

Abstract
INTRODUCTION:
Over the past two decades, continuous improvements in “omics” technologies have driven an ever-greater capacity to define the relationships between genetics, molecular pathways, and overall phenotypes. Despite this progress, the majority of genetic factors influencing complex traits remain unknown. This is exemplified by mitochondrial supercomplex assembly, a critical component of the electron transport chain, which remains poorly characterized. Recent advances in mass spectrometry have expanded the scope and reliability of proteomics and metabolomics measurements. These tools are now capable of identifying thousands of factors driving diverse molecular pathways, their mechanisms, and consequent phenotypes and thus substantially contribute toward the understanding of complex systems.
RATIONALE:
Genome-wide association studies (GWAS) have revealed many causal loci associated with specific phenotypes, yet the identification of such genetic variants has been generally insufficient to elucidate the molecular mechanisms linking these genetic variants with specific phenotypes. A multitude of control mechanisms differentially affect the cellular concentrations of different classes of biomolecules. Therefore, the identification of the causal mechanisms underlying complex trait variation requires quantitative and comprehensive measurements of multiple layers of data—principally of transcripts, proteins, and metabolites and the integration of the resulting data. Recent technological developments now support such multiple layers of measurements with a high degree of reproducibility across diverse sample or patient cohorts. In this study, we applied a multilayered approach to analyze metabolic phenotypes associated with mitochondrial metabolism.
RESULTS:
We profiled metabolic fitness in 386 individuals from 80 cohorts of the BXD mouse genetic reference population across two environmental states. Specifically, this extensive phenotyping program included the analysis of metabolism, mitochondrial function, and cardiovascular function. To understand the variation in these phenotypes, we quantified multiple, detailed layers of systems-scale measurements in the livers of the entire population: the transcriptome (25,136 transcripts), proteome (2622 proteins), and metabolome (981 metabolites). Together with full genomic coverage of the BXDs, these layers provide a comprehensive view on overall variances induced by genetics and environment regarding metabolic activity and mitochondrial function in the BXDs. Among the 2600 transcript-protein pairs identified, 85% of observed quantitative trait loci uniquely influenced either the transcript or protein level. The transomic integration of molecular data established multiple causal links between genotype and phenotype that could not be characterized by any individual data set. Examples include the link between D2HGDH protein and the metabolite D-2-hydroxyglutarate, the BCKDHA protein mapping to the gene Bckdhb, the identification of two isoforms of ECI2, and mapping mitochondrial supercomplex assembly to the protein COX7A2L. These respective measured variants in these mitochondrial proteins were in turn associated with varied complex metabolic phenotypes, such as heart rate, cholesterol synthesis, and branched-chain amino acid metabolism. Of note, our transomics approach clarified the contested role of COX7A2L in mitochondrial supercomplex formation and identified and validated Echdc1 and Mmab as involved in the cholesterol pathway.
CONCLUSION:
Overall, these findings indicate that data generated by next-generation proteomics and metabolomics techniques have reached a quality and scope to complement transcriptomics, genomics, and phenomics for transomic analyses of complex traits. Using mitochondria as a case in point, we show that the integrated analysis of these systems provides more insights into the emergence of the observed phenotypes than any layer can by itself, highlighting the complementarity of a multilayered approach. The increasing implementation of these omics technologies as complements, rather than as replacements, will together move us forward in the integrative analysis of complex traits
Recent improvements in quantitative proteomics approaches, including Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH-MS), permit reproducible large-scale protein measurements across diverse cohorts. Together with genomics, transcriptomics, and other technologies, transomic data sets can be generated that permit detailed analyses across broad molecular interaction networks. Here, we examine mitochondrial links to liver metabolism through the genome, transcriptome, proteome, and metabolome of 386 individuals in the BXD mouse reference population. Several links were validated between genetic variants toward transcripts, proteins, metabolites, and phenotypes. Among these, sequence variants in Cox7a2l alter its protein’s activity, which in turn leads to downstream differences in mitochondrial supercomplex formation. This data set demonstrates that the proteome can now be quantified comprehensively, serving as a key complement to transcriptomics, genomics, and metabolomics —a combination moving us forward in complex trait analysis.
