

Abstract
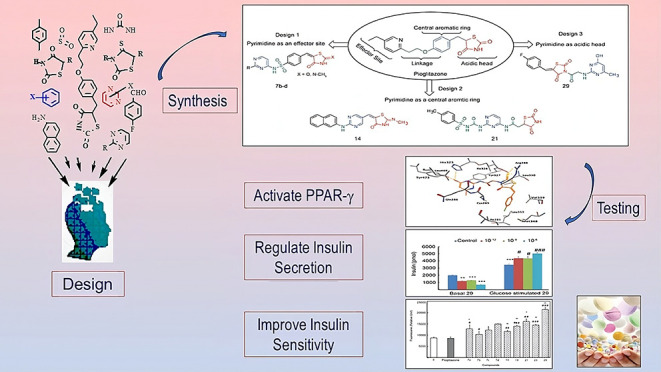
Sixteen novel thiazolidinedione (TZD) and rhodanine (RD) derivatives were designed and synthesized by introducing a pyrimidine moiety at different sites of pioglitazone’s structure. The effects of synthesized compounds on regulating glucose metabolism, improving insulin sensitivity, and activating the peroxisome proliferator-activated γ receptor (PPAR-γ) were evaluated in βTC6 cells. Compounds TZDs # 7a, 7b, 7c, and 29 reduced the basal insulin secretion by ∼20.0–67.0% and increased insulin secretion stimulated by glucose by ∼25.0–50.0% compared to control. Compounds TZDs # 14 and 21 and RDs # 33a–b and 33d–f increased basal insulin secretion by ∼20.0–100.0%, while its glucose-stimulated secretion remained unchanged. These findings suggested that the former compounds can act as antihypoglycemic during fasting and antihyperglycemic during postprandial conditions. The latter compounds should be administered before meals to avoid their hypoglycemic effect. Additionally, both TZDs and RDs improved insulin sensitivity by increasing glucose uptake by 17.0–155.0% relative to control. In silico molecular docking of synthesized drugs onto the PPAR-γ structure revealed exothermic binding modes through hydrogen bonding, van der Waals forces, and π–π stacking with binding affinities of −6.02 to −9.70 kcal/mol. Insights into the structure–activity relationship revealed that the introduction of pyrimidine linked to sulfonyl or peptide groups accounted for increased antidiabetic activity. These results demonstrated novel TZDs and RDs with high potency in stimulating insulin secretion, enhancing insulin sensitivity, and activating PPAR-γ relative to pioglitazone. They are recommended for further development as potential antidiabetic agents.
1. Introduction
Diabetes mellitus (DM) is a chronic glucose metabolic disorder caused by various genetic, environmental, and social factors such as a sedentary lifestyle and obesity. It involves long-term persistent hyperglycemia that can lead consequently to cardiovascular, renal, neurological, ocular, and other organ-related disorders.1,2 DM has become one of the world’s most serious health issues. In 2021, the international diabetes federation reported that approximately 537 million (20–79 years) are diabetic. This number is projected to rise to 643 million by 2030 and to 783 million by 2045. Additionally, 541 million are prediabetic and at increased risk of developing type 2 diabetes. In 2021, DM cost approximately 966 billion US dollars (9.0% of total world spending on adults) and caused 6.7 million deaths.3
Modification of lifestyle, use of insulin, and glucose-lowering medications are used to manage diabetes. However, there is a need to develop new medications with a long-acting metabolic effect, β-cell function protection, and optimized safety. Developed reagents should also be able to eliminate the complications of currently used ones, such as hepatic, cardiovascular, and hematological disorders.4
Thiazolidinedione (TZD) is a heterocyclic compound consisting of a five-membered C3NS ring. Rhodanine (RD) is a thiazolidine derivative in which the carbonyl group at position 4 is replaced by the thiocarbonyl group (Figure 1). Rhodanine is structurally related to thiazolidine-2,4-dione, 4- thioxothiazolidin-2-one, and 2-iminothiazolidine-4-one scaffolds.5−7 TZD and RD are bioisosteres with similarities in biological activity.4,7−10 The structural diversities and antidiabetic properties of TZDs and RDs have made them attractive targets for developing new potent antidiabetic drugs that can also treat insulin resistance. Several TZDs and RDs were reported as potential antidiabetic agents.
Figure 1.

Structural formula of thiazolidinedione and rhodanine.
TZDs are highly interesting due to their antihyperglycemic, anti-inflammatory, antimalarial, antioxidant, antibacterial, cytotoxic, and antiproliferative properties.11−13 TZDs have been shown to improve insulin sensitivity in insulin-resistant states, including obesity, impaired glucose tolerance (IGT), and polycystic ovary syndrome (PCOS). They also decrease hepatic gluconeogenesis and increase adiponectin, which in turn increases insulin sensitivity and fatty acid oxidation.14
Pioglitazone and rosiglitazone are the two TZDs approved by the US FDA as mono- or combined therapy with metformin or sulfonylureas for managing diabetes mellitus type 2 (DM2). They act by synthesizing insulin and enhancing its sensitivity in critical tissues. They are not hypoglycemic when administrated as monotherapy and not contraindicated in patients with renal disease. In addition, they reduce hepatic fats and improve liver fibrosis in patients with nonalcoholic steatohepatitis.15−21 TZDs also regulate gene expression activated by PPAR-γ.20 These genes are found in muscle, fat, and liver tissues. Subsequently, TZDs regulate glucose metabolism, fatty acid storage, and adipocyte differentiation.21 Alterations in metabolic regulation pathways of lipoprotein lipase, glucokinase, and fatty acyl-CoA synthase were also reported.14 Improvement of insulin resistance by PPAR-γ agonists has been attributed to increasing adiponectin and GLUT4 expression and opposing the effect of TNF-α in adipocytes. Increased GLUT4 expression increases the glucose uptake in adipocytes and skeletal muscle cells in response to insulin.21 Activation of PPAR-γ receptors could also induce apoptosis for cancer cells, slow the progression of medial intimal thickening, and decrease coronary intimal hyperplasia.15 Thus, functioning as a PPAR-γ agonist, TZDs can improve insulin sensitivity in the liver and peripheral tissues, enhance glucose uptake, and diminish glucose withdrawal from the liver.22 However, hepatotoxicity and risk of cardiac failure have been observed with some TZDs, such as rosiglitazone pioglitazone.23,24
Meanwhile, a plethora of pharmacological activities were reported for RDs. These included antidiabetic, antiviral, anti-inflammatory, antimicrobial, antitumor, anti-Alzheimer properties, anticancer, cholesterol esterase inhibitors, tyrosinase inhibitors, HIV-1 integrase inhibitors, antimicrobial activity, HCV NS5B polymerase inhibitors, antileukemia agents, topoisomerase II inhibitors, β- lactamase inhibitors, and tens of other effects.4,25
The antidiabetic properties of RDs were tested using different molecular targets as underlying mechanisms. These included inhibition of aldose and aldehyde reductase, α-glucosidase, α-amylase, protein tyrosine phosphatase (PTP1B), pancreatic lipase, and as PPAR-γ agonists.4,26,27
Synthetic agonists for PPAR-γ were shown to promote adipocyte differentiation, making it more insulin sensitive, improving glucose homeostasis, reducing free fatty acid, and improving peripheral tissue insulin sensitivity. PPAR-γ agonists can also prevent β-cell apoptosis and restore its function.28,29 Two factors affecting the agonistic activity of the TZD and RD compounds are the chain length (n) and the R substituents. Compounds with longer chain length (n ≥ 3) and R substituted with halogens have shown better PPAR-γ agonistic activity.4
The use of cell lines has become essential in diabetic research since it gives a better understanding of the disease and helps to discover novel therapies.30 Various cell lines such as C2C12, L6, 3T3-L, RIN5F, and HepG2 were used. Several RDs that were tested against hepatocyte cells, hemidiaphragm cells, and adipocyte cells with a glucose uptake assay were monitored.4
A global increase in the number of diabetic patients necessitates the development of more effective antidiabetic drugs with no or less side effects. The reported antidiabetic potencies of TZDs and RDs have made them attractive targets for developing new drugs. In this work, 16 TZDs and RDs were designed and synthesized using conventional and microwave-assisted synthesis.31 Synthesized compounds were structurally confirmed using elemental analysis, IR, and NMR. Antidiabetic activates of synthesized compounds were tested by measuring insulin secretion and glucose uptake of βTC6 cells. In silico molecular docking of synthesized drugs on PPAR-γ was used to better understand the mode and mechanism of binding. Structure–activity relationships were evaluated to assess the impact of the structure on activity. The results obtained suggest potential novel antidiabetic agents that may be further developed toward effective and safer drugs.
2. Results and Discussions
2.1. Design and Synthesis of Novel Thiazolidinediones as PPAR-γ Agonists
Thiazolidinediones, also known as glitazones, are PPAR-γ agonists that treat DM2 by reducing hyperglycemia and improving insulin sensitivity. Figure 2 shows the structural formula of pioglitazone, the most effective TZD. Glitazone molecules usually have a polar thiazolidine ring system as a head, a hydrophobic benzyloxy moiety or aniline moiety as a trunk linked by a two-carbon atoms’ linker, and a hydrophobic ring as a tail.32 The chiral center at position 5 of the thiazolidine ring creates S and R isomers of glitazone. The S isomer was found to be biologically active, while the R was not. Therefore, imposing a double bond in the thiazolidine-2,4-dione ring’s fifth position will cancel the chirality and create more conjugated stable achiral conformations.33 We showed here that such molecules are biologically active with higher antidiabetic effects.
Figure 2.

Proposed designs for modifying pioglitazone. Modifications kept the basic glitazone pharmacophore structure and introduced a pyrimidine moiety as an effector in design 1, an acidic head in design 2, and a central aromatic ring in design 3.
Based on the above structural features, we proposed three new designs in which a pyrimidine moiety was introduced into the pioglitazone structure as an effector, a hydrophobic center, or an acidic head (Figure 2). Since pioglitazone has proved nontoxic and pyrimidine is mostly nontoxic, our designed compounds are favorably expected to be nontoxic.34 Details about these design movements are outlined in Figure 2.
2.1.1. Design 1: Pyrimidine as an Effector Site (TZDs # 7a–d)
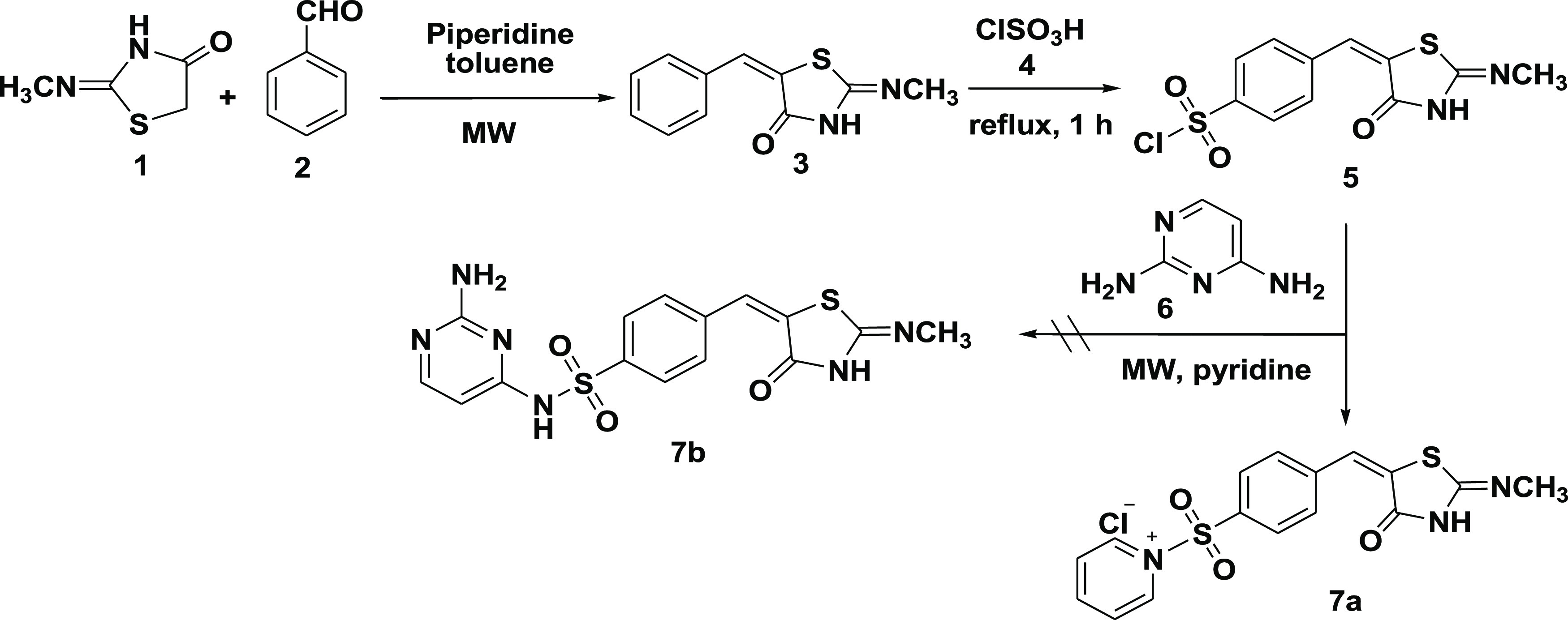
In design 1 compounds, the 2,4-TZD moiety served as the hydrophilic head, the pyrimidine served as the tail (an effector site), and the p-sulfonamidobenzylidene moiety at position 5 of 2,4-TZD served as a central aromatic ring (Figure 3). We hypothesized that the addition of the sulfonyl group as a linker would raise the lipophilicity and lower the acidity of the imino hydrogen, whereas the addition of the pyrimidine ring would enhance the relative safety profile of resultant compounds in terms of their body metabolic pathways. Glitazone compounds 7a–d were synthesized under this category. Microwave irradiation synthesis was chosen as the best synthetic route to minimize the reaction time and number of reaction steps. The retrosynthetic analysis for compounds 7a–d is shown in Figure 3.
Figure 3.
Designed template and retrosynthetic disconnection for TZD # 7a–d.
Subsequently, compound (7a) was synthesized by condensing the 2-methylimino-thiazolidin-4-one (1) with benzaldehyde (2) to produce (5E)-5-benzylidene-2-methylimino-thiazolidin-4-one (3). The latter reacted with chlorosulfonic acid (4) to form the intermediate (5) by introducing the chlorosulfonyl group into the p-position of the aromatic system.
Initially, we examined the amination of the intermediate (5) using 2,4-diaminopyrimidine (6) in the presence of dry pyridine as a catalyst.35 Interestingly, the 2,4-diaminopyrimidine did not react as a nucleophile in the presence of pyridine. Surprisingly, pyridine reacted with the chlorosulfonyl group under 10.0 min microwave irradiation to produce the pyridinium salt (7a) as a sole product. Meanwhile, 2,4-diaminopyrimdine (6) remained unreacted and was completely recovered (see Scheme 1).
Scheme 1. Formation of Pyridinium Salt TZD # 7a.
The structure of formed pyridinium chloride salt (7a) was elucidated based on elemental analysis, IR, 1H NMR, and 13C NMR spectral analyses. The IR absorption bands at 1683 cm–1 (s) and 3487 cm–1 (br) were attributed to the stretching vibrations of the C=O in the thiazolidine ring and the –NH of the TZD ring, respectively. The peaks at 3067 and 1486 cm–1 resulted from the aromatic C–H and C=N stretching vibrations, respectively [Figure S1a].
The 1H NMR [DMSO-d6, 400 MHz] spectrum showed a characteristic multiplet peak in the range of δ = 3.40–3.42 ppm, confirming the presence of an NH proton (exchangeable with D2O). The methyl proton resonated as a singlet at δ = 3.08 ppm. In contrast, the phenyl ring protons appeared as doublets at δ = 7.56 and 7.69 ppm, with an equal coupling constant of J = 8.2 Hz. The proton of the methylene group appeared as a singlet at δ = 7.88 ppm. The pyridine ring protons H-3 and H-5 appeared at δ = 7.97 ppm as a triplet with coupling constant J = 7.2 Hz. The H-4 proton gave a triplet at δ = 8.49 ppm with a coupling constant of J = 7.4 Hz, whereas the H-2 and H-6 protons gave a doublet at δ = 8.87 ppm with a coupling constant of J = 5.1 Hz. In the 13C NMR spectrum, the signal that appeared at δ = 148.4 ppm is characteristic of the olefinic carbon, while that appeared at δ = 122.9 ppm is characteristic of the TZD C5 atom. The signals that appeared at δ = 126.8–146.0, δ = 166.4, and δ = 168.2 ppm are respectively attributed to the phenyl, C=N, and C=O carbons [Figure S1a,b].
Compounds (7b–d) were obtained by microwave irradiation of aminopyrimidines mixed with p-chlorosulfonyl benzylidene-2,4-thiazolidinedione (5) in ethanol and in the presence of DIPEA as a base. The reaction proceeded smoothly to produce the corresponding 5-(4′-pyrimidine-amino-sulfonyl benzylidene)-2,4-thiazolidinediones (7b–d) with yields of 75–84%, as shown in Scheme 2. The structures of glitazones (7b–d) were confirmed using elemental, IR, 1H NMR, and 13C NMR analyses.
Scheme 2. Synthesis of 5-(4′-Substituted Benzylidene)-2,4-thiazolidinediones, TZD # 7b–d.

