

Abstract
The intestinal epithelium is a dynamic barrier that allows the selective exchange of ions, hormones, proteins, and nutrients. To accomplish this, the intestinal epithelium adopts a highly columnar morphology which is partially lost in submerged culturing systems. To achieve this, small intestinal tissue samples were utilized to obtain human intestinal crypts to form enteroids. The Transwell system was subsequently employed to form a monolayer of cells that was cultured in either the submerged condition or the air–liquid Interface (ALI) condition. We found that the human intestinal monolayer under the ALI condition exhibited morphology more similar to the normal intestinal epithelium. F‐actin localization and brush border formation were observed apically, and the integrity of the tight junctions was preserved in the ALI condition. Fewer apoptotic cells were observed in the ALI conditions as compared to the submerged conditions. The monolayer of cells expressed a higher level of secretory cell lineage genes in the ALI condition. The ALI condition positively contributes toward a more differentiated phenotype of epithelial cells. It serves as an amplifier that enhances the existing differentiation cue. The ALI system provides a more differentiated platform to study intestinal function compared to submerged conditions.
Keywords: air‐liquid interface, enteroids, intestinal epithelial cells, monolayer, submerged conditions
Small intestinal tissue samples were obtained to form enteroids in a transwell culture system. The transwell system was cultured either under submerged or ALI conditions. We found that the human intestinal monolayer exhibited similar morphology as in vivo. The brush border formation was observed apically in the ALI conditions. Tight junction was also preserved in the ALI conditions. The ALI conditions positively contribute toward a differentiated phenotype of epithelial cells.
Abbreviation
- ALI
air–liquid interface
1. INTRODUCTION
The intestinal epithelium forms a dynamic barrier between the body and the environment. Despite being a barrier, it selectively allows the exchange of certain ions, hormones, proteins, and nutrients in an organ‐specific fashion. 1 To adopt this function, the intestinal epithelial cells exhibit a highly polarized columnar morphology. However, this columnar morphology is diminished in the cultured intestinal epithelium monolayer, suggesting that an external stimulus may be missing in standard cultures where cells are submerged under a liquid medium.
The intestinal epithelial cells are exposed to fluctuating conditions of the luminal content and are subjected to different oxygen tension which alters the metabolism of the cells. Studies conducted on tracheal epithelium have suggested that the effect of an air–liquid interface (ALI) was due to cells undergoing different metabolic mechanisms under different culturing conditions. 2 In an ALI culture environment, the epithelial monolayers are fed only basally, and the apical surface is in direct contact with air. Airway epithelial cells cultured under ALI are more polarized, columnar, and differentiated. Additionally, the cells were also reported to have more differentiated structures such as cilia and secretory granules. 3 , 4 , 5 Studies conducted on gastric mucous cells from mice and pigs have also shown similarly differentiated and morphological states. 6 , 7 , 8 ALI culturing of human gastric mucosa demonstrated increased mucous production, and the monolayer was further used to investigate mucosal homeostasis and defense against infection. 9
In vitro studies on intestinal epithelium have been widely conducted using traditional cell lines like Caco‐2, heterogeneous epithelial colorectal adenocarcinoma cells, and HT‐29. The limitations of these models are due to their inadequate resemblance to the in vivo epithelium, genetic instability, and aneuploidy. In addition to this, culture is prolonged. (ca. 20 days) 10 To overcome these limitations, a 3D enteroid model was developed. 11 Enteroid models are more representative of the in vivo intestinal epithelium in terms of their heterogeneity and ability to renew and have a wide range of applications. 12 , 13 However, in a 3D model, the apical side faces inwards making it difficult to access. This makes understanding the transcellular transport, absorption and secretion a challenge. 14 , 15 In addition, the 3D enteroid may not include all the cell types, for example, secretin‐positive enteroendocrine cells. 16
Other methods used are culturing cells on an extracellular matrix coated surface (with collagen and human recombinant laminin) or using a sandwich system in which epithelial cells are in between with type I collagen gel above and a type IV collagen coating below. However, these techniques are complex. 17 , 18
To overcome these limitations, we cultured a monolayer of primary human intestinal epithelial cells under ALI conditions to generate highly polarized monolayer of cells. Our goal is to determine whether the ALI condition recapitulates the human intestinal epithelium both structurally and functionally.
2. MATERIALS AND METHODS
2.1. Isolation of human intestinal crypts
Small intestinal samples were obtained fresh as discarded, deidentified tissues. Tissue was washed multiple times with ice‐cold phosphate buffered saline (PBS) until the solution remained clear. The specimen was placed on ice with the mucosal surface facing upward. Mucosectomy was performed with surgical scissors and forceps. The mucosa was divided into approximately 1 × 1 cm pieces and washed with cold PBS via vortexing for 30 s with three 10‐s pulses until the supernatant is clear. These pieces were then incubated in 8 mM EDTA and 1 mM DTT solution in PBS for 30 min with gentle shaking at 4°C. After the incubation period, the fragments were allowed to settle and the supernatant was discarded. 30 mL of cold PBS is added to the sample and subsequently vortexed for 30 s with 3‐s pulses. 15 mL of the supernatant was removed and saved on ice. Again 15 mL of PBS was added, and the process was repeated six times. Six fractions were spun down at 100 g for 2 min. The supernatant was discarded. The contents of the pellets were examined under light microscopy. Typically, all fractions were pooled together to increase the yield of epithelial crypts. The pooled fractions were then purified using 100 μm and 70 μm pore filters. The pooled fraction after filtration would be the crypts‐rich suspended solution.
