

Abstract
Contraction of cardiomyocytes is initiated at subcellular elements called dyads, where L-type Ca2+ channels in t-tubules are located within close proximity to ryanodine receptors in the sarcoplasmic reticulum. While evidence from small rodents indicates that dyads are assembled gradually in the developing heart, it is unclear how this process occurs in large mammals. We presently examined dyadic formation in fetal and newborn sheep (Ovis aries), and the regulation of this process by fetal cardiac workload. By employing advanced imaging methods, we demonstrated that t-tubule growth and dyadic assembly proceed gradually during fetal sheep development, from 93 days of gestational age until birth (147 days). This process parallels progressive increases in fetal systolic blood pressure, and includes step-wise colocalization of L-type Ca2+ channels and the Na+/Ca2+ exchanger with ryanodine receptors. These proteins are upregulated together with the dyadic anchor junctophilin-2 during development, alongside changes in the expression of amphiphysin-2 (BIN1) and its partner proteins myotubularin and dynamin-2. Increasing fetal systolic load by infusing plasma or occluding the post-ductal aorta accelerated t-tubule growth. Conversely, reducing fetal systolic load with infusion of enalaprilat, an angiotensin converting enzyme inhibitor, blunted t-tubule formation. Interestingly, altered t-tubule densities did not relate to changes in dyadic junctions, or marked changes in the expression of dyadic regulatory proteins, indicating that distinct signals are responsible for maturation of the sarcoplasmic reticulum. In conclusion, augmenting blood pressure and workload during normal fetal development critically promotes t-tubule growth, while additional signals contribute to dyadic assembly.
Keywords: cardiomyocytes, fetal development, ryanodine receptors, t-tubule, workload
Graphical Abstract
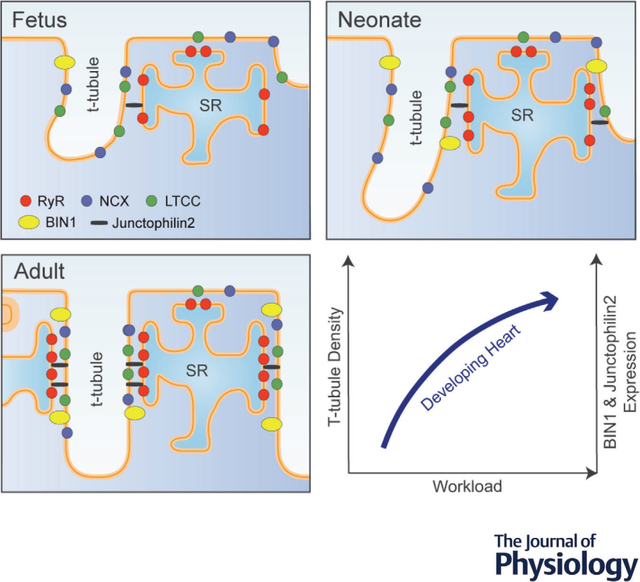
In the sheep heart, t-tubule growth begins in the fetal stage of development. Even at this early stage, growing t-tubules contain both L-type Ca2+ channels (LTCCs) and Na+/Ca2+ exchanger (NCX), which are colocalized with ryanodine receptors (RyRs) in dyadic junctions with the sarcoplasmic reticulum (SR). Progressive dyadic maturation is linked to upregulation of junctophilin-2 and BIN1, and this structural organization continues after birth. T-tubule development is highly workload sensitive. Indeed, in the healthy developing heart, gradually increasing blood pressure drives t-tubule maturation, and this process can be accelerated or blunted by interventions which increase or reduce fetal systolic load, respectively.
Introduction
Contraction of cardiac muscle is dependent on the coordinated shortening of individual cardiomyocytes, which is, in turn, dependent on a process known as excitation–contraction (EC) coupling. In healthy adult cardiomyocytes, EC coupling begins when the action potential triggers the opening of L-type Ca2+ channels (LTCCs), and the resulting influx of extracellular Ca2+ triggers additional Ca2+ release. Contraction is then elicited as released Ca2+ binds to the myofilaments. Initiation of EC coupling occurs at the interface (dyads) between LTCCs within t-tubules, which are invaginations of the surface membrane, and Ca2+ release channels called ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR) (Manfra et al., 2017). In addition to the Ca2+ entry by the LTCCs, Ca2+ release from SR via RyRs may also be triggered and modulated by the Na+/Ca2+ exchanger (NCX) if it is located nearby within the dyad (Lines et al., 2006; Sipido et al., 1997). Thus, the precise arrangement of dyadic proteins is believed to fine-tune the control of contractile function.
Emerging data from our group and others have indicated that there is surprising malleability of t-tubule and dyadic structures, which has important implications for cardiac function. In small rodents, t-tubules appear after birth, starting with a rudimentary network which is largely oriented longitudinally along the long axis of the cell (Lipsett et al., 2019). During continued maturation, overall t-tubule density increases with alignment of tubules into a predominantly transverse arrangement. Anchoring of the developing t-tubules to the SR by junctophilin 2 (JPH2) critically enables co-localization of LTCCs and RyRs required for efficient triggering of the heartbeat (Chen et al., 2013; Lipsett et al., 2019; Louch et al., 2015; Reynolds et al., 2013; Ziman et al., 2010). The t-tubule network can also undergo remodelling during conditions such as heart failure (HF), as t-tubules and dyads become lost and disorganized. This re-emergence of an immature phenotype triggers slower and dyssynchronous calcium release, which has been linked to declining contractile function in the failing heart(Heinzeletal.,2008;Kolstadetal.,2018;Lipsettetal., 2019; Louch et al., 2004, 2006).
The precise signals that control t-tubule and dyadic integrity remain unclear, and available data are unfortunately largely limited to rodent models. However, there is growing appreciation that in HF, the increased workload placed on the dilating heart is a key trigger for loss of dyadic integrity and impaired Ca2+ homeostasis (Frisk et al., 2016; Frisk et al., 2021; Ibrahim & Terracciano, 2013). Thus, the possibility is raised that t-tubules may be load-sensitive during all phases of life, including in the developing heart where maturation includes marked haemodynamic alterations. Such insight is particularly critical for advancing the field of cardiac tissue engineering, as efforts to differentiate cardiomyocytes from embryonic or pluripotent stem cells have yielded rather limited maturation of t-tubules, dyads and EC coupling (Foo et al., 2018; Parikh et al., 2017).
The fetal sheep heart is well suited for addressing such questions since, as in humans, sheep exhibit substantial cardiomyocyte differentiation in utero, which includes the appearance of t-tubules (Kim et al., 1992; Munro & Soeller, 2016; Smolich et al., 1989). Furthermore, fetal sheep can tolerate chronic instrumentation, and previous work from the group of the Thornburg and Giraud team has importantly indicated that overall cardiac development can be altered by interventions that modulate cardiac workload. Specifically, they observed that fetal hypertension in these hearts leads to hyperplastic and hypertrophic growth, accelerated cell cycle activity, and an increased fraction of binuclear cardiomyocytes (Barbera et al., 2000; Jonker, Faber et al., 2007). In contrast, they observed that decreasing workload reduced hyperplastic growth of the fetal heart (O’Tierney et al., 2010).
In the present work, we examined the formation of t-tubules and dyads in the developing sheep heart, and the effects of load-altering interventions. We hypothesized that myocardial loading conditions would accelerate the formation of t-tubules and the Ca2+ signalling apparatus, and that decreased loading would have opposite effects. To test this hypothesis, we studied fetal sheep with differential loading conditions known to alter the growth of the myocardium.
Methods
Ethical approval
All procedures and protocols were approved by the Oregon Health & Science University Institutional Animal Care and Use Committee (IACUC; license numbers: A760, IP007, IP566 and IP588), in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Animals
Healthy, timed-mated pregnant ewes (Ovis aries) of mixed Western breeds were purchased from a local vendor for all studies. Both male and female fetuses were included, and were randomly distributed between experimental groups. The work comprised three sub-studies: (1) examination of normal development of cardiomyocyte substructure spanning late gestation to the neonatal period, (2) comparison of increased and decreased systolic load in fetuses induced by infusion of plasma or enalaprilat, and (3) investigation of increased systolic load in fetuses by occlusion of the post-ductal aorta.
Sub-study 1: normal development
Four ages were studied: fetuses at 93 days, 125 days and 135 days of gestational age (term ~147 days), and neonates 1–9 days after spontaneous term delivery. For this sub-study, fetuses and lambs were not subjected to any experimental interventions or surgeries.
For fetal myocardial collection, pregnant ewes were euthanized using an intravenous injection of sodium pentobarbital (SomnaSol, 80 mg/kg, Covetrus, Columbus, OH, USA). Fetuses were exposed through a laparotomy and hysterotomy, and the umbilical vein was injected with 10,000 U of heparin (Baxter, Deerfield, IL, USA) followed by 10 ml saturated potassium chloride to arrest the heart in diastole. The umbilical cord was then tied and cut, and the fetus was weighed. The fetal heart was excised, trimmed in a standardized manner and weighed. The myocardium (left ventricular free wall) was sampled and rapidly frozen in liquid nitrogen for histological studies. After sampling, the wound edges of the heart were sealed with cyanoacrylate. The heart was then hung from a Langendorff apparatus by cannulating the aorta, and enzymatically dissociated (39°C, 160 U/ml collagenase, 0.78 U/ml protease) as previously described (Barbera et al., 2000; Jonker, Faber et al., 2007). Isolated cells were filtered and fixed in 4% paraformaldehyde.
For neonatal myocardial harvest, lambs were given 10,000 U intravenous heparin injection, and then euthanized using SomnaSol (>100 mg/kg). Body weight was recorded, and the heart was then removed, trimmed in a standardized manner and weighed. A mid-ventricular section of left ventricular myocardium was sampled and rapidly frozen in liquid nitrogen for histological studies, and the heart was dissociated for cardiomyocyte studies as described above.
Sub-studies 2 and 3: altered hemodynamic load
Ewes were accustomed to husbandry pens for several days before surgery. Sterile surgery was performed on ewes and fetuses at 120 ± 1 days as described previously (Giraud, Faber et al., 2006). In brief, anaesthesia was induced via an intravenous mixture of diazepam (10 mg) and ketamine (400 mg). The ewe was intubated and anaesthesia was maintained using 1–2% isoflurane in a carrier gas mixture of 70:30 oxygen and nitrous oxide while mechanically ventilated. The uterus was exposed via a midline abdominal incision and the superior portion of the fetus was delivered through a uterine incision. The jugular vein was cannulated with two polyvinyl catheters (1.7 mm OD, V-8, Scientific Commodities, Lake Havasu City, AZ, USA), and advanced to the right atrium for the measurement of right atrial pressure and for infusions during the experiment. The carotid artery was cannulated with two polyvinyl catheters (1.3 and 1.7 mm OD, V-5 and V-8), and the catheters were advanced to near the junction of the brachiocephalic artery and the aorta. For sub-study 3, the fetal chest was opened and a 10 mm diameter inflatable vascular occluder (In Vivo Metric Systems, Ukiah, CA, USA) was placed are around the post-ductal thoracic aorta, prior to chest closure.
For all fetuses, a 1.7 mm OD polyvinyl catheter (V-8, Scientific Commodities) was attached to the fetal skin and used to measure amniotic fluid pressure. Each fetus was then returned to the uterus, and the uterus was closed in layers to prevent amniotic fluid loss. The free ends of catheters were tunnelled subcutaneously in the ewe’s flank, and exteriorized where they were stored in a nylon pouch sutured to the skin. The abdomen was closed, and 2 million units of penicillin G (Bristol-Myers Squibb, Princeton, NJ, USA) and 2 mg ciprofloxacin were instilled into the amniotic space. Anaesthesia was terminated, and the ewe was extubated once spontaneously breathing and swallowing, and provided with ad libitum food and water. The ewes received routine postoperative pain medication (0.3 mg buprenorphine HCl s.c., twice a day for 2 days, or 0.3 mg buprenorphine HCl s.c. with 0.05 mg/kg sustained release buprenorphine s.c.) at the conclusion of surgery. Ewes were recovered in husbandry pens for 5–7 days (free access to food and water) before experiments were performed. During the experiments, the ewes were housed in stanchions in the laboratory with free access to food and water.
Fetal hemodynamics were measured continuously during the study period. Aortic and right atrial pressures were measured with a computerized data acquisition system (ADInstruments, Colorado Springs, CO, USA; Apple, Cupertino, CA, USA) and adjusted for amniotic fluid pressure. The pressure transducers (Abbott Transpac IV, Abbott Park, IL, USA) were calibrated against a mercury manometer, and zero values were checked every morning. Thereafter, data collected over the next 60-min period were averaged and recorded for later analysis. Heart rates were obtained from arterial pressure tracings.
Fetal arterial blood samples were taken daily for determination of fetal pH, blood gases, haemoglobin concentration, oxygen content, and lactate concentration at 39°C (Radiometer ABL825, Radiometer America, Westlake, OH, USA). Haematocrit was determined by centrifugation of samples in microcapillary tubes. A refractometer was used to obtain estimates of plasma protein concentrations during the experiments.
For sub-study 2, experiments commenced at 126 ± 1 days of gestational age and continued for 8 days. Five fetuses were randomized to serve as controls. Five other fetuses served as an increased systolic load group, and received plasma protein infusion to increase fetal arterial pressure (Giraud, Faber et al., 2006), and thus the systolic load of the heart. A total of 124 ± 25 g of protein was given to these fetuses during the study period, while an equal volume of lactated Ringer solution was infused into control fetuses. Comparison was made with a reduced systolic load group (n = 4), treated with infusion of the angiotensin-converting enzyme inhibitor enalaprilat (345 μg/day, i.v.) to reduce fetal arterial pressure, and thus the systolic load of the heart; in vivo data from these fetuses have already been published (O’Tierney et al., 2010). For the enalaprilat group, adequate blockade of the angiotensin converting enzyme was tested by monitoring the arterial pressure response to bolus injection of angiotensin I. This test was performed on Day 0, before initiation of enalaprilat infusion, and 8 days later. At the conclusion of the in vivo study, ewes were euthanized and fetal hearts collected as described above for the normal development series.
For sub-study 3, experiments commenced at 127 ± 1 days of gestational age and continued for 7 days. Fetuses were randomized to the control (n = 6) or to the increased biventricular load group (n = 5). In the load group, the post-ductal aortic occluder was gradually inflated to increase mean arterial pressure to >60 mmHg by the end of the experimental period. Fetal hemodynamic and blood samples were measured as described above. At the conclusion of the in vivo study, ewes were euthanized and fetal hearts collected as described above for the normal development series.
Immunohistochemistry
Frozen left ventricle heart tissue was embedded in optimal cutting temperature (OCT) compound (Thermo Fisher Scientific, Waltham, MA, USA), and cut into 10 μm-thick transverse and longitudinal cryosections using a Thermo Fisher Scientific Shandon Cryotome FE cryostat (Thermo Fisher Scientific). Sections were placed on Fisherbrand Superfrost Microscope slides (Thermo Fisher Scientific), fixed with 4% paraformaldehyde (PFA) for 30 min, and washed three times with Dulbecco’s phosphate-buffered saline (DPBS; No. 4387, BioWhittaker Inc., Walkersville, MD, USA). T-tubules were labelled with Wheat Germ Agglutinin Alexa Fluor 488 conjugate (WGA-488, 1:50; Thermo Fisher Scientific) for 30 min. Tissues were then washed with DPBS three times, mounted on cover slides in Vectashield Antifade Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA), and covered with glass coverslips.