The IR spectra showed characteristic peaks in the range of 3353–3487 cm–1, corresponding to the N–H stretching vibration on the TZD ring. The C=O stretching vibration appeared as a strong band at 1675–1698 cm–1. These two bands are essential to confirm the formation of a TZD ring [Figures S2–S4]. The 1H NMR of (7b–d) gave characteristic broad peaks at δ = 11.63 ppm integrated into two protons and assigned to the two NH protons (exchanged in D2O). Another broad signal shown in spectra of (7b–c) at δ = 8.05 ppm and equivalent to two protons were assigned to NH2 protons (exchanged in D2O). The pyrimidine protons H-5 and H-6 appeared as two doublets at δ = 5.98 ppm and δ = 7.69 ppm (J = 6.0 Hz). Additionally, the two doublets appearing at δ = 7.56 and 7.69 ppm (J = 8.0 Hz) are attributed to the aromatic protons, and the singlet appearing at δ = 7.88 ppm is attributed to the olefinic proton [Figures S2–S4]. The 13C NMR spectra showed the characteristic signal for the benzylidene methine group at δ = 141.5–142.7 ppm. The carbonyl groups appeared at around δ = 165.0 and 168.0 ppm. The C5 atom of the pyrimidine resonated around δ = 96.0–99.0 ppm, whereas the C4 and C6 atoms appeared around δ = 152.0 and δ = 158.0 ppm, respectively.
2.1.2. Design 2: Pyrimidine as the Central Aromatic Ring (TZDs # 14, 19, 21, 23, and 29)
In this design, 5-arylidene-2-imino-4-thiazolidinone (14) was prepared by replacing the central phenyl ring in pioglitazone with a pyrimidine scaffold and the hydrophobic pyridine ring end by naphthyl amine. In the resultant molecule, the thiazolidine-2,4-dione moiety served as the polar hydrophilic head, the aromatic hydrocarbon (naphthol) as the hydrophobic end, and the pyrimidine ring as the linker (Figure 4).
Figure 4.

Designed template and retrosynthetic disconnection for α-naphthyl-2-imino-4-thiazolidinone, TZD # 14.
Scheme 3 shows the synthetic mechanism of 5-arylidene-2-imino-4-thiazolidinone (14). The 5-formyl pyrimidine (11) was prepared—as an intermediate—via substitutions on C2 and C4 atoms of the pyrimidine ring. Electron-donating groups at C2 or C4 were found to be essential to activate the pyrimidine C5 atom for reactivity toward the Vilsmeier reagent. A 64% yield of intermediate 11 was obtained over 24 h.
Scheme 3. Synthesis of 5-Arylidene-2-imino-4-thiazolidinone, TZD # 14.

Our strategy aimed to have green synthesis with fewer steps by applying three components, solvent-free, and one-pot reaction. Under this condition, a mixture of 2-(α-naphthalen-1-ylamino) pyrimidine-5-carbaldehyde (11), N-methylthiourea (12), and monochloroacetic acid (13) was allowed to react under microwave irradiation for 15.0 min. The reaction resulted in the formation of compound (14) with 89% yield (Scheme 3).
The structure of compound (14) was established using elemental and spectroscopic analyses [Figure S5]. The IR spectrum showed absorption bands at 3428, 3263, 1685, and 1471 cm–1, characteristic of the NH on TZD, C=O, and C=N stretching vibrations, respectively. The Z configuration of the exocyclic C=C bond was assigned based on literature data of 1H NMR spectra of 4-TZD analogues.36,37 The olefinic proton was detected at δ = 7.59 ppm because of being deshielded by the adjacent C=O. The 13C NMR spectrum showed signals at 162.7 and 179.4 ppm corresponding to the C=N and C=O carbons connected to the TZD, respectively.
The presence of an amide linkage in the c-Jun N-terminal kinase-1 (JNK 1) inhibitors was reported beneficial in treating DM2.38 However, information on heteroaryl molecules having both 2,4-TZD and pyrimidine ring systems connected via an amide-linker has not been sufficiently reported in the literature. This encouraged us to think about bridging pyrimidine at position-6 to 2,4-TZD through an amide linkage and at position 2 to another aromatic moiety through a sulfonylurea linkage. The latter is supposed to serve as a hydrophobic end. Our hypothesis assumed that a combination of glitazone and sulfonylurea in one molecule can result in antidiabetic agents with high potency and safety (Figure 5).
Figure 5.
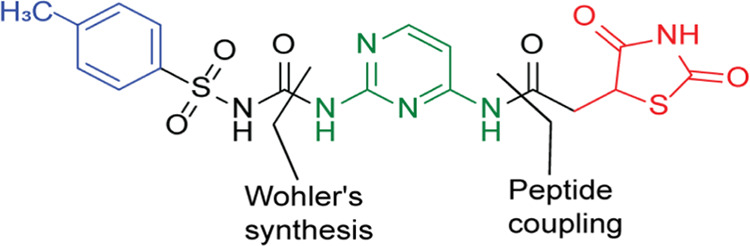
Designed template for compound TZD # 21.
Based on the above, compound # (21) was synthesized, as shown in Scheme 4. A carboxylic acid group was introduced into the 2,4-TZD via a two-step reaction in which thiourea (16) was refluxed with maleic acid anhydride (15) in water to produce imino-thiazolidinedione (17). Acid-hydrolysis of (17) resulted in 2,4-TZD acetic acid (18) with 91% yield. The structure of (18) was confirmed using 1H NMR. The H-5 atom of the TZD ring appeared as a triplet at δ = 3.38 ppm with J = 11.2 Hz. The methylene protons appeared as a doublet at δ = 2.84 ppm owing to coupling with the H-5 atom of the TZD ring.
Scheme 4. Synthesis of Compound TZD # 21.

Compound (19) was prepared by peptide coupling compound (18) with 2,4-diaminopyrimidine using N,N-dicyclohexylcarbodiimide in dry dimethylformamide (76% yield). Using elemental, IR, and NMR analyses, the structure of # (19) was confirmed [Figure S6]. The IR spectrum showed bands at 3492, 3269, 1529, and 1714 cm–1 corresponding to the NH2, NH, C=N, and C=O stretching vibrations, respectively. The 1H NMR spectrum showed signals at δ = 2.93–3.00 and 3.13–3.17 ppm attributed to the methylene protons (multiples) and at δ = 3.88 ppm (triplet with J = 8.0 Hz) attributed to the H-5 of TZD. Additionally, the 13C NMR signals at δ = 33.8 and 53.7 ppm were attributed to the methylene carbon and C5 on the TZD ring. The three signals that appeared at δ = 166.9 and 176.4 ppm were attributed to the TZD carbonyl groups. The acetamide carbonyl group was shown at δ = 173.5 ppm [Figure S6]. To produce compound # (21), Wohler’s synthesis was used to link compound (19) to p-toluene sulfonyl isocyanate (20) via urea linkage (72% yield) (Scheme 4).
Elemental and spectral analyses of compound (21) confirmed its structure. The disappearance of the IR peaks at 3492 and 3454 cm–1 indicated the conversion of the NH2 group into amide. The broad 1H NMR singlets at δ = 5.99 ppm and δ = 8.03 ppm were assigned to the protons in the urea linker (exchangeable with D2O). The pyrimidine H-5 and H-6 gave doublet signals at δ = 5.54 and 7.67 ppm, respectively, with equal coupling constants (J = 6.0 Hz). The two doublets at δ = 7.25 and 7.35 ppm were assigned to the aromatic protons on p-substituted phenyl (J = 8.0 Hz). The 13C NMR signal at δ = 151.1 ppm was attributed to the carbonyl group in urea and indicated the formation of urea linkage. All remaining carbons were observed in the expected regions [Figure S7].
In continuation of our efforts to construct further TZD-pyrimidine derivatives, compound # (23) was prepared by using the mechanism shown in Scheme 5. The Vilsmeier–Haack reaction was used to introduce a formyl group and two chlorine atoms into the TZD (1), forming the corresponding 5-formyl-2,4-dichlorothiazole (22). Subsequent reaction of (22) with 2,4-diaminopyrimine in the presence of a catalytic inorganic base (K2CO3) under microwave irradiation at 140 °C for 10 min gave the pyrimidine–thiazole derivative (23) (90% yield) (Scheme 5).
Scheme 5. Synthesis of 2-[(2-Aminopyrimidin-4-yl)amino]-4-chlorothiazole-5-carbald-ehyde, TZD # 23.
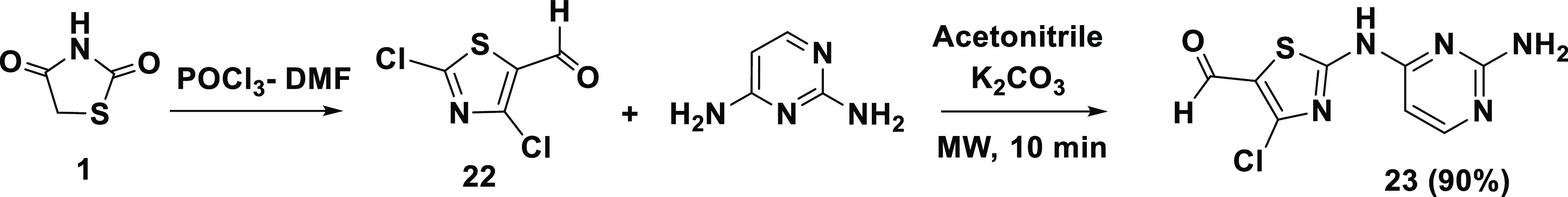
The structure of (23) was elucidated as before. The aldehydic C=O stretching vibration was shown at 1701 cm–1. The 1H NMR spectrum confirmed the presence of an aldehyde proton at δ = 9.90 ppm (singlet) and an imine proton at δ = 7.45 ppm. The amino group appeared at δ = 7.53 ppm (exchanged with D2O) [Figure S8].
2.1.3. Design 3: Pyrimidine as an Acidic Head (TZD # 29)
In this design, hydroxypyrimidine was positioned as an acidic head, the TZD ring as a central ring, and an aromatic derivative as a hydrophobic tail. This sequence in which the TZD ring is sandwiched between pyrimidine and phenyl moieties is expected to enhance the antidiabetic activity of the resultant molecule.39Figure 6 shows the designed template and the retrosynthetic disconnections for compound (29).
Figure 6.

Designed template and retrosynthetic disconnection of compound TZD # 29.
Scheme 6 shows the steps involved in the synthesis of compound (29). The reaction of TZD (1) with p-fluorobenzaldehyde (24) under microwave irradiation produced (Z)-5-(4-fluorobenzylidene) thiazolidine-2,4-dione (25). Subsequent reaction of (25) with chloroacetyl chloride (26) generated compound (27) with a linker at thiazoline nitrogen. The in situ reaction of (27) with 2-aminopyrimidine (28) in the presence of anhydrous K2CO3 produced the desired product (29) (82% yield).
Scheme 6. Synthetic mechanism of compound TZD # 29.
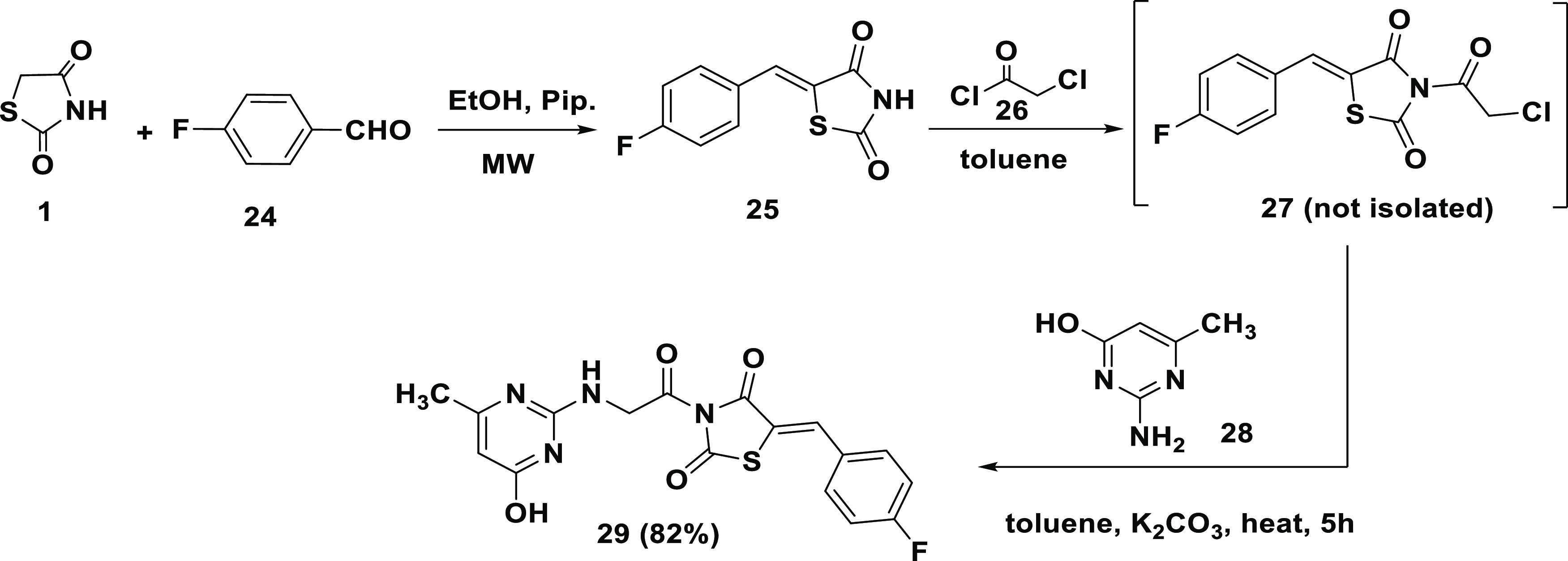
The IR spectrum of (29) showed that characteristic absorption bands at 3358, 3332, 674, and 1551 cm–1 correspond to OH, NH, C=O, and C=N vibrations, respectively [Figure S9a]. The 1H NMR spectrum showed bands at δ = 7.81 and 10.27 ppm characteristic for the NH and OH protons, respectively (exchangeable with D2O). The singlet signals at δ = 7.19 ppm and δ = 3.42 ppm are attributed to the olefinic and methylene protons, respectively. The signals at δ = 49.9, 180.9, and 166.7 and 172.8 ppm of 13C NMR spectrum are assigned to the methylene, C=S, and the carbonyl carbons, respectively (Figure S9b,c). These data confirmed the structure given in Scheme 6.
2.2. Design and Synthesis of Novel Rhodanines as PPAR-γ
Thiazolidines and rhodanines are bioisosteric with potencies for antidiabetic effects. Therefore, we aimed to design and synthesize RDs—as glitazone analogues—with enhanced efficacy. In this context, the template design of pioglitazone (Figure 2 and Section 4.1) was modified in three domains: (i) the RD moiety was replaced by RD acetic acid as a headgroup, (ii) the pyrimidine moiety was added as a central aromatic ring, and (iii) an alkyl, cycloalkyl, or aromatic ring was added as a tail (Figure 7).
Figure 7.