2.2. Maintenance of human intestinal stem cells
Every 250 crypts were suspended in 25 μL Matrigel as described in 3D Matrigel culture system developed for murine intestines. 11 The 25 μL of crypt cell/Matrigel suspension was placed in a 48‐well plate. Matrigel was allowed to polymerize in the incubator for 15 min. 250 μL/well of proliferation medium will then be added to the wells. 10 μM Y‐27632 dihydrochloride (Fisher, Pittsburgh, PA) and 2.5 μM CHIR‐99021 (Fisher, Pittsburgh, PA) were added into the proliferation media for the first 2 days after passage. The enteroids were maintained in a 37°C humidified incubator with 5% carbon dioxide until an adequate number of enteroids was reached. The medium was changed every 2 days. At the time of passage, the enteroids‐containing Matrigel were dislodged with pipettes and collected into 1.5 mL Eppendorf tubes. The pellets were acquired via microcentrifuge with three 3‐s pulses. The pellets were then digested with 500 μL of TrypLE Select disassociation reagent (Thermo Fisher, Pittsburgh, PA) in a 37°C water bath for 5 min. The disassociation reagent was then quenched with DMEM with 10% FBS and 1X AMAB (Life Technologies, Carlsbad, CA). After quenching, the enteroids were further broken down mechanically via pipetting the content with P1000 pipettes repeatedly. The pellets were then acquired via microcentrifuge with three 3‐s pulses. The pellets were then resuspended with a desirable amount of Matrigel and re‐seeded into a new tissue plate.
2.3. Generation of intestinal monolayer and ALI culture
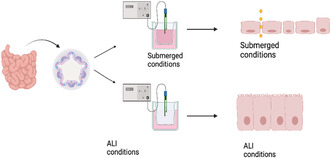
Transwell culturing method was adopted by Julie In et al. 19 The 24‐well Transwell inserts (Corning 3414, Tewsbury, MA) with polycarbonate membrane were coated with collagen IV (30 μg/mL) before cell seeding.
The enteroids‐containing Matrigel buttons were scraped off from the tissue culture plate with P1000 pipettes and washed with 0.5 mM EDTA/PBS (Sigma, Saint Louis, MO). After pelleting the contents via centrifuging at 200 rcf for 5 min, the Matrigel/enteroids were then disassociated chemically with 0.25%/0.5 mM Trypsin/EDTA in a 37°C water bath for 5 min. After quenching with DMEM/10% FBS/1X ABAM, P1000 and P200 pipettes were used to break apart the enteroids mechanically, ensuring the enteroids were disassociated into single cells. The cell‐containing solutions were then passed through a 40‐μm cell strainer before centrifuging at 200 rcf for 5 min. The cell pellets were resuspended with proliferation media supplemented with Y‐27632 dihydrochloride and CHIR‐99021. The cells were seeded into collagen IV coated Transwell inserts at a seeding density of 750,000 cells/cm2.
The monolayers would be cultured in proliferation medium with 100 μL in the insert and 600 μL in in the well with medium change every 2 days. After the monolayer has been cultured with proliferation medium for 5 days ensuring the monolayer has reached full confluency, air–liquid interface (ALI) culturing condition would be initiated. For the first 5 days of the experiment, the monolayer was cultured with 100 μL of media covering the cells on the membrane. This culturing condition was denoted as the “submerged culturing condition.” On Day 5, to start ALI culturing condition, the medium on top of the monolayer would be removed, leaving the cells exposed to the incubating air, while the medium in the outer well would be reduced to 350 μL. The medium was changed every other day.
2.4. Confocal microscopy
The confocal microscopy was done with LSM 880 Inverted Confocal Microscope with AiryScan (Zeiss, White Plains, NY) at Stanford Cell Science Imaging Facility. The increment of each scan was set at 5 μm.
2.5. Cell area and aspect ratio analysis
The aspect ratio of the monolayer was measured via ImageJ software on H&E stained tissue sections. The images of the E‐cadherin stained monolayers were used for measurement.
Images of three randomly chosen areas of each section were taken and analyzed. Three different sections/experiments were analyzed.
2.6. Immunohistochemistry
2.6.1. Sample preparation for histology
The monolayers were fixed by adding 200 μL of 10% buffered formalin (Fisher, Pittsburgh, PA) into the inserts for 30 min at room temperature. The monolayers were washed with PBS twice after the fixation. The samples needed to be embedded within Histogel (Fisher, Pittsburgh, PA) in an upright fashion. The histogel was liquified by warming it up to 65°C and was kept warm for it to remain in liquid state. The polycarbonate membrane was carefully cut off with scalpels. The membrane was then cut into four strips with razor blades and stacked one on top of the other. A lid of the 10 cm diameter petri dish was placed on an ice block and, with a transfer pipette, a couple of droplets of liquified Histogel was placed onto the lid to form a button. Once the Histogel button had solidified due to the drop in temperature, we cut a slit on the button and placed the membrane strip into the slit of the Histogel in an upright position so that when sectioning, the cross‐section of the monolayer could be observed. The Histogel button was then covered with fresh Histogel ensuring the strip was covered with Histogel. The button was then left on the ice block for 15 min before transferring to −20°C for 10 min. Lastly, the strip containing the Histogel button was then transferred to the histology cassette and stored in 70% ethanol before submitting for paraffin embedding, sectioning, and hematoxylin and eosin staining.
