

Abstract
Recent advances in single cell transcriptomics illuminate the diverse neuronal and glial cell types within the human brain. However, the regulatory programs governing cell identity and function remain unclear. Using single nucleus ATAC-seq analysis, we explore open chromatin landscapes across 1.1 million cells in 42 brain regions from three adults. Integrating this data unveils 107 distinct cell types and their specific utilization of 544,735 candidate cis-regulatory DNA elements (cCREs) in the human genome. Nearly 1/3 of the cCREs demonstrate conservation and chromatin accessibility in the mouse brain cells. We reveal strong links between specific brain cell types and neuropsychiatric disorders including schizophrenia, bipolar disorder, Alzheimer’s disease, and major depression, and develop deep learning models to predict regulatory roles of non-coding risk variants in these disorders.
Neuropsychiatric disorders and mental illnesses are the leading cause of disease burden in the United States(1). Tens of thousands of sequence variants in the human genome have been linked to the etiology of neuropsychiatric disorders(2, 3). However, interpreting the mode of action of the identified risk variants remains a daunting challenge, since the vast majority of them are non-protein-coding and remain to be annotated (4, 5). A large fraction of the non-coding risk variants might contribute to disease etiology by perturbing transcriptional regulatory sequences and target gene expression in disease-relevant cell types(6, 7). However, a lack of maps and tools to explore gene activities and their transcriptional regulatory sequences at high cellular and anatomical resolution in the brain presents a major barrier to obtaining a clearer mechanistic understanding of the broad spectrum of neuropsychiatric disorders.
The human brain is made up of hundreds of billions of neurons, which through trillions of synapses form a complex neurocircuitry to carry out diverse neurocognitive functions. The functionality of the neural circuitry is supported and maintained by an even greater number of glial cells including astrocytes, oligodendrocytes, oligodendrocyte precursor cells, and microglia, among others. Single-cell RNA-seq and high throughput imaging experiments have produced detailed cell taxonomies for mouse and human brains (8–11), leading to a comprehensive view of cell types and their molecular signatures in many brain areas(12–14). Analyses of gene expression patterns using single-cell transcriptomics and spatial transcriptomics assays (8, 10, 15–17), have further advanced our understanding of the transcriptional landscapes in different brain cell types. In comparison, analysis of the regulatory elements that drive the cell-type specific expression of genes is lagging. Current catalogs of candidate regulatory sequences in the human genome, most notably those generated by ENCODE and Epigenome Roadmap consortia (6, 7, 18, 19), still lack the information about cell-type-specific activities of each element especially those identified from brain tissues, because conventional assays performed using bulk tissue samples, unfortunately, fail to resolve cCREs in individual cell types comprising the heterogeneous tissues. Recent technological advances have enabled the analysis of open chromatin at single cell resolution (20–23) in adult mouse tissues (20, 22, 24), generating cell-type-specific maps of gene regulatory elements for a limited number of human brain cell types and brain regions (25, 26).
As part of the BRAIN Initiative Cell Census Network (BICCN), we have carried out single-cell profiling of transcriptome, chromatin accessibility, and DNA methylome across >40 regions in the human brain from multiple neurotypical adult donors. Here, we generated a single-cell chromatin accessibility atlas comprising ~1.1 million human brain cells. We used this chromatin atlas to define 107 distinct cell types, and uncover the state of chromatin accessibility at 544,735 cCREs in one or more of these brain cell types. We found nearly 1/3 of the cCREs demonstrate conservation and chromatin accessibility in the mouse brain cells. We integrated our chromatin atlas with single cell transcriptome and DNA methylome atlases to link cCREs to putative target genes. We further predicted disease relevant cell types for 19 neuropsychiatric disorders. Finally, we developed machine learning models to predict the regulatory function of disease risk variants and we created an interactive web atlas to disseminate this resource (cis-element ATLAS [CATLAS]; http://catlas.org).
A single-cell chromatin accessibility atlas of human brains
We dissected 42 brain regions from the human cortex (CTX), hippocampus (HIP), basal nuclei (BN), midbrain (MB), thalamus (THM), cerebellum (CB), and pons (PN) (according to the Allen Brain Reference Atlas (27)) from three neurotypical male donors (D1, D2, and D4), age 29, 42, and 58, respectively (Fig. 1A, Table S1). For each brain sample, we performed Single nucleus assay for transposase-accessible chromatin using sequencing (snATAC-seq) using a protocol described previously (28) (Fig. 1A, Fig. S1A–D, Table S2). Data reliability was confirmed by sequencing reads showing nucleosome-like periodicity (Fig. S1E), excellent correlation between datasets from similar brain regions (Fig. S1F), high enrichments of reads near transcription start sites (TSS), and other quality control metrics (see Methods). A total of 1,290,974 nuclei passed a set of quality control thresholds (Fig. S1G, see Methods). After removing an additional 156,614 snATAC-seq profiles that likely resulted from potential barcode collision or doublets (Fig. S1H–J, see Methods), a total of 1,134,360 nuclei were retained. Among them, 595,713 were from CTX, 72,190 from HIP, 317,480 from BN, 23,114 from MB, 50,768 from THM, 51,775 from CB and 25,459 from PN (Table S3). On average 4,970 chromatin fragments were detected in each nucleus (Table S3, Fig. S1K–M, see Methods).
Fig. 1. Single-cell analysis of chromatin accessibility in the human brain.
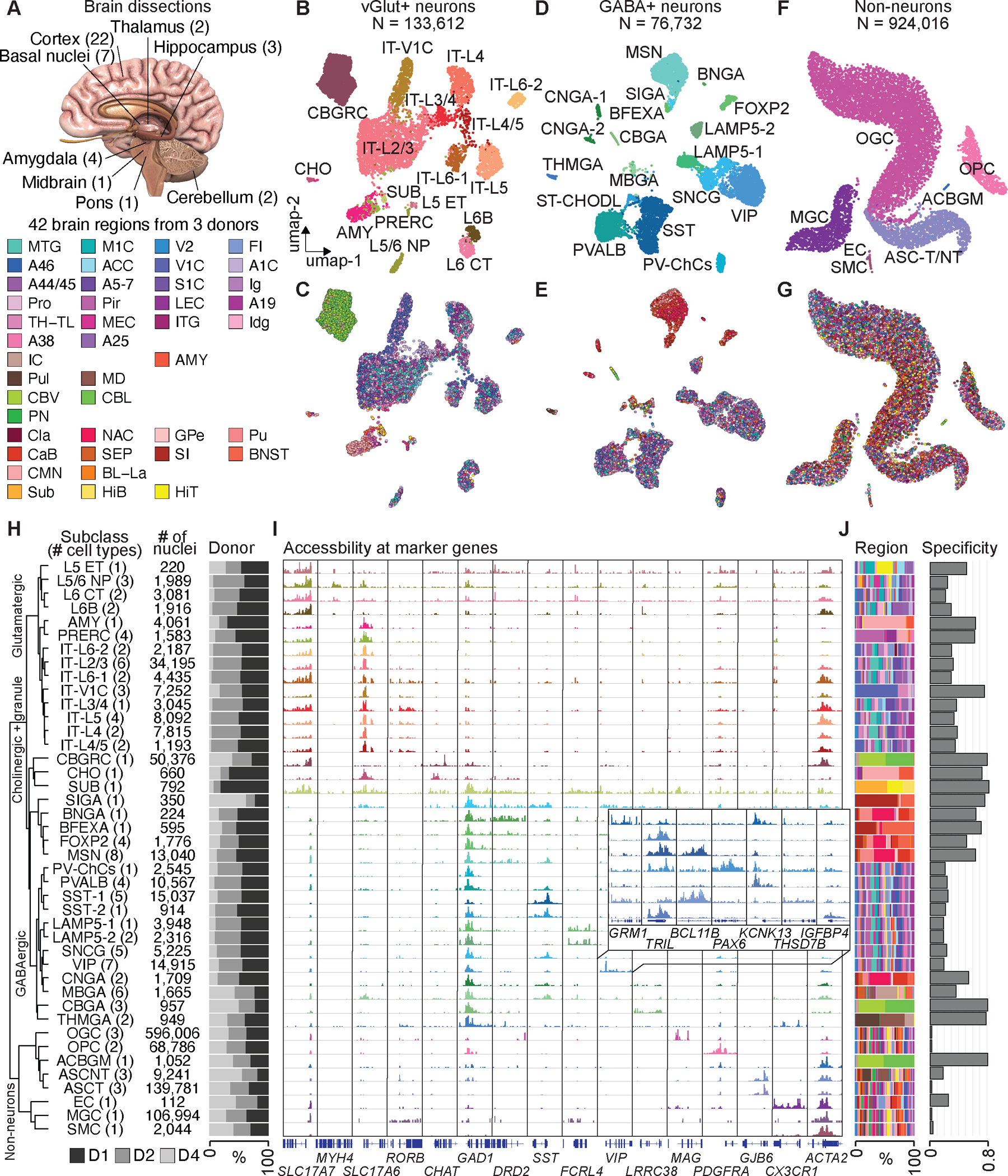
(A) Schematic of sampling strategy and 42 brain dissections. A detailed list of regions is provided in Table S1. (B) Uniform manifold approximation and projection (UMAP) embedding and clustering analysis of glutamatergic neurons from snATAC-seq data. Individual nuclei are colored and labelled by cell subclasses. A full list and description of cell subclass labels are provided in Tables S3. (C) UMAP embedding of glutamatergic neurons, colored by brain regions. (D) UMAP embedding and clustering analysis of GABAergic neurons, colored by cell subclasses. (E) UMAP embedding of GABAergic neurons, colored by brain regions. (F) UMAP embedding and clustering analysis of non- neurons, colored by cell subclasses. (G) UMAP embedding of non-neurons, colored by brain regions. (H) Left, hierarchical organization of 42 cell subclasses on chromatin accessibility. Middle, the number of nuclei in each subclass. Right, Bar chart representing the relative contribution of 3 donors to each cell subclass. (I) Genome browser tracks of aggregate chromatin accessibility profiles for each subclass at selected marker gene loci that were used for cell cluster annotation. A full list and description of subclass annotations are in Table S4. (J) Left, bar chart representing the relative contribution of brain regions to cell subclasses. Right, regional specificity scores of cell subclasses.
We next carried out iterative clustering with snATAC-seq profiles and classified them into three major classes, with class I enriched for glutamatergic (vGlut+, putatively excitatory) neurons (11.8%), class II enriched for GABAergic (GABA+, putatively inhibitory) neurons (6.8%) and class III enriched for non-neuronal cells (81.4%) (Fig. 1B, D, F, Fig. S2, Fig. S3A–B). Iterative clustering further classified the three major classes into 14 sub-classes of vGlut+ neurons, 2 sub-classes of granule cell types, 1 sub-class of cholinergic neurons, 4 sub-classes of dopaminergic neurons, 2 sub-classes of thalamic and midbrain derived neurons, 11 sub-classes of cortical GABA+ neurons, and 8 sub-classes of non-neuronal cells (Fig 1B, D, F). Each sub-class was annotated based on chromatin accessibility at promoters and gene bodies of at least three marker genes of known brain cell types, together with the brain region where the cells reside (Fig. 1C, E, G, Fig. S3C, Table S4, and S5). For each sub-class, we also conducted a third round of clustering and identified a union list of 107 distinct cell types (Fig. 1H, Fig. S4, Table S3, see Methods). To determine the optimal number of cell types within each sub-class, we evaluated the relative stability from a consensus matrix based on 100 rounds of clustering with randomized starting seeds. We then calculated the proportion of ambiguous clustering (PAC) score and dispersion coefficient (DC) to find the optimal resolution (local minimum and maximum) for cell type clustering (Fig. S2B–E). For example, vasoactive intestinal peptide-expressing (VIP) neurons were further divided into multiple cell types with distinct chromatin accessibility at multiple gene loci (Fig. 1I, Fig. S2B–E, Fig. S4). We found that the clustering result of snATAC-seq was robust to variation of sequencing depth, and signal-to-noise ratios, and most cell sub-classes showed no batch effect from at least two biological replicates using local inverse Simpson’s index (LISI) analysis, with the exception of two cell sub-classes (SUB, granule cells from subiculum, SMC, vascular smooth muscle cells) that were mostly captured from one donor (Fig. S5). To capture the relative similarity in chromatin landscapes among the 42 sub-classes we constructed a robust hierarchical dendrogram showing known organizing principles of human brain cells: the non-neuronal class is separated from the neuronal class, which were further separated based on neurotransmitter types (GABA+, dopaminergic, cholinergic, and vGlut_+) and developmental origins (Fig. 1H, Fig. S6, see Methods).
As expected, most neuronal cell types and some glial cell types were distributed in the human brain in a non-uniform fashion (Fig. 1J). We defined a regional specificity score for each sub-class based on the contribution from different brain regions. Although the majority of glial cell types were ubiquitously distributed throughout the brain, showing very low regional specificity (Fig. 1J, right), there were exceptions. For example, Bergmann glia (ACBGM), also called Golgi epithelial cells, were specialized, unipolar glial cells featuring cell bodies situated in the Purkinje cell layer and radial fibers in the cerebellum (29). This cell type was specifically found in the cerebellum (Fig. 1H, J). On the other hand, most neuronal types were characterized by regional specificity (Fig. 1J, right). We found a stark separation based on brain sub-regions for distinct neuron types including the granular cells in the cerebellum (CBGRC) and medium spiny neurons (MSN) in the basal ganglia. For vGlut+ neurons, we also observed distinct types of intra-telencephalic (IT) cortical neurons in the primary visual cortex (V1C) (IT-V1C), and excitatory neurons from the amygdala (AMY) to be highly restricted to specific brain regions or dissections. We also compared our cell clusters with the cell taxonomy defined from other modalities and attained a remarkable level of agreement. (Fig. S7, see Supplementary Text).
Mapping and characterization of human brain cCREs
As a first step towards defining the gene regulatory programs that underlie the identity and function of each brain cell type, we identified the open chromatin and cCREs in each of the 107 brain cell types. We aggregated the chromatin accessibility profiles from the nuclei comprising each cell cluster/type and identified the open chromatin regions with MACS2 (30)(Fig. S8A). We filtered the resulting accessible chromatin regions based on whether they were called in at least two donors, or pseudo-replicates (Fig. S8A–B). From our previous study, we found that read depth or cluster size can affect MACS2 peak calling scores(28). We used “score per million” (SPM)(31) to correct this bias (Fig. S8C, see Methods). About 1000 nuclei were sufficient to identify over 80% of the accessible regions in a cell type, consistent with our previous finding(28) (Fig. S8C). We iteratively merged the open chromatin regions identified from every cell type, and kept the summits with the highest MACS2 score for overlapped regions. On average, we detected 62,045 open chromatin regions per cell type (each 500 bp in length), and a union of 544,735 open chromatin regions across all 107 cell types (Fig. S8D, Table S6, and S7, see Methods). These cCREs together made up 8.8% of the human genome (hg38) (Table S8). Of these cCREs, 95.3% were located at least 2 kbp away from annotated promoter regions of protein-coding and lncRNA genes (Fig. 2A, Table S8). The promoter-distal cCREs were distributed in introns (34.8%), intergenic (27.8%), and other genomic regions. Of note, 22% of them overlap with endogenous retrotransposable elements, including the long terminal repeats classes (LTR, 6.8%), LINEs (long interspersed nuclear elements, 11.3%), SINEs (short interspersed nuclear elements, 3.9%). Several lines of evidence support the authenticity of the identified cCREs. First, both proximal and distal cCREs showed higher sequence conservation than random genomic regions with similar GC content (Fig. 2B). Second, 89.6% of cCREs overlapped with DNase hypersensitive sites (DHS) previously mapped in a broad spectrum of bulk human tissues and cell types including fetal and adult brains (32). This list further expands candidate CREs previously annotated in the human genome by the ENCODE(19) and a recent survey of chromatin accessibility in single nuclei across fetal and adult human tissues (33)(Fig. 2C).