For imaging of dissociated cardiomyocytes, cells were plated on Fisherbrand Superfrost Microscope slides (Thermo Fisher Scientific) and coated with laminin (10 μg/ml natural mouse, BD Biosciences, San Jose, CA, USA) overnight at 4°C. The cells were then washed with DPBS, fixed with 4% PFA for 10 min, quenched with 100 μmol l−1 glycine for 10 min, permeabilized with 1% Triton X-100 for 10 min, and blocked with a high blocking buffer (5% goat serum, 3% BSA and 0.02% NaN3 in DPBS) for 2 h at room temperature. Primary antibodies were diluted in a low blocking buffer (2% goat serum, 1% BSA and 0.02% NaN3 in DPBS) and applied to the permeabilized cells overnight at 4°C. Cells were then washed with DPBS and incubated with secondary antibodies diluted in the low blocking buffer described earlier, for 2 h at room temperature. The slides were then mounted in Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories), and covered with a glass cover. The following primary antibodies were used: RyR2 (Thermo Fisher Scientific cat. no. MA3-916, RRID:AB_2 183 054, 1:100), anti-CaV1.2 (CACNA1C) antibody ACC-003 (1:100, Alomone Labs, Jerusalem, Israel), and a custom rabbit anti-NCX1 antibody (1:100, Genscript Corp., Piscataway, NJ, USA, described previously; Shen et al., 2019). Secondary antibody labelling was performed with goat anti-mouse (Alexa Fluor 555, AF555, Thermo Fisher Scientific, cat. no. A21425) or goat anti-rabbit (Alexa Fluor 647, AF647, Thermo Fisher Scientific, cat. no. A-21 246) antibodies.
All images were captured on a Zeiss LSM 880 with Fast Airyscan detector (Zeiss Microscopy, Jena, Germany), using a Plan-Apochromat 63×/1.4 oil DIC M27 immersion objective. Excitation and emission wavelengths of 405/450, 488/515, 561/595 and 633/653 were used for DAPI, WGA-488, AF555 and AF647 fluorophores, respectively. Images were recorded with a pixel density of 28.3 pixels per micrometre. Fast Airyscan post-processing was conducted in Zen software (Zeiss) and applied to each image prior to export and subsequent analysis.
Image analysis
All images were processed and analysed with Fiji. For quantification of cell area, t-tubule density, or the density of calcium handling proteins (LTCC, NCX or RyR), a region of interest was first traced inside the cell membrane, selecting only the intracellular space. Airyscan images of tissue sections were thresholded to the mean fluorescence intensity of the entire cell, while the Otsu threshold method was applied on dissociated cell images. For NCX, LTCC and RyR, the fraction of overlap between two proteins (Mander’s correlation coefficient; MCC) was calculated using the Colocalization Plugin in ImageJ, JACoP. Densities were calculated as follows: Protein density = area occupied by NCX/Area of whole cell; Dyadic density = NCX density (% area) × MCCFractionNCXcontainingRyR, as described before (Lipsett et al., 2019). Transverse and longitudinal t-tubule proportions were calculated using Tubulator (Frisk et al., 2022). In brief, this program applies a built-in directionality algorithm to define transverse and longitudinal elements. Binarized images are skeletonized in order to analyse the cell organization, and transverse and longitudinal t-tubule quantities are normalized to cell area and t-tubule density of individual cells.
Western blotting
Frozen left ventricular tissues from sheep (30 mg) were homogenized in RIPA buffer (Thermo Fisher Scientific) with protease (Complete EDTA-free tablets; Roche Diagnostics, Oslo, Norway) and phosphatase inhibitors (PhosSTOP; Roche Diagnostics) using a TissueLyser II (Qiagen, Hilden, Germany). After 30 min on ice, the samples were centrifuged at 14,000 g for 10 min at 4°C. The supernatant was collected and stored at −70°C before further analysis. A Micro BCA protein assay kit (Thermo Fisher Scientific) was used to quantify protein concentrations. Protein samples were run on 12-, 18- or 26-well 4–15% Criterion TGX gels (Bio-Rad Laboratories, Hercules, CA, USA), and then transferred to 0.20 μm polyvinylidene difluoride membranes (Bio-Rad). Revert 700 Total Protein Stain (Biosciences, Lincoln, NE, USA) was used for Western blot normalization, and the signal was detected in the 700 nm channel of an Azure c600 Gel Imaging System (Azure Biosystems, Dublin, CA, USA). Blots were blocked with 5% non-fat milk or casein (1:5; Roche Diagnostics) or SEA Block blocking buffer (1:2; Thermo Fisher Scientific) in Tris-buffered saline with 0.1% Tween (TBS-T), and then incubated overnight at 4°C with primary antibody. The secondary antibody was then incubated for 1 h at room temperature. Blots were developed with enhanced chemiluminescence (ECL prime, Cytiva, MA, USA), with signals visualized using Azure c600, and quantified in AzureSpot software (Azure Biosystems). Employed primary antibodies were: RyR (1:1000; MA3-916, Thermo Fisher Scientific), NCX (1:1000, custom made; Shen et al., 2019), LTCC (1:500; ACC-003, Alomone Labs), amphiphysin-2 (BIN1; 1:2000; sc-23 918, Santa Cruz Biotechnology, Dallas, TX, USA), junctophilin-2 (1:2500; sc-51 313, Santa Cruz Biotechnology), DNM2 (1:1000; ab3457, Abcam, Cambridge, UK), MTM1 (1:250; PA5-17 972 Thermo Fisher Scientific). Secondary antibodies were anti-mouse (1:3000; NA931V, Cytiva), anti-rabbit (1:3000; NA934V, Cytiva), or anti-goat (1:3000; HAF109, R&D Systems, Minneapolis, MN, USA) IgG-horseradish peroxidase-linked whole antibody. Data were normalized to total protein and then to control values.
Quantitative RT-PCR
Approximately 15–20 mg of ventricular tissue was homogenized with a Tissue Lyser II (Qiagen). Total RNA was isolated using an RNeasy Mini kit (Qiagen) and reverse transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories). For measuring mRNA levels of different transcripts implicated in t-tubule development, gene-specific primers were designed, as indicated in Table 1. Quantitative real-time PCR reactions (25 μl) containing SYBR-green indicator (Thermo Fisher Scientific), 0.5 μM primer pair, and 5 ng/μl cDNA were performed in a 96-well reaction plate on a Quant Studio 3 Real Time PCR system (Thermo Fisher Scientific). Relative transcript levels were calculated as fold change in expression n = 2(−Ct,mean). Transcript expression levels were normalized to the housekeeping control gene tyrosine 3-monooxygenase (YWHAZ) using a previously published primer pair (McGillick et al., 2013).
Table 1.
Primer pairs employed for quantitative RT-PCR
| Target gene | Forward primer sequence | Reverse primer sequence | Original reference |
|---|---|---|---|
|
| |||
| RyR2 (exon 94–95) | CCATTCTGCACACCGTCATC | GAGCTACTTCCTTTTCTCGCT | Jung et al. (2012) |
| LTCC (Cacna1c) (exon 46–47) | CTGTCCCCAAAGAGGGGCTT | TCGCTTCAGACATTCAAGGT | Jung et al. (2012) |
| NCX1 (Slc8a1) (exon 10–11) | GAGACCTCGCTTCCCACTTC | ATGCCCAGGAAGACGTTCACC | Jung et al. (2012) |
| Jph2 (exon 4–5) | AGGCTGGGGCCAAGAAAAAG | CGATGTTCAGCAGGATCACCA | Xu et al. (2012) |
| pan-Bin1 (exon 5–5/6) | ACAATGACCTGCTCTGGATGG | GCGACTTGATGTCAGGGAACT | Gayi et al. (2018) |
| Dnm2 (exon 3–4) | ACAGGAGATCGAAGTGGAGAC | GGTCGATGAGGGTCAGGTTCAA | Gayi et al. (2018) |
| Mtm1 (exon 4–5) | GAGGCGCCACAAGTAGAGG | GAAGGCGTATCTTGTGAGGA | Gayi et al. (2018) |
Gene-specific primers were designed based on previously described primer pairs, for measuring mRNA levels of the following transcripts: RyR2, LTCC, Jph2, pan-Bin1, Dnm2 and Mtm1. Primer pair specificity was controlled via dissociation curve analysis.
Assessment of myocyte number, nucleation and cell activity
Isolated myocytes were labelled with Hoechst 33342 to enable nuclear counting (Jonker et al., 2015). The cell cycle activity was assessed with anti-Ki-67 antibody MIB-1 (Dako, Carpinteria, CA, USA), as previously described (Jonker, Zhang et al., 2007).
Statistical methods
All statistical analyses were performed in GraphPad Prism 9(GraphPad Software, San Diego, CA, USA). Data a represented as mean values ± standard deviation. P-values are indicated in all figures. Comparison between two groups was performed by Student’s unpaired t test. Multiple groups were compared using one-way or two-way ANOVA, using post hoc corrections recommended by GraphPad. Correlations were tested using linear and sigmoidal fits, as indicated.
Results
T-tubule and dyadic assembly in the fetal sheep heart
We observed that a progressive increase in heart weight in fetal sheep (Table 2) was linked to cardiomyocyte proliferation and binucleation (Table 3), as previously hypothesized (Giraud, Faber et al., 2006; Jonker et al., 2015). In contrast, the postnatal heart grows almost exclusively by cellular enlargement (Jonker et al., 2015). We examined the development of t-tubules and dyads at selected time points during these two periods of development. Figure 1A shows representative Airyscan super-resolution images of cells labelled with WGA, to stain the surface and t-tubule membranes. Two orientations are presented: sections cut longitudinally along the long axis of the cardiomyocyte and transversely. At the earliest time point examined (gestational age = 93 days; 63% of gestation), there were very few t-tubules present (Fig. 1A and B). However, by 125 days (85% of gestation), a sparse t-tubule network was visible, although cardiomyocyte size was unaltered (Fig. 1C). A continued increase in t-tubule density was observed at gestational age 135 days (92% of gestation), and cell growth was now also apparent. Notably, most cells were observed to have become binucleated by this stage, i.e. no longer undergoing cell division (Table 3) (Jonker et al., 2015). Following birth at ~147 days, a dense t-tubule network was observed, and cells were markedly hypertrophied relative to neonatal stages of development (Fig. 1A–C). More detailed analysis of tubule orientation (Fig. 1D) revealed that overall t-tubule maturation included rather proportional increases in the density of transverse and longitudinal elements (Fig. 1E). Taken together, our data show that this pattern of t-tubule growth follows an earlier developmental time course than cell growth.
Table 2.
Body and heart weights during development
| Group | Gestational age (days) | Body weight (kg) | Heart weight (g) | Heart/fetal weight ratio (g/kg) |
|---|---|---|---|---|
|
| ||||
| 93 days | 93.8 ± 1.5 | 0.6 ± 0.1 | 5.2 ± 1.3 | 8.1 ± 1.2 |
| 125 days | 125.1 ± 0.6 | 3.5 ± 0.5 (P < 0.0001) | 25.2 ± 4.8 (P < 0.0001) | 7.3 ± 0.7 (P = 0.211) |
| 135 days | 135 ± 0 | 4.1 ± 0.5 (P < 0.0001) | 26.1 ± 4.4 (P < 0.0001) | 6.4 ± 0.6 (P = 0.001) |
| Neonatal | 5.3 ± 3.9 | 4.9 ± 1.2 (P < 0.0001) | 37.2 ± 8.6 (P < 0.0001) | 7.5 ± 0.7 (P = 0.447) |
Data are means ± SD. Statistical comparisons were made vs. 93 days, by 1-way ANOVA with Dunnett’s multiple comparison test. Number of animals: 93 days, n = 4; 125 days, n = 8; 135 days, n =11; neonatal, n =10.
Table 3.
Estimated myocyte number, nucleation and cell activity during development
| Days post-conception | LV myocyte number (109) | Estimated LV nucleation (% of all myocytes) | LV cell cycle activity (% of all myocytes) | |
|---|---|---|---|---|
|
| ||||
| Mononucleated | Binucleated | |||
|
| ||||
| 93 | 0.5 | 100.0 | 0.0 | 7.7 |
| 125 | 1.3 | 74.5 | 25.4 | 2.9 |
| 135 | 1.6 | 53.6 | 46.6 | 1.9 |
| 152 (neonatal) | 0.9 | 21.4 | 76.7 | 1.4 |
Data are extrapolated from formulas and data in Jonker et al. (2015).
Figure 1. Time course of t-tubule formation and cardiomyocyte hypertrophy during development.
