

Abstract

This work presents the synthesis of glucose-based vinyl imidazolium monomers and the hydrogels produced thereof. These novel semisynthetic, cationic hydrogels were prepared by radical polymerization with different types of commercial cross-linkers such as N,N′-methylenebis(acrylamide) or poly(ethylene glycol) diacrylate. Both the type and the amount of cross-linker were investigated as influencing factors with respect to the swelling degree. It was found that the cross-linker type majorly influences the swelling degree and long-term stability of the hydrogels. Last, the influence of different anions (e.g., halogens, acetate, and triflate) in the monomer molecule on the swelling properties of the hydrogels was investigated.
Introduction
Hydrogels are hydrophilic, three-dimensional cross-linked polymers that can, depending on their type of monomer and cross-linking degree, reversibly absorb large amounts of water due to the hydrogen bond formation while retaining their shape.1 Hydrogels have very diverse properties depending on their origin (natural, synthetic, or semisynthetic), their polymer composition (homo-, co-, or multipolymeric), their charge (ionic, nonionic, zwitterionic, or amphoteric), their physical appearance (matrix, film, or microsphere), their preparation (free radical polymerization or radiation), their cross-linking method (physical or chemical), and their configuration (amorphous, crystalline, or semicrystalline).2 Hydrogels are often attributed with properties such as biocompatibility and biodegradability,3,4 either stiffness5,6 or flexibility,7 depending on the used material, and antibacterial8 and stimuli-responsive3 properties. Therefore, they are suitable for application in medicine (e.g., as a drug delivery system),9 in biotechnology,10 as an immobilization matrix for biocatalysts (e.g., Candida antarctica lipase B),11 or in agricultural industry as water storage.12
Carbohydrate-based hydrogels, as one subgroup of the abovementioned natural hydrogels, are usually produced from polysaccharides such as cellulose, hyaluronic acid, or alginate. These polysaccharides are usually cross-linked using chemical methods by forming ether or ester bonds.13 Carbohydrate-based hydrogels are already well-known in many applications in the medical field (e.g., wound dressing and drug delivery) or for enzyme encapsulation.14
In contrast to the usage of (bio)polymers as starting materials, the direct hydrogel production from a mixture of monomer, cross-linker, and polymerization initiators in water allows for a more controlled approach, enabling the user to adjust the ratios between the monomer and cross-linker more freely. This method has recently been performed with commercially available ionic liquids such as vinyl imidazolium-based ILs.8 Using polymerizable ionic liquids as starting materials furthermore leads to either anionic, cationic, or zwitterionic hydrogels, which bear interesting possibilities in the medical field due to their ion exchange-controlled drug release.9 In the case of carbohydrates, notable work has been done on glucose- and mannose-based acryl esters,15,16 on carbohydrate vinyl ethers,17 and even on cationic glucose-based monomers,18 all of which bear isopropylidene or acetyl protecting groups and have been used for copolymerizations. However, hydrogels produced from carbohydrate-based monomers have, to the best of our knowledge, not been produced and investigated before.
Our group works in the field of carbohydrate-based ionic liquids.19−21 We previously found that glucosyl imidazolium-based ionic liquids can be produced in high total yields up to 90%22 and that they exhibit a remarkably high biocompatibility in comparison to commercial imidazolium ILs.23 In this work, we synthesized a series of novel glucosyl vinyl imidazolium salts and used these as monomers to produce novel glucose-based ionic hydrogels, cross-linked by several commercially available cross-linkers (Figure 1). These hydrogels combine several properties and techniques previously not found in this combination: They are semisynthetic hydrogels (produced from a synthetically modified natural carbohydrate source) bearing a unique cationic charge not found in natural carbohydrate hydrogels and are synthesized directly from a monomer–cross-linker mixture. To characterize our novel hydrogels, we investigated the swelling behavior. Knowledge of the swelling degree of hydrogels as a function of different cross-linkers enables prediction of the behavior of these gels in potential applications. High-swelling hydrogels for example are more suited for wound dressing, while low- or nonswelling hydrogels are used in tissue engineering or drug delivery.24 The swelling behavior of our hydrogels has been investigated in different media, with different cross-linkers and at different cross-linker concentrations in each case. Furthermore, the influence of different anions on the swelling behavior was examined.
Figure 1.