Fig. 2: Identification and characterization of candidate CREs (cCREs) across human brain cell types.
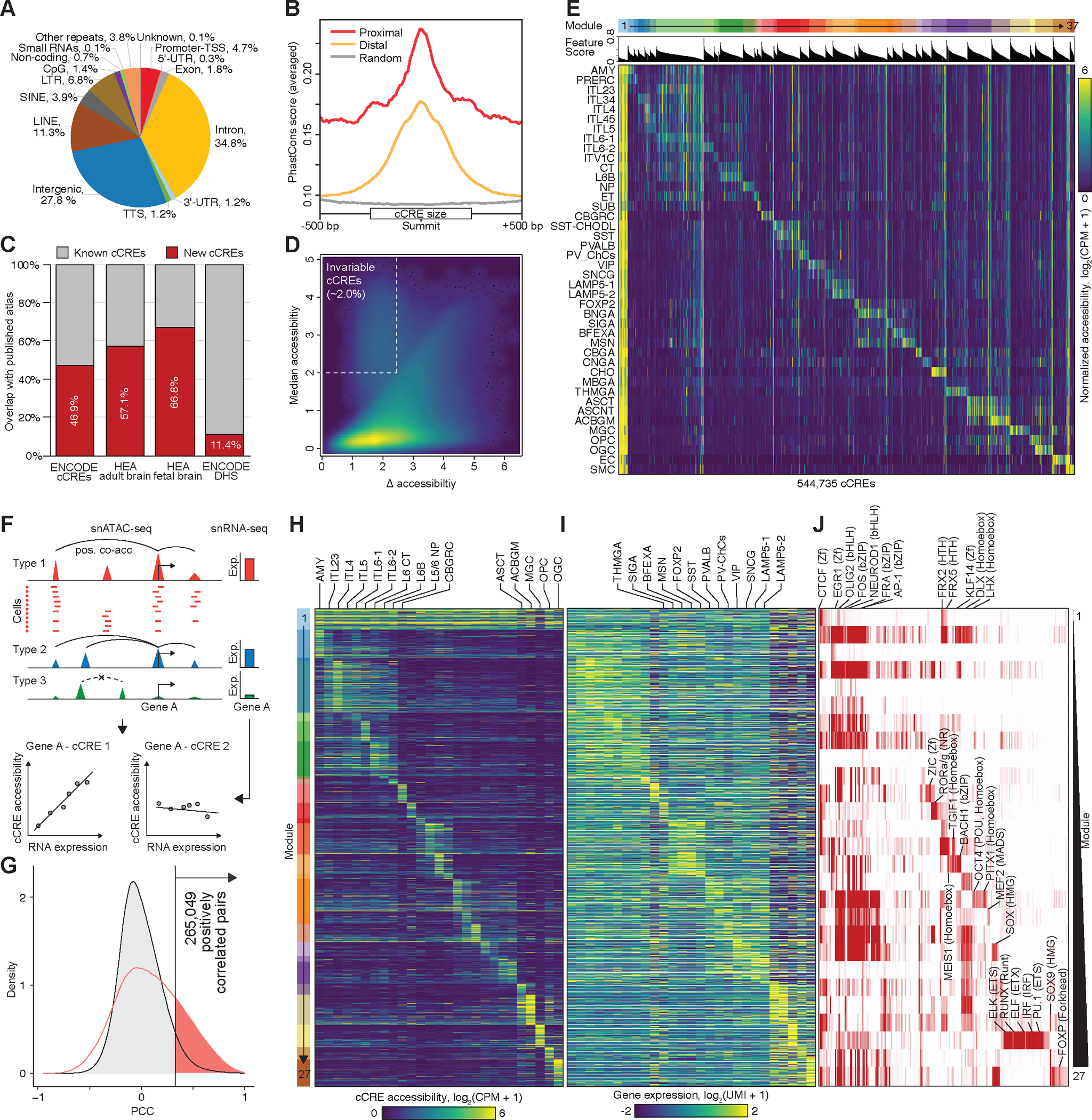
(A) Pie chart showing the fraction of cCREs that overlaps with different classes of annotated sequences in the human genome. TSS, transcription start site; TTS, transcription termination site. UTR, untranslated region. LINE, long interspersed nuclear element. SINE, short interspersed nuclear element. LTR, long terminal repeats. (B) Average phastCons conservation scores of proximal (in red) and distal cCREs (in yellow), and random genomic background is indicated in gray. (C) Stacked bar plot showing the percentage of new cCREs defined in this study (in red) and percentage of cCREs that overlapped with public recourse (in grey), including the cCREs and DHSs in the SCREEN database, cCREs identify in human enhancer atlas (HEA) fetal and adult brain. (D) Density map comparing the median and maximum variation of chromatin accessibility at each cCRE across cell types. Each dot represents a cCRE. (E) Heat map showing association of the 42 subclasses (rows) with 37 cis-regulatory modules (top, from left to right). Columns represent cCREs. A full list of subclass or module associations is in Table S9, and the association of cCREs to modules is in Table S10. CPM, counts per million. (F) Schematic overview of the computational strategy used to identify cCREs that are positively correlated with transcription of target genes. (G) In total, 265,049 pairs of positively correlated cCRE and genes (highlighted in red) were identified (FDR < 0.05). Grey filled curve shows distribution of Pearson’s correlation coefficient (PCC) for randomly shuffled cCRE–gene pairs. (H) Heat map showing chromatin accessibility of putative enhancers and (I) the expression of linked genes (right). Genes are shown for each putative enhancer separately. UMI, unique molecular identifier. (J) Enrichment of HOMER known transcription factor (TF) motifs in distinct enhancer–gene modules.
To define the cell type specificity of the cCREs, we first plotted the median levels of chromatin accessibility against the maximum variation for each element (Fig. 2D). We found that the majority of cCREs displayed highly variable chromatin accessibility across the brain cell types identified in the current study, except for a small proportion of invariable cCREs (2.0%) that showed accessibility in virtually all cell clusters, of which 87% were at proximal regions to TSS (Fig. 2D, Fig. S8E). To characterize the cell type specificity of the cCREs more explicitly, we used non-negative matrix factorization to group them into 37 modules, with elements in each module sharing similar cell type specificity profiles. Except for the first module (M1) which included mostly cell-type invariant cCREs, the remaining 36 modules displayed high cell-type restricted accessibility (Fig. 2E, Table S9, and S10). These restricted modules were enriched for distinct sets of motifs recognized by known transcriptional regulators (Table S11). For example, the NEUROG2 and ASCL1 enriched in module M4 for intratelencephalic (IT) neurons at cortical layer 2/3 (IT-L2/3) have been reported to be proneural genes, which is critical for cortical development (Table S11)(34). The SOX family factors in module M35 for oligodendrocytes (OGC) are pivotal regulators of a variety of developmental processes (Table S11)(35). These results lay a foundation for dissecting the gene regulatory programs in different brain cell types and regions.
Linking distal cCREs to target genes
To investigate the transcriptional regulatory programs that are responsible for cell-type-specific gene expression patterns in the human brain, we carried out an integrative analysis combining the snATAC-seq data collected in the current study with scRNA-seq data generated by a companion paper (Siletti, et al. 2022. in press in the same issue, #1) from matched brain regions (Fig. S7). We first connected 255,828 distal cCREs to 14,861 putative target genes by measuring the co-accessibility across single nuclei in every cell sub-class using Cicero (Fig. 2F, upper, see Methods), which resulted in a total of 1,661,975 gene–cCRE pairs within 500 kb of each other. Next, we identified the subset of cCREs the accessibility of which positively correlated with the expression of putative target genes and therefore could function as putative enhancers in neuronal or non-neuronal types (Fig. 2F, bottom). This analysis was restricted to distal cCREs and expressed genes captured from 27 matched cell sub-classes defined from integrative analysis between snATAC-seq and scRNA-seq (Fig. S7). We revealed a total of 265,049 pairs of positively correlated cCRE (putative enhancers) and genes at an empirically defined significance threshold of FDR < 0.05 (Table S12). These included 114,877 putative enhancers and 13,094 genes (Fig. 2G, Fig. S9, Table S12). The median distance between the putative enhancers and the target promoters was 176,345 bp (Fig. S9A). Each promoter region was assigned to an average of 7 putative enhancers, and each putative enhancer was assigned to two genes on average (Fig. S9B–C).
To investigate how cell-type-specific gene expression is regulated, we further classified these putative enhancers into 27 modules by using non-negative matrix factorization (Table S13, and S14). The putative enhancers in each module had a similar pattern of chromatin accessibility across cell sub-classes (Fig. 2H), and the expression of putative target genes showed a correlated pattern (Fig. 2I). This analysis revealed a large group of 5,113 putative enhancers that were linked to 4,775 genes more strongly expressed across all neuronal cell clusters than in non-neuronal cell types (module M1) (Fig. 2H, I, Table S13, and S14). These putative enhancers are strongly enriched for CTCF, and RFX binding sites (Table S15), which is consistent with what we previously found in the mouse cerebrum(28).
We also uncovered modules of enhancer–gene pairs that were active in a more restricted manner (modules M2–M27) (Fig. 2H–J). For example, we identified several modules (M2-M7) associated with cortical glutamatergic neurons (IT-L2/3, IT-L4, IT-L5, IT-L6–1, IT-L6–2), in which the putative enhancers were enriched for sequence motifs recognized by the bHLH factors NEUROD1 (Fig. 2J, Table S13–S15). Another example was module M15 associated with medium spiny neurons (MSN), in which putative enhancers were enriched for motifs of MEIS factors, which play an important role in establishing the striatal inhibitory neurons (Fig. 2J, Table S13–S15). Module M25 was associated with microglia (MGC). Genes linked to putative enhancers in this module were related to immune genes and the putative enhancers were enriched for the binding motif for ETS-factor PU.1, a known master transcriptional regulator of this cell lineage (Fig. 2J, Table S13–S15). This observation is consistent with the paradigm that cell-type-specific gene expression patterns are largely established by distal enhancer elements.
Regional specificity of glial and neuronal cell cCREs
The single-cell atlas of chromatin accessibility generated in this study provides a unique opportunity to characterize the heterogeneity of the gene regulatory programs that might underlie the specialized functions of glial and neuronal cells in each brain region. Whereas most non-neuronal cell types, including oligodendrocytes (OGCs), oligodendrocyte precursor cells (OPCs), microglia (MGC), telencephalon astrocytes (ASCTs), non-telencephalon (ASCNTs), and various vascular cells were ubiquitously distributed throughout the different brain dissections (Fig. 1J), molecular diversity has been recently reported in these cells in juvenile and adult vertebrates(36–39). We therefore leverage the largest collection of >900,000 single nuclei of non-neuronal cells and the high-resolution brain dissections in the present study (Fig. S1N), to perform in-depth characterization of the regulatory diversity of the non-neuronal populations.
The UMAP embeddings in the brain regional spaces showed a gradient among cell types of OGCs, OPCs, MGCs, and ASCTs (Fig. 3A–B, E–F, I–J, and M–N, Fig. S10). We hypothesized that these gradients may reflect heterogeneity in cCRE usage in these glial cells across brain regions. We first calculated the averaged chromatin accessibility and coefficient of variation (CV) across 42 brain regions for every cCRE identified in OGCs, OPCs, MGCs, ASCTs, respectively (Fig. 3C, G, K, and O, Fig. S10). A large number of cCREs displayed highly variable chromatin accessibility across the brain regions (Fig. 3C, G, K, and O, Fig. S10). In total 55,304 variable cCREs made up 40.1% of total cCREs identified in OGCs, 43,574 variable cCREs made up 33.0% of total cCREs identified in OPCs, 37,962 variable cCREs made up 34.5% of total cCREs identified in MGC, and 46,979 variable cCREs made up 33.1% of total cCREs identified in ASCTs. Next, using k-means clustering analysis on these variable cCREs for each glial cell population (Fig. 3D, H, L, and P), we revealed distinct open chromatin patterns in OGCs, OPCs, and ASCTs from the cerebellum (CB). A large fraction of these variable cCREs showed higher chromatin accessibility in the cerebellum exclusively (Fig. 3D, H, and P). We also observed loss of chromatin accessibility in a large number of cCREs in distinct brain structures (Fig. 3D, H, L, and P). These variable cCREs show similar regional specificities across three donors.
Fig. 3: Regional specificity of cell types correlates with chromatin accessibility.
