

Abstract
Tumor sensitivity to platinum (Pt)‐based chemotherapy and poly(adenosine diphosphate ribose) polymerase (PARP) inhibitors is increased by homologous recombination deficiency‐causing mutations; in particular, reversion mutations cause drug resistance by restoring protein function. Treatment response is predicted by breast cancer susceptibility gene 1/2 (BRCA1/2) mutations; however, BRCA1/2 reversion mutations have not been comprehensively studied in pan‐cancer cohorts. We aimed to characterize BRCA1/2 reversion mutations in a large pan‐cancer cohort of Japanese patients by retrospectively analyzing sequencing data for BRCA1/2 pathogenic/likely pathogenic mutations in 3738 patients with 32 cancer types. We identified somatic mutations in tumors or circulating cell‐free DNA that could restore the ORF of adverse alleles, including reversion mutations. We identified 12 (0.32%) patients with somatic BRCA1 (n = 3) and BRCA2 (n = 9) reversion mutations in breast (n = 4), ovarian/fallopian tube/peritoneal (n = 4), pancreatic (n = 2), prostate (n = 1), and gallbladder (n = 1) cancers. We identified 21 reversion events—BRCA1 (n = 3), BRCA2 (n = 18)—including eight pure deletions, one single‐nucleotide variant, six multinucleotide variants, and six deletion–insertions. Seven (33.3%) reversion deletions showed a microhomology length greater than 1 bp, suggesting microhomology‐mediated end‐join repair. Disease course data were obtained for all patients with reversion events: four patients acquired mutations after PARP‐inhibitor treatment failure, two showed somatic reversion mutations after disease progression, following Pt‐based treatment, five showed mutations after both treatments, one patient with pancreatic cancer and BRCA1 reversion mutations had no history of either treatment. Although reversion mutations commonly occur in BRCA‐associated cancers, our findings suggest that reversion mutations due to Pt‐chemotherapy might be correlated with BRCA1/2‐mediated tumorigenesis even in non‐BRCA‐associated histologies.
Keywords: BRCA1/2, homologous recombination deficiency, PARP inhibitor, platinum‐based chemotherapy, reversion mutation
We characterized BRCA reversion mutations in a large pan‐cancer cohort. Although reversion mutations commonly occur in BRCA‐associated cancers, our findings suggest that reversion mutations due to platinum‐based chemotherapy might be correlated with BRCA1/2‐mediated tumorigenesis even in non‐BRCA‐associated histologies.
Abbreviations
- BRCA
breast cancer susceptibility gene
- cfDNA
cell‐free DNA
- delin
deletion–insertion
- HRD
homologous recombination deficiency
- HRR
homologous recombination repair
- MMEJ
microhomology‐mediated end‐joining
- NHEJ
nonhomologous end‐joining
- PALB2
partner and localizer of BRCA2
- PARPi
poly(adenosine diphosphate ribose) polymerase (PARP) inhibitor
- POLθ
DNA polymerase θ
- Pt
platinum
- SNV
single‐nucleotide variant
1. INTRODUCTION
Platinum‐based chemotherapy and PARPi treatment cause DNA damage‐mediated cell death. Mutations in BRCA1/2 hinder the repair of this damage and predict the response to these agents. 1 , 2 , 3 , 4 , 5 Tumor lineage affects Pt‐ and PARPi‐responsive phenotypes in BRCA‐mutant tumors. BRCA mutations act as the drivers of classic BRCA‐associated tumors—including breast, ovarian, prostate, and pancreatic cancers—and exhibit zygosity dependence and selection for biallelic inactivation. These characteristics, in turn, suggest that PARP inhibition has a number of potential benefits. 6 However, BRCA mutations do not act as the drivers of oncogenesis in other noncanonical BRCA cancers, suggesting that PARP inhibition would be less beneficial in these cases. 6
It remains unclear whether selected BRCA‐mutant tumors with noncanonical histological features exhibit HRD, which is influenced by BRCA loss of function, despite the fact that overall analysis of noncanonical BRCA‐mutant tumors reveal no signs of such phenotypes. A BRCA‐dependent phenotype can be confirmed by detecting BRCA reversion mutations, subsequent to the selective pressure exerted by therapies that act through the DNA damage pathway.
In tumors exhibiting a BRCA‐mediated HRD phenotype, somatic change in BRCA1/2 could lead to resistance against Pt‐based chemotherapy or PARP inhibition. Restoration of the ORF of BRCA1/2 due to secondary reversion point mutations, insertions, or deletions not only enables BRCA genes to repair DNA damage induced by PARPi and Pt‐based therapies but also causes resistance to PARPi 5 , 7 , 8 , 9 , 10 and Pt‐based therapy. 8 , 11 , 12 , 13 , 14 , 15 Thus, Pt‐based chemotherapy and/or PARPi treatment applies a selective pressure on the original tumor with loss of BRCA function, causing secondary reversion mutations.
In this study, we used data from a cohort of 3738 patients with various tumor types, after germline genetic testing and matched tumor next‐generation sequencing, to characterize BRCA‐mediated HRD phenotypes in different types of cancers. We identified patients who showed reversion mutations indicating a BRCA‐mediated HRD phenotype, following Pt‐based chemotherapy and/or PARPi treatment.
2. MATERIALS AND METHODS
2.1. Patients and study design
We retrospectively recruited 3738 patients with solid tumors, listed in the Keio PleSSision Group Database (Keio University Hospital), based on the following criterion: patients who underwent a 324‐gene somatic genomic profiling test (FoundationOne CDx; Foundation Medicine, Inc.) (n = 3094) or a 324‐gene cfDNA‐based comprehensive genomic profiling assay (FoundationOne Liquid CDx; Foundation Medicine, Inc.) (n = 644) between August 2020 and June 2023. In total, 1320 patients (35.3%) had canonical BRCA‐associated tumors (breast, ovarian, peritoneal, pancreatic, and prostate cancers), and 2418 patients (64.7%) had noncanonical BRCA‐associated tumors (colorectal and endometrial cancers). Table 1 summarizes the patient characteristics. This study was approved by the Ethics Committee of Keio University Hospital (approval number: 20211159). Due to the retrospective nature of this study, informed consent was not required and was waived. The study procedures involving human participants complied with the Declaration of Helsinki.
