

SUMMARY
To become specialized binders, antibodies undergo a process called affinity maturation to maximize their binding affinity. Despite this process, some antibodies retain low-affinity binding to diverse epitopes in a phenomenon called polyreactivity. Here we seek to understand the molecular basis of this polyreactivity in antibodies. Our results highlight that polyreactive antigen-binding fragments (Fabs) bind their targets with low affinities, comparable to T cell receptor recognition of autologous classical major histocompatibility complex. Extensive mutagenic studies find no singular amino acid residue or biochemical property responsible for polyreactive interaction, suggesting that polyreactive antibodies use multiple strategies for engagement. Finally, our crystal structures and all-atom molecular dynamics simulations of polyreactive Fabs show increased rigidity compared to their monoreactive relatives, forming a neutral and accessible platform for diverse antigens to bind. Together, these data support a cooperative strategy of rigid neutrality in establishing the polyreactive status of an antibody molecule.
In brief
Binding promiscuity is an inherent property of immune recognition, manifested in antibodies as polyreactivity. Borowska et al. dissect this phenomenon at the molecular level and show that the binding surfaces of polyreactive antibodies use increased rigidity and neutrality in ligand engagement. This provides a key mechanism for understanding polyreactivity in antibodies.
Graphical abstract
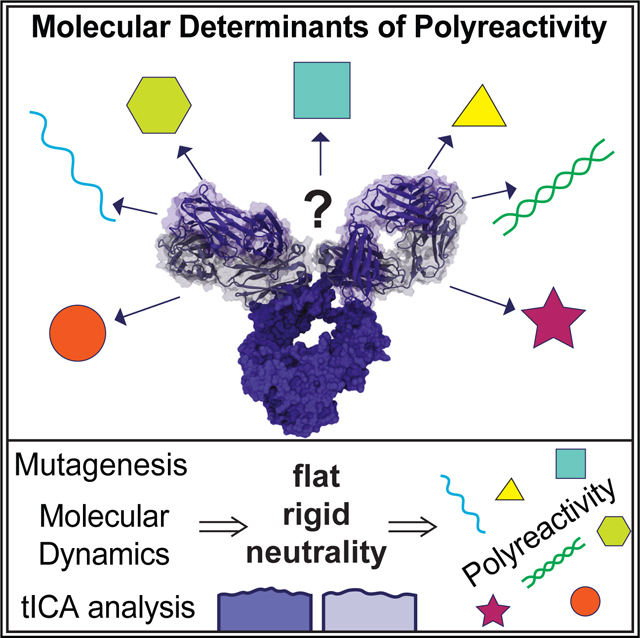
INTRODUCTION
The antigen-antibody interaction is a central reaction in the immune response of jawed vertebrates. Antibodies are generated through a process of gene shuffling (V-D-J gene recombination), and their high-affinity binding to their target, and the associated specificity, is gained through a process of somatic hypermutation where point substitutions generate amino acid changes in the antibody’s antigen-binding, complementarity-determining region (CDR) loops. This process of maturing an antibody’s affinity for antigen aids in the targeted removal of that pathogen but also comes at a cost: with increasing specificity comes a corresponding decrease in breadth, reducing the ability of the antibody to recognize mutated pathogenic targets.
In contrast to the focused specificity of antibody-antigen interactions, binding promiscuity is an inherent property of T cell receptor (TCR) recognition and is also relevant to many aspects of T cell biology.1–4 Specifically, TCRs require positive engagement of self-peptide-major histocompatibility complexes (pMHCs) during selection in the thymus (autologous TCR-pMHC interactions) in order to survey for MHC-presented non-self-peptides in the periphery. Less well appreciated is the need for promiscuity in B cell receptor (BCR) signaling and B cell biology as well as the observation that many antibodies exhibit polyreactive binding, where they recognize many structurally and chemically distinct epitopes with low affinity.5–7 This polyreactive binding is distinct from cross-reactivity, where an antibody binds an off-target epitope also with high affinity, such as what is seen in pathological autoimmune recognition. B cells expressing polyreactive BCRs and secreted polyreactive antibodies comprise 6% of the naive B cell repertoire and ~25% of the IgG memory B cell pool.5,8,9 Broadly neutralizing antibodies (bnAbs), which bind to the same epitope of multiple strains of the same virus, (i.e., against HIV and influenza viruses) are commonly polyreactive. In fact, nearly 60%–80% of bnAbs, besides recognizing their high-affinity viral target, also recognize other diverse epitopes with lower affinity.6,7,10–12
Hypotheses for the evolutionary advantage of antibody polyreactivity largely center around the recognition of pathogenic epitopes that are hard to reach, rapidly mutating, or shielded by glycosylation, allowing polyreactive antibodies to overcome extreme antigenic variation.13 A model of “heteroligation” was proposed as one advantage of polyreactivity, wherein a single antibody can bind the primary target with one Fab (fragment antigen binding) domain, while the other Fab domain binds to the surroundings (e.g., membrane or other receptors) in a polyreactive manner, thus augmenting the overall antibody binding avidity to its epitope.13 For example, the envelope protein of HIV is so sparse on the surface of the virus that only one Fab of an antibody would engage; binding of the other Fab, even at low affinity, to a nearby epitope would enhance the effective binding to the antibody’s target. Attempts to eliminate polyreactivity in broadly neutralizing antibodies come at a sacrifice of broad neutralization.13–15 This poses an interesting question of whether specificity and promiscuity in antibodies are dictated by shared residues that are involved in binding to both high- and low-affinity ligands.
Many murine homeostatic intestinal IgA antibodies are also polyreactive and have broad but defined reactivity toward gut microbiota.16 High-affinity ligands for these antibodies have not been identified. Still, they possess innate-like recognition properties that may facilitate adaptation to the diverse and dynamic array of gut dietary antigens and microbiota. The human anti-viral IgG antibodies and the murine homeostatic intestinal IgA antibodies provide two different datasets with unique origins and antigenic pressure for generation of similar polyreactivity; using these datasets, we aim to study the fundamental molecular principles that contribute to the unique and understudied phenomenon of polyreactive antibody binding.
Previously, it has been proposed that polyreactive or germline antibodies are more flexible in their CDR3 region than the classic monoreactive or antigen-experienced antibodies.15,17,18 However, this model has been challenged, including a high-throughput analysis of thousands of CDR H3 models that show no clear delineation in the flexibility between naive and antigen-experienced antibodies.19,20 Furthermore, such flexibility/plasticity may not be the only structural explanation for polyreactivity, and so far, evidence is lacking that CDR H3 alone is responsible for polyreactive interactions. In addition to conformational flexibility, other factors that have been suggested to play a role in antibody polyreactivity include CDR3 lengths, net hydrophobicity, and charged patches at the binding surface.21–28 Some studies have found an increased prevalence of arginine, glycine, valine, tyrosine, and tryptophan as both individual residues and/ or as a part of motifs in polyreactive antibodies.23–26 Much progress comes from in silico approaches that analyze, detect, and predict polyreactivity in large datasets of antibody sequences derived from natural and synthetic sources.21,28–30 In particular, one large study by Harvey et al. as well as our previous study by Boughter et al. show that amino acid contributions to polyreactivity are highly position dependent and more nuanced than previously described.21,30 Therefore, a need for a more detailed, experiment-based approach proves to be necessary.
In this work, we use experimental and in silico techniques to study polyreactivity in atomistic detail. We measure the direct affinity of polyreactive antibodies to structurally and chemically diverse ligands and, using extensive mutagenesis and polyreactivity ELISA assays, provide insight into the diverse ways a polyreactive antibody binds its many targets. Additionally, we solved crystal structures of five polyreactive and related monoreactive Fabs to investigate structural and biochemical properties involved in polyreactivity. Finally, using classical molecular dynamics (MD) simulations of solved polyreactive and monoreactive Fab crystal structures, we dissected the molecular motions inherent in these structures, and we observed increased rigidity of CDR loops of polyreactive Fabs.
