3-HA-tagged c-Cbl was a kind gift from Dr. Nicholas Carpino (Stony Brook). RIT1M90I, RIT1F82L, CBLI383M, CBLC404Y, CBLC416S, CBLC384Y, CBLY371H were generated by site-directed mutagenesis using the QuikChange Kit (Agilent). pHAGE_puro was a gift from Christopher Vakoc (Addgene plasmid # 118692; http://n2t.net/addgene:118692; RRID:Addgene_118692) 43. pHAGE-KRAS was a gift from Gordon Mills & Kenneth Scott (Addgene plasmid # 116755; http://n2t.net/addgene:116755; RRID: Addgene_116755) 44. pHAGE-PTPN11 was a gift from Gordon Mills & Kenneth Scott (Addgene plasmid # 116782; http://n2t.net/addgene:116782; RRID: Addgene_116782) 44. pHAGE-PTPN11-T73I was a gift from Gordon Mills & Kenneth Scott (Addgene plasmid # 116647; http://n2t.net/addgene:116647; RRID: Addgene_116647) 44. pDONR223_KRAS_p.A59G was a gift from Jesse Boehm & William Hahn & David Root (Addgene plasmid # 81662 ; http://n2t.net/addgene:81662 ; RRID:Addgene_81662) 45, Phage-CBL, Phage-CBLI383M, Phage-CBLC404Y, Phage-CBLC416S, Phage-RIT1, Phage-RIT1M90I and Phage-RIT1F82L and Phage-KRASA59G were generated by Azenta Life Sciences via a PCR cloning approach. pHAGE-CBLC384Y plasmid was generated at Azenta Life Science by targeted mutagenesis of pHAGE-CBL.
Generation of mutant lines.
HEK293 were transfected 24 hours after plating with 2.5 µL of Mirus Transit LT1 per µg of DNA. Cells were harvested and lysed 48 hrs after transfection using a buffer containing 25 mM Tris, pH 7.5, 1 mM EDTA, 100 mM NaCl, 1% NP-40, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 200 µM PMSF, and 0.2 mM Na3VO4. For EGF stimulation, the media was replaced 24 hours after transfection with DMEM containing 1% FBS and antibiotics. After a further 24 hours in this starvation media, the cells were stimulated with 50ng/ml EGF for 5 minutes at 37ºC. Lentiviral production and transduction of BV2 and MAC cell lines. For BV2 cell line, cells were transduced for 24 hours without the presence of Vpx VLPs and selected with 2.5 µg/mL puromycin (Fisher Scientific). For ‘MAC’ lines, Vpx-containing virus-like particles (Vpx VLPs) were produced by transfection of HEK293T cells with 4.8 ug VSV-g plasmid and 31.2 ug pSIV3/Vpx plasmid, a gift from Dr. M. Menager (Imagine Institute, Paris, France) using TransIT-293 Transfection Reagent (Mirus Bio, Fisher Scientific). Forty-eight hours after transfection, the supernatant containing Vpx VLPs was collected and used immediately for lentiviral transduction of macrophages. Viral supernatants were obtained by transfection of HEK293T cells using X-tremeGENE HP DNA Transfection Reagent (Sigma). Packaging vectors used were psPAX2 (gift from Didier Trono Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID: Addgene_12260) and pMD2.G (gift from Didier Trono, Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259). Cells were transduced for 24 h in presence of Vpx VLPs. Transduced macrophages were selected with 5 µg/mL puromycin (Fisher Scientific).
Generation of the CBL+/C404Y and isogenic WT hiPSC lines.
The CBLC404Y (c.1211 G>A) variant was inserted at the endogenous locus in the C12 WT hiPSC using Cytidine base editing (CBE) with CBE enzyme BE3-FNLS 46. Briefly, the sgRNA for CBE was designed to target the non-coding strand and introduce the position 6 “C-to-T” conversion, to create the G-to-A conversion on the coding strand. The sgRNA target sequence was cloned into the pSPgRNA (Addgene plasmid # 47108) 47 to make the gene targeting construct. To introduce the CBL C404Y variants, the WT hiPSC (C12) were dissociated using Accutase (Innovative Cell Technologies) and electroporated (1 x106 cells per reaction) with 4 µg sgRNA-construct plasmid and 4 µg CBE enzyme coding vector BE3-FNLS (Addgene plasmid # 112671) 46 using Lonza 4D-Nucleofector and the Nucleofector solution (Lonza V4XP-3034) following our previously reported protocol 48. The cells were then seeded, and 4 days later, the hiPSC were dissociated into single cells by Accutase and re-plated at a low density (4 per well in 96-well plates) to get the single-cell clones. Ten days later, individual colonies were picked, expanded and analyzed by PCR and DNA sequencing to identify the clones carried the desired CBLC404Y heterozygous variant and the isogenic WT control clones. The sgRNA target, PCR and sequencing primers are listed below.
