

Abstract
PURPOSE
Diffuse large B-cell lymphoma (DLBCL) is cured in more than 60% of patients, but outcomes remain poor for patients experiencing disease progression or relapse (refractory or relapsed DLBCL [rrDLBCL]), particularly if these events occur early. Although previous studies examining cohorts of rrDLBCL have identified features that are enriched at relapse, few have directly compared serial biopsies to uncover biological and evolutionary dynamics driving rrDLBCL. Here, we sought to confirm the relationship between relapse timing and outcomes after second-line (immuno)chemotherapy and determine the evolutionary dynamics that underpin that relationship.
PATIENTS AND METHODS
Outcomes were examined in a population-based cohort of 221 patients with DLBCL who experienced progression/relapse after frontline treatment and were treated with second-line (immuno)chemotherapy with an intention-to-treat with autologous stem-cell transplantation (ASCT). Serial DLBCL biopsies from a partially overlapping cohort of 129 patients underwent molecular characterization, including whole-genome or whole-exome sequencing in 73 patients.
RESULTS
Outcomes to second-line therapy and ASCT are superior for late relapse (>2 years postdiagnosis) versus primary refractory (<9 months) or early relapse (9-24 months). Diagnostic and relapse biopsies were mostly concordant for cell-of-origin classification and genetics-based subgroup. Despite this concordance, the number of mutations exclusive to each biopsy increased with time since diagnosis, and late relapses shared few mutations with their diagnostic counterpart, demonstrating a branching evolution pattern. In patients with highly divergent tumors, many of the same genes acquired new mutations independently in each tumor, suggesting that the earliest mutations in a shared precursor cell constrain tumor evolution toward the same genetics-based subgroups at both diagnosis and relapse.
CONCLUSION
These results suggest that late relapses commonly represent genetically distinct and chemotherapy-naïve disease and have implications for optimal patient management.
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) is an aggressive and heterogeneous lymphoma for which standard-of-care rituximab with cyclophosphamide, vincristine, doxorubicin, and prednisone (R-CHOP) immunochemotherapy results in long-term remission in 60%-70% of patients.1 However, outcomes are poor for the 30%-40% of patients with primary refractory or relapsed disease (refractory or relapsed DLBCL [rrDLBCL]) even after second-line therapy and autologous stem-cell transplantation (ASCT).2,3 The landscape of coding and noncoding somatic variants in DLBCL at diagnosis is well established,4-8 and several studies have examined the mutational landscape of cohorts of rrDLBCL, identifying somatic variants that occur more frequently in rrDLBCL.9-12 Although several of these mutations are prognostic at diagnosis for the likelihood of relapse, they are insufficient to explain the poor outcomes experienced by patients with rrDLBCL.
CONTEXT
Key Objective
What are the patterns of tumor evolution that underpin the relationship between relapse timing and outcomes to second-line (immuno)chemotherapy and autologous stem-cell transplantation in patients with diffuse large B-cell lymphoma (DLBCL)?
Knowledge Generated
Although broad molecular categories (cell-of-origin and genetic subgroups) were consistent between diagnosis and relapse, late relapses arose from serial (and potentially treatment naïve) transformations from persistent common precursor cell populations, whereas earlier relapses were more closely genetically related, implying innate (immuno)chemotherapy resistance. These observations have implications for clinical trial design and patient management, where early and late relapse patient populations should be considered separately and support additional molecular characterization of tumors at relapse to guide precision medicine approaches.
Relevance (J.W. Friedberg)
-
These results emphasize the importance of repeat biopsy and molecular analysis of relapsed DLBCL, and further our understanding of malignant B-cell evolutionary dynamics.*
*Relevance section written by JCO Editor-in-Chief Jonathan W. Friedberg, MD.
Tumor evolution is usually considered to follow one of two models: linear or branching evolution. Linear evolution is defined when the relapse tumor harbors all the variants found at diagnosis along with a set of exclusive variants, implying direct descent of the relapse from the diagnostic tumor. Branching evolution is characterized by exclusive variants in both diagnostic and relapse tumors with a variable number of shared variants (the trunk). In the transformation of follicular lymphoma (FL) to aggressive DLBCL (tFL), this branching evolution is considered evidence of a persistent common precursor cell (CPC) ancestral to both lymphomas.13-15 Previous studies of DLBCL tumor evolution have leveraged circulating tumor DNA and/or limited targeted capture space to examine the evolutionary dynamics of relapse in small cohorts, providing some evidence that branching evolution predominates.9,16-18 However, the degree to which persistent CPC populations might contribute to DLBCL relapse is not yet known.
Critically, no studies have yet, to our knowledge, examined the evolution of the mutation landscape together with gene expression profiling (GEP)-based cell-of-origin (COO)19,20 and dark-zone signature (DZsig)21,22 classification. More recently, genetics-based classifiers have been developed that leverage co-occurrence of somatic variants to identify shared biology within DLBCL. Intriguingly, the three studies that described genetics-based groups converged on 5-7 overlapping subgroups.7,8,23-25 The LymphGen algorithm allows genetics-based subgroup assignment for individual biopsies.23 These classification systems are becoming the foundation for precision medicine in DLBCL, and while the current assumption is that molecular features that underlie the classification of each tumor would be fixed over time, this requires formal testing.
Here, we examined a large population-based cohort of rrDLBCL showing that response rate and outcomes to second-line (immuno)chemotherapy and ASCT are superior for patients with late relapses relative to primary refractory or early relapse. To examine the genetic and evolutionary relationships between diagnostic and rrDLBCL underlying these clinical differences, we assembled a cohort of 129 patients with multiple DLBCL biopsies and interrogated them with a combination of fluorescence in situ hybridization (FISH) for recurrent rearrangements, GEP for COO and DZsig, and/or whole-genome or whole-exome sequencing of two or more DLBCL tumors per patient.
PATIENTS AND METHODS
Outcome analyses were performed in the outcomes cohort of 221 patients with de novo DLBCL treated with frontline R-CHOP(-like) therapy who experienced DLBCL progression/relapse (Data Supplement [Tables S1 and S2, online only]). A major inclusion criterion was treatment with second-line (immuno)chemotherapy (89% received GDP [gemcitabine, dexamethasone, and cisplatin] ± rituximab)26 with intention to treat with consolidative ASCT in patients with (immuno)chemotherapy-responsive disease (Data Supplement [Tables S1 and S2]).
The partially overlapping molecular characterization cohort comprised patients where there was sufficient material for molecular assays from multiple tumors of DLBCL morphology. Additionally, constitutional DNA was required for all patients whose tumors underwent DNA sequencing. The uniform second-line treatment approach required for the outcomes cohort was not an inclusion criterion for this cohort. Thus, patients who were not treated with intention to consolidate with ASCT were included along with patients with indolent lymphoma evident at any time in their disease course, as long as multiple DLBCL biopsies were available. This cohort is enriched for patients who experienced late relapse, reflecting historic patterns of obtaining a biopsy to confirm relapse less frequently in primary refractory disease. In total, 129 patients were identified, of whom 32 had prior indolent lymphoid neoplasms. Among the 97 patients with apparently de novo DLBCL at diagnosis, 11 had subsequent indolent lymphoma diagnoses. Pairs of biopsies were interrogated with a combination of break-apart FISH for MYC, BCL2, and/or BCL6 rearrangements, digital GEP (NanoString DLBCL90) for COO and DZsig classification,20,21 and/or whole-genome (WGS) or whole-exome sequencing (WES) (Data Supplement [Supplemental Fig 1 and Tables S3 and S4]). Further details are provided in the Data Supplement. This study was reviewed and approved by the University of British Columbia-BC Cancer Research Ethics Board in accordance with the Declaration of Helsinki. All patients provided written informed consent with the exception of patients where waiver of consent was granted by the Research Ethics Board.