Graphical Abstract:
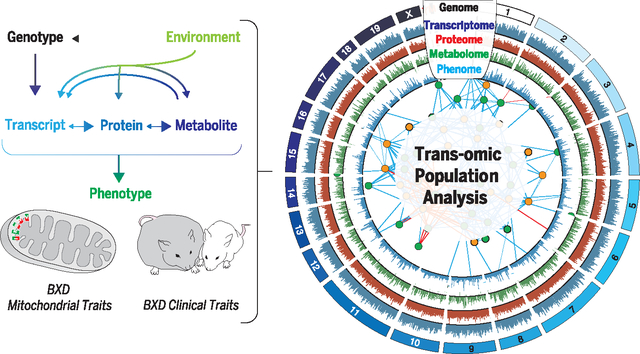
Model of the transomics analysis. A transomics approach was taken to analyze genetic and environmental variation in metabolic and mitochondrial phenotypes by measuring five distinct layers of biology in a diverse population of BXD mice. The combined analysis of all layers together provides additional information not yielded by any single omics approach.
Over the past two decades, continuous improvements in omics technologies have been driving an ever-greater capacity for quantifying relationships between genetics, the biochemical mechanisms of the cell, and overall phenotypes. Despite this progress, the majority of genetic factors determining complex trait heritability remain unknown (1). Recent advances in mass spectrometry (MS) (2–4) have expanded the scope and reliability of proteomic and metabolomic measurements. These developments in MS are permitting a leap forward in understanding complex biological systems by facilitating the accurate quantification of thousands of molecular factors involved in diverse cellular pathways—and, therefore, their mechanisms and consequent phenotypes (5). Thus far, the identification of causal genetic variants alone has been generally insufficient to characterize the underlying molecular mechanisms of action. Generating such models also requires quantitative measurements of additional layers of data, such as transcripts, proteins, and metabolites. As a multitude of control mechanisms differentially affect the cellular concentration of different classes of biomolecules, multilayered quantitative measurements on the same individuals can provide synergistic information about complex systems (6–9) [an approach also dubbed transomics or high-dimensional biology (10)].
In this study, we generated multilayered data sets to examine metabolism across 80 cohorts of the BXD genetic reference population (GRP). The BXDs are descended from C57BL/6J (B6) and DBA/2J (D2) and diverge for ~5 million sequence variants (11), similar to the number of common variants found within many human population groups (12). This population now consists of ~150 murine recombinant inbred strains with known variant susceptibility to major metabolic diseases such as diabetes (13, 14). To date, detailed biochemical analyses have established validated links to phenotypes for a few dozen gene variants. These include links between sweet taste and Tas1r3 (15), cadmium toxicity and Slc39a8 (16), and hyper-activity and Ahr (17). We have previously reported that metabolic phenotypes in the BXDs are highly variable and that this variability is highly heritable (14). Here, we have analyzed 80 BXD cohorts (composed of 386 individuals) across a battery of metabolic tests such as ad libitum running-wheel access, maximal exercise capacity, and glucose tolerance. The mice were tested over a 29-week program where they were exposed to different environmental conditions of diet: chow diet (CD) (6% kcal of fat) or high-fat diet (HFD) (60% kcal of fat). To understand the molecular basis behind the observed phenotypic variance, we quantified detailed layers of systems-scale molecular measurements in the livers of the entire population: the transcriptome (25,136 transcripts), the proteome (2622 proteins), and the metabolome (981 metabolites). Together with full coverage of genetic variants in the BXDs (18), these omics data sets provide a comprehensive platform for deconstructing the factors behind variation in clinical metabolic phenotypes. In all layers of data, trait variation could be attributed to the causal genetic loci through quantitative trait locus (QTL) analysis. These layers build on our previous research in this population (19), in which selected reaction monitoring (SRM) was used to quantify 192 proteins and targeted metabolomics approaches were used to quantify 39 metabolites in the serum and 2 metabolites in the liver. This previous study both shaped our bioinformatics procedures for transomic data sets and provided positive controls for the experimental Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH-MS) proteomics and for multilayered pathway analysis. For example, of the 13 genes with cis-pQTLs (protein) of the 192 proteins measured by SRM, 11 were identified in SWATH (all except AKR7A2 andABCB8), and 10 of these 11 also mapped to cis-pQTLs in the independent SWATH data set.
