

Abstract
Objective
We sought to investigate the role of interleukin (IL)-20 in IBD and experimental colitis.
Design
Experimental colitis was induced in mice deficient in components of the IL-20 and signal transducer and activator of transcription (STAT)2 signalling pathways. In vivo imaging, high-resolution mini-endoscopy and histology were used to assess intestinal inflammation. We further used RNA-sequencing (RNA-Seq), RNAScope and Gene Ontology analysis, western blot analysis and co-immunoprecipitation, confocal microscopy and intestinal epithelial cell (IEC)-derived three-dimensional organoids to investigate the underlying molecular mechanisms. Results were validated using samples from patients with IBD and non-IBD control subjects by a combination of RNA-Seq, organoids and immunostainings.
Results
In IBD, IL20 levels were induced during remission and were significantly higher in antitumour necrosis factor responders versus non-responders. IL-20RA and IL-20RB were present on IECs from patients with IBD and IL-20-induced STAT3 and suppressed interferon (IFN)-STAT2 signalling in these cells. In IBD, experimental dextran sulfate sodium (DSS)-induced colitis and mucosal healing, IECs were the main producers of IL-20. Compared with wildtype controls, Il20−/−, Il20ra−/− and Il20rb−/− mice were more susceptible to experimental DSS-induced colitis. IL-20 deficiency was associated with increased IFN/STAT2 activity in mice and IFN/STAT2-induced necroptotic cell death in IEC-derived organoids could be markedly blocked by IL-20. Moreover, newly generated Stat2ΔIEC mice, lacking STAT2 in IECs, were less susceptible to experimental colitis compared with wildtype controls and the administration of IL-20 suppressed colitis activity in wildtype animals.
Conclusion
IL-20 controls colitis and mucosal healing by interfering with the IFN/STAT2 death signalling pathway in IECs. These results indicate new directions for suppressing gut inflammation by modulating IL-20-controlled STAT2 signals.
Keywords: experimental colitis, inflammatory bowel disease, interferon, interleukins, signal transduction
WHAT IS ALREADY KNOWN ON THIS TOPIC
Current biological therapies still fail to induce sustained remission in most patients with IBD, underscoring the urgent need to better understand the underlying molecular mechanisms.
The balance between pro-inflammatory and anti-inflammatory cytokines plays a key role in controlling the resolution of intestinal inflammation.
WHAT THIS STUDY ADDS
Type I interferon (IFN)/signal transducer and activator of transcription (STAT)2 signalling is enhanced in patients with IBD and in mice with experimental colitis.
Type I IFN promotes necroptotic cell death in human and mouse intestinal epithelial cells in a STAT2-dependent manner.
Interleukin (IL)-20 is important in controlling gut inflammation and mucosal healing in experimental colitis.
Increased IL-20 levels in antitumour necrosis factor therapy responders might attenuate type I IFN/STAT2 signalling to protect the intestinal epithelium and favour the restitution of the intestinal mucosal integrity in patients with IBD.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Future therapeutic options for the management of patients with IBD might consider modulating IL-20 signalling in order to counterbalance detrimental type I IFN/STAT2 effects.
Introduction
IBD defines a group of chronic, relapsing and aggravating disorders, with steady increasing incidence, in which the intestinal homeostasis has been disrupted by uncontrolled immune responses against microbial and environmental factors in genetically predisposed individuals. The continuous battle between microbiota-driven inflammatory responses and the anti-inflammatory mechanisms of the mucosal immune system dictates the course of disease with alternating episodes of acute severe inflammation and phases of remission, during which the integrity of the mucosal barrier is being restored.1–4 Although biological therapies proved to be effective in limiting inflammation and promoting tissue repair, more than half of the patients with IBD do not respond to therapy thus underscoring the urgent need to better understand disease pathogenesis with the final goal of delivering optimal medical support to each and every patient.3 5–7
In 1992, the first two members of the signal transducer and activator of transcription (STAT) family, that is, STAT1 and STAT2 were discovered in the context of antiviral responses induced by interferons (IFNs).8 Five other STATs (ie, STAT3, STAT4, STAT5a, STAT5b and STAT6), four receptor-associated Janus kinases that have been initially identified as orphan tyrosine kinases (JAK1, JAK2, JAK3 and TYK2) as well as IFN regulatory factor 9 were all found to be part of the JAK-STAT signalling pathway.9 The JAK-STAT axis represents one of a handful of pleiotropic pathways that controls a daunting number of tasks by transducing information from cytokines, chemokines, growth factors and hormones in processes ranging from organism development to cell proliferation, differentiation and cell death, from inflammation and cancer to tissue remodelling and mucosal healing.9 Understanding its biology in the context of the cross-talk between microbiota and intestinal epithelial cells (IECs) at the forefront of the mucosal immune system of the gut is instrumental for the successful implementation of biological therapies in IBD.10 Of particular interest is the role of type I and type III IFN since robust production is induced by viruses and by microbial-derived ligands and metabolites, especially in dysbiosis settings.11 Nevertheless, the precise implication of discrete components of the IFN signalling pathway in IBD is incompletely understood since both the upregulation, for example, STAT1,12 13 as well as the downregulation, for example, STAT2,14 of the IFN/STAT axis have been reported. Moreover, opposing roles of type I IFN controlled the emergence and the recovery phases of colitis.15
A major player involved in anti-inflammatory responses and mucosal healing in the gut is the interleukin (IL)-10 family of cytokines.16 IL-20, one of its members, was discovered after screening expressed sequence tag databases from a human keratinocyte library by an algorithm designed to find translated sequences containing both a signal sequence and amphipathic helices.17 The transgenic overexpression of IL-20 in mice resulted in perinatal lethality with aberrant epidermal differentiation and a psoriasis-like inflammation. Two orphan receptors, the levels of which were dramatically upregulated in psoriatic skin, were identified as IL-20RA and IL-20RB, while STAT3 was described as the primary transducer of IL-20 signals.17 A couple of single nucleotide polymorphisms known to be significant susceptibility factors for psoriasis were reported in the IL20 gene of Mexican patients with UC.18 Unfortunately, neither a correlation between these genotypes and clinical characteristics of patients nor an altered IL-20 signalling in the gut were reported in that study. Another work found increased IL-20 levels in patients with active UC as compared with patients with UC in remission and to healthy non-IBD controls, respectively.19 Given the scarcity of data on the role of IL-20 in the intestine,20 we aimed in the present study to understand the biology of IL-20 in the context of gut inflammation. We found that IL-20 blocks inflammation driven by type I IFN/STAT2 signals and promotes intestinal epithelial restitution emphasising for the first time that the modulation of the IL-20/STAT2 interaction might represent a therapeutic target for the future management of IBD.
Methods
Patients
Inflammation severity and responder criteria were scored as described in the online supplemental file. Clinical characteristics of the cohorts are presented in online supplemental table 1.
gutjnl-2023-329628supp009.pdf (307.9KB, pdf)
gutjnl-2023-329628supp011.pdf (94.8KB, pdf)
Mice
Animal experiments were performed by qualified personnel in accordance with the FELASA welfare regulations. All efforts were made to reduce the total number of mice and to minimise suffering of each animal. Details of all mouse strains including the production of Stat2fl/fl mice by CRISPR/Cas9 technology (Applied StemCell, Milpitas, California, USA) and of Il20−/− and Il20ra−/− by in vitro fertilisation using sperm from the KOMP Repository and the Mouse Biology Programme at the University of California Davis are described in the online supplemental file. Knockout mice and sex-matched and age-matched C57BL/6 wildtype (WT) control mice with a body weight of around 20–30 g were used. Established criteria that were used to ensure scientific rigour in colitis experiments are described in the online supplemental file.
Statistical analysis
Applied tests are indicated in figure legends. GraphPad Prism V.8.3.0 (GraphPad Software, San Diego, California, USA) was used.
Results
IL-20 expression levels are induced in epithelial cells adjacent to mucosal lesions and correlate with antitumour necrosis factor therapy response in patients with IBD
RNA-sequencing (RNA-Seq) transcriptomic analysis of mucosal biopsies obtained from patients with IBD and non-IBD controls (raw data has not yet been released but can be made available upon request) revealed that among all IL receptors, IL20RA had the highest expression levels in patients with IBD (figure 1A). We further validated these findings on an enlarged cohort of patients by showing that IL20RA levels were markedly increased in intestinal biopsies from both patients with Crohn’s disease (CD) and UC as compared with their respective non-IBD controls (figure 1B). The analysis of a previously published microarray dataset (GSE72780)21 indicated that IL20RA levels were higher in IECs isolated from the inflamed tissue versus IECs from non-inflamed mucosa of patients with CD (online supplemental figure 1A). In line with the increased IL20RA expression, IL20 levels were also higher in patients with IBD as compared with non-IBD controls (figure 1C). The expression of IFNG and tumour necrosis factor (TNF) in patients with different degrees of inflammation is presented in online supplemental figure 1B. Whereas levels of IL20 and IL20RA did not correlate with disease characteristics like patients’ age, gender or the origin of the biopsy, we measured the highest levels of IL20 in the remission phase, that is, inflammation scores 0 and 1 as compared with active disease, that is, inflammation scores 2 and 3 (scoring criteria are described in the online supplemental file) and in both IBD groups compared with non-IBD controls (figure 1D and online supplemental figure 1C). We therefore hypothesised that IL-20 could represent an anti-inflammatory factor promoting mucosal healing in IBD. Next, we determined IL20 levels in samples from patients receiving anti-TNF therapy. The results indicated higher levels of IL20 in anti-TNF responders (criteria described in the online supplemental file) as compared with anti-TNF non-responders (figure 1E). This finding was further supported by the analysis of a previously published microarray dataset (GSE16879),22 in which IL20 levels were found to be higher in patients that would respond to anti-TNF therapy as compared with non-responders (online supplemental figure 1D).
Figure 1.