Design template for compounds RD # 33a–g.
Scheme 7 shows the synthetic mechanism for the new RD derivatives (33a–g) listed in Table 1. The two-step reaction mechanism involved the conversion of 2-substituted pyrimidines (30) into 5-formyl analogues (31) under Vilsmeier conditions. This reaction was followed by Knoevenagel condensation of (31) with RD (32) under microwave irradiation and in the presence of piperidine as a catalyst. The resultant compounds (33a–g) gave 81–89% yields (Scheme 7 and Table 1).
Scheme 7. Synthesis of 5-Substituted Rhodanine Derivatives, RD # 33a–g.

Table 1. Substituents R and R1 and 1H NMR Chemical Shifts for the Olefinic Proton in Substituted Rhodanine Derivatives (33a–g)a.
| compound (RD) | R | R1 | chemical shift (δ, ppm) | yield, % |
|---|---|---|---|---|
| 33a | N,N-dimethylamine | H | 7.43 | 89 |
| 33b | NH2 | H | 7.84 | 87 |
| 33c | N,N-dimethylamine | CH2COOH | 7.61 | 81 |
| 33d | isopropyl | CH2COOH | 7.62 | 86 |
| 33e | N-morpholine | CH2COOH | 7.64 | 88 |
| 33f | n-propylamine | CH2COOH | 7.57 | 83 |
| 33g | benzylamine | CH2COOH | 7.60 | 84 |
The table also shows the synthetic yield for each derivative.
The structures of (33a–g) were confirmed using elemental and spectral analyses [Figures S10–S16]. The IR spectra showed characteristic absorption bands around 3590 and 1713 cm–1, corresponding to the carboxylic acid OH and C=O, respectively. The bands shown at 3058 and 2960 cm–1 were attributed to the C–H stretching vibrations on aromatic and aliphatic skeletons. The 1H NMR spectra showed the olefinic proton’s signal at δ ≈ 7.64 ppm, confirming the formation of E-isomers. Olefinic protons at δ > 7.30 ppm were reported to confirm the formation of E-isomers, while at δ < 6.80 ppm confirmed the formation of E-isomers.40−42 The OH proton gave a signal at δ ≈ 11.78 ppm (exchanged with D2O), while the pyrimidine H-4 and H-6 gave a singlet at δ ≈ 8.63 ppm. The 13C NMR signal at δ = 160.2 ppm was attributed to the pyrimidine C4 and C6. The signals at δ ≈ 128.2 ppm and δ ≈ 119.1 ppm were attributed to the olefinic and C5 on the RD ring, respectively. The carbonyl carbons were shown at δ ≈ 167.0 and 167.1 ppm, while the C=S carbon was shown at δ = 192.7 ppm [Figures S10–S16].
2.3. Antidiabetic Activity
Regulation of gene’s expressions in lipid and glucose metabolisms by the PPAR transcription factors (PPAR-α, PPAR-β, and PPAR-γ) is well established.43,44 Out of the three PPAR isotypes, PPAR-γ has been extensively targeted for developing drugs that can regulate DM2, insulin sensitization, adipogenesis, cellular differentiation, atherosclerosis, and cancer.45
Troglitazone, rosiglitazone, pioglitazone, and ciglitazone are synthetic TZDs with high-affinity for activating PPAR-γ.46 Rosiglitazone and pioglitazone are marketed as PPAR-γ agonists for the treatment of DM2 that lower blood sugar by reducing insulin resistance and enhancing insulin action.47,48 However, they cause an increased risk of heart failure, bone fractures, weight gain, fluid retention, and edema.
Since activation of PPAR-γ is known to regulate insulin secretion and improve its sensitivity, synthesized TZDs and RDs will be evaluated as PPAR-γ agonists using in silico molecular docking. Binding affinities, binding modes, and binding sites toward the PPAR-γ receptor will be evaluated. The effects of synthesized compounds on insulin secretion and glucose uptake by βTC6 cells will also be evaluated. In our compounds, pyrimidine scaffolds connected to TZD or RD rings may reduce the side effects reported for commercial glitazones.
2.3.1. Effects of the Novel TZDs on Insulin Secretion by βTC6 Cells
Figure 8 shows the effect of adding pioglitazone and TZDs (10–12–10–5 M) to βTC6 cells in the absence (basal) and presence (postprandial) of 2.88 mM glucose. Insulin secreted was assayed using the high-range insulin sandwich ELISA kit (Sections 2.3.2 and 2.3.3). The addition of pioglitazone had insignificantly affected the basal insulin secretion while decreasing its glucose-stimulated secretion by ∼10% relative to the control (P < 0.001) (Figure 8A). The effect of adding TZDs # (7a–d, 14, 19, 21, 23, and 29) on insulin secretion in the absence (basal) and presence of 2.8 mM glucose (postprandial) is shown in Figure 8B–J. Compound # (7d) showed a similar behavior to pioglitazone, where insignificant effects were observed on basal and postprandial insulin secretion (Figure 8E). Compound # (23) also showed no effect on basal insulin secretion, whereas postprandial insulin secretion was reduced by 55–65% (Figure 8I).
Figure 8.

Effects of pioglitazone (A) and TZDs # 7a (B), 7b (C) 7c (D), 7d (E), 14 (F), 19 (G), 21 (H), 23 (I), and 29 (J) on insulin secretion by βTC6 cells in the absence (basal) and presence of 2.8 mM glucose. Drug concentrations of 10–12–10–6 M were used. The results are means of triplicates ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 from relative basal control and #P < 0.05, ##P < 0.01, and ###P < 0.001 from glucose at 2.8 mM.
Compounds # (7a, 7b, 7c, and 29) had significantly decreased the basal insulin secretion by ∼50, 40, 20, and 67%, and increased glucose-stimulated insulin by ∼28, 33, 25, and 50%, respectively (Figure 8B–D, 8J). These results indicate highly interesting compounds that can prevent hypoglycemia during fasting by inhibiting basal insulin secretion and prevent postprandial hyperglycemia by increasing glucose-stimulated insulin. Compound # (29) showed a superior potency with ∼67% inhibition for basal insulin and 50% increase for postprandial insulin relative to pioglitazone (Table 2).
Table 2. Effects of TZDs and RDs on Basal and Glucose-Stimulated Insulin Secretions.

A different behavior was observed for compound # (19), where both basal and glucose-stimulated insulins decreased by ∼60 and ∼12%, respectively (Figure 8G). Compounds # (14 and 21) have shown quite interesting behavior where the basal insulin release increased by ∼100 and ∼250%, respectively, whereas glucose-stimulated insulins remained almost unchanged (Figure 8F,8H).
In summary, our results showed that compound # (7d) had a similar effect on insulin secretion of βTC6 cells to pioglitazone. Compounds # (7a, 7b, 7c, and 29) reduced the basal insulin release and increased its glucose-stimulated insulin. Thus, they may act as antihypoglycemic during fasting and antihyperglycemic during postprandial. Compound # (29) showed the highest potency, with an ∼67% reduction in basal insulin and a 50% increase in postprandial insulin secretions. The results also showed that the effect of some synthesized TZDs on insulin secretion is concentration-dependent (Figure 8). These results indicated that novel TZDs and RDs are potent regulators of insulin secretion. Subsequently, animal/human testing will be required to confirm their behaviors and check for toxicity.
2.3.2. Effects of Novel RDs on Insulin Secretion by βTC6 Cells
Figure 9 shows the effects of adding pioglitazone and RDs # (33a–g) (10–12–10–5 M) on insulin secretion by βTC6 cells in the absence and presence of glucose. Pioglitazone did not affect basal insulin secretion and slightly decreased glucose-stimulated insulin by ∼10% (see Section 4.3.1). A similar trend was exhibited by compounds # (33c and 33g), where no change in basal insulin was observed, whereas stimulated insulin increased by ∼15% (Figure 9D,H and Table 2).
Figure 9.

Effects of pioglitazone (A) and RDs # 33a (B), 33b (C), 33c (D), 33d (E), 33e (F), 33f (G), and 33g (H) on insulin secretion by βTC6 cells in the absence (basal) and presence of 2.8 mM glucose. Drug concentrations of 10–12–10–6 M were used. The results are means of triplicates ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 from relative basal control, and #P < 0.05, ##P < 0.01, and ###P < 0.001 from glucose 2.8 mM.
Compounds RDs # (33a, 33b, 33d, 33e, 33f) increased the average basal insulin secretions by ∼35, 200, 20, 40, and 42%, respectively. The average stimulated insulin releases have also slightly increased (Figure 9B,C,E–G and Table 2). The effect of RD doses on insulin secretion is also shown in Figure 9. Apparently, the low 10–12 M concentrations resulted in slightly higher insulin release relative to the higher doses 10–8 and 10–6 M (Figure 9).
Thus, synthesized RDs were found to significantly increase basal insulin secretion and slightly affect its stimulated releases. Compound # (33b) was the highest potent with an ∼200% increase in basal insulin. These results suggested that RDs # 33a–f can be postprandially administered to avoid hypoglycemic effects. Though our compounds are more potent than pioglitazone, they seemed similarly capable of regulating insulin secretion and improving insulin resistance.
2.3.3. Glucose Uptake and Insulin Sensitivity
Insulin sensitivity measures the response of the body cells to insulin. Cells that are sensitive to insulin uptake glucose efficiently. Insulin resistance develops when cells become less sensitive to insulin and affects the body’s capability to absorb and use glucose. Insulin sensitivity is inversely related to insulin resistance and directly related to glucose uptake. Thus, insulin sensitivity and resistance can be measured by glucose uptake.
The effects of synthesized TZDs and RDs on the insulin sensitivity of βTC6 cells were measured by estimating the cellular glucose uptake using the Cayman’s Glucose Uptake cell-based assay kit (Section 2.3.3). A 10 μM each of TZD # (7a–d, 14, 19, 21, 23, and 29) or RD # (33a–g) was used.
Figure 10 shows the effects of TZDs (Figure 10A) and RDs (Figure 10B) on glucose uptake. All compounds increased glucose uptake/insulin sensitivity relative to the control and pioglitazone. The latter is the FDA-approved antidiabetic that enhances insulin sensitivity. Improvement in insulin sensitivity ranged between 25% for TZD # (7b) and 155% for TZD # (29) relative to the control and pioglitazone. Among all tested compounds, TZD # 29 was the most potent (P < 0.001) (Figure 10A). Improvement of insulin sensitivity by tested RDs ranged between 17% for (33c) and 70% for (33g) relative to the control and pioglitazone. Compound RD # (33g) was the most potent (P < 0.001) (Figure 10B).
Figure 10.

Effects of synthesized TZDs # 7a–d, 14, 19, 21, 23, and 29 (A) and RDs # 33a–g (B) on glucose uptake by βTC6 cells. A 10.0 μM solution each was used. The results presented are the “means of triplicates ± SEM”; *P < 0.05, **P < 0.01, and ***P < 0.001 vs control (zero concentration); and #P < 0.05, ##P < 0.01, and ###P < 0.001 vs reference drug (pioglitazone).
These results showed that synthesized TZDs and RDs have remarkable improvements in potencies for insulin sensitivity/glucose uptake by βTC6 cells relative to the control and pioglitazone.
2.4. Molecular Docking of TZDs and RDs on PPAR-γ
The PPAR-γ receptor regulates gene expression in various physiological processes such as insulin sensitivity, lipid metabolism, and inflammation. Therefore, PPAR-γ has been a target for developing various drugs that include antidiabetics.49−51 Flexibility, large size, hydrophobicity, and capability to adopt different conformations by the PPAR-γ active site have made it difficult to find a structure that is specifically and effectively activating the site. Several PPAR-γ agonists were identified using the induced-fit docking.52
In this work, activation of the PPAR-γ receptor was tested as a mechanism of the antidiabetic effect of synthesized TZDs and RDs. Tested compounds were docked into the three PPAR-γ crystal structures: 1I7I, 1WM0, and 1ZGY. Information about binding modes and affinities was collected (Section 2.5). The three structures were used to address the flexibility in the PPAR-γ receptor and its implications on ligand binding that possibly results when a single model is used. For comparison, the published scores of all previously docked ligands on the three PPAR-γ crystal structures are listed in Table S1.55
Table 3 lists the best poses docking score for TZDs # 7a, 7b, 14, 21, and 29 and RDs # 33g, 33c, and 33f, along with the corresponding PPAR-γ crystal structures. Pioglitazone is presented as a reference TZD. The compounds scored exothermic binding energies ranging from −6.02 to −9.70 kcal/mol. TZDs # (14 and 29) showed the highest exothermic affinities toward PPAR-γ with binding energy scored −8.86 and −9.70 kcal/mol, respectively. The two compounds have different scaffolds in which pyrimidine is a central aromatic ring in TZD # (14) and an acidic head in TZD # (29). Interestingly, the binding affinity of TZD # (29) is higher than reference pioglitazone (−9.25 kcal/mol). However, TZD # (14) and pioglitazone gave comparable binding affinities.
Table 3. Best Glide-XP Scores for TZD and RD Dockings on Different PPAR-γ Crystal Structures Together with Pioglitazone as a Reference Ligand.
Figure 11 shows a cartoon representation of the binding mode of TZD # (29) into the PPAR-γ active site of the 1I7I crystal structure. TZD # (29) (orange sticks) aligned on the cocrystallized ligand (blue sticks) is shown in Figure 11A. Binding interactions of TZD # (29) inside the PPAR-γ active site are shown in Figure 11B. The phenol group (polar acidic head) interacts with the side chains of Tyr473 and His323 by hydrogen bonding. Additional hydrogen bonding linking the sulfur on the TZD ring with the side chain of Arg288 is observed as a strong interaction. Moreover, multiple van der Waals interactions linking TZD # (29) with the surrounding hydrophobic residues (Ile281, Ile326, Leu330, Leu339, Meth348, and Leu469) are observed. These forces indicated that TZD # (29) had optimally fitted within the PPAR-γ active site (Figure 11B).
Figure 11.