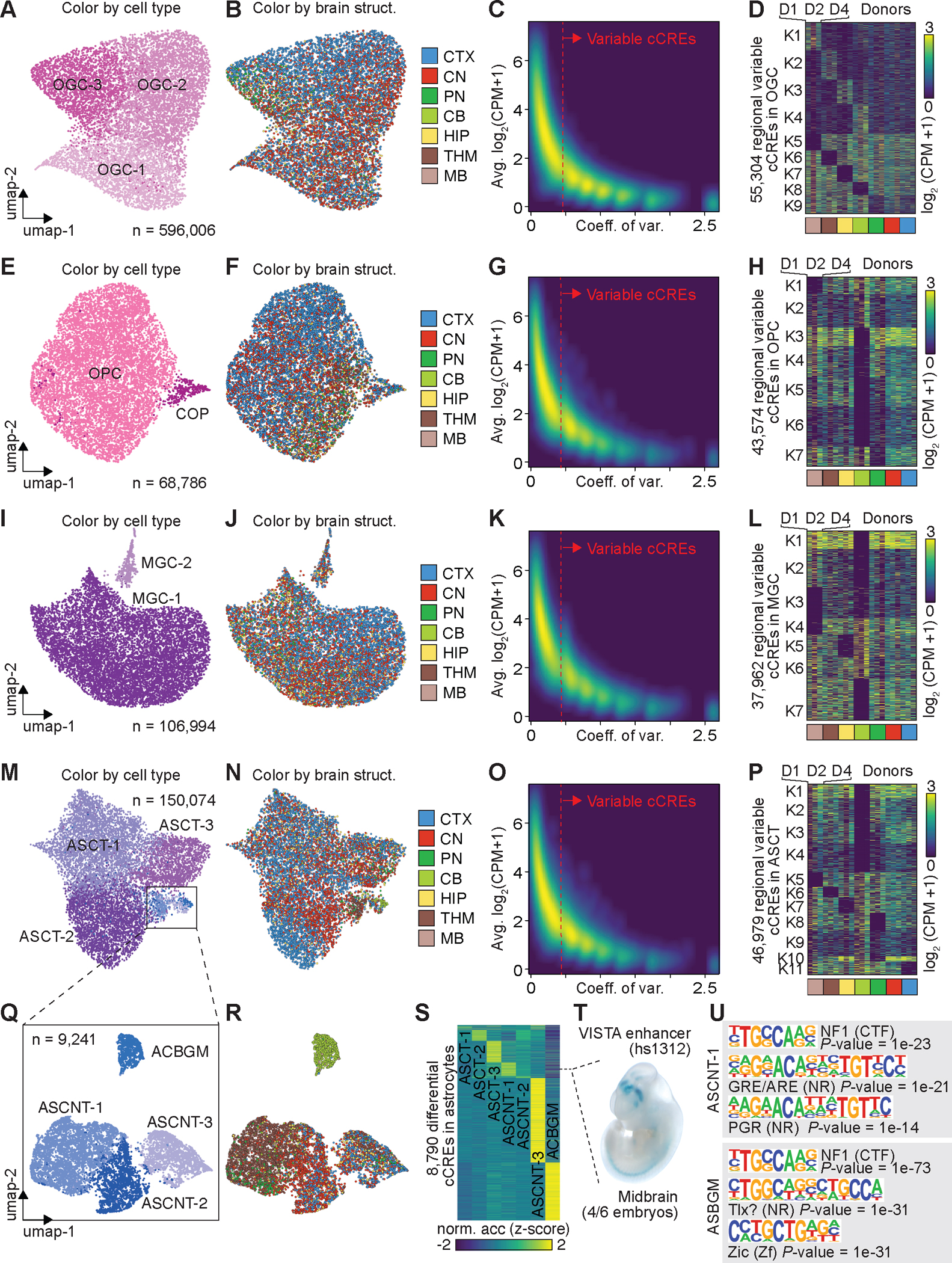
(A) UMAP embedding of cell types of oligodendrocytes (OGCs). (B) UMAP embedding of oligodendrocytes, colored by major brain structures. CTX, cortex; CN, cerebral nuclei; PN, Pons; CB, cerebellum; HIP, hippocampus; THM, thalamus; MB, midbrain. (C) Density scatter plot comparing the averaged accessibility and coefficient of variation across brain structures at each cCRE. Variable cCREs for OGCs are defined on the right side of dash line. (D) Heat map showing the normalized accessibility of variable cCREs in OGCs across major brain structures. CPM, counts per million. (E) UMAP embedding of cell types of oligodendrocyte precursor cells (OPCs). (F) UMAP embedding of OPCs, colored by major brain structures. (G) Density scatter plot comparing the averaged accessibility and coefficient of variation across brain structures at each cCRE. Variable cCREs for OPCs are defined on the right side of dash line. (H) Heat map showing the normalized accessibility of variable cCREs in OPCs. (I) UMAP embedding of cell types of microglia (MGC). (J) UMAP embedding of MGC, colored by major brain structures. (K) Density scatter plot comparing the averaged accessibility and coefficient of variation across brain structures at each cCRE. Variable cCREs for MGC are defined on the right side of dash line. (L) Heat map showing the normalized accessibility of variable cCREs in MGC. (M) UMAP embedding of cell types of astrocytes (ASCs). (N) UMAP embedding of ASCs, colored by major brain structures. (O) Density scatter plot comparing the averaged accessibility and coefficient of variation across brain structures at each cCRE. Variable cCREs for ASCs are defined on the right side of dash line. (P) Heat map showing the normalized accessibility of variable cCREs in ASCs. (Q) UMAP embedding of non-telencephalon ASCs (ASCNTs). (R) UMAP of ASCNTs, colored by major brain structures. (S) Normalized chromatin accessibility of 8,790 cell-type-specific cCREs. (T) Representative images of transgenic mouse embryos showing LacZ reporter gene expression under the control of the indicated enhancers that overlapped the differential cCRE in S (dotted line). Images were downloaded from the VISTA database (https://enhancer.lbl.gov). (U) Top enriched known motifs for astrocyte cell-type-specific cCREs.
In addition, we found a diverse population of both telencephalic (ASCT) and non-telencephalic astrocytes (ASCNT) in different major brain structures (Fig. 3M, and N). We identified three ASCNT cell types from sub-clustering of astrocytes, and one cell population restricted to the cerebellum that was annotated as Bergmann glial cell (ASBGM) (Fig. 3Q, and R). One cell type (ASCNT-1) was detected mostly in the thalamus, midbrain, and pons, whereas the other two ASCNT cell types were predominantly found in the cortex, hippocampus, and cerebral nuclei (CN). To characterize the dynamic epigenome, we compared the open chromatin landscapes among different cell types using a likelihood ratio test (Fig. 3S, Table S16, see Methods), and identified a total number of 8,790 cCREs that exhibited cell-type-restricted accessibility (range: 100–3,787) (Fig. 3S). A human enhancer, specifically accessible in the ASCNT-1 type, was previously validated by mouse transgenics to be active in the midbrain (Fig. 3T). We further performed motif analysis for differentially accessible regions in these cell types, finding enrichment of both shared and specific TF binding motifs. For example, we found CCAAT box-binding transcription factor (CTF) NF1 enriched in differential regions identified from both ASCT-1 and ASBGM, whereas TF motifs from nuclear receptors (NRs) and zinc-finger families are specifically enriched in different types (Fig. 3U, Table S17).
Lastly, we found that the cell populations of medium spiny neurons (MSNs) solved by chromatin accessibility were better separated based on the sub-regions in basal ganglia, rather than D1- and D2-types defined by the expression of two dopamine receptors DRD1 and DRD2 (Fig. S11, Table S18, Table S19, and Supplementary Text).
Epigenetic conservation of cCREs in mouse and human brain cells
To determine how conserved the gene regulatory landscapes are between human and mouse brains, we compared the human brain cCREs defined in this study with our previously published map of mouse cerebrum cCREs(28). We first performed joint clustering of 18 neuronal and glial cell sub-classes from cerebrum (each with >1,000 single nuclei) (Fig. 4A, Fig. S12)(28). We used multiple molecular features, including “gene activity scores” at homologous genes, chromatin accessibility at homologous cCREs, and transcription factor motif enrichment scores (Fig. S12A–G, see Methods). We note that clustering based on gene activity scores alone does not align brain cell types between the two species (Fig. S12A, D, and E), because of a lack of general conservation of expression patterns, as reported previously(40, 41). We identified orthologues of the human cCREs in the mouse genome by performing reciprocal homology searches, and overlapped with cCREs identified in the mouse cerebrum (Table S20, see Methods). Clustering using the chromatin accessibility at homologous cCREs alone also failed to align corresponding cell types, likely due to substantial of CRE turnovers (Fig. S12B, and F). Instead, we found that clustering based on TF motif enrichment allows for reasonable alignment of brain sub-classes between species (Fig. S12C, and G). This observation suggests that sequence motif enrichment scores are conserved molecular features that can reliably align similar cell sub-classes in the human and mouse brains (Fig. 4B, Fig. S12G). These analyses also showed that the gene regulatory programs of similar cell types share a similar grammar and syntax of gene regulation, likely in the form of combinatorial activities of conserved transcription factors.
Fig. 4: Comparative analyses of chromatin accessibility between human and mouse cerebrum.
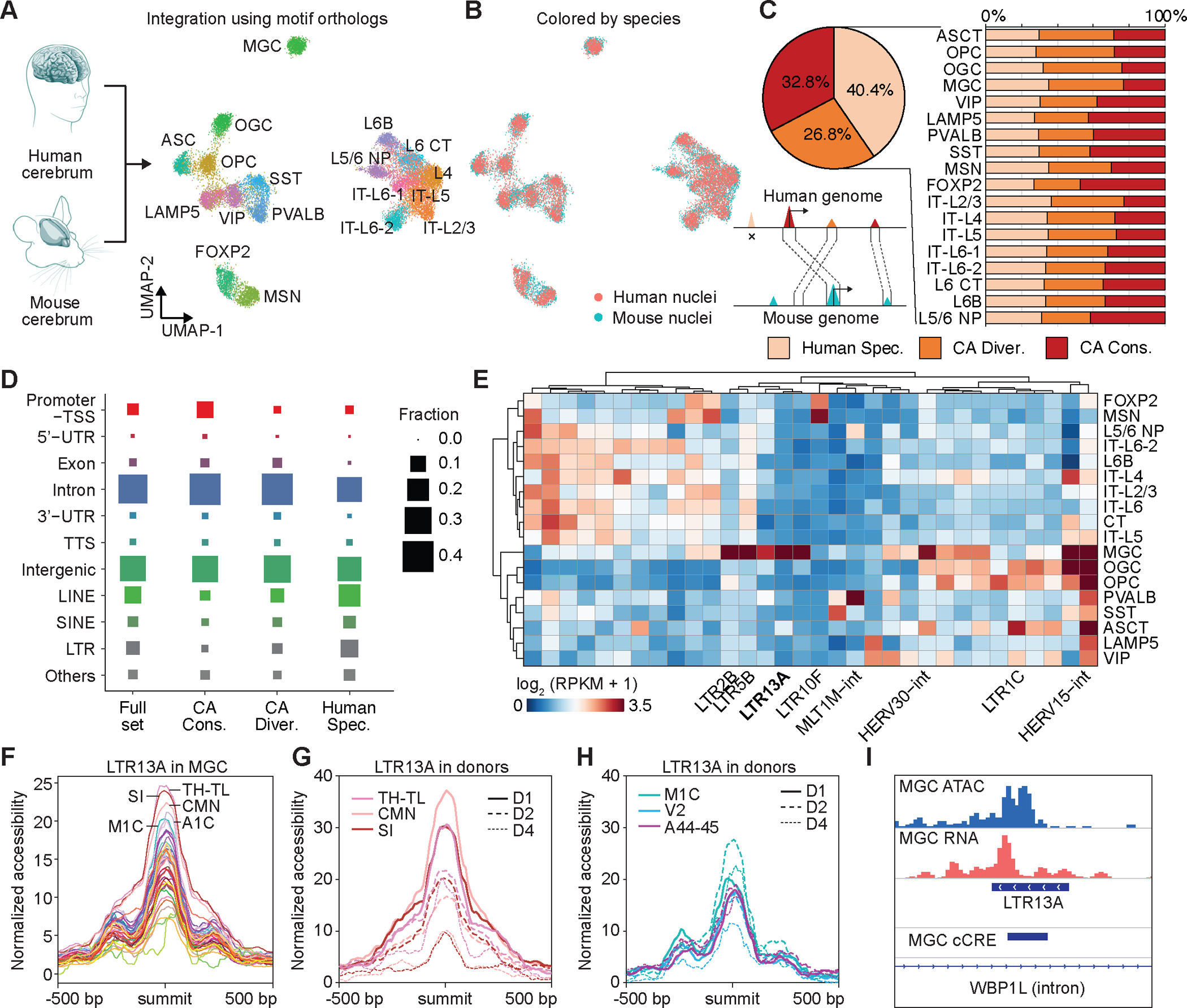
(A) UMAP co-embedding of 18 cell subclasses from both human and mouse cerebrum. (B) UMAP co-embedding of single nuclei colored by human and mouse. (C) Left, pie chart showing fraction of three categories of cCREs, including human specific, CA-divergent and CA-conserved cCREs. The CA-conserved cCREs are both DNA sequence conserved across species and have open chromatin in orthologous regions. The CA divergent cCREs are sequence conserved to orthologous regions but have not been identified as open chromatin regions in other species. Human specific cCREs are not able to find orthologous regions in the mouse genome. Right, bar plot showing three categories of cCREs in corresponding cell subclasses from human and mouse. (D) Dot plot showing fraction of genomic distribution of three categories of cCREs. (E) Normalized accessibility at variable human specific TEs in different cell subclasses. RPKM, reads per kilobase per million. (F) Average chromatin accessibility of LTR13A in microglia across different brain regions. (G) Variable chromatin accessibility of LTR13A across donors in microglia from different brain regions. (H) Invariable chromatin accessibility of LTR13A across donors in microglia from different brain regions. (L) Representative genomic locus showing chromatin accessibility and expression of LTE13A in microglia.
For ~60% of the human cCREs, mouse genome sequences with high similarity could be identified (more than 50% of bases lifted over to the human genome) (Fig. 4C, Fig. S12H–I, Table S20). Among these orthologues’ genome sequences, only half of them (32.8% of total human cCREs) were also identified as open chromatin regions in any cell sub-classes from the mouse cerebrum. We thus defined the 32.8% of human cCREs with both DNA sequence similarity and open chromatin conservation as CA (chromaitn accessibility)-conserved cCREs, and 26.8% of human cCREs with only DNA sequence similarity as CA-divergent cCREs. In addition, we defined 40.4% of human cCREs without orthologous genome sequences in the mouse genome as human-specific cCREs, although these may also include cCREs conserved in other primates or mammalian species except for the mouse (Fig. 4C, left, Table S21). This general pattern was consistent with what has been reported in other cell types between human and mouse(42). Next, we further break down and performed the same analyses on the cCREs within the corresponding cell sub-classes from both species. We observed a similar pattern and the proportion of different categories of cCREs was relatively consistent between various cell sub-classes (Fig. 4C, right, Table S21).
We further characterized the genomic distribution of different categories of cCREs. We observed that a large proportion of CA-conserved cCREs were located at or near the promoter-TSS regions in the human genome. In addition, the human-specific cCREs were enriched for transposable elements (TE), such as LINEs, SINEs, and LTRs (Fig. 4D, Table S21). Previous reports suggest that certain transposable elements are active in mammalian brains, and could hypothetically contribute to vulnerability to disease(43). In support of this hypothesis, we further characterized TE-cCREs (Fig. S13, see Supplementary Text), and our findings provide evidence that distinct TE families might be activated in specific brain cell types. For example, LTRs, including but not limited to LTR13A, LTR2B, and LTR5B display chromatin accessibility in microglia but not in other sub-classes of brain cells (Fig. 4E). The LTR13A has been reported to act as cellular gene enhancers(44). We observed that the LTR13A also has variable accessibility in microglia populations in different brain regions. For example, we identified higher accessibility in brain regions such as posterior parahippocampal gyrus (TH-TL), primary visual cortex (A1C) and primary motor cortex (M1C) from the cortex, substantia innominata and nearby nuclei (SI) and corticomedial nuclear group (CMN) from cerebral nuclei, and lower accessibility in midbrain and cerebellum (Fig. 4F). Furthermore, chromatin accessibilities at LTR13A in microglia from TH-TL, CMN and SI varied considerably among the donors, (Fig. 4G), but not in other brain regions (Fig. 4H). The chromatin accessibility of LTR13A was associated with the activation indicated by RNA expression signals (Fig. 4I), though the biological relevance of this observation requires further investigation in larger cohorts.
Taking advantage of the integration of multi-modal datasets, we next aimed to better delineate the gene regulatory programs that underlie the identity and function of each brain cell type (Fig. 5A, Fig. S14A–E). We collected single-cell genomic datasets profiled from the human primary cortex (M1C) and middle temporal gyrus (MTG), including 149,891 cells from scRNA-seq, 27,383 cells from Paired-Tag (only from M1C), 55,974 cells from snATAC-seq, 10,604 cells from snmC-seq, and 16,257 cells from snm3C-seq (Fig. 5B–C). We performed co-embedding cell clustering analysis on these datasets. (see Supplementary Text and Methods). The different single-cell assays showed excellent agreement in the same co-embedding space, which indicates the high quality of common variable features and the success of the integration strategy (Fig. 5C). The integration of multi-modal datasets allowed us to evaluate the information content, the strengths, and limitations of various assays in the prediction of potential functional enhancers. We first defined different subsets of distal cCREs by using the combination of single-cell modalities or snATAC-seq only (Fig. 5D). By comparing the different subsets of distal cCREs against validated human enhancer in the forebrain from VISTA Enhancer Browser (https://enhancer.lbl.gov)(45), we observed that the highest gain of enrichment comes with the incorporation of H3K27ac modification signals (Fig. 5D). We also found incorparing the sequence conservation information further improved the prediction of potential functional enhancers. The above observations suggest that incorporation of chromatin accessibility, histone modification information such as H3K27ac, co-accessibility between distal elements and promoters, and sequence conservation, could improve the prediction of functional enhancers.
