

Summary
Microproteins encoded by small open reading frames (sORFs) have emerged as a fascinating frontier in genomics. Traditionally overlooked due to their small size, recent technological advancements such as ribosome profiling, mass spectrometry-based strategies and advanced computational approaches have led to the annotation of more than 7000 sORFs in the human genome. Despite the vast progress, only a tiny portion of these microproteins have been characterized and an important challenge in the field lies in identifying functionally relevant microproteins and understanding their role in different cellular contexts. In this review, we explore the recent advancements in sORF research, focusing on the new methodologies and computational approaches that have facilitated their identification and functional characterization. Leveraging these new tools hold great promise for dissecting the diverse cellular roles of microproteins and will ultimately pave the way for understanding their role in the pathogenesis of diseases and identifying new therapeutic targets.
Subject areas: Biological sciences, Genetics, Molecular biology, Biotechnology
Graphical abstract
Biological sciences; Genetics; Molecular biology; Biotechnology
Introduction
The mammalian genome is composed of a vast number of uncharacterized and unannotated small open reading frames (sORFs), which are commonly misinterpreted as “junk DNA” with no defined function outside of gene regulation. With the advent of new technologies, thousands of unannotated sORFs – typically located on non-coding RNAs and untranslated regions (UTRs) of protein-coding genes - have been shown to be translated into functional proteins. New technologies, such as proteomics and ribosome profiling, in tandem with advanced bioinformatic methods, have played a critical role in driving forward the sORF field. The combination of these approaches greatly facilitated the genome-wide annotation of sORFs, thereby unraveling their involvement in various cellular functions, including those relevant to human diseases.1,2,3,4,5,6,7
Most of these novel open reading frames (ORF) defy the conventional rules for gene annotation. These rules include a minimal length of 100 codons, an in-frame AUG start and a single ORF per transcript.4,8,9 Unannotated ORFs smaller than 100 amino acids are classified as sORFs (Figure 1). These include sORFs on non-coding RNAs such as long non-coding RNAs (lncRNAs) and overlapping sequences on annotated ORFs, classified as alternative ORFs (alt-ORFs).4,10,11 Moreover, sORFs residing in the 5′ untranslated region (5′UTR) of an mRNA are referred to as upstream open reading frames (uORFs),6,12,13 while those found in the 3′UTR are known as downstream ORF (dORFs).12,13,14
Figure 1.

The classification of sORFs
Schematic representation of different open reading frames (ORFs) and their genomic location. A large fraction of the mammalian genome is composed of small open reading frames (sORFs) in the untranslated regions (red). Canonical ORFs, conventionally more than 100 amino acids long, are depicted at the top, with exons delimited with a known start and stop codon flanked by 5′ untranslated region (UTR) and 3′UTR. The mammalian genome encodes transcribed and potentially functional sORFs between 10 and 100 amino acids, which can be classified according to their genomic location. Upstream open reading frames (uORFs) are found in the 5′UTR of conventional ORFs, while downstream ORFs (dORFs) are found in the 3′UTR of conventional ORFs. In some cases, alternative ORFs arise from alternative initiation start sites within canonical ORFs and lead to shorter isoforms of a known ORF. sORFs can also be found in intronic regions of canonical ORFs and in intergenic regions between two canonical ORFs, known as intronic and intergenic sORFs, respectively. Finally, an important source of sORFs are long non-coding ORFs.
Recent endeavors to generate standardized sORF catalogs led to the annotation of more than 7000 human sORFs and suggest that sORFs form a substantial part of eukaryotic genomes.4,5,6,15 The encoded peptides or microproteins translated from sORFs are involved in a variety of cellular functions in both health and disease.2,12,16 Microproteins are involved in the downregulation of tumor angiogenesis,17 suppress tumor growth,18 or in cell proliferation.19 Furthermore, uncharacterized sORFs hold great promise as potential drug targets that drive different cellular processes underlying the pathogenesis of diseases.2,16,20
The translation of sORFs can either result in a peptide product or it may have a regulatory function, a phenomenon widely observed in the case of most known uORFs.12 About 50% of mammalian genes contain uORFs,21,22,23 which modulate ribosome access to the downstream ORF and which can reduce translational efficiency by an average of 30–48%.6 However, under stress conditions, uORF-mediated regulation allows certain genes such as ATF4 to become translationally induced to rapidly mount cellular stress responses.24,25 Genome-wide uORF translation may be subject to regulation and contribute to the translational program in embryonic stem cells or tumor initiation. Embryonic stem cells (ESCs) decrease their relative uORF translation rates when undergoing differentiation.5 Similarly, the stemness signature of muscle stem cells has been shown to be partly regulated by uORF-containing mRNAs.26 Furthermore, tumor-initiating cells increase their relative rate of uORF translation during the early stages of tumorigenesis.27
Here, we review the current methodologies for studying sORFs in the eukaryotic genome and outline emerging new techniques to study the function of sORFs and their potential involvement in disease. Although we focus on the eukaryotic genome in this review, it is worth noting that substantial progress has been made in the study of bacterial and plant sORFs and that most of the identification techniques described here can also be applied to lower organisms.
Bioinformatic approaches
New computational approaches and the availability of RNA sequencing datasets have led to better transcriptome annotations and facilitated the classification of microproteins (Figure 2). Historically, ORFs were defined as a sequence of DNA that is delimited by a start codon followed by a downstream in-frame stop codon. However, this approach was biased as it involved an artificial cutoff in annotating only proteins larger than 100 amino acids or 300 nucleotides, mainly because any sequence smaller than this cutoff was considered nonfunctional or derivative artifacts of canonical transcripts and coding sequences.9,28,29 Additionally, the 100 amino acid cutoff results from the increasing probability of artifactual ORFs and biologically meaningless sequences found in shorter ORFs.9 The likelihood of a protein-coding ORF increases with its length. Thus the reason why many algorithms had a fixed threshold of 100 amino acids was to avoid dubious non-coding ORFs.9,30,31,32 Consequently, current protein catalogs are skewed for larger proteins, which has resulted in a notable underrepresentation of microproteins.5,23 In light of the mounting evidence supporting the presence of sORFs, new algorithms have been developed to adjust the classification parameters for the annotation of ORFs.
Figure 2.

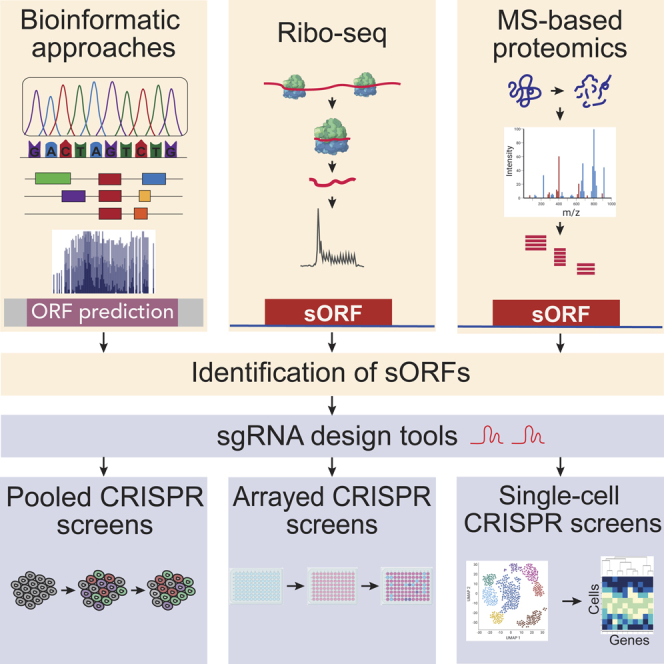
Identification of sORFs
Schematic workflow of the different methods used for the identification of small ORFs. Samples from diverse sources, human biopsies, mouse cells and cultured cells can be processed using ribosome profiling (Ribo-Seq), mass spectrometry (MS) and/or computational approaches. Ribo-Seq captures snapshots of ribosome-protected fragments that are purified and sequenced. Small ORFs showing 3-nucleotide periodicity are most likely to be translated into microproteins. Microproteins can be extracted, digested, fractionated and enriched by size selection followed by proteomics. Data are searched against custom databases containing the potential sORFs. Computational approaches to determine sORFs rely on predictions based on the conservation between species, codon bias and coding potential and transcriptomic and proteomic data analysis. The different algorithms can predict the presence of sORF based on detecting similarity to known proteins or domains, nucleotide composition, codon substitution or machine learning approaches.