Cartoon representation of docking of TZD # 29 onto the PPAR-γ binding site of PDB: 1I7I crystal structure. (A) shows TZD # 29 (orange sticks) aligned with the cocrystallized ligand (blue sticks). (B) shows the binding modes. Hydrogen bonding is shown as orange dotted lines.
A similar binding mode was observed for the interaction of TZD # (14) with the PDB: 1ZGY PPAR-γ crystal structure. Figure 12A shows that TZD # (14) conformation is well-aligned with the cocrystallized ligand (BRL) inside the PPAR-γ active site. The thiazolidinedione ring on TZD # (14) acted as a polar head and participated in hydrogen bonding with Tyr473 and His323, along with convenient physical and chemical complementarity with the PPAR-γ pocket (Figure 12B).
Figure 12.

Cartoon representation of docking of TZD # 14 onto the PPAR-γ binding site of the PDB: 1ZGY crystal structure. (A) shows TZD # 14 (orange sticks) aligned with the cocrystallized ligand (blue sticks). (B) shows the binding modes. Hydrogen bonding is shown as orange dotted lines.
Table 3 also shows the docking scores of synthesized RDs onto the PPAR-γ crystal structure of PDB: 1WM0. Exothermic binding affinities of −7.03 and −9.60 kcal/mol for RDs # (33c) and (33g) were obtained, respectively. Thus, RD # (33g) gave a higher binding affinity relative to pioglitazone as a reference PPAR-γ agonist (−9.3 kcal/mol).
Figure 13 shows a cartoon representation of docked RD # (33g) onto the PPAR-γ structure. A perfect alignment between RD # (33g) and the cocrystallized ligand (PLB) onto the binding site of 1WM0 is observed (Figure 13A).51Figure 13B shows the binding mode of RD # (33g) into the PPAR-γ active site. Binding modes—similar to the cocrystallized ligand—involving ionic interactions with Arg288 via the carboxylate group, π–π stacking interactions involving the terminal aromatic ring of RD # (33g) with the side chain of Phe246, and hydrogen bonding with the side chain of Ser289 and the backbone amide of Ile281 are observed. These multiple van der Waals interactions with the surrounding hydrophobic residues rendered strong interactions between RD # (33g) and PPAR-γ compared to the pioglitazone as a reference PPAR agonist. Similar behaviors were observed for RD # (33c) and (33f).
Figure 13.

Cartoon representation of docking of RD # 33g onto the PPAR-γ binding site of the PDB: 1WM0 crystal structure. (A) shows RD # 33g (orange sticks) aligned with the cocrystallized ligand (blue sticks). (B) shows the binding modes. Hydrogen bonding and π–π stacking interactions are shown as orange and blue dotted lines, respectively.
Taken all together, these molecular docking results support our hypothesis that synthesized TZDs and RDs function as PPAR-γ agonists. The results also support our experimental results for regulating insulin secretion and enhancing insulin resistance. They also showed the importance of structural features in the designed and synthesized scaffolds.
2.5. Molecular Dynamics of TZD and RD Complexes of PPAR-γ
To better understand the stability of the docked TZDs and RDs in the target pockets of PPAR-γ, MD simulations were applied. Figure 14 shows the RMSD plots of TZD # 14 (A), TZD # 29 (B), RD # 33g (C), and RD # 33f (D). The RMSDs for the protein backbone in complex with the four ligand molecules throughout the 100 ns MD simulation resonated between 1.0 and 2.5 Å. This strongly indicated the conformational stabilities of the protein–ligand complex molecules. Minor RMSD fluctuations implied the occurrence of minor structural perturbations in the protein backbone. Although the PPAR-TZD # 29 complex deviated from the initial docking configuration by an average of 2.5 Å, it confidently showed very stable binding during the entire course of simulation (Figure 14B).
Figure 14.

RMSD plots resulting from 100 ns MD simulations of TZD # 14 (A), TZD # 29 (B), RD # 33g (C), and RD # 33f (D), along with their corresponding protein backbone.
Figure 15 shows the cluster conformation of PPAR-TZD # 29, resulting from 100 ns MD simulations aligned on the original crystal structure with its cocrystallized ligand and its hydrogen bond plot. Interestingly, the heterocyclic ring on TZD # 29 formed hydrogen bonding with the PPAR-γ important residues, His321 and Tyr473. These two hydrogen bonds are also made by the cocrystallized ligand of the PDB: ID 1I7I (Figure 15A). These interactions seem to be constantly occurring over the course of MD simulation (Figure 15B). More notably, hydrogen bonding between the ligand hetero nitrogen and the histidine imidazole ring. Additionally, TZD # 29 seemed to be further stabilized in the PPAR active site via inserting its chlorophenyl moiety into a hydrophobic pocket through multiple van der Waals interactions with the surrounding residues such as Leu339 and Met348.
Figure 15.

