

Abstract

The incorporation of boron functionalities into transition-metal catalysts has become a promising strategy to improve catalytic performance, although their synthesis typically entails the preparation of sophisticated bifunctional ligands. We report here the facile and direct postsynthetic functionalization of rhodium(I) compound [(η5-C9H7)Rh(PPh3)2] (1) by treatment with perfluorinated boranes. Borane addition to 1 results in an unusual C(sp2)-H hydride migration from the indenyl ligand to the metal with the concomitant formation of a C–B bond. In the case of Piers’ borane [HB(C6F5)2], this is followed by a subsequent hydride migration that leads to an unprecedented 1,2-hydrogen shift reminiscent of Milstein’s cooperative dearomatization pathways. Computational investigations provide a mechanistic picture for the successive hydride-migration steps, which enriches the non-innocent chemistry of widespread indenyl ligands. Moreover, we demonstrate that the addition of Piers’ borane is highly beneficial for catalysis, increasing catalyst efficiency up to 3 orders of magnitude.
Keywords: pendant borane, hydrogenation, ligand functionalization, rhodium, σ-borane complex
Introduction
The broad concept of metal–ligand cooperation has enriched the traditional notion of an active metal site surrounded by spectator ligands.1 Among the wide variety of bifunctional ligands, those bearing a Lewis acidic site have enjoyed increasing popularity.2 Not surprisingly, ligands containing group 13 elements are the preferred choice, with boron as the more prevalent,3,4 including the recent development of ligands with multiple borane sites.5 Accordingly, catalytic applications of boron-containing transition-metal catalysts are rapidly increasing (Figure 1a). To cite some relevant examples, Peters6 and Owen7 demonstrated this positive partnership in hydrogenation reactions and Bourissou for the dehalogenation of aromatic species,8 while Szymczak has shown enhanced activity and selectivity during alkyne hydrogenation.9 Moreover, the flexibility of M–B bonds has proven crucial to accommodate challenging bond activation reactions at transition metals, as demonstrated by Peters in the context of N2 reduction.10 In addition, pendant boranes without direct M–B bonding offer great opportunities, as recently demonstrated by Werlé on the chemoselective reduction of nitroarenes.11
Figure 1.

(a) Selected examples of transition-metal catalysts that contain boron functionalities; (b) representative example of polymerization catalysts based on an indenyl/boron ligand; and (c) rhodium complexes containing borate-functionalized indenyl ligands investigated in this work.
Borane containing ligands are usually tethered to the metal by two or more supporting groups to provide sufficient stability.3 While phosphine- and nitrogen-based donors have been amply used, the functionalization of widespread indenyl ligands with boron fragments remains comparatively underdeveloped. More precisely, they have been investigated in the narrow context of olefin polymerization (Figure 1b) with early transition metals (i.e., Zr, Ti, and Hf),12 serving as self-activating catalysts13 and to develop a new class of highly tuneable donor/acceptor metallocenes.14 However, further applications have not yet widely emerged, despite the paramount position occupied by indenyl and related ligands in organometallic chemistry and homogeneous catalysis.
Besides, in terms of synthetic approaches, prior examples of indenylborane complexes require the independent and usually non-trivial preparation of the bifunctional ligand prior to metal coordination.12,13,15 This typically entails the use of hard to handle haloboranes and presents some limitations in terms of regioselectivity since the boron fragment is almost exclusively incorporated at the C1 position. Although the post-synthetic functionalization of the simpler cyclopentadienyl ligands in compounds of type (η5-C5H5)MLn is well-known,16 this more convenient approach has not been applied to indenyl complexes.
In the context of ligand post-synthetic functionalization, we recently disclosed a reversible Cp* to metal hydride migration in compound [(η5-C5Me5)Rh(PMe3)2] upon addition of bulky Au(I) fragments with concomitant formation of new (Cp*)CH2–Au bonds.17 In contrast, ligand C–H bond activation does not take place for the related indenyl compound [(η5-C9H7)Rh(PPh3)2] (1), where we could only detect the formation of Rh → Au bimetallic adducts.18 Bearing in mind the high electrophilicity of perfluorinated boranes, we hypothesized that those could succeed in the direct functionalization of the indenyl ligand in 1 and thus provide facile access to bifunctional complexes with catalytic potential. With this goal, we describe herein the reactivity of compound 1 with perfluorinated boranes B(C6F5)3 and HB(C6F5)2, demonstrating that the boron functionality readily incorporates into the indenyl moiety (Figure 1c). The resulting complexes transcend previous related systems, which are mostly based on highly acidic early transition metals. Moreover, they are genuine motifs to investigate metallic FLP-type cooperativity19 because of the contrasting Lewis basic and acidic nature of the Rh(I) and borane fragments, respectively. In this vein, we provide preliminary studies that provide evidence of enhanced catalysis by using the hydrogenation of olefins as a model reaction.
Results and Discussion
We first treated a benzene solution of compound 1 with an equimolar amount of the highly electrophilic B(C6F5)3 borane at 25 °C. This reaction rapidly leads to the activation of the C–H bond of the indenyl fragment at the C2 position toward compound 2a (Scheme 1), where the new C–B bond results in a down-shifted 11B{1H} NMR resonance at −14.5 ppm [cf. 60 ppm for B(C6F5)3]. The migration of a hydride from the C2 position of the indenyl moiety generates a Rh–H bond, with the hydride resonating at −13.08 ppm (1JHRh = 22.9, 2JHP = 20.9 Hz) in the 1H NMR spectrum. This complex remains stable in solution for prolonged periods of time under an inert atmosphere.
Scheme 1. Activation of the Indenyl Ligand in Compound 1 upon Addition of Perfluorinated Boranes B(C6F5)3 and HB(C6F5)2.

ORTEP diagrams of complexes 2a, 2b, and 3 are represented. Most hydrogen atoms are excluded for clarity, and thermal ellipsoids are set at 50% probability.
The spontaneous formation of 2a represents a rare case of electrophilic substitution of the coordinated indenyl fragment and, as we anticipated, is a very convenient route to access bifunctional ligands of this kind. This contrasts with all prior examples that required the independent synthesis of the borylated indene precursor. Moreover, placing the boron function at the C2 position of the indenyl ligand, as observed for compound 2a, has remained elusive so far, and it was only accessed in unselective and very low-yielding syntheses.13b
Aside from trisubstituted B(C6F5)3, Piers’ borane HB(C6F5)220 seems more suitable to access an active pendant borane functionality since the resulting borate moiety would contain a B–H bond susceptible to participating in chemical transformations.4c,4d,6a Despite its reduced electrophilicity, this borane similarly reacts with compound 1 to yield the corresponding indenyl activated product 2b (Scheme 1). Not surprisingly, the spectroscopic multinuclear NMR signature of 2b is analogous to that of 2a except for a new resonance in the 1H NMR spectrum at 4.70 ppm, which is attributable to the B–H terminus that sharpens upon decoupling from 11B.