Cross-linkers used in this work (MBAA = N,N′-methylenebis(acrylamide), DHEBA = N,N′-(1,2-dihydroxyethylene)bis(acrylamide), EGDMA = ethylene glycol dimethacrylate, EGDA = ethylene glycol diacrylate, and PEGDA = poly(ethylene glycol) diacrylate).
Thus, the overall goal of this work is to establish the synthesis of the abovementioned novel monomers and hydrogels and to investigate their swelling behavior, laying the groundwork for future applications.
Materials and Methods
General Information
All reagents and solvents were purchased from commercial sources and used as received without further purification, if not stated otherwise. The NMR spectra were recorded on a Bruker AVANCE 250 II, Bruker AVANCE 300 III, or 500 NEO. CDCl3 was calibrated as 7.27 (1H) and 77.00 (13C). DMSO-d6 was calibrated as 2.49 (1H) and 39.50 (13C). D2O was calibrated as 4.80 (1H). ATR IR spectra were obtained using a Thermo Fisher Scientific Nicolet 380 FT-IR at room temperature. ESI-MS was measured in an Agilent 1200/6210 Time-of-Flight LC-MS or a Thermo Scientific Exactive ESI/DART FTMS. The specific rotations were measured with a Dr. Kernchen Gyromat-HP Digital Automatic Polarimeter with concentrations given in mg per mL. The measurements of thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed on a Setaram Labsys TGA-DSC 1600 with a heating program from 25 to 500 °C and a heating rate of 10 °C/min under an argon flow of 100 mL/min. Ion chromatograms were measured on a two-channel ion chromatography system “ProfIC 940 vario” with conductivity detection from the company Metrohm. The solid-state 13C NMR experiment was carried out on a Bruker AVANCE III HD spectrometer with an operating field of 400.5 MHz proton frequency with a Bruker ASCEND DNP 9.4 T widebore (89 mm) magnet while using a 1.3 mm probe.
Experimental Section
Methyl 6-Iodo-α-d-glucopyranoside 2a
2a was prepared according to a procedure previously published by our group.22
Methyl α-d-glucopyranoside (4.855 g, 25.0 mmol), triphenylphosphine (9.836 g, 37.5 mmol), iodine (9.518 g, 37.5 mmol), and imidazole (3.404 g, 50.0 mmol) were refluxed in THF (150 mL) for 4 h. The resulting solid was filtered off, the solvent was removed, and the product was obtained as a white solid (6.486 g, 85%) after column chromatography (chloroform/methanol 12:1).
Tm =
148–149 °C. 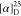 = +94.2 (c = 1.0, H2O). 1H NMR (300 MHz, DMSO-d6): δ = 2.87–2.95 (m, 1H); 3.16–3.27
(m,
3H); 3.31 (s, 3H, OCH3); 3.34–3.42 (m, 1H); 3.50–3.57
(m, 1H); 4.54 (d, 1H, 3J = 3.65 Hz, H-1);
4.78 (d, 1H, 3J = 6.43 Hz, OH); 4.86 (d,
1H, 3J = 4.99 Hz, OH); 5.17 (d, 1H, 3J = 5.83 Hz, OH). 13C NMR (75
MHz, DMSO-d6): δ = 9.5 (C-6); 54.6
(OCH3); 70.9, 71.9, 72.7, 74.1 (C-2, C-3, C-4, C-5); 99.8
(C-1). ATR-IR 3430, 3285, 2911, 2880, 2840, 1455, 1032 cm–1.
= +94.2 (c = 1.0, H2O). 1H NMR (300 MHz, DMSO-d6): δ = 2.87–2.95 (m, 1H); 3.16–3.27
(m,
3H); 3.31 (s, 3H, OCH3); 3.34–3.42 (m, 1H); 3.50–3.57
(m, 1H); 4.54 (d, 1H, 3J = 3.65 Hz, H-1);
4.78 (d, 1H, 3J = 6.43 Hz, OH); 4.86 (d,
1H, 3J = 4.99 Hz, OH); 5.17 (d, 1H, 3J = 5.83 Hz, OH). 13C NMR (75
MHz, DMSO-d6): δ = 9.5 (C-6); 54.6
(OCH3); 70.9, 71.9, 72.7, 74.1 (C-2, C-3, C-4, C-5); 99.8
(C-1). ATR-IR 3430, 3285, 2911, 2880, 2840, 1455, 1032 cm–1.
Methyl 6-Bromo-α-d-glucopyranoside 2b
2b was synthesized following the same procedure as 2a, using bromine (5.993 g, 1.92 mL, 37.5 mmol) instead of iodine. The bromine was slowly added to the THF solution at room temperature; afterward, the reaction was refluxed. The product was obtained as a white solid (4.493 g, 70%).
Tm = 127–129 °C.  = +105.6 (c = 1.2, H2O). 1H NMR (300 MHz, DMSO-d6): δ = 2.98–3.06 (m, 1H); 3.16–3.23
(m,
1H); 3.29 (s, 3H, OCH3); 3.34–3.41 (m, 1H); 3.45–3.56
(m, 2H); 3.72–3.76 (m, 1H); 4.55 (d, 1H, 3J = 3.67 Hz, H-1); 4.79 (d, 1H, 3J = 6.42 Hz, OH); 4.87 (d, 1H, 3J = 5.05
Hz, OH); 5.19 (d, 1H, 3J = 5.86 Hz, OH). 13C NMR (75 MHz, DMSO-d6): δ
= 35.2 (C-6); 54.5 (OCH3); 70.9, 71.8, 72.3, 73.0 (C-2,
C-3, C-4, C-5); 99.8 (C-1). The NMR data is according to the literature.25
= +105.6 (c = 1.2, H2O). 1H NMR (300 MHz, DMSO-d6): δ = 2.98–3.06 (m, 1H); 3.16–3.23
(m,
1H); 3.29 (s, 3H, OCH3); 3.34–3.41 (m, 1H); 3.45–3.56
(m, 2H); 3.72–3.76 (m, 1H); 4.55 (d, 1H, 3J = 3.67 Hz, H-1); 4.79 (d, 1H, 3J = 6.42 Hz, OH); 4.87 (d, 1H, 3J = 5.05
Hz, OH); 5.19 (d, 1H, 3J = 5.86 Hz, OH). 13C NMR (75 MHz, DMSO-d6): δ
= 35.2 (C-6); 54.5 (OCH3); 70.9, 71.8, 72.3, 73.0 (C-2,
C-3, C-4, C-5); 99.8 (C-1). The NMR data is according to the literature.25
Methyl 6-Chloro-α-d-glucopyranoside 2c
2c was synthesized following the same procedure as 2a, using N-chlorosuccinimide (5.007 g, 37.5 mmol) instead of iodine. The product was obtained as a white solid (1.637 g, 31%).
Tm = 110–112
°C.  = +137.8
(c = 1.2, H2O). 1H NMR (300
MHz, DMSO-d6): δ = 3.06 (ddd, 1H, 3J = 9.68 Hz, 3J = 8.62 Hz, 3J = 5.83 Hz, H-3); 3.19
(ddd, 1H, 3J = 9.75 Hz, 3J = 6.38 Hz, 3J = 3.69 Hz, H-2);
3.27 (s, 3H, OCH3); 3.37 (ddd, 1H, 3J = 9.43 Hz, 3J = 8.76 Hz, 3J = 4.96 Hz, H-4); 3.49–3.55 (m, 1H,
H-5); 3.67 (dd, 1H, 3J = 11.58 Hz, 3J = 6.39 Hz, H-6a); 3.84 (dd, 1H, 3J =
11.57 Hz, 3J = 2.17 Hz, H-6b); 4.56 (d,
1H, 3J = 3.65 Hz, H-1); 4.79 (d, 1H, 3J = 6.41 Hz, OH-2); 4.86 (d, 1H, 3J = 5.04 Hz, OH-4); 5.17 (d, 1H, 3J = 5.85 Hz, OH-3). 13C NMR (75 MHz, DMSO-d6): δ = 45.6 (C-6); 54.4 (OCH3); 71.2, 71.2, 71.8, 73.1 (C-2, C-3, C-4, C-5); 99.8 (C-1). The NMR
data is according to the literature.26
= +137.8
(c = 1.2, H2O). 1H NMR (300
MHz, DMSO-d6): δ = 3.06 (ddd, 1H, 3J = 9.68 Hz, 3J = 8.62 Hz, 3J = 5.83 Hz, H-3); 3.19
(ddd, 1H, 3J = 9.75 Hz, 3J = 6.38 Hz, 3J = 3.69 Hz, H-2);
3.27 (s, 3H, OCH3); 3.37 (ddd, 1H, 3J = 9.43 Hz, 3J = 8.76 Hz, 3J = 4.96 Hz, H-4); 3.49–3.55 (m, 1H,
H-5); 3.67 (dd, 1H, 3J = 11.58 Hz, 3J = 6.39 Hz, H-6a); 3.84 (dd, 1H, 3J =
11.57 Hz, 3J = 2.17 Hz, H-6b); 4.56 (d,
1H, 3J = 3.65 Hz, H-1); 4.79 (d, 1H, 3J = 6.41 Hz, OH-2); 4.86 (d, 1H, 3J = 5.04 Hz, OH-4); 5.17 (d, 1H, 3J = 5.85 Hz, OH-3). 13C NMR (75 MHz, DMSO-d6): δ = 45.6 (C-6); 54.4 (OCH3); 71.2, 71.2, 71.8, 73.1 (C-2, C-3, C-4, C-5); 99.8 (C-1). The NMR
data is according to the literature.26
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Iodide 3a (GVIM-I)
3a was prepared according to a procedure previously published by our group.27
2a (3.649 g, 12.0 mmol) and N-vinylimidazole (1.882 g, 20.0 mmol) were dissolved in DMF (20 mL) and stirred at 95 °C for 24 h. After cooling down, ethyl acetate (160 mL) was added and the flask was stored in a fridge overnight. The solvent was decanted, and the precipitated solid was washed with ethyl acetate (3 × 80 mL) and dried under high vacuum to achieve the product as a light-brown solid (3.959 g, 83%).
Tm = 185 °C. Td = 236 °C. 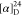 = +30.3 (c = 2.6, H2O). 1H NMR (500 MHz, D2O): δ =
3.27 (s, 3H, OCH3); 3.24–3.28 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.79 Hz, 3J = 3.82 Hz, H-2); 3.69–3.72 (m, 1H, H-3); 3.96 (ddd,
1H, 3J = 9.96 Hz, 3J = 7.47 Hz, 3J = 2.46 Hz, H-5);
4.50 (dd, 1H, 2J = 14.57 Hz, 3J = 7.46 Hz, H-6a); 4.70 (dd, 1H, 2J = 14.56 Hz, 3J = 2.49, H-6b);
4.85 (d, 1H, 3J = 3.80 Hz, H-1); 5.49
(dd, 1H, 3J = 8.67 Hz, 2J = 2.87 Hz, Vinyl-CH2); 5.86 (dd, 1H, 3J = 15.58 Hz, 2J = 2.86
Hz, Vinyl-CH2); 7.20 (dd, 1H, 3J = 15.58 Hz, 3J = 8.68 Hz, Vinyl-CH);
7.70 (d, 1H, 3J = 2.09 Hz, HAr); 7.86 (d, 1H, 3J = 2.11 Hz, HAr). 13C NMR (125 MHz, D2O): δ = 50.2 (C-6);
55.1 (OCH3); 69.2 (C-5); 70.5 (C-4); 71.0 (C-2); 72.8 (C-3);
99.3 (C-1); 109.8 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.2 (CHAr). ATR-IR 3396, 3088,
2998, 2910, 1657, 1566, 1549, 1048, 1015 cm–1. HRMS
(ESI, m/z): Calculated for C12H19N2O5+, 271.1299;
measured 271.1306. Calculated for I–, 126.9040;
measured 126.9045.
= +30.3 (c = 2.6, H2O). 1H NMR (500 MHz, D2O): δ =
3.27 (s, 3H, OCH3); 3.24–3.28 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.79 Hz, 3J = 3.82 Hz, H-2); 3.69–3.72 (m, 1H, H-3); 3.96 (ddd,
1H, 3J = 9.96 Hz, 3J = 7.47 Hz, 3J = 2.46 Hz, H-5);
4.50 (dd, 1H, 2J = 14.57 Hz, 3J = 7.46 Hz, H-6a); 4.70 (dd, 1H, 2J = 14.56 Hz, 3J = 2.49, H-6b);
4.85 (d, 1H, 3J = 3.80 Hz, H-1); 5.49
(dd, 1H, 3J = 8.67 Hz, 2J = 2.87 Hz, Vinyl-CH2); 5.86 (dd, 1H, 3J = 15.58 Hz, 2J = 2.86
Hz, Vinyl-CH2); 7.20 (dd, 1H, 3J = 15.58 Hz, 3J = 8.68 Hz, Vinyl-CH);
7.70 (d, 1H, 3J = 2.09 Hz, HAr); 7.86 (d, 1H, 3J = 2.11 Hz, HAr). 13C NMR (125 MHz, D2O): δ = 50.2 (C-6);
55.1 (OCH3); 69.2 (C-5); 70.5 (C-4); 71.0 (C-2); 72.8 (C-3);
99.3 (C-1); 109.8 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.2 (CHAr). ATR-IR 3396, 3088,
2998, 2910, 1657, 1566, 1549, 1048, 1015 cm–1. HRMS
(ESI, m/z): Calculated for C12H19N2O5+, 271.1299;
measured 271.1306. Calculated for I–, 126.9040;
measured 126.9045.
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Bromide 3b (GVIM-Br)
3b was synthesized following the same procedure as 3a but instead using 2b (1.536 g, 6.0 mmol) as a starting material and a reaction temperature of 110 °C. The product was obtained as a light-brown solid (1.659 g, 79%).
Tm = 178 °C. Td = 225 °C. 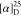 = +83.4 (c = 1.2, H2O). 1H NMR (300 MHz, D2O): δ =
3.28 (s, 3H, OCH3); 3.23–3.33 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.79 Hz, 3J = 3.79 Hz, H-2); 3.68–3.75 (m, 1H, H-3); 3.96 (ddd,
1H, 3J = 10.01 Hz, 3J = 7.47 Hz, 3J = 2.58 Hz, H-5);
4.51 (dd, 1H, 2J = 14.54 Hz, 3J = 7.40 Hz, H-6a); 4.71 (dd, 1H, 2J = 14.56 Hz, 3J = 2.56, H-6b);
4.86 (d, 1H, 3J = 3.75 Hz, H-1); 5.50
(dd, 1H, 3J = 8.68 Hz, 2J = 2.84 Hz, Vinyl-CH2); 5.87 (dd, 1H, 3J = 15.58 Hz, 2J = 2.85
Hz, Vinyl-CH2); 7.21 (dd, 1H, 3J = 15.61 Hz, 3J = 8.67 Hz, Vinyl-CH);
7.71 (d, 1H, 3J = 2.10 Hz, HAr); 7.87 (d, 1H, 3J = 2.12 Hz, HAr); 9.17 (s, 1H, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5);
70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.8 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.5
(CHAr). HRMS (ESI, m/z): Calculated for C12H19N2O5+, 271.1299; measured 271.1296. Calculated for 79Br–, 78.9183; measured 78.9183. Calculated
for 81Br–, 80.9163; measured 80.9162.
= +83.4 (c = 1.2, H2O). 1H NMR (300 MHz, D2O): δ =
3.28 (s, 3H, OCH3); 3.23–3.33 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.79 Hz, 3J = 3.79 Hz, H-2); 3.68–3.75 (m, 1H, H-3); 3.96 (ddd,
1H, 3J = 10.01 Hz, 3J = 7.47 Hz, 3J = 2.58 Hz, H-5);
4.51 (dd, 1H, 2J = 14.54 Hz, 3J = 7.40 Hz, H-6a); 4.71 (dd, 1H, 2J = 14.56 Hz, 3J = 2.56, H-6b);
4.86 (d, 1H, 3J = 3.75 Hz, H-1); 5.50
(dd, 1H, 3J = 8.68 Hz, 2J = 2.84 Hz, Vinyl-CH2); 5.87 (dd, 1H, 3J = 15.58 Hz, 2J = 2.85
Hz, Vinyl-CH2); 7.21 (dd, 1H, 3J = 15.61 Hz, 3J = 8.67 Hz, Vinyl-CH);
7.71 (d, 1H, 3J = 2.10 Hz, HAr); 7.87 (d, 1H, 3J = 2.12 Hz, HAr); 9.17 (s, 1H, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5);
70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.8 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.5
(CHAr). HRMS (ESI, m/z): Calculated for C12H19N2O5+, 271.1299; measured 271.1296. Calculated for 79Br–, 78.9183; measured 78.9183. Calculated
for 81Br–, 80.9163; measured 80.9162.
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Chloride 3c (GVIM-Cl)
3c was synthesized following the same procedure as 3a but instead using 2c (0.638 g, 3.0 mmol) as a starting material and a reaction temperature of 150 °C. The product was obtained as a brown solid (0.535 g, 58%).
Td = 201 °C. 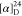 = +89.6 (c = 3.2, MeOH). 1H NMR (300 MHz, D2O): δ
= 3.27 (s, 3H, OCH3); 3.23–3.29 (m, 1H, H-4); 3.57
(dd, 1H, 3J = 9.79 Hz, 3J = 3.78
Hz, H-2); 3.68–3.74 (m, 1H, H-3); 3.96 (ddd, 1H, 3J = 9.80 Hz, 3J = 7.41
Hz, 3J = 2.60 Hz, H-5); 4.50 (dd, 1H, 2J = 14.57 Hz, 3J = 7.38 Hz, H-6a); 4.70 (dd, 1H, 2J =
14.55 Hz, 3J = 2.59, H-6b); 4.85 (d, 1H, 3J = 3.76 Hz, H-1); 5.49 (dd, 1H, 3J = 8.68 Hz, 2J = 2.85
Hz, Vinyl-CH2); 5.87 (dd, 1H, 3J = 15.58 Hz, 2J = 2.85 Hz, Vinyl-CH2); 7.21 (dd, 1H, 3J = 15.59 Hz, 3J = 8.69 Hz, Vinyl-CH); 7.71 (d, 1H, 3J = 2.12 Hz, HAr); 7.86 (d, 1H, 3J = 2.15 Hz, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5); 70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.4 (C-1);
109.7 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1
(Vinyl-CH). HRMS (ESI, m/z): Calculated
for C12H19N2O5+, 271.1299; measured 271.1291.
= +89.6 (c = 3.2, MeOH). 1H NMR (300 MHz, D2O): δ
= 3.27 (s, 3H, OCH3); 3.23–3.29 (m, 1H, H-4); 3.57
(dd, 1H, 3J = 9.79 Hz, 3J = 3.78
Hz, H-2); 3.68–3.74 (m, 1H, H-3); 3.96 (ddd, 1H, 3J = 9.80 Hz, 3J = 7.41
Hz, 3J = 2.60 Hz, H-5); 4.50 (dd, 1H, 2J = 14.57 Hz, 3J = 7.38 Hz, H-6a); 4.70 (dd, 1H, 2J =
14.55 Hz, 3J = 2.59, H-6b); 4.85 (d, 1H, 3J = 3.76 Hz, H-1); 5.49 (dd, 1H, 3J = 8.68 Hz, 2J = 2.85
Hz, Vinyl-CH2); 5.87 (dd, 1H, 3J = 15.58 Hz, 2J = 2.85 Hz, Vinyl-CH2); 7.21 (dd, 1H, 3J = 15.59 Hz, 3J = 8.69 Hz, Vinyl-CH); 7.71 (d, 1H, 3J = 2.12 Hz, HAr); 7.86 (d, 1H, 3J = 2.15 Hz, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5); 70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.4 (C-1);
109.7 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1
(Vinyl-CH). HRMS (ESI, m/z): Calculated
for C12H19N2O5+, 271.1299; measured 271.1291.
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Acetate 4a (GVIM-OAc)
3a (5.03 mmol, 2.002 g) and silver acetate (5.03 mmol, 838 mg) were suspended in water (20 mL) and stirred for 30 min under the absence of light. The yellow precipitate was removed via filtration. Activated charcoal (∼500 mg) was added to the filtrate, and the mixture was stirred for 2 h. The product 4a was achieved as a white solid (1.320 g, 79%) after filtration and removal of water.
Td = 287 °C.  = +91.6 (c = 1.3, MeOH). 1H NMR (500 MHz, D2O): δ
= 1.92 (s, 3H, CH3); 3.25 (s, 3H, OCH3); 3.21–3.25
(m, 1H,
H-4); 3.55 (dd, 1H, 3J = 9.79 Hz, 3J = 3.82 Hz, H-2); 3.67–3.71 (m, 1H,
H-3); 3.93 (ddd, 1H, 3J = 9.97 Hz, 3J = 7.47 Hz, 3J = 2.49 Hz, H-5); 4.48 (dd, 1H, 2J =
14.57 Hz, 3J = 7.46 Hz, H-6a); 4.68 (dd,
1H, 2J = 14.56 Hz, 3J = 2.51 Hz, H-6b); 4.83 (d, 1H, 3J = 3.81 Hz, H-1); 5.47 (dd, 1H, 3J =
8.68 Hz, 2J = 2.87 Hz, Vinyl-CH2); 5.84 (dd, 1H, 3J = 15.58 Hz, 2J = 2.87 Hz, Vinyl-CH2); 7.18
(dd, 1H, 3J = 15.59 Hz, 3J = 8.68 Hz, Vinyl-CH); 7.68 (d, 1H, 3J = 2.11 Hz, HAr); 7.84 (d, 1H, 3J = 2.13 Hz, HAr). 13C NMR (125 MHz,
D2O): δ = 23.2 (CH3); 50.2 (C-6); 55.0
(OCH3); 69.2 (C-5); 70.4 (C-4); 71.0 (C-2); 72.8 (C-3);
99.3 (C-1); 109.7 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.2 (CHAr); 181.3 (C=O). HRMS (ESI, m/z): Calculated for C12H19N2O5+, 271.1299; measured
271.1303.
= +91.6 (c = 1.3, MeOH). 1H NMR (500 MHz, D2O): δ
= 1.92 (s, 3H, CH3); 3.25 (s, 3H, OCH3); 3.21–3.25
(m, 1H,
H-4); 3.55 (dd, 1H, 3J = 9.79 Hz, 3J = 3.82 Hz, H-2); 3.67–3.71 (m, 1H,
H-3); 3.93 (ddd, 1H, 3J = 9.97 Hz, 3J = 7.47 Hz, 3J = 2.49 Hz, H-5); 4.48 (dd, 1H, 2J =
14.57 Hz, 3J = 7.46 Hz, H-6a); 4.68 (dd,
1H, 2J = 14.56 Hz, 3J = 2.51 Hz, H-6b); 4.83 (d, 1H, 3J = 3.81 Hz, H-1); 5.47 (dd, 1H, 3J =
8.68 Hz, 2J = 2.87 Hz, Vinyl-CH2); 5.84 (dd, 1H, 3J = 15.58 Hz, 2J = 2.87 Hz, Vinyl-CH2); 7.18
(dd, 1H, 3J = 15.59 Hz, 3J = 8.68 Hz, Vinyl-CH); 7.68 (d, 1H, 3J = 2.11 Hz, HAr); 7.84 (d, 1H, 3J = 2.13 Hz, HAr). 13C NMR (125 MHz,
D2O): δ = 23.2 (CH3); 50.2 (C-6); 55.0
(OCH3); 69.2 (C-5); 70.4 (C-4); 71.0 (C-2); 72.8 (C-3);
99.3 (C-1); 109.7 (Vinyl-CH2); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.2 (CHAr); 181.3 (C=O). HRMS (ESI, m/z): Calculated for C12H19N2O5+, 271.1299; measured
271.1303.
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Methanesulfonate 4b (GVIM-OMs)
3a (5.03 mmol, 2.002 g) and silver methanesulfonate (5.03 mmol, 1.023 g) were suspended in water (80 mL) and stirred for 30 min under the absence of light. The yellow precipitate was removed via filtration. Activated charcoal (∼500 mg) was added to the filtrate, and the mixture was stirred for 24 h. After filtration and removal of water, the remaining solid was purified via column chromatography (methanol) to achieve the product 4b as colorless wax (1.198 g, 65%).
Td = 226 °C. 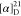 = +68.2 (c = 2.5, MeOH). 1H NMR (500 MHz, D2O): δ
= 2.82 (s, 3H, CH3); 3.25 (s, 3H, OCH3); 3.22–3.26
(m, 1H,
H-4); 3.56 (dd, 1H, 3J = 9.79 Hz, 3J = 3.83 Hz, H-2); 3.67–3.71 (m, 1H,
H-3); 3.94 (ddd, 1H, 3J = 9.97 Hz, 3J = 7.48 Hz, 3J = 2.47 Hz, H-5); 4.49 (dd, 1H, 2J =
14.57 Hz, 3J = 7.47 Hz, H-6a); 4.68 (dd,
1H, 2J = 14.56 Hz, 3J = 2.48 Hz, H-6b); 4.83 (d, 1H, 3J = 3.81 Hz, H-1); 5.48 (dd, 1H, 3J =
8.67 Hz, 2J = 2.87 Hz, Vinyl-CH2); 5.85 (dd, 1H, 3J = 15.58 Hz, 2J = 2.88 Hz, Vinyl-CH2); 7.19
(dd, 1H, 3J = 15.59 Hz, 3J = 8.69 Hz, Vinyl-CH); 7.69 (d, 1H, 3J = 1.78 Hz, HAr); 7.85 (d, 1H, 3J = 1.86 Hz, HAr); 9.14 (s, 1H, HAr). 13C NMR (125 MHz, D2O): δ = 38.5 (CH3); 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5); 70.5 (C-4);
71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.7 (Vinyl-CH2);
119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.5 (CHAr). HRMS (ESI, m/z): Calculated
for C12H19N2O5+, 271.1299; measured 271.1301. Calculated for CH3O3S–, 94.9798; measured 94.9797.
= +68.2 (c = 2.5, MeOH). 1H NMR (500 MHz, D2O): δ
= 2.82 (s, 3H, CH3); 3.25 (s, 3H, OCH3); 3.22–3.26
(m, 1H,
H-4); 3.56 (dd, 1H, 3J = 9.79 Hz, 3J = 3.83 Hz, H-2); 3.67–3.71 (m, 1H,
H-3); 3.94 (ddd, 1H, 3J = 9.97 Hz, 3J = 7.48 Hz, 3J = 2.47 Hz, H-5); 4.49 (dd, 1H, 2J =
14.57 Hz, 3J = 7.47 Hz, H-6a); 4.68 (dd,
1H, 2J = 14.56 Hz, 3J = 2.48 Hz, H-6b); 4.83 (d, 1H, 3J = 3.81 Hz, H-1); 5.48 (dd, 1H, 3J =
8.67 Hz, 2J = 2.87 Hz, Vinyl-CH2); 5.85 (dd, 1H, 3J = 15.58 Hz, 2J = 2.88 Hz, Vinyl-CH2); 7.19
(dd, 1H, 3J = 15.59 Hz, 3J = 8.69 Hz, Vinyl-CH); 7.69 (d, 1H, 3J = 1.78 Hz, HAr); 7.85 (d, 1H, 3J = 1.86 Hz, HAr); 9.14 (s, 1H, HAr). 13C NMR (125 MHz, D2O): δ = 38.5 (CH3); 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5); 70.5 (C-4);
71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.7 (Vinyl-CH2);
119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.5 (CHAr). HRMS (ESI, m/z): Calculated
for C12H19N2O5+, 271.1299; measured 271.1301. Calculated for CH3O3S–, 94.9798; measured 94.9797.
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Trifluoromethanesulfonate 4c (GVIM-OTf)
3a (5.03 mmol, 2.002 g) and silver trifluoromethanesulfonate (5.03 mmol, 1.295 g) were suspended in water (80 mL) and stirred for 2 h under the absence of light. The yellow precipitate was removed via filtration. Activated charcoal (∼500 mg) was added to the filtrate, and the mixture was stirred for 2 h. The product 4c was achieved as a colorless viscous liquid (1.850 g, 88%) after filtration and removal of water. If the black silver particles still remain in the product, a third filtration from water is necessary.
Td = 190 °C. 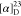 = +54.1 (c = 1.3, MeOH). 1H NMR (300 MHz, D2O): δ
= 3.27 (s, 3H, OCH3); 3.23–3.29 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.78 Hz, 3J = 3.80
Hz, H-2); 3.68–3.74 (m, 1H, H-3); 3.96 (ddd, 1H, 3J = 9.91 Hz, 3J = 7.42
Hz, 3J = 2.50 Hz, H-5); 4.50 (dd, 1H, 2J = 14.54 Hz, 3J = 7.43 Hz, H-6a); 4.70 (dd, 1H, 2J =
14.53 Hz, 3J = 2.56 Hz, H-6b); 4.85 (d,
1H, 3J = 3.78 Hz, H-1); 5.50 (dd, 1H, 3J = 8.68 Hz, 2J = 2.86 Hz, Vinyl-CH2); 5.86 (dd, 1H, 3J = 15.58 Hz, 2J = 2.86 Hz,
Vinyl-CH2); 7.20 (dd, 1H, 3J = 15.59 Hz, 3J = 8.70 Hz, Vinyl-CH);
7.71 (d, 1H, 3J = 1.82 Hz, HAr); 7.86 (d, 1H, 3J = 1.89 Hz, HAr); 9.15 (s, 1H, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5);
70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.7 (Vinyl-CH2); 119.4 (CHAr); 119.6 (q, 1J = 317.2 Hz, CF3); 123.8 (CHAr);
128.1 (Vinyl-CH); 135.4 (CHAr). 19F-NMR (282
MHz, D2O): δ = −78.8. HRMS (ESI, m/z): Calculated for C12H19N2O5+, 271.1299; measured 271.1295.
Calculated for CF3O3S–, 148.9515;
measured 148.9519.
= +54.1 (c = 1.3, MeOH). 1H NMR (300 MHz, D2O): δ
= 3.27 (s, 3H, OCH3); 3.23–3.29 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.78 Hz, 3J = 3.80
Hz, H-2); 3.68–3.74 (m, 1H, H-3); 3.96 (ddd, 1H, 3J = 9.91 Hz, 3J = 7.42
Hz, 3J = 2.50 Hz, H-5); 4.50 (dd, 1H, 2J = 14.54 Hz, 3J = 7.43 Hz, H-6a); 4.70 (dd, 1H, 2J =
14.53 Hz, 3J = 2.56 Hz, H-6b); 4.85 (d,
1H, 3J = 3.78 Hz, H-1); 5.50 (dd, 1H, 3J = 8.68 Hz, 2J = 2.86 Hz, Vinyl-CH2); 5.86 (dd, 1H, 3J = 15.58 Hz, 2J = 2.86 Hz,
Vinyl-CH2); 7.20 (dd, 1H, 3J = 15.59 Hz, 3J = 8.70 Hz, Vinyl-CH);
7.71 (d, 1H, 3J = 1.82 Hz, HAr); 7.86 (d, 1H, 3J = 1.89 Hz, HAr); 9.15 (s, 1H, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5);
70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.7 (Vinyl-CH2); 119.4 (CHAr); 119.6 (q, 1J = 317.2 Hz, CF3); 123.8 (CHAr);
128.1 (Vinyl-CH); 135.4 (CHAr). 19F-NMR (282
MHz, D2O): δ = −78.8. HRMS (ESI, m/z): Calculated for C12H19N2O5+, 271.1299; measured 271.1295.
Calculated for CF3O3S–, 148.9515;
measured 148.9519.
1-(Methyl-α-d-glucopyranosid-6-yl)-3-vinylimidazolium Bis(trifluoromethanesulfonyl)-imide 4d (GVIM-NTf2)
3a (5.03 mmol, 2.002 g) and silver bis(trifluoromethanesulfonyl)-imide (5.03 mmol, 1.948 g) were suspended in water (30 mL) and stirred for 20 h under the absence of light. The yellow precipitate was removed via filtration. Activated charcoal (∼500 mg) was added to the filtrate, and the mixture was stirred for 20 h. The product 4d was achieved as a yellow liquid (2.014 g, 73%) after filtration and removal of water. If the black silver particles still remain in the product, a third filtration from water is necessary.
Td = 286 °C. 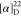 = +46.7 (c = 1.1, MeOH). 1H NMR (300 MHz, D2O): δ
= 3.27 (s, 3H, OCH3); 3.23–3.29 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.78 Hz, 3J = 3.80
Hz, H-2); 3.68–3.74 (m, 1H, H-3); 3.95 (ddd, 1H, 3J = 9.98 Hz, 3J = 7.47
Hz, 3J = 2.56 Hz, H-5); 4.50 (dd, 1H, 2J = 14.56 Hz, 3J = 7.43 Hz, H-6a); 4.70 (dd, 1H, 2J =
14.57 Hz, 3J = 2.54 Hz, H-6b); 4.85 (d,
1H, 3J = 3.77 Hz, H-1); 5.50 (dd, 1H, 3J = 8.68 Hz, 2J = 2.86 Hz, Vinyl-CH2); 5.87 (dd, 1H, 3J = 15.58 Hz, 2J = 2.87 Hz,
Vinyl-CH2); 7.20 (dd, 1H, 3J = 15.59 Hz, 3J = 8.69 Hz, Vinyl-CH);
7.71 (d, 1H, 3J = 1.82 Hz, HAr); 7.86 (d, 1H, 3J = 1.89 Hz, HAr); 9.15 (s, 1H, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5);
70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.7 (Vinyl-CH2); 119.2 (q, 1J = 319.7 Hz, CF3); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.4
(CHAr). 19F-NMR (282 MHz, D2O): δ
= −79.2. HRMS (ESI, m/z):
Calculated for C12H19N2O5+, 271.1299; measured 271.1301. Calculated for C2F6NO4S2–, 279.9168;
measured 279.9173.
= +46.7 (c = 1.1, MeOH). 1H NMR (300 MHz, D2O): δ
= 3.27 (s, 3H, OCH3); 3.23–3.29 (m, 1H, H-4); 3.58
(dd, 1H, 3J = 9.78 Hz, 3J = 3.80
Hz, H-2); 3.68–3.74 (m, 1H, H-3); 3.95 (ddd, 1H, 3J = 9.98 Hz, 3J = 7.47
Hz, 3J = 2.56 Hz, H-5); 4.50 (dd, 1H, 2J = 14.56 Hz, 3J = 7.43 Hz, H-6a); 4.70 (dd, 1H, 2J =
14.57 Hz, 3J = 2.54 Hz, H-6b); 4.85 (d,
1H, 3J = 3.77 Hz, H-1); 5.50 (dd, 1H, 3J = 8.68 Hz, 2J = 2.86 Hz, Vinyl-CH2); 5.87 (dd, 1H, 3J = 15.58 Hz, 2J = 2.87 Hz,
Vinyl-CH2); 7.20 (dd, 1H, 3J = 15.59 Hz, 3J = 8.69 Hz, Vinyl-CH);
7.71 (d, 1H, 3J = 1.82 Hz, HAr); 7.86 (d, 1H, 3J = 1.89 Hz, HAr); 9.15 (s, 1H, HAr). 13C NMR (75 MHz, D2O): δ = 50.2 (C-6); 55.0 (OCH3); 69.3 (C-5);
70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.7 (Vinyl-CH2); 119.2 (q, 1J = 319.7 Hz, CF3); 119.4, 123.8 (CHAr); 128.1 (Vinyl-CH); 135.4
(CHAr). 19F-NMR (282 MHz, D2O): δ
= −79.2. HRMS (ESI, m/z):
Calculated for C12H19N2O5+, 271.1299; measured 271.1301. Calculated for C2F6NO4S2–, 279.9168;
measured 279.9173.
Hydrogel Synthesis
3a–3c or 4a–4d were dissolved in pure water and an appropriate weight-percent of cross-linker was added. Radical polymerization (Scheme 1) was initiated by adding APS solution followed by TEMED and then immediately mixing the solution thoroughly for 10 s. The used amounts of monomer, cross-linker, water, APS, and TEMED for each hydrogel composition can be found in the Supporting Information (Tables S1–S12). After transferring the solution in cylindrical-shaped molds (10 mm diameter, 10 mm height), the gelation took place within seconds or a few minutes. After removal of the gels, they were stored in a compartment dryer at 40 °C for 3 days.
Scheme 1. Synthesis of Carbohydrate-Based Ionic Hydrogels via Radical Polymerization with PEGDA as a Cross-Linker.