RESULTS
Polyreactive Fabs engage some ligands with affinities similar to pMHC/alphabeta TCR interactions
To assess the binding affinity between polyreactive Fabs and their diverse ligands, we first selected six highly polyreactive murine IgA mAbs from a high-throughput polyreactive ELISA panel screen of hundreds of B cell clones derived from the gut mucosa.16 These six representative mouse monoclonal antibodies showed high binding toward all ELISA panel ligands (Figures 1A and S1A). Next, we extended the ELISA panel to include other well-characterized and smaller molecules that could be good targets for crystallization studies; this included lysozyme, ubiquitin, and short ssDNA. We observed similar binding to these ligands as we had seen for the polyreactive ligand panel (Figures 1B and S1B). We expressed recombinant Fabs from five of these six IgA mAb clones for monomeric binding studies and evaluated these Fabs’ binding to immobilized polyreactive ligands by biolayer interferometry (BLI). We measured binding to ssDNA (25 nt) with all selected polyreactive mouse Fab clones; no binding was present for a control monoreactive mouse IgG Fab (Figures 1C and S1C). A range of affinities between 22 and 187 μM was observed across these Fabs for ssDNA, orders of magnitude lower than typical antibody-antigen interactions and within the characteristic range of autologous TCR-MHC interactions.31,32 Binding between the polyreactive Fabs and lysozyme, ubiquitin, or even di-ubiquitin ligands could not be measured using BLI, suggesting these affinities are beyond the detection limits of this system.
Figure 1. Binding of selected polyreactive mouse IgA to polyreactive ligands.

(A) Polyreactivity ELISA OD405 values of selected mouse IgA mAbs (338E6, 43G10, and 41C10) tested at different concentrations against a polyreactivity panel of diverse antigens: DNA, insulin, lipopolysaccharide (LPS), flagellin, cardiolipin, albumin, and keyhole limpet haemocyanin (KLH). Horizontal dashed line shows cutoff OD405 for positive reactivity.
(B) ELISA OD405 measurements of additional ligands: lysozyme, ubiquitin, and 50-nt ssDNA, against the 43G10, 41C10, and 338E6 mAbs, tested as in (A).
(C) BLI binding plot between immobilized biotinylated 25-nt ssDNA and the Fabs of polyreactive mouse IgA at different concentrations (1 μM–1 mM range). Traces were reference subtracted, and kinetic parameters were calculated by fitting the data globally with Forte bio build-in analysis software (fit curves are shown as gray lines).
Single-alanine mutagenesis of polyreactive Fabs uncovers diverse binding interface residues
Next, we were interested in which residues in polyreactive antibodies are involved in binding to diverse ligands. For a comprehensive analysis, we mutated all individual CDR residues to alanine in the full-length polyreactive mAbs of the previously mentioned six Fab clones and assessed the effects of these mutations on their polyreactivity using ELISA. This analysis revealed a number of individual residues with apparent contributions to polyreactive binding (Figures 2A–2C and S2A–S2C). These include a variety of distinct amino acids and residues deriving from each of the CDR1, 2, and 3 regions of both heavy and light chains. Notably, mutations that disrupted polyreactive binding to one antigen typically resulted in decreased binding to all tested antigens, suggestive of shared mechanisms of polyreactive antibody recognition across structurally diverse antigens. Previously, several features were suggested to relate to antibody polyreactivity: the role of the individual amino acids as well as motifs in CDR loops, a bias toward contributions from the heavy chain, and long CDR3 loops.21–28 Across the six antibodies examined, we were unable to identify generalizable patterns of recognition shared across all antibodies; instead, each antibody showed a distinct pattern of contributing residues (Figures 2A–2C and S2A–S2C). Some tyrosine or arginine single mutations did indeed disrupt binding to all ligands for individual clones, but there was no general strategy shared by all Fabs for polyreactive binding. While this analysis does not test for backbone interactions of the polypeptide chain and will miss cooperative binding across multiple side chains, it does indicate that the binding to each ligand likely uses overlapping or shared epitopes on the Fab binding surface.
Figure 2. Alanine scanning of polyreactive mouse IgA mAbs and their reactivity to ligands.

(A–C) Summary plot of polyreactivity ELISA binding of three selected mouse IgA mAbs: (A) 338E6 mAb, (B) 43G10 mAb, and (C) 41C10 mAb, where selected residues of the CDR loops were mutated to alanine. All mutants were subjected to ELISAs against a panel of polyreactive antigens: DNA, insulin, LPS, flagellin, cardiolipin, albumin, and KLH. Native amino acids are shown on top, mutated to alanine (below). Results are color-coded based on a red (meaning more monoreactive) to blue (meaning more polyreactive) scale (A, on right), measured as percentage of wild-type mAb. Positions that were originally alanine were left unmutated.
Structure-based analysis of polyreactive antibodies identifies a relatively flat binding interface
Our previous bioinformatic study to characterize polyreactivity in a high-throughput manner showed promising clues: namely that on average, polyreactive antibodies are more neutral in charge and hydrophobicity and have a propensity for increased CDR-loop crosstalk.21 However, the precise physical manifestation of this crosstalk is unclear from this bioinformatic analysis. To extend our bioinformatic analysis with atomistic details, we investigated the structure of a panel of polyreactive antibodies to gain insight on how the binding interfaces were constructed at a three-dimensional level. To this end, we determined the three-dimensional structures of a selection of the aforementioned polyreactive mouse IgAs as well as human anti-influenza broadly neutralizing IgGs. One of these anti-influenza IgGs, in addition to being highly specific to hemagglutinin (HA) stalk domain on influenza A viruses, was also shown to be polyreactive.11,15 The antibodies we selected for this study come from two distinct sources (mouse/human, IgA/IgG, commensal/anti-viral) and were scored for polyreactivity using ELISA. Our aim was to focus on the most highly polyreactive clones from each group, to ensure broadness and generality while keeping the molecular detail of this study. We solved a total of five high-resolution crystal structures: three polyreactive Fabs (left) and two monoreactive Fabs (right) by X-ray crystallography (Figure 3A, Table S1).
Figure 3. Structural features of polyreactive and monoreactive Fabs.

(A) Side view of crystal structures of the polyreactive mouse 338E6 (PDB: 8FZO), mouse 43G10 (PDB: 8FZP), human 2G02 (PDB: 8G1B), and monoreactive human 3B03 (PDB: 8G1C) and human 4C05 (PDB: 8G19) Fabs, shown in cartoon representation. The variable heavy-chain (VH) and variable light-chain (VL) domains are colored in dark blue and light blue respectively.
(B and C) (B) Top view of surface representations of these five Fab structures highlighting Adaptive Poisson-Boltzmann Solver (APBS) electrostatics of their antigen-binding site with positive charges in scaled blue and negative charges in scaled red (APBS scale shown at bottom of figure). (C) Top view of surface representations of the antigen-binding regions (including the CDR loops) of these five Fab structures highlighting Eisenberg scale of hydrophobicity with low hydrophobicity shown in scaled teal and high hydrophobicity in scaled mauve (hydrophobicity scale at bottom of figure).
The Fab structures ranged from having short CDRs forming a flat antigen-binding surface (polyreactive 338E6 and mono-reactive 3B03) to longer CDRs, extending the antigen-binding site (polyreactive 43G10 and 2G02). Additionally, CDR H3 on the monoreactive 4C05 shows a structured β ribbon similar to those seen in a subset of bovine Fabs with a long stalk atop which sits a knob domain.33 To investigate previous suggestions that polyreactive Fabs have an overwhelming positive antigen-binding surface,18,23,28 we inspected the antigen-binding site of solved crystal structures for any hydrophobic or electrostatic patches in a surface view. No predominant pattern was present in charge or hydrophobicity compared between poly- and closely related monoreactive Fabs (Figures 3B and 3C). In conclusion, these structures demonstrate that polyreactivity can be achieved through a wide range of convergent structures and biochemical compositions (hydrophobicity, charges, etc.).