RESULTS
Patients With Late Relapse Have Superior Outcomes
Considering previous observations that outcomes to second-line therapies are related to progression/relapse timing,27,28 we first sought to confirm this observation in a large population-based outcomes cohort. The 221 patients were categorized into three relapse timing categories: Primary refractory disease was defined as progression or relapse within 9 months of diagnosis, approximating 3 months post-end of treatment.29 Late relapses were defined as more than 24 months after diagnosis, reflecting the definition of EFS24—a validated end point in which patients event free 24 months after immunochemotherapy collectively have superior disease-related outcomes.30 Early relapses were defined as relapse 9-24 months from diagnosis. We found significant differences in both response rates to second-line (immuno)chemotherapy (Fig 1A) and the proportion of patients who ultimately received consolidative ASCT (Fig 1B), demonstrating superior (immuno)chemosensitivity of tumors of patients experiencing late relapses. Patients experiencing late relapse had significantly superior progression-free survival and overall survival relative to patients with primary refractory or early relapse when considering either time from first progression/relapse (Figs 1C and 1D) or from ASCT (Figs 1E and 1F). Outcome differences persisted after adjusting for age at diagnosis and International Prognostic Index at relapse (Data Supplement [Supplemental Fig 2]).
FIG 1.
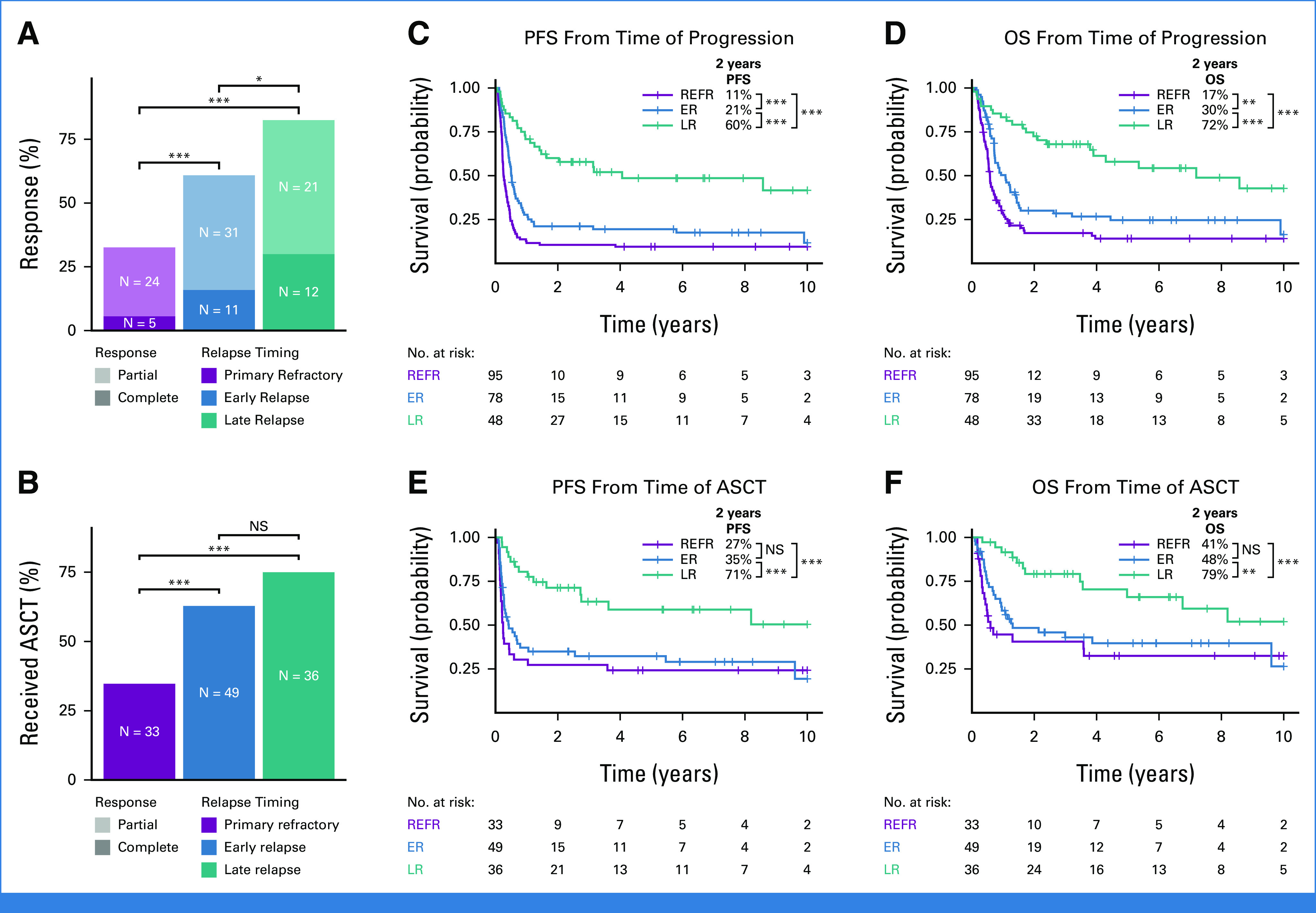
Relationship between relapse timing and outcomes to second-line therapy. (A and B) The percent of patients in each relapse timing category (A) whose relapse responded to second-line therapy and (B) who received ASCT. Groups were compared with pairwise Fisher's exact tests. (C-F) Kaplan-Meier curves showing PFS and OS from the time of progression or ASCT. P values were determined with a log-rank test. *P < .05; **P < .01; ***P < .001. ASCT, autologous stem-cell transplantation; ER, early relapse; LR, late relapse; NS, not significant; OS, overall survival; PFS, progression-free survival; REFR, primary refractory.
Late Relapses Are Highly Genetically Divergent
To examine the underlying tumor biology and patterns of evolution driving the superior outcomes observed in late relapses, we used a molecular characterization cohort of 129 patients that experienced progression/relapse with available serial DLBCL biopsies. DNA sequencing (WGS [n = 68 patients] and WES [n = 5]) was performed on multiple serial DLBCL biopsies along with constitutional DNA (Fig 2), with 21 also included in the outcomes cohort (four primary refractory, seven early relapses, and 10 late relapses).
FIG 2.