(A) Cluster conformation of the PPAR-TZD-29 resulted from 100 ns MD simulations aligned on the original crystal structure with its cocrystallized ligand (key hydrogen bonds are labeled with blue dotted lines). (B) The hydrogen bond plot of TZD # 29 made with His323 and Tyr473.
2.6. Insights into Structure–Activity Relationship
In this work, we aimed to design and synthesize novel TZDs and RDs with improved antidiabetic efficacy relative to pioglitazone, the FDA-approved antidiabetic drug that also treats insulin resistance. Designs based on modifying the molecular structure of pioglitazone by introducing pyrimidine as an effector site (tail), a central aromatic ring, or an acidic head were developed. Additionally, the ethyl ether linkage in pioglitazone was replaced by a secondary nitrogen atom between the pyrimidine and TZD or RD moiety.
Designed structures were synthesized, structurally confirmed, and tested for their antidiabetic activity. Compounds (7a and 7c) with pyrimidine as an effector site and NH2 as an electron-donating group gave better activity (increased insulin secretion) relative to (7d), having chlorine as an electron-withdrawing group. Replacing the central phenyl ring in pioglitazone with pyrimidine associated with sulfonyl and peptide linkers in TZD # (21) increased the basal insulin release by ∼250%. However, the direct connection of the effector site to the central ring in TZDs # (14 and 23) reduced their antidiabetic activity. The introduction of pyrimidine as an acidic head in TZD # (29) inhibited the basal insulin secretion by 67% while increasing its glucose-stimulated insulin by 50% relative to the reference. The reason could be attributed to the hydrogen bonding formation between the OH on pyrimidine and NH on Tyr473 and His323 of the PPAR-γ protein that subsequently resulted in higher activation of the PPAR-γ receptor (Figure 12).
Analysis of the structures–activity relationship (SAR) in RDs indicated that the introduction of acetic acid in (33c–g) played a role in improving their antidiabetic activities. The ability of this acidic center to form salt bridges with the basic protein residue (Arg288) may justify the activation of the PPAR-γ receptor. Apparently, the addition of a carbon linker between the central aromatic ring and the effector site in RD # (33g) played a role in improving its antidiabetic activity. Inversely, the introduction of the isopropyl group at position 2 in pyrimidine reduced the antidiabetic activity of RD # (33d). Replacement of isopropyl by aliphatic amine in RD # (33b) was shown to improve the overall activity. The reason could be attributed to the ability of NH2 to interact with the PPAR-γ receptor through H-bonding and/or π–π stacking (Section 4.4). These results indicated that RD ring connected to pyrimidine as an acidic center and bridged to a hydrophobic effector site via C1 or C2 linkers can render higher antidiabetic activity.
Absorption, distribution, metabolism, excretion, and metabolism (ADMET) of synthesized drugs were estimated using ADMET lab 2.0. Excellent druggable properties that include moderate molecular weights, appropriate sizes, and capability to form hydrogen bonding were obtained.
TZD # 29 and RD # 33g showed optimum permeability (<−5.15 log unit), moderate human intestinal absorption (20–30% oral bioavailability), high plasma protein binding (PPB > 93.00%) associated with optimal volume distributions (VD = 0.212–0.299 L/kg), and moderate tissue distribution. These properties suggest suitability for oral administration and potential safety in vivo. The clearance rate of 2.593–4.212 mL/min/kg and a half-life of 0.673–0.143 h also indicated rapid elimination required for therapeutic use. The metabolism of the two compounds estimated moderate inhibitory effects against CYP1A2, CYP2C9, and CYP3A4 enzymes, indicating potential involvement in other metabolic pathways. Estimation of human toxicity revealed green-yellow risks for cardiac arrhythmia, hepatotoxicity, induced liver injury, skin sensitizer, and carcinogen. These estimations indicate drug-like properties and warrant for initiating the development process. The distribution and clearance rates, limited blood–brain barrier penetration, and plasma binding suggest manageable distribution profiles. However, careful assessment of toxicity is required before any preclinical or clinical phases.
3. Conclusions
Sixteen TZDs and RDs were designed by introducing pyrimidine as an effector site, a central aromatic ring, or an acidic head into the pioglitazone structure. Resultant compounds were synthesized, structurally confirmed, and tested for regulating glucose metabolism and improving insulin sensitivity. The mechanism of action as a PPAR-γ agonist was evaluated.
TZDs # (7a, 7b, 7c, and 29) reduced the basal insulin secretion and increased its glucose-stimulated secretion. TZD # (29) showed the highest potency, with a 67% reduction and a 50% increase. Concentration-dependent patterns were noted. The results indicated compounds that could function as antihypoglycemic during fasting and as antihyperglycemic in postprandial conditions.
RDs # (33a–f) increased basal insulin secretions by 35–200%, while glucose-stimulated insulin remained almost unchanged. RD # (33b) showed the highest potency with a 200% increase. The results suggested that synthesized RDs are to be postprandially administered to avoid hypoglycemic effects.
Glucose uptake experiments indicated remarkable improvement in the insulin sensitivity of βTC6 cells. TZDs and RDs enhanced glucose’s uptake by 25–155 and 17–70%, respectively, relative to the control and pioglitazone. TZD # (29) and RD # (33g) showed the best potency.34
Molecular docking of synthesized TZDs and RDs into three PPAR-γ crystal structures scored exothermic binding energies ranging from −6.02 to – 9.70 kcal/mol. TZDs # (14) and (29) showed the highest binding affinities (−8.86 and −9.70 kcal/mol). TZD # (29) showed better binding affinity compared to pioglitazone (−9.25 kcal/mol). The two compounds were found to align with the cocrystallized ligands inside the PPAR-γ active site and interact through hydrogen bonding with Tyr473, His323, Arg288, Tyr473, or His323. Multiple van der Waals interactions with the hydrophobic residues (i.e., Ile281, Ile326, Leu330, Leu339, Meth348, and Leu469) were also observed. RD # (33g) scored better binding affinity than pioglitazone (−9.60 kcal/mol). It also showed perfect alignment with the cocrystallized ligand into the PPAR-γ binding site with binding modes involving ionic and π–π stacking interactions with Arg288 and Phe246. Multiple van der Waals interactions with hydrophobic residues and hydrogen bonding confirmed strong interactions with PPAR-γ. These results supported our experimental findings.
Insights into the structure–activity relationship indicated that introductions of the pyrimidine ring and sulfonyl and peptide linkers in pioglitazone’s structure have improved the antidiabetic activity of synthesized scaffolds. For example, TZD # (21) increased the basal insulin secretion by ∼250%, while direct connections of the pyrimidine ring (effector site) to the TZD ring in TZD # (14) increased it by 100%. Pyrimidine as an acidic head in TZD # (29) reduced the basal insulin secretion by 67% and increased glucose-stimulated insulin by 50%, relative to the reference. The RD ring system connected to the acetic acid as the acidic head enhanced the antidiabetic activity. The amino-alkyl or amino-aryl group substituted at position 2 of the pyrimidine ring improved the basal insulin secretion by 200% in RD # (33b) and by 20–40% in other RDs. Synthesized RDs were also found to increase glucose uptake relative to the control and reference (Table 2 and Figure 10).
Overall, synthesized TZDs and RDs proved more potent than pioglitazone in regulating glucose metabolism, improving insulin sensitivity, and activating the PPAR-γ receptor. TZDs # (21, 14, and 29) and RDs # (33b) showed the highest potency in regulating insulin secretion, while TZD # (29) and RD # (33g) showed the best glucose uptake and binding affinity to the PPAR-γ receptor. Synthesized TZDs and RDs seemed to be multitargeting, interacting with PPAR-γ and other proteins. As an example, TZD # (21) seemed to act by inhibiting PPAR-γ and stimulating insulin secretion from β cells. Thus, synthesized TZDs and RDs are promising candidates for further development as antidiabetic agents.
4. Experimental Section
4.1. Materials and Equipment
Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. Thin-layer chromatography (TLC) was performed on silica gel glass plates (Silica gel, 60 F254, Fluka, Merck, Darmstadt, Germany). Kieselgel S (silica gel S, 0.063–0.1 mm, Merck, Darmstadt, Germany) was used for column chromatography.
Melting points were measured using the Gallenkamp apparatus (Toledo, OH). Infrared spectra were collected in KBr pellets using Thermo Nicolet model 470 FT-IR spectrophotometers (Thermo Scientific, Waltham, MA). The 1H NMR and 13C NMR spectra were recorded using a Varian-400 MHz spectrometer (Agilent Technologies, Santa Clara, CA) using the CDCl3 solvent and tetramethyl silane (TMS) as the internal reference. Chemical shifts were stated in parts per million (δ values, ppm). Elemental analysis was performed using a Leco CHN-600 elemental analyzer (Ontario, Canada). Microwave synthesis was performed using the CEM microwave system (Matthews).
4.2. Synthesis and Spectral Characterizations of Thiazolidinedione–Pyrimidine Derivatives
4.2.1. General Procedure for the Synthesis of (7a)
The following steps were involved in the preparation of (7a):
-
(1).
Thiazolidine-2,4-dione (1)33 was prepared by mixing monochloroacetic acid (10.58 mmol, 1.00 g) with thiourea (10.6 mmol, 0.81 g) and water (2 mL) in a 10.0 mL microwave reaction vessel. The mixture was irradiated at 140 °C for 10.0 min. The resultant solid was filtered and recrystallized from hot water to yield 1.10 g (90%) of thiazolidinone, mp 124–125 °C; IR (KBr, cm–1): 1241 (CN), 1492 (CH2), 1666, 1738 (C=O), 3121 (NH); 1H NMR [CDCl3, 400 MHz]: (δ, ppm) 4.20 (s, 2H, CH2), 9.10 (brs, 1H, NH); 13C NMR [CDCl3, 100 MHz]: (δ, ppm) 35.9, 168.5, 169.2.
-
(2).
5-Benzylidene-2,4-thiazolidinedione (3)53 was prepared by mixing benzaldehyde (2) (0.188 mmol, 0.020 g) with 2,4-thiazolidinedione (1) (0.188 mmol, 0.022 g) in dry toluene (3 mL) in a 10.0 mL CEM microwave vial. A few drops of piperidine were added, and the reaction mixture was irradiated at 135 °C for 10–15 min. During cooling, the solid precipitated was filtered and washed with dry ethanol to yield 94% of compound (3), mp 242 °C.
-
(3).
4′-Chlorosulfonylbenzylidene-2,4-thiazolidinedione (5)54 was prepared by mixing benzylidene-2,4-thiazolidinedione (3) (0.0388 mol, 8.0 g) with chlorosulfonic acid (4) (0.155 mol, 18.08 g) in a 100 mL round-bottom flask equipped with a condenser and a dropping funnel at 25 °C. The reaction mixture was heated for 1 h in a water bath, then cooled and poured into crushed ice. The solid was filtered and dried to yield compound (5) (66%, mp 181 °C).
-
(4).
2-(Methylimino)-5-[4′-(N-pyridinesulfonyl) benzylidine] thiazolidine-4-one-chloride salt (7a) was prepared by mixing 2,4-diaminopyrimidine (0.1 mmol) with p-chlorosulfonyl benzylidene-2,4-thiazolidinedione (5) (0.1 mmol, 0.03 g) in 3 mL pyridine in a 10.0 mL CEM microwave vial. The mixture was heated at 100 °C for 10.0 min under microwave irradiation and then poured into 20.0 mL of ice water. The resultant solid was filtered and recrystallized from ethanol. This compound was obtained as a gray powder, with 83% yield; mp 280–281 °C; IR (KBr,cm–1): 3487 (NH TZD), 3067 (C–H, aromatic), 1683 (C=O), 1486 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.08 (s, 3H, CH3), 3.40–3.42 (m,1H, NH, exchanges with D2O), 7.56 (d, 2H, aromatic H, J = 8.2 Hz), 7.69 (d, 2H, aromatic H, J = 8.2 Hz), 7.88 (s, 1H, CH),7.97 (t, 2H, pyridine, J = 7.2 Hz), 8.49 (t, 1H, pyridine, J = 7.4 Hz), 8.87 (d, 2H, pyridine, J = 5.1 Hz); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 28.2 (CH3), 122.9 (C5-thiazolidinedione), 126.8, 127.4, 130.3, 132.3, 134.0, 142.7, 146.0 (aromatic C), 148.4 (CH carbons), 166.4 (C=N), 168.2 (C=O); anal. calcd for C16H14N3O3S2: C, 53.32; H, 3.92; N, 11.66; S, 17.79; found: 53.77; H, 3.99; N, 11.94; S, 18.07.
4.2.2. General Procedure for the Synthesis of (7b–d)
The general procedure involved mixing the required pyrimidine-based aromatic amine (0.1 mmol) with the corresponding p-chlorosulfonyl benzylidene-2,4-thiazolidinedione (5) (0.1 mmol, 0.03 g) and 0.5 mL of diisopropyl ethyl amine (DIPEA) in 3.0 mL ethanol. The mixture was heated under microwave irradiation at 100 °C for 10 min, and the reaction mixture was then poured into 20.0 mL of ice water. The resulting solid was subsequently filtered and recrystallized from ethanol.
4.2.2.1. N-(2′-Aminopyrimidin-4′-yl)-4-[2″-(methylimino)-4″-oxothiazolidin-5″-ylidene-methyl]-benzenesulfonamide (7b)
Compound (7b) was produced in the form of a light brown powder, 78% yield; mp 247–248 °C; IR (KBr, cm–1): 3554, 3524 (NH2), 3353 (NH TZD), 3029 (C–H aromatic), 1686 (C=O), 1519 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.08 (s, 3H, CH3), 5.98 (d, 1H, H5-pyrimidine, J = 6.0 Hz), 7.56 (m, 2H, aromatic), 7.58 (1H, NH, exchanges with D2O), 7.56 (d, 2H, aromatic H, J = 8.0 Hz), 7.69 (d, 1H, H6-pyrimidine, J = 6.0 Hz), 7.88 (s, 1H, olefinic H), 8.05 (brs, 2H, NH2, exchanges with D2O), 11.63 (brs, 1H, NH, exchanged with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 53.9 (CH3), 97.6 (C5-pyrimidine), 122.4 (C5-thiazolidionedione), 126.9, 130.2, 132.4, 133.4 (aromatic C), 142.7 (olefinic C), 150.3 (C4-pyrimidine), 155.6 (C=N), 165.4 (C6- pyrimidine), 166.3 (C2-pyrimidine), 167.9 (C=O); anal. calcd for C15H14N6O3S2: C, 46.14; H, 3.61; N, 21.52; S, 16.43; found: C, 46.59; H, 3.68; N, 21.80; S, 16.71.
4.2.2.2. (E)-N-(2′-Aminopyrimidin-4′-yl)-4-(2″,4″-dioxothiazolidin-5″-ylidene) Methyl-benzene-sulfonamide (7c)