Remarkably, this compound readily evolves at room temperature in solution to form the new species 3 (Scheme 1) that clearly differs from compounds 2 by NMR spectroscopy. The asymmetry of 3 is exemplified by two resonances in its 31P{1H} NMR spectrum at 42.2 (dd, 1JPRh = 179, 3JPP = 42 Hz) and 38.1 (dd, 1JPRh = 183, 3JPP = 42 Hz) ppm, contrasting with singlet resonances in the case of compounds 2a and 2b (37.8 and 41.2 ppm, respectively). 19F{1H} NMR reveals the presence of two nonequivalent fluorinated arenes that do not interconvert in the NMR time-scale, a feature that is exclusive of this system among all complexes reported in this work. Thus, two clearly separated signals due to the ortho-fluorine atoms are recorded by 19F{1H} NMR at −129.8 and −128.1 ppm. The symmetry of the indenyl ligand is broken as well. Two overlapped distinctive resonances arising from the diastereotopic geminal protons emerge at 3.78 ppm, further suggesting a loss of aromaticity. Finally, the hydridic signal in the 1H NMR spectrum exhibits a notable shift to higher frequencies from −13.72 in 2b to −7.21 ppm in 3, and now it sharpens upon 11B decoupling.
The structure of compounds 2a, 2b, and 3 was authenticated by X-ray diffraction studies (Scheme 1).21 Compounds 2a and 2b exhibit the expected structure anticipated by spectroscopic analysis, with C–B bond distances [1.650(5), 2a; 1.616(6) Å, 2b] comparable to prior examples15 and other geometric parameters ranging normal values. The structure of 3 is more intriguing and can be described as a distorted square-planar Rh(I) species with two phosphine ligands and an η2-indene coordinated as an olefin that chelates the metal through an additional interaction with a BH fragment. We anticipated the latter interaction to be described as a 3-center-2-electron σ-borate complex. Related motifs have been previously rationalized as boron-hydrides stabilized by electrophilic metals22 or as metal hydrides that interact with pendant borane functions.23 Bearing in mind the intrinsic uncertainty of locating hydrogen atoms by X-ray diffraction techniques, the structure is defined by Rh–B and Rh–H bond distances of 2.412(2) and 1.80(3) Å and an acute B–H–Rh angle of 103.55(10)°, likely imposed by its intramolecular nature in a four-membered ring heterocycle. The bridging nature of the hydride fits well with the low-frequency signal recorded by 1H NMR (δ – 7.21).24 In turn, the relatively short Rh–B distance of 2.41, only moderately above the sum of their covalent radii (2.26 Å),25 suggests the direct participation of the three elements in a σ-type complex. We further examined the bonding situation in complex 3 by computational studies. The Quantum Theory of Atoms in Molecules (QTAIM) analysis carried out (Figure S33) shows bond critical points and paths between H and both Rh and B but not between Rh and B. In addition, the Lewis-type NBO analysis describes the B–H–Rh interaction as a B–H bond delocalizing into a σ* Rh–P orbital (Figure 2). Altogether, these results fit well with a description as a σ-borate rhodium complex. It is worth noting that the formation of 3 implies the migration of a hydride from the rhodium atom to the C1 position of the indenyl ligand, which losses its C5-aromaticity, somewhat resembling the well-known dearomatization pathways described by Milstein.1a Moreover, that precise hydride ligand originates from a prior migration from the C2 position of the indenyl moiety upon addition of Piers’ borane (vide infra computational studies). Therefore, we disclose here a rare non-innocent behavior of the indenyl ligand that consists of an overall sequential two-step 1,2-H migration promoted by an electrophile, a unique process that to the best of our knowledge has remained undisclosed and that adds to the already rich chemistry of indenyl ligands.
Figure 2.

NBOs (left) and NLMOs (right) 261 (donor, BD B–H) and 262 (acceptor, BD* Rh–P) were calculated for complex 3. Most H atoms were omitted for clarity.
We naturally questioned ourselves about the precise mechanism to account for this process, which we investigated by computational means using Gaussian 09 (Revision E.01) software26 with the dispersion corrected PBE0-D3 functional (Figure 3).27 The electrophilic attack of the borane to the indenyl presents a very low barrier (TS1) to give a square planar Rh(I) diene complex (B–C), from which hydride transfer to Rh (also viewed as a C–H bond oxidative addition), concomitant with the restoration of aromaticity, is both facile (TS2) and largely exergonic. The most challenging step is the direct migration of the metal hydride to the indenyl (TS6, which also may be understood as a reductive elimination), in good agreement with the detection of intermediate 2b. Once the CH2 moiety has been formed, coordination of the olefin to Rh gives complex 3. We monitored the conversion of 2b into 3 by 31P{1H} NMR spectroscopy (Figure S33), leading to a pseudo-first-order kinetic profile (k = 0.0011 s–1) corresponding to a ΔG298‡ of 21.5 kcal/mol, in excellent agreement with the calculated barrier (22.7 kcal/mol). Nonetheless, an alternative pathway involving the formation of an agostic B–H complex via TS3, which precedes the transfer of the metal hydride (TS4, 7.1 kcal/mol) from complex 2b, was found to be only slightly higher in energy (Figure S36) and cannot therefore be ruled out.
Figure 3.

Free energy profile for the conversion of 1 and Piers’ borane into 3 at the SMD(dichloromethane)-PBE0-D3(BJ)/SDD(Rh)/6-311+G(2d,p)//SMD(dichloromethane)-PBE0-D3(BJ)/SDD(Rh)/6-31G(d,p) level of theory. The conversion of intermediate CH2* into its conformer 3 is expected to be facile (see Figure S35).
The non-innocent behavior of the indenyl ligand upon reaction with Piers’ borane encouraged us to explore the catalytic potential of this system. To do so, we carried out preliminary investigations on the catalytic hydrogenation of olefins using rhodium precursor 1 and its boron-containing derivatives 2a and 3. For convenience, we selected the hydrogenation of styrene as a benchmark reaction to gauge the effect of the pendant borates. Hydrogenation of styrene toward ethylbenzene under mild conditions (25 °C, 0.5 atm of H2, 0.5 mol % [Rh]) proceeds in good yields after 1 h of reaction (Table 1) and, more importantly, provides a first hint on the effect of incorporating boron functionalities into the indenyl ligand.
Table 1. Catalyst Screening for the Hydrogenation of Styrene.

| entry | conditionsa | cat | yield (%)b | TOF (h–1) |
|---|---|---|---|---|
| 1 | A | 1 | 50 | 100 |
| 2 | A | 2a | 63 | 126 |
| 3 | A | 3 | 81 | 162 |
| 4 | B | 3 | 100 | 58,823 |
| 5 | C | 3 | 25 | 125,000 |
| 6 | B | 1 | trace |
Conditions: (A): [Rh] 0.5 mol %, H2 (0.5 atm), 25 °C, 1 h, C6D6 (0.6 mL). (B) [Rh] 1 ppm, H2 (4 atm), 60 °C, 17 h, neat. (C) [Rh] 0.1 ppm, H2 (4 atm), 60 °C, 20 h, neat.
1H NMR yields using hexamethylbenzene as an internal standard.