IL-20 signalling is altered during remission in patients with IBD. (A) RNA-Seq analysis of the expression levels of IL receptors in an initial cohort of patients with IBD and non-IBD controls (n=10/group). (B) RNA-Seq expression levels of IL20RA in ileal samples from CD (n=100) and patients without IBD (n=29), in the upper panel, or colon samples from UC (n=46) and non-IBD control samples (n=21), in the lower panel. (C) RNA-Seq expression levels of IL20 in ileal samples from CD and patients without IBD, in the left side panel, or colon samples from UC and non-IBD control samples, in the right side panel. (D) Results of quantitative PCR for IL20 (relative to GAPDH) in non-IBD controls and patients suffering from either CD or UC. Patients were grouped according to their inflammation score into the following categories: 0, no inflammation; 1, mild inflammation; 2, moderate inflammation and 3, severe inflammation (criteria referenced in the online supplemental file). (E) IL20 expression levels (relative to ACTB) in samples from anti-TNF responders and anti-TNF non-responders. (F) Confocal imaging of cryosections from biopsies of patients with CD and UC are shown alongside non-IBD controls (n=5/group). Antibodies to IL-20RA and IL-20RB were used to identify the cells expressing these receptor chains (arrows indicate positive cells in the epithelium). (G) Fresh biopsies from six different patients with IBD were incubated for 60 min in the presence or absence of 250 ng/mL IL-20 and sections were cut and stained with anti-pSTAT3 antibodies. One representative result is shown. Arrows and arrowheads indicate positive cells in the epithelium or in the lamina propria, respectively. (H) Three-dimensional organoids generated from colon biopsies of three different patients with IBD were stained to detect the presence of IL-20RA and IL-20RB. (I) Organoids stimulated for 36 hours with 250 ng/mL IL-20 were stained for Ki67 to identify proliferating cells (arrows). (J) Localisation of IL-20+ cells at the edges of erosions in a colon biopsy of a patient with IBD. A total of four different patients with IBD were evaluated. Arrows indicate IL-20-positive cells in the epithelium and arrowheads in the wounded area. (F–J) Epithelial cell adhesion molecule (EpCAM) and 4′,6-diamidino-2-phenylindole (DAPI)/Hoechst were used to mark epithelial cells and nuclei, respectively. Scale bars, 50 µm in F and G, 100 µm in H (merged), I and J and 200 µm in H (channels). Statistics: Welch’s t-test in B and C; Kruskal-Wallis corrected for multiple comparisons in D; Mann-Whitney U test in E; B–D, mean with 95% CI is displayed. CD, Crohn’s disease; IL, interleukin; RNA-Seq, RNA-sequencing; TNF, tumour necrosis factor. *P<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
gutjnl-2023-329628supp001.pdf (166.7KB, pdf)
gutjnl-2023-329628supp002.pdf (2.2MB, pdf)
In situ, IL-20RA and IL-20RB (figure 1F) were predominantly expressed by EpCAM+ IECs (arrows) and to a lesser extent by surrounding immune cells in the lamina propria. We found higher IL-20 receptor subunit expression in both CD and UC samples compared with non-IBD controls. To provide functional evidence for the expression of IL-20RA and IL-20RB on IECs and to gain further insights into downstream signalling events, we stimulated freshly obtained colon biopsies from patients with IBD with IL-20 for 1 hour and analysed the results of immune staining in cryosections by confocal microscopy. Whereas in unstimulated biopsies, most pSTAT3+ cells were located in the lamina propria (arrowheads), IL-20 stimulation markedly induced pSTAT3 expression within IECs (arrows) indicating a direct effect of the cytokine on the IEC compartment (figure 1G). We next isolated crypts from fresh biopsies of patients with IBD and cultured them as three-dimensional organoids. IL-20RA and IL-20RB could be detected in these organoids (figure 1H) and 36 hours of stimulation with IL-20 was accompanied by enhanced proliferation as indicated by the Ki67 staining (figure 1I, arrows). This effect was associated with the induction of pSTAT3 on IL-20 stimulation, as demonstrated by western blot analysis and flow cytometry analysis (online supplemental figure 1E, F). Importantly, in the inflamed intestine of patients with IBD, IL-20+ cells were predominantly found among EpCAM+ IECs adjacent to areas with erosions or ulcerations (arrows), and to a lesser extent in non-epithelial (arrowheads) cells (figure 1J). Additional co-staining experiments validated this observation (an image from non-ulcerative IBD tissues is presented in online supplemental figure 1G) and indicated that IECs from patients with IBD expressed higher IL-20 protein levels than those from non-IBD controls (online supplemental figure 1H). The predominant epithelial cell origin of IL-20 in IBD was also confirmed by RNAScope hybridisation using IL20-specific ZZ probes (online supplemental figure 1I).
Compromised IL-20 signalling aggravates experimental gut inflammation
The analysis of RNA-Seq data23 indicated that levels of both Il20ra and Il20rb increased with the degree of inflammation and decreased at the end of the recovery phase in mice receiving DSS (online supplemental figure 2A, B, original data: ArrayExpress E-MTAB-9850), suggesting a dynamic role of IL-20 signalling in experimental colitis. We next investigated the contribution of the IL-20RA and IL-20RB in the DSS-induced colitis model using mice lacking the respective receptor chain. In steady state (without DSS), mice did not show any pathology in colon cross-sections (figure 2A). Because of the high susceptibility of knockout mice and the risk of inducing lethal inflammation, in these experiments, we used a DSS protocol that induced only mild and limited colitis in WT mice. On DSS administration, Il20ra−/− and Il20rb−/− mice developed more severe inflammation as indicated by the pronounced body weight loss, shortened colons, larger areas of tissue destruction, oedema and disturbed crypt architecture, as well as augmented infiltration of immune cells (figure 2B) and depletion of goblet cells (Ulex staining in figure 2C) as compared with WT control mice. Levels of infiltrating leucocytes (CD45+) and particularly macrophages (F4/80+) were increased in knockout mice compared with WT controls (figure 2C). RNA-Seq data analysis of colon tissue from mice receiving DSS indicated dysregulation of thousands of genes (online supplemental figure 2C, original data: ArrayExpress E-MTAB-12737). Linear regression analysis of all differentially expressed genes between the Il20ra−/− and Il20rb−/− groups (each vs WT) showed a high degree of correlation (R2=0.7898) indicating a relevant overlap in the transcriptomic changes of both receptor chain knockouts during DSS-induced colitis (figure 2D). Among the common significantly upregulated genes, most prominent categories included genes implicated in inflammation and tissue remodelling, cell junctions and cytoskeleton and the control of DNA replication and cell cycle control (figure 2E, left side). However, no clear grouping into categories was observed in the common downregulated genes which are therefore presented in alphabetical order (figure 2E, right side).
Figure 2.

Signalling through IL-20RA and IL-20RB controls experimental DSS-induced colitis. (A) Normal appearance of colon cross-sections of WT, Il20ra−/− and Il20rb−/− mice in steady state (water without (w/o) DSS). (B) Experimental colitis (three independent experiments) was induced by the administration of 2% DSS in the drinking water for 7 days in WT (n=16), Il20ra−/− mice (n=15) and Il20rb−/− mice (n=14). Body weight loss during colitis course respective to day 0 (set as 100%). High-resolution mini-endoscopy pictures indicating the degree of intestinal inflammation and histological analysis of inflammation based on H&E staining. Scores were calculated as described in the online supplemental file. Shortened colon lengths indicate aggravation of inflammation. (C) Representative colon cross-sections of mice with DSS-induced colitis that have been stained with the pan-leucocyte marker CD45, the F4/80 macrophage marker and the goblet cell marker Ulex are presented in confocal images. (D–E) RNA-Seq analysis of Il20ra−/− (n=3) and Il20rb−/− (n=3) samples respective to WT control mice (n=5). (D) Expression levels for transcripts that are co-upregulated and co-downregulated in both KO mouse strains versus WT controls from one DSS experiment. (E) Most genes that were found to be co-upregulated in the KO strains could be grouped to Gene Ontology processes that are classically involved in the progression of DSS-induced colitis (left side panel) whereas the most co-downregulated genes did not readily point out to known Gene Ontology categories and are presented in alphabetical order in the panel on the right side. Scale bars, 100 µm in A and C, 50 µm in B. Statistics: analysis of variance corrected for multiple comparisons in B, mean with SEM or 95% CI is displayed; squared Pearson’s correlation coefficient (r) of the linear regression analysis in D. DSS, dextran sulfate sodium; IL, interleukin; KO, knockout; RNA-Seq, RNA-sequencing; WT, wildtype. *P<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
gutjnl-2023-329628supp003.pdf (624.6KB, pdf)
IL-20 overexpression limits intestinal inflammation while IL-20 deficiency augments experimental DSS-induced and oxazolone-induced colitis
Whereas very few cells expressed IL-20 in untreated WT mice, increased numbers of IL-20+ cells were observed in WT mice with DSS-induced colitis, most of which were localised in the IEC compartment (figure 3A, arrows indicate IL-20+ IECs and arrowheads indicate IL-20+ lamina propria cells). To obtain a more complete picture of the nature of IL-20-producing cells, we used RNAScope with ACDBio’s custom-designed 17ZZ long probes targeting the 2-965 region of murine Il20 (NM_001311091.1). Similar to IBD, the results of this analysis confirmed the fact that most murine IL-20+ cells in DSS-induced colitis were EpCAM+ IECs (online supplemental figure 3A, arrows indicate IECs and arrowheads indicate non-epithelial cells that were positive for IL-20-specific probes). Additional immunofluorescence co-staining, some of which was conducted on consecutive slides, revealed that few immune cells (MPO+ polymorphonuclear neutrophils, F4/80+ macrophages) but no fibroblasts (Vimentin+) or glial cells (GFAP+) expressed IL-20 in the colon sections of mice with DSS-induced colitis. Confirming our initial observation of the IEC origin of IL-20 in the context of DSS-induced inflammation, EpCAM+ cells represented the vast majority of IL-20+ cells (online supplemental figure 3B), arrows indicate IL-20+ IECs and arrowheads IL-20+ non-IECs). To investigate the mechanism by which IL-20 contributes to epithelial barrier function in the gut in vivo, we placed mucosal wounds of defined size in the colon of WT mice (figure 3B). Two days later, wounds were isolated, cut, stained and imaged by confocal microscopy. We found IL-20+ cells at the very edges of the wounds (figure 3B, arrows indicate EpCAM+/IL-20+ cells; a higher magnification is presented in the inset on the left side), the site where IEC proliferation has been previously shown to be most upregulated.24
Figure 3.

