

ABSTRACT
Background
Familial renal glucosuria (FRG) is a hereditary disorder caused by variants in SLC5A2 encoding sodium-glucose cotransporter 2 (SGLT2). In this study, we aimed to characterize proximal tubule solute transport, glucagon secretion and the genotype–phenotype relationship in FRG patients.
Methods
We sequenced SLC5A2 and PDZK1IP1 in 21 FRG patients and measured the renal threshold of glucose (RTG) in 15 patients. We built an open-source online calculator of RTG, evaluated the proximal tubule transport of amino acid, uric acid and phosphate, and explored glucagon secretion after glucose ingestion in FRG patients.
Results
We identified 12 novel SLC5A2 variants (G484D, R564W, A212S, c.574+1G>C, W649*, S592Cfs*6, Q579*, Y339*, V39F, G491E, A464E and G360D) in our cohort and yielded 111 SLC5A2 variants from literature review. RTG in our cohort ranged from 1.0 to 9.2 mmol/L. Patients with two SLC5A2 variants had lower RTG (3.9 vs 6.2 mmol/L) and higher 24-h urinary glucose excretion (24hUG) than single-variant carriers (291.0 vs 40.0 mmol/1.73 m2). Patients with homozygous missense or in-frame indels had mean 24hUG of 457.2 mmol/1.73 m2, comparable to those with homozygous truncating variants (445.0 mmol/1.73 m2) and significantly more than those with homozygous splicing variants (196.6 mmol/1.73 m2). Patients with homozygous missense variants involving conservative residues (582.0 mmol/1.73 m2) had more 24hUG than those with variants at non-conservative residues (257.6 mmol/1.73 m2). Four out of 14 tested patients had mild aminoaciduria. The RTG of FRG patients had no significant correlation to phosphate reabsorption but a potential negative correlation to the fractional excretion of uric acid. Postprandial suppression of glucagon secretion was absent in most FRG patients.
Conclusions
We built a comprehensive map showing the impact of SLC5A2 variant type and variant location on glucosuria severity. Our results highlighted the role of key residues in maintaining the transport function of SGLT2 and the functional link between glucosuria and reabsorption of amino acid and uric acid in FRG patients.
Keywords: familial renal glucosuria, genotype–phenotype relationship, SGLT2, SLC5A2
KEY LEARNING POINTS.
What was known:
Familial renal glucosuria (FRG) is caused by variants in SLC5A2, the gene encoding sodium-glucose cotransporter 2 (SGLT2). The genotype–phenotype relationship in FRG patients has not been systematically evaluated.
SGLT2 inhibitors, a novel class of antidiabetic agents with cardiac and renal protective effects, have been widely used in type 2 diabetes. Detailed phenotyping of FRG patients might shed light on the long-term consequences of SGLT2 inhibition.
This study adds:
We provide a visualized summary of the genotype–phenotype relationship in FRG patients. Glucosuria severity is influenced by variant type and variant location. Homozygous missense variants, especially those involving conservative residues, can lead to severe glucosuria, highlighting the role of key residues in the transport function of SGLT2.
There is a potential functional link between SGLT2 and proximal tubule transport of solutes such as uric acid and amino acids. Postprandial suppression in glucagon secretion is absent in most FRG patients.
We built an open-source online calculator that made estimation of renal threshold of glucose (RTG) feasible in clinical settings.
Potential impact:
We might need to monitor impact of long-term SGLT2 inhibition on proximal renal tubule transport and glucagon secretion.
INTRODUCTION
Familial renal glucosuria (FRG) is a rare hereditary renal tubular disease with co-dominant inheritance [1]. Patients with FRG present with glucosuria without hyperglycemia, resulting from glucose reabsorption dysfunction in the proximal renal tubule.
Sodium-glucose cotransporter 2 (SGLT2) is a transmembrane protein expressed in the brush border of the S1 segment proximal tubule cells (PTC) [2] that mediates reabsorption of 90% of the filtered glucose load [3, 4]. Disease-causing variants in SLC5A2, the gene encoding SGLT2, lead to familial renal glucosuria [5]. A total of 111 SLC5A2 variants have been reported in FRG patients, with no apparent mutational hotspot (Supplementary data, Table S1). In FRG patients with no disease-causing SLC5A2 variants identified, mutations in PDZK1IP1, the gene encoding MAP17, a membrane protein that enhances transport activity of SGLT2, have been observed [6]. Inhibitors of SGLT2 have emerged as a novel class of antidiabetic agents with cardiac and renal protective effects [7, 8]. Phenotypes of patients with familial renal glucosuria may give us some insight into the long-term consequences of SGLT2 inhibition.
The proximal renal tubule is responsible for the reabsorption of multiple solutes, including sodium, glucose, phosphate, urate and amino acids. Although patients with disease-causing SGLT2 variants primarily present with glucosuria, it has been postulated that decreased sodium and glucose reabsorption may promote the reabsorption of phosphate [9] while inhibiting the reabsorption of urate [10]. Solute transport of proximal tubule in familial renal glucosuria has not been thoroughly characterized.
Increased glucagon level after SGLT2 inhibition has been reported in clinical studies [11, 12], and it may play a role in the development of euglycemia diabetic ketoacidosis [13]. Possible mechanisms include both carbohydrate starvation due to urinary glucose loss after SGLT2 inhibition and direct stimulation of pancreatic alpha cells due to interference of glucose sensing by SGLT2 [14, 15]. However, there are few studies on the glucagon secretion of patients with familial renal glucosuria.
In this study, we sequenced SLC5A2 and PDZK1IP1 in a cohort of 21 patients with familial renal glucosuria. We quantified their urinary glucose excretion using renal threshold of glucose (RTG), characterized solute transport in the proximal renal tubule and explored glucagon secretion in response to glucose ingestion. We reviewed all SLC5A2 variants reported in FRG patients and investigated the genotype–phenotype relationship.