By applying transomic analyses in these data sets, we observed that the levels of all four proteins composing the branched-chain ketoacid dehydrogenase (BCKD) complex in the mitochondria are, in the BXDs, tied to genetic variants in a single gene, Bckdhb. Similarly, a causal mechanistic link was observed between theD-2-hydroxyglutarate dehydrogenase (D2HGDH) protein and the metabolite D-2-hydroxyglutarate (D2HG), which in turn is linked with similar phenotypes as for humans with deficiencies in this protein, including cardiomyopathy and problems with motor control (20). Furthermore, the broad proteomics data set allowed us to identify two isoforms of the protein ECI2 that were not predicted by eQTL (expressed transcript) analysis. We examined several broad pathways in energy metabolism using a transomics approach, including lipid storage/transport, cholesterol synthesis, and the electron transport chain (ETC), all of which exhibited high levels of genetic variation at the transcript, protein, and metabolite levels. This analysis highlighted COX7A2L from the ETC—the only one of 67 quantified proteins in the ETC with consistent cis-pQTLs. Further experiments showed that this variation in protein leads to strikingly differential formation of ETC supercomplexes (SCs). In all cases, the integrated analysis of multiple omics layers provided more insight into mechanistic networks than could be gleaned from any layer by itself, highlighting the complementarity of a multilayered approach.
High-dimensional reconstruction of complex metabolic traits
To identify new genetic relationships and molecular mechanisms influencing metabolism in the BXDs, we designed an analytical pipeline to measure and combine quantitative data from five omics layers across variable environmental states: genomics, transcriptomics, proteomics, metabolomics, and phenomics (Fig. 1A). The 29-week phenotyping program includes body weight, indirect calorimetry, voluntary exercise, maximal oxygen consumption (VO2max), an oral glucose tolerance test, and spontaneous activity (Fig. 1B). All traits vary significantly due to genetic, environmental, and/or gene-by-environment (G×E) factors, including key traits such as body weight (Fig. 1C) and glucose response during an oral glucose tolerance test (Fig. 1D). At the end of the program, liver samples were used for multilayered omics analyses to serve as the platform for determining the providence and mechanism of metabolic variants across the population. Together, these data support approaches driven by prior knowledge—e.g., examining the relationships between known transcriptional and proteomic gene networks with related phenotypes—as well as data-driven approaches—e.g., the de novo identification of genes that are involved in regulation of metabolic phenotypes.
Fig. 1. Overview and validation of omics layers.