From the results depicted in Table 1, it is clear that catalysis is enhanced in the presence of the borate functionality but only to a minor extent under rather mild conditions. More precisely, catalytic performance spans from 50% yield of ethylbenzene formation for Rh(I) precursor 1 (entry 1) up to 63 and 81% for the bifunctional systems 2a and 3, respectively (entries 2 and 3). As foreseen, the best catalytic performance is achieved with compound 3, where the noninnocence behavior of the indenyl-boron ligand was already demonstrated by means of hydrogen migration events with the rhodium site. Next, we interrogated catalyst 3 and unfunctionalized 1 under harsher conditions. In particular, increasing the temperature to 60 °C and the H2 pressure to 4 atm and performing the catalysis in the absence of solvent allowed us to reach turnover numbers for compound 3 of up to 2.5 × 106, associated with turnover frequencies (TOFs) of around 1.25 × 105 h–1 (entry 5). Under the same conditions, the difference between 1 and 3 becomes dramatic as the former revealed inactive (entry 6). We exposed compound 1 to the aforesaid conditions to check whether the absence of catalyst results from limited stability, though 1 exhibited remarkable stability. Therefore, as discussed in detail in the following sections, the drastically increased activity of compound 3 derives from a more complex catalytic scenario. Besides, poisoning experiments with mercury and carbon disulfide, although not definitive, suggest the homogeneous nature of the active species (see Table S2 for further details).
We performed some preliminary investigations on the substrate scope with a series of unsaturated substrates, as shown in Scheme 2 (see the Supporting Information for experimental details). Aromatic and aliphatic alkenes, containing both electron-donating and electron-withdrawing groups, were readily hydrogenated under standard conditions (i–viii in Scheme 2). Internal olefins were also reduced (ix–x), though for the more challenging tetramethylethylene (xi), only low conversions were achieved even under harsher conditions. A vinyl (xii) and an allyl (xiii) ether could also be hydrogenated with full conversion without hydrogenolysis of the C–O bond, and similar results were obtained with an allene (xiv) and a terminal diene (xv). For other dienes (xvi–xxii), double hydrogenation of the two olefinic functions generally prevailed, though other isomers were also detected in variable amounts. α,β-unsaturated carbonyl compounds were selectively reduced at the olefinic C=C function, while the C=O bond remained intact (xxiii–xxv) unless longer reaction times and higher catalyst amounts are used (xxvi). The hydrogenation of terminal olefins attached to common ligands, as pyridine and phosphine, was also tested considering the inhibitor effect of free phosphines on other rhodium catalysts.28 Nonetheless, for this system, both vinylpyridine (xxix) and a divinyl terphenyl phosphine (xxx) could be hydrogenated in 90 and 50% yield, respectively. In the same vein, a catalytic run for styrene hydrogenation in the presence of two additional equivalents of PPh3 did not lead to catalysis decay. Finally, the hydrogenation of alkynes and α,β-unsaturated derivatives (xxxi–xxxviii) was also explored and led to reduced products in good yields, with specific regioselectivity with respect to semi- vs full-hydrogenation being controlled by tuning experimental conditions.
Scheme 2. Preliminary Studies on Substrate Scope for Complex 3 as a Hydrogenation Catalyst (in Blue are the Hydrogens Added during Hydrogenation).
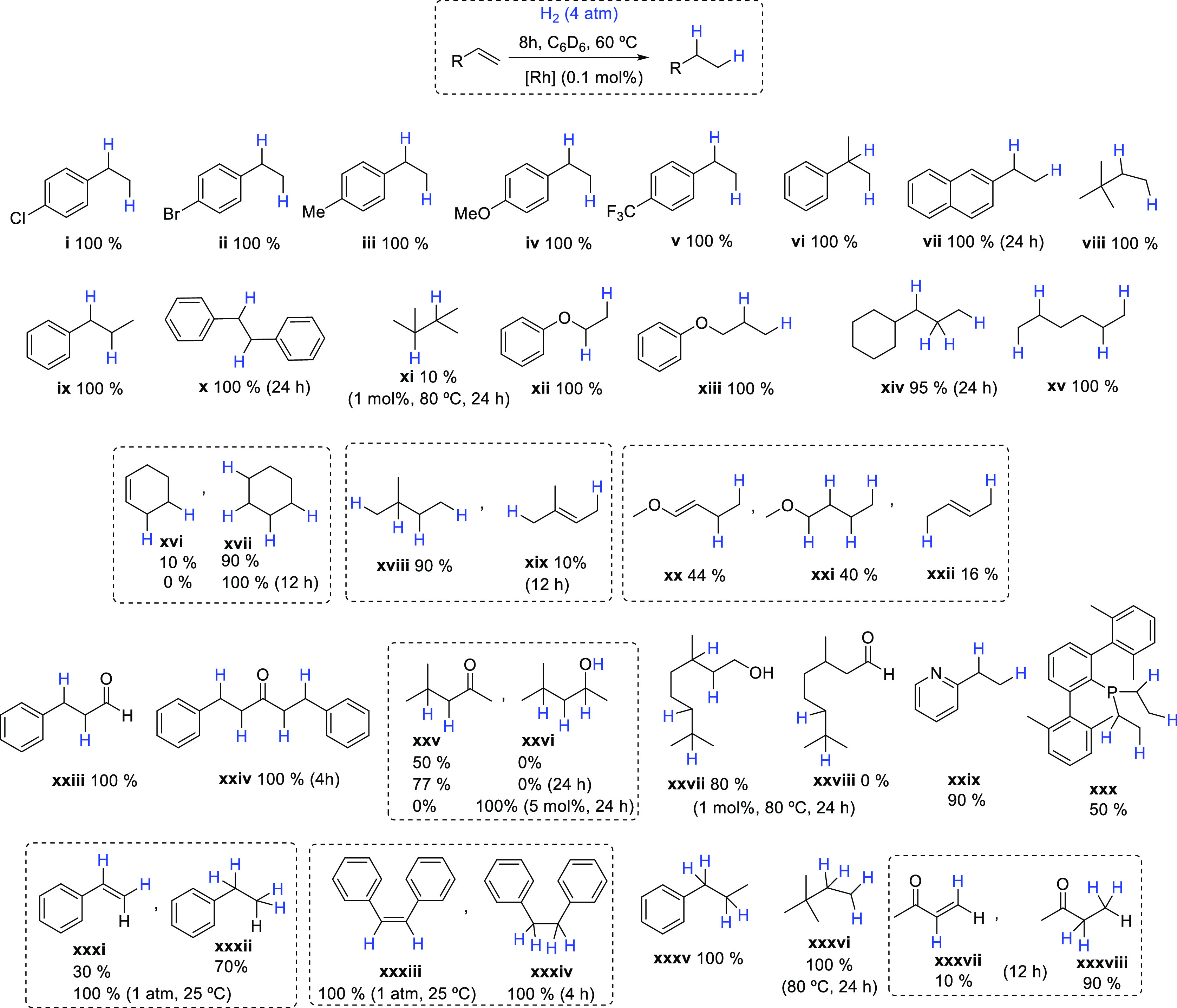
Conditions that deviate from standard are indicated in parentheses.
Aside from its direct use in catalysis, we decided to investigate the hydrogenation mechanism by both experimental and computational means in order to devise future and more challenging applications. We first carried out several stoichiometric experiments to shed some light on potential catalytic intermediates or resting states. Addition of one equivalent of styrene to a freshly prepared C6D6 solution of 3 resulted in immediate full conversion into a new species 4 (Scheme 3). Its formation is accompanied by a simplification of the corresponding 31P{1H} NMR spectrum to a single signal at 36.5 (d, 1JPRh = 145 Hz) ppm in 4. The higher symmetry of the latter species is further certified by the simpler 1H NMR pattern exhibited by the indenyl ligand (δ 6.96, 6.37, and 5.21 due to two protons each), which has recovered its aromaticity and η5-coordination. In addition, a low-frequency signal is now apparent at −13.70 ppm due to a Rh–H ligand, and no additional signals due to B–H units are visible. Instead, two signals due to two protons each at 3.03 and 2.01 ppm suggest the olefin to be inserted into the B–H bond. This occurs with concomitant regain of indenyl aromaticity, which by analogy to the DFT profile depicted in Figure 3, we attribute to the return of a hydrogen from the C1 position of the indenyl moiety to rhodium. Once more, this unusual insertion of the olefin into a borate B–H bond3e demonstrates the reversible nature of the hydrogen migrations between the metal, the indenyl ring, and the boron function, again resembling the reversible aromatization/dearomatization routes described by Milstein as a key feature for catalysis.1a
Scheme 3. Stoichiometric Reactivity of Compound 3 towards Styrene and Dihydrogen.