Fig. 5: Integration of multi-modal single cell datasets of cortical cells.
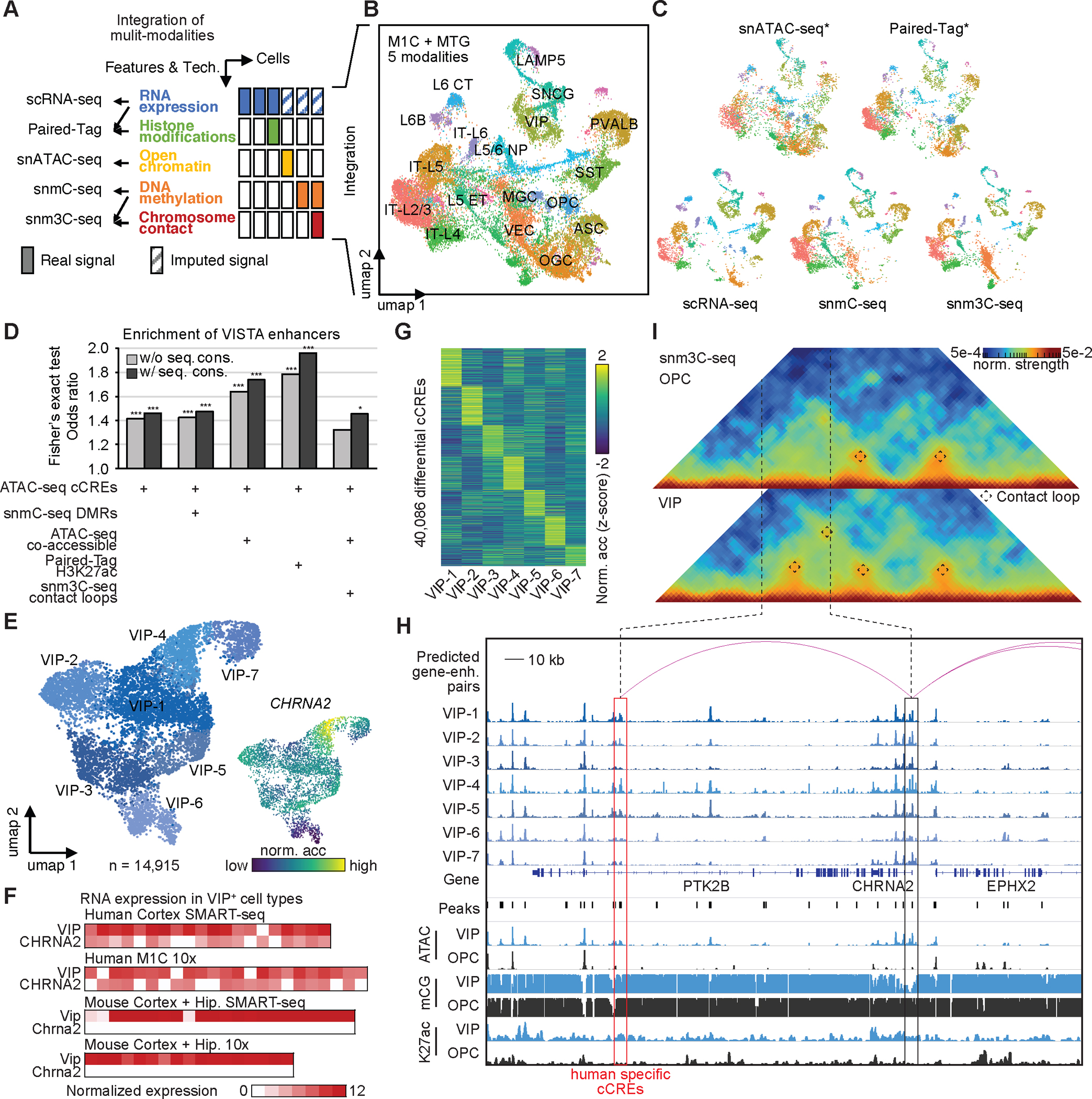
(A) Summary of single cell technologies and multi-model data integration strategies. (B) UMAP embedding and integrative clustering analysis of 18 major cell types. (C) Co-embedding of multi-model single cell datasets showing excellent agreement. * for assays using unbiased sampling strategy. (D) The UpSet plot showing the enrichment of VISTA validated enhancer in different subsets of distal cCREs, which is defined by combining information/features from single-cell modalities or snATAC-seq only. These subsets include: (1) cCREs identified from snATAC-seq only; (2) snATAC-seq cCREs overlapped with differentially methylated regions (DMRs) identified from snmC-seq; (3) snATAC-seq distal cCREs that were predicted to be co-accessible with promoter across cells; (4) snATAC-seq distal cCREs marked by H3K27ac signals from Paired-Tag, a method for joint single cell analysis of histone modification and gene expression; and (5) snATAC-seq distal cCREs predicted to be co-accessible with promoters, and linked by chromatin loops identified in snm3C-seq assays. Then, we filtered out validated human enhancers in the forebrain from VISTA enhancer browser (https://enhancer.lbl.gov). By overlapping different subsets of distal cCREs, we observed various enrichment (odds ratios from Fisher’s exact test) with combination of different assays and features. Fisher’s exact test, *, p-value < 0.05, ***, p-value < 0.001. (E) Left, UMAP embedding of VIP positive (VIP+) GABAergic cell types. Upper right, colored by donors. Bottom right, normalized accessibility at gene CHRNA2. (F) Expression of gene VIP and CHRNA2 in human VIP+ cell types, and expression of gene Vip and Chrna2 in mouse VIP+ cell types from Allen Cell Types Database: RNA-Seq Data. (G) Normalized chromatin accessibility of 40,086 VIP+ cell-type-specific cCREs. (H) Genome browser track view at the CHRNA2 locus as an example for candidate enhancers predicted from single cell multi-model datasets. Displayed chromatin accessibility profiles from snATAC-seq; DNA methylation signals (mCG) from snm3C-seq; and histone modification signals (H3K27ac) from Paired-Tag for several VIP+ neurons and oligodendrocytes precursor cells (OPC). Red Arcs represent the predicted enhancer for gene CHRNA2. (I) Triangle heat map show chromatin contacts in VIP+ neurons and OPC derived from snm3C-seq data at gene CHRNA2 locus.
We characterized the gene program in VIP-4, one cell type of VIP+ neurons, which showed distinct chromatin accessibility at gene CHRNA2 loci (Fig. 5E). The gene CHRNA2 encodes a subunit of nicotinic cholinergic receptor, which is involved in fast synaptic transmission. The co-localization of marker gene BTBD11 for VIP+ neurons and CHRNA2 from RNAscope(46) in situ hybridization experiment first validated the existence of VIP-4 type in the human cortex (Fig. S14F). We also noticed that the expression of CHRNA2 was restricted in human VIP+ cell types, but not in any VIP+ cell types identified from mouse brain (Fig. 5F) (8, 41, 47). We explored whether the human-specific expression of CHRNA2 was regulated by specific cCREs in the VIP-4 type. We identified a total of 40,086 differential cCREs between 7 VIP+ cell types (Fig. 5G, Table S22). One differential cCRE located downstream of the gene CHRNA2 showed higher accessibility in VIP-4, than in OPC (Fig. 5H, ATAC tracks for 7 VIP+ cell type and aggregated signals for VIP and OPC). This cCRE was also characterized as a human-specific cCRE in VIP+ neurons (Fig. 4C, Table S20). The specific accessibility of this cCRE and promoter of CHRNA2 in VIP+ neurons were supported by mCG signals from snmC-seq (Fig. 5H, mCG tracks). This cCRE was predicted as a putative enhancer that regulates the expression of CHRNA2 (Fig. 5H, red arcs). The potential active function of this CRE was further supported by H3K27ac modification in the cells (Fig. 5H, K27ac tracks). We additionally confirmed the chromatin interactions between this cCRE and the promoter of CHRNA2 (Fig. 5I). Taken together, the above data suggested that this human-specific cCRE could be an enhancer that regulates the distinct expression of CHRNA2 in the VIP-4 type from the human brain.
Sequence changes underlie epigenetic divergence in cCREs in distinct brain cell types
We characterized different categories of cCREs (Fig. S15–17, Table S23, see Supplementary Text), and hypothesized that the epigenetic divergence of cCREs could be partly due to evolutionary changes in DNA sequences. To test this hypothesis, we picked IT-L2/3 neurons, LAMP5+ interneurons, and MGC as representative cell sub-classes, and trained gapped-kmer SVM classifiers (gkmSVM) (48) from the DNA sequences in cCREs (Fig. 6A, B, see Methods). These models achieved excellent performance (area under receiver operating characteristic curve (AUROC) ranging from 0.856 to 0.928, and area under precision-recall curve (AUPRC) ranging from 0.850 to 0.912) in the prediction of open chromatin regions within the corresponding species (Fig. 6A, B). Next, we predicted different categories of mouse cCREs using gkmSVM models trained with human DNA sequence at cCREs in corresponding cell sub-classes (Fig. 6C). These models also achieved high accuracy (ranging from 0.83 to 0.91) in the prediction of CA-conserved mouse cCREs, and slightly lower accuracy in CA-divergent (ranging from 0.79 to 0.82) and mouse-species cCREs (ranging from 0.71 to 0.80) (Fig. 6C). Similarly, the gkmSVM models trained with mouse DNA sequences archived high accuracy in predicting human CA-conserved cCREs (ranging from 0.89 to 0.93), and slightly lower accuracy for human CA-divergent (ranging from 0.75 to 0.86) and human-specific cCREs (ranging from 0.77 to 0.84) (Fig. 6D). The human CA-divergent cCREs that failed to be predicted from mouse gkmSVM models have a potential function in regulating genes involved in specific biological processes, including glutamate receptor signaling pathway (GO:0007215), synaptic transmission - GABAergic (GO: 0051932), and various fatty acid elongation (GO: 0019367, GO:0019368, GO:0034625) in IT-L2/3 neurons, LAMP5+ interneurons, and MGC, respectively (Fig. S18, Table S24, see Supplementary Text). These results suggested that the regulatory divergence is at least in part due to evolution of DNA sequences.
Fig. 6: Epigenetic conservation and divergence of human orthologous cCREs.
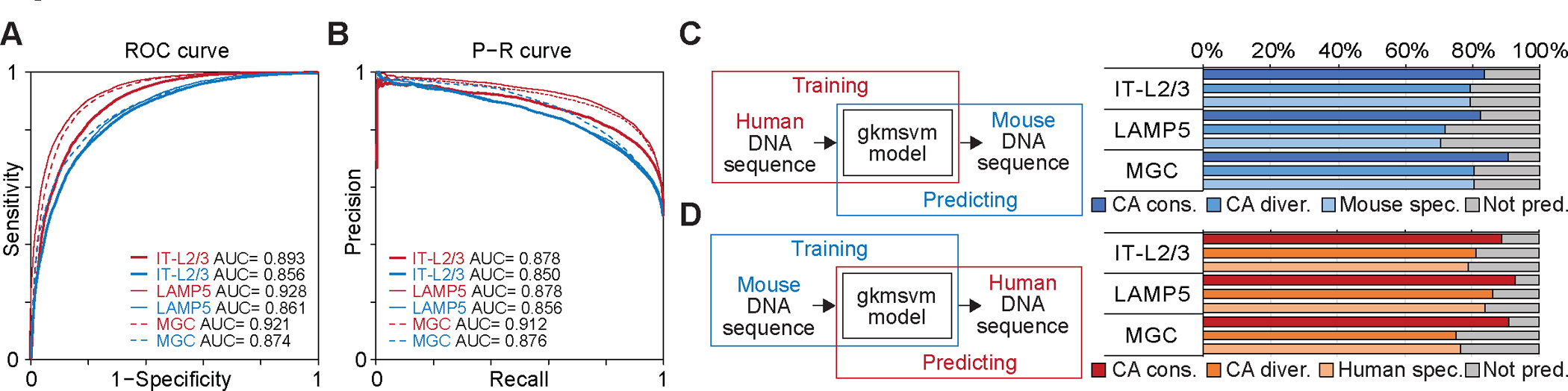
(A) Receiver operating characteristic (ROC) curve and area under curve (AUC) from gkmsvm models trained for representative human and mouse cell types. (B) Precision-recall curve (PRC) curve and area under curve (AUC) from gkmsvm models trained for representative human and mouse cell types. (C) Prediction for mouse epigenetic conserved, mouse CA-divergent, and mouse specific cCREs from gkmsvm models trained in corresponding human cell subclasses. (D) Prediction for human epigenetic conserved, human CA-divergent, and human specific cCREs from gkmsvm models trained in corresponding mouse cell subclasses.
Predicting disease relevant cell types for neuropschiatric disorders
Genome-wide association studies (GWASs) have identified genetic variants that are associated with many mental diseases and traits (Table S25), but >90% of variants are located in non-protein-coding regions of the genome (4, 5). Previous studies have shown that non-coding risk variants are enriched in cCREs active in disease relevant cell types (6, 7, 49). Leveraging the newly annotated cell-type-resolved human brain cCREs, we predict the cell types relevant to the different neuropsychiatric disorders. We performed linkage disequilibrium score regression (LDSC) analysis to determine if the genetic heritability of DNA variants associated with neuropsychiatric disorders are significantly enriched within cCREs showing chromatin accessibility in the major brain cell types in the present study (Table S25, see Methods). We found associations between 19 mental diseases and traits (Table S25, and S26) with the open chromatin landscapes in one or more cell types we identified (Fig. 7A, see Methods), and few associations for non-central nervous system traits (Fig. S19A). In particular, we observed widespread and strong enrichment of genetic variants linked to neuropsychiatric disorders such as schizophrenia (SCZ) and bipolar disorder within accessible cCREs across various neuronal cell types (Fig. 7A, Table S26). Tobacco use disorder and alcohol usage were associated with specific neuronal cell types in basal ganglia, which were previously implicated in addiction(50). Another example is neuroticism, which was restricted and associated with IT neurons from the cortex (Fig. 7A, Table S26). In addition, the risk variants from Alzheimer’s disease were significantly enriched in the cCREs found in microglia, but not in other cell types (Fig. 7A, Table S26).
Fig. 7: Interpreting noncoding risk variant of neurological disorder and traits.