Traditionally, protein annotation has heavily relied on evaluating sequence similarities and conservation across different species, which proves valuable as selection pressure is linked to functional importance.12,33 Furthermore, examining the similarity between known protein domains has provided valuable insights into the potential function of newly predicted ORFs. However, relying solely on conservation as a criterion for identifying non-canonical ORFs could limit the detection of sORFs that, although not conserved across species, may still encode functional microprotein.12,34 Short proteins are more challenging to classify than larger ones due to stricter statistical features to distinguish them from non-coding sequences.31 Sequences shorter than 100 amino acids are less likely to show conservation between species.12,35 Conserved coding sequences show a higher ratio of synonymous compared to non-synonymous substitutions (dS/dN), which can be exploited to distinguish them from non-coding regions, a difference that is less pronounced in smaller proteins.31
To address these challenges, a pipeline called PhyloCSF was developed, which can systematically resolve conservation problems by considering phylogenetic models for shorter sequences. PhyloCSF distinguishes itself from previous tools by using empirical codon models, which can compare alignments of coding regions with alignments of non-coding regions. Moreover, it incorporates genome-wide training data, taking into account codon frequencies and substitution rates to discern protein-coding from non-coding sequences.36 A recent application of PhyloCSF resulted in the identification of 144 novel coding sequences absent in existing catalogs.37 Notably, 50 of these newly discovered protein-coding genes encode microproteins containing fewer than 100 amino acids, further advocating for the utilization of PhyloCSF in sORF detection.37
Annotation of sORFs is not only challenging but also limited by computational algorithms. Many in silico approaches in the past restricted in the annotation of ORFs, including the necessity for a single coding sequence per transcript, an AUG start codon, a codon bias, a coding region longer than 100 codons and the sequence conservation.38,39,40,41 With the advent of new technologies and the general increase in sequencing data, computational pipelines have been modified to adjust these criteria. In silico approaches can now predict all possible ORF, including overlapping and shorter ORFs from existing annotations, novel predicted isoforms and novel proteins from alternative ORFs.40 OpenProt is a bioinformatic tool that uses two sources of annotations (NCBI-RefSeq and Ensembl) and publicly available ribosome profiling and mass spectrometry datasets to facilitate the annotation of predicted sORFs.40 An advantage of the technique is that it allows predicting sORFs using a minimal length of 30 amino acids and, in its latest update, has even removed the restriction for AUG start codons.42 Other annotation tools such as ORF finder,43 micPDP4 and uPEPperoni44 are also used for the detection of sORFs and are reviewed in Table 1. In recent years, sORF databases have become available such as sORFs.org 45 and smProt.46
Table 1.
Bioinformatic tools
| Methods based on sequence prediction | Reference | |
|---|---|---|
| PhyloCSF | Codon substitution and conservation elements | Sandmann et al., 202335 |
| OpenProt | Uses public available datasets to asses Ribo-seq and MS | Brunet et al., 201940 |
| RNASamba | Neuroal network to predict sORFs, recognizes Kozak sequence | Hanada et al., 201043 |
| DeepCPP | Algorithm using nucleotide bias to predict sORFs | Olexiouk et al., 201645 |
| MiPepid | Machine learning tool using for identificaton of sORFs based on known sORFs | Zhang et al., 202147 |
| csORF-finder | Uses trinucleotide deviation from expected mean to distinguish between coding sORFs from non-coding sORFs | Zhu and Gribskov, 201948 |
| ORFanage | Pseudo alignment algorithm for the detection of sORFs from RNA-seq data | Zhang et al., 202249 |
| sORF finder | Detection of sORFs according to 3-nucleotide composition bias | Brunet et al., 202142 |
| micPDP | Search for sORFs based on codon substitutions observed in whole-genome alignments | Bazzini et al., 20144 |
| uPEPperoni | Detection of uORFs based on location and transcript conservation | Skarshewski et al., 201444 |
| sORFs.org | Database of sORFs based on Ribo-seq data and integrates MS evidence and conservation searches | Li et al., 202146 |
| SmPROT | sORFs reported in literature, databases, Ribo-seq and MS data | Zhang et al., 202147 |
| Methods based on ribosome profiling | ||
| ORF-Rater | ORF Regression algorithm using Ribo-seq data to quantify translation regardless of start codon, overlap and length | Kute, et al., 202239 |
| ORFscore | Codon in-frame reads | Bazzini et al., 20144 |
| ORFquant | Annotation and quantification of tranlation level of ORFs considering multiple transcript isoforms | Ma et al., 201450 |
| RiboTaper | Identification of translated regions based on 3-nucleotide periodicity | Dunn and Weissman, 201651 |
| Ribotricer | Identification of translating ORFs based on 3-nucleotide periodicity | Oyama et al., 200752 |
| RiboWave | Uses wavelet transform and 3-nucleotide periodicity to located the P-site | Calviello et al., 201653 |
| RiboCode | De novo annotation of the translatome using 3-nucleotide periodicity | Raj et al., 201654 |
| DeepRibo | Neuronal network using ribo-seq data to determin binding patterns and translation initiation sites | Xiao et al., 201855 |
| riboHMM | Identification of coding sequences based on abundance and codon pediodicity | Xu et al., 201856 |
Compilation of different bioinformatic tools, divided according to sequence prediction and ribosome profiling methods, used for the predictions and identification of small open reading frames.
More recent tools use machine learning for the prediction of sORFs. RNASamba and DeepCPP are two pipelines that predict sORFs based on neuronal networks.47,57 RNASamba was designed to recognize non-intuitive patterns, such as the Kozak sequence, in a way that it can learn from previous sequence data to distinguish coding from non-coding sequences.57 On the other hand, DeepCPP also uses the information around the start codon, which the authors term nucleotide bias, to help predict the coding potential of RNA.47 Considering the critical role of nucleotides surrounding the start codon in translation initiation, DeepCPP evaluates the codon bias at nucleotide positions −3 to +6 from the start codon. It is interesting to point out that this nucleotide bias is not the same for non-coding mRNAs.47 MiPepid is another machine learning tool that can identify potential microproteins from the DNA sequence.48 Using a microprotein database for training, MiPepid achieved a 96% accuracy when tested on a blind dataset of high-confidence micropeptides.48 Finally, a fourth new computational tool is csORF-finder, which emerged as a tool for characterizing the translation potential of sORFs. csORF-finder aims to distinguish coding sORFs (csORFs) from non-coding sORFs in different species and thus facilitate the discovery of new functional microproteins.49
A pseudo-alignment algorithm, named ORFanage, was recently established for the detection of novel ORFs in the assembled results from RNA-seq.58 ORFanage can identify ORFs from RNA-seq data based on the similarity to the reference annotation and similarity within genes in the transcripts.58 The method relies on the assumption that protein-coding genes produced by different transcripts from the same locus should share similarities, which are then exploited to detect new microproteins.58
With growing datasets and sequencing data available, the new computational tools greatly help the identification of sORFs. While bioinformatic tools have undeniably advanced and facilitated the identification of sORFs, it is important to emphasize that a systematic characterization is essential to validate the existence of the microprotein. An overview of the computation tools can be found in Table 1.
Ribosome profiling
Ribosome profiling (Ribo-Seq) provides real-time snapshots of translation by assessing so-called ribosome-protected fragments (RPF), which indicate the mRNA portions that are being translated into proteins. Ribo-Seq is a powerful tool that allows determining ORFs at codon resolution and greatly helps the identification of previously unannotated ORFs (Figure 2).59,60 The technique was pioneered by Ingolia et al. and is based on the sequencing of the approximately 30 nucleotide-long fragments that are protected by the ribosomes after nuclease digestion.61 The reads that are obtained from the sequencing of the RPFs can be aligned to the transcriptome and provide a genome-wide overview of ribosome occupancy. Ribo-Seq allows us to determine the position of where translation is taking place and, in recent years, has helped to shed light on unconventional translation and sORF translation in regions previously thought to be non-coding.2,5,62
With the introduction of this method, Ingolia et al. showed that translation could occur in the 5′UTR of an mRNA, first in yeast and later in mouse embryonic stem cells.5,61 Ribo-Seq provided evidence that the mammalian genome undergoes substantially more translation than previously assumed. Ribo-Seq studies in Drosophila, zebrafish, mice, and humans led to the discovery of widespread translation on long non-coding RNAs (lncRNAs), upstream and downstream regions and even overlapping coding transcripts.2,6,41,63,64,65 Additionally, the translation from non-canonical start sites has helped broaden the repertoire of translated sORFs. Alternative start sites can be mapped by Ribo-Seq combined with the treatment of inhibitors such as harringtonine or lactimidomycin, which cause ribosomes to accumulate at sites of translation initiation.5,66,67 Analysis from harringtonine experiments revealed that 44% of AUG start sites are downstream of annotated proteins and represent a source of alternative ORFs, resulting in truncated proteins.5 Similarly, protein isoforms can emerge through the utilization of upstream start sites, resulting in N-terminal extensions if the start site is in frame with the main start site and lacks a stop codon in between.5 Treatment with harringtonine also revealed that translation could be initiated at near-cognate (non-AUG) start sites. Most of the near-cognate initiation sites were mapped to the 5′UTR of transcripts, suggesting that uORFs are most frequently initiated with GUG start codons.5,27
Computational tools play an important role in detecting ORFs from ribosome profiling data, enabling the identification of potential microproteins encoded by sORFs. These tools leverage the direct evidence of ribosome-protected fragments captured by ribosome profiling to pinpoint translated regions within the transcriptome in a genome-wide fashion. By analyzing ribosome footprints and especially on the basis of the characteristic three-nucleotide periodicity indicating bona fide translation, these algorithms can distinguish between coding and non-coding sequences and therefore map new ORFs. Some of the commonly used tools for sORF detection from ribosome profiling data include ORF-Rater, RiboTaper/ORF-quant, ORFscore, RiboWave, RiboCode, DeepRibo, ribotricer and riboHMM.4,41,51,53,54,55,56,68,69,70 Each of these computational tools offers distinct approaches to maximize sensitivity and specificity and together have significantly expanded our ability to annotate novel sORFs from ribosome profiling data.
Since the introduction of Ribo-Seq, the technique has allowed us to successfully identify sORFs with the potential of encoding microproteins.2,10,31,62,63,71 In recent years, numerous researchers in the field have started a joint effort to produce a standardized catalog with more than 7000 human ORFs that were identified based on Ribo-Seq. This effort to annotate sORFs in a standardized manner will facilitate future endeavors to dissect the function of these ORFs.15 Separate studies have identified sORFs over the years, however, thus far, only 3085 ORFs identified by Ribo-Seq have been found by more than one research group.15 Furthermore, despite the wealth of identified sORFs, only a select few have been further characterized, unveiling their specific roles in cellular processes. By combining standardization and systematic annotation of sORFs with the development of comprehensive tools to dissect their potential functions, the approaches will likely shed light on which sORFs impact health and disease.