MATERIALS AND METHODS
Subjects
We included 21 patients presenting with renal glucosuria, i.e. glucosuria without hyperglycemia, who visited Peking Union Medical College Hospital (PUMCH) from December 2017 to April 2022. Patients with disorders that could cause renal tubule dysfunction, such as Sjögren's syndrome, multiple myeloma, acute interstitial nephritis and chronic interstitial nephritis, were excluded. This study was approved by the ethics committee of PUMCH (JS-1233-2) with written informed consent from each participant.
Gene sequencing and variant analysis
Genomic DNA was extracted from peripheral blood. All exons of SLC5A2 and PDZK1IP1 were sequenced. Sequencing of Patients 1–10 was carried out using Tubule Panel, a custom next-generation sequencing (NGS) panel designed for hereditary tubulopathy, on an Ion Personal Genome Machine (Thermo Fisher Scientific, USA). Sequencing of Patients 11–18 was done by Sanger sequencing (Dia-up Biotech, Beijing, China). For Patients 19–21, whole-exome sequencing (BGI, China) was performed. All variants discovered by NGS were confirmed by Sanger sequencing. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines [16].
Clinical characterization and quantification of urinary glucose
Demographic data and general clinical information of the included patients were documented. Urinary glucose excretion was characterized using urinalysis, 24-h urinary glucose excretion (24hUG) and RTG. RTG was measured as previously reported [17]. Briefly, in a 4-h oral glucose tolerance test (OGTT), plasma glucose (PG) is sampled before (0 min) and 30 min, 60 min, 90 min, 120 min, 180 min and 240 min after 75 g glucose ingestion. In addition, urine samples are collected to measure urinary glucose excretion over the 4 h (UGE240min, Supplementary data, Fig. S1). From the definition of RTG,
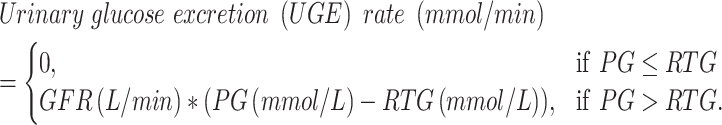 |
RTG is estimated from 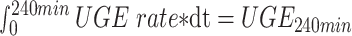 .
.
To facilitate this calculation, we built an open-source online calculator, which can be accessed at https://rtg.renaltubule.com. Protocol for using this calculator to estimate RTG and a sample data collection sheet can be found in the Supplementary data (Item S1 and S2).
Proximal renal tubule solute transport and sodium excretion
To explore possible interaction between transporters in the proximal renal tubule, we evaluated renal handling of phosphate, uric acid and amino acids. Amino acids in a morning spot urine sample were measured using chromatography (Patients 1–8, 10–13 and 16) or mass spectrometry (Patient 21). A 24-h urine sample was collected to measure urinary uric acid (UUA) and urinary creatinine (UCr). Serum uric acid (SUA) and serum creatinine (SCr) were measured on the same day. Fraction excretion of uric acid (FeUA) was calculated by UUA * SCr/(UCr * SUA) * 100%. The tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) was calculated using urinary creatine (UCr) and urinary phosphate (UP) from the second void morning urine, as well as serum creatinine (SCr) and serum phosphate (SP) measured on the same day. The calculation of TmP/GFR was carried out using the Bijvoet formula which has been described by Payne [18]. Briefly, fractional tubular reabsorption of phosphate (TRP) was calculated as 1 – [(SCr * UP)/(UCr * SP)]. TmP/GFR was calculated as TRP * SP when TRP was ≤0.86 and (0.3 * TRP * SP)/(1 – 0.8 * TRP) when TRP was >0.86. For sodium excretion, we measured 24 h urinary sodium excretion (24hUNa) and calculated fractional excretion of sodium (FeNa) using (24hUNa * SCr)/(24hUCr * SNa).
Glucagon levels
Glucagon levels were measured using blood samples taken before (0 min) and 30 min, 60 min, 90 min, 120 min, 180 min and 240 min after 75 g glucose ingestion during a 4-h oral glucose tolerance test by radioimmunoassay (North Institute of Biotechnology, Beijing, China). Each measurement was repeated twice.
Literature review and genotype–phenotype relationship
We searched PubMed with the keyword ‘Renal Glucosuria OR SGLT2 OR SLC5A2’ until May 2022. Patients’ geographic regions, SLC5A2 variants and quantification of glucosuria were summarized. We visualized variant frequency in Chinese FRG patients using the R package trackViewer [19, 20]. Homozygous variants were counted twice, and heterozygous variants were counted once. Variants in the same pedigree were counted only once. Variants were mapped on the protein domains according to protein structure deciphered by Niu et al. [2]. To explore the genotype–phenotype relationship, we compared 24-h urinary glucose in patients with different variant types and plotted 24-h urinary glucose against the variant location.
Statistical analysis
Normally distributed variables were expressed as mean ± standard deviation (SD). For variables with normal distribution and homogeneity of variance, we used the unpaired t-test for two independent groups and paired t-test for two sets of joint observations. Otherwise, the Wilcoxon rank-sum test (Mann–Whitney U test) was used to assess the difference between the two independent samples. Non-parametric Kruskal–Wallis H test was used to assess the difference among multiple independent samples. After the Kruskal–Wallis test, variations of statistical significance were further subjected to post hoc pairwise analysis using the Wilcoxon rank-sum test and Bonferroni's correction. Statistical significance was assumed when P < .05. Statistical analysis was performed with GraphPad Prism 9.4.1 or R packages ‘stats’ [19] and ‘car’ [21].
RESULTS
Genetic characterization of the FRG cohort
Among the 21 patients with renal glucosuria (Table 1), 62% were male and the mean age of presentation was 39.6 ± 10.9 years. Except for three female patients (Nos 1, 16 and 19) who complained of urinary irritation symptoms, most patients had incidental findings of glucosuria. Most patients had completely normal blood glucose (BG) levels in the oral glucose tolerance test. However, some patients had impaired fasting BG (Patients 1, 3, and 10) or slightly elevated 2hBG (Patients 1, 7, 10 and 18). Notably, Patients 3, 8, 12, 15 and 17 had 2hBG levels lower or equal to fasting BG. All the patients had normal estimated glomerular filtration rate.