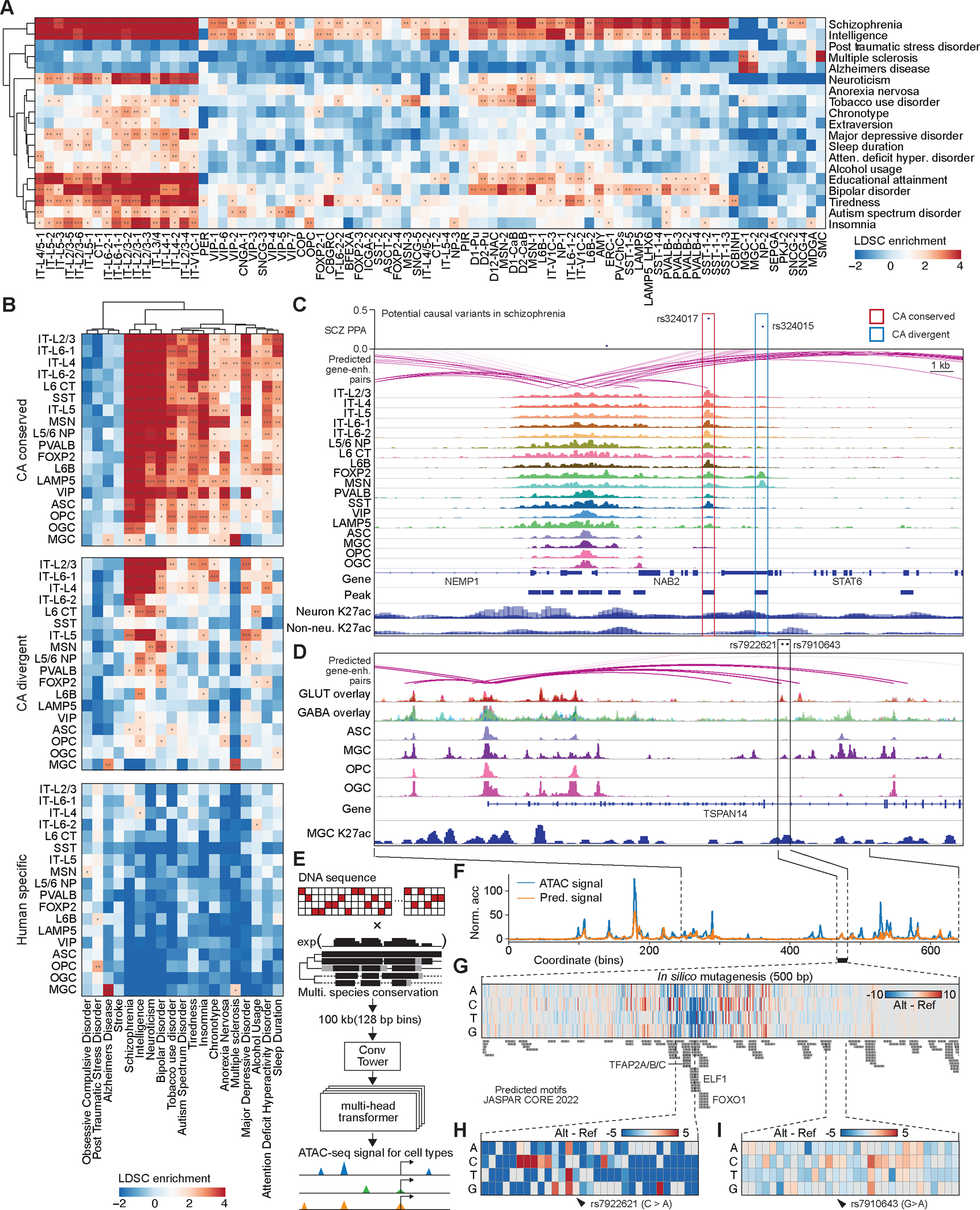
(A) Heat map showing enrichment of risk variants associated with neurological disorder and traits from genome wide association studies in human cell type-resolved cCREs. Cell-type specific linkage disequilibrium score regression (LDSC) analysis was performed using GWAS summary statistics. Total cCREs identified independently from each human cell type were used as input for analysis. P-values were corrected using the Benjamini Hochberg procedure for multiple tests. FDRs of LDSC coefficient are displayed. *, FDR < 0.05; **, FDR < 0.01; ***, FDR<0.001. Detailed results are reported in Table S26. (B) Heat map showing enrichment of risk variants associated with mental disorder and traits in three categories of cCREs. Detailed results are reported in Table S27. (C) Fine mapping and molecular characterization of schizophrenia (SCZ) risk variants in different categories of cCREs from multiple neuronal types. Genome browser tracks (GRCh38) display chromatin accessibility profiles from snATAC-seq; histone modification signals (H3K27ac) from Paired-Tag, and red arcs represent the predicted enhancer for gene TSNARE. (D) Molecular characterization of Alzheimer’s disease (AD) risk variants in microglia specific enhancer. Genome browser tracks (GRCh38) display chromatin accessibility profiles from snATAC-seq; histone modification signals (H3K27ac) from Paired-Tag, and red arcs represent the predicted enhancer for gene TSPAN14. (E) Schematic diagram of deep learning model for predicting chromatin accessibly. (C) Chromatin accessibility at TSPAN14 enhancer loci predicted in human microglia. (F) In silico nucleotide mutagenesis influenced the prediction of accessibility. Larger signals (in dark red) represent a higher accessibility prediction on altered sequence, and lower signals (in dark blue) represent lower accessibility on altered sequence. Predicted JASPAR CROE 2022 motifs were listed below. (E) Lower accessibility predicted on TSPAN14 enhancer with risk variant rs7922621 C>A. (F) Less accessibility change predicted on TSPAN14 enhancer with risk variant rs7910643 G>A.
We further provided breakout reports of LDSC analysis by using three categories of cCREs defined above (Fig. 4C). the strongest associations between cell sub-classes and GWAS traits were found in the analysis of epigenetic conserved elements (Fig. 7B, Table S27). For example, the risk variants in schizophrenia showed the most significant enrichment in epigenetic conserved elements (Fig. 7C, see Supplementary Text). Fewer associations were observed in the analysis of epigenetic divergent elements, and most of GWAS traits showed no associations when human-specific elements were used for LDSC (Fig. 7B).
Interestingly, LDSC analysis using human-specific elements revealed an association between Alzheimer’s disease (AD) and microglia (Fig. 7B, Table S27), raising the possibility that the AD-related risk variants could reside in human-specific regulatory elements, and contribute to human-specific gene regulation programs in microglia(51) (Fig. S20, see Supplementary Text). This observation suggests potential limitations of animal models of AD in revealing disease pathology in humans (52). For example, One AD risk locus contains multiple microglia-specific cCREs, which cannot find any homologous sequence in the mouse genome (Figure S19B). One of these cCREs harboring AD-risk variants rs6733839 has been predicted to be a microglia-specific enhancer that can regulate the expression of BIN1 gene, and its function was supported by both H3K27ac modification and previous validation experiment (Figure S19B) (53).
Deep learning models predict the influence of risk variants on gene regulation
To further understand how risk variants contribute to the function of regulatory elements, we used deep learning (DL) models to predict chromatin accessibility from DNA sequences (Fig. 7E, Figure S21A–C, see Methods). The deep learning model architecture was inspired by Enformer (54), which adapts attention-based architecture, Transformer, that could better capture syntactic forms (for example, the order and combination of words in a sentence) and outperforms most existing models in natural language processing tasks (55). We trained the DL model called Epiformer on the normalized pseudo bulk ATAC-seq profiles in human microglia and multiple cell subclasses. To demonstrate the utility of the resulting deep learning models, we focused on a microglia-specific cCRE that was predicted to regulate the expression of TSPAN14 gene. This cCRE harbors two Alzheimer’s disease (AD) risk variants, whose functions were investigated in a separate study (56)(Fig. 7D). The deep learning model successfully predicted the cell type-specific accessibility of cCREs at the TSPAN14 locus with a Pearson correlation coefficient of 0.72 (Fig. 7E, F). By contrast, DL models trained using ATAC-seq profiles from other cell types failed to predict the chromatin accessibility profiles of these cCREs (Fig. S21D). To predict the regulatory effects of the risk variant, we then performed in silico mutagenesis on the above microglia-specific enhancer near TSPAN14 and compared the changes of accessibility predicted from reference and altered DNA sequences. Every nucleotide within this 500 bp enhancer was mutated in silico, and the influence on accessibility was measured by assessing the difference between the predicted accessibility for the reference and altered sequences (Fig. 7G, Table S28). Among these in silico single nucleotide mutations, most in the flanking regions did not affect the predicted accessibility; however, a few nucleotide substitutions increased or decreased the predicted chromatin accessibility (Fig. 7G, Table S28). The DNA sequence most negatively associated with the predicted accessibility was predicted to contain binding motifs for transcription factors TFAP2A/B/C, ELF1, and FOXO1 (Fig. 7G). Among them, FOXO1 has been recently reported to be a critical element for the regulation of microglial cell physiology and the maintenance of the brain homeostasis(57). The model predicted that the nucleotide substitutions (C > A) of AD-risk variant rs7922621 would decrease the accessibility of microglia specific enhancer (Fig. 7H), whereas the AD-risk variant rs7910643 would barely influence chromatin accessibility (Fig. 7I). These predictions matched the experimental results obtained from microglia-like cells differentiated from two different human pluripotent stem cell lines in which the two variants were modified using PRIME editing and tested for effects on TSPAN14 expression(56). These results provide evidence that deep learning approaches might be able to capture the gene regulatory code and interpret risk variants associated with complex traits and diseases.
Discussion
In-depth knowledge of the transcriptional regulatory program in brain cells would not only improve our understanding of the molecular inner workings of neurons and non-neuronal cells, but could also shed light on the pathogenesis of a spectrum of neurological disorders. Here, we report a comprehensive profiling of chromatin accessibility at single-cell resolution in 42 human brain regions. The chromatin accessibility maps of 544,735 cCREs, were probed in >1.1 million nuclei. Taking advantage of our high-resolution brain dissections, we examined the regional specificity in chromatin accessibility of cell types in the human brain and showed that most brain cell types exhibit strong regional specificity. The described cCRE atlas (http://catlas.org) represents a rich resource for the neuroscience community to understand the molecular patterns that underlie the diversification of brain cell types in complementation to other molecular and anatomical data.
The comparsion of open chromatin landscape between human and mouse cell types uncovered a substantial degree of evolutionary changes involving both sequence turnovers and regulatory divergence. We identified ~30% of the cCREs that display sequence conservation as well as chromatin accessibility, which is likely an underestimate of the degree of conservation, since the list of brain cCREs in each species will likely be greater as more cells and brain regions are assayed. We observed that the chromatin accessibility at human-specific cCREs tend to be correlated with species-restricted gene expression, and they are enriched for TEs. Whether these TE-cCREs may serve new enhancers to drive primate specific gene expression remain to be demonstrated(58). In addition, TEs are reactivated during aging, neurodegeneration, and neuropsychiatric disorders, and their role in the disease pathology needs to be further elucidated(59) with future datasets collected from more species and donors in various conditions.
Genome-wide association studies (GWAS) have been widely used to enhance our understanding of polygenic human traits and reveal clinically relevant therapeutic targets for neuropsychiatric disorders. However, our ability to interpret the risk variants has been hampered by tissue heterogeneity and molecular functions of non-coding regulatory elements. By leveraging both epigenetic conserved, divergent, and human-specific cCRE identified from various cell types between human and mouse comparison, we prioritized likely causal variants in linkage disequilibrium, linked distal cCREs to putative target genes, and predicted motifs altered by risk variants using cutting-edge deep learning methods. We revealed hundreds of cell-type trait associations and created a framework to systematically interpret noncoding risk variants.
The present study is limited to three subjects and each brain region is surveyed at a modest depth. Future studies will be necessary to investigate the variations more deeply in chromatin landscapes across different individuals, genders, age groups and populations. Further, application of single cell multiomics as well as spatial transcriptomics tools will greatly accelerate the identification of rare human brain cell types and their gene regulatory programs.
Supplementary Material
Acknowledgments
This publication was supported by and coordinated through BICCN. This publication is part of the Human Cell Atlas- www.humancellatlas.org/publications. We would like to thank Dr. Erica Melief, Aimee Schantz, Katie Kern, Amanda Keen, and Lisa Keene of the University of Washington BioRepository and Integrated Neuropathology (BRaIN) Laboratory for outstanding technical and administrative support for tissue collection, preservation, and characterization, and the brain donors and their loved ones, without whom this research would be impossible. Dr. C. Dirk Keene from Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA, USA for brain specimen acquisition. We would like to thank the QB3 Macrolab at UC Berkeley for purification of the Tn5 transposase. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929 to Kristen Jepsen). We thank Ethan Armand for discussion and support, Qiurui Zeng for data management and transferring. We sincerely thank Dr. Yin Shen for engaging in a double blinded testing of the performance of our deep learning model related to TSPAN14 risk variants using their unpublished experimental data.
Funding:
This study was supported by National Institute of Mental Health (NIMH) grant UM1MH130994 and U01MH114812 and the Nancy and Buster Alvord Endowment (to C.D.K.). The development of deep learning models was sponsored in part by NSF Convergence Accelerator under award OIA-2040727, NIH Bridge2AI Center Program under award U54HG012510, as well as generous gifts from Google, Adobe, and Teradata (to J.S.). Zhaoning Wang is supported by the DDBrown Award from the Life Sciences Research Foundation.
Footnotes
Conflict of interst statement: B.R. is a consultant of and has equity interests in Arima Genomics, Inc. B.R. is also a co-founder of Epigenome Technologies Inc. S.L. is a paid scientific advisor to Moleculent, Combigene, and the Oslo University Center of Excellence in Immunotherapy.
Data availability
Demultiplexed data can be accessed via the NEMO archive here: https://assets.nemoarchive.org/dat-d6r90fb. Raw data are available in the NCBI Gene Expression Omnibus (GEO) under accession number GSE244618. Processed data will be available on our web portal CATLAS: http://catlas.org.
Custom code and scripts used for analysis can be accessed here: https://github.com/yal054/snATACutils and https://github.com/r3fang/SnapATAC.
The deep learning model and pre-trained model can be download from https://github.com/yal054/epiformer.