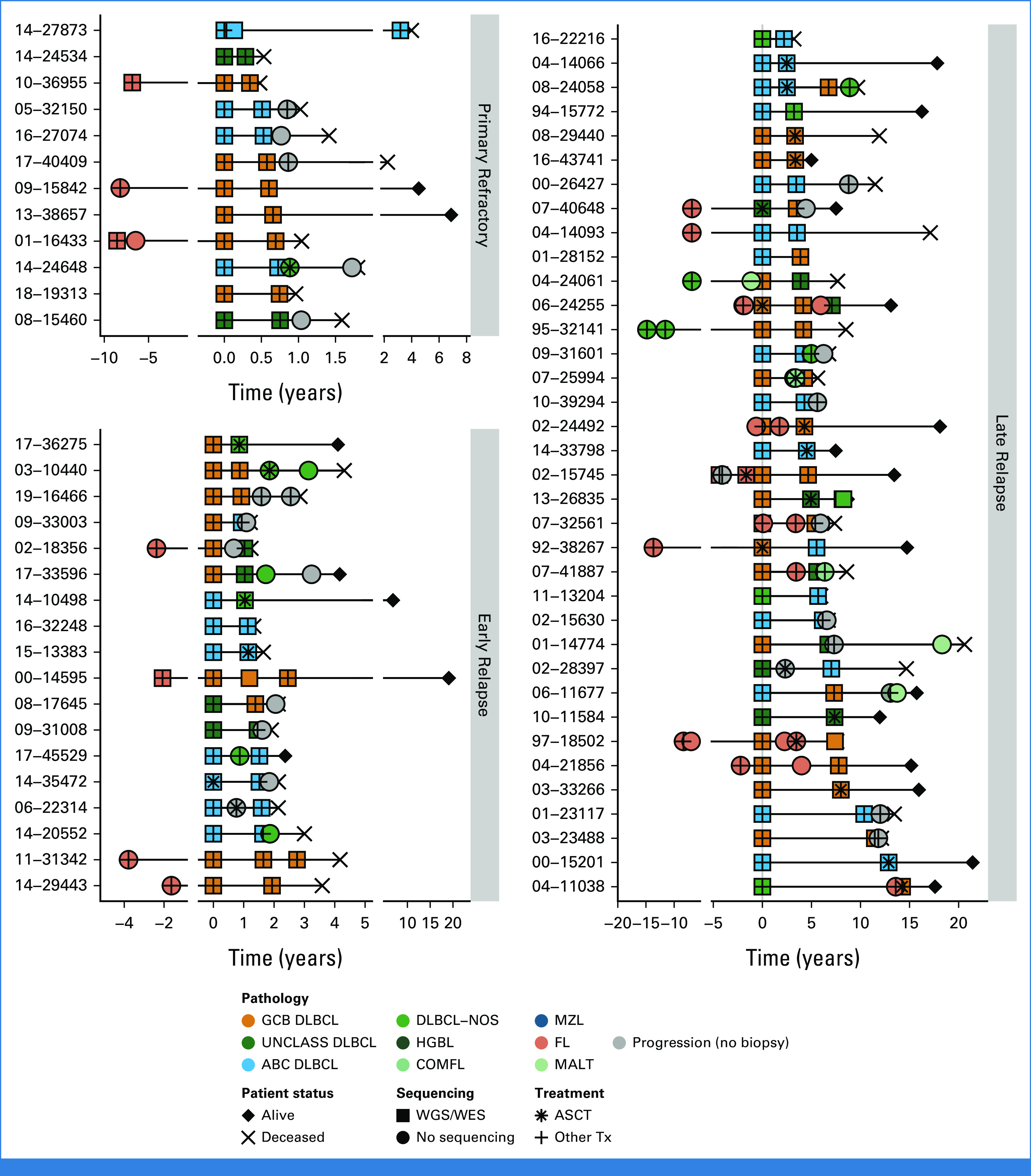
Sequencing cohort patient histories. Disease and treatment histories for all known biopsies and progression time points for patients for which WGS/WES data were generated, distributed according to relapse timing categories. Five patients were omitted because of incomplete histories. DLBCL tumors are colored according to NanoString cell-of-origin where available or are otherwise labeled DLBCL-NOS. ABC, activated B-cell-like DLBCL; ASCT, autologous stem-cell transplantation; COMFL, composite lymphoma with areas of DLBCL and FL morphology; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; GCB, germinal center B-cell-like DLBCL; HGBL, high-grade B-cell lymphoma; MALT, extranodal MZL of mucosa-associated lymphoid tissue; MZL, marginal zone lymphoma; NOS, not otherwise specified; PROG, clinical progression without a biopsy; Tx, treatment; UNCLASS, unclassified DLBCL; WES, whole-exome sequencing; WGS, whole-genome sequencing.
The use of formalin-fixed paraffin-embedded tissues for most samples resulted in variable sequencing depth (mean, 48.6X across WGS samples and 97X in exomes; Data Supplement [Supplemental Fig 3 and Table S5]). We also performed deep targeted DNA sequencing of genes relevant for LymphGen classification (LySeqST, Data Supplement [Table S6]) on multiple biopsies subjected to WGS from 47 patients and on a single biopsy from another 15 patients. The LySeqST assay achieved a mean depth of 812X across its capture space (Data Supplement [Supplemental Fig 4A and Table S7]). The lower variant allele frequencies of variants detected by LySeqST alone versus genomes demonstrates that it enhanced detection of subclonal variants that fall below the WGS limit of detection (Data Supplement [Supplemental Fig 4B]).
Next, we explored the overall divergence of mutations by comparing the number of shared (common between both biopsies) and exclusive (present in only one biopsy) mutations between the first two DLBCL biopsies in each patient. For this and all subsequent analyses, we pooled the LySeqST and WGS variant calls and only retained variants at positions with a sequencing depth of at least 10 unique molecules in all tumors from the same patient. While primary refractory and early relapse tumors have a rich landscape of variants shared between tumors, many late relapses have few, with most mutations exclusive to either the diagnostic or relapse biopsy (Fig 3A). In both primary refractory and early relapse disease, the number of mutations shared between tumors is strongly correlated with the total number of variants identified at either diagnosis or relapse with slopes nearing unity, demonstrating that most variants are shared between tumors (Fig 3B). In contrast, this correlation was weak in late relapses (Fig 3B). Comparing the percentage of exclusive variants in each tumor to the time between biopsies revealed a clear linear trend, where tumor pairs separated by many years have very few shared variants (Fig 3C and Data Supplement [Table S8]). This trend was consistent when considering time to relapse as a categorical variable (Data Supplement [Supplemental Fig 5]), when the absolute number of exclusive mutations was considered (Data Supplement [Supplemental Fig 6]) and is independent of genome coverage (Data Supplement [Table S9]). The linear relationship between exclusive variants and relapse timing was consistent when serial tFL tumors were considered separately from de novo DLBCL (Data Supplement [Supplemental Fig 7]). These results are consistent with a branching evolution model of evolution, where late relapse tumor pairs arise from a CPC harboring few lymphoma-defining mutations.
FIG 3.

Patterns of evolution in diagnostic and relapse tumor pairs. (A) Oncoplots of variants identified exclusively at diagnosis (top), relapse (bottom), or shared between biopsies (middle), highlighting the most frequently mutated genes involved in LymphGen classification. Patients are stratified by relapse timing and ordered by the mean percentage of exclusive variants. Barplots indicate the number of coding mutations present per patient in each mutation subset. (B) The relationship between total variants (all or coding only) at diagnosis or relapse versus the number of mutations shared between tumors. The dashed gray line represents the line of unity. (C) The percent of variants exclusive to either diagnostic or relapse tumors as a function of time between biopsies. R represents the Pearson correlation coefficient. (D) Concordance of heavy chain and (E) light chain V gene usage derived from RNAseq data for tumor pairs colored by V gene subgroup. Light chain rearrangements were more frequently discordant, which may suggest ongoing receptor editing.31 In all plots, alluvia connecting each tumor pair are opaque for discordant pairs and translucent for concordant pairs. N.B. Where V gene usage was discordant but both V genes belong to the same subgroup, the color is consistent across time points.
Given the high degree of divergence observed in some late relapse tumors, we used RNAseq data to identify functional expressed IG receptor rearrangements and confirm clonal relatedness of tumor pairs (Data Supplement [Table S10]). All four primary refractory and nine early relapse patients had concordant IGHV gene usage while 1 of 8 late relapses was discordant (Fig 3D). This lone patient with discordant heavy chain rearrangements also had discordant light chain rearrangements (Fig 3E), suggesting these tumors were not clonally related.
Temporal Dynamics of Structural Variants
Rearrangements of MYC, BCL2, and BCL6 are important drivers of aggressive lymphoma biology and contribute to disease and genetics-based classification.23,32,33 BCL2 rearrangement status was concordant in all 100 patients tested (Fig 4A and Data Supplement [Table S3]), consistent with the origin of BCL2 rearrangements during V(D)J recombination in early B-cell differentiation.34 In 26 patients where WGS identified BCL2 breakpoints in two or more tumors, breakpoints were always identical (Data Supplement [Table S11]).
FIG 4.