After the initial synthesis and drying, the hydrogels can be washed with pure water (3 × 10 mL, 10 min each) to remove the unreacted monomers and TEMED, as performed for the NMR studies of the hydrogels (see Results and Discussion Section.
Gravimetric Swelling Experiments
The solvent uptake was measured gravimetrically by weighing the mass of the hydrogel as a function of time. After determining the dry mass of the samples, the gels were placed in a sieve, which was in a beaker filled with PBS, water, or isotonic NaCl solution (see Supporting Information Figures S29 and S30 for the setup) and allowed to soak at 37 ± 1 °C. The sieves were removed at monitored time intervals. Both the sieves and the gels were carefully blotted with paper towel to remove the surface-bound solvent. It was then weighed and returned to solution. These swelling studies were performed in triplicate to investigate the swelling behavior.
Calculations from Gravimetric Swelling Experiments
The experimental equilibrium swelling (qt) of the hydrogels was calculated from the data by the following term
with wt being the weight of the swollen gel after time t and w0 being the weight of the initial dry gel at the time t = 0.
Results and Discussion
Monomer Synthesis
The first step of this work was to produce the envisioned starting material 3 needed for the hydrogel synthesis. We adopted the reaction conditions from our previous works on glucosyl imidazolium salts bearing different alkyl chain lengths on the imidazole,22,23 this time however focusing on vinyl imidazole (Scheme 2). Here, the vinyl group is crucial for the three-dimensional cross-linking of the resulting monomer.
Scheme 2. Synthesis of Glucosyl Vinyl Imidazolium Salts.