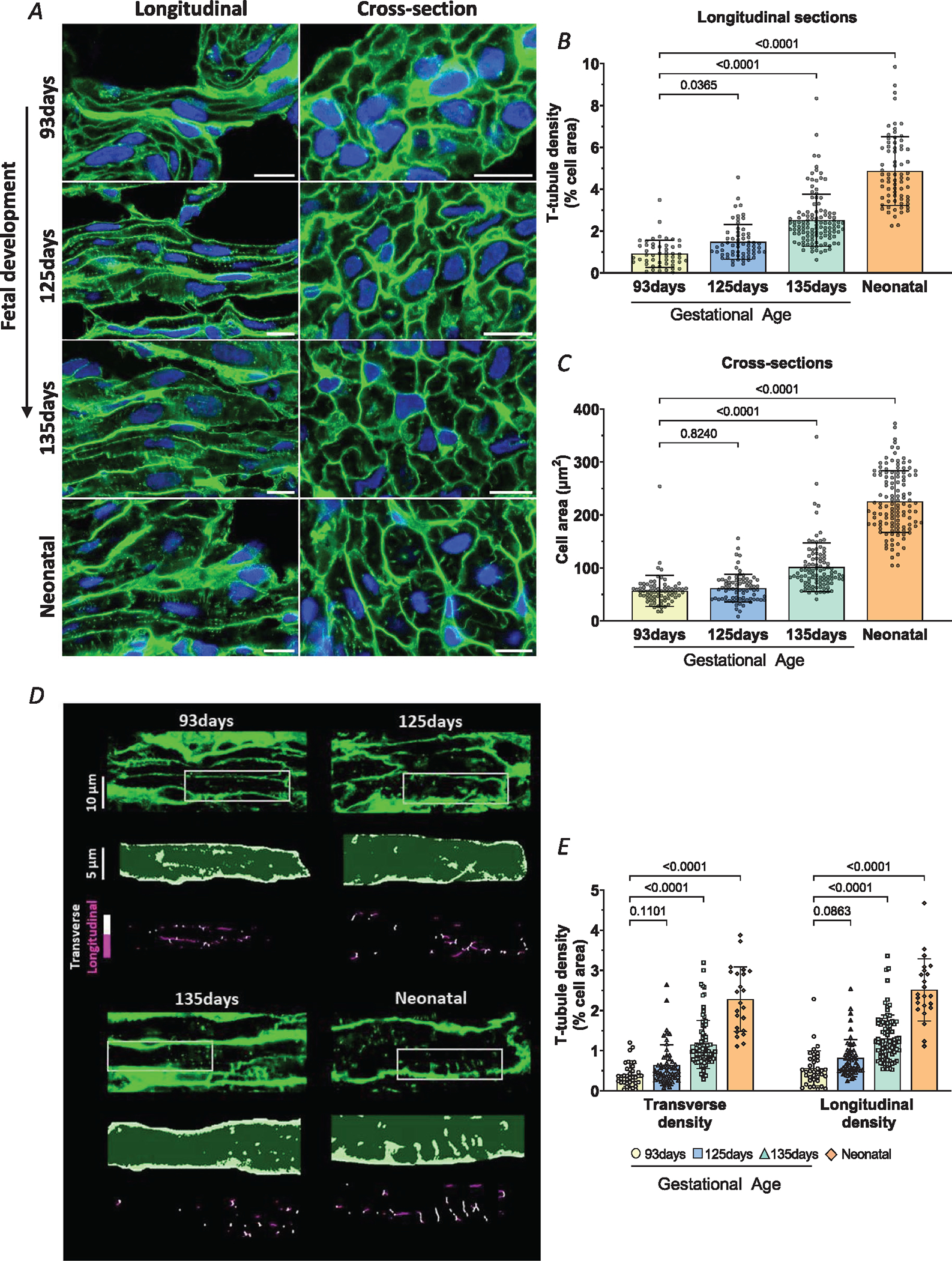
A, representative super-resolution Airyscan micrographs of t-tubules (WGA, green), and nuclei (DAPI, blue) in left ventricular myocardium from fetal (gestational ages 93, 125 and 135 days) and neonatal (8–9 days) sheep. Longitudinal and transverse sections are displayed in the left and right panels, respectively (scale bars = 10 μm). B, quantification of t-tubule signals indicated a gradual increase in density through the fetal and early neonatal periods. C, cell growth followed a somewhat later time course. D, detailed analysis of transverse and longitudinal t-tubule structures was performed in a subset of images. Representative images are presented with indicated regions enlarged and skeletonized below, with colour coding of transverse and longitudinal tubules. E, quantification of these signals revealed roughly parallel increases in the density of transverse and longitudinal t-tubule densities during maturation. Data are presented as means ± SD, with P-values indicated. Statistical comparisons were made using 1-way ANOVA versus 93 days, with Dunnett’s multiple comparison test (B, C) or 2-way ANOVA with Šidák’s Multiple comparison test (E). Number of cells (animals): t-tubule density: 93 days 50 (4), 125 days 62 (4), 135 days 114 (5), neonatal 72 (5); cell area: 93 days 73 (4), 125 days 77 (4), 135 days 99 (5), neonatal 120 (5); transverse and longitudinal densities: 93 days 37 (4), 125 days 53 (4), 135 days 87 (4), neonatal 23 (3).
Next, we investigated dyadic assembly in the developing heart by co-staining LTCCs in the t-tubule membrane and RyRs in the SR. Airyscan super-resolution imaging revealed a robust, striated RyR distribution during early fetal development (gestational age 125 days; Fig. 2Aa; quantification in Fig. 2Ab). Mirroring a sparse t-tubule presence at this stage of development, LTCC staining was also observed to be rather diffuse within the cell interior (Fig. 2Aa and 2Ac), and colocalization with RyRs was limited (Fig. 2Ad and 2Ae). During further maturation, the staining density of both RyRs and LTCC increased, as did the colocalization between the two proteins. These data suggest that RyRs are arranged in the junctional SR early during fetal development in anticipation of arriving t-tubules and LTCCs. The resulting dyadic formation appears to continue through later stages of fetal development and into the neonate.
Figure 2. Cardiac dyads are progressively assembled during the fetal period.
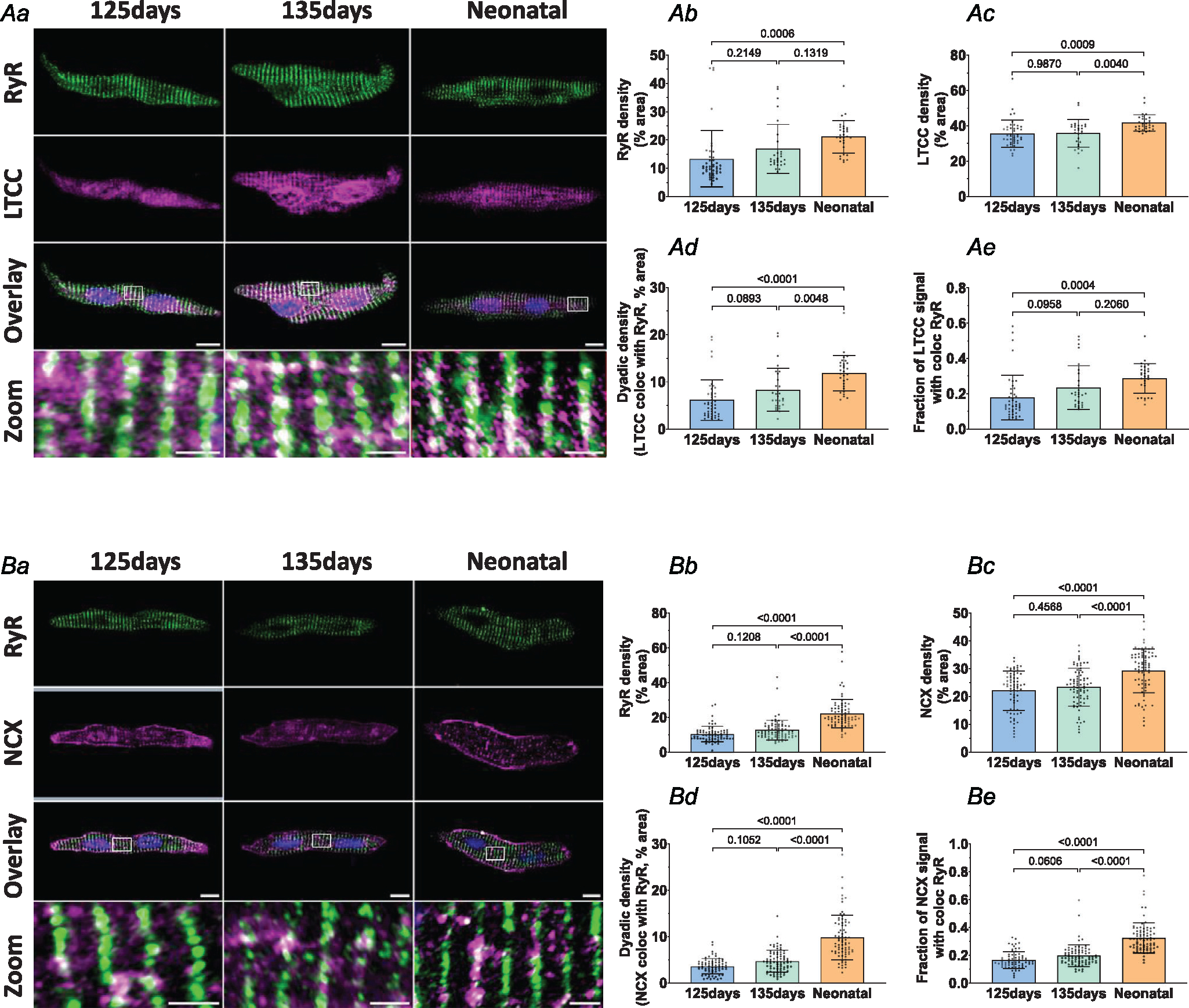
Representative super-resolution Airyscan micrographs of isolated cardiomyocytes labelled with antibodies against RyR in combination with either LTCC or NCX (A and B, respectively, scale bars: 10 μm). DAPI was used to visualize the nuclei. For the indicated regions in the overlay images, enlargements are presented in the lower panels (scale bars: 2 μm). Image analysis revealed progressive increases in RyR density (Ab and Bb), LTCC density (Ac), and NCX density (Bc) in the developing heart. Gradual assembly of dyads was confirmed by increasing density of sites where either LTCC (Ad) or NCX (Bd) colocalized with RyRs. The proportion of LTCC (Ae) and NCX (Be) signals colocalized with RyRs showed a similar pattern of increase during development. Data are presented as mean ± SD, with P-values indicated. Statistical comparisons were made using 1-way ANOVA with Tukey’s multiple comparison test. Number of cells (animals): A: 125 days 43 (4), 135 days 29 (4), 1–5 days neonatal 30 (3); B: 125 days 69 (4), 135 days 77 (6), 1–5 days neonatal 77 (5).
We then examined NCX localization in the fetal and neonatal sheep heart, as previous work in rodents has indicated that it may provide an important role in Ca2+ cycling in developing cardiomyocytes (Louch et al., 2015). Interestingly, we noted that NCX is localized both in the cell surface and intracellularly from early stages of ovine development (Fig. 2Ba). Quantitative analysis within the cell interior revealed a step-wise increase in NCX density between 125 days of gestation and the neonatal stage (Fig. 2Bc). Similar to observations made with LTCCs (Fig. 2A), paired increases in NCX and RyR signals were associated with augmenting colocalization between the proteins (Fig. 2Bd and 2Be). These observations indicate that dyadic junctions are carefully assembled to allow close positioning of LTCCs, NCX and RyRs from early fetal stages.
We further investigated these Ca2+ handling proteins in the developing heart by examining their expression at the mRNA and protein levels. In keeping with imaging of RyR signals, we observed significant, step-wise increases in both RyR mRNA and protein during fetal maturation (93, 125, 135 days) until the neonatal stage (Fig. 3A). In contrast, LTCC protein expression was relatively stable during embryonic stages, and only significantly increased in the postnatal period (Fig. 3A). NCX protein expression, on the other hand, increased during fetal development from 93 days to 135 days, but then slightly decreased after birth. Here, the apparent mismatch between total protein levels and internal staining quantification may reflect changing distribution of the protein between the cell surface and interior. Interestingly, the time course of LTCC and NCX protein expression did not closely parallel mRNA levels, which is suggestive of changing patterns of protein translation during development.
Figure 3. Expression of dyadic Ca2+ handling proteins and t-tubule regulators in the developing sheep heart.
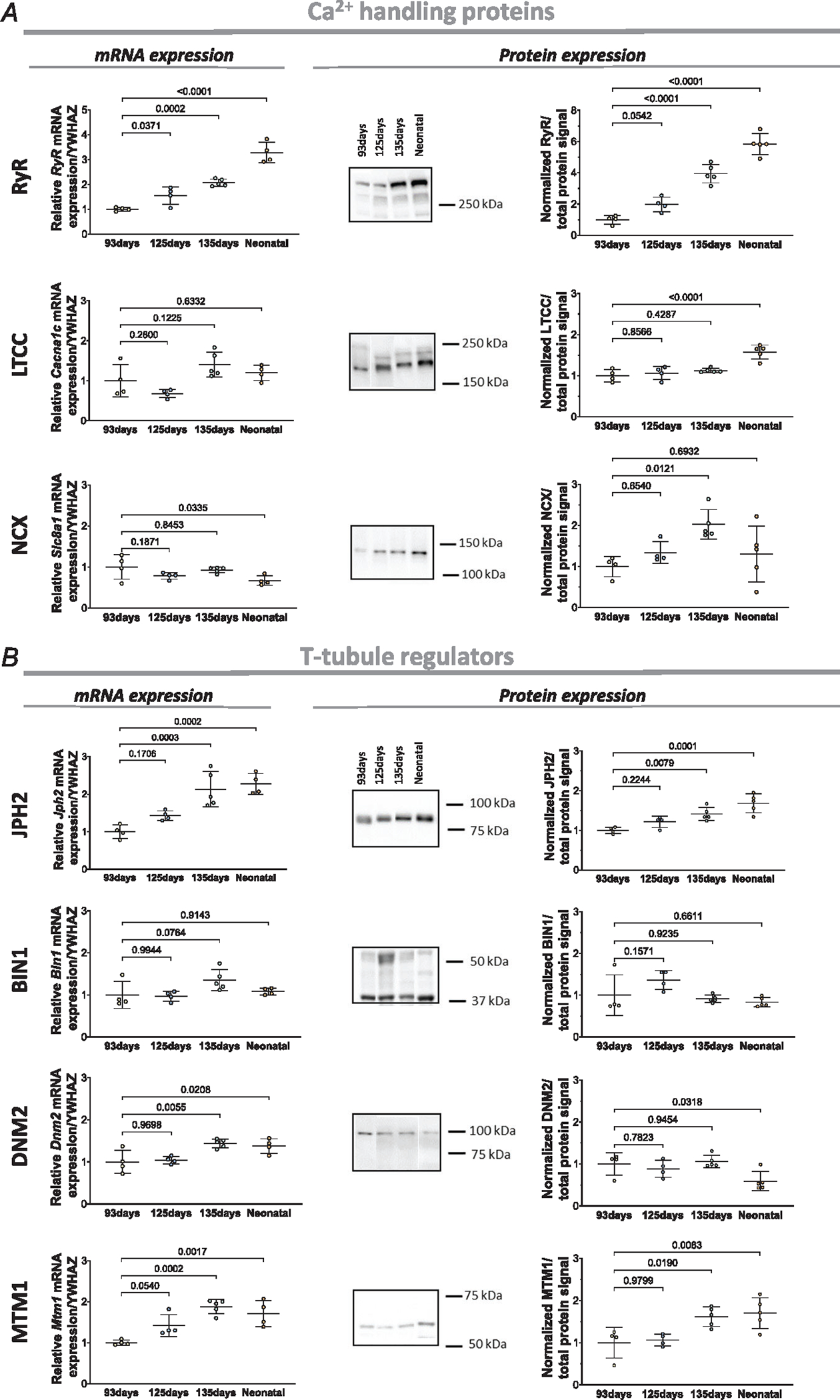
A, left panels: left ventricular mRNA levels of RYR, LTCC, and NCX during development. The targeted genes for LTCC and NCX were Cacna1c and Slc8a1, respectively. A, right panels: representative Western blots and mean data revealed progressive increases in the expression of all three proteins during the fetal period. B, changes in the expression of the t-tubule regulators JPH2, BIN1, DNM2 and MTM1 during development, with mRNA levels at left and protein levels at right. JPH2 and MTM1 protein showed significant increases during development, while BIN1 tended to increase at 125 days when t-tubule growth was initiated. DNM2 protein levels decreased after birth. Data are presented as means ± SD, with P-values indicated. Statistical comparisons were made vs. 93 days, using 1-way ANOVA with Dunnett’s multiple comparison test. For qPCR, n = 4 hearts for 93 days, 125 days and neonatal, n = 5 hearts for 135 days. For western blots (all proteins), n = 4 hearts for 93 days, 125 days, n = 5 hearts for 135 days and neonatal.