Exacerbated colitis in mice lacking IL-20 is linked to increased IFN responses. (A) Changes in the pattern of IL-20 staining between colon samples from WT mice in steady state (left side panel) compared with mice receiving DSS in the drinking water (right side panel). Arrows indicate positive staining in the IECs and arrowheads in the lamina propria, respectively. (B) Wounds of defined size were placed by a standardised protocol in the colons of mice using a biopsy forceps and the wound areas were collected 2 days thereafter, stained for IL-20 and imaged by confocal microscopy. The appearance of the wound bed at days 0 and 2 is presented in the mini-endoscopy images and one representative wound is presented in the immunofluorescence staining on the right side. Arrows point at positive cells in the epithelial cell compartment that are immediately adjacent to the wound. Magnification is shown in the inset to the left. (C) Experimental colitis (two independent experiments) was induced in WT (n=10) and Il20−/− mice (n=10) by the administration of DSS in the drinking water for 7 days after which the mice received water without DSS. Body weight loss is presented relative to day 0. High-resolution mini-endoscopy pictures indicating the degree of intestinal inflammation and the histological analysis of inflammation based on H&E staining. Scores were calculated as described in the online supplemental file. (D) Colon cryosections of mice with DSS-induced colitis were stained with the late apoptosis marker TUNEL (arrows) and the goblet cell marker Ulex as well as the pan-leucocyte marker CD45 and the specific macrophage cell marker F4/80. (E) RNA-Seq expression levels of the top 20 most upregulated and downregulated genes in the colonic samples from Il20−/− mice (n=4) as compared with WT controls (n=5) are presented with genes linked to IBD/experimental colitis in red font. (F) Selection of some of the significantly altered biological processes from the Gene Ontology analysis performed on the upregulated genes (criteria described in the ‘Results’ section) are presented. (G) Two of the most significant terms of the KEGG pathway analysis linked high colitis activity to antiviral responses (marked in turquois). (H) Differential RNA-Seq expression levels for a typical panel of type I IFN signalling-related genes. Scale bars, 50 µm in A, B and C, 100 µm in D. Statistics: Welch’s t-test in C and D, mean with SEM or 95% CI is displayed. Benjamini-Hochberg for pdaj in E, F and G; the dashed red line indicates pdaj=0.05 in F and G. DSS, dextran sulfate sodium; HPF, high power field; IEC, intestinal epithelial cell; IFN, interferon; IL, interleukin; RNA-Seq, RNA-sequencing; WT, wildtype. *P<0.05; **p<0.01; ***p<0.001.
gutjnl-2023-329628supp004.pdf (1.9MB, pdf)
To determine whether IL-20 can modulate the course of experimental gut inflammation in a preventive fashion, we constructed an expression vector encoding Il20, using a strategy that has been already successfully used to study the systemic effects of cytokines in experimental colitis.12 25 WT mice received either DNA vector coding for murine Il20 or an empty control vector by hydrodynamic tail vein injection. Two days later, all mice received DSS in the drinking water for the subsequent 7 days followed by water without DSS. Compared with the empty vector control group, mice receiving the Il20 vector lost less body weight and showed reduced intestinal inflammation both during the moderate (day 5) and the full-blown (day 10) phases of colitis as revealed by endoscopic examination and confirmed by the assessment of colitis severity on histology (online supplemental figure 4A).
gutjnl-2023-329628supp005.pdf (1.2MB, pdf)
To explore the role of IL-20 in experimental colitis in vivo, WT and Il20−/− mice received DSS for 7 days. Whereas WT mice developed mild inflammation, Il20−/− animals rapidly lost body weight. Importantly, the body weight loss could not be regained by the end of the experiment (figure 3C) thus supporting the observation made in our patients with IBD, that is, lower levels of IL-20 were present in subjects with higher inflammation scores (see figure 1D). By colonoscopy, Il20−/− mice presented looser stool and more vascular damage, less transparent and highly infiltrated mucosa indicating a more aggressive colitis which was confirmed by the shortened colon lengths and the histological analysis of colon cross-sections (figure 3C). Immunofluorescent staining indicated a significantly higher number of TUNEL+ apoptotic cells (arrows) in Il20−/− mice, whereas the number of goblet cells (Ulex+) was diminished in these mice (figure 3D). Furthermore, susceptible Il20−/− mice had increased levels of infiltrating immune cells in general (CD45+) and specifically of macrophages (F4/80+) as compared with WT controls (figure 3D). RNA-Seq data analysis of WT and Il20−/− mice with DSS-induced colitis (online supplemental figure 4B) revealed that many genes that have been previously linked to the pathogenesis of IBD (eg, Selp, Il11, Ngp, Il6, Csf3) or have been identified as markers for predicting therapy failure (eg, Lcn2) were among the top 20 most upregulated genes in susceptible Il20−/− mice (figure 3E, genes of interest for the present study are marked in red, original data: ArrayExpress E-MTAB-12737). Gene Ontology analysis of the highest significantly upregulated genes (log2fold change≥3, padj<0.05, Benjamini-Hochberg) in Il20−/− compared with WT mice indicated a significant increase in a plethora of IBD-related inflammatory processes including: inflammatory responses, neutrophil chemotaxis, positive regulation of angiogenesis, innate immune responses, negative regulation of cell proliferation or cellular responses to lipopolysaccharide (LPS), IL-1 and TNF (figure 3F). Cell differentiation was significantly downregulated in susceptible Il20−/− mice as indicated by Gene Ontology analysis of the downregulated genes (log2fold change≤−1, padj<0.05, not shown). The top KEGG pathway revealed by analysis of all differentially expressed genes with |log2fold change|≥1 and padj<0.05 was viral protein interaction with cytokine and cytokine receptor whereas other three virus-related pathways were also significantly upregulated in Il20−/− mice as compared with WT mice (ie, human T cell leukaemia virus 1 infection, viral carcinogenesis, human papillomavirus infection, not shown). Importantly, viral-related pathways including COVID-19 were also significantly upregulated in the KEGG pathway analysis of the highest upregulated genes (those that were used for the assessment of biological processes in the Gene Ontology analysis presented in figure 3F, ie, log2fold change≥3, padj<0.05) suggesting a possible link between IL-20 deficiency and the antiviral immune response (figure 3G, marked in turquois). Similar processes were also upregulated in patients as indicated by the KEGG analysis of our human IBDome cohorts (online supplemental figure 4C). Since antiviral immunity heavily relies on type I IFN responses, many IFN targets were present among the significantly upregulated genes in Il20−/− mice, some of which are presented in figure 3H.
In a final set of experiments, we aimed to analyse the role of IL-20 in a second, independent model of colitis which is based on an immune response to sensitisation and challenge with a hapten reagent. Using the two-step (presensitisation and challenge) model of oxazolone-induced colitis, we found that Il20−/− mice were more susceptible as compared with WT controls (online supplemental figure 4D), providing evidence for a broader role of IL-20 in mediating protective effects in the context of intestinal inflammation.
IL-20 interferes with epithelial STAT2 signalling to limit IFN-β-induced IEC necroptotic death
Since our RNA-Seq analysis indicated that experimental colitis in Il20−/− mice was linked to an antiviral response, we next analysed the effect of type I IFN on WT mouse organoids in the presence or absence of IL-20. We found that 1 hour pre-incubation of intestinal organoids with IL-20 reduced the phosphorylation of STAT2 after IFN-β exposure (figure 4A). In another assay, colon organoids derived from WT mice with DSS-induced colitis were pre-incubated with IL-20 for 20 hours prior to the incubation with IFN-β for additional 6 hours. Our quantitative PCR (qPCR) results indicated a dramatic decrease in the levels of typical type I IFN target genes, that is, Cxcl10 and Mx1 (online supplemental figure 5A), strengthening the idea of an interaction between IL-20-dependent and IFN-dependent signalling in the inflamed colon.
Figure 4.

The IFN-β/STAT2 axis induces necroptosis in murine IEC and IL-20 can interfere with the actions of IFN-β. Organoids from the small intestine and colon of mice were cultured as described in the online supplemental file. (A) Western blot analysis of the levels of pSTAT2 and STAT2 in WT organoids pre-incubated for 60 min with 250 ng/mL IL-20 prior to stimulation with 25 ng/mL IFN-β for 30 min. (B) Organoids from mice with DSS-induced colitis were stimulated with either IFN-β or a combination of IL-20 and IFN-β or were left untreated and cell extracts were then Co-IP with anti-pSTAT1, anti-pSTAT3 or with monoclonal isotype control antibodies. Western blot analysis was performed to detect pSTAT2. Lysates not subjected to Co-IP served as input controls for the detection of pSTAT2, STAT2 and GAPDH levels. (C) Murine colon organoids from mice with DSS-induced colitis (n=5 mice, technical duplicates or triplicates) were stimulated with IFN-β in the presence or absence of the Nec-1 and stained with PI to assess cell death and Hoechst for normalisation purposes. (D) Quantification of the results. (E) Survival of WT and Stat2−/− organoids stimulated for 36 hours with 100 ng/mL IFN-β was followed by microscopy on addition of PI (red indicates dead areas, green is autofluorescence, areas with living cells are indicated by arrows). (F) Quantification of the results in n=3 mice/group with technical triplicates. (G) Small intestine organoids from WT and Stat2−/− mice (n=3/group) were either left untreated or were stimulated for 20 hours with 25 ng/mL IFN-β and then subjected to RNA-Seq (three biological replicates/condition, pairwise design). The top 20 upregulated and downregulated genes are displayed in WT organoids in the left side panel. The vast majority of these genes that are missing in the right-side panel (ie, top 20 upregulated genes in Stat2−/− organoids after IFN-β stimulation) are marked in red. (H) Differences between levels of Mlkl and Casp8 in the RNA-Seq profiling of results from G. Scale bars, 500 µm in E. Statistics: analysis of variance in D; Welch’s t-test in F; Benjamini-Hochberg for pdaj in G and H. Co-IP, co-immunoprecipitation; DSS, dextran sulfate sodium; IEC, intestinal epithelial cell; IFN, interferon; IL, interleukin; Nec-1, necrostatin-1; ns, not significant; PI, propidium iodide; RFU, relative fluorescence units; RNA-Seq, RNA-sequencing; WT, wildtype. **p<0.01; ***p<0.001; ****p<0.0001.