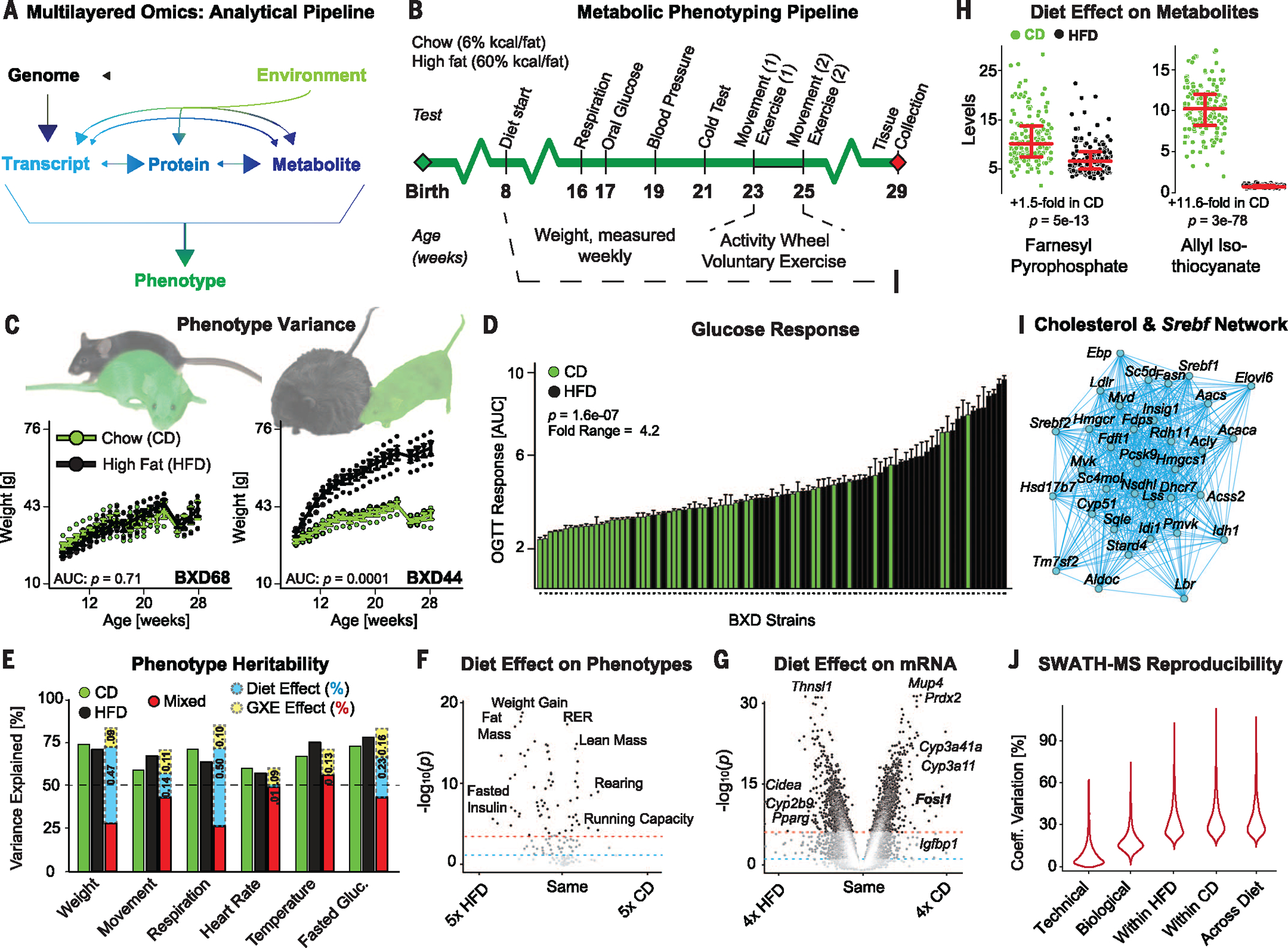
(A) General model of the multilayered approach. Arrows indicate causality between metabolic layers. HFD should not modify DNA, although other environmental factors can (i.e., mutagens). (B) Phenotyping pipeline for all individuals. (See the methods section for details on each experiment.) (C) Body weight in two strains of BXD for both diets over the full phenotyping experiment. (D) Area under the curve (AUC) of glucose excursion during a 3-hour oral glucose tolerance test for all cohorts. Bars represent mean ±SEM. (E) Heritability for several phenotypes, calculated by one-way (CD/HFD) or two-way (Mixed) analysis of variance. Some traits are affected by diet (weight and fasted glucose), others are not (heart rate and body temperature), and G×E contributions vary. (F) Volcano plot of diet effect on clinical phenotypes. (G) Volcano plot of diet effect on all transcripts. (H) Dot plot showing two example hepatic metabolites affected by diet. (I) An enriched Spearman correlation transcript network using the cholesterol biosynthesis and SREBF targets gene set. Edges indicate P ≤ 0.001. All correlations are positive. (J) Error in SWATH measurements due to different factors: technical (median CV = 6.5%), biological (CV = 17.0%), across strain (within diet) (CV = 29.6% HFD, 31.4% CD), and across all measurements (CV = 30.8%). Reported P values between diets (panels C–D, F–H) are all for Welch’s t-test.