Table 1:
General characteristics and SLC5A2 variants of 21 FRG patients.
| No. | Age (years) | Sex | BMI (kg/m2) | FBG (mmol/L) | 2hBG (mmol/L) | HbA1c (%) | eGFR (mL/min/1.73 m2) | RTG (mmol/L) | UG (mmol/L) | 24hUG (mmol) | SLC5A2 variants |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 22 | F | 27.34 | 5.9 | 8.1 | 5.0 | 129.4 | 6.3 | ≥55 | 126.39 | c.886–31_886–10del, c.1540C>T(p.P514S) |
| 2 | 58 | M | / | 5.5 | / | 5.5 | 116.5 | / | 5.5 | / | c.1451G>A(p.G484D) |
| 3 | 33 | M | 20.74 | 5.9 | 5.7 | 5.6 | 125.6 | 3.8 | ≥55 | 323.00 | c.1540C>T(p.P514S), c.1690C>T(p.R564W) |
| 4 | 41 | M | 26.56 | 4.8 | 5.5 | 5.4 | 111.1 | / | 28 | / | c.1665+1G>A |
| 5 | 43 | F | / | / | / | 5.6 | 93.0 | / | 28 | / | c.410G>A(p.R137H) |
| 6 | 33 | M | 25.71 | 5.2 | 6.7 | 4.8 | 114.0 | 6.2 | 14 | 22.00 | c.634G>T(p.A212S), c.574+1G>C |
| 7 | 59 | M | 25.95 | 5.4 | 7.8 | 4.8 | 106.1 | 1.0 | 14 | 676.50 | c.395G>A(p.R132H), c.1947G>A(p.W649*) |
| 8 | 40 | F | 19.00 | 5.0 | 5.0 | 5.6 | 117.4 | 2.1 | ≥55 | 328.78 | c.1152_1163del(p.V385_A388del), c.1774_1775del(p.S592Cfs*6) |
| 9 | 33 | F | / | 3.7 | 5.8 | 5.5 | 119.1 | / | ++ | / | c.1499T>G(p.L500R) |
| 10 | 54 | M | 27.04 | 6.2 | 8.8 | 5.7 | 89.1 | 9.2 | 28 | 42.89 | c.1420G>C(p.A474P) |
| 11 | 41 | M | 24.34 | / | / | / | 96.9 | 6.4 | ≥55 | 30.39 | c.1735C>T(p.Q579*) |
| 12 | 42 | M | 21.79 | 5.5 | 4.8 | 5.3 | 101.9 | 8.0 | NEG | 11.78 | c.265G>A(p.A89T) |
| 13 | 22 | M | 25.06 | 5.0 | 6.1 | 4.5 | 125.1 | 4.3 | ≥55 | / | c.1343A>G(p.Q448R) |
| 14 | 43 | F | 21.26 | 4.9 | 7.2 | 5.3 | 100.3 | 3.6 | ≥55 | 307.56 | c.1017C>A(p.Y339*) |
| 15 | 18 | M | 22.38 | 5.0 | 5.0 | / | 136.4 | 6.4 | TRACE | / | c.115G>T(p.V39F) |
| 16 | 43 | F | 25.22 | 4.9 | 7.7 | 5.1 | 107.5 | 8.2 | NEG | 19.06 | c.1472G>A(p.G491E) |
| 17 | 44 | F | 22.72 | 4.8 | 4.4 | 4.9 | 85.8 | 4.2 | TRACE | / | c.1435C>G(p.R479G) |
| 18 | 42 | M | 24.65 | 5.1 | 8.1 | 4.9 | 108.7 | 4.0 | ≥55 | 170.33 | c.736C>T(p.P246S), c.1391C>A(p.A464E) |
| 19 | 47 | F | 20.77 | 4.8 | 6.2 | 5.4 | 103.7 | / | ≥55 | / | c.1346G>A(p.G449D), c.127–16C>A |
| 20 | 44 | M | 21.80 | / | / | 5.1 | 109.5 | / | ≥55 | / | c.1079G>A(p.G360D), c.877A>T(p.S293C) |
| 21 | 30 | M | 24.39 | 5.4 | 6.4 | 5.2 | 128.0 | 5.1 | ≥55 | / | c.127–16C>A |
Variants in bold: novel variants.
BMI, body mass index; FBG, fasting blood glucose; eGFR, estimated glomerular filtration rate.
Sequencing of SLC5A2 yielded 27 rare variants (Table 1), including 18 missense variants, four splicing variants, three nonsense variants, one frameshift deletion and one in-frame deletion. Twelve of these variants were novel: G484D, R564W, A212S, c.574+1G>C, W649*, S592Cfs*6, Q579*, Y339*, V39F, G491E, A464E and G360D. Thirteen patients were heterozygous, and eight patients carried two variants. According to the ACMG criteria, six variants (c.127–16C>A, c.574+1G>C, Y339*, c.1665+1G>A, Q579* and S592Cfs*6) were classified as pathogenic, eight variants (A89T, R132H, R137H, c.886–31_886–10del, V385_A388del, A474P, R479G and R564W) were classified as likely pathogenic and the rest of the variants were classified as variants of uncertain significance (Supplementary data, Table S2).
Sequencing of PDZK1IP1 yielded two variants: one likely pathogenic variant p.N26fs*39 in Patient 4 and one variant of uncertain significance (V44I) in Patients 15 and 18.
Literature review and visualization of SLC5A2 variant distribution
Our literature review yielded 111 SLC5A2 variants in 157 FRG patients up to May 2022, including 85 missense variants, 4 nonsense variants, 10 frameshift indels, 3 in-frame indels, 8 splicing variants and 1 duplication. We mapped the distribution of missense and nonsense variants onto the protein structure in Fig. 1, and summarized the SLC5A2 variants and quantification of glucosuria (24-h urinary glucose, spot urine glucose concentration, or spot urine glucose–creatinine ratio) in Supplementary data, Table S1. Fifty-three variants identified in 65 Chinese FRG patients, including 27 variants found in this study, were mapped onto the SLC5A2 gene structure in Fig. 2. The variants were highly diverse, with 90.6% of the variants occurring only once or twice. Variants with relatively high frequency included c.886–31_886–10del (10.8%), c.1540C>T (6.2%) and c.127–16C>A (3.8%).