In addition, we examined the expression of proteins with reported roles in t-tubule formation and maintenance, including JPH2, BIN1, DNM2 and MTM1 (Fig. 3B). During development, we observed a significant increase in protein and mRNA expression for JPH2, a protein reported to be critical for dyadic membrane anchoring, and both anchoring and regulation of dyadic proteins (Chen et al., 2013; Guo et al., 2014; Han et al., 2013; Munro et al., 2016; Ziman et al., 2010). We did not detect significant changes in BIN1 expression at the time points examined, although a tendency to higher protein levels at 125 days of fetal development may be linked to early initiation of t-tubule growth. However, levels of the reported BIN1 partners DNM2 and MTM1 were significantly altered. Declining DNM2 protein level during early development is consistent with reports of an inhibitory effect of this protein on t-tubule growth (Gibbs et al., 2014; Gonzalez-Jamett et al., 2013; Picas et al., 2014), while increasing MTM1 expression is consistent with its suggested importance in tubulogenesis (Al-Qusairi & Laporte, 2011; Marat & Haucke, 2016; Royer et al., 2013).
Altered cardiac load by plasma or enalaprilat infusion modulates t-tubule development
Having established the normal time course for t-tubule and dyadic formation in the developing sheep heart, we next investigated how these processes are regulated by cardiac workload. Near-term fetal sheep hearts (135 days) were treated with chronic (8 day), intravascular infusion of plasma, enalaprilat or lactated Ringer solution to elevate, reduce or maintain systolic arterial pressure, respectively (Giraud et al., 2005; Giraud, Faber et al., 2006; O’Tierney et al., 2010). Obtained changes in arterial pressure are presented in Table 4, as recorded on the final day of the experiment. Plasma infusion resulted in an increase in arterial pressure, from a control value of 41.6 ± 2.1 to 61.0 ± 4.5 mmHg, while enalaprilat infusion reduced arterial pressure from 40.6 ± 3.1 in control to 30.2 ± 7.4 mmHg. Neither plasma nor enalaprilat infusion altered fetal body weights in comparison with control hearts (lactated Ringer infusion; Table 5). However, fetal heart weights were increased in the plasma treated group, but unaltered by enalaprilat treatment (Table 5).
Table 4.
Arterial blood and haemodynamic values in the plasma and enalaprilat infusion fetal heart model
| Group | Day 0 | Day 8 | P vs. Day 8 control | |
|---|---|---|---|---|
|
| ||||
| pH | Control | 7.36 ± 0.02 | 7.36 ± 0.02 | |
| Enalaprilat | 7.35 ± 0.01 | 7.21 ± 0.09 | 0.64 | |
| Plasma | 7.37 ± 0.02 | 7.36 ± 0.01 | 0.999 | |
| PCO2 (mmHg) | Control | 52.6 ± 2.4 | 52.0 ± 1.2 | |
| Enalaprilat | 47.2 ± 2.1 | 52.9 ± 5.1 | 0.879 | |
| Plasma | 51.5 ± 3.0 | 51.3 ± 2.5 | 0.924 | |
| PO2 (mmHg) | Control | 22.8 ± 1.1 | 23.1 ± 3.2 | |
| Enalaprilat | 21.3 ± 3.4 | 16.8 ± 1.5 | 0.013 | |
| Plasma | 23.5 ± 2.0 | 20.3 ± 4.0 | 0.292 | |
| Haematocrit (%) | Control | 31.3 ± 2.8 | 30.1 ± 2.2 | |
| Enalaprilat | 32.4 ± 3.1 | 33.5 ± 4.5 | 0.3219 | |
| Plasma | 35.4 ± 5.6 | 34.1 ± 4.6 | 0.225 | |
| Total haemoglobin (g/dl) | Control | 9.8 ± 1.1 | 8.9 ± 0.5 | |
| Enalaprilat | 8.9 ± 1.7 | 9.2 ± 2.1 | 0.920 | |
| Plasma | 10.5 ± 1.6 | 10.0 ± 1.1 | <0.0001 | |
| O2 content (ml/100ml) | Control | 7.3 ± 0.7 | 6.5 ± 0.5 | |
| Enalaprilat | 7.3 ± 2.3 | 6.2 ± 2.2 | 0.916 | |
| Plasma | 7.3 ± 0.9 | 5.1 ± 0.7 | 0.221 | |
| Protein (g/100ml) | Control | 3.4 ± 0.2 | 3.6 ± 0.1 | |
| Enalaprilat | 3.2 ± 0.2 | 3.3 ± 0.1 | 0.052 | |
| Plasma | 3.3 ± 0.2 | 7.4 ± 0.3 | < 0.0001 | |
| Aortic pressure (mmHg) | Control | 41.3 ± 0.9 | 41.3 ± 1.9 | |
| Enalaprilat | 40.9 ± 3.1 | 30.2 ± 7.4 | 0.009 | |
| Plasma | 41.6 ± 2.1 | 61.0 ± 4.5 | < 0.0001 | |
| Atrial pressure (mmHg) | Control | 2.0 ± 0.7 | 2.5 ± 0.9 | |
| Enalaprilat | 2.1 ± 0.8 | 1.9 ± 0.5 | 0.3745 | |
| Plasma | 1.9 ± 0.6 | 3.0 ± 0.8 | 0.4908 | |
| Heart rate (beats/min) | Control | 167 ± 7 | 143 ± 13 | |
| Enalaprilat | 163 ± 15 | 156 ± 29 | 0.486 | |
| Plasma | 171 ± 12 | 157 ± 11 | 0.439 | |
Values are presented at Day 0 and Day 8 of the intervention. Data presented as means ± SD, with P-values as indicated. Statistical comparisons were made vs. control using 1-way ANOVA with Dunnett’s multiple comparison test. n = 5 animals in all groups.
Table 5.
Body and heart weights of fetal sheep in the infusion model
| Group | Gestational age (days) | Body weight (kg) | Heart weight (g) | Heart/fetal weight ratio (g/kg) |
|---|---|---|---|---|
|
| ||||
| Control | 135 ± 0 | 3.9 ± 0.5 | 24.1 ± 4.8 | 6.2 ± 0.8 |
| Enalaprilat | 133.5 ± 0.6 | 3.7 ± 0.7 (P = 0.783) | 21.9 ± 2.7 (P = 0.728) | 6.1 ± 1.0 (P = 0.982) |
| Plasma | 135 ± 0 | 4.4 ± 0.2 (P = 0.239) | 31.6 ± 5.5 (P = 0.0466) | 7.2 ± 0.9 (P = 0.181) |
Data are presented as mean ± SD, with P-values as indicated. Statistical comparisons were made vs. control by 1-way ANOVA with Dunnett’s multiple comparison. n animals = 4 for enalaprilat and 5 for plasma and control groups.
Cell membranes were imaged in fetal left ventricular samples collected from the three infusion treatment groups. Representative images are shown in Fig. 4A, while mean measurements of t-tubule density and cell area are presented in Fig. 4B and C. Image analysis revealed that increased load (plasma infusion) markedly promoted t-tubule growth, while decreased load (enalaprilat infusion) slowed t-tubule development compared to the control group (Fig. 4B). The observed increase in t-tubule density for the plasma group resulted from an increase in both transverse and longitudinally oriented t-tubule densities (Fig. 4E and F). For the enalaprilat group, the induced reduction in t-tubule signal stemmed largely from lower density of transverse t-tubules, as the proportion of the longitudinal t-tubule was not significantly changed (Fig. 4E and F). At the molecular level, no changes were found in the expression of the dyadic Ca2+ handling proteins RyRs, LTCCs and NCX (Fig. 5A). In addition, among t-tubule regulatory proteins described earlier, only BIN1 was found to be altered. Specifically, increased BIN1 expression was noted in the plasma treatment group compared to the control group, which is consistent with the reported role of this protein in tubulogenesis (Fig. 5B).
Figure 4. T-tubule growth is accelerated in plasma-infused fetal hearts but slowed by enalaprilat infusion.
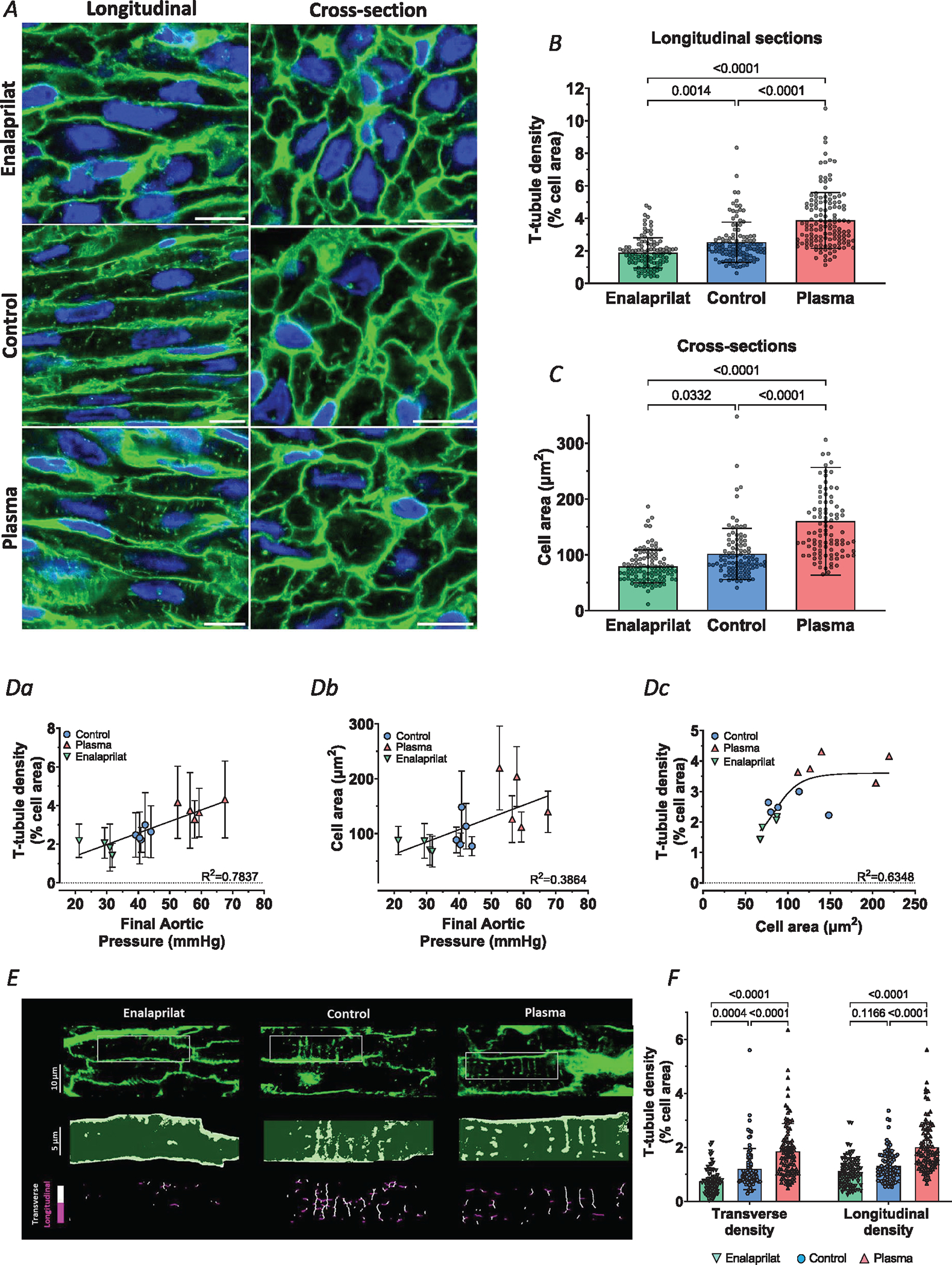
A, representative super-resolution Airyscan micrographs of t-tubules (WGA, green), and nuclei (DAPI, blue) in fetal myocardium at gestational of 135 days. Effects of increasing load were tested by plasma infusion in the fetal sheep, while blood pressure lowering was examined by enalaprilat infusion. Comparison was made with control hearts infused with lactated Ringer solution to maintain normal aortic pressure. Images for longitudinal and transverse sections are presented in the left and right panels, respectively. B and C, quantification of t-tubule density (B) and cell area (C) revealed that plasma infusion accelerated t-tubule growth and hypertrophy, while enalaprilat had opposite effects. D, correlations between final aortic pressure with either t-tubule density (Da) or cell area (Db). Dc, t-tubule density and cell area measurements were also correlated, but best fit by a sigmoidal curve. E, detailed analysis of transverse and longitudinal t-tubule structures was performed in a subset of images. Representative images are presented with indicated regions enlarged and skeletonized below, with colour coding of transverse and longitudinal tubules. F, quantification of these signals revealed that changes in workload induced roughly parallel alterations in the density of transverse and longitudinal t-tubules. Data are presented as means ± SD, with P-values indicated. Statistical comparisons were made using 2-way ANOVA with Šidák’s multiple comparison test (B and C) or 1-way ANOVA with Tukey’s multiple comparison test (F). Number of cells (animals): t-tubule density: enalaprilat 107 (4), controls 114 (5), plasma 131 (5); cell area: enalaprilat 104 (4), controls 99 (5), plasma 97 (5); transverse and longitudinal densities: enalaprilat 80 (4), controls 87 (4), plasma 94 (4).
Figure 5. Left ventricular expression of dyadic Ca2+ handling proteins and t-tubule regulators in the infusion animal model.