The synthesis starts with commercially available methyl α-d-glucopyranoside 1. The methyl glycoside is necessary to block the anomeric center of the carbohydrate, which would otherwise hinder the following two reaction steps. 1 was chemoselectively converted into the 6-halogenated glucopyranosides 2a–2c by using Appel reaction conditions. Previously, our group only worked with the 6-iodo product 2a, which is synthesized in a high yield of 85%.22 This time we also produced the 6-bromo product 2b (70% yield) and the 6-chloro product 2c (31% yield) by using bromine or N-chlorosuccinimide as halogen sources.
As the next step, vinyl imidazole was added to the 6-halogenated glucopyranosides 2a–2c. Here, the reaction temperature needed to be adjusted for each material. While a reaction temperature of 110 °C leads to 99% yield in the case of the glucosyl vinyl imidazolium iodide (GVIM-I) 3a, this product also turned to a dark brown color at this temperature. Though the NMR analysis remains seemingly pure, this dark color would negatively impact the later hydrogel studies. Lowering the reaction temperature to 95 °C leads to a visibly cleaner product with only a slight drop in yield to 83%. GVIM-Br 3b was produced at 110 °C in 79% yield without any dark coloration and GVIM-Cl 3c needs a high temperature of 150 °C to even reach a mediocre yield of 58% and also exhibits the aforementioned dark color. Furthermore, product 3a has also been individually confirmed by X-ray analysis (Figure 2).
Figure 2.