Figure 4 depicts the X-ray diffraction structure of 4 that corroborates our NMR-based formulation. The structure is analogous to that of 2b according to all of the geometric parameters. However, as opposed to the transformation of 2b into 3, in this case the absence of a stabilizing B–H unit seems to prevent the migration of the rhodium hydride toward the indenyl ring, which would otherwise result in a highly unsaturated metal site.
Figure 4.

ORTEP diagram of complex 4. Hydrogen atoms are excluded for clarity, and thermal ellipsoids are set at 50% probability.
Contrarily, exposure of C6D6 solutions of 3 to a H2 atmosphere under catalytically relevant conditions (6 atm, 25 °C) did not provoke the immediate disappearance of the complex, as was observed after styrene addition. In this case, it required longer times (ca. 4 h) to produce a new species 5 in around 70% spectroscopic yield (Scheme 3). Its formation is accompanied by the appearance of indane (C9H10) in equimolar amounts [representative signals at 1H NMR, δ = 2.69 (t, 3JHH = 7.4 Hz), 1.77 (q, 3JHH = 7.4 Hz); and 13C{1H} NMR, δ = 32.7, 25.3] due to hydrogenation and C–B bond cleavage at the indenyl ligand. In turn, complex 5 is characterized by 31P{1H} and 11B{1H} resonances at 38.2 (1JPRh = 115 Hz) and −1.2 (br) ppm, respectively. Nonetheless, the most distinctive feature of this compound is the presence of two low-frequency signals in the 1H NMR spectrum at −1.23 and −16.33 due to one proton each. Decoupling from either 11B or 31P causes the respective aforesaid resonances to clearly sharpen (Figure S21), suggesting that the lower-field resonance is directly coupled to the boron center as a B–H unit, while the higher-field signal is more influenced by the phosphine ligands. The chemical shift of the BH unit along with the geometrical parameters commented on below is indicative of some degree of σ-borane complex character, as later discussed in the context of computational studies. Besides, it has been recognized that the separation between meta and para fluorine atoms (Δm,p) of perfluorinated boranes is indicative of the coordination mode of the borane.29 For compound 5, Δm,p equals 3.3 [δF – 164.1 (Fm), −160,8 (Fp)], a slightly higher value than that in compounds 2b (Δm,p = 2.5) and 4 (Δm,p = 2.8), in agreement with a less anionic character of the boron atom. However, this value is mildly lower than that for 3 (Δm,p = 4.6), as expected for a stronger coordination of the boron center to the metal in complex 5. In solution, compound 5 exhibits dynamic behavior that accounts for the exchange of the Rh–H hydride with free dihydrogen and, at a lower rate, the intramolecular exchange between the two hydrides. Both processes were investigated by 2D-EXSY experiments, and the details are discussed in the Supporting Information (Figures S27 and S32).
The molecular structure of 5 was again corroborated by X-ray diffraction studies (Figure 5), revealing the absence of the indenyl ligand, as deduced from NMR analysis, and the formation of a highly unsaturated Rh → borane adduct. The departure of the indenyl ligand has facilitated the rearrangement of the phosphines, which are now located in a trans disposition defined by a P–Rh–P angle of 156.58(5)°. The two hydride-type ligands were located in the Fourier electron density map, with the lowest-energy configuration found by DFT matching well the experimental geometry and suggesting that they are also located trans to each other (Figure 5, up; H–Rh···H angle of 164.65°, H–Rh distances of 1.56 and 1.84 Å, the latter presenting a B–H bond distance of 1.31 Å). The crystallographic B–Rh distance is considerably shorter than that in 3, with a value of 2.316(5) Å, only slightly above of the sum of the corresponding covalent radii (2.26 Å)25 and in principle consistent with a strong dative interaction. The geometry around the boron center (not accounting for its hydride) is perfectly planar, as evinced by the sum of its three angles with Rh, C37, and C43 that accounts for an ideal 360°.
Figure 5.

ORTEP diagram of complex 5. Most hydrogen atoms are excluded for clarity, and thermal ellipsoids are set at 50% probability (up, left) and its DFT-optimized structure (up, right). Contour plots of NOCV deformation densities Δρ and associated energies ΔE(ρ) in 5 (down). Electron-density charge flows in the direction red → blue.
In principle, two extreme bonding scenarios could be envisioned for species 5: a Rh(III) dihydride that features a B···H interaction with a boryl ligand, or a Rh(I) hydride with a Z-type borane ligand (BR2H) that engages in Rh → B and (B–H) → Rh interactions. EDA-NOCV indicates that the Rh–B bond stems from donation from the electron-rich Rh center to the B atom as the major contribution [ΔE(ρ1) = −75.6 kcal/mol] (Figure 5, down). The delocalization of the B–H bond onto the terminal Rh–H σ* orbital finds support by NBO (Figure S37) and EDA-NOCV (Figure 5, bottom), although it contributes to the stabilization of the Rh → B adduct to a lesser extent [ΔE(ρ2) = −13.4 kcal/mol]. In contrast, QTAIM analysis reveals bond critical points and paths for the B–H, B–Rh, and terminal Rh–H bonds, but not between the B–H hydrogen atom and the Rh center (Figure S34), which can still be consistent with a chemical bond between two atoms.30 Therefore, we propose that structure 5 may be better described as a virtually unsupported transition metal-borane adduct. Braunschweig recently described a related unsupported Pt → borane adduct that could be presumably identified in solution but which readily evolved through B–F bond activation toward zwitterionic species.31 The instability of these sought-after motifs has been partly attributed to the required deformation energies to access pyramidal conformations at boron32 which, in our case, is likely compensated by the weak (B–H) → Rh interaction.
Next, we tested the catalytic competence of compounds 4 and 5 compared to 3 and the initial precursor 1, although the high instability of the former complexes precluded a fully precise evaluation. In fact, while compound 4 could be isolated in acceptable purity and tested as a catalyst, the isolation of 5 in the pure form escaped our efforts. As such, we generated compound 5in situ under a hydrogen atmosphere and then added to the resulting solution styrene and fresh H2 gas to monitor the catalytic reduction of the olefin by 1H NMR. The resulting kinetic profile for catalysts 3, 4, and 5 is comparatively similar (Figure 6), only differing in a slightly reduced induction period for the freshly prepared solution of 5. It is possible that Piers’ borane acts as an activating agent, facilitating the complete reduction and release of the indenyl ligand to yield 5, whose remarkable apparent simplicity may unlock the rather enhanced catalytic performance. Thus, it is possible that the active species in this system does not contain a pendant borane function but rather the cooperative action of a highly unsaturated Rh fragment and Piers’ borane and that the role of in situ-generated weakly coordinating borate anions is also important.