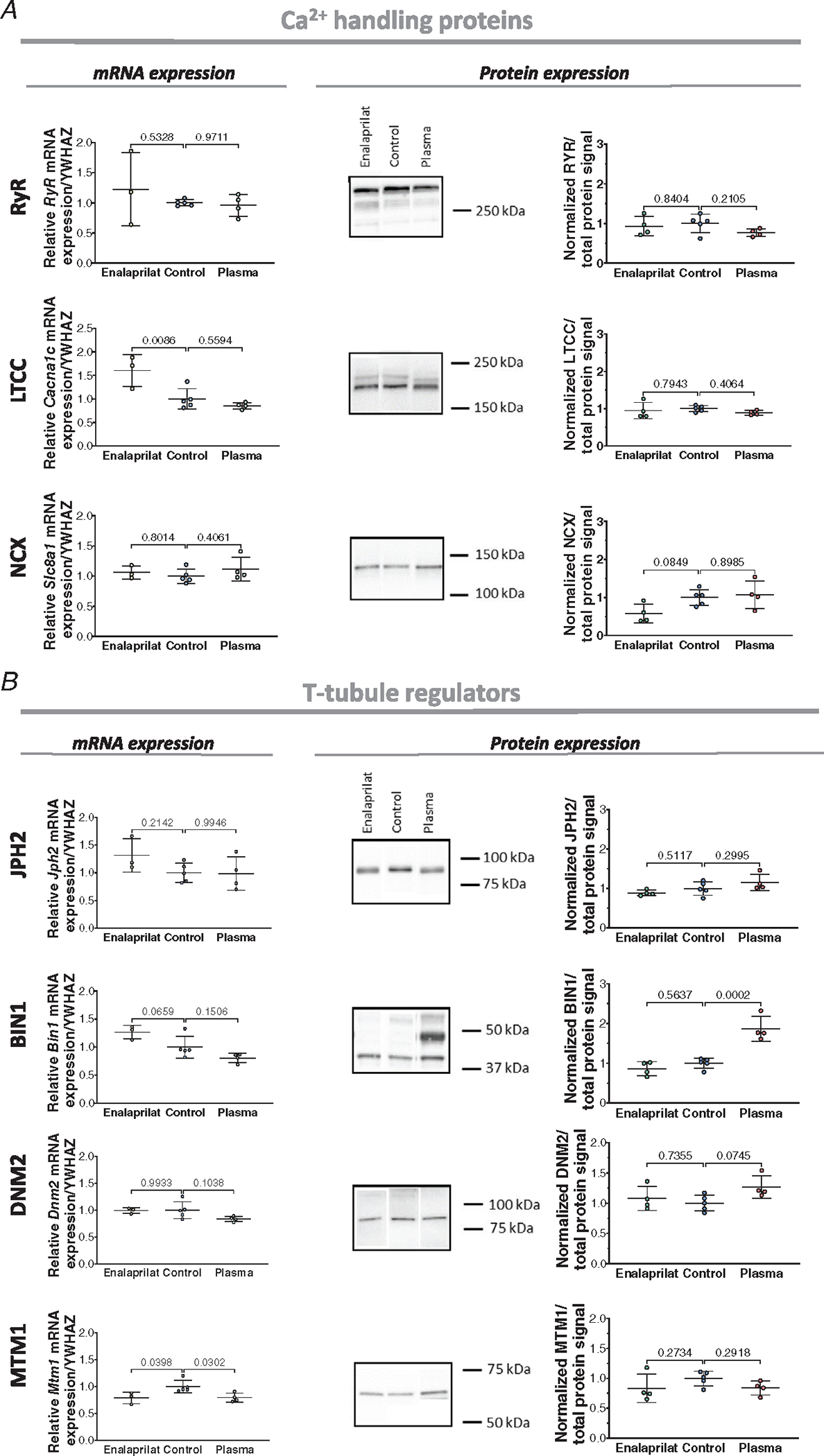
A, expression of RyR, LTCC and NCX in enalaprilat, control and plasma-infused hearts at the mRNA (left panels) and protein level (right panels; with representative blots). B, expression of the t-tubule regulators JPH2, BIN1, DNM2 and MTM1 in the three experimental groups at the mRNA (left panels) and protein levels (right panels). Data are presented as means ± SD, with P-values indicated. Statistical comparisons vs. control were made by 1-way ANOVA with Dunnett’s multiple comparison test. For qPCR (all genes), n = 3 hearts for enalaprilat, n = 3 for plasma, n = 5 hearts for control group. For western blots (all proteins), n = 4 hearts for enalaprilat and plasma groups, n = 5 hearts for controls.
Of note, plasma-infused fetal hearts showed an increase in cell area, while we found a decrease in cell area for the enalaprilat treatment group (Fig. 4C). We observed linear correlations for both t-tubule density and cell area with arterial pressure (R2 = 0.7837 and 0.3864, respectively, Fig. 4a and b). However, it should be noted that the former relationship is steeper, indicating that t-tubule density is more load-sensitive than cell size. Indeed, the best fit for the correlation between the t-tubule density and cell area was sigmoidal (Fig. 4Dc; R2 = 0.6348 for sigmoidal versus 0.5288 for linear relation), indicating that t-tubule densities can increase markedly while cellular hypertrophy is modest.
Altered cardiac load by fetal aortic constriction promotes t-tubule growth
We further investigated the load-dependence of t-tubule and dyadic formation in developing cardiomyocytes by employing an aortic occlusion procedure, to increase systolic pressure in a controlled manner. This in vivo method involves placing a silicone rubber inflatable occluder (10 mm) on the post-ductal aorta of the fetal sheep heart, to pressure load the heart for a period of 7 days. The treated fetuses exhibited increased fetal arterial pressure from the control value of 47.5 ± 2.1 to 64.9 ± 9.7 mmHg on the last day of the experiment (Table 6). Fetal body weights were not altered by the intervention, but the heart weight and heart-to-body weight ratios of the treated fetuses were higher than the control group (Table 7).
Table 6.
Arterial blood and hemodynamic values in the aortic constriction model
| Group | Day 0 | Day 7 | P vs. Day 7 control | |
|---|---|---|---|---|
|
| ||||
| pH | Control | 7.37 ± 0.03 | 7.37 ± 0.03 | |
| Aortic constriction | 7.35 ± 0.021 | 7.35 ± 0.02 | 0.179 | |
| PCO2 (mmHg) | Control | 49.9 ± 3.6 | 50.1 ± 3.8 | |
| Aortic constriction | 53.1 ± 3.0 | 52.8 ± 1.6 | 0.181 | |
| PO2 (mmHg) | Control | 20.9 ± 3.4 | 18.7 ± 4.0 | |
| Aortic constriction | 19.8 ± 2.6 | 21.6 ± 3.0 | 0.231 | |
| Haematocrit (%) | Control | 36.4 ± 3.6 | 38.1 ± 6.1 | |
| Aortic constriction | 36.8 ± 2.4 | 38 ± 5.4 | 0.979 | |
| Total haemoglobin (g/dl) | Control | 10.9 ± 0.9 | 11.6 ± 1.7 | |
| Aortic constriction | 11.1 ± 0.9 | 11.4 ± 1.5 | 0.848 | |
| O2 content (ml/100 ml) | Control | 8.6 ± 0.9 | 7.9 ± 1.3 | |
| Aortic constriction | 7.7 ± 1.7 | 8.4 ± 2.1 | 0.663 | |
| Aortic pressure (mmHg) | Control | 45.0 ± 2.9 | 47.5 ± 2.1 | |
| Aortic constriction | 44.0 ± 2.1 | 64.9 ± 9.7 | 0.004 | |
| Atrial pressure (mmHg) | Control | 2.0 ± 1.0 | 3.0 ± 1.1 | |
| Aortic constriction | 2.7 ± 0.8 | 2.8 ± 1.0 | 0.771 | |
| Heart rate (beats/min) | Control | 162 ± 5 | 152 ± 8 | |
| Aortic constriction | 170 ± 11 | 160 ± 12 | 0.250 | |
Values are presented at Day 0 and Day 7 of the intervention. Data presented as mean ± SD, with P values as indicated. Statistical comparisons were made vs. Controls using unpaired t test. n = 5 animals for both groups.
Table 7.
Body and heart weights of fetal sheep in the aortic constriction model
| Gestational age (days) | Body weight (kg) | Heart weight (g) | Heart/fetal weight ratio (g/kg) | |
|---|---|---|---|---|
|
| ||||
| Control | 134.8 ± 0.4 | 4.8 ± 0.9 | 31.4 ± 7.6 | 6.5 ± 0.6 |
| Aortic constriction | 134.6 ± 0.5 | 4.4 ± 0.9 (P = 0.458) | 33.9 ± 8.7 (P = 0.611) | 7.7 ± 0.7 (P = 0.012) |
Data are presented as means ± SD, with P-values as indicated. Statistical comparisons were made vs. control using unpaired t tests. n animals = 6 for the control group and 5 for the aortic constriction group.
Left ventricular tissues were stained with WGA to allow imaging of the cell surface and t-tubular membranes, as shown in the representative images (Fig. 6). Both t-tubule density and cell size measurements were markedly increased in the aortic occlusion treatment group (Fig. 6B and C). The increased t-tubule density in the treatment group stemmed from higher densities of transverse and longitudinal elements compared to controls (Fig. 6D and E).
Figure 6. Increasing systolic pressure load by aortic constriction accelerates t-tubule growth.
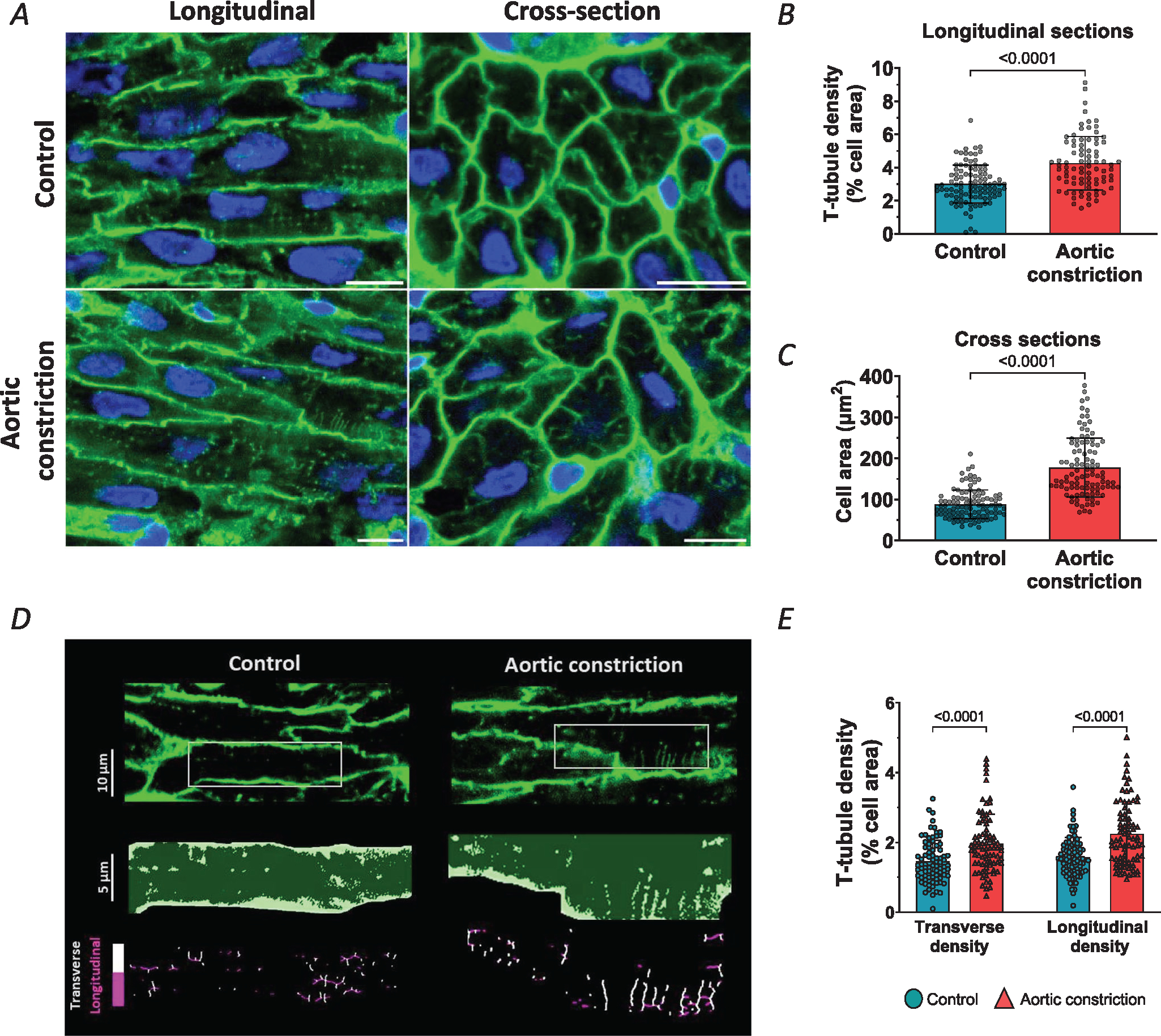
A, representative super resolution Airyscan micrographs of t-tubules (WGA, green), and nuclei (DAPI, blue) in fetal myocardium at gestational of 135 days. Fetal sheep treated with an occluder in the post-ductal aorta (aortic constriction) were compared with control animals. Images for longitudinal and transverse sections are presented in the left and right panels, respectively (scale bars: 10 μm). B and C, aortic occlusion induced increases in mean t-tubule density (B) and cell area (C) compared to controls. D, analysis of transverse and longitudinal t-tubule structures. Representative images are presented with indicated regions enlarged and skeletonized below, with colour coding of transverse and longitudinal tubules. E, quantification of these signals revealed that aortic occlusion induced a roughly parallel increase in the density of the two types of t-tubules. Data are presented as means ± SD, with P-values indicated. Statistical comparisons vs. control were made by unpaired t test (B and C), or vs. other groups by 2-way ANOVA with Šidák’s multiple comparison test. Number of cells (animals): t-tubule density: controls 100 (5), aortic constriction 83 (4); cell area: controls 103 (5), aortic constriction 99 (4); transverse and longitudinal densities: controls 81 (5), aortic constriction 80 (4).
We further examined the effects of altered workload on dyadic assembly, by labelling RyRs, LTCCs and NCX in dissociated cells (Fig. 7). Mirroring changes in t-tubule density, we observed increased LTCC staining density following aortic occlusion (Fig. 7Ac). However, RyR density and LTCC-RyR colocalization (i.e. dyadic density) remained unchanged (Fig. 7Ab, Ad, Ae). Furthermore, in cells stained for RyR and NCX, single protein densities and protein co-localization were also not modified by increased load (Fig. 7Ba–e. Consistently, the protein and mRNA expression levels of RyRs and NCX were not altered in the treated group vs. control (Fig. 8). Expression of most t-tubule and dyadic protein regulators examined was also not affected by the increased load (Fig. 8B). Thus, our data indicate that although t-tubule growth is highly load-sensitive, the precise assembly of dyads and their constituent proteins in the developing heart appears to be regulated by additional signals.
Figure 7. Cardiac dyads are not remodelled by increasing systolic load in the fetal heart.