TABLE 1.
Characteristics of Japanese patients with solid tumors who underwent 324‐gene somatic genomic profiling test (FoundationOne CDx) or 324‐gene cell‐free DNA‐based comprehensive genomic profiling assay (FoundationOne Liquid CDx)
| FoundationOne CDx | FoundationOne Liquid CDx | Total | ||
|---|---|---|---|---|
| Patients, n | 3094 | 644 | 3738 | |
| Sex, n (%) | ||||
| Female | 1611 | 272 | 1883 | |
| Male | 1483 | 372 | 1855 | |
| Cancer type | ||||
| BRCA‐associated cancer | Breast | 196 | 72 | 268 |
| Ovary/fallopian tube/peritoneal | 253 | 21 | 274 | |
| Pancreas | 263 | 114 | 377 | |
| Prostate | 254 | 147 | 401 | |
| Total | 966 | 354 | 1320 | |
| Non‐BRCA‐associated cancer | Colorectal | 539 | 72 | 611 |
| Biliary | 229 | 55 | 284 | |
| Central nervous system | 152 | 2 | 154 | |
| Sarcoma | 138 | 12 | 150 | |
| Uterus | 115 | 3 | 118 | |
| Uterine cervix | 114 | 10 | 124 | |
| NSCLC | 105 | 35 | 140 | |
| Head and neck | 99 | 12 | 111 | |
| Stomach | 85 | 15 | 100 | |
| Thyroid | 78 | 4 | 82 | |
| Skin | 75 | 1 | 76 | |
| Unknown primary | 66 | 13 | 79 | |
| Esophagus | 63 | 10 | 73 | |
| Urinary | 63 | 8 | 71 | |
| Kidney | 23 | 6 | 29 | |
| Thymus | 14 | 2 | 16 | |
| Adrenal gland | 11 | 1 | 12 | |
| Duodenum | 11 | 2 | 13 | |
| SCLC | 11 | 2 | 13 | |
| Neuroendocrine carcinoma | 9 | 2 | 11 | |
| GIST | 8 | 2 | 10 | |
| Neuroendocrine tumor | 8 | 0 | 8 | |
| Mesothelioma | 7 | 0 | 7 | |
| Liver | 6 | 3 | 9 | |
| Soft tissue | 6 | 3 | 9 | |
| Vagina | 4 | 0 | 4 | |
| Testis | 2 | 1 | 3 | |
| Others | 87 | 14 | 101 | |
| Total | 2128 | 290 | 2418 | |
Abbreviations: GIST, gastrointestinal stromal tumor; NSCLC, non‐small‐cell lung cancer; SCLC, small‐cell lung cancer.
2.2. Next‐generation sequencing analysis
After obtaining written informed consent from patients, formalin‐fixed, paraffin‐embedded samples or plasma samples were collected and subsequently assessed using FoundationOne CDx or FoundationOne Liquid CDx, 16 , 17
2.3. Analysis of BRCA1/2 pathogenic mutations
The pathogenicity of the BRCA1/2 variants was assessed according to the ClinVar classification (http://www.ncbi.nlm.nih.gov/clinvar/). Variants that had not been submitted to either database, and therefore were not defined as “likely to be pathogenic” or “pathogenic” using the Ambry five‐tier variant classification protocol (https://www.ambrygen.com/science/variant‐classification), were considered pathogenic based on American College of Medical Genetics and Genomics, Association for Molecular Pathology, and International Agency for Research on Cancer guidelines. 18 , 19 , 20
2.4. Confirmation of germline variants
For patients with potential reversion mutations in BRCA1/2, we performed genetic counseling and checked whether the primary BRCA1/2 mutations were somatic or germline events. Germline BRCA1/2 testing was carried out on the blood samples using single‐site germline variant tests based on the somatic event or potentially comprehensive analysis of BRCA1/2 using BRACAnalysis CDx (Myriad Genetics).
2.5. Identification of BRCA1/2 reversion mutations in tumor and cfDNA
We defined BRCA reversion mutations as follows: (i) a base substitution of a nonsense mutation that converted it to a missense mutation, or (ii) an insertion/deletion that restored the ORF. Confirmation that the insertion/deletion restored the ORF, and therefore could be considered a reversion mutation, required that the primary deleterious mutation in combination with the secondary mutation produced a nucleotide change divisible by 3. For example, if the primary mutation was a deletion of 2 bp and the secondary mutation was a deletion of 4 bp, either upstream or downstream, the two mutations would together constitute a net deletion of 6 bp, which is divisible by 3. Open reading frame restoration could also be defined as the loss of the primary deleterious mutation, due to a large intragenic deletion.
2.6. Identification of the MMEJ signature
For determining the types of reversion mutations, we examined microhomologies in deletions according to the method outlined by Taheri‐Ghahfarokhi et al. 21 Briefly, when checking for pure deletion mutations, we adjusted the position of the deleted sequence in the full sequence to determine whether the nucleotides before the deletion matched the last nucleotides of the deleted sequence. Next, we determined the number of the contiguous nucleotides at the beginning of the deleted sequence that matched after the deletion. If the number of contiguously matched nucleotides matches the length of the microhomology, the maximum length was considered equal to the length of the deletion. Microhomologies of at least 2 bp were considered candidates for MMEJ repair (Figure 1) and were used to define the MMEJ signature.
FIGURE 1.
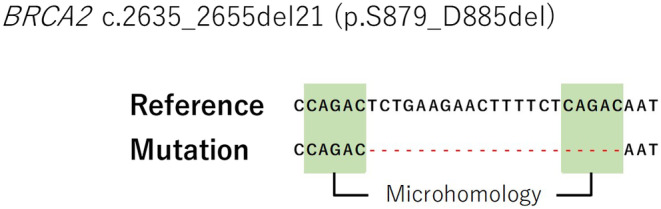
Identification of the microhomology‐mediated end‐joining (MMEJ) signature of the BRCA2 gene. The reported mutation (deletion) was aligned with the reference sequence and, if necessary, the position of the deletion was recalibrated. For instance, if labeling the deletion as either “c.2635_2655del” or “c.2630_2650del” yielded an identical deleted sequence, it would cause an error; therefore, we opted to redefine the precise location of the deletion event. This adjustment allowed us to identify and report a microhomology of length 3, as opposed to being constrained to reporting a microhomology length of 2 in the absence of this procedural refinement.