Compound (7c) was obtained in the form of a light brown powder, yield 84%; mp 299 °C; IR (KBr, cm–1): 3513, 3476 (NH2), 3353 (NH), 3130 (C–H aromatic), 1675 (C=O), 1450 (C=C); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 4.33 (brs, 1H, NH, exchanges with D2O), 5.84 (d, 1H, H5-pyrimidine, J = 6.0 Hz), 6.49 (brs, 2H, NH2, exchanges with D2O), 7.11 (m, 2H, aromatic), 7.36 (d, 2H, aromatic H, J = 8.0 Hz), 7.60 (d, 2H, H6-pyrimidine, J = 6.0 Hz), 7.88 (s, 1H, olefinic H), 11.65 (brs, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 96.7 (C5-pyrimdine), 122.9 (C5-thiazolidion), 126.8, 127.4, 132.3, 134.0 (aromatic C), 142.7 (olefinic C), 152.6 (C4-pyrimidine), 158.6 (C6-pyrimidine), 163.2 (C2-pyrimidine), 165.0 (C=O), 168.2 (C=O); anal. calcd for C14H11N5O4S2: C, 44.56; H, 2.94; N, 18.56; S, 16.99; found: C, 45.01; H, 3.01; N, 18.84; S, 17.27.
4.2.2.3. (E)-N-(2′-Chloropyrimidin-4′-yl)-4-(2″,4″-dioxothiazolidin-5″-ylidene) Methyl-benzene-sulfonamide (7d)
Compound (7d) was obtained in the form of a pale yellow powder, yield 75%; mp 312 °C; IR (KBr, cm–1): 3431 (NH TZD), 3249 (NH-sec.amine linkage), 3077 (C–H, aromatic), 1698 (C=O), 1491 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 5.87 (d,1H, H5-pyrimidine, J = 7.0 Hz), 7.10 (d, 2H, aromatic, J = 8.0 Hz), 7.18 (d, 1H, H6-pyrimidine, J = 7.0 Hz), 7.26 (d, 2H, aromatic, J = 8.0 Hz), 7.31 (s, 1H, olefinic H), 8.34 (br, NH, exchanges with D2O), 11.91 (brs, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 99.8 (C5-pyrimdine), 119.8 (C5- thiazolidinedione), 127.1, 128.3, 133.0, 134.7 (aromatic C), 141.5 (olefinic C), 152.4 (C4-pyrimidine), 158.6 (C6-pyrimidine), 163.9 (C2-pyrimdine), 165.0 (C=O), 169.1 (C=O); anal. calcd for C14H9ClN4O4S2: C, 42.37; H, 2.29; N, 14.12; S, 16.16; found: C, 42.82; H, 2.36; N, 14.40; S, 16.44.
4.2.2.4. (E)-2-(Methylimino)-5-[2′-(naphthalen-1″-ylamino)pyrimidin-5′-yl] Methylene-thiazolidin-4-one (14)
Compound (14) was prepared by mixing methylthiourea (12) (3.0 mmol, 0.27 g) with chloroacetic acid (13) (3.6 mmol, 0.2 mL) and aldehyde (11) (3.0 mmol, 0.74 g). The mixture was heated at 90–110 °C for 10–20 min under microwave irradiation, then let to cooled to room temperature and extracted with CH2Cl2. The organic layer was subsequently washed using aqueous NaHCO3 and water and then dried over anhydrous Na2SO4. The solvent was evaporated under vacuum. The resultant residue was recrystallized from EtOH/water to obtain compound (14) as a pale yellow powder.
The yield of (14) was 89%; mp 296–297 °C; IR (KBr, cm–1): 3428 (NH TZD), 3263 (NH-sec.amine linkage), 3000 (C–H, aromatic), 1685 (C=O), 1471 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.09 (s, 3H, CH3), 7.59 (s, 1H, olefinic H), 7.64 (m, 2H, naphthalene), 7.97 (s, 1H, NH, exchanges with D2O), 8.06–8.11 (m, 3H, naphthalene), 8.49 (s, 1H, naphthalene), 9.09 (s, 2H, H4,6-pyrimidine), 9.14 (m, 1H, naphthalene), 9.77 (brs, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 31.5 (CH3), 122.7 (C5-pyrimidine), 124.9 (C4-thiazolidionedione), 126.9, 127.3, 128.1, 128.3, 128.9, 128.93, 129.6, 132.6, 133.2, 134.2 (naphthalene C), 134.8 (olefinic C), 157.9 (C4,6-pyrimidine), 162.7 (C=N), 173.4 (C2-pyrimidine), 179.4 (C=O); anal. calcd for C19H15N5OS: C, 63.14; H, 4.18; N, 19.38; S, 8.87; found: C, 63.59; H, 4.25; N, 19.66; S, 9.15; anal. calcd for C19H15N5OS.2C4H6O6: C, 49.02; H, 4.11; N, 10.59; S, 4.85; found: C, 49.21; H, 3.91; N, 10.61; S, 4.84.
4.2.2.5. N-(2′-Aminopyrimidin-4′-yl)-2-(2″,4″-dioxothiazolidin-5″-yl) Acetamide (19)
Compound (19) was prepared by mixing 2,4-thiazolidinedione acetic acid (18) (10 mmol, 1.77 g) with N,N’-dicyclohexylcarbodiimide (11.0 mmol, 2.26 g) in DMF (30 mL). The mixture was stirred at 0 °C for 30 min and then mixed with 2,4-diaminopyrimidine (10.0 mmol, 1.1 g) while stirring at room temperature. The progress of the reaction was monitored by TLC, and it was complete after 6.5 h. The reaction mixture was diluted with ethyl acetate (60.0 mL), resulting in a solid mass, which was subsequently filtered. The filtrate was subsequently washed with HCl (2.0 M), aqueous sodium carbonate, and brine solution. The organic layer was removed under vacuum, and the residue was purified using a column employing a mobile phase comprised of ethyl acetate and petroleum ether (7:3) to afford (19) as a light-yellow powder.
A yield of 76% was obtained for (19) with mp 209 °C; IR (KBr, cm–1): 3492, 3454 (NH2), 3344 (NH TZD), 3269 (NH-sec.amine linkage), 3088 (C–H, aromatic), 2931 (C–H aliphatic), 1714 (C=O), 1529 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.93–3.00 (m, 1H, CH2), 3.13–3.17 (m, 1H, CH2), 3.88 (t, 1H, CH- thiazolidionedione), 4.58 (d, 1H, H5-pyrimidine, J = 6.0 Hz), 5.60 (2H, NH2, exchanges with D2O), 6.93 (brs, 1H, NH, exchanges with D2O), 8.34 (d, 1H, H6-pyrimidine, J = 6.0 Hz), 11.94 (s, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 33.8 (CH2), 53.7 (CH2), 95.9 (C5-pyrimidine), 153.1 (C4-pyrimidine), 157.0 (C6-pyrimidine), 161.0 (C2-pyrimidine), 166.9 (C=O), 173.5 (C=O), 176.4 (C=O); anal. calcd for C9H9N5O3S: C, 40.45; H, 3.39; N, 26.20; S, 12.00; found: C, 40.90; H, 3.46; N, 26.48; S, 12.28; anal. calcd for C9H9N5O3S.2C4H6O6: C, 35.98; H, 3.73; N, 12.34; S, 5.65; found: C, 36.17; H, 3.53; N, 12.33; S, 5.66.
4.2.2.6. 2-(2′,4′-Dioxothiazolidin-5-yl)-N-[2″-(p-tosylureido)pyrimidin-4″-yl] Acetamide (21)
Compound (21) was obtained by mixing N-(2′-aminopyrimidin-4′-yl)-2-(2″,4″-dioxothiazolidin-5″-yl) acetamide (19) (10.0 mmol, 5.6 g) with p-toluene sulfonyl isocyanate (20) (10.0 mmol, 1.52 mL) in absolute ethanol (30 mL). The resulting mixture was stirred at room temperature for 30.0 min. The formed precipitate was filtered, dried, and crystallized using the appropriate solvent to yield (21) as a pure off-white powder product. The (21) yield was 72%; mp 202 °C; IR (KBr, cm–1): 3536 (NH TZD), 3325 (NH-sec.amine linkage), 3032 (C–H, aromatic), 928 (C–H aliphatic), 1717 (C=O), 1573 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 2.34 (s, 1H, CH3), 2.70 (m, 1H, CH2), 2.99 (m, 1H, CH2), 4.15 (m, 1H, H5-thiazolidionedione), 5.54–5.56 (d, 1H, H5-pyrimidine, J = 6.0 Hz), 5.99 (brs, 1H, NH, exchanges with D2O), 7.25 (d, 2H, aromatic, J = 8.0 Hz),7.35 (d, 2H, aromatic, J = 8.0 Hz), 7.67 (d, 1H, H6-pyrimidine, J = 6.0 Hz), 8.03 (brs, 1H, NH, exchanges with D2O), 10.20 (s, 1H, NH, exchanges with D2O), 11.90 (s, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 25.7 (CH3), 33.8 (CH2), 47.9 (CH2), 104.3 (C5-pyrimidine), 126.0, 129.7, 141.8, 142.3 (aromatic carbon), 148.1 (C4-pyrimidine), 151.1 (C=O), 156.3 (C6-pyrimidine), 157.0 (C2-pyrimidine),166.6 (C=O), 170.6 (C=O), 179.9 (C=O); anal. calcd for C17H16N6O6S2: C, 43.96; H, 3.47; N, 18.09; S, 13.81; found: C, 44.41; H, 3.54; N, 18.37; S, 14.09.
4.2.3. General Procedure for the Synthesis of (23)
-
(1).
2,4-Dichlorothiazole-5-carbaldehyde (22) was synthesized by mixing N,N-dimethylformamide (0.042 mol, 3.2 mL) with a suspension of 2,4-thiazolidinone (0.042 mol, 4.9 g) in phosphoryl chloride (0.258 mol, 24 mL). The reaction mixture was irradiated in a microwave reactor at 100 °C for 20 min. A 64% yield of (22) with mp 49 °C was obtained.
-
(2).
2-[(2′-Aminopyrimidin-4′-yl) amino]-4-chlorothiazole-5-carbaldehyde (23) was synthesized by adding piperidine (0.001 mol, 0.09 mL) to a stirred suspension of 2,4-dichlorothiazole-5-carbaldehyde (22) (0.001 mol, 0.18 g) mixed with potassium carbonate (0.001 mol, 0.14 g) in acetonitrile (5.0 mL). The mixture was stirred at room temperature for 3 h. The formed product was purified by silica gel column chromatography using hexane:dichloromethane (1:1) as an eluent to produce (23) as a brown powder. The yield was 74%; mp 310 °C; IR (KBr, cm–1): 3463, 3415 (NH2), 3344 (NH TZD), 3207 (NH-sec.amine linkage), 3058 (C–H, aromatic), 1673 (C=O), 1499 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 5.92 (d, 1H, H5-pyrimidine, J = 6.0 Hz), 7.45 (brs, 1H, NH, exchanges with D2O), 7.53–7.59 (brs, 2H, NH2, exchanges with D2O), 8.43 (d, 1H, H6-pyrimidine, J = 6.0 Hz), 9.90 (s, 1H, CHO); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 99.6 (C5-pyrimidine), 126.7 (C2′-thiazole), 136.3 (C3′-thiazole), 143.8 (C6-pyrimidine), 153.4 (C5′-thiazole), C4-pyrimidine, 160.1 (C2-pyrimidine), 161.3 (C4-pyrimidine), 183.9 (CHO); anal. calcd for C8H6ClN5OS: C, 37.58; H, 2.37; N, 27.39; S, 12.54; found: C, 38.03; H, 2.44; N, 27.67; S, 12.82.
4.2.4. General Procedure for the Synthesis of (29)
4.2.4.1. (E)-5-(p-Fluorobenzylidene)-thiazolidin-2,4-dione (25)
Compound 25 was prepared in a way similar to that of compound (3). A yellow powder was obtained with 82% yield; 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.36–7.40 (2H, m, aromatic H), 7.65–7.68 (2H, m, aromatic H), 7.80 (s, 1H, olefinic H), 9.77 (brs, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 115.4 (aromatic Cs), 115.6 (C5-thiazolidione), 131.1, 131.3 (aromatic Cs), 142.3 (olefinic C), 163.9 (C–F), 165.4 (C=O), 176.9 (C = S).
4.2.4.2. (E)-2-[5′-(p-Fluorobenzylidene)-4′-oxo-2′-thioxothiazolidin-3′-yl]-N-(4″-hydroxy-6″-methylpyrimidin-2″-yl) Acetamide (29)
A solution made by dissolving 5-(p-fluorobenzylidene)-thiazolidin-2,4-dione (24) (0.02 mol, 4.78 g) into 50.0 mL of dry toluene was cooled to 15 °C. Chloroacetyl chloride (26) (0.02 mol, 1.59 mL) was added dropwise under stirring and allowed to cool slowly to room temperature. The solution was then refluxed for 4 h. The progress of the reaction was monitored on silica gel 60 F254 precoated TLC plates by using ethyl acetate, petroleum ether, and methanol (1:1:0.3). Excess toluene was removed under reduced pressure, and the resultant solid was filtered, washed, and recrystallized from ethanol to produce (27). Without isolation, resultant compound (27) was dissolved in 50 mL of dry toluene containing freshly dried anhydrous potassium carbonate (0.0065 mol, 0.89 g) and 2-amino-6-methylpyrimidin-4-ol 28 (0.0067 mol, 0.84 g). The mixture was refluxed for 4–5 h, followed by removing the excess toluene. The resultant solid was washed with petroleum ether, dried, and recrystallized from ethanol to afford TZD # (29) as a yellow powder in 82% yield with mp 292 °C.
Spectral analyses gave IR (KBr, cm–1): 3358 (OH), 3332 (NH), 3013 (C–H, aromatic), 1674 (C=O), 1551 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.19 (s, 3H, CH3), 3.42 (s, 2H, CH2), 5.94 (s, 1H, H5-pyrimidine), 7.90–7.11 (m, 2H, aromatic), 7.19 (olefinic H), 7.43–7.46 (m, 2H, aromatic H), 7.81 (brs, 1H, NH, exchanges with D2O), 10.27 (brs, 1H, OH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 26.9 (CH3), 49.9 (CH2), 107.1 (C5-pyrimidine), 115.4 (aromatic C), 115.7 (C5-thiazolidionedione), 131.3- 131.5 (aromatic Cs), 144.9 (C2-pyrimidine), 148.7 (olefinic C), 161.4 (C4-pyrimidine), 163.9 (C6-pyrimidine), 165.4 (C–F), 166.7 (C=O), 172.8 (C=O), 180.9 (C=S); anal. calcd for C17H13FN4O3S2: C, 50.49; H, 3.24; N, 13.85; S, 15.86; found: C, 50.94; H, 3.31; N, 14.31; S, 16.14.
4.2.5. Salt Formation
Tartrate salts of compounds (19) and (29) were prepared by adding L-tartaric acid solution (0.0215 g, 1.0 equiv) dissolved in MeOH (0.2 mL) to either (19) or (29) (0.1 g, 1.0 equiv) in EtOAc (1.5 mL). The mixtures were continuously stirred for 1 h. The reaction mixture was stirred at room temperature for an additional 1 h. The obtained salts were collected by filtration and washed with EtOAc.
4.3. Synthesis of Rhodanine–Pyrimidine Derivatives
4.3.1. 2-(4′-Oxo-2′-thioxothiazolidin-3′-yl) Acetic Acid (32)
Compound (32) was prepared by dissolving rhodanine (0.02 mol, 4.78 g) in 50.0 mL of dry toluene and then cooled to 15 °C. The solution was mixed with chloroacetic acid (0.05 M; 5.2 g in 10.0 mL water) dropwise and stirred at 40 °C for 24 h. The excess toluene was removed under reduced pressure. The resultant solid was recrystallized to afford a pale yellow powder with 81% yield and 145–148 °C mp (32); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 4.35 (s, 2H, CH2, rhodanine), 4.53 (s, 2H, CH2, acetyl), 7.84 (brs, 1H, OH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 36.5 (C5-rhodanine), 45.2 (CH2, acetyl group), 167.8 (COOH), 174.2 (C=O), 172.3 (C=O), 203.2 (C=S).54
4.3.2. General Procedure for the Synthesis of (33a–g)
A mixture of pyrimidine aldehyde (31) (0.10 mmol, 1.0 equiv) and rhodanine/rhodanine acetic acid (32) (0.10 mmol, 1.0 equiv) was prepared in dry CH2Cl2 (10.0 mL). Piperidine (0.20 mmol, 0.2 mL) was added dropwise to the mixture while stirring at room temperature for 10–15 min. The resultant solid products were filtered and recrystallized from the acetone/water solvent (2:1). The following compounds were obtained:
-
(1).
(Z)-5-[2′-(Dimethylamino)pyrimidin-5′-yl-methylene]-2-thioxothiazolidin-4-one (33a), a yellow powder with 89% yield and 227–228 °C mp. Spectral analyses gave IR (KBr, cm–1): 3375 (NH), 3108 (C–H, aromatic), 2938 (aliphatic C–H), 1674 (C=O), 1599 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.15 (s, 6H, CH3), 7.43 (s, 1H, olefinic H), 8.57 (s, 2H, H4,6-pyrimidine), 11.07 (brs, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 43.8 (CH3), 115.9 (C5-pyrimidine), 118.1 (C5-rhodanine), 128.7 (olefinic C), 160.3 (C4,6-pyrimidine), 160.8 (C2-pyrimidine) 167.1 (C=O), 192.7 (C=S); anal. calcd for C10H10N4OS2: C, 45.09; H, 3.78; N, 21.04; S, 24.08; found: C, 45.56; H, 3.85; N, 21.32; S, 24.36; anal. calcd: C10H10N4OS2.CH3I: C, 32.36; H, 3.21; N, 13.72; S, 15.71; found: C, 32.55; H, 3.01; N, 13.73; S, 15.70
-
(2).
(Z)-5-[(2′-Aminopyrimidin-5′-yl)methylene]-2-thioxothiazolidin-4-one (33b), a yellow-brown powder with 87% yield and 268 °C mp. Spectral analyses gave IR (KBr, cm–1): 3513, 3480 (NH2), 3362 (NH), 3086 (C–H, aromatic), 1721 (C=O), 1583 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 7.07 (s, 2H, NH2, exchanges with D2O), 7.84 (s, 1H, olefinic H), 8.62 (s, 2H, H4,6-pyrimidine), 11.30 (brs, 1H, NH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 117.5 (C5-pyrimidine), 122.01 (C5-rhodanine),138.9 (olefinic C), 157.8 (C4,6-pyrimidine), 158.7 (C2-pyrimidine), 164.0 (C=O), 177.9 (C=S); anal. calcd for C8H6N4OS2: C, 40.32; H, 2.54; N, 23.51; S, 26.91; found: C, 40.77; H, 2.61; N, 23.78; S, 27.19; anal. calcd: C8H6N4OS2.C4H6O6: C, 37.11; H, 3.11; N, 14.43; S, 16.51; found: C, 37.30; H, 2.91; N, 14.44; S, 16.53.
-
(3).
(Z)-2-[2′-(Dimethylamino) pyrimidin-5′-yl-methylene-4-oxo-2-thioxothiazolidin-3-yl] acetic acid (33c), a yellow powder with 81% yield and 234 °C mp. Spectral analyses showed IR (KBr, cm–1): 3290 (OH), 3048 (C–H, aromatic), 2936 (aliphatic C–H), 1705 (C=O), 1409 (C=N). The 1H NMR [DMSO-d6, 400 MHz] showed (δ, ppm) 3.19 (s, 6H, CH3), 4.33 (s, 2H, CH2), 7.61 (s, 1H, olefinic H), 8.58 (s, 2H, H4,6-pyrimidine), 9.60 (brs, 1H, OH, exchanges with D2O). The 13C NMR [DMSO-d6, 100 MHz] showed (δ, ppm) 43.8 (CH3), 48.4 (CH2), 115.9 (C5-pyrimidine), 118.1 (C5-rhodanine), 128.7 (olefinic C), 160.3 (C4,6-pyrimidine), 160.8 (C2-pyrimidine) 167.1 (C=O), 167.8 (C=O, carboxylic acid), 192.7 (C=S); anal. calcd: C12H12N4O3S2: C, 44.43; H, 3.73; N, 17.27; S, 19.77; found: C, 44.88; H, 3.80; N, 17.55; S, 20.05; calcd: C12H12N4O3S2.CH3I: C, 33.48; H, 3.24; N, 12.01; S, 13.75; found: C, 33.67; H, 3.03; N, 12.03; S, 13.77.