Figure 6.
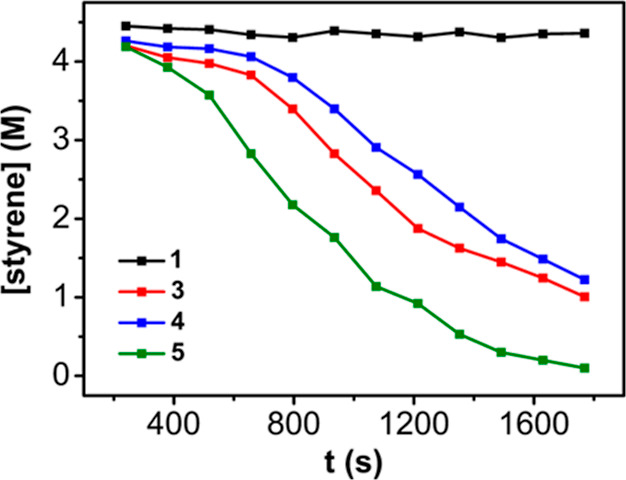
Comparison of the kinetic profiles for the hydrogenation of styrene for complexes 1, 3, 4, and 5. Conditions: [Rh] 0.001 mol %, H2 (6 atm), 25 °C, C6D6 (0.6 mL).
In this regard, we computationally investigated the reactivity of complexes 3 and 5 toward H2. While for complex 3 both the coordination and oxidative addition of H2 are endergonic (Figures S37 and 38), the more unsaturated 5 displays only mildly endergonic formation of a σ–H2 complex, from which oxidative addition leads to the exergonic generation of borate-bound species 5(H)2 (Figures S39 and 40). Based on these results and the superior catalytic profile of 5 compared to 3 (Figure 6), we focused on exploring pathways for hydrogenating styrene based on 5 and 5(H2). We have found that the latter species can effectively hydrogenate styrene (Figure 7): a shift to the κ1 coordination mode from the borate opens a vacant site that allows styrene coordination [TS5(H)2-Bbsty, 17.0 kcal/mol], which is followed by a barrierless olefin insertion into a Rh–hydride. The resulting agostic complex evolves through reductive elimination via TS5(H)CH3CHPh-H2 (19.3 kcal/mol), yielding the exergonic formation of ethylbenzene (EB) and 5′, a square planar species with greater stability than its isomer 5.
Figure 7.

Free energy profile for the hydrogenation of styrene from 5(H)2–B at the SMD(dichloromethane)-PBE0-D3(BJ)/SDD(Rh)/6-311+G(2d,p)//SMD(dichloromethane)-PBE0-D3(BJ)/SDD(Rh)/6-31G(d,p) level of theory.
Additionally, we investigated whether complex 5 could be catalytically competent through an alternative route involving direct transfer of its two metal-bound H atoms to styrene without the prior addition of H2. Indeed, Figure 8 shows that after styrene coordination (10.9 kcal/mol), both hydrides can be sequentially transferred via TS5styH1-A and TS5styH2-A, at 21.4 and 22.0 kcal/mol, respectively, giving the exergonic formation of ethylbenzene and the strongly unsaturated complex Ar2B–Rh(PPh3)2 (6), featuring a short Rh–B bond (1.91 Å). An alternative pathway is provided in Figure S42. NBO describes species 6 as Rh(I), according to 4 LPs on Rh, with the BAr2 fragment behaving as an anionic boryl ligand. Unsaturation at boron is compensated for by strong back-donation from Rh to the empty valence orbital of B (Figure S43). Contrary to 5, EDA-NOCV indicates that for 6, the electrostatic contribution is the major component of the bonding. However, the smallest value for the orbital interaction ΔEorb is obtained for the open-shell neutral fragments [Rh(PPh3)2]• and [B(C6F5)2]•, which provide a bonding scenario consistent on a covalent σ bond and Rh → B π backdonation (Figure S44B). Importantly for catalysis, species 6 can facilely coordinate and oxidatively add H2 (13.4 kcal/mol), reforming complex 5 (Figure S45). Based on this, we attempted the reverse reaction, that is, dehydrogenation of 5 toward the unsaturated formally boryl complex 6. Although our efforts to isolate and fully characterize such a compound have so far been unfruitful due to the high reactivity of these species, we could identify by NMR a major compound assigned to 6 after exposing solutions of 5 to dynamic vacuum. As expected, this species does not exhibit low-frequency 1H NMR hydride signals, while a 31P{1H} resonance is recorded at 43.9 Hz (1JPRh = 211 Hz).
Figure 8.

Free energy profile for the hydrogenation of styrene from 5 at the SMD(dichloromethane)-PBE0-D3(BJ)/SDD(Rh)/6-311+G(2d,p)//SMD(dichloromethane)-PBE0-D3(BJ)/SDD(Rh)/6-31G(d,p) level of theory.
Overall, it is rather clear that catalyst 3 containing the boron functionality is remarkably more efficient than its borane-free precursor 1 (Figure 6). Thus, while 3 reduces styrene with full conversion even at catalyst loadings as low as 0.001 mol %, the parent complex 1 only produces trace amounts of ethylbenzene under otherwise identical conditions. The above mechanistic investigations point to multiple roles of Piers’ borane to enhance catalysis. This is better exemplified in the overall mechanistic picture proposed in Scheme 4. On one hand, the borane functionalizes the indenyl ligand, facilitating its release as indane and giving access to a more reactive active species (5), characterized by a dative bond from rhodium to the borane. Thus, a second role of the borane entails the stabilization of the highly unsaturated Rh(I) fragment [Rh(H)(PPh3)2]. In addition, it also provides several plausible routes for Rh/B cooperation during the catalytic hydrogenation of styrene, the two main pathways being depicted in Scheme 4. Route A involves the initial oxidative addition of H2 onto 5, while route B entails the initial coordination of the olefin, being both associated with Figures 7 and 8, respectively. In route A, the borane moiety directly participates in H–H bond cleavage across the Rh → B bond. Besides, the flexibility of the bridging hydrides enables the generation of a vacant site at the metal assisted by boron. In turn, the main intermediates in route B evince an active participation of the boron fragment both to stabilize the low coordinate fragment [Rh(PPh3)2] by means of formal boryl compound 6 and to activate dihydrogen across the Rh=B bond.
Scheme 4. Proposed Competing Mechanisms for the Hydrogenation of Styrene Using the Combination of Precatalyst 1 and Piers’ Borane HB(C6F5)2 through Common Active Species 5.

Conclusions
The synthesis of boron containing transition-metal complexes has become a frontier approach toward innovative catalysts. At variance to the currently more extended methods involving the synthesis of sophisticated bifunctional ligands containing N- and P-donors, we exploit here a convenient postsynthetic functionalization procedure. This entails the direct treatment of electron-rich compound [(η5-C9H7)Rh(PPh3)2] with perfluorinated boranes, an approach that had been almost exclusively examined in the context of early transition-metal polymerization catalysis for simpler C5H5-based systems. We demonstrate that borate functions can be easily installed on the indenyl ligand through the formation of new C–B bonds. Importantly, this triggers an unprecedented stepwise 1,2-hydride shift at the indenyl moiety that evinces potential for catalysis. We investigated this potential for the benchmark hydrogenation of olefins. In this case, the catalyst efficiency is boosted up to 3 orders of magnitude compared to the boron-free precursor. Our mechanistic investigations suggest a complex role for the boron function, first as an activating fragment to access a highly unsaturated active species through indane release and, second, as a cooperative partner to accomplish catalytic hydrogenation by synergistic action with the rhodium center.