Reference
- 1.Murray CJL, Atkinson C, Bhalla K, Birbeck G, Burstein R, Chou D, Dellavalle R, Danaei G, Ezzati M, Fahimi A, Flaxman D, Foreman, Gabriel S, Gakidou E, Kassebaum N, Khatibzadeh S, Lim S, Lipshultz SE, London S, Lopez, MacIntyre MF, Mokdad AH, Moran A, Moran AE, Mozaffarian D, Murphy T, Naghavi M, Pope C, Roberts T, Salomon J, Schwebel DC, Shahraz S, Sleet DA, Murray J Abraham M Ali K, Atkinson C, Bartels DH, Bhalla K, Birbeck G, Burstein R, Chen H, Criqui MH, Dahodwala, Jarlais, Ding EL, Dorsey ER, Ebel BE, Ezzati M, Fahami S Flaxman, Flaxman AD, Gonzalez-Medina D, Grant B, Hagan H, Hoffman H, Kassebaum N, Khatibzadeh S, Leasher JL, Lin J, Lipshultz SE, Lozano R, Lu Y, Mallinger L, McDermott MM, Micha R, Miller TR, Mokdad AA, Mokdad AH, Mozaffarian D, Naghavi M, Narayan KMV, Omer SB, Pelizzari PM, Phillips D, Ranganathan D, Rivara FP, Roberts T, Sampson U, Sanman E, Sapkota A, Schwebel DC, Sharaz S, Shivakoti R, Singh GM, Singh D, Tavakkoli M, Towbin JA, Wilkinson JD, Zabetian A, Murray J Abraham, Ali MK, Alvardo M, Atkinson C, Baddour LM, Benjamin EJ, Bhalla K, Birbeck G, Bolliger I, Burstein R, Carnahan E, Chou D, Chugh SS, Cohen A, Colson KE, Cooper LT, Couser W, Criqui MH, Dabhadkar KC, Dellavalle RP, Jarlais, Dicker D, Dorsey ER, Duber H, Ebel BE, Engell RE, Ezzati M, Felson DT, Finucane MM, Flaxman S, Flaxman AD, Fleming T, Foreman, Forouzanfar MH, Freedman G, Freeman MK, Gakidou E, Gillum RF, Gonzalez-Medina D, Gosselin R, Gutierrez HR, Hagan H, Havmoeller R, Hoffman H, Jacobsen KH, James SL Jasrasaria R, Jayarman S, Johns N, Kassebaum N, Khatibzadeh S, Lan Q, Leasher JL, Lim S, Lipshultz SE, London S, Lopez, Lozano R, Lu Y, Mallinger L, Meltzer M, Mensah GA, Michaud C, Miller TR, Mock C, Moffitt TE, Mokdad AA, Mokdad AH, Moran A, Naghavi M, Narayan KMV, Nelson RG, Olives C, Omer SB, Ortblad K, Ostro B, Pelizzari PM, Phillips D, Raju M, Razavi H, Ritz B, Roberts T, Sacco RL, Salomon J, Sampson U, Schwebel DC, Shahraz S, Shibuya K, Silberberg D, Singh JA, Steenland K, Taylor JA, Thurston GD, Vavilala MS, Vos T, Wagner GR, Weinstock MA, Weisskopf MG, Wulf S, Murray USB of D. Collaborators, The State of US Health, 1990–2010: Burden of Diseases, Injuries, and Risk Factors. Jama. 310, 591–606 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H, The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, Kathiresan S, Kenny EE, Lindgren CM, MacArthur DG, North KN, Plon SE, Rehm HL, Risch N, Rotimi CN, Shendure J, Soranzo N, McCarthy MI, A brief history of human disease genetics. Nature. 577, 179–189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frydas A, Wauters E, van der Zee J, Broeckhoven CV, Uncovering the impact of noncoding variants in neurodegenerative brain diseases. Trends Genet. 38, 258–272 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Liu B, Montgomery SB, Identifying causal variants and genes using functional genomics in specialized cell types and contexts. Hum Genet. 139, 95–102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee B-K, Pauli F, Rosenbloom KR, Sabo P, Safi A, Sanyal A, Shoresh N, Simon JM, Song L, Trinklein ND, Altshuler RC, Birney E, Brown JB, Cheng C, Djebali S, Dong X, Dunham I, Ernst J, Furey TS, Gerstein M, Giardine B, Greven M, Hardison RC, Harris RS, Herrero J, Hoffman MM, Iyer S, Kellis M, Khatun J, Kheradpour P, Kundaje A, Lassmann T, Li Q, Lin X, Marinov GK, Merkel A, Mortazavi A, Parker SCJ, Reddy TE, Rozowsky J, Schlesinger F, Thurman RE, Wang J, Ward LD, Whitfield TW, Wilder SP, Wu W, Xi HS, Yip KY, Zhuang J, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M, Pazin MJ, Lowdon RF, Dillon LAL, Adams LB, Kelly CJ, Zhang J, Wexler JR, Green ED, Good PJ, Feingold EA, Bernstein BE, Birney E, Crawford GE, Dekker J, Elnitski L, Farnham PJ, Gerstein M, Giddings MC, Gingeras TR, Green ED, Guigó R, Hardison RC, Hubbard TJ, Kellis M, Kent WJ, Lieb JD, Margulies EH, Myers RM, Snyder M, Stamatoyannopoulos JA, Tenenbaum SA, Weng Z, White KP, Wold B, Khatun J, Yu Y, Wrobel J, Risk BA, Gunawardena HP, Kuiper HC, Maier CW, Xie L, Chen X, Giddings MC, Bernstein BE, Epstein CB, Shoresh N, Ernst J, Kheradpour P, Mikkelsen TS, Gillespie S, Goren A, Ram O, Zhang X, Wang L, Issner R, Coyne MJ, Durham T, Ku M, Truong T, Ward LD, Altshuler RC, Eaton ML, Kellis M, Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Batut P, Bell I, Bell K, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena HP, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Li G, Luo OJ, Park E, Preall JB, Presaud K, Ribeca P, Risk BA, Robyr D, Ruan X, Sammeth M, Sandhu KS, Schaeffer L, See L-H, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Hayashizaki Y, Harrow J, Gerstein M, Hubbard TJ, Reymond A, Antonarakis SE, Hannon GJ, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR, Rosenbloom KR, Sloan CA, Learned K, Malladi VS, Wong MC, Barber GP, Cline MS, Dreszer TR, Heitner SG, Karolchik D, Kent WJ, Kirkup VM, Meyer LR, Long JC, Maddren M, Raney BJ, Furey TS, Song L, Grasfeder LL, Giresi PG, Lee B-K, Battenhouse A, Sheffield NC, Simon JM, Showers KA, Safi A, London D, Bhinge AA, Shestak C, Schaner MR, Kim SK, Zhang ZZ, Mieczkowski PA, Mieczkowska JO, Liu Z, McDaniell RM, Ni Y, Rashid NU, Kim MJ, Adar S, Zhang Z, Wang T, Winter D, Keefe D, Birney E, Iyer VR, Lieb JD, Crawford GE, Li G, Sandhu KS, Zheng M, Wang P, Luo OJ, Shahab A, Fullwood MJ, Ruan X, Ruan Y, Myers RM, Pauli F, Williams BA, Gertz J, Marinov GK, Reddy TE, Vielmetter J, Partridge E, Trout D, Varley KE, Gasper C, Bansal A, Pepke S, Jain P, Amrhein H, Bowling KM, Anaya M, Cross MK, King B, Muratet MA, Antoshechkin I, Newberry KM, McCue K, Nesmith AS, Fisher-Aylor KI, Pusey B, DeSalvo G, Parker SL, Balasubramanian S, Davis NS, Meadows SK, Eggleston T, Gunter C, Newberry JS, Levy SE, Absher DM, Mortazavi A, Wong WH, Wold B, Blow MJ, Visel A, Pennachio LA, Elnitski L, Margulies EH, Parker SCJ, Petrykowska HM, Abyzov A, Aken B, Barrell D, Barson G, Berry A, Bignell A, Boychenko V, Bussotti G, Chrast J, Davidson C, Derrien T, Despacio-Reyes G, Diekhans M, Ezkurdia I, Frankish A, Gilbert J, Gonzalez JM, Griffiths E, Harte R, Hendrix DA, Howald C, Hunt T, Jungreis I, Kay M, Khurana E, Kokocinski F, Leng J, Lin MF, Loveland J, Lu Z, Manthravadi D, Mariotti M, Mudge J, Mukherjee G, Notredame C, Pei B, Rodriguez JM, Saunders G, Sboner A, Searle S, Sisu C, Snow C, Steward C, Tanzer A, Tapanari E, Tress ML, van Baren MJ, Walters N, Washietl S, Wilming L, Zadissa A, Zhang Z, Brent M, Haussler D, Kellis M, Valencia A, Gerstein M, Reymond A, Guigó R, Harrow J, Hubbard TJ, Landt SG, Frietze S, Abyzov A, Addleman N, Alexander RP, Auerbach RK, Balasubramanian S, Bettinger K, Bhardwaj N, Boyle AP, Cao AR, Cayting P, Charos A, Cheng Y, Cheng C, Eastman C, Euskirchen G, Fleming JD, Grubert F, Habegger L, Hariharan M, Harmanci A, Iyengar S, Jin VX, Karczewski KJ, Kasowski M, Lacroute P, Lam H, Lamarre-Vincent N, Leng J, Lian J, Lindahl-Allen M, Min R, Miotto B, Monahan H, Moqtaderi Z, Mu XJ, O’Geen H, Ouyang Z, Patacsil D, Pei B, Raha D, Ramirez L, Reed B, Rozowsky J, Sboner A, Shi M, Sisu C, Slifer T, Witt H, Wu L, Xu X, Yan K-K, Yang X, Yip KY, Zhang Z, Struhl K, Weissman SM, Gerstein M, Farnham PJ, Snyder M, Tenenbaum SA, Penalva LO, Doyle F, Karmakar S, Landt SG, Bhanvadia RR, Choudhury A, Domanus M, Ma L, Moran J, Patacsil D, Slifer T, Victorsen A, Yang X, Snyder M, White KP, Auer T, Centanin L, Eichenlaub M, Gruhl F, Heermann S, Hoeckendorf B, Inoue D, Kellner T, Kirchmaier S, Mueller C, Reinhardt R, Schertel L, Schneider S, Sinn R, Wittbrodt B, Wittbrodt J, Weng Z, Whitfield TW, Wang J, Collins PJ, Aldred SF, Trinklein ND, Partridge EC, Myers RM, Dekker J, Jain G, Lajoie BR, Sanyal A, Balasundaram G, Bates DL, Byron R, Canfield TK, Diegel MJ, Dunn D, Ebersol AK, Frum T, Garg K, Gist E, Hansen RS, Boatman L, Haugen E, Humbert R, Jain G, Johnson AK, Johnson EM, Kutyavin TV, Lajoie BR, Lee K, Lotakis D, Maurano MT, Neph SJ, Neri FV, Nguyen ED, Qu H, Reynolds AP, Roach V, Rynes E, Sabo P, Sanchez ME, Sandstrom RS, Sanyal A, Shafer AO, Stergachis AB, Thomas S, Thurman RE, Vernot B, Vierstra J, Vong S, Wang H, Weaver MA, Yan Y, Zhang M, Akey JM, Bender M, Dorschner MO, Groudine M, MacCoss MJ, Navas P, Stamatoyannopoulos G, Kaul R, Dekker J, Stamatoyannopoulos JA, Dunham I, Beal K, Brazma A, Flicek P, Herrero J, Johnson N, Keefe D, Lukk M, Luscombe NM, Sobral D, Vaquerizas JM, Wilder SP, Batzoglou S, Sidow A, Hussami N, Kyriazopoulou-Panagiotopoulou S, Libbrecht MW, Schaub MA, Kundaje A, Hardison RC, Miller W, Giardine B, Harris RS, Wu W, Bickel PJ, Banfai B, Boley NP, Brown JB, Huang H, Li Q, Li JJ, Noble WS, Bilmes JA, Buske OJ, Hoffman MM, Sahu AD, Kharchenko PV, Park PJ, Baker D, Taylor J, Weng Z, Iyer S, Dong X, Greven M, Lin X, Wang J, Xi HS, Zhuang J, Gerstein M, Alexander RP, Balasubramanian S, Cheng C, Harmanci A, Lochovsky L, Min R, Mu XJ, Rozowsky J, Yan K-K, Yip KY, Birney E, An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature. 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu Y-C, Pfenning A, Wang X, Liu MC, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh K-H, Feizi S, Karlic R, Kim A-R, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Abdennur N, Adli M, Akerman M, Barrera L, Antosiewicz-Bourget J, Ballinger T, Barnes MJ, Bates D, Bell RJA, Bennett DA, Bianco K, Bock C, Boyle P, Brinchmann J, Caballero-Campo P, Camahort R, Carrasco-Alfonso MJ, Charnecki T, Chen H, Chen Z, Cheng JB, Cho S, Chu A, Chung W-Y, Cowan C, Deng QA, Deshpande V, Diegel M, Ding B, Durham T, Echipare L, Edsall L, Flowers D, Genbacev-Krtolica O, Gifford C, Gillespie S, Giste E, Glass IA, Gnirke A, Gormley M, Gu H, Gu J, Hafler DA, Hangauer MJ, Hariharan M, Hatan M, Haugen E, He Y, Heimfeld S, Herlofsen S, Hou Z, Humbert R, Issner R, Jackson AR, Jia H, Jiang P, Johnson AK, Kadlecek T, Kamoh B, Kapidzic M, Kent J, Kim A, Kleinewietfeld M, Klugman S, Krishnan J, Kuan S, Kutyavin T, Lee A-Y, Lee K, Li J, Li N, Li Y, Ligon KL, Lin S, Lin Y, Liu J, Liu Y, Luckey CJ, Ma YP, Maire C, Marson A, Mattick JS, Mayo M, McMaster M, Metsky H, Mikkelsen T, Miller D, Miri M, Mukame E, Nagarajan RP, Neri F, Nery J, Nguyen T, O’Geen H, Paithankar S, Papayannopoulou T, Pelizzola M, Plettner P, Propson NE, Raghuraman S, Raney BJ, Raubitschek A, Reynolds AP, Richards H, Riehle K, Rinaudo P, Robinson JF, Rockweiler NB, Rosen E, Rynes E, Schein J, Sears R, Sejnowski T, Shafer A, Shen L, Shoemaker R, Sigaroudinia M, Slukvin I, Stehling-Sun S, Stewart R, Subramanian SL, Suknuntha K, Swanson S, Tian S, Tilden H, Tsai L, Urich M, Vaughn I, Vierstra J, Vong S, Wagner U, Wang H, Wang T, Wang Y, Weiss A, Whitton H, Wildberg A, Witt H, Won K-J, Xie M, Xing X, Xu I, Xuan Z, Ye Z, Yen C, Yu P, Zhang X, Zhang X, Zhao J, Zhou Y, Zhu J, Zhu Y, Ziegler S, Beaudet AE, Boyer LA, Jager PLD, Farnham PJ, Fisher SJ, Haussler D, Jones SJM, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai L-H, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu Y-C, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh K-H, Feizi S, Karlic R, Kim A-R, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, Jager PLD, Farnham PJ, Fisher SJ, Haussler D, Jones SJM, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai L-H, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Integrative analysis of 111 reference human epigenomes. Nature. 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding S-L, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, McGraw M, Nguyen TN, Nyhus J, Ojemann JG, Oldre A, Parry S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, Sunkin SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES, Conserved cell types with divergent features in human versus mouse cortex. Nature. 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ecker JR, Geschwind DH, Kriegstein AR, Ngai J, Osten P, Polioudakis D, Regev A, Sestan N, Wickersham IR, Zeng H, The BRAIN Initiative Cell Census Consortium: Lessons Learned toward Generating a Comprehensive Brain Cell Atlas. Neuron. 96, 542–557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng C-HL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan G-C, Cai L, Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature. 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao Z, Liu H, Xie F, Fischer S, Adkins RS, Aldridge AI, Ament SA, Bartlett A, Behrens MM, den Berge KV, Bertagnolli D, de Bézieux HR, Biancalani T, Booeshaghi AS, Bravo HC, Casper T, Colantuoni C, Crabtree J, Creasy H, Crichton K, Crow M, Dee N, Dougherty EL, Doyle WI, Dudoit S, Fang R, Felix V, Fong O, Giglio M, Goldy J, Hawrylycz M, Herb BR, Hertzano R, Hou X, Hu Q, Kancherla J, Kroll M, Lathia K, Li YE, Lucero JD, Luo C, Mahurkar A, McMillen D, Nadaf NM, Nery JR, Nguyen TN, Niu S-Y, Ntranos V, Orvis J, Osteen JK, Pham T, Pinto-Duarte A, Poirion O, Preissl S, Purdom E, Rimorin C, Risso D, Rivkin AC, Smith K, Street K, Sulc J, Svensson V, Tieu M, Torkelson A, Tung H, Vaishnav ED, Vanderburg CR, van Velthoven C, Wang X, White OR, Huang ZJ, Kharchenko PV, Pachter L, Ngai J, Regev A, Tasic B, Welch JD, Gillis J, Macosko EZ, Ren B, Ecker JR, Zeng H, Mukamel EA, A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature. 598, 103–110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kepecs A, Fishell G, Interneuron cell types are fit to function. Nature. 