gutjnl-2023-329628supp006.pdf (585.2KB, pdf)
To test how IL-20 could interfere with IFN-β signalling, we stimulated colon organoids from WT mice with DSS-induced colitis with IL-20 prior to the stimulation with IFN-β. We then used sorting on protein A-coated magnetic beads and subjected cell extracts to co-immunoprecipitation with monoclonal antibodies against either pSTAT1 or pSTAT3 or with a monoclonal rabbit antibody that served as isotype control for both anti-pSTAT1 and anti-pSTAT3, all three adjusted to the same concentration. Corresponding cell lysates that were not co-immunoprecipitated served as input in the western blot analysis. Our results indicated that IL-20 pre-incubation was able to partly block the co-immunoprecipitation of pSTAT2 by the anti-pSTAT1 antibodies after IFN-β signalling (figure 4B). In contrast, pSTAT2 was not co-immunoprecipitated by anti-pSTAT3 antibodies (figure 4B). Hence, a possible consequence could be the downregulation of IFN-β-triggered/STAT1:STAT2-mediated cell death by IL-20. To directly test how IFN-β stimulation affects IEC survival, we stimulated WT colon organoids for 36 hours with IFN-β. We used the methylthiazolyldiphenyl-tetrazolium bromide (MTT)-formazan assay26 to determine the amount of dark formazan precipitates, which is proportional to the mitochondrial dehydrogenase activity of the cells, and thus represents an indirect means for assessing the amount of cell death. IFN-β-treated IECs showed markedly more bright areas under the microscope, indicating metabolically inactive, dead cells, while untreated organoids showed more dark areas, representing living cells with active mitochondrial respiration (online supplemental figure 5B, C). In another set of experiments, the treatment of colitis mouse organoids with IFN-β plus IL-20 or IL-10 suppressed cell death compared with IFN-β treatment alone (online supplemental figure 5D). To directly quantify cell death in IFN-β-treated organoids, we used Hoechst and propidium iodide staining.27 We included necrostatin-1,28 a selective and potent inhibitor of the necroptosis executioner RIPK1, to clarify whether necroptosis is responsible for the IFN-β-induced cell death.29 These studies demonstrated that IFN-β treatment resulted in cell death that could be blocked by necrostatin-1 (figure 4C, D).
Since STAT1 has been previously linked to proliferative rather than cell death signals in the context of IFN signalling in colon IECs, we turned our attention to STAT2 which partners up with STAT1 to transmit signals from the type I IFN receptor complex.30 First, we compared the growth of intestinal organoids generated from WT and Stat2−/− mice over a period of 10 days and could observe that Stat2−/− organoids grew faster and bigger (online supplemental figure 5E, arrows indicate buds) as assessed by the quantification of bud numbers (online supplemental figure 5F) on two different days. Next, the survival of WT and Stat2−/− organoids stimulated with IFN-β was followed by microscopy on addition of propidium iodide which stains dead cells. Whereas small intestine and colon organoids from WT mice were susceptible to cell death, organoids derived from Stat2−/− were highly resistant (figure 4E,F). Taken together, these results indicated that type I IFN-triggered IEC death is mediated by STAT2. To further dissect the molecular basis of these observations, small intestine organoids derived from WT and Stat2−/− mice were stimulated with IFN-β or were left untreated for 20 hours and the extracted RNA was subjected to RNA-Seq (original data: ArrayExpress E-MTAB-12662). The experimental setup included three independent biological replicates/condition in a pairwise design (online supplemental figure 6A, left-side panel). According to our criteria for differential expression (|log2fold change|≥1, padj<0.05), IFN-β predominantly induced the upregulation over the downregulation of hundreds of genes in WT organoids whereas only a total of 20 genes were upregulated and none were significantly downregulated in Stat2−/− organoids (online supplemental figure 6A, middle-side and right-side panels). Importantly, only 5 (marked in grey) of the top 20 most upregulated genes in WT+IFN-β were also present in Stat2−/− +IFN-β, suggesting that the expression of most IFN-signature genes in the context of IECs is dependent on a functional STAT2 rather than STAT1, since Stat2−/− organoids did express STAT1 (figure 4G, genes marked in red). Noteworthy is the fact that Reg3b, a factor that is responsible for mucosal protection, was present among the top 20 upregulated genes in the Stat2−/− +IFN-β group (figure 4G right side panel, gene marked in blue). It is conceivable that in the absence of STAT2, IFN-β is capable of inducing protective effects by the formation of alternative IFN-stimulated complexes (eg, STAT1:STAT1, STAT1:STAT3, STAT3:STAT3). Gene Ontology analysis of the upregulated genes (log2fold change≥1, padj<0.05) revealed that most of the biological processes induced by IFN-β in WT organoids were linked to a functional STAT2 given the fact that they did not reach statistical significance in Stat2−/− organoids stimulated with IFN-β (online supplemental figure 6B; processes of bars marked in red in the second panel). Importantly, the dramatic increase in levels of the necroptosis-inducing protein Mlkl (but only moderate increase in the apoptosis mediator Casp8) in WT organoids stimulated with IFN-β was not present in Stat2−/− organoids undergoing the same stimulation strengthening the idea that the necroptotic effect of type I IFN in organoids is exquisitely reliant on STAT2 rather than STAT1 (figure 4H). Taken together, these results clearly indicated that the IFN-β/STAT2 axis promotes necroptotic cell death and inflammation, an effect that could be interfered by IL-20 signals.
gutjnl-2023-329628supp007.pdf (824.8KB, pdf)
IEC-specific STAT2 activity controls experimental colitis in mice
RNA-Seq data analysis indicated that both chains of the type I IFN receptor were upregulated (figure 5A), as were various IFN-target genes (figure 5B), during the course of DSS-induced colitis in WT mice (raw data has not yet been released but can be made available upon request). To address the functional role of STAT2-mediated IFN signals in vivo, we challenged Stat2−/− mice with DSS. Compared with WT, Stat2−/− mice showed significantly less body weight loss and reduced intestinal inflammation on endoscopy (figure 5C). Inflammation appeared earlier and was more severe at any time point in WT as compared with Stat2−/− mice. The extent of mucosal erosions, the disruption of normal vessel architecture, diarrhoea and bleeding were more prominent in WT animals. Likewise, neutrophil infiltration recorded by in vivo luminescence imaging was more abundant in WT mice. The significant difference in the activity disease scores between groups was also confirmed by the shortened colon lengths of WT mice as measured at the end of the experiment. Histology score analysis of colon cross-sections indicated an overall reduced number of crypts, more crypt distortions, erosions and inflammatory infiltrates in WT mice as compared with Stat2 −/− animals (figure 5C).
Figure 5.

Stat2−/− mice are resistant to experimental DSS-induced and oxazolone-induced colitis. (A, B) RNA-Seq data analysis of the levels of type I IFN receptor chains (A) and different type I IFN target genes (B) in mice receiving DSS in the drinking water versus mice receiving water without (w/o) DSS (n=5/group). (C) Experimental colitis (two independent experiments) was induced in WT (n=11) and Stat2−/− mice (n=12) by the administration of 2% DSS in the drinking water for 7 days. Body weight loss during colitis course respective to day 0. The degree of inflammation was further assessed by the colon length shortening, high-resolution mini-endoscopy, in vivo imaging of neutrophil infiltration and histology. (D) Representative staining of immune cell markers (CD11c, F4/80, MPO), the proliferation marker Ki67, the epithelial cell marker EpCAM and the late apoptosis marker TUNEL. Arrows indicate positive staining in IECs. Arrowheads in the top panels indicate positive Ki67 staining in the non-IEC compartment. (E) Top 20 most upregulated and downregulated genes in the RNA-Seq analysis of Stat2−/− vs WT mice from one DSS experiment (n=4/group). Upregulated genes that have been previously linked to beneficial outcomes in IBD/experimental colitis are marked in green whereas downregulated genes marked in blue have been previously linked to disease pathogenesis. (F) Experimental colitis was induced by cutaneous sensitisation of mice on day 0 and 1 week later by the intrarectal application of oxazolone. Significant upregulation of Ifnar2 as wells as typical IFN target genes in RNA-Seq analysis of WT mice receiving oxazolone versus WT mice w/o oxazolone treatment (n=5/group, right side panel). Representative endoscopy pictures from WT and Stat2−/− mice in the oxazolone-induced model showing fibrin deposits and altered mucosal appearance are shown alongside in vivo imaging of neutrophil infiltration and histology pictures confirming the infiltration of the gut and the destruction of the epithelial architecture. Scale bars, all 100 µm. Statistics: Benjamini-Hochberg for padj in A, B, E and F, box and whiskers with min to max are displayed in A, B and F. Welch’s t-test in C and F, mean with SEM or 95% CI is displayed. DSS, dextran sulfate sodium; IEC, intestinal epithelial cell; IFN, interferon; RNA-Seq, RNA-sequencing; WT, wildtype. *P<0.05; **p<0.01; ****p<0.0001.