Molecular dynamics analysis of polyreactive antibodies reveals a more rigid CDR landscape
An additional feature proposed to align with polyreactive antibodies in contrast to their monoreactive counterparts is an increase in conformational flexibility of their proposed binding sites.17–20 These studies limited their focus to the CDR3 loop in the heavy chain, their antibodies derived from a single source (i.e., therapeutic or human), or their MD relied solely on in silico predicted structural models in lieu of experimentally derived structures determined by methods such as X-ray crystallography or nuclear magnetic resonance (NMR). To assess the flexibility of polyreactive antibodies, we used the three-dimensional structures of the polyreactive Fabs we determined as starting points for all-atom MD simulations. We generated simulations for the three polyreactive and three monoreactive antibodies in triplicate, generating 12 ms of simulated data. Additionally, we incorporate four previously crystallized antibodies (CH65, PDB: 4WUK; 1F02, PDB: 6B3M; CR9114, PDB: 4FQH; and F16, PDB: 3ZTJ)34–37 that have been deemed to be polyreactive to expand our comparison set. Our analysis for each Fab follows a customized pyEMMA and MDtraj workflow that includes backbone rootmean-square fluctuation (RMSF) or root-mean-square deviation (RMSD), and time-lagged independent component analysis (tICA), followed by visual inspection of all distinct structural conformations of a simulated Fab (Figure 4A). Additional simulation and analysis details can be found in the STAR Methods.
Figure 4. All-atom MD simulations and analysis on example human anti-flu polyreactive vs. monoreactive IgG.

(A) Workflow of all-atom MD analysis (500 ns), including clustering of CDR conformations via time-lagged independent component analysis (tICA) (1), visual inspection of CDR clusters (2), and RMSD (Å) of Cα backbone movements in CDR loops (3).
(B) From left to right: side view of the crystal structure of the polyreactive human 2G02 Fab shown in cartoon representations; tICA analysis based on 500 ns of all-atom MD simulations, where color dots represent the distinct cluster centers identified in tICA space using a K-means algorithm; representative CDR loop structures from the MD analysis, colored according to the cluster dot color in the tICA analysis; and RMSD (Å) of the Cα backbone movements across the 500-ns MD trajectory of each of the CDR loops.
(C) Same analysis as in (B) of the monoreactive human anti-flu 3B03 Fab structure.
In our dynamic analysis, we observed that the polyreactive Fab structures are overall relatively rigid compared with monoreactive Fabs. tICA analysis for polyreactive Fabs shows consistently fewer clusters of distinct conformations suggesting overall more static behavior (Figures 4B and 4C). Occasionally, a global shift is present in polyreactive Fabs, which comes from global scaffold movements of the Fab, but the CDR loops themselves do not change much in conformation. For example, a single conformational change is present in polyreactive 2G02 Fab in the CDR L1 that persists for the remainder of the simulations based on RMSD and visualizations (Figure 4B, gray RMSD plot). However, that loop’s overall shape is conserved, suggesting that the original state captured in the crystal structure might differ from the relaxed version of the protein in solution. These trends persist over all polyreactive antibodies tested and across simulated triplicates for each Fab (Figures S3–S5).
Conversely, the monoreactive Fabs show more distinct CDR conformations and higher RMSD. The CDR H2 in the monoreactive 3B03 Fab adopts distinct conformations and many exchange events between these conformations, indicative of increased flexibility of the heavy chain relative to polyreactive Fabs (Figure 4C, green RMSD plot). Similarly, CDR H3 in the monoreactive 3B03 Fab adopts many conformations as opposed to the polyreactive 2G02 Fab. This observation is especially intriguing, given that both Fabs are also highly specific for the central stalk epitope of HA and use CDR H3 to mediate that high-affinity interaction. Across all tested antibodies, these patterns persist, with monoreactive antibodies consistently demonstrating higher CDR flexibility (Figures S6–S8). These local dynamic differences reveal overall a more rigid polyreactive binding interface, suggestive of a polyreactive binding mode that takes advantage of low entropic penalties to binding.
To expand upon our initial dataset of crystallized structures, we added the polyreactive antibodies 1F02, CH65, CR9114, and F16 to our set of simulated structures, generating an additional 3.6 μs of simulated trajectories. Antibodies CH65 and CR9114 appear to be nearly as rigid as other polyreactive antibodies, while F16 and 1F02 appear slightly more flexible at first glance (Figure S9). However, comparing the RMSF of all tested antibodies (Figure S10), we see that antibodies 1F02 and F16 are each more rigid than the least flexible monoreactive antibody (3B03). These results highlight the need to utilize a range of flexibility metrics to characterize the precise nature of antibody flexibility. While RMSD may capture large global conformational changes, it is largely incapable of identifying rapid fluctuations of specific residues. RMSF, on the other hand, can identify these fluctuating residues, but it is incapable of discerning whether these fluctuations generate meaningfully different interfaces for antigen recognition. Only those antibodies that display both a high RMSD and RMSF, as in the case of the monoreactive antibodies 4C05 and 3B03, exhibit meaningful flexibility that will play a crucial role in target recognition. Taking these simulated trajectories of antibody CDR loops as an ensemble of the most flexible regions of both polyreactive and monoreactive antibodies, we find that overall, the CDR3H and CDR3L regions of polyreactive antibodies display increased conformational rigidity when compared to their monoreactive counterparts (Figure S11).
Extensive three-dimensional bond network present in the rigid CDR loops of polyreactive Fabs
To further dissect the molecular basis of the observed polyreactivity of these Fabs, we focused on determining which factors contribute to CDR rigidity in polyreactive antibodies versus CDRs with increased flexibility in monoreactive Fabs. We were particularly interested in how the extended CDR L1 in the polyreactive 43G10 maintains its rigidity despite no stabilizing secondary structures and how the CDR H3 in monoreactive 4C05 shows flexibility despite forming a structured β ribbon (Figures 5A and 5B). Upon closer inspection of the intrachain interactions of these two Fabs CDRs, both showed striking hydrogen bond networks mediated by CDR residues. A long, unstructured CDR L1 loop in the polyreactive 43G10 is extensively supported by seven side-chain H-bonds, which together form a stable three-dimensional network. In contrast, the structured CDR H3 of the monoreactive 4C05 forms its vast H-bonds between the β ribbon’s backbones (except two H-bonds at the tip, between Ser104 and Asp106), allowing only for two-dimensional stability. This observation raises an interesting hypothesis that side chains in 43G10 CDR L1 could engage in the stability of the loop rather than in formation of specific interactions with the ligand. Conversely, the exposed side chains in the monoreactive 4C05 CDR H3, along with their biochemical properties, could infer specificity in ligand binding and might not favor promiscuity.
Figure 5. Examples of hydrogen bond network within CDR loops of polyreactive vs. monoreactive Fabs.

(A) Highlighted regions of the polyreactive 43G10 Fab structure showing the CDR1 loop of the light chain. This CDR loop maintains an extensive hydrogen bond network, shown as yellow dotted lines, mostly mediated by side chains throughout 1 μs of MD simulations. Red atoms indicate oxygens, and blue indicate nitrogens. Shown beneath the structure is the RMSD in Å of the Cα backbone movements across a 1-μs trajectory.
(B) Same analysis as in (A) but with the monoreactive 4C05 Fab structure, specifically highlighting the CDR H3 loop, which forms a β ribbon with the H-bond network (shown in dashed yellow) mostly mediated by backbone throughout 1 μs of MD simulations.
(C) Highlight of the polyreactive 2G02 CDR H3 loop, showing the mostly side-chain-mediated hydrogen-bonding network that maintains its structural stability and the two arginines at positions 100 and 101, which when mutated to lysines reduce polyreactivity.15
(D) Root-mean-square fluctuation (RMSF) of positions 97–113 of the wild-type (polyreactive, shown in blue) and RR/KK mutant (reduced polyreactivity, shown in purple) 2G02 H3 loop. The presented RMSF is a bootstrapped average over triplicate trajectories for each antibody, with the standard deviation given as shaded regions about this average. Asterisks identify statistically significant differences (p < 0.05) calculated using a non-parametric permutation test (STAR Methods).
We looked for other examples of rigidity in the CDRs of polyreactive antibodies and noticed a similar, primarily side-chain-mediated, network in CDR H3 of polyreactive 2G02 (Figure 5C). Here, seven H-bonds stabilize this loop, where the positively charged Arg100 faces inward in the loop. Interestingly, this same Arg100 and neighboring Arg101, when both mutated to Lys (along with five other germline residues) in 2G02, resulted in reduced polyreactivity as measured by the polyreactivity ELISA.15 Concurrent with the reduced polyreactivity, we also see increased conformational dynamics in the CDR H3 of the 2G02 double Lys mutant in our dynamic studies compared with wild-type 2G02 (Figure 5D). This change in rigidity could come from losing the guanidinium group on Arg that can form a three-dimensional H-bond network in place of the two-dimensional ε-ammonium group in Lys. When comparing the RMSF across all tested antibodies, we see that the 2G02 mutant is significantly more rigid than the other monoreactive antibodies but more flexible than nearly all polyreactive antibodies (Figure S10). These subtle observations suggest that the maintenance of overall rigidity in polyreactive antibodies may be a sensitive feature, with even slight deviations resulting in a loss of polyreactivity.