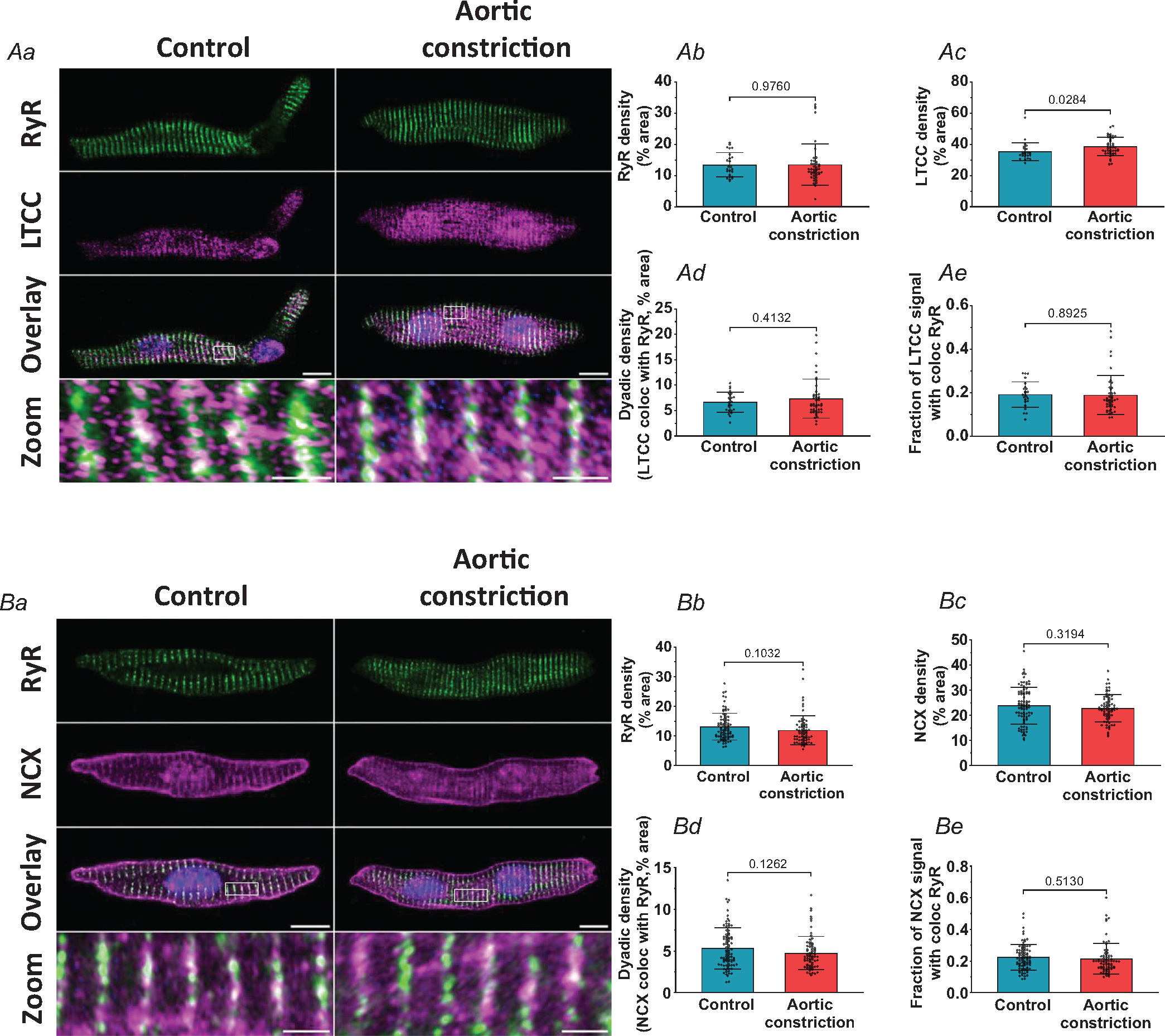
Representative super resolution Airyscan micrographs of isolated cardiomyocytes labelled with antibodies against RyR in combination with either LTCC or NCX (A and B, respectively, scale bars: 10 μm). DAPI was used to visualize the nuclei. For the indicated regions in the overlay images, enlargements are presented in the lower panels (scale bars: 2 μm). Although t-tubule growth in the aortic constriction hearts was linked to increased LTCC density (Ac), no changes were detected in RyR density (Ab and Bb), NCX density (Bc), or dyadic density, as defined by the association of either LTCC (Ad) or NCX (Bd) with RyRs. The fraction of LTCC or NCX signal co-localized with RYR was also unchanged (Ae and Be). Data are presented as means ± SD, with P-values indicated. Statistical comparisons vs. control were made by unpaired t test. Number of cells (animals): A: controls 24 (3), aortic constriction 44 (4); B: controls 85 (6), aortic constriction 70 (5).
Figure 8. Left ventricular expression of dyadic Ca2+ handling proteins and t-tubule regulators in the aortic constriction model.
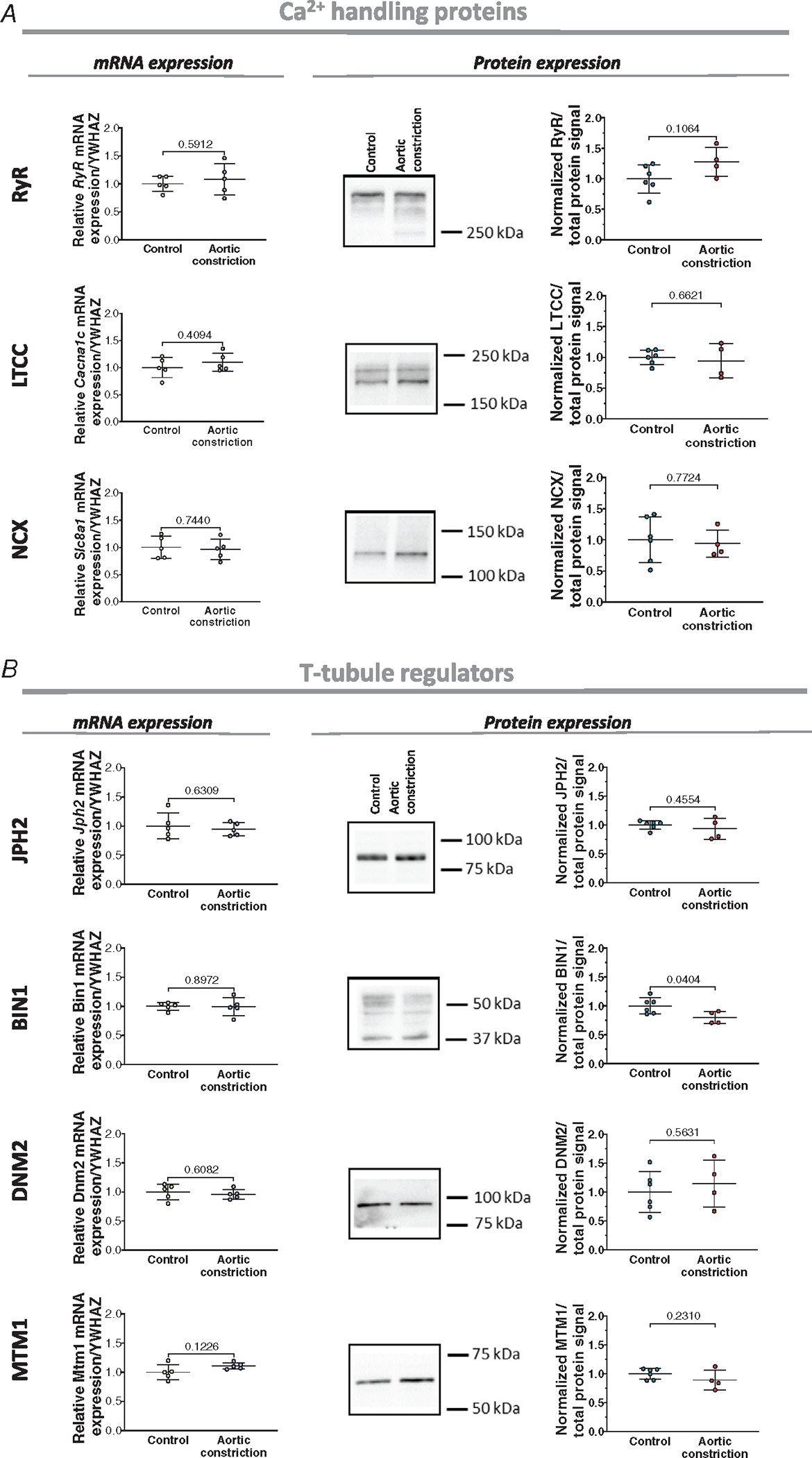
A, left panels: left ventricular mRNA levels of RyR, LTCC, and NCX in control and post-aortic constriction hearts. A, right panels: Representative Western blots and mean measurements of protein expression. B, expression of the t-tubule regulators JPH2, BIN1, DNM2, and MTM1 in the two experimental groups at the mRNA (left panels) and protein levels (right panels). Data are presented as mean ± SD, with P values indicated. Statistical comparisons vs. control were made by unpaired t test. For qPCR (all genes), n = 5 hearts for aortic constriction and control groups. For Western blots (all proteins), n = 6 hearts for the control group and n = 5 for aortic constriction group.
Discussion
Our present findings show that dyads are carefully assembled during the fetal period in the developing sheep heart and that their developmental pattern is load sensitive. These structures are formed by transverse positioning of RyRs in the junctional SR, which precedes the arrival of developing t-tubules. Dyadic assembly includes progressive colocalization of both LTCCs and NCX with RyRs, mirroring gradual increases in the expression of known dyadic regulators, such as JPH2 and BIN1. Our data show that t-tubule growth is highly load sensitive, as increasing or decreasing fetal systolic blood pressure speeds or retards t-tubule formation, respectively. However, dyadic formation was not altered by these interventions, indicating that gradually increasing cardiac workload in the healthy fetus is only one signal that contributes to assembly of the Ca2+ cycling apparatus.
Normal development of fetal sheep cardiac myocytes
Prior to the present work, most of our knowledge of fetal cardiomyocyte development stemmed from studies in small rodents, where significant maturation occurs in the first weeks after birth (Hamaguchi et al., 2013; Ziman et al., 2010). These previous studies showed that neonatal rat cardiomyocytes contain rudimentary SR terminals where RyRs are positioned at regular intervals along Z-lines (Lipsett et al., 2019; Ziman et al., 2010). This RyR arrangement occurs in anticipation of the inward growth of t-tubules, which starts at around 10 days after birth (Lipsett et al., 2019; Ziman et al., 2010). As these t-tubules mature, LTCCs progressively colocalize with RyRs in dyads, increasing the efficiency of Ca2+-induced Ca2+ release to strengthen the heartbeat (Lipsett et al., 2019).
Previous reports have suggested that larger mammals including sheep and humans appear to have much earlier t-tubule development than small rodents (Brook et al., 1983; Munro & Soeller, 2016; Sheldon et al., 1976; Smolich et al., 1989). Our current data support this view, as we observed significant t-tubule growth in fetal sheep. We found that roughly proportional increases in transverse and longitudinal tubule densities during this period (Fig. 1D and E) are linked to dyadic assembly, as growing t-tubules form junctions with the junctional SR (Fig. 2). We observed that dyadic formation is preceded by a striated pattern of RyRs present along Z-lines as early as gestational age 125 days, prior to the arrival of LTCCs and NCX in developing t-tubules between 135 days and birth (Fig. 2). This pattern of RyR, NCX and LTCC positioning closely paralleled the overall time course for the expression of these proteins (Fig. 3A). Thus, although dyadic formation starts early in sheep, the order of events resembles that reported postnatally in small rodents (Lipsett et al., 2019).
The observed apposition of LTCCs and RyRs in the fetal sheep heart is consistent with the early establishment of triggering of SR Ca2+ release by L-type Ca2+ current soon after t-tubules are grown. Interestingly, close proximity of NCX and RyRs, which is well established in adult cardiomyocytes (Jayasinghe et al., 2009; Mohler et al., 2005; Schulson et al., 2011; Wang et al., 2014), was also apparent in fetal cardiomyocytes. While such positioning of NCX near release sites is expected to favour efficient Ca2+ extrusion, it may also facilitate triggering of Ca2+ release when the exchanger operates in ‘reverse mode’ to enable Ca2+ influx (Lines et al., 2006; Sipido et al., 1997). This mode of NCX function is encouraged by high cytosolic Na+ levels and depolarized resting membrane potentials, both of which have been previously reported in immature cells (reviewed in Louch et al., 2015). Notably, we observed that although NCX protein levels increase during the fetal period, they decline again in the neonate (Fig. 3A). This pattern of expression is reminiscent of reports in developing rodent hearts, and has been proposed to parallel a gradual dominance of forward-mode NCX activity, as Na+ levels decline and membrane potentials become more negative (Artman et al., 2000; Huynh et al., 1992; Reppel et al., 2007).
In addition to proteins involved with Ca2+ homeostasis, we measured the expression of a number of reported t-tubule and dyadic regulators in the developing sheep heart. First we examined JPH2, which is a well-known anchor protein linking t-tubules to junctional SR (reviewed in Manfra et al., 2017; Setterberg et al., 2021). In agreement with previous data from rats and sheep (Munro et al., 2016; Ziman et al., 2010), we observed increasing JPH2 expression in the developing sheep heart, which paralleled observed increases in t-tubule density (compare Figs. 3B and 1B). Interestingly, recent data indicate that JPH2 also anchors LTCCs within t-tubules (Gross et al., 2021), and critically interacts with RyRs in the junctional SR (Beavers et al., 2014). Thus, our observation that increasing JPH2 expression also mirrors the time courses for LTCC and RyR deposition (Fig. 2A) suggests that dyadic assembly and function are carefully coordinated in developing cardiomyocytes.
We also examined changes in BIN1 and its partner proteins in the developing sheep heart (Fig. 3B). BIN1 has been reported to critically regulate t-tubule structure in mice (Farsad et al., 2001; Lee et al., 2002; Takei et al., 1999), sheep (Caldwell et al., 2014) and human embryonic stem cell-derived cardiomyocytes (De La Mata et al., 2019). Interestingly, BIN1 is also able to drive membrane invaginations in cells that do not normally express t-tubules (Hong et al., 2014). Recent data suggest that BIN1 may additionally facilitate dyadic formation by gathering L-type channels and phosphorylated RyRs (Fu et al., 2016; Hong et al., 2010; Hong et al., 2012). In our developmental study, we observed a tendency to higher BIN1 protein expression between gestational ages 93 days and 125 days (Fig. 3B), when t-tubule growth is in an active early phase. Notably, we did not observe an overall significant correlation between BIN1 expression and t-tubule density across developmental time points, consistent with the notion that this protein rather supports timely initiation of tubulation. Interestingly, BIN1 function is linked to its capacity to cluster specific phosphoinositides (Lee et al., 2002), for which precursors are synthesized by the phosphatidylinositol 3-phosphatase myotubularin (MTM1). In skeletal muscle, MTM1 has been shown to direct endolysosomal sorting and trafficking, and thereby BIN1-induced tubulogenesis (Al-Qusairi & Laporte, 2011; Royer et al., 2013). Assuming that a similar role exists in cardiomyocytes, an observed increase in MTM1 expression during development (Fig. 3B) may coordinate t-tubule growth together with BIN1. Other work in skeletal muscle has indicated that DNM2 interacts with BIN1 to inhibit tubulogenesis (Lee et al., 2002; Picas et al., 2014; Tasfaout et al., 2017). Thus, an observed decline in DNM2 levels during development (Fig. 3B) is also consistent with well-coordinated, BIN1-directed t-tubule growth in the normal fetal sheep heart.
The above description of dyadic assembly in maturing cardiomyocytes may give the impression that there is a targeted delivery of RyRs, LTCCs and NCX to the developing SR and t-tubular membranes. This view is, however, biased by the fact that we have observed ‘snapshots’ of the developmental process, which do not convey the true, rapid fluctuations in the positions of these membranes and their constituent proteins. For example, the jSR is highly dynamic, which allows it to rapidly fuse with or withdraw from nearby t-tubules (Drum et al., 2020), and the RyRs within the jSR are also highly mobile (Shen et al., 2022). An intriguing hypothesis presented by the Santana group is that dyad formation is initiated by the random insertion of LTCCs (Guarina et al., 2022). Since LTCCs bind to both JPH2 (Gross et al., 2021) and RyRs (van Oort et al., 2011), they propose that random sites of interaction between these proteins are stabilized. It follows then that sites with more RyRs, and presumably more JPH2, would be preserved as stable dyads, while dyads with fewer RyRs would be short-lived. Indeed, our own work has shown that non-dyadic RyR clusters are consistently smaller than those present in dyads (Shen et al., 2019). Thus, Santana and colleagues suggest that there may be a ‘left behind’ paradigm that allows maintenance of randomly assembled dyadic junctions. Full investigation of this hypothesis will likely require high-resolution, real-time imaging of developing cardiomyocytes.