Figure 1:

Variant distribution and glucosuria severity mapped onto SGLT2 protein domain structure (modified from Niu et al. [2]). Protein domains are plotted as deciphered by Niu et al. Alpha helices are shown as rectangles, and unmodeled residues are shown as dashed lines. Conserved residues are colored in different shades of blue. Residues that form disulfide bonds to stabilize the extracellular structure are highlighted in yellow. Residues that interact with MAP17 are marked with a blue box. Residues that bind Na+ and glucose are shown in red and green boxes, respectively. Missense and nonsense variants are marked with * and #, respectively. Black symbols (* and #) indicate variants previously reported in the literature, and red symbols indicate novel variants identified by this study. Allele count is listed below each variant and coloured in shades of green. 24hUG of homozygous and heterozygous carriers of each variant are visualized in different shades of gold and red, respectively. Both 24hUG corrected and not corrected by body surface area (in mmol/1.73 m2/day and mmol/day) are included in this figure.
Figure 2:

SLC5A2 variants in Chinese FRG patients. Genetic variants found in Chinese FRG patients, represented by dots in different colours, were visualized on the SLC5A2 gene structure. The height of each node corresponds to the frequency of the variant. Magenta dots: variants from the literature review; yellow dots: variants identified in this study; grey dots: variants found both in our cohort and in the literature.
Quantification of glucosuria and genotype–phenotype relationship
Estimated RTG ranged from 1.0 to 9.2 mmol/L, corresponding to the spectrum from severe to slight impairment of glucose reabsorption (Table 1). RTG was inversely correlated to lg(24hUG) (R2 = 0.765, Fig. 3A). In our cohort, patients with two SLC5A2 variants had lower mean RTG than those with a single variant (3.9 vs 6.2 mmol/L, P = .057, Fig. 3B). Similarly, our literature review showed that patients carrying two variants had higher mean 24hUG than single variant carriers (291.0 vs 40.0 mmol/1.73 m2, P < .0001, Fig. 3D). Patients with homozygous missense variants or in-frame indels had mean 24hUG comparable to those with homozygous truncating variants (457.2 vs 445.0 mmol/1.73 m2), and much higher than carriers of homozygous splicing variants (196.6 mmol/1.73 m2, P < .05, Fig. 3F). Patients with homozygous missense variants involving conservative residues had significantly higher mean 24hUG than carriers of homozygous missense variants at non-conservative residues (582.0 vs 257.6 mmol/1.73 m2, P < .01, Fig. 3G). To visualize the genotype–phenotype relationship, we built comprehensive maps including variant type, zygosity, variant location and 24hUG on the SLC5A2 gene structure (Fig. 3H) and on the SGLT2 protein domains (Fig. 1).
Figure 3:

Genotype–phenotype relationship in FRG patients. (A) 24hUG is inversely related to RTG [lg(24hUG) = −0.1881 * RTG + 2.252, R2 = 0.7650]. (B) In our cohort, patients with two SLC5A2 variants had lower RTG than those with only one (mean RTG 3.9 ± 2.1 vs 6.2 ± 2.0 mmol/L, P = .057). (C) In our cohort, the mean RTG of patients carrying heterozygous pathogenic/likely pathogenic (P/LP) variants was not significantly different from that of patients with heterozygous variants of uncertain significance (VUS) (6.08 ± 2.20 vs 6.30 ± 1.95 mmol/L, P = .8897). (D) Literature review showed that 24hUG corrected by body surface area of patients carrying a single SLC5A2 variant were significantly lower than those carrying two variants (40.0 ± 51.5 vs 291.0 ± 248.0 mmol/1.73 m2, P < .0001). (E) Literature review showed that 24hUG of patients with P/LP variants were not significantly different from those with VUS variants (homozygous: 387.3 ± 202.7 vs 409.8 ± 234.6 mmol/1.73 m2, P = .7599; heterozygous: 36.27 ± 34.20 vs 44.01 ± 66.55 mmol/1.73 m2, P = .7924). Homo = homozygous, hetero = heterozygous. (F) Patients from the literature were divided according to the type of variant they carried: mis + in-frame (missense variants and in-frame indels), truncating (nonsense variants and frameshift variants) and splicing variants. Patients with homozygous splicing variants had significantly less 24hUG (196.6 ± 150.6 mmol/1.73 m2) than those with homozygous truncating (445.0 ± 182.5 mmol/1.73 m2) or mis + in-frame variants (457.2 ± 224.1 mmol/1.73m2, P < .05). (G) Patients carrying missense variants were divided according to the conservativeness of the involved amino acid. Patients with homozygous variants at conservative residues had more 24hUG (582.0 ± 176.1 mmol/1.73 m2) than those with variants involving non-conservative residues (257.6 ± 124.3 mmol/1.73 m2, P < 0.01). (H) 24hUG mapped on the SLC5A2 gene structure. Each node represents one variant, and the node's height corresponds to an average of 24hUG of FRG patients carrying this variant. Nodes on the top: heterozygotes; nodes on the bottom: homozygotes. The colour of the nodes represents variant types (black: missense variants; pink: in-frame indels; green: frameshift indels; blue: nonsense variants; cyan: splicing variants). Circles: 24hUG normalized by body surface area (mmol/1.73 m2/day); squares: 24hUG in mmol/day.
Solute reabsorption of the proximal tubule and sodium excretion
Chromatography showed suspiciously positive aminoaciduria in Patient 5 and weakly positive results in three patients (Nos 2, 3 and 16) among 13 tested patients. For Patient 21, mass spectrometry revealed slightly elevated (1–2× upper limit of normal range) alanine, citrulline, lysine, methionine, phenylalanine, tryptophan, tyrosine and valine levels (Table 2). We observed low-normal mean serum uric acid (UA) levels (279 ± 70μmol/L) and normal mean 24-h urinary UA excretion (3.56 ± 0.70 mmol) in FRG patients (Table 2). The mean FeUA was 6.94 ± 2.28%, with a potential negative correlation between FeUA and RTG (R2 = 0.5150, Fig. 4A). FRG patients had a mean serum phosphate level of 1.14 mmol/L and a mean TmP/GFR of 1.03 mmol/L. Most patients had normal TmP/GFR, except for Patients 14 and 8, who had a slightly decreased or increased TmP/GFR, respectively. We did not observe any correlation between TmP/GFR and RTG (Fig. 4B).