ORTEP of GVIM-I 3a. Reproduced with permission from ref (22). Copyright 2022 IUCrData.
As expected, the product yield for both the 6-halogenated glucopyranosides 2a–2c as well as for the glucosyl vinyl imidazolium halogenides 3a–3c is in the order of I > Br ≫ Cl, mirroring the general reactivities of the halogens.
Since our glucosyl vinyl imidazolium products are salts with a cationic carbohydrate core, which will also turn into a cationic polymer network with freely movable anions in the hydrogels, we were also interested in the impact of different anions onto the swelling behavior of the hydrogels. Thus, besides the already produced salts 3a–3c with halogenide anions, we also synthesized the products 4a–4d in good yields from 65 to 88% by using anion exchange reactions with silver salts (Scheme 3). It should be noted that the anion exchange itself is achieved in full conversion; however, the several filtration steps necessary to remove all silver particles from the products lead to lower yields.
Scheme 3. Anion Exchange Reactions.

These products contain anions that are similar to typical, commercially available imidazolium-based ionic liquids, and thus, unsurprisingly, all four products GVIM-OAc 4a, GVIM-OMs 4b, GVIM-OTf 4c, and GVIM-NTf24d are viscous liquids or waxes at room temperature.
Hydrogel Synthesis
Initially, GVIM-I 3a as a monomer and 5.0 wt % MBAA (Figure 1), relative to the monomer weight, as a cross-linker were investigated for hydrogel synthesis. For the cross-linking polymerization between 3a and MBAA, the radical initiators ammonium persulfate (APS) and tetramethyl ethylenediamine (TEMED) were used as this system has previously also been used for dialkyl substituted vinyl imidazolium ionic liquids.28 The appropriate amounts of APS and TEMED in relation to the monomer or cross-linker usually differ for each monomer/cross-linker system,29−31 and in our case, a molar ratio (cross-linker:APS:TEMED) of 1:1.5:5.6 was found to lead to a successful gelation for 3a and 5.0 wt % MBAA. This molar ratio was used as a starting point for all of the different monomers, cross-linkers, and cross-linker concentrations investigated in this work; however, individual optimization toward higher or lower amounts of APS/TEMED was still necessary in some cases. The used amounts of monomer, cross-linker, water, APS, and TEMED for each hydrogel composition can be found in the Supporting Information (Tables S1–S12).
A first characterization of the hydrogels was performed via ATR-IR. The IR spectra of the GVIM-MBAA and GVIM-PEGDA575 hydrogels are mostly a direct combination of the spectrum of GVIM-I 3a and the corresponding cross-linker. The fingerprint area of the carbohydrate monomer is also present in the spectra of the hydrogels (see Supporting Information Figures S25–S28 for the IR spectra).
As a second characterization method, we also performed a solid-state 13C NMR of a GVIM-MBAA hydrogel. Solid-state NMR is necessary due to the insolubility of the hydrogels in any common solvent. For sample preparation, a dried hydrogel was milled into a powder and the said hydrogel powder was washed (three times with pure water, 10 min each) and dried again. The washing water was analyzed by NMR, and a mixture of unreacted GVIM-I and TEMED was found (see Supporting Information Figure S23). The solid-state 13C NMR of the GVIM-MBAA hydrogel shows broad signals as expected for polymers. In comparison to GVIM-I, a new broad signal of the polymeric backbone in the aliphatic region (∼25 to 50 ppm) and a new signal at ∼175 ppm for the amide group of MBAA can be seen, while the characteristic vinyl-CH2 signal at ∼110 ppm disappeared, thus proving the formation of the expected polymeric structure (see Supporting Information Figure S24).
Hydrogels: Swelling Behavior
A key property of hydrogels is their ability to absorb large amounts of water reversibly, increasing in mass and volume without losing their structure or shape. Hydrogel swelling consists of two separate transport processes. First, the solvent converges through the pores of the gel. Then, the solvent diffuses between the struts of the polymer network.28,32 Several parameters affect the swelling of the hydrogels. In addition to solvent motion and interaction with the polymeric network, these include thermodynamic compatibility, the nature of the cross-linker, its chain length, and the degree of swelling.28
Our initial hydrogel of 3a with 5.0 wt % MBAA was first investigated in ultrapure water, isotonic sodium chloride solution, and phosphate buffered saline (PBS, pH = 7.4). Figure 3 shows that, independent of the medium, the mass of the hydrogels increased continuously over time until a maximum was reached. The hydrogels swollen in ultrapure water reach a plateau in their weight, while the mass of the hydrogels swollen in isotonic sodium chloride or PBS solutions decreased after their maximum. After some time, these gels also reached an equilibrium. The reason for this is an ion exchange of the hydrogels (I–) with the medium (Cl– or phosphate). The anion exchange could be proven by performing ion chromatography of the medium before and after swelling (see Supporting Information Figures S31–33 for the ion chromatograms).
Figure 3.