Before delving into multilayered data sets for the analysis of complex molecular networks, we assessed the quality and significance of each omics layer individually. For phenotypes, heritability was calculated for all traits within dietary groups (green and black bars, Fig. 1E), then across all cohorts combined (red bars). The known environmental factor, diet, furthermore allowed us to calculate the independent environmental effect (blue) and the strain-dependent G×E influence (yellow). As expected, HFD feeding strikingly worsens parameters of metabolic health in most strains, particularly for traits such as body weight, glucose response, and running capacity (Fig. 1F). However, we observed a tremendous range in HFD response among BXD strains: The body weight in some strains is unchanged (e.g., BXD68), whereas others nearly double in size (BXD44) (Fig. 1C). Next, we examined the transcriptome data (Affymetrix Mouse Gene 1.0 ST microarrays), in which 25,099 annotated probe sets were quantified. Of these, 21,970 were designated as protein coding, with the unaligned transcripts corresponding to non-coding genes such as open reading frames and putative Riken cDNAs or genes that only have unreviewed UniProt identifiers. When transcript levels were compared across diets, many of those most strongly modulated conform to expectations from the literature—e.g., Fosl1 is down-regulated in HFD (21) and Pparg is up-regulated in HFD (22) (Fig. 1G)—although the cause of variation in other top genes is less clear (e.g., Thnsl1). Likewise for metabolites; some were observed to be strongly affected by dietary and genetic factors in both diets [e.g., farnesyl pyrophosphate (FPP)], and others fluctuated in only one diet (e.g., allyl isothiocyanate) (Fig. 1H). Network analysis of large and metabolically relevant gene sets in these omics layers, such as cholesterol biosynthesis (Fig. 1I), showed high levels of enrichment in transcript covariation compared with noise, indicating that the variant transcript levels are functionally linked and physiologically relevant (described in more detail later). We then assessed the data generated by SWATH-MS proteomics (3). As SWATH-MS is an emerging technology, we performed several additional checks, including the technical error, the biological error within cohort, and the errors within diet and across all samples (Fig. 1J). Reproducibility was excellent, with median technical error being ~8% of overall variation in protein levels. Similarly, variation within biological replicates was much lower than variation across the genotypes or dietary conditions (Fig. 1J).
Next, we examined transcript-protein relationships. Of the 2622 unique proteins quantified in all cohorts, 2600 aligned to measured transcripts. Spearman correlation analysis was performed for all pairs in both diets independently. The data indicated that 1004 transcript-protein pairs correlate at nominal significance in CD (raw P < 0.05) (Fig. 2A) and 938 in HFD cohorts. Of these, 637 pairs (~25%) correlated at least nominally significantly in both diets (Fig. 2B, green, purple, and red points). This moderate—although still highly significant—correlation between genes’ transcripts and proteins is in line with previous population studies, which examined smaller numbers (hundreds) of such pairs (19, 23). Variation in any given transcript or protein’s expression within a single tissue in normal population data sets can be highly variable. Among the 2600 transcript-protein pairs, 90% of transcripts vary between 1.4- and 4.0-fold across all samples. The magnitude of this variance strongly influences cross-layer correlation. When the 2600 paired transcripts are binned into 10 groups based on expression range across CD cohorts, the highest bin has 260 transcripts with ≥2.8-fold range, whereas the lowest bin has 260 transcripts with ≤1.4-fold range. Correspondingly, 56% of transcript-protein pairs correlate at ρ ≥ |0.32| (raw P ≤ 0.05) in the top bin, versus only 20% in the lowest bin (Fig. 2C). Fundamentally, a protein cannot exist if there is no corresponding transcript; thus in some sense, 100% of transcript-protein pairs could be considered correlated. Conversely, we observe that only ~15 to 20% of transcript-protein pairs are reactive to small changes in the other’s expression (i.e., the lower bins) (Fig. 2C).