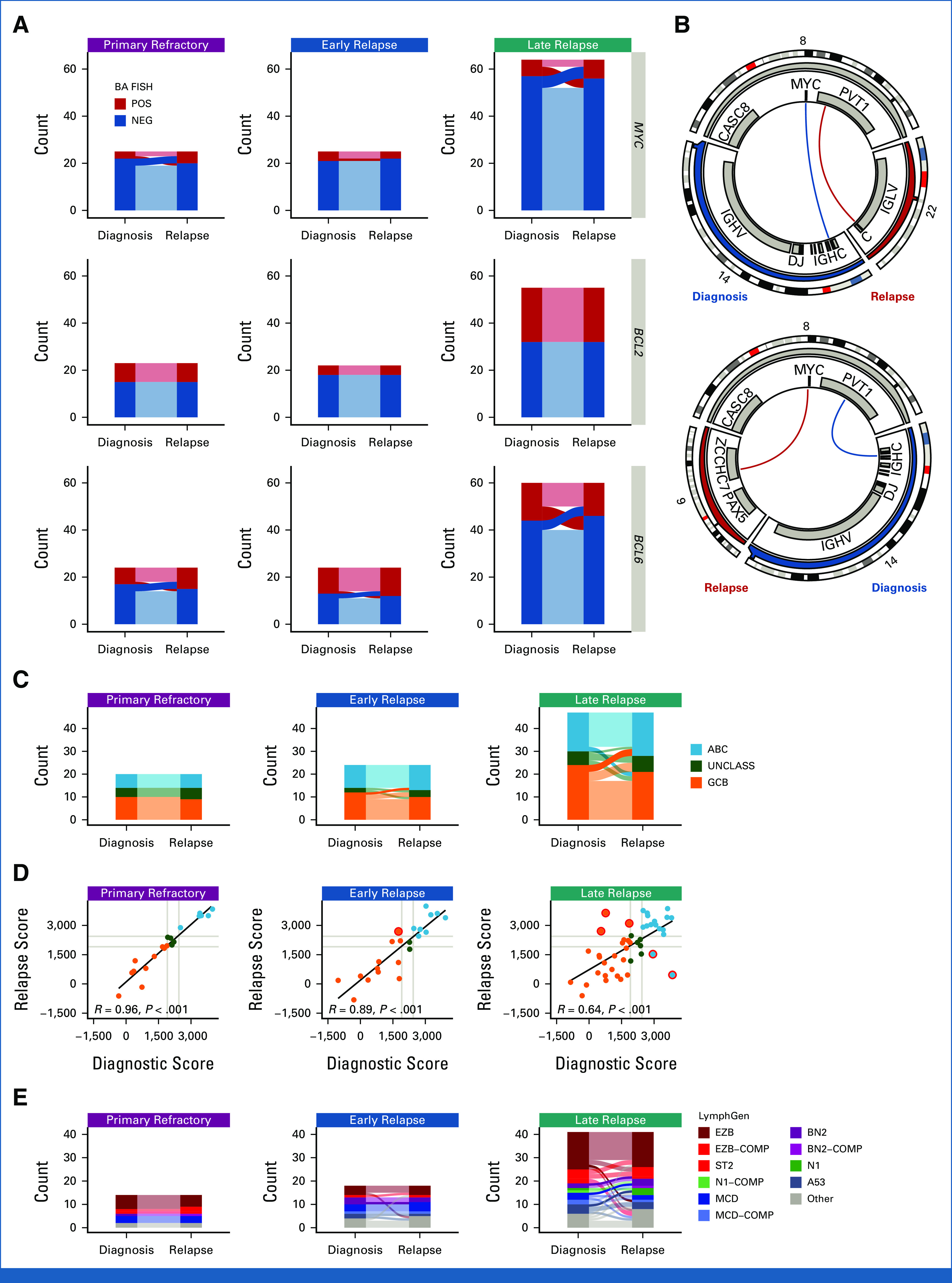
Comparison of structural variants and gene expression profiling and genetic classifications between biopsies. (A) Concordance of BA-FISH results between diagnosis and first relapse for MYC, BCL2, and BCL6 translocations. (B) Circos plots showing discordant MYC translocations in two patients who experienced late relapse. Top: a tumor pair that was positive for BA-FISH at both timepoints; bottom: a tumor pair that was BA-FISH positive at diagnosis and negative at relapse. (C) Alluvial comparison of COO classifications in diagnostic/relapse pairs stratified by relapse timing. Frank discordance (ABC to GCB or vice versa) is indicated by opaque alluvia. (D) A scatter plot comparing DLBCL90 COO scores across tumor pairs. Red circles highlight frank discordance in COO classification. R values indicate Pearson correlation coefficient. (E) Comparison of LymphGen classifications between tumor pairs. Frank discordance (a switch between two mutually exclusive non-other classifications) is emphasized with opaque alluvia. ABC, activated B-cell-like DLBCL; BA, break-apart; COMP, composite; COO, cell-of-origin; FISH, fluorescence in situ hybridization; GCB, germinal center B-cell-like DLBCL; NEG, negative; POS, positive; UNCLASS, unclassified DLBCL.
MYC and BCL6 rearrangements were detected in 20 of 114 and 26 of 108 patients, respectively. In contrast to BCL2 rearrangements, the rate of discordance in rearrangement status between biopsies was substantial at 70% of MYC-rearranged patients and 65% of BCL6-rearranged patients. Interestingly, in all 10 patients where BCL6 rearrangements were identified by WGS at multiple time points, the breakpoints were identical. MYC breakpoints were identified by WGS in multiple tumors from six patients, one of which was cryptic to FISH.35 One patient with primary refractory disease, two with early relapse, and one late relapse had identical MYC breakpoints in both tumors. However, two patients with late relapses (both high-grade B-cell lymphoma with MYC and BCL2) had different MYC translocation partners at diagnosis and relapse (Fig 3B and Data Supplement [Table S11]). These findings suggest that in some patients experiencing late relapse, the original MYC-translocated aggressive lymphoma is effectively eradicated by treatment, whereas the indolent CPC harboring a BCL2 translocation or other variants can persist for many years with new MYC translocations arising in a subsequent aggressive lymphoma.
Biological Consistency of Tumor Pairs
We next evaluated the consistency of molecular subgroups using GEP and LymphGen. We observed high levels of concordance of COO subtype between diagnosis and relapse in the 91 patients tested (Data Supplement [Table S3]). None of 20 primary refractory patients, only 1 of 24 early relapse patients (4.2%), and only 5 of 47 late relapses (10.6%) were frankly discordant (ie, activated B-cell-like (ABC) to germinal center B-cell-like (GCB) DLBCL or vice versa; Fig 4C). Comparison of the NanoString linear predictor scores between time points revealed a weaker correlation in late relapse patients (Fig 4D). A similar trend was observed in DZsig scores (Data Supplement [Supplemental Fig 8]).
To evaluate consistency in genetic subgroup assignment, we compared LymphGen classifications across 73 diagnostic/relapse tumor pairs, yielding a genetic classification for 80% of tumors. LymphGen classifications were highly concordant in all relapse timing categories, with discordance mainly occurring in patients with overlapping composite or Other (not assigned to any subgroup with sufficient confidence) tumors (Fig 4E, Data Supplement [Table S12]). A single early relapse patient of 18 (5.6%) patients had frank discordance (BN2 to MCD) and 4 of 41 (9.8%) among late relapses.