Acknowledgments
We thank Grant PID2019-110856GA-I00 funded by MCIN/AEI/10.13039/501100011033, Junta de Andalucía (P18-FR-4688), and US/JUNTA/FEDER, UE (US-1380849). J.J.M. thanks Junta de Andalucía for the postdoctoral program “Personal Investigador Doctor” (ref DOC_00153). The authors acknowledge the use of CESGA computational facilities. Drs. López-Serrano and Roselló are gratefully acknowledged for his assistance on EXSY studies and for general discussions, respectively.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c02764.
Experimental procedures, NMR spectra, crystal structure determinations, exchange EXSY experiments, details on catalytic studies, and computational details (PDF)
Crystallographic information (CIF)
Crystallographic information (CIF)
Crystallographic information (CIF)
Crystallographic information (CIF)
Crystallographic information (CIF)
Computational data (XYZ)
Author Contributions
M.G.A. synthesized and characterized all compounds. J.J.M. and M.A.G. carried out computational studies. M.G.A. and C.M. carried out XRD studies. J.C. wrote the original draft. J.C. supervised the overall project. All authors contributed to review and editing. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Gunanathan C.; Milstein D. Metal–Ligand Cooperation by Aromatization Dearomatization: A New Paradigm in Bond Activation and “Green” Catalysis. Acc. Chem. Res. 2011, 44, 588–602. 10.1021/ar2000265. [DOI] [PubMed] [Google Scholar]; b Khusnutdinova J. R.; Milstein D. Metal–Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54 (42), 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]; c Elsby M. R.; Baker R. T. Strategies and mechanisms of metal–ligand cooperativity in first-row transition metal complex catalysts. Chem. Soc. Rev. 2020, 49 (24), 8933–8987. 10.1039/D0CS00509F. [DOI] [PubMed] [Google Scholar]
- a Kuzu I.; Krummenacher I.; Meyer J.; Armbruster F.; Breher F. Multidentate ligand systems featuring dual functionality. Dalton Trans. 2008, 5836–5865. 10.1039/b808347a. [DOI] [PubMed] [Google Scholar]; b Amgoune A.; Bourissou D. σ-Acceptor Z-type ligands for transition metals. Chem. Commun. 2011, 47, 859–871. 10.1039/C0CC04109B. [DOI] [PubMed] [Google Scholar]; c Devillard M.; Bouhadir G.; Bourissou D. Cooperation between Transition Metals and Lewis Acids: A Way To Activate H2 and H–E bonds. Angew. Chem., Int. Ed. 2015, 54, 730–732. 10.1002/anie.201410781. [DOI] [PubMed] [Google Scholar]; d Jones J. S.; Gabbaï F. P. Coordination- and Redox-Noninnocent Behavior of Ambiphilic Ligands Containing Antimony. Acc. Chem. Res. 2016, 49 (5), 857–867. 10.1021/acs.accounts.5b00543. [DOI] [PubMed] [Google Scholar]; e Guan W.; Zeng G.; Kameo H.; Nakao Y.; Sakaki S. Cooperative Catalysis of Combined Systems of Transition-Metal Complexes with Lewis Acids: Theoretical Understanding. Chem. Rec. 2016, 16, 2405–2425. 10.1002/tcr.201600086. [DOI] [PubMed] [Google Scholar]; f You D.; Gabbaï F. P. Tunable σ-Accepting, Z-Type Ligands for Organometallic Catalysis. Trends Chem. 2019, 1 (5), 485–496. 10.1016/j.trechm.2019.03.011. [DOI] [Google Scholar]
- a Braunschweig H.; Dewhurst R. D.; Schneider A. Electron-Precise Coordination Modes of Boron-Centered Ligands. Chem. Rev. 2010, 110 (7), 3924–3957. 10.1021/cr900333n. [DOI] [PubMed] [Google Scholar]; b Owen G. R. Hydrogen atom storage upon Z-class borane ligand functions: an alternative approach to ligand cooperation. Chem. Soc. Rev. 2012, 41, 3535–3546. 10.1039/c2cs15346g. [DOI] [PubMed] [Google Scholar]; c Kameo H.; Nakazawa H. Recent Developments in the Coordination Chemistry of Multidentate Ligands Featuring a Boron Moiety. Chem.—Asian J. 2013, 8, 1720–1734. 10.1002/asia.201300184. [DOI] [PubMed] [Google Scholar]; d Owen G. R. Functional group migrations between boron and metal centres within transition metal–borane and –boryl complexes and cleavage of H–H, E–H and E–E′ bonds. Chem. Commun. 2016, 52, 10712–10726. 10.1039/C6CC03817D. [DOI] [PubMed] [Google Scholar]; e Saha K.; Ghosh S. Hydroboration reactions using transition metal borane and borate complexes: an overview. Dalton Trans. 2022, 51, 2631–2640. 10.1039/D1DT04289K. [DOI] [PubMed] [Google Scholar]
- To see some relevant examples:; a Miller A. J. M.; Labinger J. A.; Bercaw J. E. Reductive Coupling of Carbon Monoxide in a Rhenium Carbonyl Complex with Pendant Lewis Acids. J. Am. Chem. Soc. 2008, 130 (36), 11874–11875. 10.1021/ja805108z. [DOI] [PubMed] [Google Scholar]; b Kiernicki J. J.; Zeller M.; Szymczak N. K. Hydrazine Capture and N–N Bond Cleavage at Iron Enabled by Flexible Appended Lewis Acids. J. Am. Chem. Soc. 2017, 139 (50), 18194–18197. 10.1021/jacs.7b11465. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Iannetelli A.; Tizzard G.; Coles S. J.; Owen G. R. Sequential Migrations between Boron and Rhodium Centers: A Cooperative Process between Rhodium and a Monosubstituted Borohydride Unit. Inorg. Chem. 2018, 57 (1), 446–456. 10.1021/acs.inorgchem.7b02700. [DOI] [PubMed] [Google Scholar]; d Iannetelli A.; Da Costa R. C.; Guwy A. J.; Tizzard G. J.; Coles S. J.; Owen G. R. Transformation of a Norbornadiene Unit to Ethylenylcyclopentene Requiring Cooperation between Boron and Rhodium Centers. Organometallics 2020, 39 (10), 1976–1988. 10.1021/acs.organomet.0c00155. [DOI] [Google Scholar]