Fig. 2. Multilayer analysis of associations and causality.
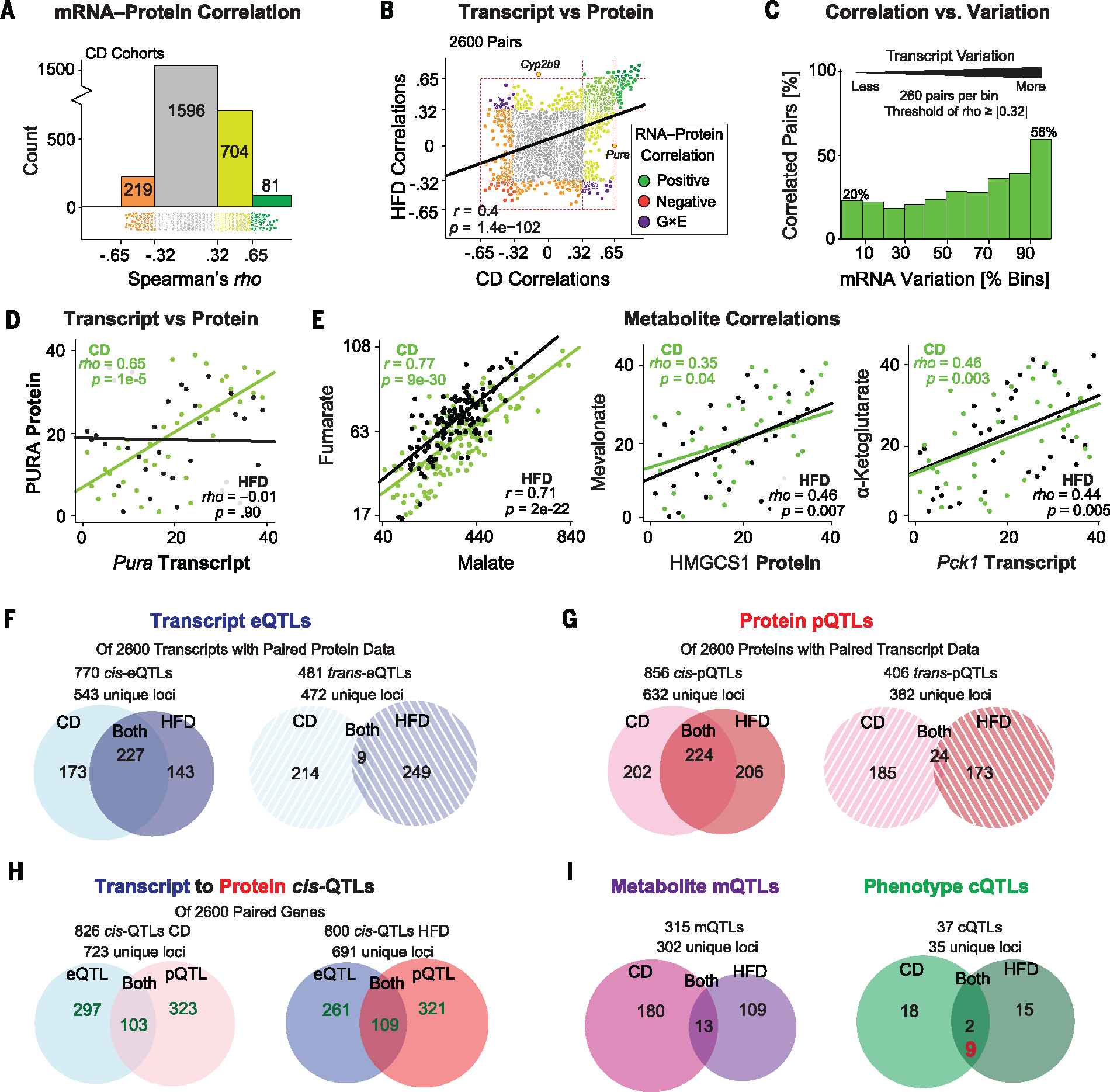
(A) Histogram of 2600 transcript-protein pair Spearman correlations in CD. ρ = 0.32 corresponds to a nominal P < 0.05. ρ = 0.65 corresponds to Bonferroni-corrected significance. (B) Correlation plot of transcript-peptide Spearman correlation coefficients in CD against HFD. (C) Transcript-protein correlation prevalence in CD cohorts, binned by transcript variation. Among the top 10% most variable transcripts (260 pairs), 56% of pairs correlate, in contrast to only 20% of pairs in the lowest bin. Nominal significance cutoffs are used, so ~5% of matches in each bin are false positives. (D) The transcript Pura correlates significantly with its protein in CD but not in HFD. (E) Malate and fumarate, two adjacent metabolites in the TCA cycle, correlate strongly. Several other cross-layer correlations are observed between metabolites and their adjacent enzymes in major metabolic pathways. (F) Venn diagram and count of all cis- and trans-eQTLs across diets for the 2600 transcripts with matching protein measurements. (G) Venn diagram and count of all cis- and trans-pQTLs for the same 2600 proteins. (H) Overlap between cis-eQTLs and cis-pQTLs in both diets. Fifty-nine genes map to cis-QTLs in all four data sets (intersection not shown). (I) Venn diagram of all mQTLs and cQTLs in both diets. In red for cQTLs: overlapping cQTLs that are genome-wide significant in one diet (LRS ≥ 18) and locally significant in the other (LRS ≥ 12).