Mass spectrometry-based proteomics
Although Ribo-Seq demonstrates the translatability of sORFs, it does not provide direct evidence that these microproteins are present in a cell. Theoretically, RNA-binding proteins could also protect mRNA fragments of similar size to ribosome footprints and would end up in the Ribo-Seq library. In addition, it has been argued that some RPF could also result from stalled ribosomes not actively translating RNA. As outlined above, to discern true translation from other types of protected mRNA fragments, the characteristic three-nucleotide periodicity of Ribo-Seq datasets can be assessed by computational methods. The three-nucleotide periodicity greatly helps detect bona fide translation of longer ORFs. However, the triplet periodicity is challenging to detect on very small ORFs such as certain uORFs.
Mass spectrometry (MS) can detect and quantify proteins and, therefore, verify the presence of the microproteins. In that sense, MS based proteomics is currently the only experimental technique able to provide evidence of the existence of a microprotein. Nevertheless, the identification of microproteins by MS methods can be challenging and is hampered by the fact that sORFs are commonly excluded from protein databases, initiate with near-cognate start sites and produce a few unique tryptic peptides.7 In recent years, however, MS strategies have been further optimized for the identification of sORF (Figure 2).4,7,13,50
Mass spectrometry is the gold standard used to characterize the proteome.72 Originally based on the observations of four peptides with less than 150 amino acids made by Oyama et al.,52 an MS technique for the detection of microproteins was developed and further optimized. By combining peptidomics with RNA sequencing, Slavoff et al. detected a total of 90 sORF-encoded peptides (SEPs), 86 of them being newly discovered.7
Due to their small size and abundance, MS-based detection of microproteins requires previous fractionation and enrichment approaches.29 In MS studies, peptide mapping allows direct identification and quantification of proteins. Proteins are fragmented by tryptic digestion and the molecular weight of the peptides is measured and compared with reference databases. Tryptic digestion can present a problem since the smaller-sized proteins contain very few and sometimes even no tryptic peptide fragments, which biases mapping to more stable and abundant proteins.29,73,74 Replacing trypsin with different proteases can enhance microprotein detection.73,75,76 Size exclusion approaches are used to enhance the detection of low molecular weight peptides.77,78,79,80
Mass spectrometry can also be combined with separation techniques such as liquid chromatography to help with the identification of microproteins. In recent years, MS has become important, not only because of its ability to identify proteins (whether small or large), but also for its power to quantify and molecularly characterize them via the identification of posttranslational modifications.78,81
The challenge of the different MS strategies to detect sORFs is the requirement of reference databases from which the peptides can be identified.7,50 In most MS experiments, a custom database is generated that contains all potential peptides translated from the transcriptome.7,50,78,82 The absence of sORF repositories and catalogs encourages the coupling of MS with genomic or transcriptomic data.83 Many new sORF have been identified by mapping MS data to RNA sequencing and Ribo-Seq data.7,84 Using three- or six-frame translation to generate an expanded reference protein database can improve the detection of sORFs.84,85,86 Other challenges include the small size of the encoded peptides and the lack of conservation of sORF sequences between organisms.
To ensure that the peptide identified via MS is not a result of false positive proteomic profiling, it is critical to validate newly discovered microproteins. This is especially true for microproteins for which only 1 peptide has been detected. A widely adopted validation technique involves the use of isotopically labeled standards, which are chemically indistinguishable.78 The synthetic peptide should show similar MS spectra profiles except for a mass shift introduced from the isotopic label of the synthetic peptide.78,81 A second method used for the validation of small encoded peptides is the siRNA-based silencing of the transcript together with targeted MS and peptide standards, assessed by using RT-qPCR.78
Mass spectrometry has become a powerful tool to validate the expression of microproteins. Additionally, MS can help infer protein function via interaction partners, as recently shown using the so-called MicroID approach.87 Proximity biotinylation-based techniques have the potential to systematically map the interaction partners of microproteins and serve as an attractive method for characterizing sORF-encoded microproteins. In the MicroID approach, the authors developed an elegant high-throughput technique by which novel sORFs were identified and mapped to different subcellular localizations such as the nucleus or nucleolus. Furthermore, they also determined functional information based on molecular interactors accessed via transcriptome data.87 Another powerful example for MS-based identification of microproteins is major human leukocyte antigen class I (HLA-I) peptidomics. Using such a strategy in induced pluripotent stem cells (iPSCs), 240 non-canonical peptides from uORFs but also sORFs on lncRNAs could be identified, indicating that a portion of sORFs can enter the HLA-I presentation pathway to become part of the antigen repertoire.2 They also employed an elegant, minimally disruptive mNeonGreen split-fluorescent tagging strategy to visualize select microproteins, which is based on a 16 amino acid tag fused to the microprotein and can be detected once it complements with the remainder of the split mNG protein.2,88,89
Given the high number of potential microproteins identified by these different methods, it is critical to validate their expression by orthogonal assays. The predicted microproteins can be validated using different tools, including reporter assays, epitope tagging and loss of function assays. In a recent study using comparative proteomics, the authors annotated differentially expressed novel sORFs in leukemia cells and reported their subcellular localization by FLAG-tagged microprotein overexpression and subsequent visualization by immunofluorescence.90 A potential issue for microproteins is that common fluorescent tags often exceed the size of microproteins, which can affect their biophysical and biochemical properties. To this end, an elegant new technique introduces a single non-canonical amino acid either at the N- or C-terminus of microproteins. This technique, called single-residue terminal labels (STELLA), can be exploited for minimal tagging of microproteins without disturbing the physical or biochemical properties of microproteins.91 Microproteins can also be validated by chemical labeling coupled with proteomic identification, as previously reviewed.92 Protein interactions can help elucidate the putative role of a new microprotein. PRISMA, a protein interaction screen on a peptide matrix, was developed to identify the interactome of evolutionary young microproteins via sequence motifs.35
With the improvement in omics technologies, the combination of mass spectrometry, ribosome profiling and bioinformatic tools has enabled a better and more comprehensive discovery of sORFs and their corresponding microproteins.2,62,86,93 The concurrent identification of sORFs through both Ribo-Seq and MS can offer robust evidence for the existence of microproteins, laying the necessary foundation for their subsequent characterization. While integrating these different technologies enabled the annotation of new microproteins, it remains a challenge to characterize and validate these newly discovered sORFs and determine their function in different cellular contexts.
Translation initiation and regulation of gene expression
Similar to conventional genes, the control of sORF expression involves tight regulation at various stages of the gene expression cascade, including transcriptional and translational control mechanisms. For uORFs, mRNA isoforms can include or exclude uORFs, achieved through alternative transcription start site selection or by alternative splicing, which enables the cell to elegantly regulate uORF-mediated cellular function.94,95,96,97,98 Long-read sequencing methods, such as those offered by the PacBio and Nanopore platforms, enable comprehensive assessment of mRNA isoforms, including the presence or absence of uORFs, along with the assessment of the full transcript.99,100,101 Additional methods geared toward uORF detection and transcription start site selection include the 5′ cap capture methods such as CAGE-seq (Cap Analysis of Gene Expression) to generate snapshots of the 5′ end and the 5′UTR.102,103
In addition to transcriptional control, translational regulation significantly impacts uORF expression due to the critical role of translation start site recognition by scanning ribosomes.104,105 Decades of research, particularly focusing on the arguably best-studied uORF-containing gene ATF4, have highlighted how cellular context and changes in the translational machinery influence the recognition of uORFs.24,104
Translation can be divided into three main steps: initiation, elongation and termination, which includes ribosome recycling for a new round of protein synthesis. Translation initiation is the rate-limiting phase of translation.106,107,108 It begins with the formation of the ternary complex (eIF2-GTP-Met-tRNAi), which assembles with the 40S ribosomal subunit to form the preinitiation complex (PIC) to scan the 5′ untranslated region (5′UTR).108,109 When the PIC reaches the start codon, the GTP in the ternary complex is hydrolyzed and with the release of eIF2-GDP, the 60S ribosomal subunit joins the PIC complex to form the 80S complex and initiate translation.108
The 5′UTR plays therefore a critical role in ribosome recruitment to mRNA, influencing translation start site selection and initiation.23,104,110,111 Different structures and elements in the 5′UTR, such as RNA secondary structures, Internal Ribosome Entry Sites (IRES), motifs for RNA binding proteins, single or multiple upstream initiation sites and uORFs can shape the translation of the downstream main ORF.104,110,111,112,113 In the case of the uORFs, translation initiates within the 5′UTR and can represent competition for the PIC to detect the start codon of the main coding sequence, consequently negatively regulating translation of the CDS.67,104,114,115 Nonetheless, it is important to point out that uORFs do not always repress the translation of the main ORF, as specific conditions such as stress may allow the re-initiation of translation at the main ORF.104,115,116,117,118 Thus, uORFs can add an additional layer of regulation to rapidly boost downstream translation to changes in the cellular environment. Over the last decades, luciferase-based assays in cultured cells have greatly helped in elucidating the regulatory function of uORFs. These approaches could be combined with translation-competent lysates for in vitro translation, which enables recapitulation of the key steps of uORF translation and the regulatory role of uORFs with regard to main ORF translation.119
The sequence context surrounding the initiation codon is important for the translation of uORFs and protein-coding sequences.120 The PIC recognizes preferentially the correct start codon – usually AUG – in an optimal context known as the Kozak consensus sequence.