| sgRNA target | PCR-Forward primer | PCR-Reverse primer (used for sequencing) | |
|---|---|---|---|
| CBLC404Y | TAAGACAGGATGTGCACATG | TGGGCTCCACATTCCAACTA | GCCCTGACCTTCTGATTCCT |
Western Blotting
For HEK293 cells, lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with the appropriate antibodies. Horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) and Western blotting substrate (Thermo) were used for detection. For anti-Cdc42 immunoprecipitation experiments, cell lysates (1 mg total protein) were incubated overnight with 1 µg of anti-Cdc42 antibody (Santa Cruz) and 25 µL of protein A agarose (Roche) at 4°C. Anti-Flag immunoprecipitations were done with anti-Flag M2 affinity resin (Sigma). The beads were washed three times with lysis buffer, then eluted with SDS-PAGE buffer and resolved by SDS-PAGE. The proteins were transferred to PVDF membrane for Western blot analysis. Antibodies used are Phospho-p44/42 MAPK (pErk 1/2) (Thr202/Tyr204) is from Cell Signaling #4370, total p44/42 MAPK (Erk1/2) is from Cell Signaling #9102, HA tag from Millipore # 05–904, Flag antibody is from Sigma (#A8592), pCHEK2 (T383) antibody is from Abcam, #ab59408, Cdc42 antibody is from Santa Cruz (#sc87) and Anti-γ-Tubulin antibody (Sigma T6557). For Immunoprecipitation Kinase assay in HEK293T cells, cell lysates (1 mg protein) were incubated overnight with 30 µL of anti-Flag M2 affinity resin on a rotator at 4°C, then washed three times with Tris-buffered saline (TBS). A portion of each sample was eluted with SDS-PAGE sample buffer and analyzed by anti-Flag Western blotting. The remaining sample was used for a radioactive kinase assay. The immunoprecipitated proteins were incubated with 25 µL of reaction buffer (30 mM Tris, pH 7.5, 20 mM MgCl2, 1 mg/mL BSA, 400 µM ATP), 650 µM CHKtide peptide (KKKVRSGLYRSPSMPENLNRPR, SignalChem), and 50 – 100 cpm/pmol of [γ32-P] ATP at 30°C for 15 minutes. The reactions were quenched using 45 µL of 10% trichloroacetic acid. The samples were centrifuged and 30 µL of the reaction was spotted onto Whatman P81 cellulose phosphate paper. After washing with 0.5% phosphoric acid, incorporation of radioactive phosphate into the peptide was measured by scintillation counting. For MAC lines and hiPSC-derived cells, cell lysates obtained with RIPA buffer + 1:1000 Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific) were sonicated 3 times for 30sec at 4°C (Bioruptor, Diagenode). Protein quantification of supernatant was done with Precision Red Advanced Protein Assay (Cytoskeleton). Proteins were boiled for 5 min at 95°C in NuPAGE LDS sample buffer (Invitrogen) and separated in NuPAGE 4%–12% Bis-Tris Protein Gel (Invitrogen) in NuPAGE MES SDS Running Buffer (Invitrogen). Electrophoretic transfer to a nitrocellulose membrane (ThermoFisher Scientific) was done in NuPAGE Transfer Buffer (Invitrogen). Blocking was performed for 60 min in TBS-T + 5% nonfat milk (Cell Signaling) and incubated with primary antibodies at 4°C: rabbit anti-p44/42 MAPK (ERK1/2) (Cell Signaling; 1:1000); rabbit anti-P-p44/42 MAPK (Cell Signaling, 1:1000); rabbit anti-c-CBL (Cell signaling, 1:1000); rabbit anti-RIT1 (Abcam, 1:1000); mouse anti-KRAS (clone 3B10–2F2, Sigma, 1 μg/mL); mouse anti-Actin (clone MAB1501, Sigma, 1:10,000). Primary antibodies were detected using the secondary anti-rabbit IgG HRP-linked (Cell Signaling, 1:1000) or the anti-mouse IgG HRP-linked (Cell Signaling, 1:1000) were used to detect primary antibodies, with SuperSignal™ West Femto Chemiluminescent Substrate (ThermoFischer Scientific) using a ChemiDoc MP Imaging System (Bio-Rad). pERK/ERK ratios were measured with ImageJ software.
DNA/ RNA isolation, dd-PCR and RTqPCR in MAC lines and hiPSC-derived cells.