DISCUSSION
In this work, we aimed to understand the molecular basis of polyreactivity in antibodies and to determine any unifying patterns that dictate whether a Fab will be monoreactive (highly specific) versus polyreactive (binding to diverse targets with low affinity). We were able to determine the affinity of multiple polyreactive Fabs to one of the polyreactive ligands, ssDNA (KD = 22–187 μM), which is on the lower affinity end of measurable protein-protein interactions, however in agreement with previously published values by Ausserwoger et al. using a microfluidic approach.27 To put this in context, these KDs are within the range of those measured for conventional αβ TCRs to their cognate agonist ligands presented by MHC molecules.31,32 Furthermore, many self-pMHC-TCR interactions, such as those that occur during positive selection in the thymus, are even weaker and difficult to measure with conventional direct binding methods. Considering that antibodies have increased valences and can form higher-order self-complexes depending on the Fc region utilized, the final avidity of a polyreactive interaction can be significantly amplified; therefore, valency likely plays an important role in polyreactive associations. This property can be especially beneficial in therapeutic antibodies; i.e., once in plasma or administered by subcutaneous (SC), higher valency can increase its target engagement, longer half-life, and therefore enhanced clearance of antigens. However, it is essential to strike a balance between binding affinity and other factors, as there could also be some risks associated with high affinities that can lead to off-target effects or reduced tissue penetration. All these factors should be carefully considered in the development of therapeutic mAbs.
Additionally, despite other groups reporting specific motifs and patterns characteristic of polyreactive antibodies,14,17–20,28,29 no single generalizable pattern in sequence or structures between polyreactive and monoreactive Fabs were identified in our own work and other large bioinformatic analyses.21,30 This supports a recognition strategy model akin to evolutionarily convergence whereby polyreactive antibodies utilize different molecular strategies to achieve a similar outcome: binding to their polyreactive ligands, and polyreactive binding in general, can be clone specific rather than ligand specific. Along these lines, single alanine substitutions often reduce binding across multiple ligands, suggesting that common residues on a particular Fab may be used to engage diverse ligands.
Our crystal structures of polyreactive Fabs provide an experimentally determined starting structure for our MD simulations. In this analysis, we observed that polyreactive antibodies tend to be more rigid than their monoreactive counterparts, as they adopt fewer conformations in their CDRs. We also identified multiple examples of events that enhance rigidity in the CDRs of polyreactive Fabs, including the formation of stabilizing structures like hairpins and an increased network of side-chain-mediated stabilizing hydrogen bonds. While this contrasts with the model whereby enhanced dynamics can provide an adaptive “induced fit” for diverse targets, we propose that a more rigid, flat surface with exposed backbone atoms may provide an “inoffensive” surface that can provide a large, inclusive binding surface for many different types of biochemical engagements. Furthermore, examples from other receptor systems suggest that the binding of structurally diverse ligands does not always require significant conformational changes of the receptor. For example, NKG2D, a conserved activating receptor, binds to several highly divergent ligands including, in humans, the MHC-class-I-polypeptide-related sequence A and B and the cytomegalovirus UL16-binding proteins using similar binding sites.38 NKG2D uses a set of tyrosine residues in its binding site, which through different rotamer conformations, form intermolecular interactions with distinct residues on these divergent ligands in a “rigid adaptation” model of engagement.39
Our previous research focused on developing a bioinformatic tool to identify biophysical characteristics of polyreactivity, and we have built a classifier to identify polyreactivity based on amino acid sequence.21 These previous results, also independently validated by Harvey et al., agree well with our findings here, namely that there are few, if any, uniform sequence features that determine antibody polyreactivity.21,30 In large sequence databases and in the structures presented here, there do not appear to be strong enrichments for specific amino acids, large patches of hydrophobic or positively charged amino acids, or biases toward longer CDR loops. Instead, polyreactivity appears to be far more nuanced than any single metric. Our previous bioinformatic results suggest that there is a move toward neutrality of the binding interface, neither strongly hydrophobic nor hydrophilic, nor excessively charged, consistent with our present observations of side chains of CDRs being engaged in stabilization rather than exposed to solvent where they could participate in direct ligand binding. Moreover, we also found an increase in inter- and intra-loop crosstalk between residues in polyreactive antibodies, quantified using mutual information, which may physically manifest as the increased rigidity that we observed in our all-atom MD simulations.
Interestingly, this increased rigidity and loop crosstalk is somewhat at odds with what has been suggested in the field previously. While multiple studies have speculated that flexibility may be a key feature of polyreactive antibodies, two notable studies stand out in their efforts to quantitatively characterize this flexibility.15,17 Prigent and Jarossay et al. utilize a careful thermodynamic approach and classical MD simulations initiated from crystal structures and determine that polyreactive anti-HIV-1 antibodies are more flexible than their monoreactive counterparts. The authors find that the activation entropy, which importantly is distinct from the net change in entropy due to binding, is positive for monoreactive antibodies and negative for polyreactive antibodies. However, the absolute magnitude of these entropic changes is important to consider. While the activation entropy of the monoreactive antibodies is favorable, it is large in magnitude (~60 kJ/mol), strongly contrasting with the much smaller unfavorable activation entropy of the polyreactive antibodies (~10 kJ/mol). This may be reinterpreted to suggest that a favorable change in entropy is less critical for polyreactive binding, suggestive of rigid CDR loops. While the simulations reported alongside these thermodynamic experiments were rigorously analyzed, we believe our extended analysis, with trajectories three orders of magnitude longer, may more effectively capture the dynamics of these polyreactive antibodies.
Guthmiller et al. utilize a computational enhanced sampling protocol to assess flexibility, finding that modeled structures of polyreactive antibodies sweep out a broader region in a projected space, with more shallow energetic wells.18 This is interpreted as an increase in flexibility of these polyreactive antibodies; however it is unclear that these projected landscapes necessarily correlate with antibody flexibility. As we found, a similar projection strategy (Figures S3 and S6) can show multiple distinct conformational islands that are largely dependent on framework region flexibility, while the CDR loop itself remains rigid (as in the case of 2G02 and CH65). Further, the use of modeled structures may be inappropriate for careful study of TCR and antibody structures. As we have found, the difference between polyreactive and monoreactive antibodies may lie in the intricate side-chain networks formed between CDR loops. While structural modeling has progressed significantly in its ability to predict backbone conformations, side-chain modeling remains a challenge.40 Inaccurate side-chain prediction could completely alter the relevant dynamics of the antibodies discussed in this study. Lastly, it is unclear if enhanced sampling is a proper tool to assess the extent of antibody flexibility. While enhanced sampling is a powerful approach to explore the possible conformational space of a protein, it is unclear whether these conformations would ever be sampled in a physiological setting. Enhanced sampling effectively lowers energy barriers between conformational states, but in vivo, the barriers between these states may be practically insurmountable. In further support of our findings of rigidity in polyreactive antibodies, some of the longest antibody simulations to date of the polyreactive antibody CH65 likewise find a rigid binding interface, despite long CDR loops.41
Our results suggest that alternative binding models for polyreactivity should be explored. While most binding models are geared toward high-affinity interactions, antibody polyreactivity may be best explained by a comparison to TCR engagement of classical MHC, which shares similarities in the inherent low-affinity and promiscuous engagement of multiple ligands. MHC-TCR complexes present a spectrum of dynamic behaviors whereby their responses are based on affinity, on and off rates, and ligand concentrations. It has been demonstrated that pairs with slower off rates and lower ligand concentrations fit the conformational selection model, and those with fast on rates are better explained by the induced-fit model.42–44 Some MHC-TCR pairs also display a combination of both models proposed as conformational melding.45 As we observed with our polyreactive Fabs, there is no single model to explain their ability to engage at low-affinity, multiple chemically and structurally distinct epitopes. These models we have referenced will be more appropriately applied to antibody polyreactivity when structural data of these complexes are achieved, perhaps through methods more sensitive and appropriate for low-affinity interactions such as NMR.
