

Abstract
Keratocyst is a developmental odontogenic cyst arising from remnants/rests of the dental lamina with biologic behavior similar to benign neoplasm. The presence of multiple odontogenic keratocysts is rare and seen in Gorlin-Goltz syndrome (GGS). GGS syndrome presents with multisystem involvement and the classical triad of multiple basocellular epitheliomas, keratocysts in the jaws, and bifid ribs; that characterize the diagnosis of this syndrome. Multiple odontogenic keratocyst are the most consistent features of the syndrome in 65%–100% of affected individuals and are generally diagnosed at a very early age. Early diagnosis and proper counseling of the parent and patient might help to reduce the morbidity, encourage follow-up for timely treatment, and help in avoiding ionizing radiation that would lead to the development of malignancies.
Keywords: Gorlin-Goltz syndrome, multiple basal cell carcinoma, multiple odontogenic keratocyst, multi-system disorder
Introduction
Odontogenic keratocyst (OKC) is a developmental odontogenic cyst arising from odontogenic primordium or remnants/rests of the dental lamina and has biological behavior similar to benign neoplasm. Solitary OKC is the second most common developmental odontogenic cyst but the presence of multiple OKC is rare. One of the conditions that commonly present with multiple OKC is Gorlin-Goltz syndrome (GGS).
GGS is a rare multisystem disorder that is inherited as autosomal dominant with variable expressivity and high penetrance. GGS is also known as basal cell nevus syndrome, GGS, Gorlin syndrome, Nevoid basal-cell carcinoma (BCC) syndrome, multiple BCC syndrome, multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib syndrome, hereditary cutaneous-mandibular polyonocosis, and Hermans–Herzberg phakomatosis.[1,2]
The condition is very rare with an incidence as low as 1 in 50,000–150,000[3] in the general population and prevalence varying from 1/57,000 to 1/256,000.[4] The expressions may vary from region to region. It is found that both genders are affected equally.[3]
Pathogenesis of GGS is frequently associated with abnormality in the long arm of chromosome 9 (q22.3 – q31) and loss of/mutation of a human patched gene (PTCH 1). PTCH 1 is a tumor suppressor gene which plays an important role in the cell cycle and embryonic structuring.[5,6] 60% of the patients with the syndrome have no known affected family members which may occur because of spontaneous mutations.[7,8]
The syndrome presents with multisystem involvement, including the integumentary, nervous, endocrinal, and skeletal systems.[8] Literature suggests a continued expansion of the range of symptoms presented by the syndrome and now; >100 different recognized features are identified.[1] Evans et al. in 1993 established the major and minor criteria for the diagnosis of the syndrome,[9] which was modified by Kimonis et al. in 1997.[10]
However, in the Indian scenario, GGS is rarely reported and cases of sporadic mutations are sparse. Hence, the present report is a rare case wherein we attempt to expand the spectrum of the syndrome for better understanding and emphasize on the early diagnosis of the condition which will help in improving the quality of life of the patient.
Case Report
A 17-year-old boy, the only child of nonconsanguineous parents, born after an uncomplicated pregnancy, presented with irregularly arranged upper front teeth and few unerupted teeth. On examination, it was noted that 51, 52, 62, and 63 were over–retained, and 12, 22, and 23 were clinically missing [Figure 1]. The patient seemed to have a lower Intelligent Quotient (IQ) and followed instructions late (mental age of 11 years and IQ of 64.5). His head appeared larger compared to his face and showed frontal bossing, hypertelorism (38 mm), and mandibular retrognathism [Figure 2].
Figure 1.
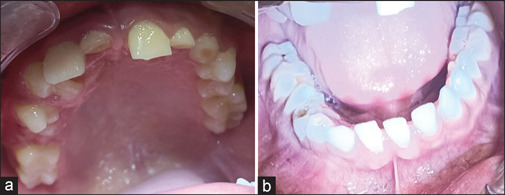
Intraoral photograph showing, (a) Maxillary teeth and, (b) Mandibular teeth
Figure 2.