Monitoring the start codon selection is feasible through various techniques, including Global Translation Initiation Sequencing (GTI-seq), which employs a similar principle as the translation start site inhibitor Harringtonine.67,121 These methods enable genome-wide mapping of translation start sites, which are based on blocking initiating ribosomes while allowing the elongating ribosomes to run off. Coupled with the ribosome profiling protocol, GTI-seq or Harringtonine treatment solely results in translation start site peaks without the reads from elongating ribosomes, therefore providing strong evidence that the start sites of potential sORFs are indeed recognized by the translational machinery. Global translation initiation sequencing (GTI-seq) indicates that approximately 74% of the upstream translation initiation sites (TIS) are non-AUG start codons. CUG is the predominant start codon for uORF, showing a frequency of ∼30% compared to ∼25% frequency of the conventional AUG TIS.67 Additionally, conventional ribosome profiling studies similarly suggest that uORFs show a preference for near-cognate start codons, with CUG and GUG being the most frequent TIS.5,27
Genome editing using CRISPR
A major challenge in the sORF field is distinguishing functionally relevant sORFs from mere sORF expression. As outlined above, there are multiple approaches to detect and annotate sORFs experimentally and computationally. Consequently, the field will have to progress beyond the essential task of cataloging sORFs and transition toward comprehensive genome-wide analyses to unravel the functional significance of sORFs. For the majority of sORFs, their functions remain untested, necessitating the use of genome editing techniques to explore loss-of-function and gain-of-function effects and analyses of the resulting phenotypes in cell culture studies and in vivo. These strategies will be essential in shedding light on the functional roles of sORFs.
The most commonly used and rapidly evolving genome editing technique is the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) system and their associated Cas endonucleases. These short regularly spaced repeats were found in both bacteria and archaea and are part of their DNA repair system against phages and plasmids.122,123,124 Among the different types of immunity, type II can be applied to genome editing.124,125,126,127
The microbial adaptive immune response has been exploited to target any genomic location by using a single guide RNA (sgRNA) in combination with the Cas endonucleases.127 Endogenous repair mechanisms such as homologous directed repair (HDR) or non-homologous end-joining (NHEJ) are induced by the DSB.126,128,129 As a result, the DSB can be used to insert, delete, or modify a genomic target. When introducing exogenous DNA fragments as templates for recombination, specific mutations can be introduced. In the absence of a DNA template for HDR, the NHEJ pathway prevails, leading to insertions or deletions introduced to the target locus.130,131
Altering the sequence of the sgRNA will allow to target any region of interest in the genome. In recent years, optimizing the sequence of the sgRNA has led to improvements in the on-target activity and a reduction of off-target effects.132 Different tools can be used to design sgRNAs and predict their target activity, however, these tools commonly rely on canonical genes as reference, posing challenges for targeting smaller genes like microproteins. In the human genome, the overall frequency of finding a ‘GG’ is 5.21%, which means that the ‘GG’ dinucleotide, critical for the sgRNA design, is found approximately every 42 bases.133 For smaller genes, such as microproteins, this issue significantly reduces the chances of finding a PAM site and designing good sgRNAs to target them. Considering that the median size of an uORF is 48–78 nucleotides12,23,27,134,135 and the median size of a translated human sORF from a long non-coding RNA is 72 nucleotides,12 there may be, on average, only 2–4 PAM motifs per microprotein (considering both strands). This notion aligns with our own experience and the design of typically 2–3 sgRNAs per sORF. Furthermore, the design of CRISPR screens can be particularly challenging for sORFs that overlap other genetic elements, such as altORFs. It is important to note that any phenotype attributed to the disruption of an sORF should be complemented by for example reintroducing the sORF.
Over the past decade, numerous studies have demonstrated the remarkable efficiency of the CRISPR/Cas system for genome editing. As interest in the technique grew, different Cas endonucleases have been discovered, modified and used for genome editing. CRISPR interference (CRISPRi) employs a modified version of the Cas9 protein, the dead Cas9 (dCas9), which is fused with an interference domain and is used to silence the expression of a gene. On the other hand, CRISPR activation (CRISPRa) utilizes a dCas9 that is fused to an activation domain, such as for example the dCas9-SunTag system, to turn on gene expression. Additionally, the CRISPR/Cas12a and Cas12b systems function similarly to CRISPR/Cas9 but exhibit different PAM sequences and editing efficiencies, introduce sticky ends instead of blunt ends, and cut at distinct sites relative to the PAM sequence.136,137,138 These variations make Cas12a and Cas12b valuable additions to the CRISPR platform, particularly in targeting sORFs that may have been resistant to Cas9 gene editing due to low editing efficiency or missing NGG PAM sequence in the sORF locus.
CRISPR screens
CRISPR screens have emerged as a powerful tool to dissect the function of genes. The two main types of CRISPR screens, arrayed and pooled screens, have become instrumental in addressing diverse biological questions (Figure 3). Serving as an unbiased interrogation of gene function, CRISPR screens introduce perturbations into cells, which subsequently reveal cellular phenotypes.139
Figure 3.

Targeting sORFs using CRISPR
Schematic representation of the CRISPR screening workflow. Top panel: For pooled CRISPR screens, the sgRNA library is transduced into Cas9-expressing cells in vitro. Cells are harvested at the end of the experiment (e.g., following a certain number of passages or treatment) and submitted to sequencing. The enrichment and depletion of the sgRNAs is then used to infer gene function. Middle panel: Arrayed CRISPR screens are carried out in different wells, where one sgRNA is targeted per well. In an arrayed screen, the phenotype can be linked directly to the sgRNA to determine gene function. Lower panel: single-cell CRISPR screens in vitro and in vivo. Similar to pooled CRISPR screens, cells are transduced with a pooled library. Single cells are then subjected to single-cell RNA-seq to obtain the transcriptomic readout coupled to cell-type specific sgRNA representation. In an in vivo single-cell CRISPR screen, the sgRNA library is delivered, for example, directly into mouse embryos or adult mice. At a later time point, the organ of interest is collected, and cells are isolated for single-cell RNA-seq, which can determine proliferative changes and the transcriptomic consequences of the sgRNA in different cell types.
In arrayed screens, individual reagents are synthesized and distributed into multi-well plates and are therefore spatially separated. As each well introduces a distinct perturbation, the approach enables the identification of the specific sgRNA responsible for the gene perturbation without the need for sequencing. Arrayed screens can have advantages, such as the possibility to couple them with microscopy-based high-content screenings, but are also laborious and therefore result in lower throughput.132,140,141 On the other hand, pooled CRISPR screens provide a scalable and powerful platform and allow the targeting of multiple genes using a library of pooled sgRNAs. This library is delivered to cells through lentiviral transduction, resulting in cells harboring single sgRNAs that integrate into the cell’s DNA and edit the targeted gene based on the sgRNA sequence. Subsequently, these perturbed cells are subjected to selective pressure or monitored over multiple passages. At the end of the experiment, sequencing and sgRNA identification enable us to infer gene function by calculating the representation of the sgRNAs in the library.140
In a recent study, 553 non-canonical ORFs were comprehensively analyzed using a CRISPR screen coupled to single-cell RNA sequencing.142 Among the targeted ORFs, 386 were identified as sORFs with less than 100 amino acids and resided either upstream or downstream of known protein-coding genes and on long non-coding RNAs (lncRNAs). By performing a CRISPR loss-of-function screen in eight different cell lines, the authors observed viability phenotypes in 10% of the targeted ORFs.142 Subsequent analyses of the 13 top-scoring ORFs provided valuable insights into their functional role in cancer cell survival.142 This study exemplifies therefore the power of CRISPR screenings in deciphering the function of microproteins as they enable an unbiased approach to simultaneously target them and determine their function in different biological contexts.
The main advantage of CRISPR screens lies in their high throughput and scalability, which enables the simultaneous interrogation of thousands of genes or microproteins. While most of these screens have been conducted in vitro using various cell lines, one significant drawback is their inability to account for environmental factors and cellular interactions between different cell types. To address these limitations, different approaches have been taken to design in vivo screens in mouse models, which can be challenging to set up but provide the opportunity to assess the consequences of sORF perturbations within a living organism. More recent in vivo screens include for example screens to identify modulators for tumor growth or immunotherapy targets.143,144
Single-cell CRISPR screens
While pooled CRISPR screenings are undeniably powerful, they have the important limitation that they are restricted to simple readouts such as proliferation or the expression of a marker gene. To address this drawback, a suite of new tools has been developed that enable the coupling of single-cell RNA sequencing with pooled CRISPR screening (Table 2), thereby providing a high-throughput functional dissection of genes with single-cell transcriptomic readout.
Table 2.
Single-cell CRISPR methods
| Method | Reference | |
|---|---|---|
| Perturb-seq | The Perturb-seq method combines a pooled CRISPR screen with single-cell RNA-seq by a guide barcode (GBC) expressed for each perturbation. | Prensner et al., 2021142 |
| CRISP-seq: | The CRISP-seq technique allows the identification of the sgRNA that infects each individual cell by using a vector that contains the gRNA sequence and a transcribed polyadenylated unique guide index with a fluorescent selection marker. | Hanna and Doench, 2020141 |
| Mosaic-seq: mosaic single-cell analysis by indexed CRISPR sequencing | Mosaic-seq couples CRISPRi with single-cell RNA sequencing. The library of vectors targeting enhancers also carries a unique barcode that allows the identification of the sgRNA. | Braun et al., 2016143 |
| CROP-seq: CRISPR droplet sequencing | The CROP-seq method, unlike the other methods, does not pair the sgRNA with a barcode. Instead, the CROP-seq method uses the sgRNA as a barcode overlapping the Pol II transcript. | Manguso et al., 2017144 |
Summary of the different single-cell CRISPR methods currently available with a small description of the principle of the method.