Finally, a more functional understanding of the selection of polyreactivity in vivo poses an attractive area for improving antibody-based vaccines. On one side, polyreactivity in antibody-based drug candidates has long been a highly undesirable characteristic, because its binding promiscuity can elicit detrimental immune reactions in clinical practice. However, more recent evidence has emerged that antibodies with “aberrant” (polyreactive) behavior could offer novel types of therapeutics that may be applicable in distinct pathological conditions.46 Thus, understanding how to manipulate the binding interface of an antibody to manage its polyreactivity will have important applications for their therapeutic value.
Limitations of the study
In this work, we provide molecular insights into polyreactivity using biochemical and biophysical studies based on crystallographic structures of antibodies. We chose this approach because it allows us to explore the molecular basis of antibody polyreactivity. However, this detailed approach comes with sample size limitations due to the challenging nature of obtaining high-quality crystals suitable for structure determination. Additionally, to represent the breadth of polyreactivity more accurately, we sourced our antibodies from two species, having different functions and specificities; however, the limited number of antibodies examined using this approach may not fully represent the diversity and complexity of the human/mouse antibody repertoire. Furthermore, our all-atom molecular dynamic simulations (more extensive compared to previously published MD simulations of antibodies) have short timescales compared to the relevant binding (kon and koff) timescales and are based on assumptions of all-atom force field accuracy and a classical interpretation of atomistic interactions. Finally, the use of ELISA as an experimental technique has inherent limitations and may not fully capture the complexity of antibody polyreactivity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Erin J. Adams (ejadams@uchicago.edu).
Materials availability
Plasmids generated in this study are available upon request.
Data and code availability
Crystal structures were deposited in the RCSB database under following PDB IDs: 8FZO, 8FZP, 8G19, 8G1B, 8G1C.
All processed data are freely available online via a Zenodo repository linked through the GitHub page associated with this manuscript (discussed further in the next point). Raw trajectory files are too large (~10 TB) to post on publicly accessible repositories but are available from the lead contact upon request.
All relevant code for data normalization, processing, and statistical analysis are available on the GitHub page (https://github.com/ctboughter/manuscript_md_analysis). Instructions for use and relevant packages required for the analysis are provided within the ReadMe file and documentation page.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines and culture conditions for protein expression
Bacterial cultures for plasmid amplification E. coli DH5α and for protein expression BL21(DE3)pLysS were grown in Luria Broth (Thermo Fisher) media supplemented with appropriate selection antibiotic ampicillin or kanamycin (Sigma) and maintained at 37 °C at ambient CO2 with shaking at 250 rpm.
Baculovirus was generated in Spodoptera frugiperda (Sf9) cells maintained in Sf-900 III SFM (Gibco) supplemented with 10% FBS Superior (MERCK, Biochrom). For insect protein expression, Trichoplusia ni (High Five) cells grown in Insect-XPRESS Protein-free Insect Cell Medium with L-Gln (Lonza) were infected with baculovirus. Insect cells were maintained at 27 °C at ambient CO2 with shaking at 120 rpm.
Expi293 suspension cells (Thermo Fisher) were maintained in Expi293 Expression Medium (Thermo Fisher) on a 120 rpm shaking platform at 37°C in a 5% CO2 humidified incubator. Protein was produced by transient transfection using ExpiFectamine (Thermo Fisher).
METHOD DETAILS
Constructs design
For mouse Fab clones: variable domain of both heavy and kappa chains of mouse IgA clones were amplified from plasmids provided by Albert Bendelac. Similarly, both chains of modified IgG constant domain amplified from plasmids gifted by Anthony Kossiakoff were fused with corresponding variable domains by overlap extension PCR. Both chains were cloned with sequence and ligation independent (SLIC) method in a pACgp67a vector with a 3C protease sequence, either basic or acid zippers and a C-terminal 6x histidine tag.
DNA for human Fab clones were obtained and unmodified from Patrick Wilson. DNA for ubiquitin used in ELISA assay was obtained from Matthew L. Wohlever (The University of Toledo).
Protein expression and purification
Baculoviruses with all hybrid mouse Fab chains were prepared by transfecting and amplifying into Sf9 cells. Mouse Fabs were expressed in the Hi5 cells by co-infecting with baculoviruses containing both chains and incubating up to 3 days. Fabs were purified from the supernatant using Ni-NTA agarose in buffer A, o/n 4°C 3C protease cleavage, second Ni-NTA subtractive step and size exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare).
Human Fabs were transfected into suspension Expi293 cells (ThermoFisher) using Expifectamine and according to manufacturer’s protocol. Supernatant was harvested after 5 days, spun down for 15 min at 4°C/3,000 ×g and incubated o/n with 1 mL bv Ni-NTA agarose in buffer A (10mM HEPES 7.2, 150mM NaCl) with f.c. 20 mM imidazole pH 7.2. Next day after washing the column with high salt buffer A supplemented with 20 mM imidazole, then eluted in 5 mL of buffer A with 350 mM imidazole. Samples were further purified by size exclusion chromatography in buffer A (Superdex 200 10/300 GL, GE Healthcare).
Ubiquitin construct was heat shock transformed into BL21(DE3)pLysS and single colony was used to inoculate 25 mL terrific broth (TB) starter culture supplemented with ampicillin, grown overnight at 37°C and 250 rpm shaking. 1L TB/Amp culture was inoculated with 1% of starter culture, induced with 1 mM IPTG at OD600 and continued to grow for 4 h/37°C with 250 rpm. Cells were harvested by centrifugation and resuspended in 50 mL buffer A with high salt (10 mM HEPES pH 7.2, 500 mM NaCl supplemented with 1 mM PMSF and 2 mM DNase I). Cells were lysed by passing three times through a high-pressure microfluidizer (Avastin), soluble fraction was collected by centrifugation for 40 min at 4°C/18,500 ×g and 2mL bv Ni-NTA beads, f.c. 20mM imidazole 7.2 were added for o/n incubation. Next day after washing the column with high salt buffer A supplemented with 20 mM imidazole, protein was eluted in buffer A with 10 c.v. 350 mM imidazole. Typically, the protein was further purified by size exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare) in buffer A. Peak fractions were pooled, concentrated to 10 mg/mL in a 3-kDa MWCO Amicon Ultra centrifugal filter (Millipore).
All protein concentrations were determined by A280 using a calculated extinction coefficient.
ELISA
Polyreactivity assays were performed as described.5,11,13 ELISA plates (Thermo) were coated overnight with 50 mL/well antigen diluted in carbonate buffer (Bethyl) except for cardiolipin, which was coated overnight in 100% ethanol left uncovered to allow evaporation. The following antigens and concentrations were used for coating: calf thymus DNA (Life Technologies), 10 mg/mL; human insulin (Fitzgerald), 5 mg/mL; LPS from E. coli (Sigma), 10 mg/mL; flagellin from S. typhimurium (InvivoGen), 2 mg/mL; cardiolipin (Sigma), 10 mg/mL; albumin from human serum, low endotoxin (Sigma), 10 mg/mL; and KLH, endotoxin-free (Millipore), 10 mg/mL; lysozyme from chicken egg white (Sigma), 10 mg/mL; ubiquitin (inhouse) 10 mg/mL; 50 nt ssDNA (IDT) with sequence: ATACCTTGCAGAAATCGAGGCCGTTCGTTAATTCCTGTTGCATTCGTACC, 5 mg/mL. Plates were washed 4x with nanopure H2O with an ELISA plate washer (BioTek) and blocked with 150 mL/well blocking buffer [1X TBS-T (Thermo), 1 mM EDTA (Boston BioProducts)] for 1 h at 37°C. Plates were washed 4x with H2O using an ELISA plate washer (BioTek), and 50 mL/well of mAbs pre-diluted in TBS pH 7.4 were added. Different mutants of mAbs were assayed at 1 mg/mL and three additional 1:4 dilutions. Plates were incubated at 37°C for 1 h then washed 4x with H2O. 75 mL/well of goat anti-human IgG HRP (Southern Biotech) diluted in blocking buffer were added, plates were incubated 1 h at 37°C, then washed 4x with H2O. 150 mL/well of blocking buffer was added, and plates were incubated for 5 min at room temperature. Plates were washed 4x with H2O, and 100 mL/well of developing reagent was added (Super AquaBlue ELISA Substrate, eBioscience). Plates were monitored at OD405 using an ELISA plate reader (BioTek), and reading was stopped when the positive control reached an OD405 of ~3.0. Average OD405 of TBS-only wells was subtracted from mAb containing wells for analysis.