2.6.2. Immunostaining of differentiation marker
Unstained slides were sectioned at 4 μm thickness with 2 sections per slide. Slides were washed with xylene for 5 min twice to remove the paraffin wax. The sections were then rehydrated with a progressive decreasing concentration of ethanol in water from 100% to 70% for 2 min and finished with 5 min of incubation in deionized water. Antigen retrieval was done by incubating the slides in 1X Antigen Retrieval Citra Solution (Fisher, Pittsburgh, PA) for 15 min at 100°C followed by 20 min of cooling in a cold water bath. Samples were then permeabilized with 0.5% Triton‐X for 5 min and washed with PBS‐Tween twice afterward. A hydrophobic barrier was created with a PAP pen (Abcam, Waltham, MA) before covering the sections with a blocking solution containing 2% bovine serum albumin and 5% normal goat serum for 1 h at room temperature to prevent non‐specific staining. Following blocking, primary antibody working solutions with primary antibody diluted in blocking solution at a ration which is antibody specific were added on top of the designated sections. The sections were incubated at 4°C overnight. The primary antibodies used were Muc2 (Santa Cruz, Dallas, TX), E‐cadherin (Santa Cruz, Abcam), ki67 (Santa Cruz), and villin (Santa Cruz). The next day, the slides were washed with PBS‐Tween three times to wash away excess primary antibodies before adding secondary antibodies which were diluted in PBS‐Tween at a 1:50 ratio on top of the sections. For the secondary antibodies, Alexafluor 488 goat anti‐mouse, Alexafluor 488 goat anti‐rabbit, Alexafluor 594 goat anti‐mouse, or Alexafluor 594 goat anti‐rabbit were used depending on the primary antibodies used at 1:200 ratio. Excessive secondary antibodies were washed away with three PBS‐Tween washes before adding mounting media with DAPI (Fluoroshield with DAPI, Sigma, Saint Louis, MO) was added. Fluorescent images were taken with Olympus microscope with CellSens software (Olympus, Center Valley, PA).
2.6.3. TUNEL assay
The In situ BrdU‐Red DNA Fragmentation (TUNEL) assay was performed following the protocol provided by the vendor (ab66110, Abcam, Waltham, MA) except for 0.5% Triton‐X instead of protease K treatment. The samples were counterstained with DAPI at the end.
2.6.4. Quantification of ki67 and TUNEL
Immunofluorescent pictures of each sample with ki67 staining and TUNEL assay treatment were taken. Three randomly chosen areas of each slide were taken. Positive ki67 cells were only counted when ki67 and DAPI signals were colocalized. TUNEL‐positive cells were only counted when their signal was colocalized with DAPI.
2.6.5. ZO1 staining
Samples were fixed at the designated time points with cold Methanol at −20°C for 10 min followed by washing with PBS twice. The fixed sample could be stored in PBS or processed for Histology immediately afterward. The sample processing protocol before paraffin embedding, Immunostaining protocol has also been previously described. The samples would be stained against human Zo1 (Abcam, Waltham, MA).
2.7. TEER measurement
TEER measurement was done with an Epithelial Volt/Ohm meter (World Precision Instrument, Sarasota, FL). Before the measurement, 250 μL of fresh medium was added into the insert and 700 μL of fresh medium was added into the well for all the samples there were either under normal or ALI culturing conditions. STX2 electrode (World Precision Instrument, Sarasota, FL) was sterilized with 70% ethanol followed by washing with PBS. When measuring, we held the STX2 electrode straight up with the short end in the insert and the long end in the well. The reading on the meter would be in the unit of an ohm (Ω). To convert, we subtract the reading with the reading of a blank insert, then multiply by the area of the insert to acquire the unit of Ω cm2. After the measurement, the appropriate volume of the medium would be adjusted according to its culturing condition.
2.8. Differentiation media
The differentiation media consisted of Advanced DMEM/F12 (Invitrogen, Carlsbad, CA) 1 mM N‐Acetylcysteine (Sigma), 2 mM GlutaMax‐1 (Invitrogen), 1 mM HEPES buffer (Invitrogen), 1X B‐27 supplement (Thermofisher), 0.5 μM A83‐01 (Sigma), 10 nM Gastrin (Sigma), 50 ng/mL recombinant human EGF (Peprotech), 1X PSQ (Thermofisher), and 100 ng/mL Noggin (Peprotech).
2.9. Generation of L‐WRN condition media
L‐WRN condition media was generated in a large batch. L‐WRN cells were seeded into 15 cm tissue culture dishes (Corning, Tewksbury, MA) with culture media that comprised of Dulbecco's Modified Eagle's Media (DMEM, Life Technologies, Carlsbad, CA), 10% Fetal Bovine Serum (FBS, Atlanta biologicals, Flowery branch, GA), 1X Penicillin–Streptomycin‐Glutamine (PSQ, Thermo Fisher, Waltham, MA) until confluent. Once confluent, G418 (500 μg/mL, Millipore Sigma, Saint Louis, MO) and Hygromycin (500 μg/mL, Millipore Sigma, Saint Louis, MO) were added into the culture media for cell selection. After 24 h of selection, the media was switched back to culture media for the surviving cells to reach confluency again. Once the cells were confluent, the cells were disassociated via incubation with 2 mL of 0.25% Trypsin–EDTA (Fisher, Waltham, MA) at 37°C for 5 min and quenched with 5 mL of cell culture media. The cell suspended solution was then collected and centrifuged at 200 rcf for 5 min. The pellets were then expanded into 4500 cm2 Triple flasks (Fisher, Waltham, MA) and cultured with cell culture media until confluent. Once confluency was reached, the cells were cultured with collection media comprised of Advance‐DMEM/F12 (Fisher) and 10% FBS and 1X PSQ. The next day, the supernatant was collected via EMD Millipore Stericup (Fisher, Waltham, MA) and denoted as Batch 1. The cells were recovered with fresh collection media. The collection process was repeated four times for the subsequent f4 days. The whole process would result in four batches of the L‐WRN conditioned media (L‐WRN CM).