It is important that the differentiation of t-tubules and the subcellular Ca2+ signalling apparatus is understood within the context of the overall pattern of cardiomyocyte maturation. Previous work has shown that sheep cardiomyocytes terminally differentiate (binucleate) during the last third of gestation, and then grow in both length and cross-sectional dimensions, leading to continued hypertrophy of the whole fetal heart (Jonker et al., 2015). Our present data confirm that cell cycle activity decreases markedly by gestational age 135 days, and is near zero at birth (Table 3), while myocyte hypertrophy starts in utero and increases rapidly after birth (Fig. 1C; Jonker et al., 2015). Notably, we detected cell growth at gestational age 135 days, which is distinctly later than the onset of t-tubule development (Fig. 1 A–C). This observation suggests that t-tubule growth is not simply regulated by cellular hypertrophy, but by other signals such as fetal blood pressure.
Regulation of cardiomyocyte development by systolic load
The fetal heart is highly plastic, which is believed to allow maintenance of adequate blood flow during normal gestation. Amongst other signals, fetal blood pressure has been shown to be a particularly important regulator of growth and maturation (binucleation) of fetal cardiomyocytes (Barbera et al., 2000; Jonker, Faber et al., 2007; O’Tierney et al., 2010; Segar et al., 1997). In the present work, we hypothesized that systolic blood pressure (and associated workload) is also critical for maturation of the Ca2+ signalling apparatus. This proposition was based on previous work in adult hearts, showing that the t-tubule network is highly workload sensitive. Indeed, pioneering work by the Terracciano group has shown that t-tubule density is decreased by mechanical overload or underload of the healthy heart (Ibrahim et al., 2012). Furthermore, in diseases such as heart failure, t-tubule disruption in cardiomyocytes has been linked to elevated workload as the heart dilates (Frisk et al., 2016; Frisk et al., 2021; Ibrahim & Terracciano, 2013). In contrast, the developing heart is subjected to low load at early stages, but progressive increases in load during the third trimester (Struijk et al., 2008; Rudolph & Heymann, 1970). Our present data clearly link these changes in fetal systolic pressure to initiation of t-tubule growth, as we observed that further increasing systolic pressure by plasma infusion or aortic constriction increased t-tubule density in comparison with control animals of the same gestational age (Figs. 4A and B, 6A and B). It is important to note that these interventions induced relatively modest increases in systolic blood pressure, reaching values of 61.0 ± 4.5 and 64.9 ± 9.7 mmHg, respectively (compared with 41.3 ± 1.9 and 47.5 ± 2.1 mmHg in controls). These heightened blood pressures remained below the normal systolic blood pressure present in adult sheep, and were well under values required to induce t-tubule loss and heart failure (reviewed in Setterberg et al., 2021). In contrast, reducing blood pressure by enalaprilat infusion reduced the appearance of t-tubules (Fig. 4A and B). In all cases, the proportions of transversely and longitudinally oriented t-tubules were roughly maintained (Figs. 4F and 6E). While varying blood pressure also affected cardiomyocyte size, we observed that t-tubules were more steeply load-sensitive, again supporting that t-tubule growth is not simply regulated in parallel to cellular hypertrophy (Fig. 4Dc).
The renin–angiotensin system (RAS) is a significant contributor to fetal cardiac load regulation, as it is a regulator of arterial pressure and fluid volume in the fetus (Anderson et al., 1994; Binder & Anderson, 1992; Faber et al., 1998). RAS activity also regulates cardiomyocyte size, although it is not the main effector of load-dependent growth of the fetal heart (Jonker, Faber et al., 2007; Moritz et al., 2001; O’Tierney et al., 2010; Segar et al., 1997; Segar et al., 2001; Sundgren et al., 2003). While t-tubule development could conceivably be linked to RAS regulation, it is important to note that we employed two different hypertensive interventions with distinct RAS involvement. Indeed, while the aortic occlusion model is associated with high circulating levels of angiotensin II (Segar et al., 1997; Segar et al., 2001), low angiotensin II levels are generated in the plasma infusion model (Giraud et al., 2005). Since we found that elevating cardiac load with either method stimulated comparable increases in t-tubule density (compare Figs 4B and 6B), our data suggest that RAS is not centrally involved in t-tubule growth.
Our present findings, and previous work in adult heart, indicate that there is a bell-shaped relationship between t-tubule density and workload, which is centred around an optimal range of workload that favours t-tubule assembly and maintenance. We believe that the low load experienced by the developing heart and the high load placed on the failing heart place these conditions on either side of this optimal point, and result in low t-tubule density. The fact that the normal fetal heart works on the ascending limb of this t-tubule density–workload curve appears to support either compensatory or suppressive capacity. However, functional changes linked to alterations in t-tubule density require parallel changes in the expression of Ca2+ handling proteins, and we did not observe changes in the overall levels of RyR, LTCC or NCX protein (Figs 5 and 8). Nevertheless, we should consider that there are likely considerable changes in LTCC and NCX expression that occur on the surface membrane during cardiac development (Louch et al., 2015), meaning that overall protein expression may not directly parallel the localization of these proteins within t-tubules. Indeed, careful image analysis revealed increasing LTCC density within t-tubules in the aortic constriction model (Fig. 7Ac), suggesting that load-dependent assembly of t-tubules includes positioning of these channels. Importantly, no parallel changes were observed in RyR images, and colocalization of LTCCs and RyRs was unaltered. Based on these findings, we conclude that while assembly of t-tubules and their constituent LTCCs is load-sensitive in the fetal heart, assembly of RyRs in the junctional SR is not. Thus, dyadic construction and plasticity in the developing heart appears to require signals in addition to changing haemodynamics.
Our analyses of dyadic regulatory proteins may provide an indication as to why dyadic formation was not modulated by load-altering interventions. We did not observe significant changes in JPH2 expression following any of the interventions (Figs 5B and 8B). Thus, given the reported roles of JPH2 in anchoring dyads and interacting with both LTCCs and RyRs (see above), it seems that t-tubules grown during higher systolic load conditions might not effectively anchor with the SR, and their constituent LTCCs might be unable to localize with RyRs. On the other hand, we found that BIN1 increased under hypertension conditions (Fig. 5B), which would be expected to promote initiation of t-tubule growth, and possibly also dyadic formation given its reported ability of the protein to gather L-type and RyR channels (Fu et al., 2016; Hong et al., 2010, 2012). However, while BIN1 expression appears to be load sensitive, expression of its partner proteins Mtm1 and Dnm2 was unchanged. Further work will be required to understand these BIN1–MTM1–DNM2 interactions, as this topic remains in its infancy in cardiomyocytes (Setterberg et al., 2021).
Clinical significance
In a clinical context, fetal hyper- and hypotension are associated with fetal pathology, and our current findings suggest that impaired cardiac function in such conditions likely involves changes in cardiomyocyte substructure. However, our data also support the concept of fetal heart plasticity in health, which includes adaptation of cell structure to meet the changing needs of the neonatal circulation. Importantly, the condition of the prenatal heart also influences health after birth. Thus, load-dependent changes in fetal heart maturation and differentiation may affect long-term cardiovascular function (Thornburg et al., 2008).
While we aim to better understand human development, access to developing human tissue is very limited, and it is critical that we can employ animal models that accurately reflect these processes. In this regard, the present work is amongst the first to perform a detailed assessment of cardiomyocyte assembly in a large mammal. Such insight is critical to form a basis for ongoing work aimed at differentiating human stem cells into well differentiated cardiomyocytes. Unfortunately, cardiomyocytes derived from embryonic stem cells or human-induced pluripotent stem cells have historically shown poor maturation, with only rudimentary development of the Ca2+ signalling apparatus. Nevertheless, based on an improving understanding of the developmental cardiac milieu, enhanced differentiation of these cells has recently been reported (Huang et al., 2020; Parikh et al., 2017; Silbernagel et al., 2020). Our current work supports that augmenting workload is an essential component of normal t-tubule maturation, which should be fine-tuned to optimize differentiation of stem cells, although full dyadic maturation appears to require additional, unknown signals. Possible candidates include cortisol, insulin growth factor and triiodothyronine (T3) levels which are well known to be critically involved in healthy cardiac development (Chattergoon et al., 2012; Giraud, Louey et al., 2006; Jonker et al., 2020).
Supplementary Material
Key points.
T-tubule growth and dyadic assembly proceed gradually in cardiomyocytes during fetal sheep development, from 93 days of gestational age until the post-natal stage.
Increasing fetal systolic load by infusing plasma or occluding the post-ductal aorta accelerated t-tubule growth and hypertrophy.
In contrast, reducing fetal systolic load by enalaprilat infusion slowed t-tubule development and decreased cardiomyocyte size.
Load-dependent modulation of t-tubule maturation was linked to altered expression patterns of the t-tubule regulatory proteins junctophilin-2 and amphiphysin-2 (BIN1) and its protein partners.
Altered t-tubule densities did not influence dyadic formation, indicating that distinct signals are responsible for maturation of the sarcoplasmic reticulum.
Translational perspective.
The present work is amongst the first to perform a detailed assessment of cardiomyocyte assembly in a large mammal. The findings indicate that impaired cardiac function in fetal hyper- and hypo-tension conditions can involve changes in cardiomyocyte substructure influencing prenatal heart health. These load-dependent changes in fetal heart maturation and differentiation may also affect health after birth and long-term cardiovascular function. In addition to understanding pathology, the results have implications for ongoing work aimed at maturing human stem cells into well-differentiated cardiomyocytes suitable for cardiac transplantation and repair.
Acknowledgements
The authors are grateful to Loni Socha and Sarah M Alaniz for surgical and technical assistance at the Animal facility Core, and Stefanie Kaech Petrie and Crystal Chaw for the assistance at the Advanced Light Microscopy Core at OHSU. The authors also thank Hege Katrin Ugland for expert help with Western Blotting at the Institute for Experimental Research.
Funding
This research was supported by The Norwegian Research Council (Grants 287978, and 647714), the National Heart, Lung and Blood Institute (R01 HL146997) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Grant P01 HD034430-20).
Biography

Ornella Manfra is currently a postdoctoral fellow at the Institute for Experimental Medical Research in Oslo, Norway. Originally from Italy, she obtained her doctoral degree in Pharmacology at the University of Oslo. In 2016 she joined the Louch lab, where her research focuses on the structure and function of cardiac muscle cells during fetal development and disease.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supporting information
Additional supporting information can be found online in the Supporting Information section at the end of the HTML view of the article. Supporting information files available:
Statistical Summary Document
Peer Review History
Data availability statement
All data that have been included in this study are available upon reasonable request to the authors.