In a conventional pooled CRISPR screen, it is not possible to identify which sgRNA is expressed in each cell. The main issue is that the sgRNA, being processed by the human U6 RNA Polymerase III (RNAP III), will not undergo posttranscriptional modifications, including the polyadenylation, making it incompatible with the RNA-sequencing techniques.145 To solve this issue, the sgRNA in each cell can be identified via a Polymerase II transcribed barcode used in Perturb-seq, CRISP-seq and Mosaic-seq methods,146,147,148 or by detecting the sgRNA within the Pol II transcript used in the CROP-seq method.149 In the past six years, many more techniques have been developed, allowing for the integration of CRISPR with next-generation sequencing. The advent of single-cell CRISPR screens offers the opportunity to investigate gene function within the context of regulatory pathways and holds great promise for investigating microproteins at single-cell transcriptomic resolution in vitro and in vivo.146,147,148,149,150
Only a few large-scale functional characterization screens of microproteins have been performed by using single-cell CRISPR screenings. In a recent study, Chen et al. combined Ribo-Seq and mass spectrometry techniques to annotate and generate a library of non-canonical ORFs. Initially, a pooled CRISPR screen was performed, leading to the identification of over 500 potential targets exhibiting a significant proliferation phenotype. To gain deeper insights into the role of non-canonical ORFs, a second step involved a Perturb-seq screen, focusing on 83 uORFs and 80 lncRNAs, to assess the transcriptomic changes resulting from the loss-of-function of the sORF.2 These analyses revealed sORF functions in different cellular pathways suggesting that sORFs play diverse cellular roles and highlighting the power of single-cell CRISPR screenings to analyze the function of microproteins.2 Another elegant example of such a screening strategy was a study aimed at identifying regulators of zygote genome activation (ZGA)-like transcription in mouse embryonic stem cells, which exploited a modified CROP-seq vector used for a CRISPRa library coupled with single-cell transcriptomics.151 Out of the 230 genes that were assessed, 24 were identified to have a ZGA-like signature and 9 of those genes were independently validated as ZGA-like transcription regulators.151
Single-cell CRISPR screens are especially powerful when carried out in vivo as they have the potential to interrogate gene function simultaneously in different tissue cell types. To date, only a few in vivo single-cell CRISPR screens have been performed. In 2020, the Perturb-seq vector was used for a CRISPR screen in vivo targeting 35 risk genes for autism spectrum disorder and developmental delay (ASD/ND).152 The in vivo Perturb-seq method was able to target the five main different cell types in the brain and uncover common pathways targeted by multiple perturbations.152 More recently, an in vivo immune screen was performed using the CROP-seq vector to elucidate tumor immune evasion mechanisms.153 These studies expand the power of genetic screens into biological and disease models in mammals, facilitating the understanding of tissue-wide gene function. These advanced single-cell CRISPR technologies provide therefore an attractive set of tools for targeting sORFs and hold great promise in understanding sORF function in different disease contexts.
Conclusion
The rapid advancement in the field of sORFs has been driven by a variety of innovative technologies and computational approaches that emerged rather recently, offering opportunities for functional characterization and understanding of the role of microproteins. The development of various in silico tools and pipelines has revolutionized the annotation and identification of sORFs, leading to the expansion of the standardized catalog of these elusive coding sequences in eukaryotic genomes. Ribosome profiling and mass spectrometry-based approaches helped tremendously to identify and validate sORFs by providing direct experimental evidence that they are translated into stable microproteins. As demonstrated in recent studies, the utilization of CRISPR-based systems, especially when coupled with single-cell RNA sequencing, enables comprehensive and systematic analyses of sORF expression and function. We expect that large-scale CRISPR screens will continue to expand the functional repertoire of microproteins. These tools hold great promise for dissecting sORF function in different cellular contexts and unraveling their role in the pathogenesis of disease.
Nevertheless, the field faces challenges in identifying functionally relevant sORFs and sifting through the catalog of thousands of potential sORFs in the mammalian genome to tease apart relevant sORFs from those without a clear cellular function. Further investigations utilizing loss-of-function and gain-of-function approaches in relevant cellular contexts will be crucial for elucidating the functional significance of these microproteins. As sORFs continue to attract growing interest, it is evident that the continued integration of new methodologies will pave the way for new discoveries in this fascinating field of microproteins. Ultimately, unraveling the diverse cellular roles of sORFs will have profound implications, shedding light on regulatory mechanisms and uncovering new therapeutic targets for a wide range of diseases.
Acknowledgments
The Sendoel lab was supported by an SNSF Professorship grant (grant number 176825), by the European Research Council (ERC) under the European Union′s Horizon 2020 research and innovation program (grant agreement No 759006), by the Swiss Cancer Research foundation (KFS-5023-02-2020-R), the National Center of Competence in Research (NCCR) on RNA and Disease funded by the SNSF and the UZH Candoc Grant 2022 from the University of Zurich (Nr. FK-22-039).
Author contributions
Conceptualization, FVF and AS; Writing - Original Draft, FVF and AS; Writing - Review and Editing, FVF and AS; Visualization, FVF and AS; All authors contributed to the article and approved the submitted version.
Declaration of interests
The authors declare no competing financial interests.
References
- 1.Ruiz-Orera J., Albà M.M. Translation of Small Open Reading Frames: Roles in Regulation and Evolutionary Innovation. Trends Genet. 2019;35:186–198. doi: 10.1016/j.tig.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Chen J., Brunner A.-D., Cogan J.Z., Nuñez J.K., Fields A.P., Adamson B., Itzhak D.N., Li J.Y., Mann M., Leonetti M.D., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 2020;367:140–146. doi: 10.1126/science.aav5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGillivray P., Ault R., Pawashe M., Kitchen R., Balasubramanian S., Gerstein M. A comprehensive catalog of predicted functional upstream open reading frames in humans. Nucleic Acids Res. 2018;46:3326–3338. doi: 10.1093/nar/gky188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bazzini A.A., Johnstone T.G., Christiano R., MacKowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–993. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chew G.L., Pauli A., Schier A.F. Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat. Commun. 2016;7:1–10. doi: 10.1038/ncomms11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slavoff S.A., Mitchell A.J., Schwaid A.G., Cabili M.N., Ma J., Levin J.Z., Karger A.D., Budnik B.A., Rinn J.L., Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sieber P., Platzer M., Schuster S. The Definition of Open Reading Frame Revisited. Trends Genet. 2018;34:167–170. doi: 10.1016/j.tig.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Basrai M.A., Hieter P., Boeke J.D. Small Open Reading Frames: Beautiful Needles in the Haystack. Genome Res. 1997;7:768–771. doi: 10.1101/GR.7.8.768. [DOI] [PubMed] [Google Scholar]
- 10.Mackowiak S.D., Zauber H., Bielow C., Thiel D., Kutz K., Calviello L., Mastrobuoni G., Rajewsky N., Kempa S., Selbach M., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biol. 2015;16:1–21. doi: 10.1186/s13059-015-0742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Q., Medina S.G., Kushawah G., Devore M.L., Castellano L.A., Hand J.M., Wright M., Bazzini A.A. Translation affects mRNA stability in a codon-dependent manner in human cells. Elife. 2019;8:1–22. doi: 10.7554/eLife.45396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couso J.P., Patraquim P. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 2017;18:575–589. doi: 10.1038/nrm.2017.58. [DOI] [PubMed] [Google Scholar]
- 13.Pueyo J.I., Magny E.G., Couso J.P. New Peptides Under the s(ORF)ace of the Genome. Trends Biochem. Sci. 2016;41:665–678. doi: 10.1016/j.tibs.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Khitun A., Ness T.J., Slavoff S.A. Small open reading frames and cellular stress responses. Mol. Omi. 2019;15:108–116. doi: 10.1039/c8mo00283e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mudge J.M., Ruiz-Orera J., Prensner J.R., Brunet M.A., Calvet F., Jungreis I., Gonzalez J.M., Magrane M., Martinez T.F., Schulz J.F., et al. Standardized annotation of translated open reading frames. Nat. Biotechnol. 2022;40:994–999. doi: 10.1038/s41587-022-01369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silva J., Fernandes R., Romão L. In: The mRNA Metabolism in Human Disease. Romão L., editor. 2019. Translational Regulation by Upstream Open Reading Frames and Human Diseases; pp. 99–116. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y., Wu S., Zhu X., Zhang L., Deng J., Li F., Guo B., Zhang S., Wu R., Zhang Z., et al. LncRNA-encoded polypeptide ASRPS inhibits triple-negative breast cancer angiogenesis. J. Exp. Med. 2020;217 doi: 10.1084/jem_20190950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J.Z., Chen M., Chen D., Gao X.C., Zhu S., Huang H., Hu M., Zhu H., Yan G.R. A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol. Cell. 2017;68:171–184.e6. doi: 10.1016/j.molcel.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Polycarpou-Schwarz M., Groß M., Mestdagh P., Schott J., Grund S.E., Hildenbrand C., Rom J., Aulmann S., Sinn H.P., Vandesompele J., et al. The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene. 2018;37:4750–4768. doi: 10.1038/s41388-018-0281-5. [DOI] [PubMed] [Google Scholar]