2.10. Proliferation Media
The proliferation medium consisted of 50/50 mix of Advanced DMEM/F12 (Invitrogen, Carlsbad, CA) and L‐WRN conditioned media (L‐WRN CM). The mixture was supplemented with 1 mM N‐acetylcysteine (Sigma, Saint Louis, MO), 2 mM GlutaMax‐1 (Invitrogen, Carlsbad, CA), 1 mM HEPES buffer (Invitrogen, Carlsbad, CA), 10 mM nicotinamide (Sigma, Saint Louis, MO), 1X B‐27 supplement (Thermofisher, Waltham, MA), 0.5 μM A83‐01 (Sigma, Saint Louis, MO), 10 nM gastrin (Sigma, Saint Louis, MO), 50 ng/mL recombinant human EGF (Peprotech, Rocky Hill, NJ), 1X PSQ (Thermofisher, Waltham, MA), and 10 μM SB‐202190 (Sigma, Saint Louis, MO).
2.11. Analysis of RNA expression
RNA extraction was described in a previous publication. 20 Primer and probe combinations were purchased from Applied Biosystem (Taqman Expression Assay, Hs00969422_m1(LGR5), Hs00894052_g1(muc2), Hs00167206_m1(SLC11A2), Hs00426232_m1(lyz), Hs00900375_m1(Chga), Hs01031724_m1(vil1), Hs02758991_g1(GAPDH)). RT‐PCR was performed following the description in a previous publication [20]with the ABI 7900HT Fast Real‐Time PCR system. Cycle numbers of all samples were normalized to GAPDH with human intestinal whole bowel serving as control tissue.
2.12. Statistics
Differences between groups were evaluated via Student's t‐test. A p‐value less than 0.05 was considered statistically significant.
3. RESULTS
3.1. The effect of ALI on monolayer morphology
After 2 days in the ALI condition, cells in the monolayer exhibited morphological change that could be observed in cross‐sectional analysis (Figure 1). The height of the cells was significantly increased compared to those in the submerged condition as cells became more columnar in shape in the ALI condition. Although cells also had basally located nuclei when cultured in the submerged condition, these cells remained relatively flat when compared to those cultured in ALI conditions for up to 14 days. A more pronounced brush border could also be observed in cells cultured in the ALI condition. Examination of cells cultured in ALI conditions by confocal microscopy demonstrated marked f‐actin localization on the apical side of the cells (Figure 2). Such polarization effect of ALI on epithelial cells was consistently observed using enteroids derived from multiple human tissue samples (Figure 3).
FIGURE 1.
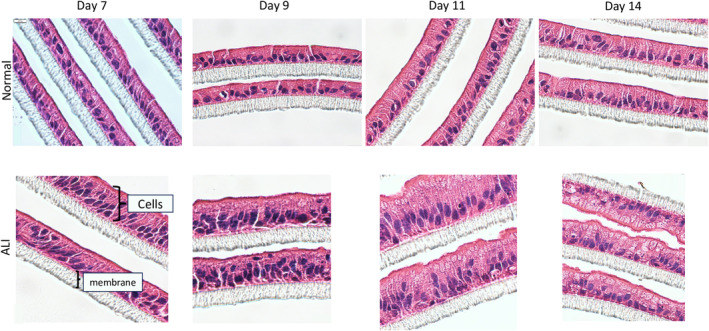
Micrographs of hematoxylin and eosin‐stained monolayers derived from human enteroids cultured in vitro under two different conditions, Normal(top), where the monolayer is submerged under proliferation media and ALI (bottom), where there is no medium on top of the monolayer. These are under bright field, under 600× magnification. Scale bar: 10 μm.
FIGURE 2.

Confocal microscopy images of the monolayer both under ALI conditions (left) and normal conditions(right) stained with FITC phalloidin against F‐actin. The cross‐section of the monolayer under ALI showed a distinct actin‐rich brush border(green) present at the apical side of the monolayer compared to the normal culturing conditions. Scale bar: 25 μm.
FIGURE 3.
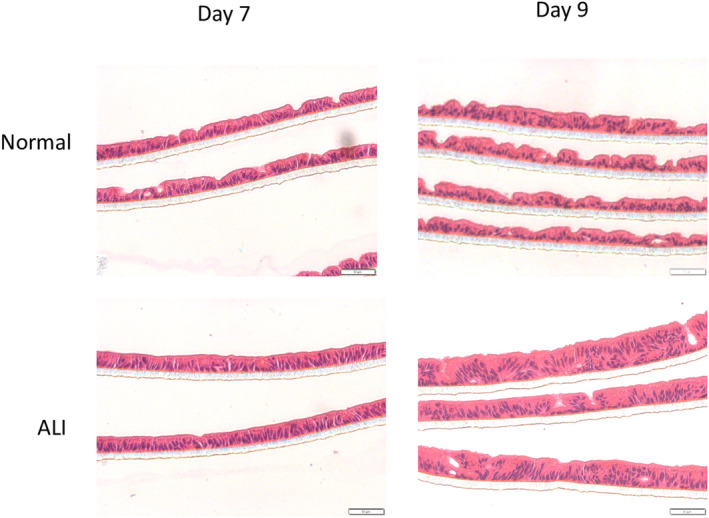
Micrographs of hematoxylin and eosin‐stained monolayers derived from different lines of human enteroids cultured in vitro under two different conditions, Normal(top), where the monolayer is submerged under the proliferation medium and ALI(bottom), where there is no medium on top of the monolayer. These are seen under bright field under 200× magnification. Scale bar: 50 μm.