2.7. Statistical analysis
Fisher's exact test was used to compare the rates of BRCA1/2 mutations in different tumor types. p < 0.05 was considered statistically significant.
3. RESULTS
3.1. Study cohort and BRCA1/2 pathogenic variants
Table 1 shows the patient characteristics and the number of patients with each cancer type. In the FoundationOne CDx cohort, the analysis involved 3094 patients, including 1611 men (52.1%) and 1483 women (47.9%). Of these, 31.2% (n = 966) patients had BRCA‐associated cancers (breast, n = 196; ovarian/peritoneal, n = 253; pancreatic, n = 263; and prostate, n = 254), and 68.8% (n = 2128) patients had non‐BRCA‐associated cancers. In the FoundationOne Liquid CDx cohort, the analysis involved 644 patients, including 272 women (42.2%) and 372 men (57.8%). Of these, 55.0% (n = 354) patients had BRCA‐associated cancers (breast, n = 72; ovarian/peritoneal, n = 21; pancreatic, n = 114; and prostate, n = 147), and 45.0% (n = 290) patients had non‐BRCA‐associated cancers. The most frequent tumors in non‐BRCA‐associated cancers were colorectal and biliary cancers in each cohort.
In all tumor types, 5.0% (n = 156) of the patients in the FoundationOne CDx cohort had pathogenic variants for BRCA1/2 (Table 2). The overall prevalence for patients with pathogenic mutations of BRCA1, BRCA2, and their comutants was 1.8% (n = 55), 3.1% (n = 97), and 0.13% (n = 4), respectively. The prevalence rates of BRCA1/2 were significantly higher in BRCA‐associated cancers (overall, 9.9% [n = 96]; breast, 8.7% [n = 17]; ovarian/peritoneal, 13.0% [n = 33]; pancreatic, 4.9% [n = 13]; and prostate, 13.0% [n = 33]) than in non‐BRCA‐associated cancers (2.8% [n = 60]) (p < 0.001).
TABLE 2.
BRCA1/2 pathogenic variants in Japanese patients with solid tumors who underwent 324‐gene somatic genomic profiling test (FoundationOne CDx)
| Total (n) | Patients with BRCA1 pathogenic mutations (n) | Patients with BRCA2 pathogenic mutations (n) | Patients with BRCA1/BRCA2 pathogenic comutations (n) | Patients with BRCA1/BRCA2 pathogenic mutations (n, %) | ||
|---|---|---|---|---|---|---|
| Patients, n | 3094 | 55 | 97 | 4 | 156 (5.0) | |
| Cancer type | ||||||
| BRCA‐associated cancer | Breast | 196 | 2 | 15 | 0 | 17 (8.7) |
| Ovary/fallopian tube/peritoneal | 253 | 26 | 7 | 0 | 33 (13.0) | |
| Pancreas | 263 | 1 | 12 | 0 | 13 (4.9) | |
| Prostate | 254 | 3 | 30 | 0 | 33 (13.0) | |
| Total | 966 | 32 | 64 | 0 | 96 (9.9) | |
| Non‐BRCA‐associated cancer | Colorectal | 539 | 4 | 6 | 1 | 11 (2.0) |
| Biliary | 229 | 3 | 4 | 2 | 9 (3.9) | |
| Central nervous system | 152 | 1 | 4 | 0 | 5 (3.3) | |
| Sarcoma | 138 | 1 | 3 | 0 | 4 (2.9) | |
| Uterus | 115 | 3 | 2 | 1 | 6 (5.2) | |
| Uterine cervix | 114 | 1 | 2 | 0 | 3 (2.6) | |
| NSCLC | 105 | 0 | 3 | 0 | 3 (2.9) | |
| Head and neck | 99 | 0 | 1 | 0 | 1 (1.0) | |
| Stomach | 85 | 2 | 2 | 0 | 4 (4.7) | |
| Thyroid | 78 | 0 | 0 | 0 | 0.0 | |
| Skin | 75 | 1 | 0 | 0 | 1 (1.3) | |
| Unknown primary | 66 | 5 | 2 | 0 | 7 (10.6) | |
| Esophagus | 63 | 0 | 3 | 0 | 3 (4.8) | |
| Urinary | 63 | 1 | 1 | 0 | 2 (3.2) | |
| Kidney | 23 | 0 | 0 | 0 | 0.0 | |
| Thymus | 14 | 0 | 0 | 0 | 0.0 | |
| Adrenal gland | 11 | 0 | 0 | 0 | 0.0 | |
| Duodenum | 11 | 1 | 0 | 0 | 1 (9.1) | |
| SCLC | 11 | 0 | 0 | 0 | 0.0 | |
| Neuroendocrine carcinoma | 9 | 0 | 0 | 0 | 0.0 | |
| GIST | 8 | 0 | 0 | 0 | 0.0 | |
| Neuroendocrine tumor | 8 | 0 | 0 | 0 | 0.0 | |
| Mesothelioma | 7 | 0 | 0 | 0 | 0.0 | |
| Liver | 6 | 0 | 0 | 0 | 0.0 | |
| Soft tissue | 6 | 0 | 0 | 0 | 0.0 | |
| Vagina | 4 | 0 | 0 | 0 | 0.0 | |
| Testis | 2 | 0 | 0 | 0 | 0.0 | |
| Others | 87 | 0 | 0 | 0 | 0.0 | |
| Total | 2128 | 23 | 33 | 4 | 60 (2.8) | |
Abbreviations: GIST, gastrointestinal stromal tumor; NSCLC, non‐small‐cell lung cancer; SCLC, small‐cell lung cancer.