Extraoral photograph showing (a) Frontal view with frontal bossing, and (b) Profile view with mandibular retrognathism and large circumference of the head, and (c) Hypertelorism with 38 mm inter inner-canthus width
Orthopantomagram (OPG) showed three lytic lesions, one in the maxillary anterior region and two in the mandible [Figure 3]. In the maxilla, the lytic lesion was associated with an impacted left canine. In the mandible, one was seen in the anterior region crossing the midline associated with the impacted right canine and the second in relation to the left impacted third molar. All three lytic lesions were multilocular with no associated calcification. A provisional diagnosis of multiple OKC was considered as there was no associated facial swelling or root resorption of adjacent teeth.
Figure 3.

Orthopantomagram showing three multilocular radiolucency, (a) The maxillary anterior region, (b) Mandibular anterior region and, (c) Mandibular left posterior region
Since multiple OKC was considered the clinical diagnosis, and correlation with the facial findings (increased head circumference and mental retardation) directed us to investigate for GGS. Palmar and plantar pits were noticed on clinical examination [Figure 4]. The bifid left 4th rib was noted on chest X-ray [Figure 5a] and skull radiograph showed calcification in falxcerebri [Figure 5b], spina bifida in the upper dorsal vertebra (D2-4 vertebrae) was also noted. However, the family history did not reveal any member with similar findings.
Figure 4.
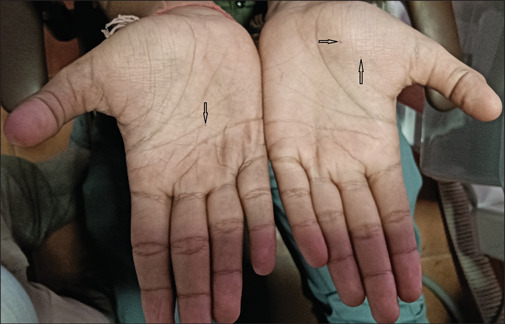
Photograph with arrows showing multiple palmar pits
Figure 5.

(a) Chest X-ray showing bifid fourth rib (arrow), Spina bifida (circle), and (b) Skull radiograph with bold white arrow showing calcification of falx cerebri
The patient was taken for surgery for the lytic lesions of the jaw and cheesy material was noted on opening into the site. Lesions were enucleated, suitably labeled, and sent for histopathological investigation. The exposed bony cavity was fixed using 5 Fluorouracil. All three tissues were fixed with 10% formalin, processed in graded alcohol, and embedded in paraffin wax. Four microns thick sections were cut and stained with hematoxylin and eosin.
Histopathology of all three jaw lesions was similar and showed cystic lumen, lining, and collagenous capsule. The cystic lining was comprised of stratified squamous parakeratinized epithelium of 6–8 layers of even thickness. Basal cell palisading and parakeratin corrugation were evident. Exfoliated parakeratin was seen in the lumen. A few areas of epithelial budding into the connective tissue capsule were also evident. Capsules consisted of thick bundles of collagen with areas of chronic inflammation. The histopathological features of all three lesions were suggestive of OKC with inflammation [Figures 6 and 7].
Figure 6.

Photomicrograph showing histopathology of odontogenic keratocyst, (a) Maxillary lesion, (b) Mandibular left posterior region, and (c) Mandibular anterior region
Figure 7.

Photomicrograph showing the budding of cystic lining into a capsule
Histopathological findings in correlation with the clinical examination and the radiographic investigations were suggestive of sporadic cases of GGS. After 1 year of follow-up, the patient was asymptomatic and the OPG revealed no recurrence with good healing of all three lesions [Figure 8].
Figure 8.

Orthopantomagram after 1-year follow-up
Discussion
GGS is dated to Dynastic Egyptian times, as per attuned findings in mummies in 1000 BC.[11,12] Pandeshwar P et al have stated that the first reported case was in 1894 in a patient with multiple basal cell carcinomas, scoliosis, and learning disability. Relationship between basal cell epitheliomas and developmental malformations were reported by few authors. In 1960 a classical triad (multiple basocellular epitheliomas, keratocysts in the jaws and bifid ribs) was established that characterizes the diagnosis of this syndrome.[4] This triad was later modified by Rayner et al.[13] who established that the diagnostic criteria would require cysts to appear in combination with calcification of the falx cerebri or palmar and plantar pits.[4] The present case report supports the Rayner et al[13] hypothesis.