Resistant Stat2−/− mice showed increased numbers of proliferating Ki67/EpCAM-double positive cells and had lower numbers of infiltrating F4/80+, CD11c+ and MPO+ cells as well as fewer TUNEL+ apoptotic cells (figure 5D, arrows). Collectively, these results clearly indicated that Stat2−/− animals were capable of controlling DSS-induced inflammation by various mechanism including a diminished recruitment of inflammatory cells and lower numbers of dead IECs. Indeed, inflammatory markers (ie, S100a8 and S100a9), members of the tissue remodelling/wound repair enzymes (ie, Mmp8), cell death factors (ie, Casp14) or genes implicated in the LPS/inflammatory response (ie, Wfdc1, Cled4d, Acod1) were all present among the most downregulated genes in resistant Stat2−/− mice from the DSS-induced colitis model (figure 5E, genes of interest for the present study are marked in blue). Importantly, Il36g, one of the top 20 most upregulated genes in susceptible IL20ra−/− mice, was present among the top 20 most downregulated genes in resistant Stat2−/− mice (underlined in figure 5E), providing further evidence for a cross-talk control mechanism between the IL-20/STAT3 and the IFN/STAT2 axis. Furthermore, resistant Stat2−/− mice had increased levels of various factors which have been previously correlated with lower inflammation levels in patients with IBD including the mouse ortholog of human CYP3A4 (ie, Cyp3a25 and Cyp3a44) or the negative regulator of monocyte/macrophage cytokine production, that is, Tmem178 31 (figure 5E, upregulated genes of interest in this context are marked in green). The heatmap clustering and Gene Ontology analysis of that experiment are presented in online supplemental figure 6C (original data: ArrayExpress E-MTAB-12655).
The analysis of RNA-Seq data from a complimentary mouse model in which colitis was induced by the administration of oxazolone in WT mice (presensitisation and challenge) revealed that the expression of type I IFN receptor 2 chain as well as of tens of IFN target genes (eg, Isg20, Ifit6, Ifit1) was significantly upregulated as compared with oxazolone-untreated WT mice (figure 5F, upper panels), supportive of type I IFN signalling in this context (raw data has not yet been released but can be made available upon request). Compared with WT animals, Stat2−/− mice were more resistant to oxazolone-induced colitis as indicated by lower levels of inflammation on endoscopy (eg, less fibrin, diarrhoea, bleeding and mucosal damage) and histology (eg, less crypt damage and oedema, fewer inflammatory cell infiltrates) providing further evidence for a broader critical role of STAT2 in the control of gut inflammation (figure 5F, middle and lower panels).
We next wondered whether the IEC-specific observations made in organoids could be translated in vivo. Accordingly, Stat2-floxed mice were generated using CRISPR/Cas9 technology (described in the online supplemental file). We crossed these mice to Villin-cre transgenic mice,32 to generate IEC-specific STAT2-deficient mice (Stat2ΔIEC ). During DSS-induced colitis, Stat2ΔIEC mice lost less body weight and developed less inflammation compared with Stat2fl/fl /Villin-cre-negative littermates by endoscopy, histology and in vivo imaging of neutrophil infiltration (figure 6A). Similarly, Stat2ΔIEC mice were more resistant in the oxazolone-induced colitis model (figure 6B) as compared with their floxed littermates, providing definitive evidence for the fact that the IEC-specific loss of STAT2 protects mice in different models of experimental colitis which are based on various pathological mechanisms.
Figure 6.
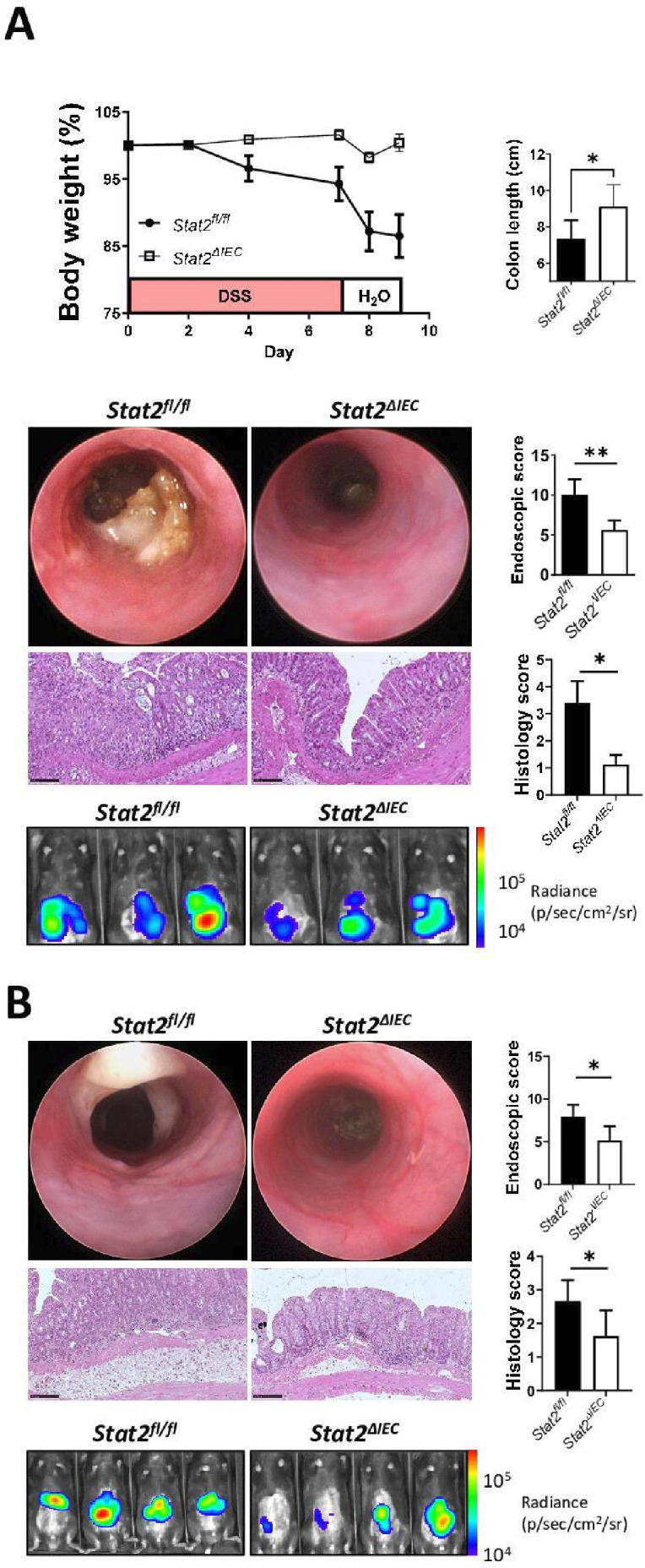
Stat2ΔIEC mice are resistant to DSS-induced and oxazolone-induced colitis. (A) Colitis (two independent experiments) was induced in mice by the administration of DSS in the drinking water for 7 days. Stat2ΔIEC mice were more resistant as compared with Stat2fl/fl littermates as indicated by the body weight curve, colon length, mini-endoscopy, histology and real-time imaging of neutrophil infiltration assessed by in vivo luminescence. (B) Compared with their Stat2fl/fl control littermates, Stat2ΔIEC developed less inflammation as defined by a similar set of criteria in the complementary model of oxazolone-induced colitis. Statistics: Welch’s t-test in A and B, mean with SEM or 95% CI is displayed. DSS, dextran sulfate sodium; IEC, intestinal epithelial cell. *P<0.05; **p<0.01.
IL-20 can interfere with the IFN-β/STAT2 epithelial cell death signalling in patients with IBD
Our extensive RNA-Seq analysis of human tissue biopsies indicated that STAT2 was upregulated in both patients with CD and UC as compared with non-IBD controls (figure 7A). Likewise, levels of the type I IFN receptor two chain as well as those of typical IFN target genes, for example, ISG20 were also upregulated in both patients with CD and UC as compared with non-IBD controls (online supplemental figure 7A, B). Moreover, we detected more activated STAT2 in samples from patients with IBD as compared with non-IBD controls (figure 7B, arrows) by immunofluorescence. Functionally, a short time incubation of IBD organoids with IFN-β induced pSTAT2 as indicated by western blot analysis (figure 7C) whereas longer incubation times resulted in organoids’ death (online supplemental figure 7C, red arrows indicate dead organoids, yellow arrows indicate distressed organoids and green arrows point to living, healthy organoids). We then stimulated IBD organoids with IFN-β for 24 hours and observed an increase in MLKL and pMLKL levels compared with unstimulated organoids by western blot analysis (figure 7D). Consistent with the observations made in mouse organoids, blocking necroptosis with the highly specific human MLKL inhibitor necrosulfonamide was able to rescue IBD-derived organoids from IFN-β-induced cell death (figure 7E). In another set of experiments, the presence of IL-20 resulted in fewer organoids being killed by IFN-β (online supplemental figure 7D). Although IL-10 was also able to partly protect organoids in the context of IFN-β co-stimulation (online supplemental figure 7D), quantification of the results from the propidium iodide/Hoechst and MTT-formazan indicated that IL-20 was more potent than IL-10 (online supplemental figure 7E, F).
Figure 7.

IL-20 interferes with IFN/STAT2-dependent necroptotic cell-death pathway in IECs from patients with IBD. (A) Levels of STAT2 expression in Crohn’s disease, UC and non-IBD controls as detected by RNA-Seq. (B) Confocal imaging of pSTAT2 in biopsies from Crohn‘s disease, UC and non-IBD controls (n=5/group) with positive IECs indicated by arrows. (C) Three-dimensional organoids generated from colon biopsies of patients with IBD were either stimulated for 30 min with 25 ng/mL IFN-β or were left untreated and protein extracts were subjected to western blot analysis with anti-pSTAT2 and STAT2 antibodies. Results from two patients are shown. Beta-actin served as loading control. (D) Results of MLKL and pMLKL levels after the incubation of IBD organoids with IFN-β for 24 hours. (E) IBD organoids (n=3 different patients) were incubated with IFN-β in the presence or absence of necrosulfonamide for 36 hours and cell death was assessed by staining with propidium iodide to mark dead cells. Hoechst was used for normalisation purposes. (F) Western blot analysis of pSTAT2 and STAT2 in IBD organoids after stimulations with IL-20 and IFN-β. Scale bars, 75 µm in B. Statistics: Welch’s t-test in A, mean with 95% CI is displayed. (G) Schematic representation of possible interactions between IL-20 and type I IFN signals in homeostasis, during the active phase and the resolution phases of intestinal inflammation. IEC, intestinal epithelial cell; IFN, interferon; IL, interleukin; RNA-Seq, RNA-sequencing. *P<0.05; ****p<0.0001.
gutjnl-2023-329628supp008.pdf (1.3MB, pdf)
IL-20 pre-incubation decreased levels of pSTAT2 after IFN-β as assessed by western blot analysis (figure 7F). Furthermore, stimulating fresh biopsies from patients with IBD suffering from active disease with IFN-β induced pSTAT2 in IECs (arrows) and in some lamina propria cells and this effect could be diminished by the pre-incubation with the STAT3 activator IL-20 prior to IFN-β addition (online supplemental figure 7G). Taken together, our results indicated that IL-20 signals are able to interfere with the pathogenic effects of type I IFN/STAT2 in IBD (figure 7G).