- a Zurakowski J. A.; Austen B. J. H.; Brown K. R.; Drover M. W. Bis(1-Bora-4-phosphorinane) Ring Closure at Cp*M (M = Fe, Co) Complexes. Chem. Commun. 2022, 58, 2500–2503. 10.1039/D1CC07060F. [DOI] [PubMed] [Google Scholar]; b Zurakowski J. A.; Austen B. J. H.; Drover M. W. Exterior Decorating: Lewis Acid Secondary Coordination Spheres for Cooperative Reactivity. Trends Chem. 2022, 4, 331–346. 10.1016/j.trechm.2022.01.007. [DOI] [Google Scholar]; c Demchuk M.; Zurakowski J. A.; Austen B. J. H.; Nelson D. J.; Drover M. W. Competitive Gold/Nickel Transmetalation. Chem. Commun. 2022, 58, 68–71. 10.1039/D1CC06064C. [DOI] [PubMed] [Google Scholar]; d Drover M. W.; Dufour M. C.; Lesperance-Nantau L. A.; Noriega R. P.; Levin K.; Schurko R. W. Octaboraneyl Complexes of Nickel: Monomers for Redox-Active Coordination Polymers. Chem.—Eur. J. 2020, 26, 11180–11186. 10.1002/chem.202001218. [DOI] [PubMed] [Google Scholar]; e Beagan D. M.; Kiernicki J. J.; Zeller M.; Szymczak N. K. A Bidentate Ligand Featuring Ditopic Lewis Acids in the Second Sphere for Selective Substrate Capture and Activation. Angew. Chem., Int. Ed. 2023, 62, e202218907 10.1002/anie.202218907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Harman W. H.; Peters J. C. Reversible H2 Addition across a Nickel–Borane Unit as a Promising Strategy for Catalysis. J. Am. Chem. Soc. 2012, 134 (11), 5080–5082. 10.1021/ja211419t. [DOI] [PubMed] [Google Scholar]; b Fong H.; Moret M.-E.; Lee Y.; Peters J. C. Heterolytic H2 Cleavage and Catalytic Hydrogenation by an Iron Metallaboratrane. Organometallics 2013, 32, 3053–3062. 10.1021/om400281v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoureas N.; Kuo Y. Y.; Haddow M. F.; Owen G. R. Double addition of H2to transition metal–borane complexes: a ‘hydride shuttle’ process between boron and transition metal centres. Chem. Commun. 2011, 47, 484–486. 10.1039/C0CC02245D. [DOI] [PubMed] [Google Scholar]
- Kameo H.; Yamamoto J.; Asada A.; Nakazawa H.; Matsuzaka H.; Bourissou D. Palladium–Borane Cooperation: Evidence for an Anionic Pathway and Its Application to Catalytic Hydro-/Deutero-dechlorination. Angew. Chem., Int. Ed. 2019, 58, 18783–18787. 10.1002/anie.201909675. [DOI] [PubMed] [Google Scholar]
- Tseng K. N. T.; Kampf J. W.; Szymczak N. K. Modular Attachment of Appended Boron Lewis Acids to a Ruthenium Pincer Catalyst: Metal–Ligand Cooperativity Enables Selective Alkyne Hydrogenation. J. Am. Chem. Soc. 2016, 138 (33), 10378–10381. 10.1021/jacs.6b03972. [DOI] [PubMed] [Google Scholar]
- See for example:; a Moret M.-E.; Peters J. C. N2 Functionalization at Iron Metallaboratranes. J. Am. Chem. Soc. 2011, 133, 18118–18121. 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fajardo J. Jr.; Peters J. C. Tripodal P3XFe–N2 Complexes (X = B, Al, Ga): Effect of the Apical Atom on Bonding, Electronic Structure, and Catalytic N2-to-NH3 Conversion. Inorg. Chem. 2021, 60, 1220–1227. 10.1021/acs.inorgchem.0c03354. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dong H. T.; Chalkley M. J.; Oyala P. H.; Zhao J.; Alp E. E.; Hu M. Y.; Peters J. C.; Lehnert N. Exploring the Limits of Dative Boratrane Bonding: Iron as a Strong Lewis Base in Low-Valent Non-Heme Iron-Nitrosyl Complexes. Inorg. Chem. 2020, 59, 14967–14982. 10.1021/acs.inorgchem.0c01686. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Matson B. D.; Peters J. C. Fe-Mediated HER vs N2RR: Exploring Factors That Contribute to Selectivity in P3EFe(N2) (E = B, Si, C) Catalyst Model Systems. ACS Catal. 2018, 8, 1448–1455. 10.1021/acscatal.7b03068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh V.; Chatterjee B.; Chang W. C.; Cramer H. H.; Hindemith C.; Randel H.; Weyhermüller T.; Farès C.; Werlé C. An Adaptive Rhodium Catalyst to Control the Hydrogenation Network of Nitroarenes. Angew. Chem., Int. Ed. 2022, 61, e202205515 10.1002/anie.202205515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Braunschweig H.; Dörfler R.; Friedrich M.; Kraft M.; Oechsner A. Catalytic Activity of [2]Borametallocenophanes. Z. Anorg. Allg. Chem. 2011, 637, 2125–2128. 10.1002/zaac.201100315. [DOI] [Google Scholar]; b Rufanov K.; Avtomonov E.; Kazennova N.; Kotov V.; Khvorost A.; Lemenovskii D.; Lorberth J. Polyelement substituted cyclopentadienes and indenes — Novel ligand precursors for organotransition metal chemistry. J. Organomet. Chem. 1997, 536–537, 361–373. 10.1016/S0022-328X(96)06757-5. [DOI] [Google Scholar]; c Sun Y.; Piers W. E.; Yap G. P. A. Acetone and Acetophenone Adducts of the Zwitterionic Zirconocene Cp*[η5-C5Me4CH2B(C6F5)3]ZrC6H5. Organometallics 1997, 16 (12), 2509–2513. 10.1021/om970066b. [DOI] [Google Scholar]
- a Sun Y.; Spence R. E. v. H.; Piers W. E.; Parvez M.; Yap G. P. A. Intramolecular Ion–Ion Interactions in Zwitterionic Metallocene Olefin Polymerization Catalysts Derived from “Tucked-In” Catalyst Precursors and the Highly Electrophilic Boranes XB(C6F5)2 (X = H, C6F5). J. Am. Chem. Soc. 1997, 119 (22), 5132–5143. 10.1021/ja970140h. [DOI] [Google Scholar]; b Duchateau R.; Lancaster S. J.; Thornton-Pett M.; Bochmann M. Synthesis of Cyclopentadienyl-Indenyl-and Fluorenylbis(pentafluorophenyl)boranes as Ligands in Titanium and Zirconium Half-Sandwich Complexes. The Crystal Structures of C13H9B(C6F5)2·t-BuNH2, C13H8SiMe3B(C6F5)2, and {η5-C5H4B(C6F5)2}TiCl3. Organometallics 1997, 16 (23), 4995–5005. 10.1021/om970433j. [DOI] [Google Scholar]; c Song X.; Bochmann M. Preparation of (Cp*) {C5Me4CH2B(C6F5)3}ZrPh, a novel zwitterionic single-component alkene polymerisation catalyst. J. Organomet. Chem. 1997, 545–546, 597–600. 10.1016/S0022-328X(97)00389-6. [DOI] [Google Scholar]; d Kohrt S.; Kehr G.; Daniliuc C. G.; Rojas R. S.; Rieger B.; Troll C.; Erker G. Borata-Alkene Derived Syntheses of (F5C6)2B-Substituted Bis(indenyl) Group 4 Metal Complexes. Organometallics 2016, 35, 2689–2693. 10.1021/acs.organomet.6b00427. [DOI] [Google Scholar]
- Ostoja Starzewski K. A.; Kelly W. M.; Stumpf A.; Freitag D. Donor/Acceptor Metallocenes: A New Structure Principle in Catalyst Design. Angew. Chem., Int. Ed. 1999, 38, 2439–2443. . [DOI] [PubMed] [Google Scholar]