The diagnostic criteria for nevoid BCC were established by Evans et al.[9] and modified by Kimonis et al. in 1997,[10] who stated that when two major or one major and two minor criteria should be present for diagnosis.
Major criteria: (i) More than two BCC or one BCC at younger than 30 years of age or more than 10 basal cell nevi, (ii) OKC (proven on histology) or polyostotic bone cyst, (iii) Three or more palmar or plantar pits, (iv) bifid, fused, or markedly splayed ribs, (v) Ectopic calcification: lamellar or early at younger than 20 years of age, (vi) Falxcerebri calcification, (vii) Positive family history of nevoid BCC. Some authors consider the plurilamellar appearance of the falx cerebri calcification as a pathognomonic symptom of GGS.
Minor criteria: (i) Macrocephaly determined after adjustment with height, (ii) Skeletal anomalies: hemivertebrae, scoliosis, syndactyly, polydactyly, and shortened 4th metacarpal, (iii) Radiological abnormalities such as bridging of sella turcica, vertebral anomalies, and modeling defect of hands and feet, (iv) Medulloblastoma, (v) Ovarian fibroma, (vi) Congenital malformations: cleft lip or palate, polydactylism, or eye anomalies (cataract, coloboma, and microphthalmus). Over 100 minor criteria have been reported in the literature for diagnosing GGS.[8]
Mutations in tumor suppressor gene PTCH (human homolog of a Drosophila segment polarity gene Patch) located in chromosome 9q22.3, the protein found in the Hedgehog signaling pathway are considered the cause for the syndrome. The gene in the absence of its ligand, acts as a cell-cycle regulator, inhibiting expression of downstream genes that control cell fate, patterning, and growth. For a tumor suppressor gene to be inactivated, two mutagenic hits are required. First involves a mutation in one allele, which can be dominantly inherited if present in a germ cell, but which is classically considered to have no phenotypic effect. The second is loss of heterozygosity, when both alleles are inactivated, tumorigenesis occurs. Loss of heterozygosity is demonstrated in BCC, OKCs, and medulloblastoma. Various physical anomalies of the brain, ribs, vertebrae, and limbs can occur apparently with one hit. The single germ cell hit may account for the malformations and their inconsistency in GGS patients.[11,12]
The most likely position for a gene is between DNA markers D9S12 and D9S53. GGS can also be caused by mutations in the PTCH 2 gene located on chromosome 1p32. Mutations in other genes, Smoothened and Sonic Hedgehog, SUFU (Chromosome 10q24.32) have been reported in isolated cases in the absence of mutation in the PTCH 1 gene.[12] Mutations in a single gene or a combination of these genes are probably responsible for the various manifestations of GGS and needs to be further studied.[6] The literature demonstrates more than 225 mutations in the PTCH 1 gene in GGS.
GGS is characterized by complete penetrance, which signifies that all individuals who inherit the gene will develop symptoms of the disorder. GGS is also characterized by variable expressivity, indicating that widely varying signs and symptoms can occur among affected individuals.[1]
Keratocystic odontogenic tumors are among the most consistent and common features of GGS. They are found in 65%–100% of affected individuals.[11,14] Clinically, the lesions are aggressive and have a tendency to recur after surgical treatment. Histopathologically, the epithelial cells of the basal layer show increased mitotic activity, together with a potential for budding and the presence of daughter cysts in the wall. Woolgar et al.[15] and Dominiguez et al.[16] found that the syndrome keratocyst had markedly increased the number of satellite cysts, solid islands of epithelial proliferation, odontogenic rests within the capsule, and mitotic figures in the cystic lining. It was also noted that syndrome keratocysts occur at a much earlier age than single keratocysts.[2]
OKC in GGS involves the mandible more frequently than the maxilla,[11,14] and similarly, the present report shows two lesions in the mandible and one in the maxilla. The posterior regions are the most commonly affected sites[11] but the present report shows two lesions in the anterior and one in the posterior region of the jaw. Multiple OKC may be a presenting feature in other conditions including Ehlers–Danlos syndrome, Noonan syndrome, and oral-facial-digital syndrome. In some rare cases, nonsyndromic multiple OKCs have also been described.[5]
Two treatment methodologies have been proposed for OKCs in GGS: conservative and aggressive. The conservative method involves enucleation (with or without curettage) and marsupialization. Aggressive methods include peripheral ostectomy, chemical curettage with Carnoy's solution, and resection.[11] Conservative approaches should be considered for young patients because an aggressive treatment can cause unfavorable effects on the development of the jaw, teeth, and the eruption process.[14] The present case was treated by enucleation following the application of 5 Flurouracil.