Discussion
The integrity of the intestinal barrier which is dictated by the perpetual cross-talk between the microbiota and the mucosal immune responses represents the key prerequisite for homeostasis in the gut. A breach of the epithelial barrier may result in dysbiosis which is characterised by an altered composition of the microbiota leading to intestinal inflammation.2–4 Our present study indicated that type I IFN/STAT2-signalling, which is activated in patients with IBD, negatively impacts intestinal homeostasis and drives epithelial cell necroptosis. Accordingly, Stat2 deficiency in IECs conferred protection in different experimental colitis mouse models. Furthermore, we identified IL-20 as a potent modulator of the type I IFN/STAT2 axis in the gut and could show that mice deficient in IL-20 signalling had increased susceptibility to experimental colitis and delayed intestinal mucosal healing. In patients with IBD, higher levels of IL20 were associated with increased rates of responsiveness to anti-TNF therapy suggesting a possible use of IL-20 as a biomarker for the management of IBD.
Our initial RNA-Seq data analysis of human biopsies indicated that among all IL receptors, IL20RA had the highest expression in patients with IBD as compared with non-IBD controls. This finding strengthened results of a microarray dataset published by López-Posadas et al who found higher IL20RA in isolated IECs from the inflamed versus the non-inflamed areas of the terminal ileum of patients with CD.21 Further extending our present RNA-Seq studies and complementing them with qPCR and immunofluorescence studies, we could confirm that IL-20RA and IL-20 and IL-20RB were upregulated in patients with IBD as compared with non-IBD controls. Moreover, IL-20 levels were higher in patients that responded to anti-TNF as compared with non-responders. We further identified that STAT3 which represents a central regulator of intestinal homeostasis can be activated by IL-20 stimulation in IECs from patients with IBD.
Members of the IL-20 subfamily of IL-10-related cytokines were shown to strengthen the epithelial barrier function by promoting the proliferation and differentiation of epithelial cells, inducing genes involved in wound healing and tissue remodelling and, with the exception of IL-26, the production of antimicrobial peptides (eg, S100A7, S100A8 and S100A9) and β-defensins.20 33 34 Moreover, IL-24, which has been recently linked to IBD,35 was also shown to induce messenger RNA (mRNA) of Muc1, Muc3, Muc4 in HT-29 cells.36 IL-19 was found to either protect from,37 or promote inflammation.38 Similarly, type III IFN (ie, IL-28) signals were protective in the colon,12 39 but promoted inflammation in the small intestine.13 A possible explanation for why IL-20 received little attention relies on the fact that IL-20 mRNA is not readily detectable under steady state conditions in the uninflamed gut. Additionally, the 3’ untranslated region of its complementary DNA contains as many as seven AUUUA and one perfect UUAUUUAUU mRNA instability motifs which might explain why the mRNA of IL-20 is extremely rare and short-lived and thus hard to detect.17 Moreover, whereas few studies indicated that colonic subepithelial myofibroblasts and epithelial cell lines do express IL-20RA and IL-20RB others found no expression of these receptor chains in the gut.37 40–42
A previous report found that IL20 mRNA levels were lower whereas those of IL20RA and IL20RB were higher in colon biopsies from patients with UC in remission compared with patients with active UC and non-inflammatory controls.19 In mice with DSS-induced colitis, we observed by immunofluorescence staining and RNAScope analysis that IL-20 is predominantly expressed by EpCAM+ IECs and by very few scattered EpCAM-negative cells, among which we identified some neutrophils and macrophages but no fibroblasts or glial cells. Similarly, highest expression of IL-20 was noted in IECs and some lamina propria cells in patients with IBD. IL-20RA and IL-20RB were predominantly expressed by IECs and to a lesser extent by lamina propria cells. These findings are in line with those from the above cited report which indicated that protein levels, unlike those of mRNA, were increased for IL-20 and IL-20RB on IECs and for IL-20, IL-20RA and IL-20RB in inflammatory cells in active lesions from patients with UC.19
On binding to its receptor, IL-20 signals primarily through the JAK-STAT pathway. Whereas the activation of STAT1 was observed with higher IL-20 concentration in most settings,17 and STAT5 was shown to be activated following IL-20 stimulation in endothelial cells,43 the major route proceeds through STAT3.33 In our study, we could demonstrate that IL-20 induced STAT3 phosphorylation in IECs from organoids and from IBD biopsies. Hence, IL-20 might coordinate mucosal healing in the context of type I IFN-induced pathogenesis by its impact on STAT3-mediated signalling. Several mechanisms might contribute to this outcome. First, the overexpression of STAT3 in cancer B cell lines was able to negatively regulate type I IFN signalling by blocking the promoter regions of critical genes of this pathway, that is, STAT1, STAT2 and IRF7 and IRF9.44 Our unpublished observations suggest that this scenario is less likely in primary IECs, since cytokine-dependent activation of pSTAT3 had only limited effects on IRF7, IRF9, STAT1 and STAT2 mRNA and protein levels. Second, STAT3 may associate with STAT2, as shown in skeletal muscle during myogenic differentiation, thus making it unavailable for mediating type I IFN signalling and thereby blocking death impulses.45 In our co-immunoprecipitation experiment, we could not observe an interaction between pSTAT3 and pSTAT2. Third, suppressor of cytokine signalling 3, downstream of STAT3 signalling, inhibits type I IFN responses.46 Fourth, STAT3 may compete against STAT2 for binding to STAT1,47 and therefore proliferation (STAT1:STAT3) might predominate over cell death (STAT1:STAT2). The latter scenario is favoured by our co-immunoprecipitation analysis showing that pSTAT1 can bind to pSTAT2 whereas pSTAT3 cannot, hence pSTAT3 could be free to interact with pSTAT1.
The presence of IL-20 at the edges of colonic wounds in our mice with DSS-induced colitis and in patients with IBD suggested that damage to epithelial tissues favours local production of IL-20, possibly due to exposure to microbial factors after barrier alteration. In support of this, CpG oligonucleotides, a well-known microbial product, as recently revised,48 was found to significantly induce IL-20 promoter activity.49 Local production of IL-20 near erosions/ulcerations may aid in the restoration of epithelium through a mechanism that depends on STAT3. A similar pattern has been reported earlier for various factors involved in the proliferation of mucosal cells and recovery processes, such as IL-22.24
In contrast to the constitutive overexpression of IL-20 in transgenic mice which showed perinatal lethality,17 our WT mice in which IL-20 overexpression was achieved by the administration of Il20 vectors did not show any pathology. Furthermore, these mice were able to better control the inflammation and showed less tissue destruction after DSS administration. The high level of IL20 in patients with IBD is likely to represent a compensatory anti-inflammatory mechanism aimed to counteract IFN-induced inflammation. Whereas the addition of IFN-β alone damaged organoids, this action could be partly blocked by IL-20 suggesting an anti-inflammatory effect of IL-20 by modulating IEC survival.
By staining biopsies from patients with IBD, we identified EpCAM+ IECs as major sources of phosphorylated STAT2. Microbiota triggers initiate immediate and very robust type I IFN signalling in experimental DSS colitis,50 and since this signalling induces STAT2-dependent IECs death and accentuates inflammation, our findings in patients sustained a pathogenic role for STAT2 in IBD. In line with this hypothesis, Stat2−/− animals presented lower levels of inflammation during the course of DSS-induced colitis. To further dwell on the importance of STAT2 signalling in IECs, we crossed our newly created Stat2-floxed mice with Villin-cre transgenic mice to specifically cut the floxed gene in the intestinal epithelium.32 The finding that conditional Stat2ΔIEC were more resistant to experimental colitis as compared with Stat2-floxed littermates was in stark contrast to the phenotype of Stat1ΔIEC and Stat3ΔIEC mice which were more sensitive to experimental colitis.12 24 To further understand the molecular basis of this specificity, we performed experiments using intestinal organoids from Stat2−/− and WT mice. Stat2−/− organoids grew faster and bigger compared with WT organoids and were extremely resistant to cytokine-induced cell death. Indeed, it is by now well established that type I IFN inhibits cell growth and induces cell death in a STAT2-dependent manner, as demonstrated by the increased resistance against type I IFN-induced cell death in STAT2-deficient Jurkat,51 and Daudi,52 cell lines. Caspase 3-mediated apoptosis and the loss of mitochondrial membrane potential were shown to be involved in this process.51 Our RNA-Seq analysis indicated that type I IFN stimulation increased Mlkl and to a lesser extent Casp8 in WT organoids whereas no change was observed in Stat2−/− organoids, suggesting that STAT2-mediated cell death is rather linked to MLKL-dependent necroptosis than to caspase 8-dependent apoptosis. Furthermore, genes that have been previously linked to the breakdown of the gut homeostasis and the immune tolerance during colitis, for example, IFIT2/ISG54 (regulates caspase 3-mediated apoptosis via Bax and Bak in the mitochondrial pathway),53 CXCL10 (inhibits IEC proliferation),54 IFI204/IFI16 (increases production of IFNs and inflammatory cytokines and regulates cell death)—whose levels were increased in IECs from patients with IBD55—were among the top most upregulated genes in WT organoids treated with IFN-β.