Genomic DNA was extracted using QIAamp DNA Micro Kit (50) (Qiagen), following the manufacturer’s instructions. Total RNA was extracted using RNeasy Mini kit (Qiagen), following the manufacturer’s instructions. cDNA was generated by reverse transcription using Invitrogen SuperScript IV Reverse Transcriptase (Invitrogen) with oligo(dT) primers. The TaqMan gene expression assays used were c-CBL FAM (Hs01011446_m1), CBLb FAM (Hs00180288_m1) and GAPDH VIC (Hs02786624_g1) (ThermoFisher Scientific). RT-qPCR was performed using Applied Biosystems TaqMan Fast Advanced Master Mix (ThermoFisher Scientific) and a QuantStudio 6 Flex Real-Time PCR System (ThermoFisher Scientific). The results were normalized to GAPDH. For droplet PCR analyses, assays specific for the detection of I383M, C384Y, C404Y and C416S in CBL and F82L and M90I in RIT1, A59G in KRAS and corresponding WT sequences (listed below) were obtained from Bio-Rad. Cycling conditions were tested to ensure optimal annealing/extension temperature as well as optimal separation of positive from empty droplets. Optimization was done with a known positive control. After PicoGreen quantification, 2.6–9 ng gDNA or cDNA were combined with locus-specific primers, FAM- and HEX-labeled probes, HaeIII, and digital PCR Supermix for probes (no dUTP). All reactions were performed on a QX200 ddPCR system (Bio-Rad catalog # 1864001) and each sample was evaluated in technical duplicates. Reactions were partitioned into a median of ~19,000 droplets per well using the QX200 droplet generator. Emulsified PCRs were run on a 96-well thermal cycler using cycling conditions identified during the optimization step (95°C 10’; 40 cycles of 94°C 30’ and 52–55°C 1’; 98°C 10’; 4°C hold). Plates were read and analyzed with the QuantaSoft software to assess the number of droplets positive for mutant or wild-type DNA.
| Assay Name | Assay ID |
|---|---|
| CBL_I383M | dHsaMDS675699482 |
| CBL_C384Y | dHsaMDS386449640 |
| CBL_C404Y | dHsaMDS437459772 |
| CBL_mRNA_C404Y | dMDS334857054 |
| CBL_C416S | dHsaMDS613275900 |
| RIT1_ F82L | dMDS959028273 |
| RIT1_M90I | dHsaMDS133045056 |
| KRAS_A59G | dHsaMDS581417660 |
Flow cytometry analyses for surface antigens CSF1-R, CD11b, MRC1, α5β3, CD11c, Tim4, HLA-DR, CD45, CD14, NGFR, EGFR, CD36 and SIRPα were performed using PE-conjugated anti-CD115 (CSF1-R) (clone 9–4D2, BD Pharmingen), PE/Cy7-conjugated anti-CD11b (clone ICRF44, Biolegend), Alexa Fluor 488-conjugated anti-CD206 (MRC1) (clone 19.2, ThermoFisher Scientific), PE-conjugated anti-integrin α5β3 (clone 23C6, R&D systems), PE/Cy5-conjugated anti-CD11c (Clone B-ly6, BD Pharmigen), APC-conjugated anti-Tim4 (Clone 9F4, BioLegend), PE/Cy7-conjugated anti-HLA-DR (clone G46–6, BD Pharmigen), BV650-conjugated anti-CD45 (clone HI30, BD Horizon), APC/Cy7-conjugated anti-CD14 (clone M5E2, Biolegend), PE-conjugated anti-NGFR (clone ME20.4, eBioscience), Alexa Fluor 647-conjugated anti-EGFR (clone EGFR.1, BD Pharmigen), APC/Cy7-conjugated anti-CD36 (clone 5–271, BioLegend) and APC-conjugated anti-CD172a (SIRPα) (Clone: 15 414, ThermoFisher Scientific) antibodies. Iba1 expression was detected following fixation and permeabilization of macrophages using BD Cytofix/Cytoperm solution (BD Pharmingen). Cells were marked with Zombie Violet Viability (Biolegend). After incubation with FcR Blocking Reagent (Miltenyi Biotec), cells were stained with Alexa Fluor 555-conjugated anti-Iba1 antibody (clone E4O4W, Cell Signaling). Flow cytometry was performed using a BD Biosciences LSR Fortessa flow cytometer with Diva software. Data were analyzed using FlowJo (BD Biosciences LLC).
Cell proliferation analyses.
For hiPSC-derived cells, cell suspension was filtered through a 100 µm nylon mesh (Corning) and marked with Zombie Violet Viability (Biolegend). After incubation with FcR Blocking Reagent (Miltenyi Biotec), surface receptors were labelled with PE/Cy7-conjugated anti-CD11b (clone ICRF44, Biolegend), Alexa Fluor 488-conjugated anti-CD206 (MRC1) (clone 19.2, ThermoFisher Scientific), BV650-conjugated anti-CD45 (clone HI30, BD Horizon), APC/Cy7-conjugated anti-CD14 (clone M5E2, Biolegend) prior to EdU detection. For proliferation studies in the mouse macrophage cell lines, macrophages were incubated with 10 µM EdU (ThermoFischer Scientific) for 2 hours at 37°C and collected by cell scraping and marked with Zombie Violet Viability (Biolegend) prior to EdU detection. EdU detection was performed using the Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (ThermoFischer Scientific), following manufacturer’s instructions. hiPSC-derived macrophages were analyzed using a BD Biosciences Aria III cell sorter and macrophages were identified as CD11b+CD45+CD14+MRC1+. The macrophage cell lines were analyzed using a BD Biosciences LSR Fortessa flow cytometer. Data were analyzed using FlowJo 10.6 (BD Biosciences LLC).