In all tumor types, 8.1% (n = 52) of patients in the FoundationOne Liquid CDx cohort had pathogenic variants in BRCA1/2 (Table 3). The overall prevalence for pathogenic variants of BRCA1, BRCA2, and their comutants was 2.2% (n = 14), 5.3% (n = 34), and 0.6% (n = 4), respectively. The prevalence rates of BRCA1/2 were significantly higher in BRCA‐associated cancers (overall, 9.9% [n = 35]; breast, 15.3% [n = 11]; ovarian/peritoneal, 28.6% [n = 6]; pancreatic, 6.1% [n = 7]; and prostate, 7.5% [n = 11]) than in non‐BRCA‐associated cancers (5.9% [n = 17]) (p < 0.001).
TABLE 3.
BRCA1/2 pathogenic variants in Japanese patients with solid tumors who underwent 324‐gene cell‐free DNA‐based comprehensive genomic profiling assay (FoundationOne Liquid CDx)
| Total (n) | Patients with BRCA1 pathogenic variants (n) | Patients with BRCA2 pathogenic variants (n) | Patients with BRCA1/BRCA2 pathogenic covariants (n) | Patients with BRCA1/BRCA2 pathogenic variants (n, %) | ||
|---|---|---|---|---|---|---|
| Patients, n | 644 | 14 | 34 | 4 | 52 (8.1) | |
| Cancer type | ||||||
| BRCA‐associated cancer | Breast | 72 | 4 | 6 | 1 | 11 (15.3) |
| Ovary/fallopian tube/peritoneal | 21 | 4 | 2 | 0 | 6 (28.6) | |
| Pancreas | 114 | 1 | 6 | 0 | 7 (6.1) | |
| Prostate | 147 | 1 | 9 | 1 | 11 (7.5) | |
| Total | 354 | 10 | 23 | 2 | 35 (9.9) | |
| Non‐BRCA‐associated cancer | Colorectal | 72 | 1 | 2 | 1 | 4 (5.6) |
| Biliary | 55 | 1 | 2 | 0 | 3 (5.5) | |
| Central nervous system | 2 | 0 | 0 | 0 | 0 (0.0) | |
| Sarcoma | 12 | 0 | 0 | 0 | 0 (0.0) | |
| Uterus | 3 | 0 | 0 | 0 | 0 (0.0) | |
| Uterine cervix | 10 | 0 | 2 | 0 | 2 (20.0) | |
| NSCLC | 35 | 0 | 2 | 0 | 2 (5.7) | |
| Head and neck | 12 | 0 | 0 | 0 | 0.0 (0.0) | |
| Stomach | 15 | 0 | 1 | 0 | 1 (6.7) | |
| Thyroid | 4 | 0 | 0 | 0 | 0 (0.0) | |
| Skin | 1 | 0 | 0 | 0 | 0 (0.0) | |
| Unknown primary | 13 | 1 | 1 | 1 | 3 (23.1) | |
| Esophagus | 10 | 0 | 1 | 0 | 1 (10.0) | |
| Urinary | 8 | 0 | 0 | 0 | 0 (0.0) | |
| Kidney | 6 | 0 | 0 | 0 | 0 (0.0) | |
| Thymus | 2 | 0 | 0 | 0 | 0 (0.0) | |
| Adrenal gland | 1 | 0 | 0 | 0 | 0 (0.0) | |
| Duodenum | 2 | 0 | 0 | 0 | 0 (0.0) | |
| SCLC | 2 | 0 | 0 | 0 | 0 (0.0) | |
| Neuroendocrine carcinoma | 2 | 0 | 0 | 0 | 0 (0.0) | |
| GIST | 2 | 0 | 0 | 0 | 0 (0.0) | |
| Liver | 3 | 0 | 0 | 0 | 0 (0.0) | |
| Soft tissue | 3 | 0 | 0 | 0 | 0 (0.0) | |
| Testis | 1 | 0 | 0 | 0 | 0 (0.0) | |
| Others | 14 | 1 | 0 | 0 | 1 (7.1) | |
| Total | 290 | 4 | 11 | 2 | 17 (5.9) | |
Abbreviations: GIST, gastrointestinal stromal tumor; NSCLC, non‐small‐cell lung cancer; SCLC, small‐cell lung cancer.
3.2. Patients with reversion mutations
Among 208 patients with BRCA1/2 pathogenic mutations, 21 reversion mutations were observed in 12 patients, including three patients with primary BRCA1 mutations and nine patients with primary BRCA2 mutations (Figure 2). Among the 12 patients with reversion mutations, five different cancer types were observed (breast, n = 4; ovarian/fallopian tube/peritoneal, n = 4; pancreatic, n = 2; prostate, n = 1; and gallbladder, n = 1); case 4 even had two BRCA2 primary mutations. Notably, one patient (case 6) did not undergo Pt‐based or PARPi therapy treatments, while disease progression was observed in 11 patients after treatment with Pt‐based or PARPi therapy. Among these 21 reversion mutations, three mutations were detected in BRCA1 carriers, and 18 mutations were detected in BRCA2 carriers. These corrected the original mutation in the tumor in some cases or re‐established the mutation‐induced disruption of the ORF of BRCA. Therefore, these secondary mutations can restore the expression of BRCA protein and are termed reversion mutations. Among the different tumor types, 5.8% (n = 12) of patients with a BRCA pathogenic mutation carried reversion mutations in all cohorts. Compared to patients with detectable reversions in BRCA2, fewer patients had detectable reversions in BRCA1—4.3% (BRCA1) versus 7.6% (BRCA2)—although the difference was not statistically significant.
FIGURE 2.

List of reversion mutations in BRCA1/2 carriers. The nucleotide and protein coding sequences for primary and reversion mutations are shown with respect to the reference genome, for patients with BRCA1 (n = 3) and BRCA2 (n = 19) reversion mutations in different types of cancers—ovarian, fallopian tubal, breast, pancreatic, gall bladder, and prostate cancers. VAF, variant allele frequency.