Mucosal healing assessed via endoscopy has emerged as a major therapeutic goal in IBD since it predicts sustained clinical remission and resection-free survival.56 Deficiency in Il20, Il20ra or Il20rb all resulted in increased susceptibility of mice to DSS-induced colitis. Additional results showing higher susceptibility to oxazolone-induced colitis in mice deficient in IL-20 signalling strengthened the hypothesis of a broader relevance of IL-20 in the context of intestinal inflammation and mucosal healing. However, none of these models is an ideal setting to mimic findings in human IBD. Future studies using immunologically mediated mouse models in combination with the use of conditional knockout models will be helpful to define the precise place of IL-20 signalling in the pathogenesis of IBD and its possible implications for therapy.
The treatment of IECs with IFN-β induced MLKL, a key regulator of a caspase-independent lytic form of cell death called necroptosis which is driven by RIPK1.57 Our functional studies indicated that the IFN-induced cell death can be downregulated by pharmacologically blocking necroptosis in both human and murine IECs. Likewise, IL-20 and to a lesser extent IL-10, another cytokine with potent anti-inflammatory effects on IECs,58 were able to block IFN-induced cell death. Since IL-10 is expressed under steady state conditions in the gut, whereas IL-20 was mainly expressed on injury of IECs in the inflamed mucosa, these findings suggest a scenario in which IL-10 controls IEC survival in intestinal homeostasis, while IL-20 may act as a danger signal to allow homeostasis to be restored. The implications of this concept for clinical therapy require further investigation. Previous attempts to limit disease by activating STAT3-inducing cytokines have not been successful in achieving remission in patients with IBD. In one study, recombinant IL-10 therapy in human clinical IBD trials had disappointing outcomes, possibly due to its low mucosal bioavailability.59 Furthermore, despite promising results in experimental colitis models that used genetically modified bacteria expressing IL-10,60 61 phase II clinical trials showed no improvement of disease activity in IBD by IL-10 administration.62 Recently, it was found that IL-10-secreting B cells that were activated by physiologically relevant bacteria did not ameliorate T cell-mediated colitis or induced Tr1 cells in the absence of T cell IL-27 signalling in vivo.63 In general, treating active IBD with recombinant cytokines has so far yielded unsatisfactory outcomes. In the future, it may be an interesting concept to focus on maintaining remission instead of inducing it as a therapeutic target in IBD for members of the IL-10 family. This notion may be of particular interest for IL-20 since it provides direct protection functions on the epithelium that could strengthen the intestinal barrier, which plays a fundamental role in the pathogenesis of recurrences and flare-ups in IBD.64 Future studies should investigate whether enhancing IL-20 signalling by itself or in combination with other cytokines like IL-10 could have potential benefits in patients with IBD.
Taken together, our results indicated that IL-20 is able to restore homeostasis by blocking type I IFN/STAT2-mediated death signals and to promote tissue repair during intestinal inflammation. Thus, modulation of the interactions between IL-20 and STAT2-dependent signalling emerges as a potentially novel approach to improve epithelial integrity in IBD. Further understanding of the complex interactions between IL-20- and STAT2-dependent signalling will be critical for identifying innovative therapeutic targets for intervention in patients with IBD.
gutjnl-2023-329628supp010.pdf (1MB, pdf)
Acknowledgments
We would like to thank our technicians for excellent assistance: Alexandra Wandersee (sample preparation during the initial phase of the project), Ludmilla Sologub and Angelika Wilfer (preparation of human samples), Astrid Taut (preparation of the Il20 and control vectors). We would also like to thank Martina Döhler and Dieter Engelkamp (Transgenic Facility, Biology Department, Faculty of Sciences, University of Erlangen-Nuremberg, Erlangen, Germany) for the generation of the Il20−/− and IL20ra−/− mouse strains by in vitro fertilisation.
Footnotes
Correction notice: This article has been corrected since it published Online First. The funding statement has been updated.
Contributors: Conceived and designed experiments: MTC, ZH, CB, MFN. Performed experiments: MTC, ZH, CG, LW, AD, LS, SZ. Provided critical samples/materials and reagents/protocols: CG, RA, IS, LW, RG-B, LE, GH, SZ, TR, SV, SD, AAK, BS, AH, SW, JS, SF, CB, MFN. Analysed and interpreted data: MTC, ZH, MGA, GS, ZT, AAK, BS, CB, MFN. Wrote the paper: MTC and MFN. All authors critically read and approved the final version of the manuscript. MTC is the guarantor.
Funding: GS was supported by a DOC fellowship of the Austrian Academy of Sciences. ZT was supported by the Austrian Science Fund project I-6057. DFG sequencing grant - project number 418055832 (BS and CB). This work received funding from the Interdisciplinary Center for Clinical Research Erlangen: IZKF A78 (MTC and MFN) and from the German Research Council: DFG CH1428/2-1 (MTC), DFG CH1428/2-2 (MTC), DFG NE490/13-1 (MFN), DFG NE490/13-2 (MFN), CRC TRR241-Project-ID 375876048 (CG, RA, SZ, ZT, AAK, BS, SW, CB, MFN), CRC 1181 (CB, MFN), FOR 2438 (CB, MFN), KFO 257 (RA, SW,CB, MFN), SPP 1656 (CG, SW, MFN).
Competing interests: MFN has served as an advisor for Pentax, Giuliani, PPM, BMS, Janssen, MSD, Takeda and Boehringer. MFN has served as an Associate Editor of the journal/Editorial Board Member. BS has served as consultant for AbbVie, Arena, BMS, Boehringer, Celgene, Falk, Galapagos, Janssen, Lilly, Pfizer, Prometheus and Takeda and received speaker’s fees from AbbVie, CED Service, Falk, Ferring, Janssen, Novartis, Pfizer, Takeda (served as representative of the Charité). All other authors have nothing to disclose.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Open access data relevant for the present study is available from https://www.ebi.ac.uk/biostudies/arrayexpress: E-MTAB-12655, E-MTAB-12662, E-MTAB-12737 or upon request.
Ethics statements
Patient consent for publication
Consent obtained directly from patient(s).
Ethics approval
The collection of human samples was approved by the local Ethics Committee and Review Board of the University of Erlangen-Nuremberg, the Charité—Universitätsmedizin Berlin and the Humanitas Research Hospital, Milan. Animal experiments were approved by local authorities. Each patient gave written informed consent.
References
- 1. Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science 2005;307:1920–5. 10.1126/science.1106442 [DOI] [PubMed] [Google Scholar]
- 2. Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest 2007;117:514–21. 10.1172/JCI30587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010;28:573–621. 10.1146/annurev-immunol-030409-101225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. MacDonald TT, Monteleone I, Fantini MC, et al. Regulation of homeostasis and inflammation in the intestine. Gastroenterology 2011;140:1768–75. 10.1053/j.gastro.2011.02.047 [DOI] [PubMed] [Google Scholar]
- 5. Buckley CD, Gilroy DW, Serhan CN, et al. The resolution of inflammation. Nat Rev Immunol 2013;13:59–66. 10.1038/nri3362 [DOI] [PubMed] [Google Scholar]
- 6. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 2017;14:269–78. 10.1038/nrgastro.2016.208 [DOI] [PubMed] [Google Scholar]
- 7. Kaser A, Tilg H. Novel therapeutic targets in the treatment of IBD. Expert Opin Ther Targets 2008;12:553–63. 10.1517/14728222.12.5.553 [DOI] [PubMed] [Google Scholar]
- 8. Schindler C, Shuai K, Prezioso VR, et al. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 1992;257:809–13. 10.1126/science.1496401 [DOI] [PubMed] [Google Scholar]
- 9. Philips RL, Wang Y, Cheon H, et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell 2022;185:3857–76. 10.1016/j.cell.2022.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol 2019;20:970–9. 10.1038/s41590-019-0415-0 [DOI] [PubMed] [Google Scholar]
- 11. Wirusanti NI, Baldridge MT, Harris VC. Microbiota regulation of viral infections through interferon signaling. Trends Microbiol 2022;30:778–92. 10.1016/j.tim.2022.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiriac MT, Buchen B, Wandersee A, et al. Activation of epithelial signal transducer and activator of transcription 1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology 2017;153:123–38. 10.1053/j.gastro.2017.03.015 [DOI] [PubMed] [Google Scholar]
- 13. Günther C, Ruder B, Stolzer I, et al. Interferon lambda promotes paneth cell death via STAT1 signaling in mice and is increased in inflamed Ileal tissues of patients with Crohn’s disease. Gastroenterology 2019;157:1310–22. 10.1053/j.gastro.2019.07.031 [DOI] [PubMed] [Google Scholar]
- 14. Mudter J, Weigmann B, Bartsch B, et al. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am J Gastroenterol 2005;100:64–72. 10.1111/j.1572-0241.2005.40615.x [DOI] [PubMed] [Google Scholar]
- 15. Rauch I, Hainzl E, Rosebrock F, et al. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur J Immunol 2014;44:2749–60. 10.1002/eji.201344401 [DOI] [PubMed] [Google Scholar]
- 16. Ouyang W, Rutz S, Crellin NK, et al. Regulation and functions of the IL-10 family of Cytokines in inflammation and disease. Annu Rev Immunol 2011;29:71–109. 10.1146/annurev-immunol-031210-101312 [DOI] [PubMed] [Google Scholar]
- 17. Blumberg H, Conklin D, Xu WF, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell 2001;104:9–19. 10.1016/s0092-8674(01)00187-8 [DOI] [PubMed] [Google Scholar]
- 18. Yamamoto-Furusho JK, De-León-Rendón JL, de la Torre MG, et al. Genetic polymorphisms of interleukin 20 (IL-20) in patients with ulcerative colitis. Immunol Lett 2013;149:50–3. 10.1016/j.imlet.2012.11.008 [DOI] [PubMed] [Google Scholar]
- 19. Fonseca-Camarillo G, Furuzawa-Carballeda J, Llorente L, et al. IL-10-- and IL-20--expressing epithelial and inflammatory cells are increased in patients with ulcerative colitis. J Clin Immunol 2013;33:640–8. 10.1007/s10875-012-9843-4 [DOI] [PubMed] [Google Scholar]