Binding studies
All the binding studies were carried out using Bio-layer interferometry (BLI) (either Forte bio or Octet K2 from Pall Life Sciences) at room temperature (22°C). Biotinylated 25 nt long ssDNA (from IDT, sequence ATACCTTGCAGAAATCGAGGCCGTT) was immobilized on a Streptavidin sensor to reach ~2–3 RU for binding with mouse IgA Fab clones at different concentrations (1 μM-1 mM range), and responses (in nm) were recorded. HEPES buffer saline (10 mM HEPES pH 7.2, 150 mM NaCl) was used in all the measurements, and sensor tips were recharged with 0.1 M Glycine, pH 2.5 after each round. Traces were reference subtracted, and kinetic parameters (KD, kon, koff) were calculated by fitting the data globally with Forte bio build-in analysis software (BLItz Pro 1.1.0.25).
Crystallography
Crystals of different Fab clones and single-chain ligand-Fab complexes were grown at room temperature by sitting drop vapor diffusion. Equal molar volumes of a protein solution containing 10–15 mg/mL complex were mixed with a reservoir solution containing different salts and PEGs. Crystallization conditions and cryoprotection are shown in the table below.
Crystallization conditions and data collection
| Crystal | Crystallization condition | Cryoprotection |
|---|---|---|
| 338E6 Fab | 29% PEG 3000, 0.1M Sodium Sulfate, 0.1M Tris pH 8.5 (optimization) | 20% Glycerol |
| 43G10 Fab | 16% PEG 4000, 0.2M Ammonium sulfate, 0.1M HEPES pH 7.5, 10% Isopropanol (optimization, seeding) | 20% Ethylene Glycol |
| 2G02 Fab | 20% PEG 3350, 0.2M Magnesium formate (directly from HT screen) | 40% PEG 3350 |
| 3B03 Fab | 25% PEG4000, 0.2M Ammonium sulfate, 0.1M Sodium acetate pH 4.6 (optimization) | 20% Glycerol |
| 4C05 Fab | 30% PEG 5000 MME, 0.2M Ammonium sulfate, 0.1M MES pH 6.5 (directly from HT screen) | 20% Ethylene Glycol |
Each dataset was collected from a single crystal region for all protein samples at 100 K at APS beamlines 23ID and 24ID (λ = 0.9791 Å) on an Eiger detector. The data were processed, scaled, and merged using iMosfilm, truncated with ccp4, and a search model using PHASER as a molecular replacement tool was done with PHENIX.48,52 Cycles of manual model building, and refinement (torsional non-crystallographic symmetry restraints applied throughout) were done with COOT and PHENIX.52,53 Data collection and processing statistics are listed in Table S1. Structure figures were generated with PyMOL (http://www.pymol.org).
All-atom MD simulations
All simulations performed were prepared using the CHARMM-GUI Input Generator.53–56 All Fab structures were fully hydrated with TIP3P water molecules and neutralized with 0.15 M KCl. All simulations were carried out in simulation boxes with periodic boundary conditions (20) using the additive PARAM36 force field from the CHARMM (Chemistry at HARvard Macromolecular Mechanics).55 Simulations ran for ~500 ns, with a 2fs time step at 300.15 K and used a combination of NAMD and AMBER.56 For all simulated systems run on the Midway Computing Cluster at the University of Chicago, at least two replicas were run to confirm the results’ independence on initial velocity assignments. One simulation was extended to a full 1 μs to search for significant deviations over time, which did not occur. Data were analyzed using a customized pyEMMA and MDtraj package installed within the Jupyter notebook. All analysis scripts are available on GitHub (https://github.com/ctboughter/manuscript_md_analysis)
QUANTIFICATION AND STATISTICAL ANALYSIS
Typically, RMSD and RMSF are not compared statistically, requiring the creation of a new statistical protocol to address this short-coming. Due to the inherent time-series dependence of the RMSD traces, a direct statistical comparison is difficult, and as such all RMSD traces are generated using standard packages within MDtraj. We can, however, take advantage of the fact that RMSF averages fluctuations over time, abrogating the time-series dependence of the data. We can calculate this RMSF in windows over the trajectory (4ns per window) and use these discrete RMSF measurements to bootstrap RMSF averages and standard deviations across replicates of the same molecular systems. We can use these same discrete RMSF measurements for statistical comparison between simulations utilizing a non-parametric permutation test to shuffle these windows across simulations in the generation of our permutations. All bootstrap and permutation calculations entail 10,000 resampling steps with or without sample replacement, respectively, to generate final values.
This protocol for assessing statistical significance incorporates an important assumption, namely that sites on a given CDR loop are exchangeable across the tested antibodies. While this assumption is likely valid for comparisons between antibody 2G02 and its mutant counterpart (as in Figure 5D), the dynamics of each individual CDR loop across distinct antibodies will be strongly dependent on its local composition and structural context. To properly normalize each RMSF for comparisons across antibodies, we first subtract the N-terminal RMSF value from the entire trace (note that all traces in Figure S10 start at RMSF = 0.0). As we are interested in loop dynamics and not framework dynamics, this step acts to normalize for these antibody framework deviations. That the C-terminal RMSF likewise converges to zero is a sign that this normalization is valid. Second, we align the most flexible regions (as measured by RMSF) of each CDR loop to a position of 0 to compare peak flexibility of each antibody, controlling for those antibodies that are not most flexible in the exact middle of the CDR loop. The analysis of Figure S11 takes this one step further, combining the discrete RMSF values of each antibody CDR loop at these central regions and the two flanking positions on either side, and pooling them in either a monoreactive or polyreactive bin. These bins are then subject to the non-parametric permutation test to generate p value estimates. Again, all scripts and data used to generate these figures and tests are available on GitHub (https://github.com/ctboughter/manuscript_md_analysis).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
|
| ||
| E. coli BL21(DE3)pLysS | Laboratory of E.J. Adams | N/A |
| E. coli DH5α | Laboratory of E.J. Adams | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| StockOptions Salt Kit | Hampton Research | Cat# HR2-245 |
| StockOptions pH | Hampton Research | Cat# HR2-941 |
| NeXtal Stock PEG 3,000 | Molecular Dimensions (previously QIAGEN) | Cat# MD2-100-8 |
| NeXtal Stock PEG 3,350 | Molecular Dimensions (previously QIAGEN) | Cat# MD2-100-9 |
| NeXtal Stock PEG 4,000 | Molecular Dimensions (previously QIAGEN) | Cat# MD2-100-11 |
| NeXtal Stock PEG 5,000MME | Molecular Dimensions (previously QIAGEN) | Cat# MD2-100-19 |
| Sf-900 II SFM | GIBCO | Cat# 10902104 |
| Ultra-low IgG FBS | Invitrogen | Cat# 16250078 |
| FBS Superior | MERCK, Biochrom | Cat# S 0615 |
| Insect-XPRESS Protein-free Insect Cell Medium with L-Gln | Lonza | Cat# 12-730F |
| E. coli biotin ligase BirA | Laboratory of E.J. Adams | N/A |
| Expi Expression System Kit | Thermo Fisher | Cat# A14635 |
| Biotin | SIGMA-ALDRICH | Cat# B4501 |
| Calf thymus DNA | Life Technologies | Cat# 15633019 |
| Human insulin | Fitzgerald | Cat# 30AI51 |
| LPS from E.coli | Millipore-Sigma | Cat# L2630 |
| Flagellin from S. typhiumurium | InvivoGen | Cat# TLRL-STFLA |
| Cardiolipin | Millipore-Sigma | Cat# C0563 |
| Albumin from human serum, low endotoxin | Millipore-Sigma | Cat# A5843 |
| KLH, endotoxin-free | Millipore-Sigma | Cat# H8283 |
| Lysozyme from chicken egg white | Millipore-Sigma | Cat# SAE0152 |
| 50nt ssDNA | IDT | ATACCTTGCAGAAATCGAGGCCGT TCGTTAATTCCTGTTGCATTCGTACC |
| Biotinylated 25 nt s DNA | IDT | ATACCTTGCAGAAATCGAGGCCGTT |
| Goat anti-human IgG HRP | Southern Biotech | Cat# 2040-05; RRID: AB_2795644 |
| Super AquaBlue ELISA Substrate | Thermo Fisher (eBioscience) | Cat# 00-4203-56 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Nickel-nitrilotriacetic acid (Ni-NTA) resin | QIAGEN | Cat# 30250 |