- 20.Sriram A., Bohlen J., Teleman A.A. Translation acrobatics: how cancer cells exploit alternate modes of translational initiation. EMBO Rep. 2018;19 doi: 10.15252/embr.201845947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Somers J., Pöyry T., Willis A.E. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013;45:1690–1700. doi: 10.1016/j.biocel.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young S.K., Wek R.C. Upstream open reading frames differentially regulate genespecific translation in the integrated stress response. J. Biol. Chem. 2016;291:16927–16935. doi: 10.1074/jbc.R116.733899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calvo S.E., Pagliarini D.J., Mootha V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vattem K.M., Wek R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zismanov V., Chichkov V., Colangelo V., Jamet S., Wang S., Syme A., Koromilas A.E., Crist C. Phosphorylation of eIF2α is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell. 2016;18:79–90. doi: 10.1016/j.stem.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 27.Sendoel A., Dunn J.G., Rodriguez E.H., Naik S., Gomez N.C., Hurwitz B., Levorse J., Dill B.D., Schramek D., Molina H., et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature. 2017;541:494–499. doi: 10.1038/nature21036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carninci P., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 29.Leong A.Z.X., Lee P.Y., Mohtar M.A., Syafruddin S.E., Pung Y.F., Low T.Y. Short open reading frames (sORFs) and microproteins: an update on their identification and validation measures. J. Biomed. Sci. 2022;29:1–15. doi: 10.1186/S12929-022-00802-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makarewich C.A., Olson E.N. Mining for Micropeptides. Trends Cell Biol. 2017;27:685–696. doi: 10.1016/j.tcb.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frith M.C., Forrest A.R., Nourbakhsh E., Pang K.C., Kai C., Kawai J., Carninci P., Hayashizaki Y., Bailey T.L., Grimmond S.M. The abundance of short proteins in the mammalian proteome. PLoS Genet. 2006;2:515–528. doi: 10.1371/journal.pgen.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skovgaard M., Jensen L.J., Brunak S., Ussery D., Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 2001;17:425–428. doi: 10.1016/s0168-9525(01)02372-1. [DOI] [PubMed] [Google Scholar]
- 33.Couso J.P. Finding smORFs: Getting closer. Genome Biol. 2015;16:15–17. doi: 10.1186/s13059-015-0765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright B.W., Yi Z., Weissman J.S., Chen J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol. 2022;32:243–258. doi: 10.1016/J.TCB.2021.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandmann C.-L., Schulz J.F., Ruiz-Orera J., Kirchner M., Ziehm M., Adami E., Marczenke M., Christ A., Liebe N., Greiner J., et al. Evolutionary origins and interactomes of human, young microproteins and small peptides translated from short open reading frames. Mol. Cell. 2023;83:994–1011.e18. doi: 10.1016/j.molcel.2023.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin M.F., Jungreis I., Kellis M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27 doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mudge J.M., Jungreis I., Hunt T., Gonzalez J.M., Wright J.C., Kay M., Davidson C., Fitzgerald S., Seal R., Tweedie S., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Res. 2019;29 doi: 10.1101/gr.246462.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng H., Chan W.S., Li Z., Wang D., Liu S., Zhou Y. Small Open Reading Frames: Current Prediction Techniques and Future Prospect. Curr. Protein Pept. Sci. 2011;12:503. doi: 10.2174/138920311796957667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kute P.M., Soukarieh O., Tjeldnes H., Trégouët D.A., Valen E. Small Open Reading Frames, How to Find Them and Determine Their Function. Front. Genet. 2022;12:2903. doi: 10.3389/fgene.2021.796060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunet M.A., Brunelle M., Lucier J.F., Delcourt V., Levesque M., Grenier F., Samandi S., Leblanc S., Aguilar J.D., Dufour P., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Res. 2019;47:D403–D410. doi: 10.1093/nar/gky936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fields A.P., Rodriguez E.H., Jovanovic M., Stern-Ginossar N., Haas B.J., Mertins P., Raychowdhury R., Hacohen N., Carr S.A., Ingolia N.T., et al. A Regression-Based Analysis of Ribosome-Profiling Data Reveals a Conserved Complexity to Mammalian Translation. Mol. Cell. 2015;60:816–827. doi: 10.1016/j.molcel.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunet M.A., Lucier J.F., Levesque M., Leblanc S., Jacques J.F., Al-Saedi H.R.H., Guilloy N., Grenier F., Avino M., Fournier I., et al. OpenProt 2021: Deeper functional annotation of the coding potential of eukaryotic genomes. Nucleic Acids Res. 2021;49:D380–D388. doi: 10.1093/nar/gkaa1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanada K., Akiyama K., Sakurai T., Toyoda T., Shinozaki K., Shiu S.-H. sORF finder: a program package to identify small open reading frames with high coding potential. Bioinformatics. 2010;26:399–400. doi: 10.1093/bioinformatics/btp688. [DOI] [PubMed] [Google Scholar]
- 44.Skarshewski A., Stanton-Cook M., Huber T., Al Mansoori S., Smith R., Beatson S.A., Rothnagel J.A. UPEPperoni: An online tool for upstream open reading frame location and analysis of transcript conservation. BMC Bioinf. 2014;15:1–6. doi: 10.1186/1471-2105-15-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olexiouk V., Crappé J., Verbruggen S., Verhegen K., Martens L., Menschaert G. SORFs.org: A repository of small ORFs identified by ribosome profiling. Nucleic Acids Res. 2016;44:D324–D329. doi: 10.1093/nar/gkv1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y., Zhou H., Chen X., Zheng Y., Kang Q., Hao D., Zhang L., Song T., Luo H., Hao Y., et al. SmProt: A Reliable Repository with Comprehensive Annotation of Small Proteins Identified from Ribosome Profiling. Dev. Reprod. Biol. 2021;19:602–610. doi: 10.1016/j.gpb.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y., Jia C., Fullwood M.J., Kwoh C.K. DeepCPP: a deep neural network based on nucleotide bias information and minimum distribution similarity feature selection for RNA coding potential prediction. Brief. Bioinform. 2021;22:2073–2084. doi: 10.1093/bib/bbaa039. [DOI] [PubMed] [Google Scholar]
- 48.Zhu M., Gribskov M. MiPepid: MicroPeptide identification tool using machine learning. BMC Bioinf. 2019;20:1–11. doi: 10.1186/s12859-019-3033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang M., Zhao J., Li C., Ge F., Wu J., Jiang B., Song J., Song X. csORF-finder: an effective ensemble learning framework for accurate identification of multi-species coding short open reading frames. Brief. Bioinform. 2022;23 doi: 10.1093/bib/bbac392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma J., Ward C.C., Jungreis I., Slavoff S.A., Schwaid A.G., Neveu J., Budnik B.A., Kellis M., Saghatelian A. Discovery of Human sORF-Encoded Polypeptides (SEPs) in Cell Lines and Tissue. J. Proteome Res. 2014;13:1757–1765. doi: 10.1021/pr401280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dunn J.G., Weissman J.S. Plastid: Nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genom. 2016;17:958. doi: 10.1186/s12864-016-3278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oyama M., Kozuka-Hata H., Suzuki Y., Semba K., Yamamoto T., Sugano S. Diversity of transplantation start sites may define increased complexity of the human short ORFeome. Mol. Cell. Proteomics. 2007;6:1000–1006. doi: 10.1074/mcp.M600297-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Calviello L., Mukherjee N., Wyler E., Zauber H., Hirsekorn A., Selbach M., Landthaler M., Obermayer B., Ohler U. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods. 2016;13:165–170. doi: 10.1038/nmeth.3688. [DOI] [PubMed] [Google Scholar]
- 54.Raj A., Wang S.H., Shim H., Harpak A., Li Y.I., Engelmann B., Stephens M., Gilad Y., Pritchard J.K. Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling. Elife. 2016;5 doi: 10.7554/eLife.13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao Z., Huang R., Xing X., Chen Y., Deng H., Yang X. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 2018;46:e61. doi: 10.1093/nar/gky179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu Z., Hu L., Shi B., Geng S., Xu L., Wang D., Lu Z.J. Ribosome elongating footprints denoised by wavelet transform comprehensively characterize dynamic cellular translation events. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Camargo A.P., Sourkov V., Pereira G.A.G., Carazzolle M.F. RNAsamba: Neural network-based assessment of the protein-coding potential of RNA sequences. NAR Genom Bioinform. 2020;2:lqz024. doi: 10.1093/nargab/lqz024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varabyou A., Erdogdu B., Salzberg S.L., Pertea M. Investigating open reading frames in known and novel transcripts using ORFanage. Nat. Comput. Sci. 2023;2023:1–9. doi: 10.1038/s43588-023-00496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ingolia N.T. Ribosome Footprint Profiling of Translation throughout the Genome. Cell. 2016;165:22–33. doi: 10.1016/j.cell.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinez T.F., Chu Q., Donaldson C., Tan D., Shokhirev M.N., Saghatelian A. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 2020;16:458–468. doi: 10.1038/S41589-019-0425-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aspden J.L., Eyre-Walker Y.C., Phillips R.J., Amin U., Mumtaz M.A.S., Brocard M., Couso J.P. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 2014;3:1–19. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji Z., Song R., Regev A., Struhl K. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife. 2015;4:1–21. doi: 10.7554/eLife.08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chothani S.P., Adami E., Widjaja A.A., Langley S.R., Viswanathan S., Pua C.J., Zhihao N.T., Harmston N., D’Agostino G., Whiffin N., et al. A high-resolution map of human RNA translation. Mol. Cell. 2022;82:2885–2899.e8. doi: 10.1016/j.molcel.2022.06.023. [DOI] [PubMed] [Google Scholar]