Table 2:
Solute transport of the proximal tubule in FRG patients.
| No. | Aminoaciduria | RTG (mmol/L) | 24hUG (mmol) | UA (μmol/L) | 24hUA excretion (mmol) | FeUA (%) | P (mmol/L) | TmP/GFR (mmol/L) |
|---|---|---|---|---|---|---|---|---|
| 1 | Negative | 6.3 | 126.39 | 246 | 2.63 | 5.20 | 1.21 | / |
| 2 | Weakly positive | / | / | 296 | / | / | 1.22 | / |
| 3 | Negative | 3.8 | 323.00 | 231 | 4.34 | 6.56 | 1.08 | / |
| 4 | Negative | / | / | 244 | 3.46 | 7.18 | 1.10 | / |
| 5 | Suspiciously positive | / | / | 209 | / | / | 1.30 | / |
| 6 | Negative | 6.2 | 22.00 | 308 | 3.18 | 6.62 | 1.02 | 1.014 |
| 7 | Negative | 1.0 | 676.50 | 194 | 4.05 | 8.93 | 0.94 | 0.974 |
| 8 | Negative | 2.1 | 328.78 | 152 | 3.73 | 8.90 | 1.30 | 1.435 |
| 9 | / | / | / | 220 | / | / | 1.21 | / |
| 10 | Negative | 9.2 | 42.89 | 379 | 3.30 | 3.21 | 1.10 | 1.090 |
| 11 | Negative | 6.4 | 30.39 | 375 | / | / | 1.12 | / |
| 12 | Negative | 8.0 | 11.78 | 283 | 3.54 | 7.45 | 1.17 | / |
| 13 | Weakly positive | 4.3 | / | 348 | 3.45 | 5.29 | 1.01 | 0.930 |
| 14 | / | 3.6 | 307.56 | 271 | 3.29 | 8.57 | 0.85 | 0.658 |
| 15 | / | 6.4 | / | 333 | / | / | 1.15 | / |
| 16 | Weakly positive | 8.2 | 19.06 | 292 | 2.55 | 5.01 | 1.29 | 1.107 |
| 17 | / | 4.2 | / | 194 | / | / | 1.04 | / |
| 18 | / | 4.0 | 170.33 | 363 | 5.22 | 5.39 | 1.16 | / |
| 19 | / | / | / | 196 | 3.58 | 12.00 | 1.21 | / |
| 20 | / | / | / | 371 | / | / | 1.19 | / |
| 21 | Positive | 5.1 | / | 350 | / | / | 1.17 | / |
| Mean ± SD | 5.2 ± 2.3 | 187.15 ± 206.91 | 279 ± 70 | 3.56 ± 0.70 | 6.94 ± 2.28 | 1.14 ± 0.12 | 1.03 ± 0.23 |
/: lab results not available; UA: blood uric acid, normal range 210–416 μmol/L (male) or 150–357 μmol/L (female); P: serum phosphate, normal range 0.81–1.45 mmol/L; TmP/GFR: the ratio of the renal tubular maximum reabsorption rate of phosphate to glomerular filtration rate, normal range 0.8–1.35 mmol/L.
Figure 4:

Phosphate and UA reabsorption in the proximal tubule and sodium excretion of patients with different RTG levels. (A) FeUA seems to negatively correlate with RTG (FeUA = −0.5057 * RTG + 9.073, R2 = 0.5150). (B) One patient had slightly decreased TmP/GFR, and one patient had slightly increased TmP/GFR. The rest of the patients had TmP/GFR within the normal range (0.8–1.35 mmol/L). No correlation was observed between TmP/GFR and RTG. (C) FeNa showed no correlation with RTG. (D, E) The data did not show any correlation between sodium excretion (24hUNa) and glucose excretion (RTG or 24hUG).
As is shown in Supplementary data, Table S4, 24hUNa ranged from 81 to 398 mmol, with mean 24hUNa of 191.93 ± 79.34 mmol. FeNa ranged from 0.34% to 1.01% and mean FeNa was 0.65 ± 0.20%. We did not observe correlation between sodium excretion (24hUNa or FeNa) and glucose excretion (24hUG or RTG, Fig. 4C–E).
Glucagon secretion and RTG
The mean fasting glucagon level (133.4 ng/L) in 12 FRG patients was lower than the mean glucagon level 30 min (174.6 ng/L, P = .005) and 120 min after glucose ingestion (165.4 ng/L, P = .038, Fig. 5B). Only Patient 15 had decreased 30 min postprandial glucagon compared with fasting. Four patients (No. 6, 7, 15 and 18) had lower 120 min postprandial glucagon than fasting. We found no apparent correlation between glucagon levels and RTG (Fig. 5C and D). The area under the curve (AUC) of the glucagon–time curve had no significant correlation with RTG levels (R2 = 0.08, Fig. 5D).
Figure 5:

The relationship between glucagon secretion and RTG. Plasma glucose (A) and glucagon (B) levels were sampled in FRG patients during a 4-h OGTT (mean ± standard error). (C) Heatmap visualizing the relationship between glucagon levels and RTG. Each row represents an FRG patient arranged in the order of RTG, and each column represents a time point in the OGTT. Glucagon levels are represented with different shades of colour. (D) No significant correlation was observed between the area under the curve (AUC) of the glucagon–time curve and RTG.
DISCUSSION
In this study, we provided a visualized overview of the SLC5A2 mutational landscape and genotype–phenotype relationship, and identified the crucial role of missense and in-frame indels, especially those involving conservative residues. We developed an open-source online calculator to facilitate the estimation of the RTG for clinicians. We systematically evaluated alterations in proximal renal tubule solute transport and plasma glucagon levels in FRG patients to delineate long-term effects of SGLT2 inhibition.