As with phenotypes and transcripts, G×E effects were observed in transcript-protein correlations, such as for the several dozen transcript-protein pairs whose correlation segregated depending on diet (Fig. 2B, purple dots). For the 137 most significant correlations—those that met the Bonferroni-corrected significance threshold (corresponding to ρ = ±0.65) in at least one diet—135 correlated at least nominally significantly (P < 0.05) in the other diet with the same directionality. For the other two, Cyp2b9 and Pura, strong correlation was observed in one diet and no correlation in the other (e.g., Fig. 2D). This frequent discrepancy between variation in transcript and protein levels indicates that reanalyzing metabolic pathways using more comprehensive proteomic coverage can identify unknown biological mechanisms. Last, we examined the metabolomics layer. Here, metabolite signatures of 979 unique mass-to-charge ratios (m/z) were measured in 357 liver samples using a time-of-flight MS approach (24). These 979 features were then aligned to specific chemical signatures using previously assembled reference libraries (24). Initial data quality checks were performed by analyzing successive metabolites within pathways, such as the tricarboxylic acid (TCA) cycle and glycolysis. Clear connections were frequently observed between consecutive metabolites of a given pathway, such as between malate and fumarate (Fig. 2E). Although metabolites do not fit as neatly into the direct relationships of gene-transcript-protein, the measurement quality and the physiological relevance of metabolite variation may be examined through relationships between the transcript and protein levels of different enzymes with their up- or downstream metabolites. In this analysis, cross-dimensional correlations between known factors were frequently observed, including for cholesterol biosynthesis, glycolysis, and the TCA cycle (25) (Fig. 2E). Together, these validation steps confirm the general data quality and reliability and the potential of a multilayered analytical approach.