Recurrence in odontogenic cysts (12%–62.5%)[14] requires repeated surgical excisions. Enblock resection can be considered in cases of: (i) Recurrence (with previous enucleation with an adjunctive procedure); (ii) Recurence (with previous marsupialization followed by enucleation with an adjunctive procedure); (iii) Multilocular aggressive lesions; (iv) Multiple nonsyndromic and syndromic OKCs; and (v) OKC exhibiting aggressive clinical behavior like the destruction of adjacent tissues. In children who have unerupted teeth, conservative management should be considered because an aggressive procedure can have an adverse effect on teeth development, eruption process, and development of the jaw.[11,14]
Appropriate management after diagnosing the case as GGS depends on early recognition of the disease, a detailed family history, and a thorough evaluation of signs and symptoms. A team consisting of various specialists is the prime requisite for successful management.
Development of multiple BCC, especially in the head-and-neck region, is a significant feature of the syndrome[12] and usually develops among puberty and mid-30s.[1] However, can occur at almost any age, with literature suggesting occurrence in children as young as 2 years.[1] It is noted that about 10% of the patients may present with a malignant neoplasm or a BCC.[8] Fortunately, our patient was not suffering from BCC but presented with multiple nevi on the face. Once diagnosed early, the patient would benefit from early treatment and follow-up (up to 80% of the white population and 38% of Blacks develop BCCs).[5,7] Literature reported a case where BCCs did not develop until after 50 years of age. Approximately 10% of individuals with GGS, especially those with dark skin and limited sunlight exposure, do not develop any BCCs.[1]
5%–10% of the patients may develop brain medulloblastoma, a potential cause of early death. Although survival in Gorlin-Goltz patients is not affected significantly, morbidity from complications is momentous.
GGS is known for its varied features. Palmar and plantar pits occur in 35%-87% of the patients and the literature review suggests that 80% develop by the age of 15 years and 85% after the age of 20 years. Ectopic calcifications of falx cerebri (21.2%–92%); relative macroencephaly with an increased occipito-frontal circumference of >55 cm (5%–80%); hypertelorism (6%–78%); bifid, fused, wide, partially missing, or underdeveloped ribs (16%–58%) are generally noted in patients worldwide. The present case report demonstrated all these features. Epidermoid cysts occur in 50% and cleft lip and palate and Sprengel's deformity are rare in this syndrome with 0%–9% and 4%–22% occurrence.
In cases without a positive family history of the disorder, the gene mutation may occur spontaneously for no noticeable reason, which represents a new mutation and is known as a sporadic case. Approximately 35%–50% of affected individuals are because of sporadic mutation.[1] The present case is an example of a sporadic mutation as the family history was not positive.
Recall visits and follow-ups of these cases shall be of utmost importance as these patients show an increased propensity to multiple malignant neoplasms and are also sensitive to ionizing radiation, including ultraviolet radiation.