Enzyme-linked immunosorbent assay.
Supernatants of iPSC-derived microglia-like cells were analyzed for human inflammatory cytokines IL-6, TNFα, IL-1β, IFNγ and for the complement C3 and complement Factor H by Enzyme-linked immunosorbent assay (ELISA) at Eve Technologies (Calgary, AB).
Bulk RNA sequencing (RNAseq).
Three biological replicates were processed for each condition/cell line. In view of RNA sequencing, phase separation in cells lysed in 1 mL TRIzol Reagent (ThermoFisher Scientific) was induced with 200 µL chloroform and RNA was extracted from the aqueous phase using the miRNeasy Mini Kit (Qiagen) on the QIAcube Connect (Qiagen) according to the manufacturer’s protocol with 350 µL input, or using the MagMAX mirVana Total RNA Isolation Kit (ThermoFisher Scientific) on the KingFisher Flex Magnetic Particle Processor (ThermoFisher Scientific) according to the manufacturer’s protocol with 350 µL input. Samples were eluted in 30 µL RNase-free water. After RiboGreen quantification and quality control by Agilent BioAnalyzer, 231–500 ng of total RNA with RIN values of 9.4–10 underwent polyA selection and TruSeq library preparation according to instructions provided by Illumina (TruSeq Stranded mRNA LT Kit, Illumina), with 8 cycles of PCR. Samples were barcoded and run on a NovaSeq 6000 in a PE100 run, using the NovaSeq 6000 S4 Reagent Kit (200 Cycles) (Illumina). An average of 90 million paired reads was generated per sample. Ribosomal reads represented 0–1.6% of the total reads generated and the percent of mRNA bases averaged 79%.
Bulk RNAseq analysis.
FastQ files of 2x100bp paired-end reads were quality checked using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, 2012). Samples with high quality reads (Phred score >= 30) were aligned to the Mus musculus genome (GRCm38.80) for the MAC lines or Homo sapiens (assembly GRCh38.p14) for the IPSCs lines using STAR aligner. For the MAC lines, we computed the expression count matrix from the mapped reads using HTSeq (www-huber.embl.de/users/anders/HTSeq) and one of several possible gene model databases. The raw count matrix generated by HTSeq are then be processed using the R/Bioconductor package DESeq (www-huber.embl.de/users/anders/DESeq) which is used to both normalize the full dataset and analyze differential expression between sample groups. For the ISPCs line dataset, gene quantification was performed using feature counts from the Subread package in R. Gene expression levels were normalized and log2 transformed using the Trimmed Mean of M-values (TMM) method and differential expression analysis was performed using the edgeR package in R. For hiPSC derived cells, gene-set enrichment analysis (GSEA) (Hallmark, KEGG, GO, REACTOME) were performed using the fgsea package in R on a pre-ranked list (formula: sign(ogFC) * -log10(PValue)) on all expressed genes in the dataset. For the MAC cell lines dataset, GSEA was performed using gsea4.3.2 for KEGG and HALLMARK canonical pathways in MSigDB v 7.5.1. Significant genesets were selected based on an FDR <= 0.25. For lists of differentially expressed genes, genes were selected with controlled False Positive Rate (B&H method) at 5% (FDR <= 0.05). Genes were considered upregulated/downregulated for log2 fold change> 1.5 or <-1.5.
Statistical analysis.
Statistical methods are detailed in the corresponding sections above (Quantification of mutational load and statistics, Bulk RNAseq analysis, Sn-RNAseq analysis) and in the Fig. legends. P values of 0.05 and adj. P values (FDR) of 0.25 are considered significant unless otherwise specified.
Supplementary Material
Acknowledgements:
This study was supported by grants from NIH: P30 CA008748 MSKCC core grant, 1R01NS115715-01, 1 R01 HL138090-01, and 1 R01 AI130345-01 to FG, and Basic and Translational Immunology Grants from Ludwig Center for Cancer Immunotherapy and from Cycle for Survival to FG. RV was supported by the 2018 AACR-Bristol-Myers Squibb Fellowship for Young Investigators in Translational Immuno-oncology, Grant Number 18-40-15-VICA. LW was supported by NYSTEM training award C32559GG and a Charles H Revson fellowship. Sequencing costs and analysis were covered in part by a SRA between Third Rock venture and MSKCC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. MAC mouse cell lines were kindly provided by Dr Richard E Stanley. Code for Shearwater ML and for single cell mRNA genotyping were provided by Dr Inigo Martincorena by Dr Noor Sohail respectively.