References
- Al-Qusairi L, & Laporte J (2011). T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skeletal Muscle, 1(1), 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DF, Barbera A, & Faber JJ (1994). Substantial reductions in blood pressure after bilateral nephrectomy in fetal sheep. The American Journal of Physiology, 266(1 Pt 2), H17–20. [DOI] [PubMed] [Google Scholar]
- Artman M, Henry G, & Coetzee WA (2000). Cellular basis for age-related differences in cardiac excitation-contraction coupling. Progress in Pediatric Cardiology, 11(3), 185–194. [DOI] [PubMed] [Google Scholar]
- Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, & Thornburg KL (2000). Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 279(4), R1157–R1164. [DOI] [PubMed] [Google Scholar]
- Beavers DL, Landstrom AP, Chiang DY, & Wehrens XH (2014). Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovascular Research, 103(2), 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder ND, & Anderson DF (1992). Plasma renin activity responses to graded decreases in renal perfusion pressure in fetal and newborn lambs. The American Journal of Physiology, 262(3 Pt 2), R524–529. [DOI] [PubMed] [Google Scholar]
- Brook WH, Connell S, Cannata J, Maloney JE, & Walker AM (1983). Ultrastructure of the myocardium during development from early fetal life to adult life in sheep. Journal of Anatomy, 137(Pt 4), 729–741. [PMC free article] [PubMed] [Google Scholar]
- Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, Dibb KM, & Trafford AW (2014). Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circulation Research, 115(12), 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, & Thornburg KL (2012). Thyroid hormone drives fetal cardiomyocyte maturation. FASEB Journal, 26(1), 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Guo A, Zhang C, Chen R, Zhu Y, Hong J, Kutschke W, Zimmerman K, Weiss RM, Zingman L, Anderson ME, Wehrens XH, & Song LS (2013). Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovascular Research, 100(1), 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Mata A, Tajada S, O’Dwyer S, Matsumoto C, Dixon RE, Hariharan N, Moreno CM, & Santana LF (2019). BIN1 Induces the Formation of T-Tubules and Adult-Like Ca2+ Release Units in Developing Cardiomyocytes. Stem Cells, 37(1), 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drum BM, Yuan C, de la Mata A, Grainger N, & Santana LF (2020). Junctional sarcoplasmic reticulum motility in adult mouse ventricular myocytes. American Journal of Physiology. Cell Physiology, 318(3), C598–C604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber JJ, Anderson DF, & Binder ND (1998). Delayed vasoconstriction of the umbilico-placental circulation by angiotensin in fetal sheep. Placenta, 19(8), 675–676. [DOI] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, & De Camilli P (2001). Generation of high curvature membranes mediated by direct endophilin bilayer interactions. The Journal of Cell Biology, 155(2), 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo KS, Lehtinen ML, Leung CY, Lian X, Xu J, Keung W, Geng L, Kolstad TRS, Thams S, Wong AO, Wong N, Bylund K, Zhou C, He X, Jin SB, Clarke J, Lendahl U, Li RA, Louch WE, & Chien KR (2018). Human ISL1(+) ventricular progenitors self-assemble into an in vivo functional heart patch and preserve cardiac function post infarction. Molecular Therapy, 26(7), 1644–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk M, Le C, Shen X, Roe AT, Hou Y, Manfra O, Silva GJJ, van Hout I, Norden ES, Aronsen JM, Laasmaa M, Espe EKS, Zouein FA, Lambert RR, Dahl CP, Sjaastad I, Lunde IG, Coffey S, Cataliotti A, Gullestad L, Tonnessen T, Jones PP, Altara R, & Louch WE (2021). Etiology-dependent impairment of diastolic cardiomyocyte calcium homeostasis in heart failure with preserved ejection fraction. Journal of the American College of Cardiology, 77(4), 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk M, Norseng PA, Stenersen Espe EK, & Louch WE (2022). Tubulator: an automated approach to analysis of t-tubule and dyadic organization in cardiomyocytes. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 377(1864), 20210468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk M, Ruud M, Espe EK, Aronsen JM, Roe AT, Zhang L, Norseng PA, Sejersted OM, Christensen GA, Sjaastad I, & Louch WE (2016). Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovascular Research, 112(1), 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Shaw SA, Naami R, Vuong CL, Basheer WA, Guo X, & Hong T (2016). Isoproterenol promotes rapid ryanodine receptor movement to bridging integrator 1 (BIN1)-organized dyads. Circulation, 133(4), 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayi E, Neff LA, Massana Munoz X, Ismail HM, Sierra M, Mercier T, Decosterd LA, Laporte J, Cowling BS, Dorchies OM, & Scapozza L (2018). Tamoxifen prolongs survival and alleviates symptoms in mice with fatal X-linked myotubular myopathy. Nature Communications, 9(1), 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs EM, Davidson AE, Telfer WR, Feldman EL, & Dowling JJ (2014). The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Disease Models and Mechanisms, 7(1), 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud GD, Faber JJ, Jonker S, Davis L, & Anderson DF (2005). Intravascular infusions of plasma into fetal sheep cause arterial and venous hypertension. Journal of Applied Physiology, 99(3), 884–889. [DOI] [PubMed] [Google Scholar]
- Giraud GD, Faber JJ, Jonker SS, Davis L, & Anderson DF (2006). Effects of intravascular infusions of plasma on placental and systemic blood flow in fetal sheep. American Journal of Physiology. Heart and Circulatory Physiology, 291(6), H2884–H2888. [DOI] [PubMed] [Google Scholar]
- Giraud GD, Louey S, Jonker S, Schultz J, & Thornburg KL (2006). Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology, 147(8), 3643–3649. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Jamett AM, Momboisse F, Haro-Acuna V, Bevilacqua JA, Caviedes P, & Cardenas AM (2013). Dynamin-2 function and dysfunction along the secretory pathway. Frontiers in Endocrinology, 4, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross P, Johnson J, Romero CM, Eaton DM, Poulet C, Sanchez-Alonso J, Lucarelli C, Ross J, Gibb AA, Garbincius JF, Lambert J, Varol E, Yang Y, Wallner M, Feldsott EA, Kubo H, Berretta RM, Yu D, Rizzo V, Elrod J, Sabri A, Gorelik J, Chen X, & Houser SR (2021). Interaction of the joining region in junctophilin-2 with the L-Type Ca2+ channel is pivotal for cardiac dyad assembly and intracellular Ca2+ Dynamics. Circulation Research, 128(1), 92–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarina L, Moghbel AN, Pourhosseinzadeh MS, Cudmore RH, Sato D, Clancy CE, & Santana LF (2022). Biological noise is a key determinant of the reproducibility and adaptability of cardiac pacemaking and EC coupling. Journal of General Physiology, 154(9), e202012613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Zhang X, Iyer VR, Chen B, Zhang C, Kutschke WJ, Weiss RM, Franzini-Armstrong C, & Song LS (2014). Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proceedings of the National Academy of Sciences, USA, 111(33), 12240–12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi S, Kawakami Y, Honda Y, Nemoto K, Sano A, Namekata I, & Tanaka H (2013). Developmental changes in excitation-contraction mechanisms of the mouse ventricular myocardium as revealed by functional and confocal imaging analyses. Journal of Pharmacological Sciences, 123(2), 167–175. [DOI] [PubMed] [Google Scholar]
- Han J, Wu H, Wang Q, & Wang S (2013). Morphogenesis of T-tubules in heart cells: the role of junctophilin-2. Science China Life Sciences, 56(7), 647–652. [DOI] [PubMed] [Google Scholar]
- Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D’Hooge J, & Sipido K (2008). Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circulation Research, 102(3), 338–346. [DOI] [PubMed] [Google Scholar]
- Hong T, Yang H, Zhang SS, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J, Gorelik J, Marban E, Jan LY, & Shaw RM (2014). Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nature Medicine, 20(6), 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong TT, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, Fang K, Halushka MK, Russell SD, Colecraft H, Hoopes CW, Ocorr K, Chi NC, & Shaw RM (2012). BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm, 9(5), 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, & Shaw RM (2010). BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biology, 8(2), e1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Peres Moreno Maia-Joca R, Ong CS, Wilson I, DiSilvestre D, Tomaselli GF, & Reich DH (2020). Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. Journal of Molecular and Cellular Cardiology, 138, 1–11. [DOI] [PubMed] [Google Scholar]
- Huynh TV, Chen F, Wetzel GT, Friedman WF, & Klitzner TS (1992). Developmental changes in membrane Ca2+ and K+ currents in fetal, neonatal, and adult rabbit ventricular myocytes. Circulation Research, 70(3), 508–515. [DOI] [PubMed] [Google Scholar]
- Ibrahim M, Kukadia P, Siedlecka U, Cartledge JE, Navaratnarajah M, Tokar S, van Doorn C, Tsang VT, Gorelik J, Yacoub MH, & Terracciano CM (2012). Cardiomyocyte Ca2+ handling and structure is regulated by degree and duration of mechanical load variation. Journal of Cellular and Molecular Medicine, 16(12), 2910–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M, & Terracciano CM (2013). Reversibility of T-tubule remodelling in heart failure: mechanical load as a dynamic regulator of the T-tubules. Cardiovascular Research, 98(2), 225–232. [DOI] [PubMed] [Google Scholar]
- Jayasinghe ID, Cannell MB, & Soeller C (2009). Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophysical Journal, 97(10), 2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, & Giraud GD (2007). Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 292(2), R913–R919. [DOI] [PubMed] [Google Scholar]
- Jonker SS, Giraud GD, Chang EI, Elman MR, & Louey S (2020). Coronary vascular growth matches IGF-1-stimulated cardiac growth in fetal sheep. FASEB Journal, 34(8), 10041–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker SS, Louey S, Giraud GD, Thornburg KL, & Faber JJ (2015). Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB Journal, 29(10), 4346–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, & Faber JJ (2007). Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. Journal of Applied Physiology, 102(3), 1130–1142. [DOI] [PubMed] [Google Scholar]
- Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, Dirschinger RJ, Seyfarth M, Lam JT, Sinnecker D, Gudermann T, Lipp P, & Laugwitz KL (2012). Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Molecular Medicine, 4(3), 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Kim DJ, Lee IJ, Rah BJ, Sawa Y, & Schaper J (1992). Human fetal heart development after mid-term: morphometry and ultrastructural study. Journal of Molecular and Cellular Cardiology, 24(9), 949–965. [DOI] [PubMed] [Google Scholar]
- Kolstad TR, van den Brink J, MacQuaide N, Lunde PK, Frisk M, Aronsen JM, Norden ES, Cataliotti A, Sjaastad I, Sejersted OM, Edwards AG, Lines GT, & Louch WE (2018). Ryanodine receptor dispersion disrupts Ca2+ release in failing cardiac myocytes. eLife, 7, e39427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, & De Camilli P (2002). Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science, 297(5584), 1193–1196. [DOI] [PubMed] [Google Scholar]
- Lines GT, Sande JB, Louch WE, Mork HK, Grottum P, & Sejersted OM (2006). Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophysical Journal, 91(3), 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsett DB, Frisk M, Aronsen JM, Norden ES, Buonarati OR, Cataliotti A, Hell JW, Sjaastad I, Christensen G, & Louch WE (2019). Cardiomyocyte substructure reverts to an immature phenotype during heart failure. The Journal of Physiology, 597(7), 1833–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, & Sipido KR (2004). Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovascular Research, 62(1), 63–73. [DOI] [PubMed] [Google Scholar]
- Louch WE, Koivumaki JT, & Tavi P (2015). Calcium signalling in developing cardiomyocytes: Implications for model systems and disease. The Journal of Physiology, 593(5), 1047–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, & Sejersted OM (2006). T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. The Journal of Physiology, 574(2), 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfra O, Frisk M, & Louch WE (2017). Regulation of cardiomyocyte T-tubular structure: opportunities for therapy. Current Heart Failure Reports, 14(3), 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marat AL., & Haucke V. (2016). Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO Journal, 35(6), 561–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S, McMillen IC, & Morrison JL (2013). The fetal sheep lung does not respond to cortisol infusion during the late canalicular phase of development. Physiological Reports, 1(6), e00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Davis JQ, & Bennett V (2005). Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biology, 3(12), e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz KM, Campbell DJ, & Wintour EM (2001). Angiotensin-(1–7) in the ovine fetus. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 280(2), R404–R409. [DOI] [PubMed] [Google Scholar]
- Munro ML, Jayasinghe ID, Wang Q, Quick A, Wang W, Baddeley D, Wehrens XH, & Soeller C (2016). Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. Journal of Cell Science, 129(23), 4388–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro ML, & Soeller C (2016). Early transverse tubule development begins in utero in the sheep heart. Journal of Muscle Research and Cell Motility, 37(6), 195–202. [DOI] [PubMed] [Google Scholar]
- O’Tierney PF, Anderson DF, Faber JJ, Louey S, Thornburg KL, & Giraud GD (2010). Reduced systolic pressure load decreases cell-cycle activity in the fetal sheep heart. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 299(2), R573–R578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tonnessen T, Kryshtal DO, Louch WE, & Knollmann BC (2017). Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circulation Research, 121(12), 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picas L, Viaud J, Schauer K, Vanni S, Hnia K, Fraisier V, Roux A, Bassereau P, Gaits-Iacovoni F, Payrastre B, Laporte J, Manneville JB, & Goud B (2014). BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nature Communications, 5(1), 5647. [DOI] [PubMed] [Google Scholar]
- Reppel M, Sasse P, Malan D, Nguemo F, Reuter H, Bloch W, Hescheler J, & Fleischmann BK (2007). Functional expression of the Na+/Ca2+ exchanger in the embryonic mouse heart. Journal of Molecular and Cellular Cardiology, 42(1), 121–132. [DOI] [PubMed] [Google Scholar]
- Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, Landstrom AP, Song LS, Ackerman MJ, & Wehrens XH (2013). Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovascular Research, 100(1), 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer B, Hnia K, Gavriilidis C, Tronchere H, Tosch V, & Laporte J (2013). The myotubularin-amphiphysin 2 complex in membrane tubulation and centronuclear myopathies. Embo Reports, 14(10), 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph AM, & Heymann MA (1970). Circulatory changes during growth in the fetal lamb. Circulation Research, 26(3), 289–299. [DOI] [PubMed] [Google Scholar]
- Schulson MN, Scriven DR, Fletcher P, & Moore ED (2011). Couplons in rat atria form distinct subgroups defined by their molecular partners. Journal of Cell Science, 124(7), 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segar JL, Dalshaug GB, Bedell KA, Smith OM, & Scholz TD (2001). Angiotensin II in cardiac pressure-overload hypertrophy in fetal sheep. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 281(6), R2037–R2047. [DOI] [PubMed] [Google Scholar]
- Segar JL, Scholz TD, Bedell KA, Smith OM, Huss DJ, & Guillery EN (1997). Angiotensin AT1 receptor blockade fails to attenuate pressure-overload cardiac hypertrophy in fetal sheep. The American Journal of Physiology, 273(4), R1501–1508. [DOI] [PubMed] [Google Scholar]
- Setterberg IE, Le C, Frisk M, Li J, & Louch WE (2021). The Physiology and Pathophysiology of T-Tubules in the Heart. Frontiers in Physiology, 12, 718404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon CA, Friedman WF, & Sybers HD (1976). Scanning electron microscopy of fetal and neonatal lamb cardiac cells. Journal of Molecular and Cellular Cardiology, 8(11), 853–854. [DOI] [PubMed] [Google Scholar]
- Shen X, van den Brink J, Bergan-Dahl A, Kolstad TR, Norden ES, Hou Y, Laasmaa M, Aguilar-Sanchez Y, Quick AP, Espe EKS, Sjaastad I, Wehrens XHT, Edwards AG, Soeller C, & Louch WE (2022). Prolonged beta-adrenergic stimulation disperses ryanodine receptor clusters in cardiomyocytes and has implications for heart failure. eLife, 11, e77725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, van den Brink J, Hou Y, Colli D, Le C, Kolstad TR, MacQuaide N, Carlson CR, Kekenes-Huskey PM, Edwards AG, Soeller C, & Louch WE (2019). 3D dSTORM imaging reveals novel detail of ryanodine receptor localization in rat cardiac myocytes. The Journal of Physiology, 597(2), 399–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbernagel N, Korner A, Balitzki J, Jaggy M, Bertels S, Richter B, Hippler M, Hellwig A, Hecker M, Bastmeyer M, & Ullrich ND (2020). Shaping the heart: Structural and functional maturation of iPSC-cardiomyocytes in 3D-micro-scaffolds. Biomaterials, 227, 119551. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Maes M, & van de Werf F (1997). Low efficiency of Ca2+ entry through the Na+-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na+-Ca2+ exchange. Circulation Research, 81(6), 1034–1044. [DOI] [PubMed] [Google Scholar]
- Smolich JJ, Walker AM, Campbell GR, & Adamson TM (1989). Left and right ventricular myocardial morphometry in fetal, neonatal, and adult sheep. The American Journal of Physiology, 257(1 Pt 2), H1–9. [DOI] [PubMed] [Google Scholar]
- Struijk PC, Mathews VJ, Loupas T, Stewart PA, Clark EB, Steegers EA, & Wladimiroff JW (2008). Blood pressure estimation in the human fetal descending aorta. Ultrasound in Obstetrics & Gynecology, 32(5), 673–681. [DOI] [PubMed] [Google Scholar]
- Sundgren NC, Giraud GD, Stork PJ, Maylie JG, & Thornburg KL (2003). Angiotensin II stimulates hyperplasia but not hypertrophy in immature ovine cardiomyocytes. The Journal of Physiology, 548(3), 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K, Slepnev VI, Haucke V, & De Camilli P (1999). Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nature Cell Biology, 1(1), 33–39. [DOI] [PubMed] [Google Scholar]
- Tasfaout H, Buono S, Guo S, Kretz C, Messaddeq N, Booten S, Greenlee S, Monia BP, Cowling BS, & Laporte J (2017). Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nature Communications, 8(1), 15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg KL, Louey S, & Giraud GD (2008). The role of growth in heart development. Nestlé Nutrition Institute Workshop Series: Pediatric Program, 61, 39–51. [DOI] [PubMed] [Google Scholar]
- van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, & Wehrens XH (2011). Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation, 123(9), 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Landstrom AP, Wang Q, Munro ML, Beavers D, Ackerman MJ, Soeller C, & Wehrens XH (2014). Reduced junctional Na+/Ca2+-exchanger activity contributes to sarcoplasmic reticulum Ca2+ leak in junctophilin-2-deficient mice. American Journal of physiology Heart and Circulatory Physiology, 307(9), H1317–H1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, Feng XH, Guo YB, Li SF, Lai ST, Zhou P, Li LL, Yang HQ, Luo GZ, Bai Y, Xi JJ, Gao W, Han QD, Zhang YY, Wang XJ, Meng X, & Wang SQ (2012). Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circulation Research, 111(7), 837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziman AP, Gomez-Viquez NL, Bloch RJ, & Lederer WJ (2010). Excitation-contraction coupling changes during postnatal cardiac development. Journal of Molecular and Cellular Cardiology, 48(2), 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that have been included in this study are available upon reasonable request to the authors.