Swelling tests of GVIM-I hydrogels with 5.0 wt % MBAA in water, isotonic NaCl, and PBS (pH = 7.4) (37 ± 1 °C; n = 3).
After this initial test, we next tested the influence of different anions present in 3a–3c (I = iodide, Br = bromide, Cl = chloride) and 4a–4d (OAc = acetate, OMs = mesylate, OTf = triflate, and NTf2 = bistriflimide) on the swelling behavior (Figure 4). While 3a–3c where all suitable monomers and easily lead to hydrogels under our radical polymerization conditions, the production of hydrogels from 4a–4d proved to be more difficult, as even small amounts of remaining silver from the anion exchange step (Scheme 3) will prematurely catalyze the radical polymerization.33 This can be avoided by thoroughly purifying the products 4a–4d from any remaining silver with activated charcoal.
Figure 4.

Swelling tests of GVIM hydrogels with varying anions with 5.0 wt % MBAA in water (37 ± 1 °C; n = 3; the standard deviations were omitted for more clarity, see Supporting Information Figure S34 including standard deviations).
To directly compare the influence of the different anions on the swelling behavior without factors like anion exchange, these swelling curves were measured in ultrapure water. It can be discerned from Figure 4 that the overall chemical behavior of each anion directly correlates with the swelling degrees of the hydrogels. More hydrophobic anions, like the fluorinated OTf and NTf2 anions, lead to comparatively lower swelling degrees, while the nonfluorinated counterpart of OTf, the mesylate OMs, reaches a higher swelling degree. Interestingly, the chemically similar halogenides exhibit notably different swelling degrees, with around 2.2 for chloride, 1.7 for bromide, and 1.0 for iodide. This may be explained with the increasing atom size from chloride to iodide, which then leads to less space for the water to occupy in the polymeric hydrogel network. Alternatively, the presence of the different anions during radical polymerization could potentially affect the polymerization process itself, thus leading the structurally changed networks.
For the following experiments with different cross-linker types and concentrations, we decided to continue with only GVIM-I 3a as a monomer since this monomer has the most yield-efficient synthesis of all products (Scheme 2) and it leads to the most form-stable, flexible, and reproducible hydrogels from all of the tested GVIM monomers 3a–3c and 4a–4d. We furthermore decided to continue all further swelling experiments in PBS (pH = 7.4) since we aim to study the use of our hydrogels as drug delivery systems in the future. PBS provides a constant pH value and is comparable to the osmolarity and ion concentration of the human organism.34
The swelling behavior of MBAA was next tested in cross-linker concentrations between 4.5 and 7.0 wt % in relation to the weight of monomer (Figure 5). These concentrations are the upper and lower limit for MBAA in the hydrogel synthesis as no gelation takes place below 4.5 wt % and the gelation process becomes too fast for handling above 7.0 wt %. Independent of the cross-linker content, the mass of the hydrogels initially increased continuously over time until they reached a maximum weight value. After 35 min, the gels with 5.5 wt % MBAA were the first to reach their maximum swelling degree of 0.79. All other gels reached the maximum value between 50 and 60 min. The gel with 4.5 wt % MBAA swelled the most, and the maximum degree of swelling was 0.90. It was expected that the gels with the lowest amount of cross-linker would swell the most and that the degree of swelling would decrease with an increase in amount of cross-linker since a higher degree of cross-linking leads to smaller meshes in the polymer network for the water to occupy.35 In principle, this trend can also be seen in Figure 5 with the gels with 4.5 wt % MBAA, the lowest cross-linker concentration, exhibiting the highest swelling degree. However, the swelling degree of 6.5 wt % is unexpectedly the lowest, and not 7.0 wt %.
Figure 5.

Swelling tests of GVIM-I hydrogels with different MBAA amounts in PBS, pH = 7.4 (37 ± 1 °C; n = 3).
In addition to MBAA, DHEBA (Figure 1) was also investigated as a structurally similar acrylamide-based cross-linker (Figure 6). The use of DHEBA as a cross-linker led to a 2.65-fold increase in the degree of swelling compared to the swelling degree of MBAA-gels. This was expected due to the additional hydroxy groups present in this cross-linker. Furthermore, Figure 6 shows clearly that at 7.5 wt % DHEBA, the gels absorbed more water at their maximum swelling degree than the gels with 10 wt %, as expected. Interestingly, the gels degraded completely within a few hours. Just like the MBAA-gels, the DHEBA gels initially increased until they reached their maximum degree of swelling. After the maximum, the masses decreased steadily until they reached their dry weight and subsequently lost even more mass until they finally completely degraded. It is assumed that the degradation is caused by splitting of the polymer network. Since the cross-linking between GVIM-I 3a and the acrylamide groups of MBAA leads to stable hydrogels, it can be assumed that the dihydroxy ethyl functionality of DHEBA, its only difference from MBAA, is the cause of this degradation. No further DHEBA concentrations were investigated due to their instability and degradation.
Figure 6.

Swelling tests of GVIM-I hydrogels with two different DHEBA amounts in PBS, pH = 7.4 (37 ± 1 °C; n ≥ 2).
Besides acrylamides, the second type of cross-linker studied were ethylene glycol diacrylates with different chain lengths. PEGDA and EGDMA (Figure 1) are well-known in the literature as cross-linking agents for a number of hydrogels of different origins and show high biocompatibility in the 3D cross-linked state.36−38 This is of major importance for subsequent applications in the fields of medicine or biotechnology.39−42
Since EGDMA was known and successfully used in the literature, this dimethacrylate cross-linker was started with. However, it was found that this cross-linker did not form reasonable hydrogels with GVIM-I 3a. They either gelled inhomogeneously or were not stable in shape. On the other hand, EGDA (Figure 1), which has the same structure as EGDMA, but without the additional methyl groups on the acrylate, was suitable for hydrogel synthesis. The EGDA hydrogels were firm and retained their shape, which made it possible to investigate their swelling behavior (Figure 7).
Figure 7.

Swelling tests of GVIM-I hydrogels with different EGDA amounts in PBS, pH = 7.4 (37 ± 1 °C; n = 3).
The EGDA hydrogels were investigated with cross-linker concentrations between 10 wt %, which was the lowest possible concentration for gelation to take place, and 30 wt %. Gelation above 30 wt % was possible but lead to more brittle hydrogels. On average, the EGDA cross-linked gels swelled more than the MBAA gels, although the cross-linker concentration was significantly higher. The highest swelling degree reached by the MBAA gels was 0.9 for 4.5 wt % MBAA, while the highest swelling degree of the EGDA gels was around 1.6 with 10 wt % EGDA. Interestingly, while the MBAA gels show a weight loss after their maximum, which was attributed to anion exchange processes, this effect cannot, or only slightly, be seen for the EGDA gels. Thus, the hydrogel composition of GVIM-I 3a and EDGA seems to lead to a decreased anion exchange in comparison to MBAA as a cross-linker. Last, the EDGA gels show a notable increase in their mass after 20 h of swelling. This indicates that the gels did not reach their equilibrium yet (see Figure 13 for long-term swelling experiments).
Figure 13.

Long-term swelling of the hydrogels with 5 wt % MBAA and 15 wt % diacrylate cross-linkers in PBS, pH = 7.4 (37 ± 1 °C; n = 3).
Since PEGDA is a widely used cross-linker in addition to EGDA, PEGDA with three different chain lengths (Mn = 250, 575, and 700 g·mol–1) was used in this work (Figure 1).
Figure 8 shows the swelling behavior of different PEGDA250 concentrations. Like EDGA, the investigated cross-linker concentrations were between 10 and 30 wt %, which was found to be the optimal range for gelation. As with all gels considered so far, the mass of the PEGDA250 gels initially increased quickly, regardless of their cross-linker concentration. After the initial exponential swelling, they reached a plateau around their maximum weight. The hydrogel with the lowest cross-linker concentration (10 wt %) swelled the most and reached a degree of swelling of 3.04, while the gel with the highest PEGDA250 concentration (30 wt %) reached the lowest degree of swelling with 2.18, thus confirming again that lower cross-linker concentrations lead to higher swelling degrees. Compared to the previously used cross-linkers, the PEGDA250 hydrogels swelled the most.
Figure 8.

Swelling tests of GVIM-I hydrogels with different PEGDA250 amounts in PBS, pH = 7.4 (37 ± 1 °C; n = 3).
After PEGDA250, PEGDA575 was used as the next cross-linker. Figure 9 shows examples of the freshly synthesized gel (I), which has a diameter of 10 mm due to the mold used for synthesis, the dried gel (II), which shrinks in size due to the removal of water, and the swollen gel (III), which increases in size by 1.5 to 15 mm.
Figure 9.