- a Arndt P.; Jäger-Fiedler U.; Klahn M.; Baumann W.; Spannenberg A.; Burlakov V.; Rosenthal U. Formation of Zirconocene Fluoro Complexes: No Deactivation in the Polymerization of Olefins by the Contact-Ion-Pair Catalysts [Cp′2ZrR]+[RB(C6F5)3]–†. Angew. Chem., Int. Ed. 2006, 45 (25), 4195–4198. 10.1002/anie.200600361. [DOI] [PubMed] [Google Scholar]; b Swarnakar A. K.; Ferguson M. J.; McDonald R.; Rivard E. Transition metal-mediated donor–acceptor coordination of low-oxidation state Group 14 element halides. Dalton Trans. 2016, 45, 6071–6078. 10.1039/C5DT03018H. [DOI] [PubMed] [Google Scholar]; c Tao X.; Daniliuc C. G.; Soloviova K.; Strassert C. A.; Kehr G.; Erker G. Arylallenes and the halogeno-B(C6F5)2 reagents: facile formation of 2-borylindenes. Chem. Commun. 2019, 55, 10166–10169. 10.1039/C9CC04199K. [DOI] [PubMed] [Google Scholar]
- See the following review work describing examples of borane addition to cyclopentadienyl ligands and references cited thereinAldridge S.; Bresner C. The coordination chemistry of boryl and borate substituted cyclopentadienyl ligands. Coord. Chem. Rev. 2003, 244, 71–92. 10.1016/S0010-8545(03)00100-0. [DOI] [Google Scholar]
- Alférez M. G.; Moreno J. J.; Hidalgo N.; Campos J. Reversible Hydride Migration from C5Me5 to RhI Revealed by a Cooperative Bimetallic Approach. Angew. Chem., Int. Ed. 2020, 59, 20863–20867. 10.1002/anie.202008442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alférez M. G.; Moreno J. J.; Maya C.; Campos J. Polarized Au(i)/Rh(i) bimetallic pairs cooperatively trigger ligand non-innocence and bond activation. Dalton Trans. 2023, 52, 3835–3845. 10.1039/D3DT00410D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alférez M. G.; Hidalgo N.; Campos J.. Frustated Lewis Pairs based on Transition Metals. Frustrated Lewis Pairs; Slootweg C., Jupp A., Eds.; Springer, 2020. [Google Scholar]; b Navarro M.; Campos J. Bimetallic frustrated Lewis pairs. Adv. Organomet. Chem. 2021, 75, 95–148. 10.1016/bs.adomc.2021.01.001. [DOI] [Google Scholar]; c Navarro M.; Moreno J. J.; Pérez-Jiménez M.; Campos J. Small molecule activation with bimetallic systems: a landscape of cooperative reactivity. Chem. Commun. 2022, 58, 11220–11235. 10.1039/D2CC04296G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D. J.; von H Spence R. E.; Piers W. E.; Piers W. E. Bis(pentafluorophenyl)borane: Synthesis, Properties, and Hydroboration Chemistry of a Highly Electrophilic Borane Reagent. Angew. Chem., Int. Ed. Engl. 1995, 34, 809–811. 10.1002/anie.199508091. [DOI] [Google Scholar]
- Deposition numbers 2239975 (for 2a), 2239977 (for 5), 2239979 (for 2b), 2239980 (for 4), and 2239981 (for 3) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Center and Fachinformationszentrum Karlsruhe Access Structures service.2020.
- a Drover M. W.; Bowes E. G.; Love J. A.; Schafer L. L. Accessing δ-B–H Coordinated Complexes of Rh(I) and Ir(I) Using Mono- and Dihydroboranes: Cooperative Stabilization by a Phosphoramidate Coligand. Organometallics 2017, 36, 331–341. 10.1021/acs.organomet.6b00784. [DOI] [Google Scholar]; b Drover M. W.; Johnson H. C.; Schafer L. L.; Love J. A.; Weller A. S. Reactivity of an Unsaturated Iridium(III) Phosphoramidate Complex, [Cp*Ir{κ2-N,O}] [BArF4]. Organometallics 2015, 34, 3849–3856. 10.1021/acs.organomet.5b00397. [DOI] [Google Scholar]; c Drover M. W.; Schafer L. L.; Love J. A. Capturing HBCy2: Using N,O-Chelated Complexes of Rhodium(I) and Iridium(I) for Chemoselective Hydroboration. Angew. Chem., Int. Ed. 2016, 55, 3181–3186. 10.1002/anie.201511448. [DOI] [PubMed] [Google Scholar]
- Cowie B. E.; Emslie D. J. H. M–H–BR3 and M–Br–BR3 interactions in rhodium and nickel complexes of an ambiphilic phosphine–thioether–borane ligand. Can. J. Chem. 2018, 96, 484–491. 10.1139/cjc-2017-0758. [DOI] [Google Scholar]
- Douglas T. M.; Chaplin A. B.; Weller A. S.; Yang X.; Hall M. B. Monomeric and Oligomeric Amine–Borane σ-Complexes of Rhodium. Intermediates in the Catalytic Dehydrogenation of Amine–Boranes. J. Am. Chem. Soc. 2009, 131 (42), 15440–15456. 10.1021/ja906070r. [DOI] [PubMed] [Google Scholar]
- Cordero B.; Gómez V.; Platero-Prats A. E.; Revés M.; Echeverría J.; Cremades E.; Barragán F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. J.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision E.01; Gaussian, Inc., Wallingford CT, 2013.
- a Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]; b Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- a Luo J.; Oliver A. G.; Scott McIndoe J. A detailed kinetic analysis of rhodium-catalyzed alkyne hydrogenation. Dalton Trans. 2013, 42, 11312–11318. 10.1039/c3dt51212f. [DOI] [PubMed] [Google Scholar]; b Crabtree R. H. Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure. Chem. Rev. 2015, 115, 127–150. 10.1021/cr5004375. [DOI] [PubMed] [Google Scholar]
- Beringhelli T.; Donghi D.; Maggioni D.; D’Alfonso G. Solution structure, dynamics and speciation of perfluoroaryl boranes through 1H, 11B and 19F NMR spectroscopy. Coord. Chem. Rev. 2008, 252, 2292–2313. 10.1016/j.ccr.2008.01.018. [DOI] [Google Scholar]
- Foroutan-Nejad C.; Shahbazian S.; Marek R. Toward a Consistent Interpretation of the QTAIM: Tortuous Link between Chemical Bonds, Interactions, and Bond/Line Paths. Chem.—Eur. J. 2014, 20, 10140–10152. 10.1002/chem.201402177. [DOI] [PubMed] [Google Scholar]
- Bauer J.; Braunschweig H.; Dewhurst R. D.; Radacki K. Reactivity of Lewis Basic Platinum Complexes Towards Fluoroboranes. Chem.—Eur. J. 2013, 19, 8797–8805. 10.1002/chem.201301056. [DOI] [PubMed] [Google Scholar]
- a Borthakur B.; Das S.; Phukan A. K. Strategies toward realization of unsupported transition metal–boron donor–acceptor complexes: an insight from theory. Chem. Commun. 2018, 54, 4975–4978. 10.1039/C8CC02027B. [DOI] [PubMed] [Google Scholar]; b Chval Z.; Dvořáčková O.; Chvalová D.; Burda J. V. Square-Planar Pt(II) and Ir(I) Complexes as the Lewis Bases: Donor–Acceptor Adducts with Group 13 Trihalides and Trihydrides. Inorg. Chem. 2019, 58, 3616. 10.1021/acs.inorgchem.8b02765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.