| Superdex S200 | (GE Healthcare) | Cat# 17517501 |
| Blitz Biolayer Interferometry instrument (Forte Bio and Octet K2) | PALL ForteBio | N/A |
| Streptavidin biosensor tips | PALL ForteBio | Cat# 18-5020 |
| Amicon Ultra-4 Centrifugal Filter Units | Millipore Sigma | Cat# UFC803024 |
| ELISA plate washer | BioTek | N/A |
| ELISA plate reader | BioTek | N/A |
|
| ||
| Deposited data | ||
|
| ||
| 338E6 Fab structure | This paper | PDB ID: 8FZO |
| 43G10 Fab structure | This paper | PDB ID: 8FZP |
| 2G02 Fab structure | This paper | PDB ID: 8G1B |
| 3B03 Fab structure | This paper | PDB ID: 8G1C |
| 4C05 Fab structure | This paper | PDB ID: 8G19 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| S. frugiperda: Sf9 cell line | Laboratory of E.J. Adams | CLS Cat# 604328/p700_Sf9, RRID: CVCL_0549 |
| T. ni: High Five cell line | Laboratory of E.J. Adams | CCLV Cat# CCLV-RIE 0350, RRID: CVCL_C190 |
| Expi293 | Thermo Fisher | Cat# A14635 |
|
| ||
| Recombinant DNA | ||
|
| ||
| All mouse Fab clones | Laboratory of A. Bendelac | N/A |
| All human Fab clones | Laboratory of P. Wilson | N/A |
| Humanized Fab scaffold | Laboratory of A. Kossiakoff | N/A |
| Insect cell expression vector pACgp67A | Laboratory of E.J. Adams | N/A |
| E. coli expression vector pET28a | Laboratory of E.J. Adams | N/A |
| BestBac 2.0 Δ v-cath/chiA Linearized Baculovirus DNA | Expression Systems | Cat# 91-002 |
| Human ubiquitin | Laboratory of R. Keenan | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism (v5) | GraphPad | http://www.graphpad.com |
| CCP4 (v7.0.032) | Winn et al., 2011 | http://www.ccp4.ac.uk |
| Phenix (v1.11.1-2575) | Adams et al., 2010 | http://www.phenix-online.org |
| Coot (v0.8.7, part of CCP4 package) | Emsley and Cowtan, 2004 | http://www.ccp4.ac.uk |
| Pymol (v1.7.6.6) | PyMOL | https://www.pymol.org |
| Phaser (part of CCP4 package) | McCoy et al., 2007 | www.ccp4.ac.uk/html/phaser.html |
| PDBePISA | Krissinel & Henrick 200747 | http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver |
| iMosflm (7.2.2) | Battyeetal. 201148 | https://www.mrc-lmb.cam.ac.uk/harry/imosflm/ver740/introduction.html |
| BLItzPro (1.1.0.25) | PALL ForteBio | N/A |
| VMD (1.9.3) | Humphrey et al., 199649 | www.ks.uiuc.edu/Research/vmd/ |
| MDtraj (1.9.4) | McGibbon et al., 201550 | https://www.mdtraj.org/ |
| PyEmma (2.5.7) | Scherer et al., 201551 | http://emma-project.org/ |
Highlights.
Polyreactive antibodies show increased rigidity and neutrality in their binding surfaces
Polyreactive antibodies engage with low affinities akin to autologous TCR-MHC interactions
ACKNOWLEDGMENTS
This work is based on research conducted at the Northeastern Collaborative Access Team beamlines (NE-CAT), funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on the 24-ID-E beamline is funded by an NIH-ORIP HEI grant (S10OD021527). This research used the Advanced Photon Source resources, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the APS staff at GM/CA CAT (23IDB-C) and NE-CAT (24ID-E) for their use and assistance with X-ray beamlines and help and advice during data collection. We thank Dr. Kristof Nolan and Dr. Sobhan Roy for their help and advice on solving the crystal structure. We would also like to acknowledge the University of Chicago’s Research Computing Center Midway resource, where most all-atom MD simulations were carried out. This work was supported by NIH grants R01AI115471 and R01AI155984 (to E.J.A.) and U01AI125250, R01AI144094, and R01AI038339 (to A.B.); T32GM007281 and F30AI124476 (to J.J.B.); NSF grant MCB-1517221 (to B.R. and C.T.B.); and NIH NIBIB Training Grant T32 EB009412 (C.T.B.). The authors would like to acknowledge the recent passing of our co-author, friend, and colleague Dr. Albert Bendelac; he was a pillar in the immunology field and will be dearly missed.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113190.
REFERENCES
- 1.Chen J, Eisen HN, and Kranz DM (2003). A model T-cell receptor system for studying memory T-cell development. Microbes Infect. 5, 233–240. [DOI] [PubMed] [Google Scholar]
- 2.Udaka K, Tsomides TJ, and Eisen HN (1992). A naturally occurring peptide recognized by alloreactive CD8+ cytotoxic T lymphocytes in association with a class I MHC protein. Cell 69, 989–998. [DOI] [PubMed] [Google Scholar]
- 3.Wucherpfennig KW, and Strominger JL (1995). Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 80, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wucherpfennig KW, Sette A, Southwood S, Oseroff C, Matsui M, Strominger JL, and Hafler DA (1994). Structural requirements for binding of an immunodominant myelin basic protein peptide to DR2 isotypes and for its recognition by human T cell clones. J. Exp. Med 179, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC (2003). Predominant Autoantibody Production by Early Human B Cell Precursors. Science 301, 1374–1377. [DOI] [PubMed] [Google Scholar]
- 6.Dennison SM, Anasti K, Scearce RM, Sutherland L, Parks R, Xia S-M, Liao H-X, Gorny MK, Zolla-Pazner S, Haynes BF, and Alam SM (2011). Nonneutralizing HIV-1 gp41 Envelope Cluster II Human Monoclonal Antibodies Show Polyreactivity for Binding to Phospholipids and Protein Autoantigens. J. Virol 85, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, et al. (2005). Cardiolipin Polyspecific Autoreactivity in Two Broadly Neutralizing HIV-1 Antibodies. Science 308, 1906–1908. [DOI] [PubMed] [Google Scholar]
- 8.Koelsch K, Zheng N-Y, Zhang Q, Duty A, Helms C, Mathias MD, Jared M, Smith K, Capra JD, and Wilson PC (2007). Mature B cells class switched to IgD are autoreactive in healthy individuals. J. Clin. Invest 117, 1558–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, and Wardemann H. (2008). Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Frey G, Peng H, Rits-Volloch S, Garrity J, Seaman MS, and Chen B. (2014). Mechanism of HIV-1 Neutralization by Antibodies Targeting a Membrane-Proximal Region of gp41. J. Virol 88, 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrews SF, Huang Y, Kaur K, Popova LI, Ho IY, Pauli NT, Henry Dunand CJ, Taylor WM, Lim S, Huang M, et al. (2015). Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med 7, 316ra192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bajic G, van der Poel CE, Kuraoka M, Schmidt AG, Carroll MC, Kelsoe G, and Harrison SC (2019). Autoreactivity profiles of influenza hemagglutinin broadly neutralizing antibodies. Sci. Rep 9, 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mouquet H, Scheid JF, Zoller MJ, Krogsgaard M, Ott RG, Shukair S, Artyomov MN, Pietzsch J, Connors M, Pereyra F, et al. (2010). Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prigent J, Lorin V, Kök A, Hieu T, Bourgeau S, and Mouquet H. (2016). Scarcity of autoreactive human blood IgA(+) memory B cells. Eur. J. Immunol 46, 2340–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guthmiller JJ, Lan LYL, Fernández-Quintero ML, Han J, Utset HA, Bitar DJ, Hamel NJ, Stovicek O, Li L, Tepora M, et al. (2020). Polyreactive Broadly Neutralizing B cells Are Selected to Provide Defense against Pandemic Threat Influenza Viruses. Immunity 53, 1230–1244.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, and Bendelac A. (2017). Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358, eaan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prigent J, Jarossay A, Planchais C, Eden C, Dufloo J, Kök A, Lorin V, Vratskikh O, Couderc T, Bruel T, et al. (2018). Conformational Plasticity in Broadly Neutralizing HIV-1 Antibodies Triggers Polyreactivity. Cell Rep. 23, 2568–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernández-Quintero ML, Loeffler JR, Bacher LM, Waibl F, Seidler CA, and Liedl KR (2020). Local and Global Rigidification Upon Antibody Affinity Maturation. Front. Mol. Biosci 7, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeliazkov JR, Sljoka A, Kuroda D, Tsuchimura N, Katoh N, Tsumoto K, and Gray JJ (2018). Repertoire Analysis of Antibody CDR-H3 Loops Suggests Affinity Maturation Does Not Typically Result in Rigidification. Front. Immunol 9, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burnett DL, Schofield P, Langley DB, Jackson J, Bourne K, Wilson E, Porebski BT, Buckle AM, Brink R, Goodnow CC, and Christ D. (2020). Conformational diversity facilitates antibody mutation trajectories and discrimination between foreign and self-antigens. Proc. Natl. Acad. Sci. USA 117, 22341–22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boughter CT, Borowska MT, Guthmiller JJ, Bendelac A, Wilson PC, Roux B, and Adams EJ (2020). Biochemical patterns of antibody polyreactivity revealed through a bioinformatics-based analysis of CDR loops. Elife 9, e61393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lecerf M, Kanyavuz A, Lacroix-Desmazes S, and Dimitrov JD (2019). Sequence features of variable region determining physicochemical properties and polyreactivity of therapeutic antibodies. Mol. Immunol 112, 338–346. [DOI] [PubMed] [Google Scholar]