PEGDA575 gels in three different stadiums. (I = fresh synthesized, II = dried for 3 days at room temperature, III = swollen gel).
The swelling behavior of PEGDA 575 hydrogels is shown in Figure 10. In difference to every other cross-linker investigated in this work, all PEGDA575 hydrogels, independent of their cross-linker concentration, swelled at the same rate and reached the same maximum degrees of swelling of 1.68. The only outlier here is the hydrogel produced with 15 wt % PEGDA575, which achieved a degree of swelling of 2.29. To rule out any mistakes during measurement, this particular concentration was produced and measured six times instead of the usual three times.
Figure 10.

Swelling tests of GVIM-I hydrogels with different PEGDA575 amounts in PBS, pH = 7.4 (37 ± 1 °C; n = 3).
As the third and final candidate in the PEGDA series, PEGDA700 was also used as a cross-linker (Figure 11).
Figure 11.

Swelling tests of GVIM-I hydrogels with different PEGDA700 amounts in PBS, pH = 7.4 (37 ± 1 °C; n = 3).
Similar to PEGDA575, the different concentrations of PEGDA700 show only a very small influence on the swelling degree, though the already discussed trend of lower cross-linker concentrations leading to higher swelling degrees can also be seen with PEGDA700. The swelling degrees here are between 2.20 and 2.45.
To achieve a direct comparison between the three similar PEGDA cross-linkers, 10 wt % PEGDA250, 25 wt % PEGDA575, and 30 wt % PEGDA700 are plotted together in Figure 12, as these three swelling curves correlate to roughly the same mol % of cross-linker (16–17 mol %). In this comparison, PEGDA700 as the longest of the three cross-linkers would be expected to lead to the highest swelling degrees (due to bigger pore sizes available for water to occupy) followed by PEGDA575 and PEGDA250. This trend however cannot be seen in Figure 12, as in our case, PEGDA250 leads to the highest swelling degree in the direct comparison followed by PEGDA700 and then PEGDA 575. It is possible that other effects, like additional van der Waals forces, influence this unsuspected trend.
Figure 12.