- 66.Michel A.M., Baranov P.V. Ribosome profiling: a Hi-Def monitor for protein synthesis at the genome-wide scale. Wiley Interdiscip. Rev. RNA. 2013;4:473–490. doi: 10.1002/wrna.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee S., Liu B., Lee S., Huang S.-X., Shen B., Qian S.-B. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA. 2012;109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clauwaert J., Menschaert G., Waegeman W. DeepRibo: a neural network for precise gene annotation of prokaryotes by combining ribosome profiling signal and binding site patterns. Nucleic Acids Res. 2019;47:e36. doi: 10.1093/nar/gkz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Calviello L., Hirsekorn A., Ohler U. Quantification of translation uncovers the functions of the alternative transcriptome. Nat. Struct. Mol. Biol. 2020;27:717–725. doi: 10.1038/s41594-020-0450-4. [DOI] [PubMed] [Google Scholar]
- 70.Choudhary S., Li W., Smith A.D. Accurate detection of short and long active ORFs using Ribo-seq data. Bioinformatics. 2020;36:2053–2059. doi: 10.1093/bioinformatics/btz878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruiz-Orera J., Messeguer X., Subirana J.A., Alba M.M. Long non-coding RNAs as a source of new peptides. Elife. 2014;3 doi: 10.7554/ELIFE.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S., et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bartel J., Varadarajan A.R., Sura T., Ahrens C.H., Maaß S., Becher D. Optimized proteomics workflow for the detection of small proteins. J. Proteome Res. 2020;19:4004–4018. doi: 10.1021/acs.jproteome.0c00286. [DOI] [PubMed] [Google Scholar]
- 74.Müller S.A., Kohajda T., Findeiß S., Stadler P.F., Washietl S., Kellis M., von Bergen M., Kalkhof S. Optimization of parameters for coverage of low molecular weight proteins. Anal. Bioanal. Chem. 2010;398:2867–2881. doi: 10.1007/s00216-010-4093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaulich P.T., Cassidy L., Bartel J., Schmitz R.A., Tholey A. Multi-protease Approach for the Improved Identification and Molecular Characterization of Small Proteins and Short Open Reading Frame-Encoded Peptides. J. Proteome Res. 2021;20:2895–2903. doi: 10.1021/acs.jproteome.1c00115. [DOI] [PubMed] [Google Scholar]
- 76.Cassidy L., Kaulich P.T., Maaß S., Bartel J., Becher D., Tholey A. Bottom-up and top-down proteomic approaches for the identification, characterization, and quantification of the low molecular weight proteome with focus on short open reading frame-encoded peptides. Proteomics. 2021;21 doi: 10.1002/pmic.202100008. [DOI] [PubMed] [Google Scholar]
- 77.Ma J., Diedrich J.K., Jungreis I., Donaldson C., Vaughan J., Kellis M., Yates J.R., Saghatelian A. Improved Identification and Analysis of Small Open Reading Frame Encoded Polypeptides. Anal. Chem. 2016;88:3967–3975. doi: 10.1021/acs.analchem.6b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khitun A., Slavoff S.A. Proteomic Detection and Validation of Translated Small Open Reading Frames. Curr. Protoc. Chem. Biol. 2019;11:e77. doi: 10.1002/cpch.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He C., Jia C., Zhang Y., Xu P. Enrichment-Based Proteogenomics Identifies Microproteins, Missing Proteins, and Novel smORFs in Saccharomyces cerevisiae. J. Proteome Res. 2018;17:2335–2344. doi: 10.1021/acs.jproteome.8b00032. [DOI] [PubMed] [Google Scholar]
- 80.Ahrens C.H., Wade J.T., Champion M.M., Langer J.D. A Practical Guide to Small Protein Discovery and Characterization Using Mass Spectrometry. J. Bacteriol. 2022;204 doi: 10.1128/jb.00353-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cassidy L., Kaulich P.T., Tholey A. Proteoforms expand the world of microproteins and short open reading frame-encoded peptides. iScience. 2023;26:106069. doi: 10.1016/J.ISCI.2023.106069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vanderperre B., Lucier J.F., Bissonnette C., Motard J., Tremblay G., Vanderperre S., Wisztorski M., Salzet M., Boisvert F.M., Roucou X. Direct Detection of Alternative Open Reading Frames Translation Products in Human Significantly Expands the Proteome. PLoS One. 2013;8:e70698. doi: 10.1371/journal.pone.0070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schlesinger D., Elsässer S.J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. FEBS J. 2021;289:53–74. doi: 10.1111/febs.15769. [DOI] [PubMed] [Google Scholar]
- 84.Lu S., Zhang J., Lian X., Sun L., Meng K., Chen Y., Sun Z., Yin X., Li Y., Zhao J., et al. A hidden human proteome encoded by “non-coding” genes. Nucleic Acids Res. 2019;47:8111–8125. doi: 10.1093/nar/gkz646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Crappé J., Ndah E., Koch A., Steyaert S., Gawron D., De Keulenaer S., De Meester E., De Meyer T., Van Criekinge W., Van Damme P., et al. PROTEOFORMER: deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Res. 2015;43:e29. doi: 10.1093/NAR/GKU1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tharakan R., Kreimer S., Ubaida-Mohien C., Lavoie J., Olexiouk V., Menschaert G., Ingolia N.T., Cole R.N., Ishizuka K., Sawa A., et al. A methodology for discovering novel brain-relevant peptides: Combination of ribosome profiling and peptidomics. Neurosci. Res. 2020;151:31–37. doi: 10.1016/j.neures.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 87.Na Z., Dai X., Zheng S.-J., Bryant C.J., Loh K.H., Su H., Luo Y., Buhagiar A.F., Cao X., Baserga S.J., et al. Mapping subcellular localizations of unannotated microproteins and alternative proteins with MicroID. Mol. Cell. 2022;82:2900–2911.e7. doi: 10.1016/j.molcel.2022.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kamiyama D., Sekine S., Barsi-Rhyne B., Hu J., Chen B., Gilbert L.A., Ishikawa H., Leonetti M.D., Marshall W.F., Weissman J.S., et al. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016;7 doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feng S., Varshney A., Coto Villa D., Modavi C., Kohler J., Farah F., Zhou S., Ali N., Müller J.D., Van Hoven M.K., et al. Bright split red fluorescent proteins for the visualization of endogenous proteins and synapses. Commun. Biol. 2019;21:1–12. doi: 10.1038/s42003-019-0589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cao X., Khitun A., Na Z., Dumitrescu D.G., Kubica M., Olatunji E., Slavoff S.A. Comparative Proteomic Profiling of Unannotated Microproteins and Alternative Proteins in Human Cell Lines. J. Proteome Res. 2020;19:3418–3426. doi: 10.1021/acs.jproteome.0c00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lafranchi L., Schlesinger D., Kimler K.J., Elsässer S.J. Universal Single-Residue Terminal Labels for Fluorescent Live Cell Imaging of Microproteins. J. Am. Chem. Soc. 2020;142:20080–20087. doi: 10.1021/jacs.0c09574. [DOI] [PubMed] [Google Scholar]
- 92.Chen Y., Cao X., Loh K.H., Slavoff S.A. Chemical labeling and proteomics for characterization of unannotated small and alternative open reading frame-encoded polypeptides. Biochem. Soc. Trans. 2023;51:1071–1082. doi: 10.1042/BST20221074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Heesch S., Witte F., Schneider-Lunitz V., Schulz J.F., Adami E., Faber A.B., Kirchner M., Maatz H., Blachut S., Sandmann C.L., et al. The Translational Landscape of the Human Heart. Cell. 2019;178:242–260. doi: 10.1016/j.cell.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 94.Cheng Z., Otto G.M., Powers E.N., Keskin A., Mertins P., Carr S.A., Jovanovic M., Brar G.A. Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell. 2018;172:910–923. doi: 10.1016/j.cell.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weber R., Ghoshdastider U., Spies D., Duré C., Valdivia-Francia F., Forny M., Ormiston M., Renz P.F., Taborsky D., Yigit M., et al. Monitoring the 5’UTR landscape reveals isoform switches to drive translational efficiencies in cancer. Oncogene. 2023;42:638–650. doi: 10.1038/S41388-022-02578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hollerer I., Barker J.C., Jorgensen V., Tresenrider A., Dugast-Darzacq C., Chan L.Y., Darzacq X., Tjian R., Ünal E., Brar G.A. Evidence for an integrated gene repression mechanism based on mRNA isoform toggling in human cells. G3. 2019;9:1045–1053. doi: 10.1534/g3.118.200802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Floor S.N., Doudna J.A. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016;5 doi: 10.7554/eLife.10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arribere J.A., Gilbert W.V. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res. 2013;23:977–987. doi: 10.1101/GR.150342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rhoads A., Au K.F. PacBio Sequencing and Its Applications. Dev. Reprod. Biol. 2015;13:278–289. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kono N., Arakawa K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019;61:316–326. doi: 10.1111/DGD.12608. [DOI] [PubMed] [Google Scholar]
- 101.De Coster W., Weissensteiner M.H., Sedlazeck F.J. Towards population-scale long-read sequencing. Nat. Rev. Genet. 2021;22:572–587. doi: 10.1038/S41576-021-00367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haberle V., Forrest A.R.R., Hayashizaki Y., Carninci P., Lenhard B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Res. 2015;43:e51. doi: 10.1093/NAR/GKV054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Murata M., Nishiyori-Sueki H., Kojima-Ishiyama M., Carninci P., Hayashizaki Y., Itoh M. Detecting expressed genes using CAGE. Methods Mol. Biol. 2014;1164:67–85. doi: 10.1007/978-1-4939-0805-9_7. [DOI] [PubMed] [Google Scholar]