We built the most comprehensive genotype–phenotype dataset in FRG, including 12 novel SLC5A2 variants identified in this study and 111 variants from the literature review. Projection of the variants onto the SLC5A2 gene structure showed that the variants are distributed throughout the gene without obvious mutational hotspot. Patients carrying two SLC5A2 variants had lower RTG and larger amounts of urinary glucose excretion than those carrying a single variant, consistent with the previously reported co-dominant inheritance pattern [1]. Notably, homozygous missense or in-frame indel carriers can have glucosuria as severe as patients with homozygous truncating variants, highlighting the role of key residues in maintaining the structure and the transport function of SGLT2. This is echoed by the finding that patients with homozygous missense variants affecting conservative residues have significantly more urinary glucose excretion than those with variants involving non-conservative residues.
One prominent example is that homozygous missense variant K321R at the glucose binding site leads to severe glucosuria (mean 24hUG 635.2 mmol/1.73 m2/day). Another interesting finding is that patients with homozygous nonsense variants with predicted nonsense-mediated decay (NMD), i.e. variants located at least 50 nucleotides upstream of the last exon junction (Y128*, W440*), had 24hUG ranging from 342.2 to 403.9 mmol/1.73 m2/day, lower than what would be expected if NMD occurred. Compensatory glucose reabsorption mediated by SGLT1 [22] might play a role in this discrepancy, yet the exact mechanism warrants further study.
MAP17, encoded by PDZK1IP1, can interact with the 13th transmembrane helix of SGLT2 [2] and enhance its transport activity [6]. There have been reports of homozygous carriers of splicing variant c.176+1G>A in PDZK1IP1 to present with renal glucosuria without detectable variants in SLC5A2 [6]. In our cohort, Patient 4 carried both a heterozygous SLC5A2 variant c.1665+1G>A and a heterozygous PDZK1IP1 variant N26fs*39. Both variants were inherited from the proband's father, making it hard to delineate the impact of each variant from the pedigree analysis. Both Patients 15 and 18 carried a heterozygous missense PDZK1IP1 variant V44I besides SLC5A2 variants, which affects a residue that interacts with SGLT2 [2] but has a relatively high minor allele frequency at 0.0494 in East Asian populations in gnomAD. Further data are still needed to reveal the frequency of PDZK1IP1 variants in FRG patients and whether they can act as a modifier to SLC5A2 variants.
The RTG is defined as the plasma glucose level at which the filtered glucose load meets the maximum reabsorption capacity of the renal tubule, and begins to leak from the urine. RTG in healthy subjects has been reported to be 9.5–11.0 mmol/L [23, 24]. The gold standard for the determination of RTG is the stepwise hyperglycemic clamp procedure (SHCP). However, this is a complicated procedure requiring special equipment, limiting its application in clinical settings. Polidori et al. validated a novel method to estimate RTG from urinary glucose excretion and blood glucose–time curve and proved its consistency with the SHCP method [17]. Aires et al. first applied this method in familial renal glucosuria patients and found that homozygous variant carriers had much lower mean RTG (0.95 mmol/L) than heterozygous variant carriers (4.91 mmol/L) [25]. Yet this method requires complicated calculation, which is difficult to carry out in clinical settings. We adopted this method to estimate RTG in a 4-h OGTT and developed an open-source online calculator to facilitate the calculation for clinicians. For plasma glucose levels sampled at time points other than 0 h, 0.5 h, 1 h, 1.5 h, 2 h, 3 h and 4 h, we also developed a ‘Use Other Timepoints’ option to allow greater flexibility.
We observed that FRG patients had mild alterations in proximal renal tubule transport of solutes such as UA and amino acids. We found that FRG patients had slightly decreased serum UA levels and that FeUA correlates with glucosuria severity. This is consistent with our previous findings that SGLT2 inhibitors induced a dose-dependent reduction in serum UA levels [10]. Indeed, urinary UA excretion increased with glucosuria in patients with type 1 [26] and type 2 diabetes [10, 27], regardless of whether the glucosuria was caused by hyperglycemia or SGLT2 inhibition. Although the mechanism is not entirely clear, Chino et al. observed that extracellular glucose could stimulate the secretion of UA by GLUT9 in Xenopus oocytes [28] and suggested that glucose-stimulating GLUT9-mediated excretion of UA at the proximal tubule might explain the uricosuric effect of glucosuria. Our finding that 35.7% of FRG patients had mild aminoaciduria indicated that aminoaciduria might be more prevalent in FRG patients than previously reported [29]. Aminoaciduria has also been reported in type 1 and type 2 diabetic patients with glucosuria [30]. Although the exact mechanism remains to be elucidated, our observations suggest a functional link between transporters in PTC.
As both SGLT2 and Na+/phosphate cotransporters utilize the Na+ gradient at the brush border in PTC, it has been proposed that reduction in Na+/glucose transport might increase Na+/phosphate reabsorption, raising concerns over long-term bone health with SGLT2 inhibition. Previous clinical studies showed that 5-day treatment of canagliflozin induced a 16% increase in serum phosphate in healthy volunteers [9] and 6-week treatment of dapagliflozin increased serum phosphate by 9% in type 2 diabetes patients [31]. Although Sweet Pee mice carrying homozygous nonsense SLC5A2 variant had elevated fractional excretion of phosphate compared with wild-type, there was no significant change in 24-h phosphate excretion or serum phosphate levels [32]. In our cohort, five out of seven FRG patients had normal TmP/GFR. Unlike UA, we did not observe a correlation between phosphate handling (TmP/GFR) and glucose handling (RTG).
Sodium wasting and activation of renin–angiotensin–aldosterone system were reported in some FRG patients [33, 34]. In our FRG cohort, the mean 24hUNa (191.93 mmol) was comparable to the level previously reported in Chinese population (mean 24hUNa 189.07 mmol) [35]. FeNa in our cohort (0.65 ± 0.20%) was comparable to FeNa previously reported in control group [36]. We did not find evidence of correlation between sodium excretion and glucose excretion. In line with previous findings that the natriuretic effect of SGLT2 inhibitors was evident in the acute phase and blunted in the chronic phase [36–39], our observations might reflect adaptation to chronic SGLT2 deficiency.