-
(4).
(Z)-2-[5′-(2″-(Isopropylamino)pyrimidin-5″-yl)methylene-4′-oxo-2′-thioxothiazolidin-3′-yl] acetic acid (33d), a yellow powder with 86% yield and 257 °C mp. Spectral analyses gave IR (KBr, cm–1): 3442 (OH), 3242 (NH), 3080 (C–H, aromatic), 2865 (aliphatic C–H), 1716 (C=O), 1448 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 1.19 (s, 6H, CH3, isopropyl group), 4.12–4.17 (m, CH-isopropyl group), 4.39 (s, 1H, CH2, rhodanine), 7.62 (s, 1H, olefinic H), 8.13 (d, 1H, NH, J = 6.0 Hz), 8.58 (s, 2H, H4,6-pyrimidine), 11.04 (brs, 1H, OH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 22.6 (CH3, isopropyl), 43.9 (CH, isopropyl), 45.4 (CH2, rhodanine), 116.4 (C5-pyrimidine), 117.6 (C5-rhodanine), 129.1 (olefinic C), 160.3 (C4,6-pyrimidine), 160.9 (C2-pyrimidine) 167.1 (C=O), 167.2 (C=O, carboxylic acid), 192.7 (C=S). Anal. calcd: C13H14N4O3S2: C, 46.14; H, 4.17; N, 16.56; S, 18.95; found: C, 46.59; H, 4.24; N, 16.84; S, 19.23; calcd: C13H14N4O3S2.C4H6O6: C, 41.80; H, 4.13; N, 11.47; S, 13.13; found: C, 41.99; H, 3.93; N, 11.48; S, 13.15.
-
(5).
(Z)-2-[5′-(2″-Morpholinopyrimidin-5″-yl)methylene-4′-oxo-2′-thioxothiazolidin-3′-yl] acetic acid (33e), a yellow powder with 88% yield and 248 °C mp. Spectral analyses gave IR (KBr, cm–1): 3590 (OH), 3058 (C–H, aromatic), 2960 (aliphatic C–H), 1713 (C=O), 1508 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 3.64–3.65 (m, 4H, morpholine), 3.81–3.83 (m, 4H, morpholine), 4.32 (s, 2H, CH2), 7.64 (s, 1H, olefinic H), 8.63 (s, 2H, H4,6-pyrimidine), 11.78 (brs, 1H, OH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 44.5 (CH2), 48.4, 66.3 (morpholine), 116.8 (C5-pyrimidine), 119.1 (C5- rhodanine), 128.2 (olefinic C), 160.2 (C4,6-pyrimidine), 160.4 (C2-pyrimidine), 167.0 (C=O), 167.1 (C=O, carboxylic acid), 192.7 (C=S); anal. calcd: C14H14N4O4S2: C, 45.89; H, 3.85; N, 15.29; S, 17.50; found: C, 46.34; H, 3.92; N, 15.57; S, 17.78; calcd: C14H14N4O4S2.CH3I: C, 35.44; H, 3.37; N, 11.02; S, 12.62; found: C, 35.63; H, 3.18; N, 11.05; S, 12.64.
-
(6).
(Z)-2-[4′-Oxo-5′-[2″-(n-propylamino)pyrimidin-5″-yl-methylene]-2′-thioxothiazolidin-3′-yl-] acetic acid (33f), a white powder with 83% yield and 244 °C mp. Spectral analyses gave IR (KBr, cm–1): 3545 (OH), 3253 (NH), 3120 (C–H, aromatic), 2951 (aliphatic C–H), 1715 (C=O), 1450 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 0.87 (s, 3H, CH3, propyl group), 2.89 (s, 4H, 2CH2, propyl group), 4.34 (s, CH2, rhodanine), 7.57 (s, 1H, olefinic H), 8.20 (s, 1H, NH, exchanges with D2O), 8.50 (s, 2H, H4,6-pyrimidine), 9.23 (brs, 1H, OH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 11.8 (CH3, propyl), 22.7 (CH2, propyl), 43.8 (CH2, propyl), 48.3 (CH2, rhodanine), 116.5 (C5-pyrimidine), 117.8 (C5-rhodanine), 128.9 (olefinic C), 160.8 (C4,6-pyrimidine), 161.8 (C2-pyrimidine), 167.1 (C=O), 167.8 (C=O, carboxylic acid), 192.7 (C=S). Anal. calcd: C13H14N4O3S2: C, 46.14; H, 4.17; N, 16.56; S, 18.95; found: C, 46.59; H, 4.24; N, 16.84; S, 19.23; calcd: C13H14N4O3S2.C4H6O6: C, 41.80; H, 4.13; N, 11.47; S, 13.13; found: C, 41.98; H, 3.94; N, 11.48; S, 13.14.
-
(7).
(Z)-2-[5′-[2″-(Benzylamino)pyrimidin-5″-yl]methylene-4′-oxo-2′-thioxothiazolidin-3′-yl] acetic acid (33g), a yellow powder with 84% yield and 233 °C mp. Spectral analyses gave IR (KBr, cm–1): 3581 (OH), 3244 (NH), 3111 (C–H, aromatic), 2944 (aliphatic C–H), 1708 (C=O), 1524 (C=N); 1H NMR [DMSO-d6, 400 MHz]: (δ, ppm) 4.31 (s, 2H, CH2, rhodanine), 4.57 (d, 2H, CH2, benzylamine, J = 6.3 Hz), 7.20–7.29 (m, 5H, aromatic H), 7.60 (s, 1H, olefinic H), 8.58 (s, 2H, H4,6-pyrimidine), 8.70 (s, 1H, NH, exchanges with D2O), 9.13 (brs, 1H, OH, exchanges with D2O); 13C NMR [DMSO-d6, 100 MHz]: (δ, ppm) 3.9 (CH2, rhodanine), 48.5 (CH2, benzylamine), 117.1 (C5-pyrimidine), 118.4 (C5- rhodanine), 127.3, 127.5, 128.6, 128.8 (aromatic), 139.8 (olefinic C), 160.8 (C4,6-pyrimidine), 161.8 (C2-pyrimidine), 167.2 (C=O), 167.4 (C=O, carboxylic acid), 192.7 (C=S); anal. calcd: C17H14N4O3S2: C, 52.84; H, 3.65; N, 14.50; S, 16.59; found: C, 53.29; H, 3.72; N, 14.78; S, 16.87; calcd: C17H14N4O3S2.C4H6O6: C, 47.01; H, 3.76; N, 10.44; S, 11.95; found: C, 47.20; H, 3.56; N, 10.46; S, 11.97.
4.3.3. Salt Formation
Tartrate salts of synthesized rhodanines were prepared using the following two methods:
Method 1. To a solution of 33b, 33d, 33f, or 33g (0.1 g, 1.0 equiv) in EtOAc (1.5 mL), a methanolic solution of l-tartaric acid (0.0215, 1.0 equiv) (0.2 mL) was added over 1 h at room temperature with continuous stirring. Stirring was continued for an additional 1 h. Tartrate salts were collected by filtration and washed with EtOAc.
Method 2. To a solution of 33a, 33c, or 33e (4.4 mmol, 1.0 g) in acetonitrile (5.0 mL), iodomethane (4.4 mmol, 0.5 mL) was added with continuous stirring for 1 h at room temperature. Excess solvent was removed under vacuum, and solid residue was washed with diethyl ether (2 × 10 mL) to give pure tartrate salts.
4.4. Antidiabetic Activity
4.4.1. Cell Culture
The βTC6 mouse immortalized insulin-secreting pancreatic β cell line (T-SV40) was grown in Dulbecco’s Modified Eagle’s Medium (DMEM) in a 5.0% CO2 incubator at 37 °C. The medium contained 25.0 mM glucose, 1.0 mM sodium pyruvate, 4.0 mM l-glutamine, 44.0 mM sodium bicarbonate, 15.0% (v/v) fetal bovine serum (FBS), and 50.0 μg/mL gentamicin. The medium was replaced by a fresh culture medium every 48 h, and the cells were subcultured as necessary to prevent overconfluence. The cells’ passage was done by treatment with 0.25% trypsin and 0.91 mM EDTA at passages 6–8.
4.4.2. Insulin Secretion Assay
The βTC6 cells (1.0 × 105 cells/mL) were cultured for 48 h in a 24-well plate in a 5% CO2 incubator at 37 °C. The cells were then preincubated for 30 min in modified Krebs/Ringer buffer (KRB) (118.5 mM NaCl, 25 mM NaHCO3, 4.74 mM KCl, 1.19 mM MgSO4, 2.54 mM CaCl2, 10.0 mM HEPES, 1.19 mM KH2PO4, 0.1% bovine serum albumin (BSA), pH 7.4). The resultant cells were washed and incubated for another 30 min with fresh buffer.
Solutions of tested compounds (10–6–10–12 M) in 0.000004% DMSO were obtained by diluting the stock standard solutions with KRB. Each well of βTC6 cells was treated with 250.0 μL of test compounds at different concentrations and then incubated in a 5% CO2 incubator at 37 °C for 120 min in the absence and presence of 2.80 mM glucose solution.
The total reaction volume/cell was kept at 1.0 mL by adding either 750 or 500 μL of KRB first, followed by 250 μL of 4-times concentrated dose of the compound and glucose (basal group: 750 μL of KRB + 4 × 250 μL of test drug; glucose-stimulated group: 500 μL of KRB + 4 × 250 μL of glucose (2.8 mM) + 4 × 250 μL of test drug).
Incubation of resultant cells was followed by collecting the supernatant layers, which were subjected to sandwich ELISA using the high-range insulin assay kit (Mercodia, Sylveniusgatan 8A, SE-75450, Uppsala, Sweden). As per the manufacturer’s instructions, 10.0 μL of the sample was incubated with enzyme conjugate solutions on a shaker plate for 2.0 h at room temperature. The plates were then washed, and 3,3′,5,5′-tetramethylbenzidine (TMB) was added for 15.0 min. The absorbance of the resultant solution at 450 nm was measured using a Tecan microplate reader (see Strasse 103, 8708, Männedorf, Switzerland). The sensitivity of the insulin ELISA kit was 216 pmol/L. The average intra- and interday assay’s coefficients of variation were 3.37 and 2.29%, respectively. The levels of insulin were expressed as pmol/L.
4.4.3. Glucose Uptake Assay
The cellular glucose uptake by βTC6 cells was evaluated using Cayman’s Glucose Uptake cell-based assay kit. The kit uses a fluorescently labeled deoxyglucose analogue (2-[N-(7-nitrobenz-2-oxa-1,3-diaxol-4-yl) amino]-2-deoxyglucose; 2-NBDG) as a probe for detecting glucose taken up by cultured cells.
A 5 × 104 βTC6 cells/mL were seeded onto a 96-well clear, flat-bottom, black plate and allowed to adhere overnight at 37 °C and 5% CO2 in the tissue culture medium (DMEM). The medium contained 25 mM glucose, 1 mM sodium pyruvate, 4 mM l-glutamine, 44.05 mM NaHCO3, 15% (v/v) FBS, and 50 μg/mL gentamicin. The medium was then removed, the cells were washed with KRB (118.5 mM NaCl, 25 mM NaHCO3, 4.74 mM KCl, 1.19 mM MgSO4, 2.54 mM CaCl2, 10 mM HEPES, 1.19 mM KH2PO4, 0.1% BSA, pH 7.4), and the cells were conditioned at 37 °C and 5% CO2 for 30 min, two times. The conditioning buffer was replaced by 450 μM of 2-NBDG along with either a 10.0 μM test compound or a 10.0 μM reference drug in KRB. The cells were then incubated further at 37 °C, 5% CO2 for 10.0 min to allow them to endocytose the glucose analogue. At the end of the treatment, the plate was centrifuged for 5 min at 400g at room temperature. Subsequently, the 2-NBDG in the basal medium was then removed, the cells were washed with 200 μL of the cell-based assay buffer, and the plate was centrifuged for 5 min at 400g at room temperature. A 100 μL of cell-based assay buffer was added to each well after aspiration of the supernatant layer. Using the TECAN infinite M200 microplate reader, amounts of 2-NBDG taken up by cells were measured by following fluorescence emission at excitation and emission wavelengths of 485 and 535 nm, respectively.
4.5. In Silico Molecular Docking and Dynamics of TZDs and RDs onto PPAR-γ
Three PPAR-γ structures were obtained from the protein data bank: PDB: 1ZGY, PPDB: 1I7I, and PDB: 1WM0(55−59) and used in this docking study. The PPAR-γ structures were checked for any missing atoms using the protein preparation module in the molecular operating environment (MOE).60 Next, they were processed via the Protein Preparation Wizard61 in Maestro software62 to assign protonation states on ionizable groups, set up partial charges on the protein atoms, and relieve any existing clashes. The binding pocket of each PPAR-γ structure was identified by the cocrystallized ligand. Next, a grid box was created using the receptor grid generation module in Glide.63 To validate the accuracy of the above docking algorithm, a self-docking experiment was carried out against the cocrystallized ligands. RSMD values less than 1.0 Å (PDB: 1ZGY = 0.27 Å, PDB: 1I7I = 0.43 Å, and PDB: 1WM0 = 0.30 Å) were obtained.
The MOE was used to create the tested compounds (ligands) and assign the protonation state to each ionizable group.64 Ligands were then prepared using the LigPrep module65 in the Maestro program66 to assign partial charges to the ligand atoms and generate a single, low-energy conformation for each ligand using the OPLS force field.
The prepared ligands were docked into the three PPAR-γ structures using Glide software with the extra-precision (XP) Algorithm employed for conformational sampling.67 Subsequently, docked poses were scored via the Glide-XP scoring function. The docking output contained three poses for each ligand that represent its binding mode with each PPAR-γ structure. The ligand pose with the best score was selected for the final docking list. All PDB data are provided in the Supporting Information, along with their docking scores in Table S1.
To better understand the ligand′s binding affinity toward the target pocket, MD simulations were conducted using the AMBER18 package.68 Partial charges were assigned as per the ff19SB force field. The leap module of AmberTools was employed to prepare the solvated system of each protein–ligand complex with a truncated octahedral box of TIP3P water. Afterward, energy was minimized in a two-step process using the PMEMD program in the AMBER18 package.68 Initially, all solute atoms were restrained by 500 kcal mol–1 force constant during the first minimization, and then the entire system was subjected to minimization without any constraints. The system was gradually heated from 0.0 to 300.0 K using the NVT ensemble. The SHAKE algorithm was applied to all hydrogen atoms that contain bonds with a collision frequency of 1.0–1.66 ps using the Langevin thermostat. Each protein–ligand complex was then run for 100 ns in the NPT ensemble with the system temperature set to 300 K and pressure set to 1.01 × 105 Pa.
4.6. Statistical Analysis
Statistical analyses were performed using SPSS-20 software. The effects of test compounds compared to the control were evaluated using the Student’s t-test. The results were presented as mean ± SEM.
Acknowledgments
The financial support of the UAE University research affairs under Grant Numbers 31S030 and G00001322 is greatly acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c07149.
Author Present Address
∥ Department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, Khalifa University, 127788 Abu Dhabi, United Arab Emirates
The authors declare no competing financial interest.
Supplementary Material
References
- Pattan S. R.; Kekare P.; Dighe N. S.; Nirmal S. A.; Musmade D. S.; Parjane S. K.; Daithankar A. V. Synthesis, and Biological Evaluation of Some 1, 3, 4-Thiadiazoles. J. Chem. Pharm. Res. 2009, 1, 191–198. [Google Scholar]
- Goyal R.; Jialal I.. Diabetes Mellitus Type 2; StatPearls Publishing: Treasure Island, FL, 2023. [Google Scholar]
- International Diabetes Federation. IDF Diabetes Atlas, 10th edn. Brussels, Belgium: 2021. Available at: www.diabetesatlas.org.
- Kamar A. K. D. A.; Yin L. J.; Liang C. T.; Fung G. T.; Avupati V. R. Rhodanine Scaffold: A review of Antidiabetic Potential and Structure Activity Relationships (SAR). Med. Drug Discovery 2022, 15, 100131 10.1016/j.medidd.2022.100131. [DOI] [Google Scholar]
- Tripathi A. C.; Gupta S. J.; Fatima G. N.; Sonar P. K.; Verma A.; Saraf S. K. 4- Thiazolidinones: The Advances Continue. Eur. J. Med. Chem. 2014, 72, 52–77. 10.1016/j.ejmech.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Abhinit M.; Ghodke M.; Pratima N. A. Exploring Potential of 4-Thiazolidinone: A Brief Review. Int. J. Pharm. Sci. 2009, 1, 47–64. [Google Scholar]
- Tomašić T.; Peterlin M. L. Rhodanine as a Scaffold in Drug Discovery: A Critical Review of its Biological Activities and Mechanisms of Target Modulation. Expert Opin. Drug Discovery 2012, 7, 549–560. 10.1517/17460441.2012.688743. [DOI] [PubMed] [Google Scholar]