- 20. Niess JH, Hruz P, Kaymak T. The interleukin-20 cytokines in intestinal diseases. Front Immunol 2018;9:1373. 10.3389/fimmu.2018.01373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. López-Posadas R, Becker C, Günther C, et al. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation. J Clin Invest 2016;126:611–26. 10.1172/JCI80997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arijs I, De Hertogh G, Lemaire K, et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first Infliximab treatment. PLoS One 2009;4:e7984. 10.1371/journal.pone.0007984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patankar JV, Müller TM, Kantham S, et al. E-type prostanoid receptor 4 drives resolution of intestinal inflammation by blocking epithelial necroptosis. Nat Cell Biol 2021;23:796–807. 10.1038/s41556-021-00708-8 [DOI] [PubMed] [Google Scholar]
- 24. Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 2009;206:1465–72. 10.1084/jem.20082683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013;39:357–71. 10.1016/j.immuni.2013.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grabinger T, Luks L, Kostadinova F, et al. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis 2014;5:e1228. 10.1038/cddis.2014.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bode KJ, Mueller S, Schweinlin M, et al. A fast and simple fluorometric method to detect cell death in 3d intestinal organoids. Biotechniques 2019;67:23–8. 10.2144/btn-2019-0023 [DOI] [PubMed] [Google Scholar]
- 28. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112–9. 10.1038/nchembio711 [DOI] [PubMed] [Google Scholar]
- 29. Stolzer I, Ruder B, Neurath MF, et al. Interferons at the crossroad of cell death pathways during gastrointestinal inflammation and infection. Int J Med Microbiol 2021;311:151491. 10.1016/j.ijmm.2021.151491 [DOI] [PubMed] [Google Scholar]
- 30. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity 2012;36:503–14. 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mahajan S, Decker CE, Yang Z, et al. Plcγ2/Tmem178 dependent pathway in myeloid cells modulates the pathogenesis of cytokine storm syndrome. J Autoimmun 2019;100:62–74. 10.1016/j.jaut.2019.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Madison BB, Dunbar L, Qiao XT, et al. Cis elements of the Villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 2002;277:33275–83. 10.1074/jbc.M204935200 [DOI] [PubMed] [Google Scholar]
- 33. Rutz S, Wang X, Ouyang W. The IL-20 subfamily of cytokines--from host defence to tissue homeostasis. Nat Rev Immunol 2014;14:783–95. 10.1038/nri3766 [DOI] [PubMed] [Google Scholar]
- 34. Wegenka UM. IL-20: biological functions mediated through two types of receptor complexes. Cytokine Growth Factor Rev 2010;21:353–63. 10.1016/j.cytogfr.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 35. Ónody A, Veres-Székely A, Pap D, et al. Interleukin-24 regulates mucosal remodeling in inflammatory bowel diseases. J Transl Med 2021;19:237. 10.1186/s12967-021-02890-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andoh A, Shioya M, Nishida A, et al. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J Immunol 2009;183:687–95. 10.4049/jimmunol.0804169 [DOI] [PubMed] [Google Scholar]
- 37. Azuma Y-T, Matsuo Y, Kuwamura M, et al. Interleukin-19 protects mice from innate-mediated colonic inflammation. Inflamm Bowel Dis 2010;16:1017–28. 10.1002/ibd.21151 [DOI] [PubMed] [Google Scholar]
- 38. Steinert A, Linas I, Kaya B, et al. The stimulation of macrophages with TLR ligands supports increased IL-19 expression in inflammatory bowel disease patients and in colitis models. J Immunol 2017;199:2570–84. 10.4049/jimmunol.1700350 [DOI] [PubMed] [Google Scholar]
- 39. Broggi A, Tan Y, Granucci F, et al. IFN-Lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 2017;18:1084–93. 10.1038/ni.3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andoh A, Zhang Z, Inatomi O, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005;129:969–84. 10.1053/j.gastro.2005.06.071 [DOI] [PubMed] [Google Scholar]
- 41. Dambacher J, Beigel F, Zitzmann K, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut 2009;58:1207–17. 10.1136/gut.2007.130112 [DOI] [PubMed] [Google Scholar]
- 42. Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 2008;14:282–9. 10.1038/nm1720 [DOI] [PubMed] [Google Scholar]
- 43. Tritsaris K, Myren M, Ditlev SB, et al. IL-20 is an arteriogenic cytokine that remodels collateral networks and improves functions of ischemic hind limbs. Proc Natl Acad Sci USA 2007;104:15364–9. 10.1073/pnas.0707302104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu L, Zhu F, Zhang M, et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc Natl Acad Sci USA 2018;115. 10.1073/pnas.1715118115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang K, Wang C, Xiao F, et al. JAK2/STAT2/STAT3 are required for myogenic differentiation. Journal of Biological Chemistry 2008;283:34029–36. 10.1074/jbc.M803012200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sakai I, Takeuchi K, Yamauchi H, et al. Constitutive expression of SOCS3 confers resistance to IFN-alpha in chronic myelogenous leukemia cells. Blood 2002;100:2926–31. 10.1182/blood-2002-01-0073 [DOI] [PubMed] [Google Scholar]
- 47. Stancato LF, David M, Carter-Su C, et al. Preassociation of STAT1 with STAT2 and STAT3 in separate signalling complexes prior to cytokine stimulation. J Biol Chem 1996;271:4134–7. 10.1074/jbc.271.8.4134 [DOI] [PubMed] [Google Scholar]
- 48. Lau HCH, Sung J-Y, Yu J. Gut microbiota: impacts on gastrointestinal cancer immunotherapy. Gut Microbes 2021;13:1–21. 10.1080/19490976.2020.1869504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang L, Li K, Xu Q, et al. Potential synergy between SNP and CpG-A or IL-1Β in regulating transcriptional activity of IL-20 promoter. J Invest Dermatol 2014;134:389–95. 10.1038/jid.2013.314 [DOI] [PubMed] [Google Scholar]
- 50. Czarnewski P, Parigi SM, Sorini C, et al. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat Commun 2019;10:2892. 10.1038/s41467-019-10769-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Romero-Weaver AL, Wang H-W, Steen HC, et al. Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol Cancer Res 2010;8:80–92. 10.1158/1541-7786.MCR-08-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Du Z, Fan M, Kim J-G, et al. Interferon-resistant daudi cell line with a STAT2 defect is resistant to apoptosis induced by chemotherapeutic agents. J Biol Chem 2009;284:27808–15. 10.1074/jbc.M109.028324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stawowczyk M, Van Scoy S, Kumar KP, et al. The interferon stimulated gene 54 promotes apoptosis. J Biol Chem 2011;286:7257–66. 10.1074/jbc.M110.207068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sasaki S, Yoneyama H, Suzuki K, et al. Blockade of CXCL10 protects mice from acute colitis and enhances crypt cell survival. Eur J Immunol 2002;32:3197–205. [DOI] [PubMed] [Google Scholar]
- 55. Caneparo V, Pastorelli L, Pisani LF, et al. Distinct anti-IFI16 and anti-GP2 antibodies in inflammatory bowel disease and their variation with infliximab therapy. Inflamm Bowel Dis 2016;22:2977–87. 10.1097/MIB.0000000000000926 [DOI] [PubMed] [Google Scholar]
- 56. Neurath MF, Travis SPL. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut 2012;61:1619–35. 10.1136/gutjnl-2012-302830 [DOI] [PubMed] [Google Scholar]
- 57. Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008;4:313–21. 10.1038/nchembio.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nguyen HD, Aljamaei HM, Stadnyk AW. The production and function of endogenous interleukin-10 in intestinal epithelial cells and gut homeostasis. Cell Mol Gastroenterol Hepatol 2021;12:1343–52. 10.1016/j.jcmgh.2021.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marlow GJ, van Gent D, Ferguson LR. Why interleukin-10 supplementation does not work in Crohn’s disease patients. World J Gastroenterol 2013;19:3931–41. 10.3748/wjg.v19.i25.3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Steidler L, Neirynck S, Huyghebaert N, et al. Biological containment of genetically modified lactococcus lactis for intestinal delivery of human interleukin 10. Nat Biotechnol 2003;21:785–9. 10.1038/nbt840 [DOI] [PubMed] [Google Scholar]
- 61. Luerce TD, Gomes-Santos AC, Rocha CS, et al. Anti-inflammatory effects of lactococcus Lactis NCDO 2118 during the remission period of chemically induced colitis. Gut Pathog 2014;6:33. 10.1186/1757-4749-6-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Braat H, Rottiers P, Hommes DW, et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin Gastroenterol Hepatol 2006;4:754–9. 10.1016/j.cgh.2006.03.028 [DOI] [PubMed] [Google Scholar]
- 63. Mishima Y, Liu B, Hansen JJ, et al. Resident bacteria-stimulated IL-10-secreting B cells ameliorate T cell-mediated colitis by inducing Tr-1 cells that require IL-27-signaling. Cell Mol Gastroenterol Hepatol 2015;1:295–310. 10.1016/j.jcmgh.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rath T, Atreya R, Bodenschatz J, et al. Intestinal barrier healing is superior to endoscopic and histologic remission for predicting major adverse outcomes in inflammatory bowel disease: the prospective Erica trial. Gastroenterology 2023;164:241–55. 10.1053/j.gastro.2022.10.014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
gutjnl-2023-329628supp009.pdf (307.9KB, pdf)
gutjnl-2023-329628supp011.pdf (94.8KB, pdf)
gutjnl-2023-329628supp001.pdf (166.7KB, pdf)
gutjnl-2023-329628supp002.pdf (2.2MB, pdf)
gutjnl-2023-329628supp003.pdf (624.6KB, pdf)
gutjnl-2023-329628supp004.pdf (1.9MB, pdf)
gutjnl-2023-329628supp005.pdf (1.2MB, pdf)
gutjnl-2023-329628supp006.pdf (585.2KB, pdf)
gutjnl-2023-329628supp007.pdf (824.8KB, pdf)
gutjnl-2023-329628supp008.pdf (1.3MB, pdf)
gutjnl-2023-329628supp010.pdf (1MB, pdf)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information. Open access data relevant for the present study is available from https://www.ebi.ac.uk/biostudies/arrayexpress: E-MTAB-12655, E-MTAB-12662, E-MTAB-12737 or upon request.