505, 318–326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, Zilionis R, Ratner A, Borges-Monroy R, Klein AM, Sabatini BL, Greenberg ME, Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci. 21, 120–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yap E-L, Greenberg ME, Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron. 100, 330–348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, Manno GL, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S, Molecular Architecture of the Mouse Nervous System. Cell. 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, Penn O, Bakken T, Menon V, Miller J, Fong O, Hirokawa KE, Lathia K, Rimorin C, Tieu M, Larsen R, Casper T, Barkan E, Kroll M, Parry S, Shapovalova NV, Hirschstein D, Pendergraft J, Sullivan HA, Kim TK, Szafer A, Dee N, Groblewski P, Wickersham I, Cetin A, Harris JA, Levi BP, Sunkin SM, Madisen L, Daigle TL, Looger L, Bernard A, Phillips J, Lein E, Hawrylycz M, Svoboda K, Jones AR, Koch C, Zeng H, Shared and distinct transcriptomic cell types across neocortical areas. Nature. 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang R, Xia C, Close JL, Zhang M, He J, Huang Z, Halpern AR, Long B, Miller JA, Lein ES, Zhuang X, Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science. 377, 56–62 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B, A map of the cis-regulatory sequences in the mouse genome. Nature. 488, 116–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abascal F, Acosta R, Addleman NJ, Adrian J, Afzal V, Ai R, Aken B, Akiyama JA, Jammal OA, Amrhein H, Anderson SM, Andrews GR, Antoshechkin I, Ardlie KG, Armstrong J, Astley M, Banerjee B, Barkal AA, Barnes IHA, Barozzi I, Barrell D, Barson G, Bates D, Baymuradov UK, Bazile C, Beer MA, Beik S, Bender MA, Bennett R, Bouvrette LPB, Bernstein BE, Berry A, Bhaskar A, Bignell A, Blue SM, Bodine DM, Boix C, Boley N, Borrman T, Borsari B, Boyle AP, Brandsmeier LA, Breschi A, Bresnick EH, Brooks JA, Buckley M, Burge CB, Byron R, Cahill E, Cai L, Cao L, Carty M, Castanon RG, Castillo A, Chaib H, Chan ET, Chee DR, Chee S, Chen H, Chen H, Chen J-Y, Chen S, Cherry JM, Chhetri SB, Choudhary JS, Chrast J, Chung D, Clarke D, Cody NAL, Coppola CJ, Coursen J, D’Ippolito AM, Dalton S, Danyko C, Davidson C, Davila-Velderrain J, Davis CA, Dekker J, Deran A, DeSalvo G, Despacio-Reyes G, Dewey CN, Dickel DE, Diegel M, Diekhans M, Dileep V, Ding B, Djebali S, Dobin A, Dominguez D, Donaldson S, Drenkow J, Dreszer TR, Drier Y, Duff MO, Dunn D, Eastman C, Ecker JR, Edwards MD, El-Ali N, Elhajjajy SI, Elkins K, Emili A, Epstein CB, Evans RC, Ezkurdia I, Fan K, Farnham PJ, Farrell NP, Feingold EA, Ferreira A-M, Fisher-Aylor K, Fitzgerald S, Flicek P, Foo CS, Fortier K, Frankish A, Freese P, Fu S, Fu X-D, Fu Y, Fukuda-Yuzawa Y, Fulciniti M, Funnell APW, Gabdank I, Galeev T, Gao M, Giron CG, Garvin TH, Gelboin-Burkhart CA, Georgolopoulos G, Gerstein MB, Giardine BM, Gifford DK, Gilbert DM, Gilchrist DA, Gillespie S, Gingeras TR, Gong P, Gonzalez A, Gonzalez JM, Good P, Goren A, Gorkin DU, Graveley BR, Gray M, Greenblatt JF, Griffiths E, Groudine MT, Grubert F, Gu M, Guigó R, Guo H, Guo Y, Guo Y, Gursoy G, Gutierrez-Arcelus M, Halow J, Hardison RC, Hardy M, Hariharan M, Harmanci A, Harrington A, Harrow JL, Hashimoto TB, Hasz RD, Hatan M, Haugen E, Hayes JE, He P, He Y, Heidari N, Hendrickson D, Heuston EF, Hilton JA, Hitz BC, Hochman A, Holgren C, Hou L, Hou S, Hsiao Y-HE, Hsu S, Huang H, Hubbard TJ, Huey J, Hughes TR, Hunt T, Ibarrientos S, Issner R, Iwata M, Izuogu O, Jaakkola T, Jameel N, Jansen C, Jiang L, Jiang P, Johnson A, Johnson R, Jungreis I, Kadaba M, Kasowski M, Kasparian M, Kato M, Kaul R, Kawli T, Kay M, Keen JC, Keles S, Keller CA, Kelley D, Kellis M, Kheradpour P, Kim DS, Kirilusha A, Klein RJ, Knoechel B, Kuan S, Kulik MJ, Kumar S, Kundaje A, Kutyavin T, Lagarde J, Lajoie BR, Lambert NJ, Lazar J, Lee AY, Lee D, Lee E, Lee JW, Lee K, Leslie CS, Levy S, Li B, Li H, Li N, Li S, Li X, Li YI, Li Y, Li Y, Li Y, Lian J, Libbrecht MW, Lin S, Lin Y, Liu D, Liu J, Liu P, Liu T, Liu XS, Liu Y, Liu Y, Long M, Lou S, Loveland J, Lu A, Lu Y, Lécuyer E, Ma L, Mackiewicz M, Mannion BJ, Mannstadt M, Manthravadi D, Marinov GK, Martin FJ, Mattei E, McCue K, McEown M, McVicker G, Meadows SK, Meissner A, Mendenhall EM, Messer CL, Meuleman W, Meyer C, Miller S, Milton MG, Mishra T, Moore DE, Moore HM, Moore JE, Moore SH, Moran J, Mortazavi A, Mudge JM, Munshi N, Murad R, Myers RM, Nandakumar V, Nandi P, Narasimha AM, Narayanan AK, Naughton H, Navarro FCP, Navas P, Nazarovs J, Nelson J, Neph S, Neri FJ, Nery JR, Nesmith AR, Newberry JS, Newberry KM, Ngo V, Nguyen R, Nguyen TB, Nguyen T, Nishida A, Noble WS, Novak CS, Novoa EM, Nuñez B, O’Donnell CW, Olson S, Onate KC, Otterman E, Ozadam H, Pagan M, Palden T, Pan X, Park Y, Partridge EC, Paten B, Pauli-Behn F, Pazin MJ, Pei B, Pennacchio LA, Perez AR, Perry EH, Pervouchine DD, Phalke NN, Pham Q, Phanstiel DH, Plajzer-Frick I, Pratt GA, Pratt HE, Preissl S, Pritchard JK, Pritykin Y, Purcaro MJ, Qin Q, Quinones-Valdez G, Rabano I, Radovani E, Raj A, Rajagopal N, Ram O, Ramirez L, Ramirez RN, Rausch D, Raychaudhuri S, Raymond J, Razavi R, Reddy TE, Reimonn TM, Ren B, Reymond A, Reynolds A, Rhie SK, Rinn J, Rivera M, Rivera-Mulia JC, Roberts BS, Rodriguez JM, Rozowsky J, Ryan R, Rynes E, Salins DN, Sandstrom R, Sasaki T, Sathe S, Savic D, Scavelli A, Scheiman J, Schlaffner C, Schloss JA, Schmitges FW, See LH, Sethi A, Setty M, Shafer A, Shan S, Sharon E, Shen Q, Shen Y, Sherwood RI, Shi M, Shin S, Shoresh N, Siebenthall K, Sisu C, Slifer T, Sloan CA, Smith A, Snetkova V, Snyder MP, Spacek DV, Srinivasan S, Srivas R, Stamatoyannopoulos G, Stamatoyannopoulos JA, Stanton R, Steffan D, Stehling-Sun S, Strattan JS, Su A, Sundararaman B, Suner M-M, Syed T, Szynkarek M, Tanaka FY, Tenen D, Teng M, Thomas JA, Toffey D, Tress ML, Trout DE, Trynka G, Tsuji J, Upchurch SA, Ursu O, Uszczynska-Ratajczak B, Uziel MC, Valencia A, Biber BV, van der Velde AG, Nostrand ELV, Vaydylevich Y, Vazquez J, Victorsen A, Vielmetter J, Vierstra J, Visel A, Vlasova A, Vockley CM, Volpi S, Vong S, Wang H, Wang M, Wang Q, Wang R, Wang T, Wang W, Wang X, Wang Y, Watson NK, Wei X, Wei Z, Weisser H, Weissman SM, Welch R, Welikson RE, Weng Z, Westra H-J, Whitaker JW, White C, White KP, Wildberg A, Williams BA, Wine D, Witt HN, Wold B, Wolf M, Wright J, Xiao R, Xiao X, Xu J, Xu J, Yan K-K, Yan Y, Yang H, Yang X, Yang Y-W, Yardımcı GG, Yee BA, Yeo GW, Young T, Yu T, Yue F, Zaleski C, Zang C, Zeng H, Zeng W, Zerbino DR, Zhai J, Zhan L, Zhan Y, Zhang B, Zhang J, Zhang J, Zhang K, Zhang L, Zhang P, Zhang Q, Zhang X-O, Zhang Y, Zhang Z, Zhao Y, Zheng Y, Zhong G, Zhou X-Q, Zhu Y, Zimmerman J, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, Halow J, Nostrand ELV, Freese P, Gorkin DU, Shen Y, He Y, Mackiewicz M, Pauli-Behn F, Williams BA, Mortazavi A, Keller CA, Zhang X-O, Elhajjajy SI, Huey J, Dickel DE, Snetkova V, Wei X, Wang X, Rivera-Mulia JC, Rozowsky J, Zhang J, Chhetri SB, Zhang J, Victorsen A, White KP, Visel A, Yeo GW, Burge CB, Lécuyer E, Gilbert DM, Dekker J, Rinn J, Mendenhall EM, Ecker JR, Kellis M, Klein RJ, Noble WS, Kundaje A, Guigó R, Farnham PJ, Cherry JM, Myers RM, Ren B, Graveley BR, Gerstein MB, Pennacchio LA, Snyder MP, Bernstein BE, Wold B, Hardison RC, Gingeras TR, Stamatoyannopoulos JA, Weng Z, Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, Lee C, Regalado SG, Read DF, Steemers FJ, Disteche CM, Trapnell C, Shendure J, A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell. 174, 1309–1324.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ, Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, Pokholok D, Aryee MJ, Steemers FJ, Lebofsky R, Buenrostro JD, Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol. 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, Zhang K, Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Preissl S, Fang R, Huang H, Zhao Y, Raviram R, Gorkin DU, Zhang Y, Sos BC, Afzal V, Dickel DE, Kuan S, Visel A, Pennacchio LA, Zhang K, Ren B, Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci. 21, 432–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corces MR, Shcherbina A, Kundu S, Gloudemans MJ, Frésard L, Granja JM, Louie BH, Eulalio T, Shams S, Bagdatli ST, Mumbach MR, Liu B, Montine KS, Greenleaf WJ, Kundaje A, Montgomery SB, Chang HY, Montine TJ, Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat Genet. 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, Aldinger KA, Pokholok D, Zhang F, Milbank JH, Zager MA, Glass IA, Steemers FJ, Doherty D, Trapnell C, Cusanovich DA, Shendure J, A human cell atlas of fetal chromatin accessibility. Science. 370 (2020), doi: 10.1126/science.aba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding S-L, Royall JJ, Sunkin SM, Ng L, Facer BAC, Lesnar P, Guillozet-Bongaarts A, McMurray B, Szafer A, Dolbeare TA, Stevens A, Tirrell L, Benner T, Caldejon S, Dalley RA, Dee N, Lau C, Nyhus J, Reding M, Riley ZL, Sandman D, Shen E, van der Kouwe A, Varjabedian A, Write M, Zollei L, Dang C, Knowles JA, Koch C, Phillips JW, Sestan N, Wohnoutka P, Zielke HR, Hohmann JG, Jones AR, Bernard A, Hawrylycz MJ, Hof PR, Fischl B, Lein ES, Comprehensive cellular-resolution atlas of the adult human brain: Adult human brain atlas. J Comp Neurol. 524, Spc1–Spc1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li YE, Preissl S, Hou X, Zhang Z, Zhang K, Qiu Y, Poirion OB, Li B, Chiou J, Liu H, Pinto-Duarte A, Kubo N, Yang X, Fang R, Wang X, Han JY, Lucero J, Yan Y, Miller M, Kuan S, Gorkin D, Gaulton KJ, Shen Y, Nunn M, Mukamel EA, Behrens MM, Ecker JR, Ren B, An atlas of gene regulatory elements in adult mouse cerebrum. Nature. 598, 129–136 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leung AW, Li JYH, The Molecular Pathway Regulating Bergmann Glia and Folia Generation in the Cerebellum. Cerebellum. 17, 42–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, Satpathy AT, Mumbach MR, Hoadley KA, Robertson AG, Sheffield NC, Felau I, Castro MAA, Berman BP, Staudt LM, Zenklusen JC, Laird PW, Curtis C, Network TCGAA, Greenleaf WJ, Chang HY, Akbani R, Benz CC, Boyle EA, Broom BM, Cherniack AD, Craft B, Demchok JA, Doane AS, Elemento O, Ferguson ML, Goldman MJ, Hayes DN, He J, Hinoue T, Imielinski M, Jones SJM, Kemal A, Knijnenburg TA, Korkut A, Lin D-C, Liu Y, Mensah MKA, Mills GB, Reuter VP, Schultz A, Shen H, Smith JP, Tarnuzzer R, Trefflich S, Wang Z, Weinstein JN, Westlake LC, Xu J, Yang L, Yau C, Zhao Y, Zhu J, The chromatin accessibility landscape of primary human cancers. Science. 362 (2018), doi: 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meuleman W, Muratov A, Rynes E, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Teodosiadis A, Reynolds A, Haugen E, Nelson J, Johnson A, Frerker M, Buckley M, Sandstrom R, Vierstra J, Kaul R, Stamatoyannopoulos J, Index and biological spectrum of human DNase I hypersensitive sites. Nature. 584, 244–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K, Hocker JD, Miller M, Hou X, Chiou J, Poirion OB, Qiu Y, Li YE, Gaulton KJ, Wang A, Preissl S, Ren B, A single-cell atlas of chromatin accessibility in the human genome. Cell. 184, 5985–6001.e19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oproescu A-M, Han S, Schuurmans C, New Insights Into the Intricacies of Proneural Gene Regulation in the Embryonic and Adult Cerebral Cortex. Front Mol Neurosci. 14, 642016 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wittstatt J, Reiprich S, Küspert M, Crazy Little Thing Called Sox—New Insights in Oligodendroglial Sox Protein Function. Int J Mol Sci. 20, 2713 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, Koeppen J, Viana JF, Oliveira JF, Voet T, Ponting CP, Belgard TG, Holt MG, Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun. 11, 1220 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marques S, Zeisel A, Codeluppi S, van Bruggen D, Falcão AM, Xiao L, Li H, Häring M, Hochgerner H, Romanov RA, Gyllborg D, Muñoz-Manchado AB, Manno GL, Lönnerberg P, Floriddia EM, Rezayee F, Ernfors P, Arenas E, Hjerling-Leffler J, Harkany T, Richardson WD, Linnarsson S, Castelo-Branco G, Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 352, 1326–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan Y-L, Yuan Y, Tian L, Microglial regional heterogeneity and its role in the brain. Mol Psychiatr. 25, 351–367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, Toland A, Calcuttawala K, Shin H, Pálovics R, Shin A, Wang EY, Luo J, Gate D, Schulz-Schaeffer WJ, Chu P, Siegenthaler JA, McNerney MW, Keller A, Wyss-Coray T, A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 603, 885–892 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, Shen Y, Pervouchine DD, Djebali S, Thurman RE, Kaul R, Rynes E, Kirilusha A, Marinov GK, Williams BA, Trout D, Amrhein H, Fisher-Aylor K, Antoshechkin I, DeSalvo G, See L-H, Fastuca M, Drenkow J, Zaleski C, Dobin A, Prieto P, Lagarde J, Bussotti G, Tanzer A, Denas O, Li K, Bender MA, Zhang M, Byron R, Groudine MT, McCleary D, Pham L, Ye Z, Kuan S, Edsall L, Wu Y-C, Rasmussen MD, Bansal MS, Kellis M, Keller CA, Morrissey CS, Mishra T, Jain D, Dogan N, Harris RS, Cayting P, Kawli T, Boyle AP, Euskirchen G, Kundaje A, Lin S, Lin Y, Jansen C, Malladi VS, Cline MS, Erickson DT, Kirkup VM, Learned K, Sloan CA, Rosenbloom KR, de Sousa BL, Beal K, Pignatelli M, Flicek P, Lian J, Kahveci T, Lee D, Kent WJ, Santos MR, Herrero J, Notredame C, Johnson A, Vong S, Lee K, Bates D, Neri F, Diegel M, Canfield T, Sabo PJ, Wilken MS, Reh TA, Giste E, Shafer A, Kutyavin T, Haugen E, Dunn D, Reynolds AP, Neph S, Humbert R, Hansen RS, Bruijn MD, Selleri L, Rudensky A, Josefowicz S, Samstein R, Eichler EE, Orkin SH, Levasseur D, Papayannopoulou T, Chang K-H, Skoultchi A, Gosh S, Disteche C, Treuting P, Wang Y, Weiss MJ, Blobel GA, Cao X, Zhong S, Wang T, Good PJ, Lowdon RF, Adams LB, Zhou X-Q, Pazin MJ, Feingold EA, Wold B, Taylor J, Mortazavi A, Weissman SM, Stamatoyannopoulos JA, Snyder MP, Guigo R, Gingeras TR, Gilbert DM, Hardison RC, Beer MA, Ren B, Consortium ME, A comparative encyclopedia of DNA elements in the mouse genome. Nature. 