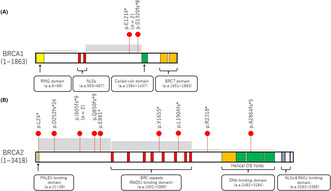
Almost all primary BRCA1/2 mutations in these patients were of germline origin (3/3 cases for BRCA1 and 9/10 for BRCA2) and were predominantly detected in hotspot mutated regions within exon 11 of the gene (Figure 3). Additionally, for BRCA2, they were commonly located in regions that encode the DNA‐binding domain, BRC repeats, or the N‐terminal part of the protein, all of which occur between the PALB2‐interacting domain and DNA‐binding domains (Figure 3). 22 Additionally, to facilitate comparison with the findings of previous studies on BRCA reversion mutations, specifically those reported by Pettitt et al., 12 we have annotated the positions of the reversion mutations outlined in Pettitt et al.'s study within the domains of Figure 3. An analysis of these annotations revealed that the 13 reversion mutations identified in our study generally follow the same trends as those previously reported. Intriguingly, although reversion mutations within the PALB2‐binding domain in BRCA2 have not been previously documented, we observed such a mutation in case 5 with breast cancer, specifically a reversion at BRCA2 p.L24* within the PALB2‐binding domain. These reversions were all found in tumors wherein the primary mutation produced a frameshift consisting of a secondary deletion, multinucleotide variant, or delin event, which thereby restored the ORF. In cases of reversion in which the primary mutation was a nonsense SNV, these occurred not only through secondary mutation, which involved another SNV, but also through deletions or delins.
FIGURE 3.
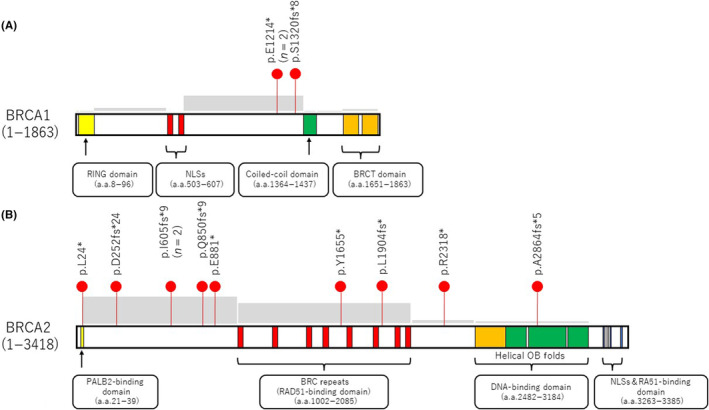
Distribution of reversion mutations on the various domain structures of BRCA1/2. (A) Reversion mutations in the RING, nuclear localization signal (NLS), coiled‐coil, and BRCA1 C‐terminal (BRCT) domains of BRCA1. (B) Reversion mutations in the partner and localizer of BRCA2 (PALB2)‐binding, RAD51‐binding, DNA‐binding, and NLS and RA51‐binding domains of BRCA2. Red dots represent patients. n denotes the number of patients showing each reversion mutation. Gray boxes indicate the frequency of BRCA reversion mutations, as reported by Pettitt et al. 12
Notably, three cases had multiple BRCA reversion mutations. Each reversion mutation showed frequency changes in different alleles, suggesting the potential expansion of certain clonal fractions and indicating the ability of BRCA reversion mutations to capture multiclonal heterogeneity. Reversion mutations were not dependent on treatment type and tended to accumulate in specific regions of either the BRCA1 or BRCA2 protein.
Detailed clinical data were available for all 12 patients with reversion mutations. Seven of the 12 patients received first‐line Pt therapy with an overall median time of 6.0 months (range, 4–30 months). The remaining four patients who did not receive first‐line Pt therapy but received PARPi therapy instead and had an overall median time of 11.5 months (range, 8–13 months). Reversion mutation was also identified in one patient with pancreatic cancer who had not received prior treatment with a PARPi or Pt agent. The patient with gallbladder cancer (case 7) was on first‐line Pt therapy for 10 months (Figure 4).
FIGURE 4.
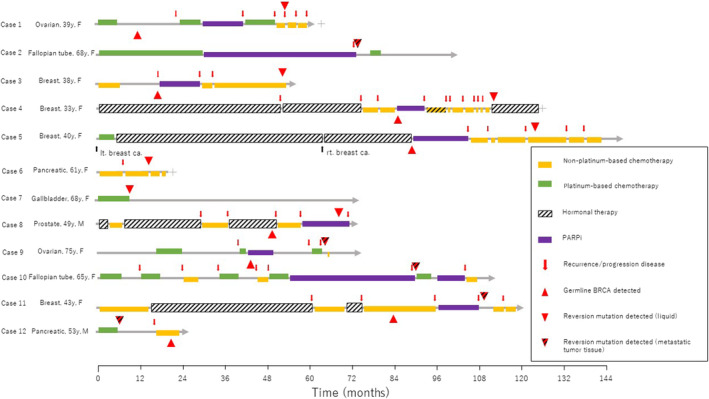
Treatment timeline of 12 patients whose solid tumors developed reversion mutations. ca., cancer; F, female; lt., left; M, male; PARPi, poly(adenosine diphosphate ribose) polymerase (PARP) inhibitor; rt., right; y, years.
3.3. Reversion mutations with large deletions and the MMEJ repair mechanism
Eight secondary “deletions” were identified in BRCA2. No secondary “deletions” were identified in BRCA1. Most deletion events were accounted for by the error‐prone DNA repair mechanisms of end‐joining, commonly by classical NHEJ, or alternative end‐joining.
Of the multiple resistance mechanisms reported preclinically, MMEJ has been associated with BRCA reversion mutations. 10 Microhomology‐mediated end‐joining, involving POLθ, is an alternative DNA damage repair pathway for end‐joining of small sequence microhomologies located closely around the breakpoint. Upon microhomology analysis on surrounding secondary deletions, there were eight “deletions” in four patients that had microhomologies of greater than 1 bp (cases 4, 7, 10, and 12), suggesting MMEJ repair. 23 These deletions varied from small deletions of 2 bp up to large deletions of 2493 bp. Anywhere from 2 to 5 bp of microhomology were identified around the breakpoints, without any correlations between the deletion and microhomology lengths (Figure 5).
FIGURE 5.