Convergent Evolution in Divergent Pairs
The relative consistency of molecular subgroups as proxies for tumor biology is at odds with our observation that late relapses share relatively few mutations with the diagnostic tumor. To reconcile these disparities, we performed phylogenetic analyses for each patient, leveraging all coding mutations alongside noncoding mutations in regions known to be affected by aberrant somatic hypermutation (aSHM; Data Supplement [Table S13]).6 In primary refractory tumors, most somatic variants are found in the shared phylogenetic trunk (clonal in both tumors; Fig 5A and Data Supplement [Supplemental Fig 9]). In early relapse tumors, the trunk is comparatively shorter with branching evolution giving rise to exclusive mutations in both diagnostic and relapse tumors (Fig 5B). In late relapses, few mutations are in the trunk, with substantial divergence on each branch (Figs 5C and 5D). In the patient with discordant IGHV usage described earlier, the trunk comprised a single shared coding mutation (Fig 5E) providing further evidence that these tumors were not clonally related, arising independently.
FIG 5.

Representative phylogenetic reconstructions. (A-E) Each row of plots displays data for a single patient. Tumors are labeled according to order of occurrence and LymphGen classification. Subclones are colored consistently across all plots for each patient. From left to right: CCF of subclones estimated by PhyClone; VAF of each variant as a scatter plot with the diagnostic tumor on the x-axis and relapse tumor on the y-axis with selected genes labeled; the fraction of mutations shared between both tumors (ie, all mutations in a cluster with a CCF > 0.1); and the inferred phylogenetic relationship between tumors. Hotspot mutations at MYD88 L265P and CD79B Y179 and missense mutations in the CREBBP lysine acetyltransferase (KAT) domain are indicated with an asterisk. The VAF scatter plots without gene labels are also presented in the Data Supplement (Supplemental Fig 9). CCF, cancer cell fraction; VAF, variant allele frequency.
Each of the genetic subgroups of DLBCL have distinct hallmark mutations. We examined patterns of evolution involving these hallmark mutations to determine whether they are present in the inferred CPC population. Variant calls from 28 patients with divergent patterns of evolution were used, defined as having at least 25% of mutations exclusive to each tumor. We then identified truncal (shared among all tumors from the same patient) and exclusive mutations. In total, 28 genes had truncal mutations detected in two or more patients (Fig 6A and Data Supplement [Table S14]), including MYD88L265P (4 of 5 mutated patients) and CREBBP KAT domain mutations (5 of 5 mutated patients), which are genetic subgroup hallmark mutations. In contrast, other genetic subgroup-defining mutations were less frequently truncal, including NOTCH2 PEST domain truncating mutations (1 of 3), EZH2Y646 (0 of 2), and TET2 mutations (2 of 7). As individual mutations, for example EZH2Y646, may be considered for treatment selection or prognosis, this finding underscores the importance of recharacterizing late relapses. Loci affected by aSHM, including BCL2, IGLL5, and BTG2, had a high number of both truncal and exclusive mutations, suggesting that aSHM can be an early shared event but continues after divergence.
FIG 6.
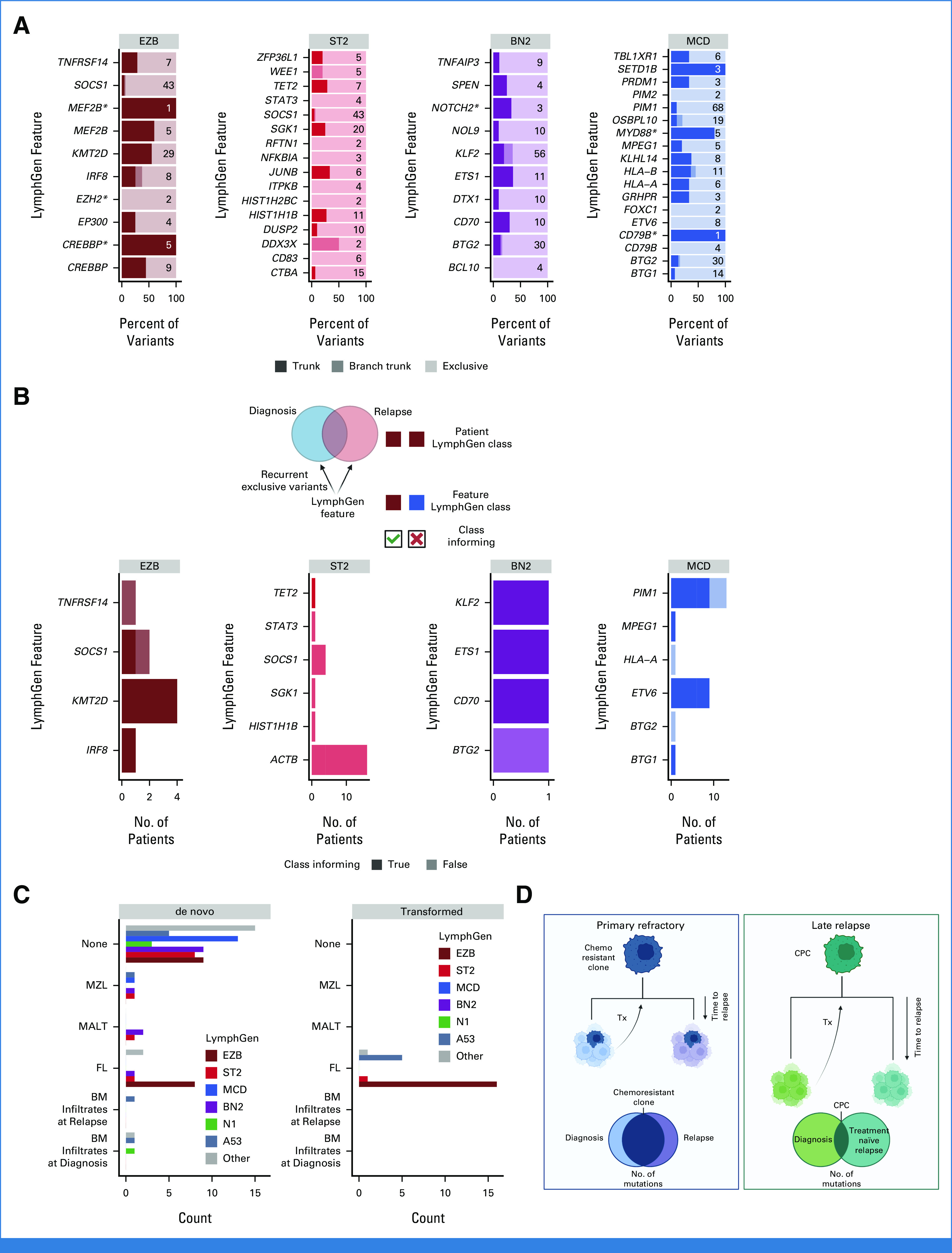
Classification features in divergent tumor pairs. (A) LymphGen classification features that were mutated in two or more tumors from the same patient. Trunk variants (darkest) are identical in all tumors from the same patient; branch trunk are variants not shared across all tumors but are common between at least two; and exclusive are those found in only one tumor. Numbers on each bar represent the total number of variants considered in each feature. (B) LymphGen classification features that acquired exclusive variants in two or more tumors from the same patient. Class informing indicates that the mutations arose in patients in which LymphGen classification matched the class association of the acquired variants. (C) Patient LymphGen classifications stratified according to associated low-grade lymphoma entities. Transformed indicates low-grade disease preceded the first DLBCL diagnosis while de novo indicates that the low-grade diagnosis was made after DLBCL diagnosis. (D) A model of the relationship between relapse timing, evolutionary patterns, and outcomes. BM, bone marrow; CPC, common precursor cell; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MALT, extranodal MZL of mucosa-associated lymphoid tissue; MZL, marginal zone lymphoma; Tx, treatment. Created with BioRender.com.