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bakken TE, Jorstad NL, Hu Q, Lake BB, Tian W, Kalmbach BE, Crow M, Hodge RD, Krienen FM, Sorensen SA, Eggermont J, Yao Z, Aevermann BD, Aldridge AI, Bartlett A, Bertagnolli D, Casper T, Castanon RG, Crichton K, Daigle TL, Dalley R, Dee N, Dembrow N, Diep D, Ding S-L, Dong W, Fang R, Fischer S, Goldman M, Goldy J, Graybuck LT, Herb BR, Hou X, Kancherla J, Kroll M, Lathia K, van Lew B, Li YE, Liu CS, Liu H, Lucero JD, Mahurkar A, McMillen D, Miller JA, Moussa M, Nery JR, Nicovich PR, Niu S-Y, Orvis J, Osteen JK, Owen S, Palmer CR, Pham T, Plongthongkum N, Poirion O, Reed NM, Rimorin C, Rivkin A, Romanow WJ, Sedeño-Cortés AE, Siletti K, Somasundaram S, Sulc J, Tieu M, Torkelson A, Tung H, Wang X, Xie F, Yanny AM, Zhang R, Ament SA, Behrens MM, Bravo HC, Chun J, Dobin A, Gillis J, Hertzano R, Hof PR, Höllt T, Horwitz GD, Keene CD, Kharchenko PV, Ko AL, Lelieveldt BP, Luo C, Mukamel EA, Pinto-Duarte A, Preissl S, Regev A, Ren B, Scheuermann RH, Smith K, Spain WJ, White OR, Koch C, Hawrylycz M, Tasic B, Macosko EZ, McCarroll SA, Ting JT, Zeng H, Zhang K, Feng G, Ecker JR, Linnarsson S, Lein ES, Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 598, 111–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minnoye L, Taskiran II, Mauduit D, Fazio M, Aerschot LV, Hulselmans G, Christiaens V, Makhzami S, Seltenhammer M, Karras P, Primot A, Cadieu E, van Rooijen E, Marine J-C, Egidy G, Ghanem GE, Zon L, Wouters J, Aerts S, Genome Res, in press, doi: 10.1101/gr.260844.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reilly MT, Faulkner GJ, Dubnau J, Ponomarev I, Gage FH, The Role of Transposable Elements in Health and Diseases of the Central Nervous System. J Neurosci. 33, 17577–17586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deniz Ö, Ahmed M, Todd CD, Rio-Machin A, Dawson MA, Branco MR, Endogenous retroviruses are a source of enhancers with oncogenic potential in acute myeloid leukaemia. Nat Commun. 11, 3506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Visel A, Minovitsky S, Dubchak I, Pennacchio LA, VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang F, Flanagan J, Su N, Wang L-C, Bui S, Nielson A, Wu X, Vo H-T, Ma X-J, Luo Y, RNAscope A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. J Mol Diagnostics. 14, 22–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yao Z, van Velthoven CTJ, Nguyen TN, Goldy J, Sedeno-Cortes AE, Baftizadeh F, Bertagnolli D, Casper T, Chiang M, Crichton K, Ding S-L, Fong O, Garren E, Glandon A, Gouwens NW, Gray J, Graybuck LT, Hawrylycz MJ, Hirschstein D, Kroll M, Lathia K, Lee C, Levi B, McMillen D, Mok S, Pham T, Ren Q, Rimorin C, Shapovalova N, Sulc J, Sunkin SM, Tieu M, Torkelson A, Tung H, Ward K, Dee N, Smith KA, Tasic B, Zeng H, A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell. 184, 3222–3241.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghandi M, Mohammad-Noori M, Ghareghani N, Lee D, Garraway L, Beer MA, gkmSVM: an R package for gapped-kmer SVM. Bioinform Oxf Engl. 32, 2205–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA, Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science. 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson J, Bundoc-Baronia R, Comiskey G, McGovern TF, Facing Addiction in America: The Surgeon General’s Report on Alcohol, Drugs, and Health: A Commentary. Alcohol Treat Q. 35, 445–454 (2017). [Google Scholar]
- 51.Kosoy R, Fullard JF, Zeng B, Bendl J, Dong P, Rahman S, Kleopoulos SP, Shao Z, Girdhar K, Humphrey J, Lopes K. de P., Charney AW, Kopell BH, Raj T, Bennett D, Kellner CP, Haroutunian V, Hoffman GE, Roussos P, Genetics of the human microglia regulome refines Alzheimer’s disease risk loci. Nat Genet. 54, 1145–1154 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drummond E, Wisniewski T, Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O’Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK, Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science. 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, Assael Y, Jumper J, Kohli P, Kelley DR, Effective gene expression prediction from sequence by integrating long-range interactions. Nat Methods. 18, 1196–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser L, Polosukhin I, Attention Is All You Need. Arxiv (2017), doi: 10.48550/arxiv.1706.03762. [DOI] [Google Scholar]
- 56.Yang X, Wen J, Yang H, Jones IR, Zhu X, Liu W, Li B, Clelland CD, Luo W, Wong MY, Ren X, Cui X, Song M, Liu H, Chen C, Nicolas Eng, Ravichandran M, Sun Y, Lee D, Buren EV, Jiang M-Z, Chan CSY, Ye CJ, Perera R, Gan L, Li Y, Shen Y, Characterize Alzheimer’s disease genetic association with molecular and cellular function in Microglia. Nature Genetics, in press. doi: 10.1038/s41588-023-01506-8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seong C, Kim HJ, Byun J-S, Kim Y, Kim D-Y, FoxO1 Controls Redox Regulation and Cellular Physiology of BV-2 Microglial Cells. Inflammation. 46, 752–762 (2023). [DOI] [PubMed] [Google Scholar]
- 58.Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, Turner JMA, Bertelsen MF, Murchison EP, Flicek P, Odom DT, Enhancer Evolution across 20 Mammalian Species. Cell. 160, 554–566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Bree EJ, Guimarães RLFP, Lundberg M, Blujdea ER, Rosenkrantz JL, White FTG, Poppinga J, Ferrer-Raventós P, Schneider A-FE, Clayton I, Haussler D, Reinders MJT, Holstege H, Ewing AD, Moses C, Jacobs FMJ, A hidden layer of structural variation in transposable elements reveals potential genetic modifiers in human disease-risk loci. Genome Res. 32, 656–670 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Durbin R, Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ou J, Liu H, Yu J, Kelliher MA, Castilla LH, Lawson ND, Zhu LJ, ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. Bmc Genomics. 19, 169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, Mukamel EA, Zhang K, Zhang Y, Behrens MM, Ecker JR, Ren B, Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat Commun. 12, 1337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, Sisu C, Wright JC, Armstrong J, Barnes I, Berry A, Bignell A, Boix C, Carbonell Sala S, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, García Girón C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Howe KL, Hunt T, Izuogu OG, Johnson R, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Riera FC, Ruffier M, Schmitt BM, Stapleton E, Suner M-M, Sycheva I, Uszczynska-Ratajczak B, Wolf MY, Xu J, Yang YT, Yates A, Zerbino D, Zhang Y, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Tress ML, Flicek P, GENCODE 2021. Nucleic Acids Res. 49, gkaa1087- (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolock SL, Lopez R, Klein AM, Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benaglia T, Chauveau D, Hunter DR, Young D, mixtools : An R Package for Analyzing Finite Mixture Models. J Stat Softw. 32 (2009), doi: 10.18637/jss.v032.i06. [DOI] [Google Scholar]
- 66.Amemiya HM, Kundaje A, Boyle AP, The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep-uk. 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell. 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Traag VA, Waltman L, van Eck NJ, From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep-uk. 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McInnes L, Healy J, Saul N, Großberger L, UMAP: Uniform Manifold Approximation and Projection. J Open Source Softw. 3, 861 (2018). [Google Scholar]
- 71.Suzuki R, Shimodaira H, Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 22, 1540–1542 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Drost H-G, Philentropy: Information Theory and Distance Quantification with R. J Open Source Softw. 3, 765 (2018). [Google Scholar]
- 73.Quinlan AR, Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Pe’er D, Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 174, 716–729.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding S-L, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, McGraw M, Nguyen TN, Nyhus J, Ojemann JG, Oldre A, Parry S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, Sunkin SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES, Conserved cell types with divergent features in human versus mouse cortex. Nature. 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li YE, Xiao M, Shi B, Yang Y-CT, Wang D, Wang F, Marcia M, Lu ZJ, Identification of high-confidence RNA regulatory elements by combinatorial classification of RNA–protein binding sites. Genome Biol. 18, 169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Müller A, Nothman J, Louppe G, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay É, Scikit-learn: Machine Learning in Python. Arxiv (2012), doi: 10.48550/arxiv.1201.0490. [DOI] [Google Scholar]
- 78.Hoyer PO, Non-negative matrix factorization with sparseness constraints. Arxiv (2004), doi: 10.48550/arxiv.cs/0408058. [DOI] [Google Scholar]
- 79.Kim H, Park H, Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics. 23, 1495–1502 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, Adey AC, Steemers FJ, Shendure J, Trapnell C, Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell. 71, 858–871.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Delignette-Muller ML, Dutang C, fitdistrplus : An R Package for Fitting Distributions. J Stat Softw. 64 (2015), doi: 10.18637/jss.v064.i04. [DOI] [Google Scholar]
- 83.Zhu C, Zhang Y, Li YE, Lucero J, Behrens MM, Ren B, Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods. 18, 283–292 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schep AN, Wu B, Buenrostro JD, Greenleaf WJ, chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods. 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T, deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shaw P, Uszkoreit J, Vaswani A, Self-Attention with Relative Position Representations. Arxiv (2018), doi: 10.48550/arxiv.1803.02155. [DOI] [Google Scholar]
- 87.Shazeer N, GLU Variants Improve Transformer. Arxiv (2020), doi: 10.48550/arxiv.2002.05202. [DOI] [Google Scholar]
- 88.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell. 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. Bmc Bioinformatics. 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee BT, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee CM, Muthuraman P, Nassar LR, Nguy B, Pereira T, Perez G, Raney BJ, Rosenbloom KR, Schmelter D, Speir ML, Wick BD, Zweig AS, Haussler D, Kuhn RM, Haeussler M, Kent WJ, The UCSC Genome Browser database: 2022 update. Nucleic Acids Res. 50, D1115–D1122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.S. W. G. of the P. G. Consortium, Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, Daly MJ, Price AL, Neale BM, LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barakat TS, Halbritter F, Zhang M, Rendeiro AF, Perenthaler E, Bock C, Chambers I, Functional Dissection of the Enhancer Repertoire in Human Embryonic Stem Cells. Cell Stem Cell. 23, 276–288.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perdomo-Sabogal Á, Nowick K, Genetic variation in human gene regulatory factors uncovers regulatory roles in local adaptation and disease. Genome Biol Evol. 11, 2178–2193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Playfoot CJ, Duc J, Sheppard S, Dind S, Coudray A, Planet E, Trono D, Transposable elements and their KZFP controllers are drivers of transcriptional innovation in the developing human brain. Genome Res. 31, 1531–1545 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Öztürk Z, O’Kane CJ, Pérez-Moreno JJ, Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front Neurosci-switz. 14, 48 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marballi KK, Gallitano AL, Immediate Early Genes Anchor a Biological Pathway of Proteins Required for Memory Formation, Long-Term Depression and Risk for Schizophrenia. Front Behav Neurosci. 12, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agrawal S, Baulch JE, Madan S, Salah S, Cheeks SN, Krattli RP, Subramanian VS, Acharya MM, Agrawal A, Impact of IL-21-associated peripheral and brain crosstalk on the Alzheimer’s disease neuropathology. Cell Mol Life Sci. 79, 331 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Colton C, Wilt S, Gilbert D, Chernyshev O, Snell J, Dubois-Dalcq M, Species differences in the generation of reactive oxygen species by microglia. Mol Chem Neuropathol. 28, 15–20 (1996). [DOI] [PubMed] [Google Scholar]
- 100.Kim YR, Kim YM, Lee J, Park J, Lee JE, Hyun Y-M, Neutrophils Return to Bloodstream Through the Brain Blood Vessel After Crosstalk With Microglia During LPS-Induced Neuroinflammation. Frontiers Cell Dev Biology. 8, 613733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Demultiplexed data can be accessed via the NEMO archive here: https://assets.nemoarchive.org/dat-d6r90fb. Raw data are available in the NCBI Gene Expression Omnibus (GEO) under accession number GSE244618. Processed data will be available on our web portal CATLAS: http://catlas.org.
Custom code and scripts used for analysis can be accessed here: https://github.com/yal054/snATACutils and https://github.com/r3fang/SnapATAC.
The deep learning model and pre-trained model can be download from https://github.com/yal054/epiformer.