To determine whether the taller cells were different in size, we measured the height and width of cultured cells in both submerged and ALI conditions (Figure 4). The aspect ratio of cells in the ALI condition was significantly higher than those cultured in the submerged condition and was more similar to that in the normal human intestinal epithelium. In contrast, the cross‐sectional area of the cells was similar to that in normal human intestinal epithelium in both conditions.
FIGURE 4.
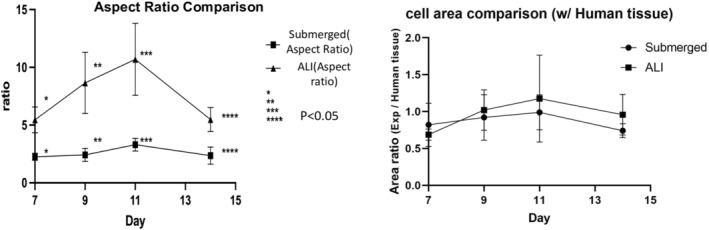
(A) Aspect ratio of the cells was compared between Normal and ALI conditions. The aspect ratio is the ratio of height to width of the cells, which is increased in columnar morphology as seen in the ALI culturing conditions. There was a significant difference in the aspect ratio of the cells between the two conditions. Aspect ratio = H/W. (n = 3). (B) The cross‐sectional cell area comparison ratio between in vitro monolayer derived from human enteroids under the 2 culturing conditions, normal and ALI with the cross‐sectional cell area of human intestinal epithelium.
3.2. Functional barrier of the monolayer
Tight junction development is an essential characteristic of an epithelial layer that possess the integrity to function as a barrier. The distribution of Zonula occludens‐1 (Zo1) at the apical side of cells was observed when cultured in ALI condition, whereas there was little to no Zo1 expression in cells cultured in the submerged condition on Day 14 (Figure 5). Transepithelial resistance of the two culturing conditions was determined by the TEER value measurement. The TEER value of cells in the submerged conditions increased steadily over the first 5 days in the submerged condition (Figure 6). This upward trend continued for cells in the submerged condition. When the monolayer was switched to the ALI condition on Day 5, the TEER value of the monolayer remained at a relatively stable level around 100 Ωcm2, which was similar to the TEER value of the human intestine.
FIGURE 5.
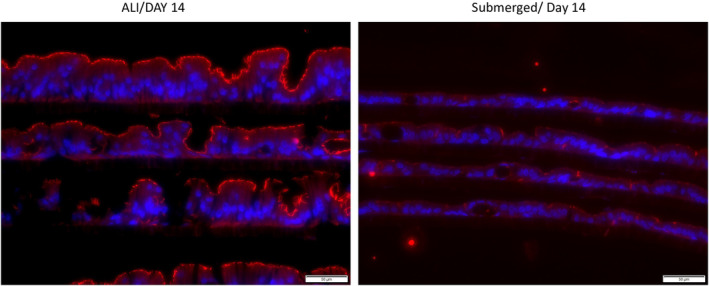
Zo1(Zonula Occludens‐1) staining (red) is seen distinctly on the apical aspect of the monolayers cultured under ALI(left) conditions, with little to no expression under normal(right) culturing conditions. Scale bar: 50 μm.
FIGURE 6.
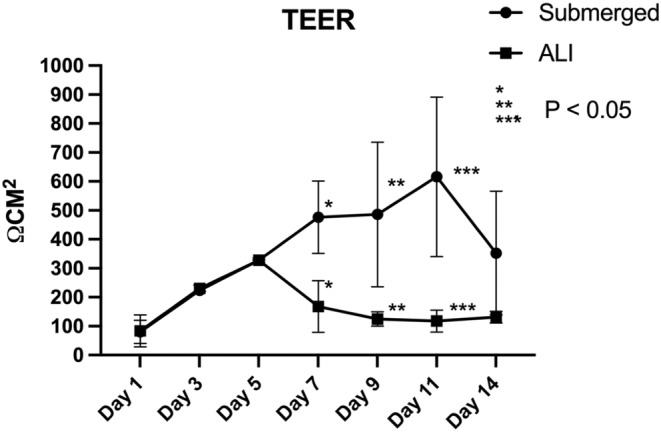
The measurement of TEER (transepithelial electrical resistance) value on monolayers cultured under Normal and ALI culturing conditions. A significant difference can be seen after the ALI culturing was initiated on Day 5. (n = 3).
3.3. Ki67 / TUNEL analysis
Cellular turnover in the two monolayer culture conditions was compared to that in the normal human intestinal epithelium. The percentage of the ki67‐positive cells was fairly constant in the submerged condition, whereas the percentage of such cells decreased significantly in the ALI condition over 9 days (Figure 7). Compared to the human intestinal epithelium, the percentage of proliferating cells was similar to that in the submerged condition. There were fewer cells that were TUNEL‐positive when cultured in the ALI condition. Compared to the human intestinal epithelium, the percentage of the cells that underwent apoptotic event was similar to that in the ALI condition but was higher in the submerged condition (Figure 8).
FIGURE 7.