As clinical studies in diabetic patients demonstrated increased glucagon levels after the use of SGLT2 inhibitors [11, 12], we measured glucagon levels in 12 FRG patients who underwent OGTT-based RTG measurement. We observed that many FRG patients had higher plasma glucagon levels 30 min and 120 min after glucose ingestion compared with fasting. Despite the classical belief of postprandial suppression of glucagon, results from clinical studies are discrepant. Færch et al. observed that most diabetic and prediabetic subjects had lower glucagon levels 120 min after glucose intake than at 0 min, and that those with non-suppressive 30-min glucagon had higher insulin resistance [40]. On the other hand, Wagner et al. reported that 21%–34% of subjects with normal glucose tolerance or prediabetes had increased glucagon at 120 min and that non-suppressed glucagon was associated with a lower risk of impaired glucose tolerance [41]. This controversy likely reflects the complexity of glucagon secretion regulation and the diversity of the included population. Nonsuppressive postprandial glucagon observed in our FRG cohort might either be due to altered glucose sensing of pancreatic alpha cells [15] or lower plasma glucose levels due to urinary glucose loss. Further studies characterizing glucagon and insulin responses during euglycemic glucose clamps in both FRG patients and healthy controls will shed light on this topic.
Limitations
This study has several limitations. As FRG is a rare disease, statistical power is limited by the relatively small sample size at a single institute. We hope to conduct multicentre studies with a larger sample size in the future based on collaborative networks such as the National Rare Diseases Registry System of China. For patients carrying two SLC5A2 variants, we did not test whether the two alleles were in cis or in trans. We hope that development of long-read sequencing techniques will facilitate variant phasing. In this study, we did not carry out in vitro characterization of the SLC5A2 variants. We hope to conduct systematic evaluation of the impact of variants on the expression and transport function of SGLT2 in future studies.
CONCLUSION
We built a comprehensive genotype–phenotype correlation map in FRG showing the impact of SLC5A2 variant type and variant location on glucosuria severity, and highlighted the role of key residues in maintaining the transport function of SGLT2. We identified a functional link between glucosuria and transport of other solutes, such as amino acids and UA, in the proximal renal tubule in FRG patients.
Supplementary Material
Contributor Information
Lubin Xu, Department of Nephrology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Ruohuan Zhao, Department of Nephrology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China; 4+4 M.D. Program, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Yumo Zhao, Department of Nephrology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Xueqing Tang, Department of Nephrology, Shandong Qianfoshan Hospital, Jinan, Shandong Province, China.
Nuo Si, McKusick-Zhang Center for Genetic Medicine, State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences Chinese Academy of Medical Sciences, School of Basic Medicine Peking Union Medical College, Beijing, China.
Xiuzhi Guo, Department of Laboratory Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Cai Yue, Department of Nephrology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Min Nie, Department of Endocrinology &Key Laboratory of Endocrinology, National Health, and Family Planning Commission; Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Limeng Chen, Department of Nephrology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
FUNDING
This work was partially supported by grants from the National Key R&D Program of China (2022YFC2703901 to C.Y.); National Natural Scientific Foundation of China (82 170 709, 81 970 607 to L.C.); CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-003 to L.C.); Beijing Natural Science Foundation (L202035 to L.C.); National High Level Hospital Clinical Research Funding (2022-PUMCH-B-019, 2022-PUMCH-D-002 to L.C.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
DATA AVAILABILITY STATEMENT
The data underlying this article will be shared on reasonable request to the corresponding author.
CONFLICT OF INTEREST STATEMENT
All authors declare no conflicts of interest.
REFERENCES
- 1. Santer R, Kinner M, Lassen CLet al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol 2003;14:2873–82. 10.1097/01.ASN.0000092790.89332.D2 [DOI] [PubMed] [Google Scholar]
- 2. Niu Y, Liu R, Guan Cet al. Structural basis of inhibition of the human SGLT2-MAP17 glucose transporter. Nature 2022;601:280–4. 10.1038/s41586-021-04212-9 [DOI] [PubMed] [Google Scholar]
- 3. Wright EM, Loo DD, Hirayama BA.. Biology of human sodium glucose transporters. Physiol Rev 2011;91:733–94. 10.1152/physrev.00055.2009 [DOI] [PubMed] [Google Scholar]
- 4. Ghezzi C, Loo DDF, Wright EM.. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018;61:2087–97. 10.1007/s00125-018-4656-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van den Heuvel LP, Assink K, Willemsen Met al. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet 2002;111:544–7. 10.1007/s00439-002-0820-5 [DOI] [PubMed] [Google Scholar]
- 6. Coady MJ, El Tarazi A, Santer Ret al. MAP17 Is a necessary activator of renal Na+/glucose cotransporter SGLT2. J Am Soc Nephrol 2017;28:85–93. 10.1681/ASN.2015111282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braunwald E. Gliflozins in the management of cardiovascular disease. N Engl J Med 2022;386:2024–34. 10.1056/NEJMra2115011 [DOI] [PubMed] [Google Scholar]
- 8. Perkovic V, Jardine MJ, Neal Bet al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 2019;380:2295–306. 10.1056/NEJMoa1811744 [DOI] [PubMed] [Google Scholar]
- 9. Blau JE, Bauman V, Conway EMet al. Canagliflozin triggers the FGF23/1,25-dihydroxyvitamin D/PTH axis in healthy volunteers in a randomized crossover study. JCI Insight 2018;3:e99123. 10.1172/jci.insight.99123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y, Xu L, Tian Det al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obes Metab 2018;20:458–62. 10.1111/dom.13101 [DOI] [PubMed] [Google Scholar]