In addition, we noted that divergent tumor pairs harbored exclusive mutations in the same genes—an example of convergent evolution, where the tumors arrive at the same biology. In the representative MCD-classified early relapse tumor pair, each tumor had independently acquired class-defining mutations in BTG1, PIM1, and ETV6 (Fig 5B); similarly the representative late relapse BN2 tumor pair in CD70 (Fig 5C); and the representative late relapse EZB tumor pair in FOXO1 and MYC (Fig 5D). This pattern of recurrent, independent mutation of the same genes was observed in 16 of 28 patients with divergent tumor evolution (Data Supplement [Tables S15 and S16]). We hypothesized that subgroup-defining mutations would be recurrent and concordant with the defined subgroup. Although this was observed in some features, such as MCD-related PIM1 and ETV6 in patients with MCD-classified tumors, others, such as ACTB, typically associated with the ST2 LymphGen class only recurred in patients without ST2-classified tumors (Fig 6B).
It has been speculated that the shared hallmark mutations between individual LymphGen subgroups and specific indolent lymphomas reflects shared evolutionary history and CPC features.23,25 As expected on the basis of this model, patients with FL at any time in their disease course had DLBCL tumors predominantly classified as EZB while the few marginal zone lymphoma/extranodal MZL of mucosa-associated lymphoid tissue lymphomas occurred in patients with BN2- and ST2-classified DLBCLs (Fig 6C).
DISCUSSION
Leveraging multiple metrics of tumor evolution including cytogenetics, GEP, and unbiased genome- and exome-wide sequencing, we have established distinct patterns of tumor evolution that correlate strongly with the timing of DLBCL progression/relapse. The high rate of mutations exclusive to both diagnostic and relapse biopsies shows that branching evolution predominates in late relapses, strongly supporting the existence of persistent CPC populations capable of giving rise to multiple DLBCL manifestations over time. GEP- and genetics-based classifications remain consistent, suggesting that the earliest mutations in a CPC constrain the biology of subsequent DLBCL(s). This constrained evolution may be the basis for the remarkable convergence of the three studies that defined genetic subgroups of DLBCL.7,8,24,25 Subgroup-defining mutations in the LymphGen classification were sometimes among the inferred CPC mutations identified while others were not consistently clonal, suggesting that additional genomic aberrations or other nongenetic features, such as DNA methylation or tumor microenvironment, are still to be discovered. Furthermore, these CPC mutations appear to constrain the set of loci that acquire mutations during tumorigenesis. Larger cohorts and model systems will be required to determine the mechanisms of these observed constraints. Patterns of aSHM, known to reflect highly actively transcribed regions, are strongly associated with genetic subgroup23 and thus may reflect B-cell differentiation states36,37 that have been occupied during the evolution from CPC to DLBCL.
The patterns of DLBCL tumor evolution observed here help explain the responses to second-line (immuno)chemotherapy observed at disease relapse in DLBCL. In primary refractory disease, the pattern of tumor evolution suggests that innate chemoresistance is present at diagnosis, with little change in the composition of mutations on treatment (Fig 6D). The population of primary refractory patients should, therefore, be the focus in identifying both genetic and nongenetic mechanisms of resistance to frontline immunochemotherapy. In this study and others, these tumors do not typically respond to (immuno)chemotherapy-based second-line regimens, and outcomes are poor3 while alternatives to chemotherapy result in superior outcomes in this patient population.38,39
In contrast, our observations of the biology of late relapse are consistent with elimination of the original DLBCL but persistence of a CPC harboring a very small number of mutations. These CPC populations subsequently give rise to a genetically divergent DLBCL with a large number of newly acquired mutations (Fig 6D). Although they share genetic features, the repertoire of driver mutations in the relapse is not preserved. As these late relapses are effectively chemotherapy naïve, immunochemotherapy-based regimens may remain a rational treatment option.
This divergence in evolutionary patterns and subsequent (immuno)chemotherapy sensitivity associated with relapse timing suggests that clinical trials and routine management should consider patients with early versus late relapse as distinct groups. The trials demonstrating the superiority of second-line chimeric antigen receptor T-cell therapy compared with immunochemotherapy and ASCT were performed in patients experiencing progression/relapse within 12 months of end of frontline therapy.38,39 Larger cohorts than those examined in this study will be required to determine whether a specific time-to-relapse interval can be defined that reliably differentiates patients where the disease represents true rrDLBCL as opposed to new DLBCL arising from a CPC population. Furthermore, particularly when precision medicine approaches are being considered, molecular characterization of the tumor at relapse is recommended as mutations and specific pathway perturbations may differ from those observed at the time of diagnosis.
Henry S. Ngu
Honoraria: AstraZeneca
Christopher K. Rushton
Employment: SAGA Diagnostics
Graham W. Slack
Honoraria: Seagen
Consulting or Advisory Role: Seagen
Jeffrey W. Craig
Honoraria: BeiGene
Expert Testimony: Bayer
Alina S. Gerrie
Honoraria: Janssen, AbbVie, AstraZeneca, BeiGene
Consulting or Advisory Role: Janssen, AbbVie, AstraZeneca, BeiGene
Research Funding: AbbVie (Inst), Roche Canada (Inst), Janssen (Inst), AstraZeneca (Inst)
Travel, Accommodations, Expenses: Janssen
Ciara L. Freeman
Consulting or Advisory Role: Bristol Myers Squibb/Celgene, ONK Therapeutics
Research Funding: Bristol Myers Squibb/Celgene
Diego Villa
Honoraria: Roche Canada, Janssen, Gilead Sciences, Acerta Pharma/AstraZeneca, Celgene, AbbVie, BeiGene, Kyowa Kirin International, Sandoz, Merck
Consulting or Advisory Role: Roche Canada, Janssen, Gilead Sciences, Acerta Pharma/AstraZeneca, Celgene, AbbVie, BeiGene, Kyowa Kirin International, Sandoz, Merck
Research Funding: Roche (Inst), AstraZeneca Canada (Inst)
Kevin Song
Honoraria: Janssen, Bristol Myers Squibb/Celgene, Amgen, Sanofi
Consulting or Advisory Role: Janssen, Bristol Myers Squibb/Celgene, Amgen, Sanofi
Michael Crump
Consulting or Advisory Role: Gilead Sciences, Novartis Canada Pharmaceuticals Inc
Research Funding: Roche Canada
Annette E. Hay
Research Funding: Karyopharm Therapeutics (Inst), Roche/Genentech (Inst), Merck (Inst), Seagen (Inst), Janssen (Inst), AbbVie (Inst)
John Kuruvilla
Honoraria: AbbVie, Bristol Myers Squibb, Amgen, AstraZeneca, Celgene, Gilead Sciences, Janssen, Karyopharm Therapeutics, Merck, Novartis, Roche, Seagen, Pfizer, BeiGene
Consulting or Advisory Role: AbbVie, Bristol Myers Squibb, Gilead Sciences, Karyopharm Therapeutics, Merck, Roche, Seagen, Monte Rosa Therapeutics
Research Funding: Roche, AstraZeneca, Merck
Other Relationship: Karyopharm Therapeutics
Kerry J. Savage
Honoraria: Bristol Myers Squibb, Janssen Oncology, AbbVie, Seagen
Consulting or Advisory Role: Bristol Myers Squibb, Seagen
Research Funding: Roche (Inst), Bristol Myers Squibb (Inst)
Uncompensated Relationships: Beigene, Regeneron
Robert Kridel
Research Funding: Roche, AbbVie
Travel, Accommodations, Expenses: Eisai
Aly Karsan
Honoraria: Jazz Pharmaceuticals
Research Funding: AstraZeneca, Pfizer
Laurie H. Sehn
Honoraria: Amgen, AbbVie, Gilead Sciences, Janssen-Ortho, Kite, a Gilead Company, Merck, Roche/Genentech, Seagen, Teva, AstraZeneca, Incyte, Sandoz-Novartis, Genmab, Celgene/Bristol Myers Squibb, BeiGene
Consulting or Advisory Role: AbbVie, Seagen, Janssen, Amgen, Roche/Genentech, Gilead Sciences, Kite, a Gilead Company, Merck, Teva, TG therapeutics, AstraZeneca, Incyte, Sandoz-Novartis, Genmab, Celgene/Bristol Myers Squibb, Beigene
Research Funding: Roche/Genentech (Inst), Teva (Inst)
Christian Steidl
Consulting or Advisory Role: Seagen, Roche, Bayer, Curis, AbbVie
Research Funding: Bristol Myers Squibb (Inst), Trillium Therapeutics (Inst), Epizyme (Inst)
Patents, Royalties, Other Intellectual Property: Holder of a patent “Method for Determining Lymphoma Type” using the Nanostring platform
Expert Testimony: Bayer
Ryan D. Morin
Consulting or Advisory Role: Epizyme
Patents, Royalties, Other Intellectual Property: Inventor on patent relating to mutation-based classification of DLBCL
David W. Scott
Consulting or Advisory Role: Janssen, AbbVie, AstraZeneca, Incyte
Research Funding: Janssen, Roche/Genentech
Patents, Royalties, Other Intellectual Property: Named inventor on a pending patent describing gene expression profiling in prognostication in classical Hodgkin lymphoma. Named inventor on a patent on the use of gene expression profiling to assign cell-of-origin in diffuse large B-cell lymphoma. Named inventor on a pending patent on the use of gene expression profiling to determine the proliferation signature in mantle cell lymphoma. Named inventor on a pending patent describing using gene expression profiling to identify molecular subtypes of GCB-DLBCL
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the International Conference for Malignant Lymphoma, Lugano, Switzerland, June 18-22, 2021 and at the American Society of Hematology annual meeting, Atlanta, GA, December 10-13, 2021. Presented at the American Society of Hematology annual meeting, New Orleans, LA, December 11, 2022.