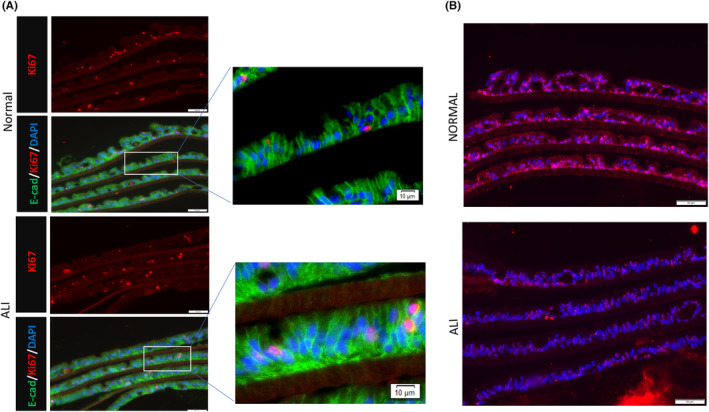
(A) Immunofluorescence of cultured human intestinal epithelial monolayer under both normal(top) and ALI(bottom) culture conditions. Ki67‐positive cells (red) could be seen in both culturing conditions. Scale bar: 50 μm; (B) TUNEL assay on the monolayers culturing under two culturing conditions, Normal(top) and ALI(bottom). There are fewer TUNEL‐positive apoptotic cells(red) in the ALI culturing conditions. Scale bar: 50 μm.
FIGURE 8.
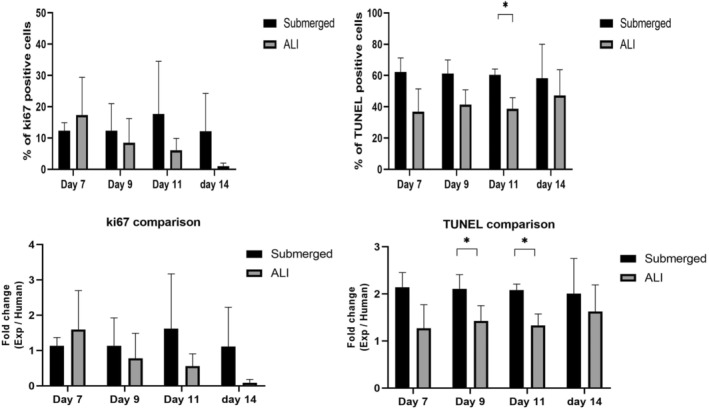
Quantitative comparison of Ki67 positive cells and TUNEL‐positive cells between in vitro cultured monolayer and human intestinal epithelium. No significant difference was observed in ki67 positive cells. Under ALI culturing conditions, the percentage of apoptotic cells decreased to a similar level as on human intestinal epithelium. (n = 3). *p < 0.05.
3.4. Differentiation of the monolayer
Cellular differentiation was examined in the two culture conditions. Villin was localized on the apical side of the cells in the ALI condition while there was less apical localization in the submerged condition (Figure 9A). Ferroportin (FPN) is an iron transporter located in the apical region of duodenal villus. Cells derived from the duodenum cultured in the ALI condition exhibited FPN positive signal on the apical surface, where little FPN expression was observed in the submerged condition (Figure 9B).
FIGURE 9.
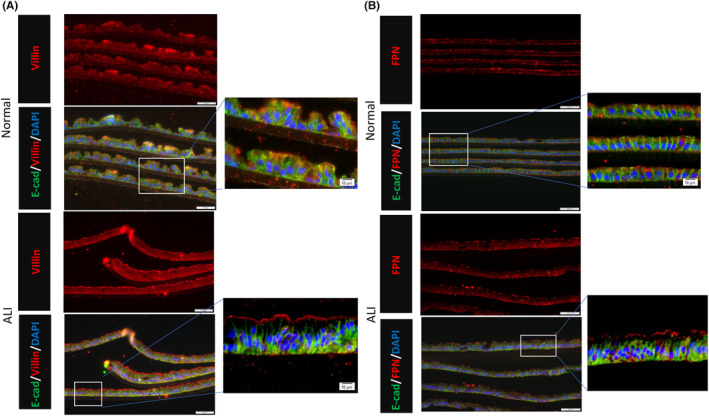
(A) Immunofluorescence of cultured human intestinal epithelial monolayer under both Normal(top) and ALI(bottom) culture conditions. Villin signal (red) can be seen localized at the apical side of the monolayer under ALI conditions. Scale bar: 50 μm. (B) Immunofluorescence of cultured human intestinal epithelial monolayer under both normal(top) and ALI(bottom) culture conditions. A positive Ferroportin(FPN) signal (red) can be seen localized at the apical side of the monolayer under ALI conditions. Scale bar: 50 μm.
Other intestinal differentiation markers were examined via quantitative RT‐PCR (Figure 10). Across a panel of markers, the mRNA levels in the ALI condition were consistently higher than those in the submerged condition. Compared to the normal human intestine, lysozyme, villin, and SLC11a2 mRNA were high in the ALI condition and lower in the submerged condition, whereas MUC2 and CHGA showed lower expression in both culture conditions.
FIGURE 10.
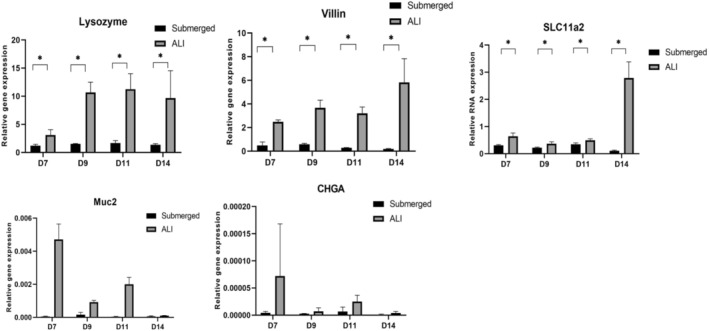
RT‐qPCR analysis on multiple differentiation markers for the human intestinal epithelial monolayers. The expression of Lysozyme, Villin, and SLC11a2 showed significantly higher expression in the monolayer under ALI conditions. The other two markers (Muc2 and CHgA) also showed the same trend; however, overall expression remained low). The human intestine was used as control. (n = 3). *p < 0.05.