- 11. Ferrannini E, Muscelli E, Frascerra Set al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 2014;124:499–508. 10.1172/JCI72227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merovci A, Solis-Herrera C, Daniele Get al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest 2014;124:509–14. 10.1172/JCI70704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peters AL, Buschur EO, Buse JBet al. Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care 2015;38:1687–93. 10.2337/dc15-0843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pedersen MG, Ahlstedt I, El Hachmane MFet al. Dapagliflozin stimulates glucagon secretion at high glucose: experiments and mathematical simulations of human A-cells. Sci Rep 2016;6:31214. 10.1038/srep31214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonner C, Kerr-Conte J, Gmyr Vet al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 2015;21:512–7. 10.1038/nm.3828 [DOI] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale Set al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Polidori D, Sha S, Ghosh Aet al. Validation of a novel method for determining the renal threshold for glucose excretion in untreated and canagliflozin-treated subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab 2013;98:E867–71. 10.1210/jc.2012-4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem 1998;35:201–6. 10.1177/000456329803500203 [DOI] [PubMed] [Google Scholar]
- 19. R Core Team . R: a Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2022. [Google Scholar]
- 20. Ou J, Zhu LJ.. trackViewer: a bioconductor package for interactive and integrative visualization of multi-omics data. Nat Methods 2019;16:453–4. 10.1038/s41592-019-0430-y [DOI] [PubMed] [Google Scholar]
- 21. Fox J, Werisberg S. An {R} Companion to Applied Regression, 3rd edn. Thousand Oaks CA: Sage, 2019. https://socialsciences.mcmaster.ca/jfox/Books/Companion/ [Google Scholar]
- 22. Rieg T, Masuda T, Gerasimova Met al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Ren Physiol 2014;306:F188–93. 10.1152/ajprenal.00518.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeFronzo RA, Hompesch M, Kasichayanula Set al. Characterization of renal glucose reabsorption in response to Dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013;36:3169–76. 10.2337/dc13-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rave K, Nosek L, Posner Jet al. Renal glucose excretion as a function of blood glucose concentration in subjects with type 2 diabetes—results of a hyperglycaemic glucose clamp study. Nephrol Dial Transplant 2006;21:2166–71. 10.1093/ndt/gfl175 [DOI] [PubMed] [Google Scholar]
- 25. Aires I, Fila M, Polidori Det al. Determination of the renal threshold for glucose excretion in familial renal glucosuria. Nephron 2015;129:300–4. 10.1159/000381677 [DOI] [PubMed] [Google Scholar]
- 26. Lytvyn Y, Škrtić M, Yang GKet al. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am J Physiol Renal Physiol 2015;308:F77–83. 10.1152/ajprenal.00555.2014 [DOI] [PubMed] [Google Scholar]
- 27. Qin Y, Zhang S, Cui Set al. High urinary excretion rate of glucose attenuates serum uric acid level in type 2 diabetes with normal renal function. J Endocrinol Invest 2021;44:1981–8. 10.1007/s40618-021-01513-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chino Y, Samukawa Y, Sakai Set al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos 2014;35:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Magen D, Sprecher E, Zelikovic Iet al. A novel missense mutation in SLC5A2 encoding SGLT2 underlies autosomal-recessive renal glucosuria and aminoaciduria. Kidney Int 2005;67:34–41. 10.1111/j.1523-1755.2005.00053.x [DOI] [PubMed] [Google Scholar]
- 30. Bingham C, Ellard S, Nicholls AJet al. The generalized aminoaciduria seen in patients with hepatocyte nuclear factor-1α mutations is a feature of all patients with diabetes and is associated with glucosuria. Diabetes 2001;50:2047–52. 10.2337/diabetes.50.9.2047 [DOI] [PubMed] [Google Scholar]
- 31. de Jong MA, Petrykiv SI, Laverman GDet al. Effects of dapagliflozin on circulating markers of phosphate homeostasis. Clin J Am Soc Nephrol 2019;14:66–73. 10.2215/CJN.04530418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerber C, Wang X, David Vet al. Long-term effects of Sglt2 deletion on bone and mineral metabolism in mice. JBMR Plus 2021;5:e10526. 10.1002/jbm4.10526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Calado J, Loeffler J, Sakallioglu Oet al. Familial renal glucosuria: SLC5A2 mutation analysis and evidence of salt-wasting. Kidney Int 2006;69:852–5. 10.1038/sj.ki.5000194 [DOI] [PubMed] [Google Scholar]
- 34. Calado J, Sznajer Y, Metzger Det al. Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant 2008;23:3874–9. 10.1093/ndt/gfn386 [DOI] [PubMed] [Google Scholar]
- 35. Tan M, He FJ, Wang Cet al. Twenty-four-hour urinary sodium and potassium excretion in China: a systematic review and meta-analysis. J Am Heart Assoc 2019;8:e012923. 10.1161/JAHA.119.012923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griffin M, Rao VS, Ivey-Miranda Jet al. Empagliflozin in heart failure: diuretic and cardiorenal effects. Circulation 2020;142:1028–39. 10.1161/CIRCULATIONAHA.120.045691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Opingari E, Verma S, Connelly KAet al. The impact of empagliflozin on kidney injury molecule-1: a subanalysis of the effects of empagliflozin on cardiac structure, function, and circulating biomarkers in patients with type 2 diabetes CardioLink-6 trial. Nephrol Dial Transplant 2020;35:895–7. 10.1093/ndt/gfz294 [DOI] [PubMed] [Google Scholar]
- 38. Zanchi A, Burnier M, Muller MEet al. Acute and chronic effects of SGLT2 inhibitor empagliflozin on renal oxygenation and blood pressure control in nondiabetic normotensive subjects: a randomized, placebo-controlled trial. J Am Heart Assoc 2020;9:e016173. 10.1161/JAHA.119.016173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thomson SC, Rieg T, Miracle Cet al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 2012;302:R75–83. 10.1152/ajpregu.00357.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Færch K, Vistisen D, Pacini Get al. Insulin resistance is accompanied by increased fasting glucagon and delayed glucagon suppression in individuals with normal and impaired glucose regulation. Diabetes 2016;65:3473–81. 10.2337/db16-0240 [DOI] [PubMed] [Google Scholar]
- 41. Wagner R, Hakaste LH, Ahlqvist Eet al. Nonsuppressed glucagon after glucose challenge as a potential predictor for glucose tolerance. Diabetes 2016;66:1373–9. 10.2337/db16-0354 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.