SUPPORT
Supported by a Large Scale Applied Research Project funded by Genome Canada (13124), Genome BC (271LYM), Canadian Institutes of Health Research (CIHR; GP1-155873), the British Columbia Cancer Foundation (BCCF) and the Provincial Health Services Authority (PHSA). It was also supported by Terry Fox Research Institute (TFRI) Program Project Grant Nos. (1061, 1108), TFRI Marathon of Hope Cancer Centre Network, the Genome BC Marathon of Hope Cancer Centre program (MOH001), and Grant No. 1P01CA229100 from the National Cancer Institute. R.D.M. and C.S. are supported by Michael Smith Foundation for Health Research, Career Investigator Awards. D.W.S. is supported by a Michael Smith Foundation for Health Research, Health Professional Investigator Award. B.C. is supported by a CIHR Canada Graduate Scholarship Doctoral Award.
PREPRINT VERSION
Preprint version available on medRxiv.
DATA SHARING STATEMENT
Complete sequencing data including WGS/WES, RNAseq, and targeted sequencing will be deposited at the European Genome/Phenome Archive (EGA) under study ID EGAS00001007053. All code for variant calling and phylogenetic analysis are available as part of LCR-modules. An extensive repository of code for statistical analysis is available at https://github.com/LCR-BCCRC/DLBCL-tumor-evolution.
AUTHOR CONTRIBUTIONS
Conception and design: Laura K. Hilton, Robert Kridel, Christian Steidl, Ryan D. Morin, David W. Scott
Financial support: Marco A. Marra, Christian Steidl, Ryan D. Morin, David W. Scott
Administrative support: David W. Scott
Provision of study materials or patients: Pedro Farinha, Alina S. Gerrie, Judith A. Rodrigo, Kevin Song, Michael Crump, Lois Shepherd, Annette E. Hay, John Kuruvilla, Robert Kridel, David W. Scott
Collection and assembly of data: Laura K. Hilton, Henry S. Ngu, Brett Collinge, Kostiantyn Dreval, Merrill Boyle, Barbara Meissner, Graham W. Slack, Pedro Farinha, Jeffrey W. Craig, Ciara L. Freeman, Judith A. Rodrigo, Kevin Song, Lois Shepherd, Annette E. Hay, John Kuruvilla, Kerry J. Savage, Robert Kridel, Marco A. Marra, Laurie H. Sehn, Christian Steidl, Ryan D. Morin, David W. Scott
Data analysis and interpretation: Laura K. Hilton, Kostiantyn Dreval, Susana Ben-Neriah, Christopher K. Rushton, Jasper C.H. Wong, Manuela Cruz, Andrew Roth, Alina S. Gerrie, Diego Villa, Michael Crump, John Kuruvilla, Kerry J. Savage, Aly Karsan, Laurie H. Sehn, Christian Steidl, Ryan D. Morin, David W. Scott
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Relapse Timing Is Associated With Distinct Evolutionary Dynamics in Diffuse Large B-Cell Lymphoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Henry S. Ngu
Honoraria: AstraZeneca
Christopher K. Rushton
Employment: SAGA Diagnostics
Graham W. Slack
Honoraria: Seagen
Consulting or Advisory Role: Seagen
Jeffrey W. Craig
Honoraria: BeiGene
Expert Testimony: Bayer
Alina S. Gerrie
Honoraria: Janssen, AbbVie, AstraZeneca, BeiGene
Consulting or Advisory Role: Janssen, AbbVie, AstraZeneca, BeiGene
Research Funding: AbbVie (Inst), Roche Canada (Inst), Janssen (Inst), AstraZeneca (Inst)
Travel, Accommodations, Expenses: Janssen
Ciara L. Freeman
Consulting or Advisory Role: Bristol Myers Squibb/Celgene, ONK Therapeutics
Research Funding: Bristol Myers Squibb/Celgene
Diego Villa
Honoraria: Roche Canada, Janssen, Gilead Sciences, Acerta Pharma/AstraZeneca, Celgene, AbbVie, BeiGene, Kyowa Kirin International, Sandoz, Merck
Consulting or Advisory Role: Roche Canada, Janssen, Gilead Sciences, Acerta Pharma/AstraZeneca, Celgene, AbbVie, BeiGene, Kyowa Kirin International, Sandoz, Merck
Research Funding: Roche (Inst), AstraZeneca Canada (Inst)
Kevin Song
Honoraria: Janssen, Bristol Myers Squibb/Celgene, Amgen, Sanofi
Consulting or Advisory Role: Janssen, Bristol Myers Squibb/Celgene, Amgen, Sanofi
Michael Crump
Consulting or Advisory Role: Gilead Sciences, Novartis Canada Pharmaceuticals Inc
Research Funding: Roche Canada
Annette E. Hay
Research Funding: Karyopharm Therapeutics (Inst), Roche/Genentech (Inst), Merck (Inst), Seagen (Inst), Janssen (Inst), AbbVie (Inst)
John Kuruvilla
Honoraria: AbbVie, Bristol Myers Squibb, Amgen, AstraZeneca, Celgene, Gilead Sciences, Janssen, Karyopharm Therapeutics, Merck, Novartis, Roche, Seagen, Pfizer, BeiGene
Consulting or Advisory Role: AbbVie, Bristol Myers Squibb, Gilead Sciences, Karyopharm Therapeutics, Merck, Roche, Seagen, Monte Rosa Therapeutics
Research Funding: Roche, AstraZeneca, Merck
Other Relationship: Karyopharm Therapeutics
Kerry J. Savage
Honoraria: Bristol Myers Squibb, Janssen Oncology, AbbVie, Seagen
Consulting or Advisory Role: Bristol Myers Squibb, Seagen
Research Funding: Roche (Inst), Bristol Myers Squibb (Inst)
Uncompensated Relationships: Beigene, Regeneron
Robert Kridel
Research Funding: Roche, AbbVie
Travel, Accommodations, Expenses: Eisai
Aly Karsan
Honoraria: Jazz Pharmaceuticals
Research Funding: AstraZeneca, Pfizer
Laurie H. Sehn
Honoraria: Amgen, AbbVie, Gilead Sciences, Janssen-Ortho, Kite, a Gilead Company, Merck, Roche/Genentech, Seagen, Teva, AstraZeneca, Incyte, Sandoz-Novartis, Genmab, Celgene/Bristol Myers Squibb, BeiGene
Consulting or Advisory Role: AbbVie, Seagen, Janssen, Amgen, Roche/Genentech, Gilead Sciences, Kite, a Gilead Company, Merck, Teva, TG therapeutics, AstraZeneca, Incyte, Sandoz-Novartis, Genmab, Celgene/Bristol Myers Squibb, Beigene
Research Funding: Roche/Genentech (Inst), Teva (Inst)
Christian Steidl
Consulting or Advisory Role: Seagen, Roche, Bayer, Curis, AbbVie
Research Funding: Bristol Myers Squibb (Inst), Trillium Therapeutics (Inst), Epizyme (Inst)
Patents, Royalties, Other Intellectual Property: Holder of a patent “Method for Determining Lymphoma Type” using the Nanostring platform
Expert Testimony: Bayer
Ryan D. Morin
Consulting or Advisory Role: Epizyme
Patents, Royalties, Other Intellectual Property: Inventor on patent relating to mutation-based classification of DLBCL
David W. Scott
Consulting or Advisory Role: Janssen, AbbVie, AstraZeneca, Incyte
Research Funding: Janssen, Roche/Genentech
Patents, Royalties, Other Intellectual Property: Named inventor on a pending patent describing gene expression profiling in prognostication in classical Hodgkin lymphoma. Named inventor on a patent on the use of gene expression profiling to assign cell-of-origin in diffuse large B-cell lymphoma. Named inventor on a pending patent on the use of gene expression profiling to determine the proliferation signature in mantle cell lymphoma. Named inventor on a pending patent describing using gene expression profiling to identify molecular subtypes of GCB-DLBCL
No other potential conflicts of interest were reported.