- 23.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. (2017). Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 114, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly RL, Le D, Zhao J, and Wittrup KD (2018). Reduction of Non-specificity Motifs in Synthetic Antibody Libraries. J. Mol. Biol 430, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, and Sidhu SS (2008). The Intrinsic Contributions of Tyrosine, Serine, Glycine and Arginine to the Affinity and Specificity of Antibodies. J. Mol. Biol 377, 1518–1528. [DOI] [PubMed] [Google Scholar]
- 26.Rabia LA, Zhang Y, Ludwig SD, Julian MC, and Tessier PM (2018). Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng. Des. Sel 31, 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ausserwöger H, Krainer G, Welsh TJ, Thorsteinson N, de Csilléry E, Sneideris T, Schneider MM, Egebjerg T, Invernizzi G, Herling TW, et al. (2023). Surface patches induce nonspecific binding and phase separation of antibodies. Proc. Natl. Acad. Sci. USA 120, e2210332120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, and Deane CM (2019). Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 116, 4025–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim H, and No KT (2022). Prediction of polyreactive and nonspecific single-chain fragment variables through structural biochemical features and protein language-based descriptors. BMC Bioinf. 23, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harvey EP, Shin J-E, Skiba MA, Nemeth GR, Hurley JD, Wellner A, Shaw AY, Miranda VG, Min JK, Liu CC, et al. (2022). An in silico method to assess antibody fragment polyreactivity. Nat. Commun 13, 7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corr M, Slanetz AE, Boyd LF, Jelonek MT, Khilko S, al-Ramadi BK, Kim YS, Maher SE, Bothwell AL, and Margulies DH (1994). T cell receptor-MHC class I peptide interactions: affinity, kinetics, and specificity. Science 265, 946–949. [DOI] [PubMed] [Google Scholar]
- 32.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, and Gascoigne NR (1996). T-cell-receptor affinity and thymocyte positive selection. Nature 381, 616–620. [DOI] [PubMed] [Google Scholar]
- 33.Stanfield RL, Wilson IA, and Smider VV (2016). Conservation and diversity in the ultralong third heavy-chain complementarity-determining region of bovine antibodies. Sci. Immunol 1, aaf7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee PS, Arnell AJ, and Wilson IA (2015). Structure of the apo anti-influenza CH65 Fab. Acta Crystallogr. F Struct. Biol. Commun F71, 145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nachbagauer R, Shore D, Yang H, Johnson SK, Gabbard JD, Tompkins SM, Wrammert J, Wilson PC, Stevens J, Ahmed R, et al. (2018). Broadly Reactive Human Monoclonal Antibodies Elicited following Pandemic H1N1 Influenza Virus Exposure Protect Mice against Highly Pathogenic H5N1 Challenge. J. Virol 92, e00949–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, et al. (2012). ighly Conserved Protective Epitopes on Influenza B Viruses. Science 337, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, et al. (2011). A Neutralizing Antibody Selected from Plasma Cells That Binds to Group 1 and Group 2 Influenza A Hemagglutinins. Science 333, 850–856. [DOI] [PubMed] [Google Scholar]
- 38.Eagle RA, and Trowsdale J. (2007). Promiscuity and the single receptor: NKG2D. Nat. Rev. Immunol 7, 737–744. [DOI] [PubMed] [Google Scholar]
- 39.McFarland BJ, and Strong RK (2003). Thermodynamic Analysis of Degenerate Recognition by the NKG2D Immunoreceptor Not Induced Fit but Rigid Adaptation. Immunity 19, 803–812. [DOI] [PubMed] [Google Scholar]
- 40.Xu G, Wang Q, and Ma J. (2020). Improving Protein Side-Chain Modeling by Deep Neural Networks and Ensemble Methods. J. Chem. Inf. Model 60, 6691–6697. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt AG, Xu H, Khan AR, O’Donnell T, Khurana S, King LR, Manischewitz J, Golding H, Suphaphiphat P, Carfi A, et al. (2013). Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc. Natl. Acad. Sci. USA 110, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Govern CC, Paczosa MK, Chakraborty AK, and Huseby ES (2010). Fast on-rates allow short dwell time ligands to activate T cells. Proc. Natl. Acad. Sci. USA 107, 8724–8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crooks JE, Boughter CT, Scott LR, and Adams EJ (2018). The hypervariable loops of free TCRs sample multiple distinct metastable conformations in solution. Front. Mol. Biosci 5, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greives N, and Zhou HX (2014). Both protein dynamics and ligand concentration can shift the binding mechanism between conformational selection and induced fit. Proc. Natl. Acad. Sci. USA 111, 10197–10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borbulevych OY, Piepenbrink KH, and Baker BM (2011). Conformational melding permits a conserved binding geometry in TCR recognition of foreign and self molecular mimics. J. Immunol 186, 2950–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dimitrov JD (2020). Harnessing the therapeutic potential of “rogue” antibodies. Trends Pharmacol. Sci 41, 409–417. [DOI] [PubMed] [Google Scholar]
- 47.Krissinel E, and Henrick K. (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- 48.Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011). A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey W, Dalke A, and Schulten K. (1996). VMD: visual molecular dynamics. J Mol Graph. 14, 33–38. 27–28. [DOI] [PubMed] [Google Scholar]
- 50.McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang LP, Lane TJ, and Pande VS (2015). MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys J 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherer MK, Trendelkamp-Schroer B, Paul F, Pérez-Hernández G, Hoffmann M, Plattner N, Wehmeyer C, Prinz JH, and Noé F. (2015). PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J Chem Theory Comput 11, 5525–5542. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG, and Cowtan K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jo S, Kim T, and Im W. (2007). Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS One 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jo S, Kim T, Iyer VG, and Im W. (2008). CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 56.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. (2016). CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/ OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theor. Comput 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystal structures were deposited in the RCSB database under following PDB IDs: 8FZO, 8FZP, 8G19, 8G1B, 8G1C.
All processed data are freely available online via a Zenodo repository linked through the GitHub page associated with this manuscript (discussed further in the next point). Raw trajectory files are too large (~10 TB) to post on publicly accessible repositories but are available from the lead contact upon request.
All relevant code for data normalization, processing, and statistical analysis are available on the GitHub page (https://github.com/ctboughter/manuscript_md_analysis). Instructions for use and relevant packages required for the analysis are provided within the ReadMe file and documentation page.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.