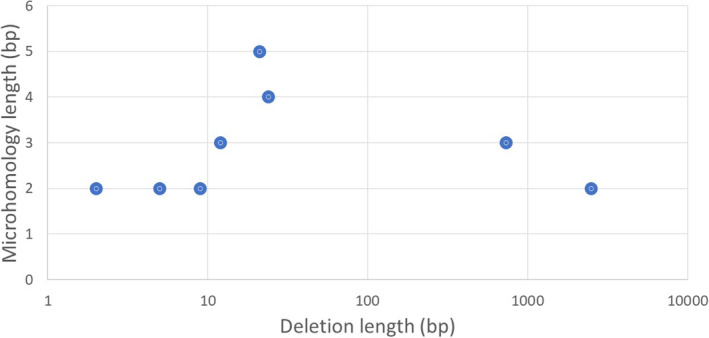
Correlation between deletion and microhomology lengths in BRCA1 and BRCA2 reversion mutations.
4. DISCUSSION
In this genomic analysis of 3738 patients with various cancer histologies, we identified BRCA1/2 reversion mutations across BRCA‐associated tumor types. Furthermore, clinical data helped identify rare cases in which Pt‐based therapy resulted in selective pressure, and consequent reversion mutations, in tumors showing noncanonical histologies—an initial phenotype associated with the loss of BRCA function. The BRCA‐mediated HRD phenotype has been shown to be lineage‐specific and is frequently identified in BRCA‐associated tumor types. 6 Although the incidence of the BRCA‐mediated HRD phenotype in noncanonical cancer has not been determined, a previous study on BRCA reversion mutations in a large pan‐cancer cohort reported two patients with BRCA reversion mutations with tumors showing noncanonical histologies involving esophagogastric and lung cancer. 24
Here, we analyzed a large database for BRCA1/2 alterations and identified a BRCA reversion mutation in a patient with gallbladder cancer. Although gallbladder cancer is not considered a canonical BRCA‐driven tumor, patients with gallbladder cancer have been shown to carry BRCA mutations. 25 A large registry‐based study in Japan suggested that pathogenic variants of BRCA1 and BRCA2 were associated with the increased risk of cancers of the biliary tract, stomach, and esophagus, in addition to BRCA‐associated cancers (breast, ovarian/fallopian tube/peritoneal, pancreatic, and prostate cancers). 26 The identification of mechanisms linking pathogenic variants and these cancer types for the potential efficacy of PARPi requires further investigation, given that HRR defects have been identified in cases of biliary and esophageal cancer. 27 Although it is unclear whether BRCA is, in fact, a pathogenic driver in these cancers, few studies have investigated BRCA reversion mutations in these cancers. 24 The patient with gallbladder cancer responded positively to Pt‐based therapy before the detection of a reversion mutation, suggesting that the initial phenotype was BRCA‐mediated HRD, with the reversion mutation restoring the DNA repair function of the tumor, thereby inducing resistance. This patient showed rapid clinical deterioration following testing with PARPi after developing the reversion mutation. This might be attributable to the effects of reversion in restoring WT BRCA function.
The functionally important roles of different protein domains in inducing resistance to treatment are well illustrated by mapping the consequences of reversion mutations in BRCA proteins. Reversion mutations in exon 11 of BRCA2 produce very long deletions involving a considerable amino acid length. This region encodes BRC repeats and the binding domains for RAD51 recombinase that are essential in HRR for BRCA2 function. 28 Two extreme cases resulted in the deletions of 732 bp (244 a.a.; deletion in eight BRC repeats) and 2493 bp (831 a.a.; deletion in eight BRC repeats) in patients with gallbladder cancer progression after Pt therapy. However, no reversion reported to date has deleted all BRCA repeats, which suggests that a minimum of two is required to ensure the preservation of BRCA2 function and conferral of resistance to Pt or PARPi. 29 Consistent with this indication, mice with a homozygous deletion of BRCA2 exon 11 have been found to be nonviable. 30 Accumulation of reversions involving the N‐terminal or DNA‐binding regions of BRCA2 is limited to smaller deletions or mutations, suggesting greater constraints around the amino acid changes in these regions. This is despite the finding that some in vitro reversion events involve complete loss of the DNA‐binding region of BRCA2. 7
Notably, in our cohort, more deletions were observed among the BRCA2 secondary mutations, especially for mutations greater than 1 bp. Although the limited number of cases prevented any focus on the mutational mechanisms driving reversion mutations through SNVs or delins, understanding the contribution of other DNA repair pathways, such as translesion synthesis, nucleotide excision repair, base excision repair, and mismatch repair, to such outcomes would be interesting.
Deletion is the primary mechanism underlying reversion mutations in BRCA. The prevalence of deletions suggests the existence of a DNA double‐strand break intermediate, which is repaired by end‐joining mechanisms. Regarding NHEJ, its mutational signature commonly involves small delins that are generated by limited DNA‐end resection. This resection is itself imposed by the presence of Ku heterodimers bound to the ends of the DNA break. Therapeutic implications of this effect may follow, given the recent introduction of compounds that inhibit DNA‐PK, a key protein kinase involved in NHEJ, in clinical settings. 29 In contrast, MMEJ repair signatures involve more extensive DNA‐end resection and microhomologies (2–5 bp) flanking the break site. These can occur at distances of several hundred base pairs, resulting in large deletions and chromosomal translocations. 31 Notably, we identified significant microhomology in deletions affecting BRCA genes regardless of therapeutic regimen, which provides evidence of MMEJ repair mechanism, particularly in BRCA2. A critical component in MMEJ repair is POLθ, which is encoded by the POLQ gene. 32 POLθ is essential to cell survival in BRCA‐deficient cell lines and competes for similar DNA repair substrates with HRR proteins. Preclinical efforts to generate POLQ inhibitors are now underway, and we look forward to learning whether the blockage of NHEJ and/or MMEJ repair in BRCA‐mutant backgrounds potentially prevents the accumulation of reversion mutations and accordingly resistance to drug treatments.
Interestingly, case 4 had two BRCA2 primary mutations (one germline and one somatic), and both mutations were genetically reverted in the tumor sample. To the best of our knowledge, this is the first case showing individual reversion mutations against two BRCA primary mutations. Notably, one patient (case 6) did not undergo either Pt‐based or PARPi treatments. It has been previously reported that a patient with Pt‐resistant primary ovarian cancer and a BRCA1 reversion mutation had a history of breast cancer treated with an interstrand DNA cross‐linking agent, cyclophosphamide, which could exert selection pressure for the correction of DNA repair defects. 33 However, in our cohort, this patient had no history of another cancer treated with such a drug.