REFERENCES
- 1. Sehn LH, Salles G. Diffuse large B-cell lymphoma. N Engl J Med. 2021;384:842–858. doi: 10.1056/NEJMra2027612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rovira J, Valera A, Colomo L, et al. Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann Hematol. 2015;94:803–812. doi: 10.1007/s00277-014-2271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood. 2017;130:1800–1808. doi: 10.1182/blood-2017-03-769620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arthur SE, Jiang A, Grande BM, et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun. 2018;9:4001. doi: 10.1038/s41467-018-06354-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378:1396–1407. doi: 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chapuy B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–690. doi: 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rushton CK, Arthur SE, Alcaide M, et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020;4:2886–2898. doi: 10.1182/bloodadvances.2020001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morin RD, Assouline S, Alcaide M, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22:2290–2300. doi: 10.1158/1078-0432.CCR-15-2123. [DOI] [PubMed] [Google Scholar]
- 11. Mareschal S, Dubois S, Viailly PJ, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosom Cancer. 2016;55:251–267. doi: 10.1002/gcc.22328. [DOI] [PubMed] [Google Scholar]
- 12. Trinh DL, Scott DW, Morin RD, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013;121:3666–3674. doi: 10.1182/blood-2013-01-479865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kridel R, Chan FC, Mottok A, et al. Histological transformation and progression in follicular lymphoma: A clonal evolution study. PLoS Med. 2016;13:e1002197. doi: 10.1371/journal.pmed.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okosun J, Bodor C, Wang J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–181. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pasqualucci L, Khiabanian H, Fangazio M, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–140. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:364ra155. doi: 10.1126/scitranslmed.aai8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee B, Lee H, Cho J, et al. Mutational profile and clonal evolution of relapsed/refractory diffuse large B-cell lymphoma. Front Oncol. 2021;11:628807. doi: 10.3389/fonc.2021.628807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Juskevicius D, Lorber T, Gsponer J, et al. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia. 2016;30:2385–2395. doi: 10.1038/leu.2016.135. [DOI] [PubMed] [Google Scholar]
- 19. Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 20. Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123:1214–1217. doi: 10.1182/blood-2013-11-536433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ennishi D, Jiang A, Boyle M, et al. Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol. 2019;37:190–201. doi: 10.1200/JCO.18.01583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alduaij W, Collinge BJ, Ben-Neriah S, et al. Molecular determinants of clinical outcomes in a real-world diffuse large B-cell lymphoma population. Blood. 2023;141:2493–2507. doi: 10.1182/blood.2022018248. [DOI] [PubMed] [Google Scholar]
- 23. Wright GW, Huang DW, Phelan JD, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37:551–568.e14. doi: 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lacy SE, Barrans SL, Beer PA, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood. 2020;135:1759–1771. doi: 10.1182/blood.2019003535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morin RD, Arthur SE, Hodson DJ. Molecular profiling in diffuse large B-cell lymphoma: Why so many types of subtypes? Br J Haematol. 2022;196:814–829. doi: 10.1111/bjh.17811. [DOI] [PubMed] [Google Scholar]
- 26. Crump M, Kuruvilla J, Couban S, et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32:3490–3496. doi: 10.1200/JCO.2013.53.9593. [DOI] [PubMed] [Google Scholar]
- 27. Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–4190. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y, Farooq U, Link BK, et al. Late relapses in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2019;37:1819–1827. doi: 10.1200/JCO.19.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hitz F, Connors JM, Gascoyne RD, et al. Outcome of patients with primary refractory diffuse large B cell lymphoma after R-CHOP treatment. Ann Hematol. 2015;94:1839–1843. doi: 10.1007/s00277-015-2467-z. [DOI] [PubMed] [Google Scholar]
- 30. Maurer MJ, Ghesquières H, Jais JP, et al. Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2014;32:1066–1073. doi: 10.1200/JCO.2013.51.5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collins AM, Watson CT. Immunoglobulin light chain gene rearrangements, receptor editing and the development of a self-tolerant antibody repertoire. Front Immunol. 2018;9:2249. doi: 10.3389/fimmu.2018.02249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia. 2022;36:1720–1748. doi: 10.1038/s41375-022-01620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of mature lymphoid neoplasms: A report from the Clinical Advisory Committee. Blood. 2022;140:1229–1253. doi: 10.1182/blood.2022015851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsujimoto Y, Gorham J, Cossman J, et al. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 35. Hilton LK, Tang J, Ben-Neriah S, et al. The double-hit signature identifies double-hit diffuse large B-cell lymphoma with genetic events cryptic to FISH. Blood. 2019;134:1528–1532. doi: 10.1182/blood.2019002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Machado HE, Mitchell E, Øbro NF, et al. Diverse mutational landscapes in human lymphocytes. Nature. 2022;608:724–732. doi: 10.1038/s41586-022-05072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Álvarez-Prado ÁF, Pérez-Durán P, Pérez-García A, et al. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J Exp Med. 2018;215:761–771. doi: 10.1084/jem.20171738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. 2022;386:640–654. doi: 10.1056/NEJMoa2116133. [DOI] [PubMed] [Google Scholar]
- 39. Abramson JS, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: Primary analysis of the phase 3 TRANSFORM study. Blood. 2023;141:1675–1684. doi: 10.1182/blood.2022018730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Complete sequencing data including WGS/WES, RNAseq, and targeted sequencing will be deposited at the European Genome/Phenome Archive (EGA) under study ID EGAS00001007053. All code for variant calling and phylogenetic analysis are available as part of LCR-modules. An extensive repository of code for statistical analysis is available at https://github.com/LCR-BCCRC/DLBCL-tumor-evolution.