Swelling diagram of all three PEGDA cross-linkers with 16 to 17 mol % of PEGDA.
Overall, the tested cross-linkers MBAA, EGDA, PEGDA250, PEGDA575, and PEGDA700 all exhibited the same swelling behavior of reaching a clear plateau, an equilibrium, after an initial exponential swelling phase. However, some hydrogels, most clearly the ones produced with EDGA and PEGDA250, continued to gain weight after a total of 19 to 20 h in PBS. To assess the long-term behavior of the gels, a long-term swelling was carried out. It was decided to use 5 wt % MBAA and 15 wt % cross-linker concentrations for all diacrylate cross-linkers. The hydrogels were prepared and evaluated in the same way as the other swelling tests. The hydrogels were placed in PBS (37 ± 1 °C, pH = 7.4), and the mass was taken every 24 h (Figure 13).
The MBAA gels already reached their maximum degree of swelling after 24 h and then remained in equilibrium. Their shape was stable over the entire time, and there was no visible degradation. In contrast, the degree of swelling of all (poly)ethylene glycol diacrylate gels increased with each measurement. During the increase in mass, the diacrylate gels gradually lost their shape until a complete degradation took place. The PEGDA700 gels were the first to degrade after 7 days, the PEGDA250 and EGDA gels followed on days 8 and 9, respectively, and after a total of 14 days, the PEGDA575 gels were also completely degraded. The maximum degrees of swelling achieved by the gels in this long-term experiment ranged from 0.68 (5 wt % MBAA) to 12.96 (15 wt % PEGDA575).
Conclusions
In this work, we synthesized seven novel glucose-based vinyl imidazolium (GVIM) monomers with varying anions. These GVIM monomers have been used for the production of novel semisynthetic hydrogels with a unique cationic carbohydrate-based polymeric network, which cannot be found in previously known natural carbohydrate-based hydrogels, which are either neutral (e.g., cellulose and chitosan) or anionic (e.g., alginate).
We overall evaluated the influence of the counteranion as well as the type and concentration of each tested commercially available cross-linker (MBAA, DHEBA, EGDMA, EGDA, and PEGDA) on the swelling behavior of the GVIM hydrogels. In the case of the anions, both the anion size and their hydrophilicity or hydrophobicity have been found to influence the swelling behavior. GVIM iodide, which can be produced in a simple two-step process with a high total yield of 71% and which was recently evaluated by our group as remarkably biocompatible in comparison to the dialkyl imidazolium ionic liquids, was found to be the most promising of the GVIM monomers.
The bis(acrylamide) cross-linker MBAA leads to overall lower swelling degrees than the (poly)ethylene glycol cross-linkers EGDA and PEGDA; however, the GVIM-MBAA hydrogels exhibit a high long-term stability, while the GVIM-(P)EDGA hydrogels all degrade over time. This opens up unique applications of each hydrogel depending on the cross-linker. In the case of the different cross-linker concentrations, we found that the swelling degree of the hydrogel decreases with increasing cross-linker amounts. Both DHEBA and EGDMA were found to be unsuitable as cross-linkers for our monomer.
As the next steps, we aim to measure the biocompatibility and antimicrobial properties of our hydrogels and, assuming a high biocompatibility, to evaluate our GVIM hydrogels as drug delivery systems, where we expect interesting ion controlled properties.
Acknowledgments
Financial support by the NFDI4Cat (DFG project number 441926934) is gratefully acknowledged by the authors. We also thank the “European Regional Development Fund” (ERDF) for the financial support for the ion chromatography. Last, we thank Florian Taube (group of Prof. Corzilius, Institute of Chemistry, University of Rostock) for measuring the solid-state 13C NMR.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c06804.
All 1H, 13C, and 19F NMR spectra of the products described in this work, experimental setup of the hydrogel synthesis and swelling degree measurements, ion chromatograms of the PBS buffer before and after swelling, IR spectra of selected starting materials and hydrogels, and composition of the hydrogels (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ahmed E. M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. 10.1016/j.jare.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek B.; Nadolna K.; Owczarek A.. The physical and chemical properties of hydrogels based on natural polymers. In: Hydrogels Based on Natural Polymers; Elsevier, 2020, 151–172. 10.1016/B978-0-12-816421-1.00006-9. [DOI] [Google Scholar]
- Ding H.; Li B.; Liu Z.; Liu G.; Pu S.; Feng Y.; Jia D.; Zhou Y. Decoupled pH- and Thermo-Responsive Injectable Chitosan/PNIPAM Hydrogel via Thiol-Ene Click Chemistry for Potential Applications in Tissue Engineering. Adv. Healthcare Mater. 2020, 9, e2000454 10.1002/adhm.202000454. [DOI] [PubMed] [Google Scholar]
- Bingol B.; Altuncu S.; Duman F. D.; Ak A.; Gulyuz U.; Acar H. Y.; Okay O.; Avci D. One-Step Injectable and Bioreducible Poly(β-Amino Ester) Hydrogels as Controlled Drug Delivery Platforms. ACS Appl. Polym. Mater. 2019, 1, 1724–1734. 10.1021/acsapm.9b00287. [DOI] [Google Scholar]
- Jastram A.; Claus J.; Janmey P. A.; Kragl U. Rheological properties of hydrogels based on ionic liquids. Polym. Test. 2021, 93, 106943 10.1016/j.polymertesting.2020.106943. [DOI] [Google Scholar]
- Großeheilmann J.; Bandomir J.; Kragl U. Preparation of Poly(ionic liquid)s-Supported Recyclable Organocatalysts for the Asymmetric Nitroaldol (Henry) Reaction. Chem. - Eur. J. 2015, 21, 18957–18960. 10.1002/chem.201504290. [DOI] [PubMed] [Google Scholar]
- Wang F.; Li Z.; Khan M.; Tamama K.; Kuppusamy P.; Wagner W. R.; Sen C. K.; Guan J. Injectable, rapid gelling and highly flexible hydrogel composites as growth factor and cell carriers. Acta Biomater. 2010, 6, 1978–1991. 10.1016/j.actbio.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Claus J.; Jastram A.; Piktel E.; Bucki R.; Janmey P. A.; Kragl U. Polymerized ionic l iquids-based hydrogels with intrinsic antibacterial activity: Modern weapons against antibiotic-resistant infections. J. Appl. Polym. Sci. 2021, 138, 50222. 10.1002/app.50222. [DOI] [Google Scholar]
- Claus J.; Eickner T.; Grabow N.; Kragl U.; Oschatz S. Ion Exchange Controlled Drug Release from Polymerized Ionic Liquids. Macromol. Biosci. 2020, 20, e2000152 10.1002/mabi.202000152. [DOI] [PubMed] [Google Scholar]
- Lin S.; Yuk H.; Zhang T.; Parada G. A.; Koo H.; Yu C.; Zhao X. Stretchable Hydrogel Electronics and Devices. Adv. Mater. 2016, 28, 4497–4505. 10.1002/adma.201504152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollmisch A.; Kragl U.; Großeheilmann J. Enzyme Immobilization in Polymerized Ionic Liquids-based Hydrogels for Active and Reusable Biocatalysts. SynOpen 2018, 2, 192–199. 10.1055/s-0037-1610144. [DOI] [Google Scholar]
- Li S.; Chen G. Agricultural waste-derived superabsorbent hydrogels: Preparation, performance, and socioeconomic impacts. J. Cleaner Prod. 2020, 251, 119669 10.1016/j.jclepro.2019.119669. [DOI] [Google Scholar]
- Zhu T.; Mao J.; Cheng Y.; Liu H.; Lv L.; Ge M.; Li S.; Huang J.; Chen Z.; Li H.; Yang L.; Lai Y. Recent Progress of Polysaccharide-Based Hydrogel Interfaces for Wound Healing and Tissue Engineering. Adv. Mater. Interfaces 2019, 6, 1900761. 10.1002/admi.201900761. [DOI] [Google Scholar]
- Gholamali I. Stimuli-Responsive Polysaccharide Hydrogels for Biomedical Applications: A Review. Regener. Eng. Transl. Med. 2021, 7, 91–114. 10.1007/s40883-019-00134-1. [DOI] [Google Scholar]
- Nasiri Oskooie M.; Pooresmaeil M.; Namazi H. Design and synthesis of vinylic glycomonomers and glycopolymer based on α-D-glucofuranose moieties. J. Polym. Res. 2019, 26, 268. 10.1007/s10965-019-1969-0. [DOI] [Google Scholar]
- Namazi H.; Pooresmaeil M.; Oskooie M. N. New glyco-copolymers containing α-D-glucofuranose and α-D-mannofuranose groups synthesized by free-radical polymerization of sugar-based monomers. Polym. Bull. 2022, 79, 4891–4903. 10.1007/s00289-021-03731-9. [DOI] [Google Scholar]
- Rodygin K. S.; Werner I.; Ananikov V. P. A Green and Sustainable Route to Carbohydrate Vinyl Ethers for Accessing Bioinspired Materials with a Unique Microspherical Morphology. ChemSusChem 2018, 11, 292–298. 10.1002/cssc.201701489. [DOI] [PubMed] [Google Scholar]
- Bielas R.; Maksym P.; Erfurt K.; Hachula B.; Gawecki R.; Tarnacka M.; Waskiewicz S.; Mielanczyk L.; Mrozek-Wilczkiewicz A.; Chrobok A.; Paluch M.; Kaminski K. Sugar decorated star-shaped (co)polymers with resveratrol-based core – physicochemical and biological properties. J. Mater. Sci. 2022, 57, 2257–2276. 10.1007/s10853-021-06755-8. [DOI] [Google Scholar]
- Jopp S. Carbohydrate Based Ionic Liquids (CHILs): Synthesis and Applications. Eur. J. Org. Chem. 2020, 2020, 6418–6428. 10.1002/ejoc.202000714. [DOI] [Google Scholar]
- Gaida B.; Brzeczek-Szafran A. Insights into the Properties and Potential Applications of Renewable Carbohydrate-Based Ionic Liquids: A Review. Molecules 2020, 25, 3285. 10.3390/molecules25143285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo V.; Iuliano A.; Guazzelli L. Sugar-Based Ionic Liquids: Multifaceted Challenges and Intriguing Potential. Molecules 2021, 26, 2052. 10.3390/molecules26072052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann P.; Jopp S. Novel Glucosylimidazolium Ionic-Liquid-Supported Novozym 435 Catalysts - A Proof of Concept for an Acrylation Reaction. ChemistryOpen 2022, 11, e202200135 10.1002/open.202200135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopp S.; Fleischhammer T.; Lavrentieva A.; Kara S.; Meyer J. Synthesis, biocompatibility, and antimicrobial properties of glucose-based ionic liquids. RSC Sustain. 2023, 1, 1751–1764. 10.1039/D3SU00191A. [DOI] [Google Scholar]
- Feng W.; Wang Z. Tailoring the Swelling-Shrinkable Behavior of Hydrogels for Biomedical Applications. Adv. Sci. 2023, 10, 2303326. 10.1002/advs.202303326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Ruaud P.; Plusquellec D. Synthese de nouveaux tensioactifs glycosidiques par l’intermediaire de sucres halogenes. Thioethers et sulfones derives de l’α-d-glucoside et l’α-d-mannoside de methyle. Tetrahedron 1991, 47, 5185–5192. 10.1016/S0040-4020(01)87130-1. [DOI] [Google Scholar]
- Wheatley D. E.; Fontenelle C. Q.; Kuppala R.; Szpera R.; Briggs E. L.; Vendeville J.-B.; Wells N. J.; Light M. E.; Linclau B. Synthesis and Structural Characteristics of all Mono- and Difluorinated 4,6-Dideoxy-d-xylo-hexopyranoses. J. Org. Chem. 2021, 86, 7725–7756. 10.1021/acs.joc.1c00796. [DOI] [PubMed] [Google Scholar]
- Lambrecht S.; Villinger A.; Jopp S. 1-(Methyl-α-d-gluco-pyran-osid-6-yl)-3-vinyl-imidazolium iodide di-methyl-formamide monosolvate. IUCrData 2022, 7, x220265. 10.1107/S2414314622002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastram A.; Lindner T.; Luebbert C.; Sadowski G.; Kragl U. Swelling and Diffusion in Polymerized Ionic Liquids-Based Hydrogels. Polymers 2021, 13, 1834. 10.3390/polym13111834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul V.; Mohamed R.; Kuckling D.; Adler H.-J. P.; Choudhary V. Interpenetrating polymer network (IPN) nanogels based on gelatin and poly(acrylic acid) by inverse miniemulsion technique: Synthesis and characterization. Colloids Surf., B 2011, 83, 204–213. 10.1016/j.colsurfb.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Orakdogen N.; Okay O. Influence of the Initiator System on the Spatial Inhomogeneity in Acrylamide-Based Hydrogels. J. Appl. Polym. Sci. 2007, 103, 3228–3237. 10.1002/app.24977. [DOI] [Google Scholar]
- Qi X.-M.; Chen G.-G.; Gong X.-D.; Fu G.-Q.; Niu Y.-S.; Bian J.; Peng F.; Sun R.-C. Enhanced mechanical performance of biocompatible hemicelluloses based hydrogel via chain extension. Sci. Rep. 2016, 6, 33603. 10.1038/srep33603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goycoolea F. M.; Fernández-Valle M. E.; Aranaz I.; Heras Á. pH- and Temperature-Sensitive Chitosan Hydrogels: Swelling and MRI Studies. Macromol. Chem. Phys. 2011, 212, 887–895. 10.1002/macp.201000301. [DOI] [Google Scholar]
- Ji H.; Song X.; Cheng H.; Luo L.; Huang J.; He C.; Yin J.; Zhao W.; Qiu L.; Zhao C. Biocompatible In Situ Polymerization of Multipurpose Polyacrylamide-Based Hydrogels on Skin via Silver Ion Catalyzation. ACS Appl. Mater. Interfaces 2020, 12, 31079–31089. 10.1021/acsami.0c02495. [DOI] [PubMed] [Google Scholar]
- Son M.; Lee Y. S.; Lee M. J.; Park Y.; Bae H.-R.; Lee S. Y.; Shin M.-G.; Yang S. Effects of osmolality and solutes on the morphology of red blood cells according to three-dimensional refractive index tomography. PloS One 2021, 16, e0262106 10.1371/journal.pone.0262106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavda H.; Patel C. Effect of crosslinker concentration on characteristics of superporous hydrogel. Int. J. Pharm. Investig. 2011, 1, 17–21. 10.4103/2230-973X.76724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You B.; Li Q.; Dong H.; Huang T.; Cao X.; Liao H. Bilayered HA/CS/PEGDA hydrogel with good biocompatibility and self-healing property for potential application in osteochondral defect repair. J. Mater. Sci. Technol. 2018, 34, 1016–1025. 10.1016/j.jmst.2017.11.016. [DOI] [Google Scholar]
- Testore D.; Zoso A.; Kortaberria G.; Sangermano M.; Chiono V. Electroconductive Photo-Curable PEGDA-Gelatin/PEDOT:PSS Hydrogels for Prospective Cardiac Tissue Engineering Application. Front. Bioeng. Biotechnol. 2022, 10, 897575 10.3389/fbioe.2022.897575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirf D.; Higginbotham C. L.; Rowan N. J.; Devery S. M. Cyto- and genotoxicological assessment and functional characterization of N-vinyl-2-pyrrolidone-acrylic acid-based copolymeric hydrogels with potential for future use in wound healing applications. Biomed. Mater. 2010, 5, 35002. 10.1088/1748-6041/5/3/035002. [DOI] [PubMed] [Google Scholar]
- Mei Q.; Yuen H.-Y.; Zhao X. Mechanical stretching of 3D hydrogels for neural stem cell differentiation. Bio-des. Manuf. 2022, 5, 714–728. 10.1007/s42242-022-00209-z. [DOI] [Google Scholar]
- Liang J.; Zhang X.; Chen Z.; Li S.; Yan C. Thiol-Ene Click Reaction Initiated Rapid Gelation of PEGDA/Silk Fibroin Hydrogels. Polymers 2019, 11, 2102. 10.3390/polym11122102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra P.; Rameshbabu A. P.; Das D.; Dhara S.; Panda A. B.; Pal S. Stimuli-responsive, biocompatible hydrogel derived from glycogen and poly(N-isopropylacrylamide) for colon targeted delivery of ornidazole and 5-amino salicylic acid. Polym. Chem. 2016, 7, 5426–5435. 10.1039/C6PY01128D. [DOI] [Google Scholar]
- Cocarta A.-I.; Hobzova R.; Trchova M.; Svojgr K.; Kodetova M.; Pochop P.; Uhlik J.; Sirc J. 2-Hydroxyethyl Methacrylate Hydrogels for Local Drug Delivery: Study of Topotecan and Vincristine Sorption/Desorption Kinetics and Polymer-Drug Interaction by ATR-FTIR Spectroscopy. Macromol. Chem. Phys. 2021, 222, 2100086. 10.1002/macp.202100086. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.