- 104.Hinnebusch A.G., Ivanov I.P., Sonenberg N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016;352:1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hinnebusch A.G. Structural Insights into the Mechanism of Scanning and Start Codon Recognition in Eukaryotic Translation Initiation. Trends Biochem. Sci. 2017;42:589–611. doi: 10.1016/j.tibs.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 106.Andreev D.E., O’Connor P.B.F., Loughran G., Dmitriev S.E., Baranov P.V., Shatsky I.N. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2017;45:513–526. doi: 10.1093/NAR/GKW1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hershey J.W.B., Sonenberg N., Mathews M.B. Principles of translational control: An overview. Cold Spring Harb. Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sonenberg N., Hinnebusch A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hao P., Yu J., Ward R., Liu Y., Hao Q., An S., Xu T. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun. Signal. 2020;18:1–20. doi: 10.1186/s12964-020-00607-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang H., Wang Y., Lu J. Function and Evolution of Upstream ORFs in Eukaryotes. Trends Biochem. Sci. 2019:1–13. doi: 10.1016/j.tibs.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 111.Schuster S.L., Hsieh A.C. The Untranslated Regions of mRNAs in Cancer. Trends Cancer. 2019;5:245–262. doi: 10.1016/j.trecan.2019.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sajjanar B., Deb R., Raina S.K., Pawar S., Brahmane M.P., Nirmale A.V., Kurade N.P., Manjunathareddy G.B., Bal S.K., Singh N.P. Untranslated regions (UTRs) orchestrate translation reprogramming in cellular stress responses. J. Therm. Biol. 2017;65:69–75. doi: 10.1016/j.jtherbio.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 113.Lacerda R., Menezes J., Romão L. More than just scanning: the importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell. Mol. Life Sci. 2017;74:1659–1680. doi: 10.1007/s00018-016-2428-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morris D.R., Geballe A.P. Upstream Open Reading Frames as Regulators of mRNA Translation. Mol. Cell Biol. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wethmar K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip. Rev. RNA. 2014;5:765–778. doi: 10.1002/wrna.1245. [DOI] [PubMed] [Google Scholar]
- 116.Silva J., Fernandes R., Romão L. Gene expression regulation by upstream open reading frames in rare diseases. J. Rare Dis. Res. Treat. 2017;2:33–38. doi: 10.29245/2572-9411/2017/4.1121. [DOI] [Google Scholar]
- 117.Araujo P.R., Yoon K., Ko D., Smith A.D., Qiao M., Suresh U., Burns S.C., Penalva L.O.F. Before it gets started: Regulating translation at the 5′; UTR. Comp. Funct. Genom. 2012;2012:475731. doi: 10.1155/2012/475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jackson R.J., Hellen C.U.T., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gurzeler L.A., Ziegelmüller J., Mühlemann O., Karousis E.D. Production of human translation-competent lysates using dual centrifugation. RNA Biol. 2022;19:78–88. doi: 10.1080/15476286.2021.2014695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fresno M., Jiménez A., Vázquez D. Inhibition of Translation in Eukaryotic Systems by Harringtonine. Eur. J. Biochem. 1977;72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- 122.Mojica F.J.M., Díez-Villaseñor C., Soria E., Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000;36:244–246. doi: 10.1046/J.1365-2958.2000.01838.X. [DOI] [PubMed] [Google Scholar]
- 123.Makarova K.S., Aravind L., Grishin N.V., Rogozin I.B., Koonin E.V. A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res. 2002;30:482–496. doi: 10.1093/NAR/30.2.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Barrangou R., Marraffini L.A. CRISPR-Cas Systems: Prokaryotes Upgrade to Adaptive Immunity. Mol. Cell. 2014;54:234–244. doi: 10.1016/J.MOLCEL.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Makarova K.S., Haft D.H., Barrangou R., Brouns S.J.J., Charpentier E., Horvath P., Moineau S., Mojica F.J.M., Wolf Y.I., Yakunin A.F., et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011;96:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wright A.V., Nuñez J.K., Doudna J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell. 2016;164:29–44. doi: 10.1016/J.CELL.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 127.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Doudna J.A., Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 129.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/NPROT.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weterings E., Chen D.J. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 131.Jiang W., Marraffini L.A. CRISPR-Cas: New Tools for Genetic Manipulations from Bacterial Immunity Systems. Annu Rev Microbiol. 2015;69:209–228. doi: 10.1146/annurev-micro-091014-104441. [DOI] [PubMed] [Google Scholar]
- 132.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scherer S. Cold Spring Harbor Laboratory Press; 2008. A Short Guide to the Human Genome. [Google Scholar]
- 134.Rodriguez C.M., Chun S.Y., Mills R.E., Todd P.K. Translation of upstream open reading frames in a model of neuronal differentiation. BMC Genom. 2019;20:1–18. doi: 10.1186/s12864-019-5775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Renz P.F., Valdivia Francia F., Sendoel A. Some like it translated: small ORFs in the 5′UTR. Exp. Cell Res. 2020;396:112229. doi: 10.1016/j.yexcr.2020.112229. [DOI] [PubMed] [Google Scholar]
- 136.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strecker J., Jones S., Koopal B., Schmid-Burgk J., Zetsche B., Gao L., Makarova K.S., Koonin E.V., Zhang F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019;10 doi: 10.1038/S41467-018-08224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Paul B., Montoya G. CRISPR-Cas12a: Functional overview and applications. Biomed. J. 2020;43:8–17. doi: 10.1016/J.BJ.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shalem O., Sanjana N.E., Zhang F. High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bock C., Datlinger P., Chardon F., Coelho M.A., Dong M.B., Lawson K.A., Lu T., Maroc L., Norman T.M., Song B., et al. High-content CRISPR screening. Nat. Rev. Methods Prim. 2022;2:1–23. doi: 10.1038/s43586-021-00093-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hanna R.E., Doench J.G. Design and analysis of CRISPR-Cas experiments. Nat. Biotechnol. 2020;38:813–823. doi: 10.1038/S41587-020-0490-7. [DOI] [PubMed] [Google Scholar]
- 142.Prensner J.R., Enache O.M., Luria V., Krug K., Clauser K.R., Dempster J.M., Karger A., Wang L., Stumbraite K., Wang V.M., et al. Noncanonical open reading frames encode functional proteins essential for cancer cell survival. Nat. Biotechnol. 2021;39:697–704. doi: 10.1038/s41587-020-00806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Braun C.J., Bruno P.M., Horlbeck M.A., Gilbert L.A., Weissman J.S., Hemann M.T. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc. Natl. Acad. Sci. USA. 2016;113:E3892–E3900. doi: 10.1073/pnas.1600582113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Manguso R.T., Pope H.W., Zimmer M.D., Brown F.D., Yates K.B., Miller B.C., Collins N.B., Bi K., La Fleur M.W., Juneja V.R., et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nowak C.M., Lawson S., Zerez M., Bleris L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Res. 2016;44:9555–9564. doi: 10.1093/nar/gkw908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dixit A., Parnas O., Li B., Chen J., Fulco C.P., Jerby-Arnon L., Marjanovic N.D., Dionne D., Burks T., Raychowdhury R., et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016;167:1853–1866.e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jaitin D.A., Weiner A., Yofe I., Lara-Astiaso D., Keren-Shaul H., David E., Salame T.M., Tanay A., van Oudenaarden A., Amit I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 2016;167:1883–1896.e15. doi: 10.1016/J.CELL.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 148.Xie S., Duan J., Li B., Zhou P., Hon G.C. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell. 2017;66:285–299.e5. doi: 10.1016/J.MOLCEL.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 149.Datlinger P., Rendeiro A.F., Schmidl C., Krausgruber T., Traxler P., Klughammer J., Schuster L.C., Kuchler A., Alpar D., Bock C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods. 2017;14:297–301. doi: 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cheng J., Lin G., Wang T., Wang Y.Y., Guo W., Liao J., Yang P., Chen J., Shao X., Lu X., et al. Massively Parallel CRISPR-Based Genetic Perturbation Screening at Single-Cell Resolution. Adv. Sci. 2022;10:2204484. doi: 10.1002/ADVS.202204484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alda-Catalinas C., Bredikhin D., Hernando-Herraez I., Santos F., Kubinyecz O., Eckersley-Maslin M.A., Stegle O., Reik W. A Single-Cell Transcriptomics CRISPR-Activation Screen Identifies Epigenetic Regulators of the Zygotic Genome Activation Program. Cell Syst. 2020;11:25–41.e9. doi: 10.1016/j.cels.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jin X., Simmons S.K., Guo A., Shetty A.S., Ko M., Nguyen L., Jokhi V., Robinson E., Oyler P., Curry N., et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science. 2020:370. doi: 10.1126/science.aaz6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hou J., Liang S., Xu C., Wei Y., Wang Y., Tan Y., Sahni N., McGrail D.J., Bernatchez C., Davies M., et al. Single-cell CRISPR immune screens reveal immunological roles of tumor intrinsic factors. NAR Cancer. 2022;4 doi: 10.1093/narcan/zcac038. [DOI] [PMC free article] [PubMed] [Google Scholar]