Our study has a few limitations. First, although BRCA reversion mutations resulting from selective pressure from DNA‐damaging agents suggest a BRCA‐mediated phenotype, a possibility remains that reversion mutations in some tumors are random events. In this regard, one of our patients with pancreatic cancer and a reversion mutation in BRCA1 had no history of either PARPi or Pt‐based therapy, which are generally associated with reversion mutations due to their targeting of the DNA damage repair processes. This patient had been treated with gemcitabine (a DNA synthesis‐inhibiting agent) and paclitaxel (an antimicrotubular agent) and with secondary chemotherapy with irinotecan (a DNA topoisomerase I inhibitor) and 5‐fluorouracil (pyrimidine analog antineoplastic chemotherapy agent). However, whether the reversion mutation in this patient with pancreatic cancer emerged spontaneously or due to resistance to these chemotherapies remains unknown. This patient had a high mutational burden due to MLH1 methylation, and it is possible that the BRCA2 mutation was a passenger mutation. Second, given the small sample size, the data should be interpreted carefully. Third, we could not verify our results with any public databases because, to the best of our knowledge, there are no public databases exclusively on BRCA reversion mutations. Even if multiple mutations were reported in BRCA, we cannot determine whether these are reversion mutations as we do not have information on the treatment course or timing of sample collection for sequencing.
Another limitation of our analysis was that we did not investigate the “back mutations to wild type” BRCA reversion mutation. Many previous studies have reported BRCA1/2 reversion mutations as back mutations to WT. 33 , 34 Therefore, to distinguish the occurrence of back mutations from retention of WT alleles or contamination of cells with WT alleles, examination of intragenic SNPs is essential. Furthermore, regarding liquid biopsy, if the cfDNA amount was small, in such cases during chemotherapy, the somatic mutation was not detected, which produced false negative results. Therefore, our analysis likely underestimates the prevalence of reversion mutations.
In conclusion, although most BRCA‐driven tumors occur in breast, ovarian, pancreatic, or prostate cancers, we analyzed BRCA reversion mutations in a large pan‐cancer cohort and identified rare cases of gallbladder cancer mediated through the loss of BRCA function. Reversion mutations due to Pt therapy could reflect a rare BRCA‐mediated phenotype in tumors showing noncanonical histologies. Therefore, monitoring the HRR mutation status along the course of disease progression and treatment could be beneficial in identifying resistance mechanisms and guiding the choice of subsequent therapies.
AUTHOR CONTRIBUTIONS
Kohei Nakamura: Conceptualization; writing – original draft. Hideyuki Hayashi: Data curation. Ryutaro Kawano: Data curation. Marin Ishikawa: Data curation. Eriko Aimono: Data curation. Takaaki Mizuno: Data curation. Hajime Kuroda: Formal analysis; investigation; resources. Yasuyuki Kojima: Formal analysis; investigation; resources. Naoki NIIKURA: Formal analysis; investigation; resources. Aya Kawanishi: Formal analysis; investigation; resources. Kei Takeshita: Formal analysis; investigation; resources. Shinsuke Suzuki: Formal analysis; investigation; resources. Shinichi Ueno: Formal analysis; investigation; resources. Kosuke Okuwaki: Formal analysis; investigation; resources. Jiichiro Sasaki: Formal analysis; investigation; resources. Masatoshi Yamaguchi: Formal analysis; investigation; resources. Kenta Masuda: Formal analysis; investigation; resources. Tatsuyuki Chiyoda: Formal analysis; investigation; resources. Wataru Yamagami: Formal analysis; investigation; resources. Chihiro Okada: Data curation. Sachio Nohara: Data curation. Shigeki Tanishima: Data curation. Hiroshi Nishihara: Conceptualization; writing – review and editing.
FUNDING INFORMATION
This study was supported by the Japan Agency for Medical Research and Development (AMED) (Grant Number JP22ck0106872), JSPS KAKENHI (Grant‐in‐Aid for Scientific Research (C); grant number 23 K08829), Takeda Science Foundation (Medical Research Grants), Cell Science Research Foundation, Kanzawa Medical Research Foundation, and the Uehara Memorial Foundation.
CONFLICT OF INTEREST STATEMENT
Hiroshi Nishihara is a member of the Cancer Science Editorial Board. Sachio Nohara, Chihiro Okada, Shigeki Tanishima, and Hiroshi Nishihara are employees of Mitsubishi Electric Software Corporation. Sawa Nohara (wife of Sachio Nohara) is an employee of Kaina Home Care Station. Naomi Tanishima (wife of Shigeki Tanishima) is an employee of INOUE Co., Ltd. Hideaki Tanishima (child of Shigeki Tanishima) is an employee of MARUI & Co., Ltd. Mai Tanishima (child of Shigeki Tanishima) is an employee of Konan Medical Center. Takaaki Mizuno is employee of Rakuten Medical K.K. Takaaki Mizuno from CLINIAL Co., Ltd (corporate stocks). Tatsuyuki Chiyoda received a research grant from Takeda Pharmaceutical Company. The other authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: This study was part of a research project approved by the Ethics Committee of Keio University Hospital (approval number: 20211159). The study procedures involving human participants complied with the principles of the Declaration of Helsinki.
Informed consent: All patients in this study provided opt‐out consent.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
ACKNOWLEDGMENTS
The authors are grateful to Hiroshi Yamada and Tomoka Fujikura (Keio University Hospital) for their technical assistance.
Nakamura K, Hayashi H, Kawano R, et al. BRCA1/2 reversion mutations in a pan‐cancer cohort. Cancer Sci. 2024;115:635‐647. doi: 10.1111/cas.16033
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are not available as the study participants did not consent to public sharing of their data.