Conclusion
Early diagnosis and treatment may reduce the severity of the long-term sequelae, including malignancy and maxillofacial deformation and destruction. Regular follow-up (3–4 times a year or more) is crucial for such patients to detect new odontogenic cysts and BCC that occur continuously. Genetic counseling to family members helps to detect necessary diagnostic criteria and thus improves their survival through well-directed treatment.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.NORD (National Organization of Rare Disease) Nevoid Basal Cell Carcinoma Syndrome. [Last accessed on 2023 Jan 18]; Available from: https://rarediseases.org/rare-diseases/nevoid-basal-cell-carcinoma-syndrome/ [Google Scholar]
- 2.Agrawal A, Murari A, Vutukuri S, Singh A. Gorlin-Goltz syndrome: Case report of a rare hereditary disorder. Case Rep Dent. 2012;2012:475439. doi: 10.1155/2012/475439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joshi PS, Deshmukh V, Golgire S. Gorlin-Goltz syndrome. Dent Res J (Isfahan) 2012;9:100–6. doi: 10.4103/1735-3327.92963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pandeshwar P, Jayanthi K, Mahesh D. Gorlin-Goltz Syndrome. Case Reports in Dentistry. 2012:4. doi: 10.1155/2012/247239. Article ID 247239. pages, doi:10.1155/2012/247239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maheshwari S, Dudhal R, Rajesh U, Prateek Imaging in Gorlin-Goltz syndrome with emphasis on diffusion-weighted imaging. Indian J Musculoskelet Radiol. 2020;2:128–32. [Google Scholar]
- 6.Lata J, Verma N, Kaur A. Gorlin-Goltz syndrome: A case series of 5 patients in North Indian population with comparative analysis of literature. Contemp Clin Dent. 2015;6:S192–201. doi: 10.4103/0976-237X.166813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hegde S, Shetty SR. Radiological features of familial Gorlin-Goltz syndrome. Imaging Sci Dent. 2012;42:55–60. doi: 10.5624/isd.2012.42.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Eshanth R, Indiran V, Mariappan K, Maduraimuthu P. A rare association of Gorlin-Goltz syndrome. Neurol India. 2018;66:847–9. doi: 10.4103/0028-3886.232340. [DOI] [PubMed] [Google Scholar]
- 9.Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA. Complications of the naevoid basal cell carcinoma syndrome: Results of a population based study. J Med Genet. 1993;30:460–4. doi: 10.1136/jmg.30.6.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299. [PubMed] [Google Scholar]
- 11. [Last accessed on 2023 Jan 18]; Available from: https://gorlinsyndrome.org/diagnosis/#:~:text=Minor%20criteria%20(symptoms)%20of%20Gorlin%20syndrome%3A&text=Large%20head%20size%20and%20protruding,or%20tumors%20in%20the%20iris . [Google Scholar]
- 12.Casaroto AR, Loures DR, Moreschi E, Veltrini VC, Trento CL, Gottardo VD, Lara VS. Early diagnosis of Gorlin-Goltz syndrome: Case report. Head Face Med. 2011;7:2. doi: 10.1186/1746-160X-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rayner CRW, Towers JF, Wilson JSP. What is Gorlin's syndrome? The Diagnosis and management of the Basal Cell Naevus syndrome, based on a study of thirty seven patients. British Journal of Plastic surgery. 1976;30:62–7. doi: 10.1016/s0007-1226(77)90037-6. [DOI] [PubMed] [Google Scholar]
- 14.Al-Jarboua MN, Al-Husayni AH, Al-Mgran M, Al-Omar AF. Gorlin-Goltz syndrome: A case report and literature review. Cureus. 2019;11:e3849. doi: 10.7759/cureus.3849. DOI 10.7759/cureus.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woolgar JA, Rippin JW, Browne RM. A comparative histological study of odontogenic keratocysts in basal cell naevus syndrome and control patients. J Oral Pathol Med. 1978;16:75–80. doi: 10.1111/j.1600-0714.1987.tb00691.x. [DOI] [PubMed] [Google Scholar]
- 16.Dominguez FV, Keszler A. Comparative study of keratocysts, associated and non-associated with nevoid basal cell carcinoma syndrome. J Oral Pathol Med. 1988;17:39–42. doi: 10.1111/j.1600-0714.1988.tb01503.x. [DOI] [PubMed] [Google Scholar]