Footnotes
Competing interests. FG has been a paid consultant (no equity) to Third Rock Ventures from 2018 to 2020. Sequencing costs and analysis in this study were covered in part by a SRA between Third Rock venture and MSKCC. This work led to patents PCT/US2022/037893/WO2023004054A1 ‘Methods and compositions for the treatment of alzheimer’s disease’ by MSKCC and PCT/US2018/047964 ‘Kinase mutation-associated neurodegenerative disorders by MSKCC’.
Code availability.
All code used in this study has been previously published as referenced in the method section above.
Data availability.
DNA sequencing data processed for selection of somatic variants are available for all patients and samples in Supplementary Table S3. Raw DNA sequencing data (FASTQ files) from targeted-deep sequencing are deposited in dbGaP under project accession number phs002213.v1.p1, for samples where patient-informed consent for public deposition of DNA sequencing data was obtained. Bulk RNAseq raw data are deposited in GEO (number pending). Sn-RNAseq raw data are deposited in GEO (number pending) and as an interactive analysis web tool accessible at https://weillcornellmed.shinyapps.io/Human_brain/.
REFERENCES
- 1.Hebert L. E., Weuve J., Scherr P. A. & Evans D. A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80, 1778–1783 (2013). 10.1212/WNL.0b013e31828726f5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Association, A. s. 2019. Alzheimer’s Disease Facts and Figures. https://www.alz.org/media/documents/alzheimers-facts-and-figures-2019-r.pdf (2019). [Google Scholar]
- 3.Lanoiselee H. M. et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 14, e1002270 (2017). 10.1371/journal.pmed.1002270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goate A. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 (1991). 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- 5.Chartier-Harlin M. C. et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844–846 (1991). 10.1038/353844a0 [DOI] [PubMed] [Google Scholar]
- 6.Levy-Lahad E. et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977 (1995). 10.1126/science.7638622 [DOI] [PubMed] [Google Scholar]
- 7.Levy-Lahad E. et al. A familial Alzheimer’s disease locus on chromosome 1. Science 269, 970–973 (1995). 10.1126/science.7638621 [DOI] [PubMed] [Google Scholar]
- 8.Rogaev E. I. et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778 (1995). 10.1038/376775a0 [DOI] [PubMed] [Google Scholar]
- 9.Saunders A. M. et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472 (1993). 10.1212/wnl.43.8.1467 [DOI] [PubMed] [Google Scholar]
- 10.Murrell J. R. et al. Association of apolipoprotein E genotype and Alzheimer disease in African Americans. Arch Neurol 63, 431–434 (2006). 10.1001/archneur.63.3.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sando S. B. et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol 8, 9 (2008). 10.1186/1471-2377-8-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lumsden A. L., Mulugeta A., Zhou A. & Hyppönen E. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine 59, 102954 (2020). 10.1016/j.ebiom.2020.102954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonsson T. et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368, 107–116 (2013). 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerreiro R. et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127 (2013). 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McQuade A. & Blurton-Jones M. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J Mol Biol 431, 1805–1817 (2019). 10.1016/j.jmb.2019.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnaud L. et al. APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-kappaB activation. Cell Rep 40, 111200 (2022). 10.1016/j.celrep.2022.111200 [DOI] [PubMed] [Google Scholar]
- 17.Serrano-Pozo A. et al. Effect of APOE alleles on the glial transcriptome in normal aging and Alzheimer’s disease. Nature Aging 1, 919–931 (2021). 10.1038/s43587-021-00123-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez G. A., Tai L. M., LaDu M. J. & Rebeck G. W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. Journal of Neuroinflammation 11, 111 (2014). 10.1186/1742-2094-11-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nott A. et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). 10.1126/science.aay0793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krasemann S. et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581. e569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathys H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keren-Shaul H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290. e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Miller M. B., Reed H. C. & Walsh C. A. Brain Somatic Mutation in Aging and Alzheimer’s Disease. Annu Rev Genomics Hum Genet 22, 239–256 (2021). 10.1146/annurev-genom-121520-081242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martincorena I. et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015). 10.1126/science.aaa6806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martincorena I. & Campbell P. J. Somatic mutation in cancer and normal cells. Science 349, 1483–1489 (2015). 10.1126/science.aab4082 [DOI] [PubMed] [Google Scholar]
- 26.Behjati S. et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425 (2014). 10.1038/nature13448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McConnell M. J. et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 356 (2017). 10.1126/science.aal1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keogh M. J. et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat Commun 9, 4257 (2018). 10.1038/s41467-018-06331-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei W. et al. Frequency and signature of somatic variants in 1461 human brain exomes. Genet Med 21, 904–912 (2019). 10.1038/s41436-018-0274-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park J. S. et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat Commun 10, 3090 (2019). 10.1038/s41467-019-11000-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Gama A. M. et al. Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep 21, 3754–3766 (2017). 10.1016/j.celrep.2017.11.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khoshkhoo S. et al. Contribution of Somatic Ras/Raf/Mitogen-Activated Protein Kinase Variants in the Hippocampus in Drug-Resistant Mesial Temporal Lobe Epilepsy. JAMA Neurol 80, 578–587 (2023). 10.1001/jamaneurol.2023.0473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh H. Y. et al. BRAF somatic mutation contributes to intrinsic epileptogenicity in pediatric brain tumors. Nat Med 24, 1662–1668 (2018). 10.1038/s41591-018-0172-x [DOI] [PubMed] [Google Scholar]