REFERENCES
- 1. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. doi: 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- 2. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐917. doi: 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- 3. Tan DSP, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case‐control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26(34):5530‐5536. doi: 10.1200/JCO.2008.16.1703 [DOI] [PubMed] [Google Scholar]
- 4. Vencken PMLH, Kriege M, Hoogwerf D, et al. Chemosensitivity and outcome of BRCA1− and BRCA2‐associated ovarian cancer patients after first‐line chemotherapy compared with sporadic ovarian cancer patients. Ann Oncol. 2011;22(6):1346‐1352. doi: 10.1093/annonc/mdq628 [DOI] [PubMed] [Google Scholar]
- 5. Noordermeer SM, van Attikum H. PARP inhibitor resistance: a tug‐of‐war in BRCA‐mutated cells. Trends Cell Biol. 2019;29(10):820‐834. doi: 10.1016/j.tcb.2019.07.008 [DOI] [PubMed] [Google Scholar]
- 6. Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA‐mediated phenotypes. Nature. 2019;571(7766):576‐579. doi: 10.1038/s41586-019-1382-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111‐1115. doi: 10.1038/nature06548 [DOI] [PubMed] [Google Scholar]
- 8. Lin KK, Harrell MI, Oza AM, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high‐grade ovarian carcinoma. Cancer Discov. 2019;9(2):210‐219. doi: 10.1158/2159-8290.CD-18-0715 [DOI] [PubMed] [Google Scholar]
- 9. Dréan A, Williamson CT, Brough R, et al. Modeling therapy resistance in BRCA1/2‐mutant cancers. Mol Cancer Ther. 2017;16(9):2022‐2034. doi: 10.1158/1535-7163.MCT-17-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tobalina L, Armenia J, Irving E, O'Connor MJ, Forment JV. A meta‐analysis of reversion mutations in BRCA genes identifies signatures of DNA end‐joining repair mechanisms driving therapy resistance. Ann Oncol. 2021;32(1):103‐112. doi: 10.1016/j.annonc.2020.10.470 [DOI] [PubMed] [Google Scholar]
- 11. Simmons AD, Nguyen M, Pintus E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: a case report. BMC Cancer. 2020;20(1):215. doi: 10.1186/s12885-020-6657-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pettitt SJ, Frankum JR, Punta M, et al. Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. 2020;10(10):1475‐1488. doi: 10.1158/2159-8290.CD-19-1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vidula N, Rich TA, Sartor O, et al. Routine plasma‐based genotyping to comprehensively detect germline, somatic, and reversion BRCA mutations among patients with advanced solid tumors. Clin Cancer Res. 2020;26(11):2546‐2555. doi: 10.1158/1078-0432.CCR-19-2933 [DOI] [PubMed] [Google Scholar]
- 14. Domchek SM. Reversion mutations with clinical use of PARP inhibitors: many genes, many versions. Cancer Discov. 2017;7(9):937‐939. doi: 10.1158/2159-8290.CD-17-0734 [DOI] [PubMed] [Google Scholar]
- 15. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2‐mutated cancers. Nature. 2008;451(7182):1116‐1120. doi: 10.1038/nature06633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. U.S. Food and Drug Administration . FoundationOne CDx‐ P170019/S014 technical information. Accessed February 18, 2022 https://www.accessdata.fda.gov/cdrh_docs/pdf17/P170019S006C.pdf
- 17. U.S. Food and Drug Administration . FoundationOne Liquid CDx (F1 LiquidCDx)‐ P190032/S010 technical information. Accessed June 08, 2023 https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032S010C.pdf
- 18. Pesaran T, Karam R, Huether R, et al. Beyond DNA: an integrated and functional approach for classifying germline variants in breast cancer genes. Int J Breast Cancer. 2016;2016:2469523. doi: 10.1155/2016/2469523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29(11):1282‐1291. doi: 10.1002/humu.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics. 2014;30(7):1015‐1016. doi: 10.1093/bioinformatics/btt755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taheri‐Ghahfarokhi A, Taylor BJM, Nitsch R, et al. Decoding non‐random mutational signatures at Cas9 targeted sites. Nucleic Acids Res. 2018;46(16):8417‐8434. doi: 10.1093/nar/gky653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rebbeck TR, Mitra N, Wan F, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. Jama. 2015;313(13):1347‐1361. doi: 10.1001/jama.2014.5985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ceccaldi R, Liu JC, Amunugama R, et al. Homologous‐recombination‐deficient tumours are dependent on Polθ‐mediated repair. Nature. 2015;518(7538):258‐262. doi: 10.1038/nature14184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murciano‐Goroff YR, Schram AM, Rosen EY, et al. Reversion mutations in germline BRCA1/2‐mutant tumors reveal a BRCA‐mediated phenotype in non‐canonical histologies. Nat Commun. 2022;13(1):7182. doi: 10.1038/s41467-022-34109-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Breast Cancer Linkage Consortium . Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310‐1316. doi: 10.1093/jnci/91.15.1310 [DOI] [PubMed] [Google Scholar]
- 26. Momozawa Y, Sasai R, Usui Y, et al. Expansion of cancer risk profile for BRCA1 and BRCA2 pathogenic variants. JAMA Oncol. 2022;8(6):871‐878. doi: 10.1001/jamaoncol.2022.0476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen LWM, Martens J, Van Hoeck A, Cuppen E. Pan‐cancer landscape of homologous recombination deficiency. Nat Commun. 2020;11(1):5584. doi: 10.1038/s41467-020-19406-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pellegrini L, Yu DS, Lo T, et al. Insights into DNA recombination from the structure of a RAD51‐BRCA2 complex. Nature. 2002;420(6913):287‐293. doi: 10.1038/nature01230 [DOI] [PubMed] [Google Scholar]
- 29. Siaud N, Barbera MA, Egashira A, et al. Plasticity of BRCA2 function in homologous recombination: genetic interactions of the PALB2 and DNA binding domains. PLoS Genet. 2011;7(12):e1002409. doi: 10.1371/journal.pgen.1002409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418‐425. doi: 10.1038/ng747 [DOI] [PubMed] [Google Scholar]
- 31. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495‐506. doi: 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maga G, Shevelev I, Ramadan K, Spadari S, Hübscher U. DNA polymerase θ purified from human cells is a high‐fidelity enzyme. J Mol Biol. 2002;319(2):359‐369. doi: 10.1016/S0022-2836(02)00325-X [DOI] [PubMed] [Google Scholar]
- 33. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1‐mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581‐2586. doi: 10.1158/0008-5472.CAN-08-0088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008‐3015. doi: 10.1200/JCO.2010.34.2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are not available as the study participants did not consent to public sharing of their data.