- Kaminskyy D.; Kryshchyshyn A.; Lesyk R. Recent Developments with Rhodanine as a Scaffold for Drug Discovery. Expert Opin. Drug Discovery 2017, 12, 1233–1252. 10.1080/17460441.2017.1388370. [DOI] [PubMed] [Google Scholar]
- Singh S. J.; Chauhan S. M. S. Potassium Carbonate Catalyzed One Pot Four-Component Synthesis of Rhodanine Derivatives. Tetrahedron Lett. 2013, 54, 2484–2488. 10.1016/j.tetlet.2013.03.004. [DOI] [Google Scholar]
- Nitsche C.; Klein C. D. Aqueous Microwave-Assisted One-Pot Synthesis of N-substituted Rhodanines. Tetrahedron Lett. 2012, 53, 5197–5201. 10.1016/j.tetlet.2012.07.002. [DOI] [Google Scholar]
- Joshi H.; Pal T.; Ramaa C. S. A New Dawn for the Use of Thiazolidinediones in Cancer Therapy. Expert Opin. Invest. Drugs 2014, 23, 501–510. 10.1517/13543784.2014.884708. [DOI] [PubMed] [Google Scholar]
- Raveendran A. V.; Fernandez C. J.; Jacob K. Efficacy and Cardiovascular Safety of Thiazolidinediones. Curr. Drug Saf. 2021, 16, 233–249. 10.2174/1574886315666201026125530. [DOI] [PubMed] [Google Scholar]
- Rashid M.; Shrivastava N.; Husain A. Synthesis and SAR Strategy of Thiazolidinedione: A Novel Approach for Cancer Treatment. J. Chil. Chem. Soc. 2020, 65, 4817–4832. 10.4067/S0717-97072020000204817. [DOI] [Google Scholar]
- Day C. T. Thiazolidinediones: A New Class of Antidiabetic Drugs. Diabetic Med. 1999, 16, 179–192. 10.1046/j.1464-5491.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- Yau H.; Rivera K.; Lomonaco R.; Cusi K. The Future of Thiazolidinedione Therapy in the Management of Type 2 Diabetes Mellitus. Curr. Diabetes Rep. 2013, 13 (3), 329–341. 10.1007/s11892-013-0378-8. [DOI] [PubMed] [Google Scholar]
- Musso G.; Cassader M.; Paschetta E.; Gambino R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177 (5), 633–640. 10.1001/jamainternmed.2016.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Liu X.; Wang L.; Yang Z. Thiazolidinediones for Nonalcoholic Steatohepatitis: A Meta Analysis of Randomized Clinical Trials. Medicine 2016, 95 (42), e4947 10.1097/MD.0000000000004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanouchi T. Concomitant Therapy with Pioglitazone and Insulin for the Treatment of Type 2 Diabetes. Vasc. Health Risk Manage. 2010, 6, 189–197. 10.2147/VHRM.S5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liver Tox: Clinical and Research Information on Drug-Induced Liver Injury National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD; 2017.
- Vieira R.; Souto S. B.; Sánchez-López E.; Machado A. L.; Severino P.; Jose S.; Santini A.; Fortuna A.; García M. L.; Silva A. M.; Souto E. B. Sugar Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome - Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12 (4), 152 10.3390/ph12040152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gor D.; Gerber B. S.; Walton S. M.; Lee T. A.; Nutescu E. A.; Touchette D. R. Antidiabetic Drug Use Trends in Patients with Type 2 Diabetes Mellitus and Chronic Kidney disease: A cross-sectional analysis of the National Health and Nutrition Examination Survey. J. Diabetes 2020, 12 (5), 385–395. 10.1111/1753-0407.13003. [DOI] [PubMed] [Google Scholar]
- Tyagi S.; Gupta P.; Saini A. S.; Kaushal C.; Sharma S. The Peroxisome Proliferator-Activated Receptor: A Family of Nuclear Receptors Role in Various Diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Haluzik M.; Gavrilova O.; Yakar S.; Portas J.; Sun H.; Pajvani U. B.; Scherer P. E.; Le Roith D. Thiazolidinediones Improve Insulin Sensitivity in Adipose Tissue and Reduce the Hyperlipidemia Without Affecting the Hyperglycaemia in A Transgenic Model of Type 2 Diabetes. Diabetologia 2004, 47, 2215–2225. 10.1007/s00125-004-1581-6. [DOI] [PubMed] [Google Scholar]
- Kahn S. E.; Haffner S. M.; Heise M. A.; Herman W. H.; Holman R. R.; Jones N. P.; Kravitz B. G.; Lachin J. M.; Neill M. C. O.; Zinman B.; Viberti G. Glycemic Durability of Rosiglitazone, Metformin, or Glyburide Monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wu Y.; Piao H.; Zhao X.; Zhang W.; Wang Y.; Liu M. A Comprehensive Review on the Biological and Pharmacological Activities of Rhodanine Based Compounds for Research and Development of Drugs. Mini-Rev. Med. Chem. 2018, 18, 948–961. 10.2174/1389557516666160928162724. [DOI] [PubMed] [Google Scholar]
- Cheng A. Y. Y.; Fantus I. G. Oral Antihyperglycemic Therapy for Type 2 Diabetes Mellitus. Can. Med. Assoc. J. 2005, 172 (2), 213–226. 10.1503/cmaj.1031414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S.; Fantus I. G. Inhibition of The Protein Tyrosine Phosphatase PTP1B: Potential Therapy for Obesity, Insulin Resistance and Type-2 Diabetes Mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. 10.1016/j.beem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Kim H. I.; Ahn Y. H. Role of Peroxisome Proliferator-Activated Receptor-γ in the Glucose-Sensing Apparatus of Liver and β-Cells. Diabetes 2004, 53, S60–S65. 10.2337/diabetes.53.2007.S60. [DOI] [PubMed] [Google Scholar]
- Janani C.; Ranjitha Kumari B. D. PPAR Gamma Gene–A Review. Diabetes Metab. Syndrome 2015, 9, 46–50. 10.1016/j.dsx.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Ramya R.; Dhamotharan R. In-vitro and In-vivo Animal Model for Screening Antidiabetic Activity of Hellenia Speciosa (j. koenig) S. R. Dutta. Int. J. Pharm. Sci. Res. 2019, 26, 5016–5024. [Google Scholar]
- National Center for Biotechnology Information . PubChem Patent Summary for US-10689374-B1, Pyrimidine-Thiazolidinone Derivatives, 2021.
- Nanjan M. J.; Praveen T. K.; Suresh B.; Vergheese J.; Desai B. J.; Kumar B. R. P. CoMFA Study on Thiazolidine-2, 4-dones for their Antihyperglycemic Activity. Lett. Drug Des. Discovery 2008, 5, 79–87. 10.2174/157018008783928409. [DOI] [Google Scholar]
- Kumar B. R. P.; Nanjan M. J.; Suresh B.; Karvekar M. D.; Adhikary L. Microwave Induced Synthesis of the Thiazolidine-2,4-dione Motif and the Efficient Solvent Free-Solid Phase Parallel Syntheses of 5-Benzylidene-Thiazolidine-2,4-dione and 5-Benzylidene-2-Thioxo-Thiazolidine-4-one Compounds. J. Heterocycl. Chem. 2006, 43, 897–903. 10.1002/jhet.5570430413. [DOI] [Google Scholar]
- Sharma V.; Chitranshi N.; Kumar Agarwal A. Significance and Biological Importance of Pyrimidine in the Microbial World. Int. J. Med. Chem. 2014, 2014, 202784 10.1155/2014/202784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kołaczek A.; Fusiarz C.; Ławecka J.; Branowska D. Biological Activity and Synthesis of Sulfonamide Derivatives: A Brief Review. Chemik 2014, 68, 620–628. [Google Scholar]
- Ottanà R.; Maccari R.; Barreca M. L.; Bruno G.; Rotondo A.; Rossi A.; Chiricosta A.; Di-Paola R.; Sautebin L.; Cuzzocrea S.; Vigorita M. G. 5-Arylidene-2-imino-4-thiazolidinones: Design and Synthesis of Novel Anti-inflammatory Agents. Bioorg. Med. Chem. 2005, 13, 4243–4252. 10.1016/j.bmc.2005.04.058. [DOI] [PubMed] [Google Scholar]
- Kasmi S.; Hamelin J.; Benhaoua H. Microwave-Assisted Solvent Free Synthesis of Imino-thiazolines. Tetrahedron Lett. 1998, 39, 8093–8096. 10.1016/S0040-4039(98)01836-X. [DOI] [Google Scholar]
- Jawale D. W.; Pratap U. R.; Netankar P.; Mane R. A. Synthesis of New Acetamides Encompassing Thiazolidinedione and Pyrimidines. Chem.–Biol. Interface 2012, 2, 420–425. [Google Scholar]
- Jain A. K.; Vaidya A.; Ravichandran V.; Kashaw S. K.; Agrawal R. K. Recent Developments, and Biological Activities of Thiazolidinone Derivatives: A Review. Bioorg. Med. Chem. 2012, 20, 3378–3395. 10.1016/j.bmc.2012.03.069. [DOI] [PubMed] [Google Scholar]
- Sing W. T.; Lee C. L.; Yeo S. L.; Lim S. P.; Sim M. M. Arylalkylidene Rhodanine with Bulky and Hydrophobic Functional Group as Selective HCV NS3 protease inhibitor. Bioorg. Med. Chem. Lett. 2001, 11, 91–94. 10.1016/S0960-894X(00)00610-7. [DOI] [PubMed] [Google Scholar]
- Khodair A. I. A Convenient Synthesis of 2-Arylidene-5H-thiazolo[2,3-b] quinazoline-3,5[2H]-diones and their Benzoquinazoline Derivatives. J. Heterocycl. Chem. 2002, 39, 1153–1160. 10.1002/jhet.5570390607. [DOI] [Google Scholar]
- Ohishi Y.; Mukai T.; Nagahara M.; Yajima M.; Kajikawa N.; Miahara K.; Takano T. Preparations of 5-alkylmethylidene-3-carboxymethyl Rhodanine Derivatives and their Aldose Reductase Inhibitory Activity. Chem. Pharm. Bull. 1990, 38, 1911–1919. 10.1248/cpb.38.1911. [DOI] [PubMed] [Google Scholar]
- Hédou D.; Guillon R.; Lecointe C.; Loge C.; Chosson E.; Besson T. Novel Synthesis of Angular Thiazolo[5,4-f] and [4,5-h] quinazolines, Preparation of their Linear Thiazolo[4,5-g] and [5,4-g] Quinazoline Analogs. Tetrahedron 2013, 69, 3182–3191. 10.1016/j.tet.2013.02.066. [DOI] [Google Scholar]
- Sánchez A. I.; Martinez-Barrasa V.; Burgos C.; Vaquero J. J.; Alvarez-Builla J.; Terricabras E.; Segarra V. Synthesis and Evaluation of Quinazoline Derivatives as Phosphodiesterase 7 Inhibitors. Bioorg. Med. Chem. 2013, 21, 2370–2378. 10.1016/j.bmc.2013.01.067. [DOI] [PubMed] [Google Scholar]
- Walpole C.; Liu Z.; Lee E. E.; Yang H.; Zhou R.; Mackintosh N.; Sjogren M.; Taylor D.; Shen J.; Batey R. A. Diastereoselective Synthesis of Fluorinated Piperidine Quinazoline Spirocycles as iNOS Selective Inhibitors. Tetrahedron Lett. 2012, 53, 2942–2947. 10.1016/j.tetlet.2012.03.050. [DOI] [Google Scholar]
- Abbott P. A.; Bonnert R. V.; Caffrey M. V.; Cage P. A.; Cooke A. J.; Donald D. K.; Furber M.; Hill S.; Withnall J. Fused Mesoionic Heterocycles: Synthesis of [1,2,3]triazolo[1,5-a]quinoline, [1,2,3]triazolo[1,5-a]quinazoline, [1,2,3]triazolo[1,5-a]quinoxaline and [1,2,3]triazolo[5,1-c]benzotriazine Derivatives. Tetrahedron 2002, 58, 3185–3198. 10.1016/S0040-4020(02)00269-7. [DOI] [Google Scholar]
- Harayama T.; Hori A.; Serban G.; Morikami Y.; Matsumoto T.; Abe H.; Takeuchi Y. Concise Synthesis of Quinazoline Alkaloids, Luotonins A and B, and Rutaecarpine. Tetrahedron 2004, 60, 10645–10649. 10.1016/j.tet.2004.09.016. [DOI] [Google Scholar]
- Cleves A. E.; Jain A. N. Knowledge-Guided Docking: Accurate Prospective Prediction of Bound Configurations of Novel Ligands using Surflex-Dock. J. Comput.-Aided Mol. Des. 2015, 29, 485–509. 10.1007/s10822-015-9846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T.; Fairall L.; Amin K.; Inaba Y.; Szanto A.; Balint B. L.; Nagy L.; Yamamoto K.; Schwabe J. W. R. Structural Basis for the Activation of PPARÎ3 by Oxidized Fatty Acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan H. J.; Wang X.; Nogle S.; Liao S.; Wu C. To Probe Full and Partial Activation of Human Peroxisome Proliferator-Activated Receptors by Pan-Agonist Chiglitazar using Molecular Dynamics Simulations. PPAR Res. 2020, 2020, 5314187 10.1155/2020/5314187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östberg T.; Svensson S.; Selen G.; Uppenberg J.; Thor M.; Sundbom M.; Sydow-Bäckman M.; Gustavsson A. L.; Jendeberg L. A New Class of Peroxisome Proliferator-Activated Receptor Agonists with a Novel Binding Epitope Shows Antidiabetic Effects. J. Biol. Chem. 2004, 279, 41124–41130. 10.1074/jbc.M401552200. [DOI] [PubMed] [Google Scholar]
- Salam N. K.; Huang T. H. W.; Kota B. P.; Kim M. S.; Li Y.; Hibbs D. E. Novel PPAR-Gamma Agonists Identified From a Natural Product Library: A Virtual Screening, Induced-Fit Docking and Biological Assay Study. Chem. Biol. Drug Des. 2008, 71, 57–70. 10.1111/j.1747-0285.2007.00606.x. [DOI] [PubMed] [Google Scholar]
- Srikanth Kumar K.; Lakshmana A. R.; Basaveswara Rao M. V. Design, Synthesis, Biological Evaluation and Molecular Docking Studies of Novel 3-Substituted-5-[indol-3-yl) methylene]-thiazolidine-2,4-dione Derivatives. Heliyon 2018, 4, e00807 10.1016/j.heliyon.2018.e00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapalli N. R.; Karumudi B. S.; Rangisetti S. K.; Oleti S. M.; Naga S. R.; Rama R. N. Synthesis, Characterization and Antimicrobial Evaluation of some Azole Derivatives. World J. Pharm. Pharm. Sci. 2015, 4, 865–875. [Google Scholar]
- RCSB . Protein Data Bank 2015http://www.pdb.org/.
- Li Y.; Mihwa C.; Suino K.; Kovach A.; Daugherty J.; Kliewer S. A.; Xu H. E. Structural and Biochemical Basis for Selective Repression of the Orphan Nuclear Receptor Liver Receptor Homolog 1 by Small Heterodimer Partner. Proc. Natl. Acad. Sci. U.S.A. 2005, 27, 9505–9510. 10.1073/pnas.0501204102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronet P.; Petersen J. F.; Folmer R.; Blomberg N.; Sjoblom K.; Karlsson U.; Lindstedt E. L.; Bamberg K. Structure of the PPARg and -Ligand Binding Domain in Complex with AZ 242; Ligand Selectivity and Agonist Activation in the PPAR Family. Structure 2001, 9 (8), 699–706. 10.1016/S0969-2126(01)00634-7. [DOI] [PubMed] [Google Scholar]
- Östberg T.; Svensson S.; Selén S. G.; Selén G.; Uppenberg J.; Uppenberg J.; Thor M.; Thor M.; Sundbom M.; Sundbom M.; Sydow-Bäckman M.; Sydow-Bäckman M.; Gustavsson A. L.; Gustavsson A. L.; Jendeberg L. A New Class of Peroxisome Proliferator-Activated Receptor Agonists with a Novel Binding Epitope Shows Antidiabetic Effects. J. Biol. Chem. 2004, 279, 41124–41130. 10.1074/jbc.M401552200. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC: Montreal, Canada, 2023http://www.chemcomp.com.
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Maestro 9.9.013; Schrodinger LLC: Portland, OR, 2015http://www.schrodinger.com/.
- Small-Molecule Drug Discovery Suite 2015-2: Glide, v; Schrödinger, LLC: New York, NY, 2015.
- MOE, Molecular Operating Environment; Chemical Computing Group: Montreal, Canada, 2015http://www.chemcomp.com.
- Xu M.; Ke X.; Shaozhong W.; Zhu-Jun Y. Assembly of Indolo[1,2-c] Quinazolines using ZnBr2- Promoted Domino Hydroamination–Cyclization. Tetrahedron Lett. 2013, 54, 4675–4678. 10.1016/j.tetlet.2013.06.079. [DOI] [Google Scholar]
- Shi F.; Youquan D.; Tianlong S.; Hongzhou Y. A novel ZrO2 – SO42– Supported Palladium Catalyst for Syntheses of Disubstituted Ureas from Amines by Oxidative Carbonylation. Tetrahedron Lett. 2001, 42, 2161–2163. 10.1016/S0040-4039(01)00124-1. [DOI] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Yu B.; Tang L. D.; Li Y. L.; Song S. H.; Ji X. L.; Lin M. S.; Wu C. F. Design, Synthesis, and Antitumor Activity of 4-Aminoquinazoline Derivatives Targeting VEGFR-2 Tyrosine Kinase. Bioorg. Med. Chem. Lett. 2012, 22, 110–114. 10.1016/j.bmcl.2011.11.061. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graphics Model. 2006, 25, 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.