- 34.Lim J. S. et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nature Medicine 21, 395–400 (2015). 10.1038/nm.3824 [DOI] [PubMed] [Google Scholar]
- 35.Poduri A. et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48 (2012). 10.1016/j.neuron.2012.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolaev S. I. et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N Engl J Med 378, 250–261 (2018). 10.1056/NEJMoa1709449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mass E. et al. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 549, 389–393 (2017). 10.1038/nature23672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Askew K. et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep 18, 391–405 (2017). 10.1016/j.celrep.2016.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reu P. et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep 20, 779–784 (2017). 10.1016/j.celrep.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bian Z. et al. Deciphering human macrophage development at single-cell resolution. Nature (2020). 10.1038/s41586-020-2316-7 [DOI] [PubMed] [Google Scholar]
- 41.Kim J.-S. et al. Monocyte-derived microglia with Dnmt3a mutation cause motor pathology in aging mice. bioRxiv, 2023.2011.2016.567402 (2023). 10.1101/2023.11.16.567402 [DOI] [Google Scholar]
- 42.Frank S. A. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci U S A 107 Suppl 1, 1725–1730 (2010). 10.1073/pnas.0909343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evrony G. D. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012). 10.1016/j.cell.2012.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng D. T. et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17, 251–264 (2015). 10.1016/j.jmoldx.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durham B. H. et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 25, 1839–1842 (2019). 10.1038/s41591-019-0653-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bras J., Guerreiro R. & Hardy J. Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci 13, 453–464 (2012). 10.1038/nrn3271 [DOI] [PubMed] [Google Scholar]
- 47.Renton A. E., Chiò A. & Traynor B. J. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17, 17–23 (2014). 10.1038/nn.3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karch C. M., Cruchaga C. & Goate A. M. Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26 (2014). 10.1016/j.neuron.2014.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karch C. M. & Goate A. M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77, 43–51 (2015). 10.1016/j.biopsych.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner M. R. et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 12, 310–322 (2013). 10.1016/s1474-4422(13)70036-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrari R. et al. A genome-wide screening and SNPs-to-genes approach to identify novel genetic risk factors associated with frontotemporal dementia. Neurobiol Aging 36, 2904.e2913-2926 (2015). 10.1016/j.neurobiolaging.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kouri N. et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun 6, 7247 (2015). 10.1038/ncomms8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scholz S. W. & Bras J. Genetics Underlying Atypical Parkinsonism and Related Neurodegenerative Disorders. Int J Mol Sci 16, 24629–24655 (2015). 10.3390/ijms161024629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nalls M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46, 989–993 (2014). 10.1038/ng.3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chakravarty D. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017 (2017). 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landrum M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–985 (2014). 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martincorena I. et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). 10.1126/science.aau3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaiswal S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498 (2014). 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Genovese G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487 (2014). 10.1056/NEJMoa1409405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaiswal S. & Ebert B. L. Clonal hematopoiesis in human aging and disease. Science 366 (2019). 10.1126/science.aan4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bouzid H. et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. Nature Medicine (2023). 10.1038/s41591-023-02397-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rauen K. A. The RASopathies. Annu Rev Genomics Hum Genet 14, 355–369 (2013). 10.1146/annurev-genom-091212-153523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schnittger S. et al. Molecular analyses of 15,542 patients with suspected BCR-ABL1-negative myeloproliferative disorders allow to develop a stepwise diagnostic workflow. Haematologica 97, 1582–1585 (2012). 10.3324/haematol.2012.064683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernandes M. S. et al. Novel oncogenic mutations of CBL in human acute myeloid leukemia that activate growth and survival pathways depend on increased metabolism. J Biol Chem 285, 32596–32605 (2010). 10.1074/jbc.M110.106161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sargin B. et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 110, 1004–1012 (2007). 10.1182/blood-2007-01-066076 [DOI] [PubMed] [Google Scholar]