The low expression of Muc2 in both conditions provided a platform to observe the cellular response to other environmental cues. When Wnt signals were withdrawn from the culture medium, significantly more Muc2‐positive cells were observed (Figure 11). More Muc2‐positive cells were observed for both ALI and submerged conditions.
FIGURE 11.
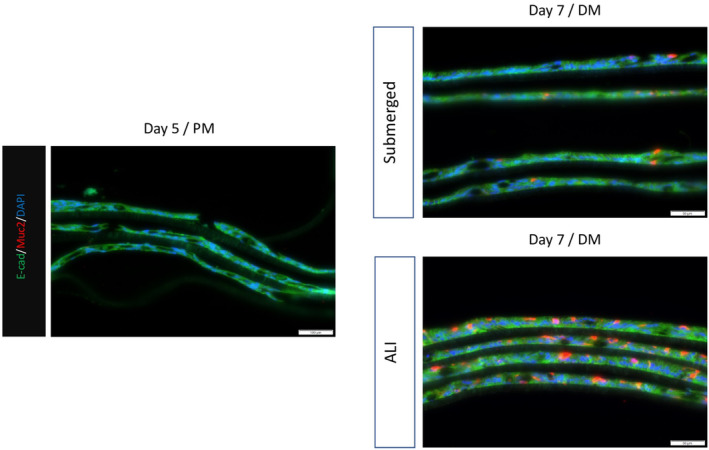
Immunofluorescence of Muc2 expression on the monolayer on Day 5 with proliferation media and Day 7 cultured with differentiation media (DM). The media was switched on Day 5 from proliferation media (PM) to differentiation media. ALI was initiated at the same time. The Muc2 expression was much higher in the monolayer cultured under ALI conditions.
4. DISCUSSION
Air–liquid interface culturing condition has been widely used in epithelial cell culture. It has proved to promote proliferation and differentiation in tracheal epithelial cells and gastric epithelial cells. 2 , 4 , 6 Under ALI culturing conditions, the primary intestinal epithelial cells not only adopted a columnar morphology, but also led to the formation of the brush border in vitro that resembled human enterocytes in vivo. We utilized a 3D enteroid model to generate the cells for the monolayer culture system. While the 3D model is easy to work with and is fairly representative of human tissue, 21 the in vivo environment is structurally complex and that niche enviroinment is not replicated through the 3D enteroid models. 16
Epithelial cells serve as a physical barrier and allow the transport of certain ions, hormones, proteins, and nutrients in an organ‐specific fashion. To perform this selective function, the epithelial cells adopt a polarity, separating each cell into different structural and functional domains. 1 This is a distinct characteristic of the epithelial cells which was demonstrated under air–liquid interface culturing by F‐actin localization on the apical surface.
The tight junction formation which is necessary for barrier function was confirmed by the presence of Zo‐1 and a stable TEER value, both of which were not seen under the submerged condition. Although submerged cultured monolayer exhibited higher TEER values, it also displayed higher variabilities compared to the monolayer under ALI culturing. Furthermore, studies have demonstrated that high TEER values are not physiologically relevant. 22 The Zo‐1 and stable TEER value positively contributed to monolayer maintenance. Loss of tight junction would result in a permeable epithelium causing various pathogens and toxins to pass through easily. Furthermore, our data suggest that there is a reduction in the number of apoptotic cells under the ALI condition which is also anticipated to be a contributing factor toward monolayer maintenance. The number of apoptotic cells was comparable to the number in vivo.
The similarities of the monolayer under ALI condition to the human intestine can also be established by the presence of villin and ferroportin on immunostaining along with RNA expression analysis which is suggestive of a more differentiated state. The localization of villin at the apical side of the monolayer indicates the presence of a brush border composed of microvilli at the apical region of the epithelial cells. In the future, electron microscopy can be performed to demonstrate the presence of villin under ALI. The expression of MUC2 and CHGA was low overall which suggests that even though there was little secretory lineage present, the monolayer of cells under ALI conditions was still more differentiated than its submerged counterpart. The low expression of MUC 2 and CHGA could be attributed to its growth in proliferation media instead of differentiation media. Other studies of intestinal monolayers conducted in submerged media have also exhibited all major intestinal cell lineages; however, the expression of MUC2 and CHGA is low overall as compared to in vivo conditions. 23
The ALI system consistently displayed a higher expression of secretory cell lineage genes as compared to the submerged media which suggests that ALI acts as an amplifier. It does not have the same effect as differentiation medium, which induces differentiation by transforming proliferative cell lineage to secretory cell lineage. A relatively higher expression of lysozyme and villin was observed in culture than in the human intestine since the expression of these two genes already existed under submerged culturing conditions. On the other hand, Muc2 and CHGA genes were not expressed under submerged culturing conditions, so even though the gene expression under the ALI condition was still heightened, the overall relative expression remained low. To confirm this hypothesis, we withdrew the Wnt signals from the culture medium, in both ALI and submerged culturing conditions, and more Muc 2 + cells formed under ALI conditions.
A study from Altay et. al achieved similar monolayer morphology on murine intestinal epithelial cells via culturing the monolayer with two different media at the same time. The medium in the well was nutrient‐rich compared to the medium in the Transwell. The nutrient gradient achieved through this method could also be achieved via ALI culturing condition since the medium would be absent in the Transwell. Since the thickness of the monolayer in this study also increased, one cannot help but wonder whether the key factor contributing to the effect of the ALI culturing condition on the monolayer development is the presence of the nutrient gradient across the apical‐basal axis. This would be an interesting next step to this study in discovering the underlying driving force of ALI culture.