- 66.Dunbar A. J. et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res 68, 10349–10357 (2008). 10.1158/0008-5472.CAN-08-2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernard V., Gebauer N., Dinh T., Stegemann J., Feller A. C. & Merz H. Applicability of next-generation sequencing to decalcified formalin-fixed and paraffin-embedded chronic myelomonocytic leukaemia samples. Int J Clin Exp Pathol 7, 1667–1676 (2014). [PMC free article] [PubMed] [Google Scholar]
- 68.Loh M. L. et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114, 1859–1863 (2009). 10.1182/blood-2009-01-198416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grand F. H. et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 113, 6182–6192 (2009). 10.1182/blood-2008-12-194548 [DOI] [PubMed] [Google Scholar]
- 70.Klampfl T. et al. Complex patterns of chromosome 11 aberrations in myeloid malignancies target CBL, MLL, DDB1 and LMO2. PLoS One 8, e77819 (2013). 10.1371/journal.pone.0077819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niemeyer C. M. et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42, 794–800 (2010). 10.1038/ng.641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Javadi M., Richmond T. D., Huang K. & Barber D. L. CBL linker region and RING finger mutations lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN. J Biol Chem 288, 19459–19470 (2013). 10.1074/jbc.M113.475087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ogawa S. Genetics of MDS. Blood 133, 1049–1059 (2019). 10.1182/blood-2018-10-844621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gomez-Segui I. et al. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia 27, 1943–1946 (2013). 10.1038/leu.2013.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim E. et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discov 6, 714–726 (2016). 10.1158/2159-8290.CD-16-0160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Niihori T. et al. Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J Hum Genet 50, 192–202 (2005). 10.1007/s10038-005-0239-7 [DOI] [PubMed] [Google Scholar]
- 77.Soblet J., Limaye N., Uebelhoer M., Boon L. M. & Vikkula M. Variable Somatic TIE2 Mutations in Half of Sporadic Venous Malformations. Mol Syndromol 4, 179–183 (2013). 10.1159/000348327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okeyo-Owuor T. et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29, 909–917 (2015). 10.1038/leu.2014.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith M. A. et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat Cell Biol 21, 640–650 (2019). 10.1038/s41556-019-0314-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choi J. et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res 27, 253–262 (2014). 10.1111/pcmr.12197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heim R. A. et al. Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Hum Mol Genet 4, 975–981 (1995). 10.1093/hmg/4.6.975 [DOI] [PubMed] [Google Scholar]
- 82.Bollag G. et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 12, 144–148 (1996). 10.1038/ng0296-144 [DOI] [PubMed] [Google Scholar]
- 83.Song X., Ma F. & Herrup K. Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J Neurosci 39, 6378–6394 (2019). 10.1523/JNEUROSCI.0774-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zannini L., Delia D. & Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 6, 442–457 (2014). 10.1093/jmcb/mju045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fang Y. et al. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J 23, 3164–3174 (2004). 10.1038/sj.emboj.7600315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Haferlach T. et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247 (2014). 10.1038/leu.2013.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ward P. S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010). 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brownlee P. M., Chambers A. L., Cloney R., Bianchi A. & Downs J. A. BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep 6, 973–981 (2014). 10.1016/j.celrep.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ku J. L. et al. Genetic alterations of the TGF-beta signaling pathway in colorectal cancer cell lines: a novel mutation in Smad3 associated with the inactivation of TGF-beta-induced transcriptional activation. Cancer Lett 247, 283–292 (2007). 10.1016/j.canlet.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 90.Bougeard G. et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33, 2345–2352 (2015). 10.1200/JCO.2014.59.5728 [DOI] [PubMed] [Google Scholar]
- 91.Liyasova M. S., Ma K. & Lipkowitz S. Molecular pathways: cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res 21, 1789–1794 (2015). 10.1158/1078-0432.CCR-13-2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brand K., Kentsch H., Glashoff C. & Rosenberger G. RASopathy-associated CBL germline mutations cause aberrant ubiquitylation and trafficking of EGFR. Hum Mutat 35, 1372–1381 (2014). 10.1002/humu.22682 [DOI] [PubMed] [Google Scholar]
- 93.Meyer Zum Buschenfelde U. et al. RIT1 controls actin dynamics via complex formation with RAC1/CDC42 and PAK1. PLoS Genet 14, e1007370 (2018). 10.1371/journal.pgen.1007370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aoki Y. et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet 93, 173–180 (2013). 10.1016/j.ajhg.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boyd L. C. et al. Neurological manifestations of Erdheim-Chester Disease. Ann Clin Transl Neurol 7, 497–506 (2020). 10.1002/acn3.51014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bhatia A. et al. Neurologic and oncologic features of Erdheim-Chester disease: a 30-patient series. Neuro Oncol (2020). 10.1093/neuonc/noaa008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Diamond E. L. et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov 6, 154–165 (2016). 10.1158/2159-8290.CD-15-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Heritier S. et al. Incidence and risk factors for clinical neurodegenerative Langerhans cell histiocytosis: a longitudinal cohort study. Br J Haematol 183, 608–617 (2018). 10.1111/bjh.15577 [DOI] [PubMed] [Google Scholar]