Our limitations include employing RNA expression as a surrogate for immunostaining since we were unable to visualize good protein expression through immunostaining in the ALI system.
The ALI monolayer technology provides an excellent method to study the intestinal platform more efficiently. It is derived from healthy human cells, unlike caco2, which is a cancer cell line. The differentiation profile of these cells can be used to study intestinal diseases that affect the villi, villus epithelial transport, and drug toxicity testing where differentiated cells behave differently from a mixture of crypt and villus cells. We can also attempt to study the impact of microbes on epithelial homeostasis. 16
5. CONCLUSION
This study has described the effect of ALI culturing on monolayer development. A more differentiated monolayer was seen under ALI in comparison with the submerged media. ALI culturing media serves as an amplifier where it could enhance the existing differentiation cue. ALI culturing condition provides a more physiologically relevant human intestinal epithelial monolayer in both morphological and differentiation perspective.
AUTHOR CONTRIBUTIONS
James Dunn conceived and designed the research; Po‐Yu Lin performed the research and acquired the data; Po‐Yu Lin analyzed and interpreted the data. All authors were involved in drafting and revising the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by NIH grant NIDDK R01 DK083319.
Sabapaty A, Lin P‐Y, Dunn JCY. Effect of air–liquid interface on cultured human intestinal epithelial cells. FASEB BioAdvances. 2024;6:41‐52. doi: 10.1096/fba.2023-00132
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
REFERENCES
- 1. Roignot J, Peng X, Mostov K. Polarity in mammalian epithelial morphogenesis. Cold Spring Harb Perspect Biol. 2013;5(2):a013789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kondo M, Tamaoki J, Sakai A, Kameyama S, Kanoh S, Konno K. Increased oxidative metabolism in cow tracheal epithelial cells cultured at air‐liquid interface. Am J Respir Cell Mol Biol. 1997;16(1):62‐68. [DOI] [PubMed] [Google Scholar]
- 3. Lin H, Li H, Cho HJ, et al. Air‐liquid interface (ALI) culture of human bronchial epithelial cell monolayers as an in vitro model for airway drug transport studies. J Pharm Sci. 2007;96(2):341‐350. [DOI] [PubMed] [Google Scholar]
- 4. Pezzulo AA, Starner TD, Scheetz TE, et al. The air‐liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2011;300(1):L25‐L31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Whitcutt MJ, Adler KB, Wu R. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In Vitro Cell Dev Biol. 1988;24(5):420‐428. [DOI] [PubMed] [Google Scholar]
- 6. Ootani A, Toda S, Fujimoto K, Sugihara H. An air‐liquid interface promotes the differentiation of gastric surface mucous cells (GSM06) in culture. Biochem Biophys Res Commun. 2000;271(3):741‐746. [DOI] [PubMed] [Google Scholar]
- 7. Yokoyama F, Sakata Y, Ootani A, et al. Differentiation of gastric surface mucous cells (GSM06) induced by air‐liquid interface is regulated partly through mitogen‐activated protein kinase pathway. J Gastroenterol Hepatol. 2007;22(12):2310‐2315. [DOI] [PubMed] [Google Scholar]
- 8. Nossol C, Diesing AK, Walk N, et al. Air‐liquid interface cultures enhance the oxygen supply and trigger the structural and functional differentiation of intestinal porcine epithelial cells (IPEC). Histochem Cell Biol. 2011;136(1):103‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boccellato F, Woelffling S, Imai‐Matsushima A, et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut. 2019;68(3):400‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moon C, VanDussen KL, Miyoshi H, Stappenbeck TS. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol. 2014;7(4):818‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262‐265. [DOI] [PubMed] [Google Scholar]
- 12. Creff J, Malaquin L, Besson A. In vitro models of intestinal epithelium: toward bioengineered systems. J Tissue Eng. 2021;12:2041731420985202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 2016;18(3):246‐254. [DOI] [PubMed] [Google Scholar]
- 14. Bein A, Shin W, Jalili‐Firoozinezhad S, et al. Microfluidic organ‐on‐a‐chip models of human intestine. Cell Mol Gastroenterol Hepatol. 2018;5(4):659‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Altay G, Larrañaga E, Tosi S, et al. Self‐organized intestinal epithelial monolayers in crypt and villus‐like domains show effective barrier function. Sci Rep. 2019;9(1):10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Chen YG. 2D‐ and 3D‐based intestinal stem cell cultures for personalized medicine. Cell. 2018;7(12):Article 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura T, Sato T. Advancing intestinal organoid technology toward regenerative medicine. Cell Mol Gastroenterol Hepatol. 2018;5(1):51‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tong Z, Martyn K, Yang A, et al. Towards a defined ECM and small molecule based monolayer culture system for the expansion of mouse and human intestinal stem cells. Biomaterials. 2018;154:60‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. In J, Foulke‐Abel J, Zachos NC, et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell‐derived colonoids. Cell Mol Gastroenterol Hepatol. 2016;2(1):48‐62. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geisbauer CL, Chapin JC, Wu BM, Dunn JC. Transplantation of enteric cells expressing p75 in the rodent stomach. J Surg Res. 2012;174(2):257‐265. [DOI] [PubMed] [Google Scholar]
- 21. Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020;21(10):571‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. J Lab Autom. 2015;20(2):107‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozuka K, He Y, Koo‐McCoy S, et al. Development and characterization of a human and mouse intestinal epithelial cell monolayer platform. Stem Cell Reports. 2017;9(6):1976‐1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.