- 99.Cybulski C. et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol 29, 3747–3752 (2011). 10.1200/JCO.2010.34.0778 [DOI] [PubMed] [Google Scholar]
- 100.Graham J. M. Jr. & Schwartz C. E. MED12 related disorders. Am J Med Genet A 161A, 2734–2740 (2013). 10.1002/ajmg.a.36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang S., Zheng X., Lu C., Li G. M., Allis C. D. & Li H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev 30, 1611–1616 (2016). 10.1101/gad.284323.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walter M. J. et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25, 1153–1158 (2011). 10.1038/leu.2011.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xi T., Jones I. M. & Mohrenweiser H. W. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics 83, 970–979 (2004). 10.1016/j.ygeno.2003.12.016 [DOI] [PubMed] [Google Scholar]
- 104.Shihab H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543 (2015). 10.1093/bioinformatics/btv009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adzhubei I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249 (2010). 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ng P. C. & Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 11, 863–874 (2001). 10.1101/gr.176601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kircher M., Witten D. M., Jain P., O’Roak B. J., Cooper G. M. & Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315 (2014). 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Itan Y. et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods 13, 109–110 (2016). 10.1038/nmeth.3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blasi E., Barluzzi R., Bocchini V., Mazzolla R. & Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol 27, 229–237 (1990). 10.1016/0165-5728(90)90073-v [DOI] [PubMed] [Google Scholar]
- 110.Henn A., Lund S., Hedtjärn M., Schrattenholz A., Pörzgen P. & Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 26, 83–94 (2009). 10.14573/altex.2009.2.83 [DOI] [PubMed] [Google Scholar]
- 111.Yu W. et al. CSF-1 receptor structure/function in MacCsf1r-/- macrophages: regulation of proliferation, differentiation, and morphology. J Leukoc Biol 84, 852–863 (2008). 10.1189/jlb.0308171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xiong Y., Song D., Cai Y., Yu W., Yeung Y. G. & Stanley E. R. A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. J Biol Chem 286, 952–960 (2011). 10.1074/jbc.M110.166702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Anzalone A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghosh S., Castillo E., Frias E. S. & Swanson R. A. Bioenergetic regulation of microglia. Glia 66, 1200–1212 (2018). 10.1002/glia.23271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu X. & Quan N. Microglia and CNS Interleukin-1: Beyond Immunological Concepts. Front Neurol 9, 8 (2018). 10.3389/fneur.2018.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Roy E. R. et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. The Journal of Clinical Investigation 130, 1912–1930 (2020). 10.1172/JCI133737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jayaraman A., Htike T. T., James R., Picon C. & Reynolds R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathologica Communications 9, 159 (2021). 10.1186/s40478-021-01264-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ou W. et al. Biologic TNF-α inhibitors reduce microgliosis, neuronal loss, and tau phosphorylation in a transgenic mouse model of tauopathy. Journal of Neuroinflammation 18, 312 (2021). 10.1186/s12974-021-02332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Thapa D. R. et al. Longitudinal analysis of peripheral blood T cell receptor diversity in patients with systemic lupus erythematosus by next-generation sequencing. Arthritis Res Ther 17, 132 (2015). 10.1186/s13075-015-0655-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lindberg O. R., Brederlau A. & Kuhn H. G. Epidermal growth factor treatment of the adult brain subventricular zone leads to focal microglia/macrophage accumulation and angiogenesis. Stem Cell Reports 2, 440–448 (2014). 10.1016/j.stemcr.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qu W. S. et al. Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J Neuroinflammation 9, 178 (2012). 10.1186/1742-2094-9-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Coniglio S. J. et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med 18, 519–527 (2012). 10.2119/molmed.2011.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scheltens P. et al. An exploratory clinical study of p38alpha kinase inhibition in Alzheimer’s disease. Ann Clin Transl Neurol 5, 464–473 (2018). 10.1002/acn3.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lachen-Montes M. et al. An early dysregulation of FAK and MEK/ERK signaling pathways precedes the beta-amyloid deposition in the olfactory bulb of APP/PS1 mouse model of Alzheimer’s disease. J Proteomics 148, 149–158 (2016). 10.1016/j.jprot.2016.07.032 [DOI] [PubMed] [Google Scholar]
- 125.Schöll M. et al. Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Scientific Reports 5, 16404 (2015). 10.1038/srep16404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kinney J. W., Bemiller S. M., Murtishaw A. S., Leisgang A. M., Salazar A. M. & Lamb B. T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 4, 575–590 (2018). 10.1016/j.trci.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequencing data processed for selection of somatic variants are available for all patients and samples in Supplementary Table S3. Raw DNA sequencing data (FASTQ files) from targeted-deep sequencing are deposited in dbGaP under project accession number phs002213.v1.p1, for samples where patient-informed consent for public deposition of DNA sequencing data was obtained. Bulk RNAseq raw data are deposited in GEO (number pending). Sn-RNAseq raw data are deposited in GEO (number pending) and as an interactive analysis web tool accessible at https://weillcornellmed.shinyapps.io/Human_brain/.