Metabolic relationships to multilayered data
We next sought to identify causal genetic mechanisms that determine molecular expression levels of any of the omics layers through QTL analysis. The cohorts in both dietary states were analyzed for eQTLs, pQTLs, mQTLs (metabolite), and cQTLs (clinical phenotype). Across all 2600 transcripts for which we generated associated protein data, 543 genes mapped to 770 significant cis-eQTLs and 472 genes mapped to 481 distinct trans-eQTLs (Fig. 2F) (QTLs detected in both diets are considered twice). Of the 543 genes with cis-eQTLs, 227 mapped consistently across both diets (41%), whereas trans-eQTLs rarely overlapped (2% overlap detected, with ~0.2% overlap expected by chance) (Fig. 2F). At the protein level, 632 distinct genes mapped to 856 cis-pQTLs, and 382 genes mapped to 406 distinct trans-pQTLs (Fig. 2G). Across diets, we observed that 35% of cis-pQTLs mapped in both diets, similar to the ratio for transcripts. Consistent trans-pQTLs were still quite rare, albeit more common than for transcripts (~6%). Roughly 4% of examined genes mapped as cis-eQTLs and cis-pQTLs in at least one diet (103 or 109 versus 2600), whereas of genes with a cis-QTL of any sort, roughly 85% were unique to the transcript or protein level (103 of 826 for CD and 109 of 800 for HFD were shared cis-QTLs) (Fig. 2H). Fifty-nine genes (2.2%) mapped to cis-QTLs in both layers and both diets. For the metabolite layer, 315 significant mQTLs (LRS ≥ 18) were detected, of which 13 mapped consistently in both dietary states (~4%). For phenotypes, we calculated 37 significant cQTLs, of which 2 mapped significantly to the same locus in both diets: movement activity [caused by Ahr (17)] and heart rate (causal gene unknown). To increase the scope of QTLs consistent across diets, one dietary data set may also be used to identify a hypothesis at system-wide significance (LRS ≥ 18), and the other diet may be used to test the hypothesis at locus-specific significance (LRS ≥ 12). In doing so, an additional 7 cQTLs are observed as consistent in both diets (Fig. 2I, red number).
Solving QTLs: Finding the quantitative trait gene
For cis-QTLs, the causal factors can be quickly identified: With few exceptions, they will be driven by variants within the gene itself or immediately adjacent. For trans-QTLs, mQTLs, and cQTLs, the identification of the causal quantitative trait gene (QTG) is challenging due to the width of the QTLs. In the BXDs, QTLs calculated using 40 strains are typically 2 to 8 Mb wide, with an average of 10 genes per Mb (14). We first examined the 24 genes with trans-pQTLs that were observed in both dietary cohorts to search for QTGs and downstream effects. One of these 24 genes is Bckdha, which encodes the E1 alpha polypeptide of the BCKD complex. The BCKDHA protein levels map to a trans-pQTL on chromosome 9 in both diets, whereas no such trans-eQTL is observed for the Bckdha transcript (Fig. 3A). Strikingly, this locus contains the E1 beta polypeptide, Bckdhb, which itself has cis-pQTLs and cis-eQTLs in the BXDs (19). Bckdha and Bckdhb encode the E1 subunit of the BCKD complex, which, together with the E2 (Dbt) and E3 (Dld) subunits, regulates the breakdown of branched-chain amino acids (BCAAs). Variants in either E1 subunit can lead to an inborn error of metabolism called type 1 maple syrup urine disease (MSUD) (26), the biochemical features of which some strains of the BXDs are known to exhibit (19). Notably, the transcript levels of these four genes coding for the complex components have no strong association, whereas the expression levels of the proteins are strongly coupled (Fig. 3B). That is, genetic variants in Bckdhb are causal for protein level variation in BCKDHA and BCKDHB, which in turn correlate strongly positively with dihydrolipoamide branched chain transacylase E2 (DBT) and dihydrolipoamide dehydrogenase (DLD) levels in both diets: the levels of all four proteins are linked. Conversely, although Bckdhb gene variants cause transcriptional changes in Bckdhb mRNA (a cis-eQTL), there is no effect on Bckdha mRNA, nor any correlation with Dbt or Dld. Although a significant difference in the BCAA/alanine ratio across the BXDs has been observed and linked to the Bckdhb allele (19), we observe no association with other metabolic hallmarks of MSUD, such as insulin or glucose levels (Fig. 3C). However, such a link may only be observed in more exacerbated states, such as in cohorts fed diets high in BCAAs, as suggested by prior literature (27). This example highlights the importance of examining protein levels in the diagnosis and elucidation of other metabolic diseases and underlines the possibility of using the BXDs as a MSUD model. Additionally, this finding highlights the possibility that the expression levels of proteins within multigene complexes may be more tightly coregulated than are their transcripts (e.g., Fig. 3B).
Fig. 3. Identifying the QTGs and causal mechanisms driving QTLs.
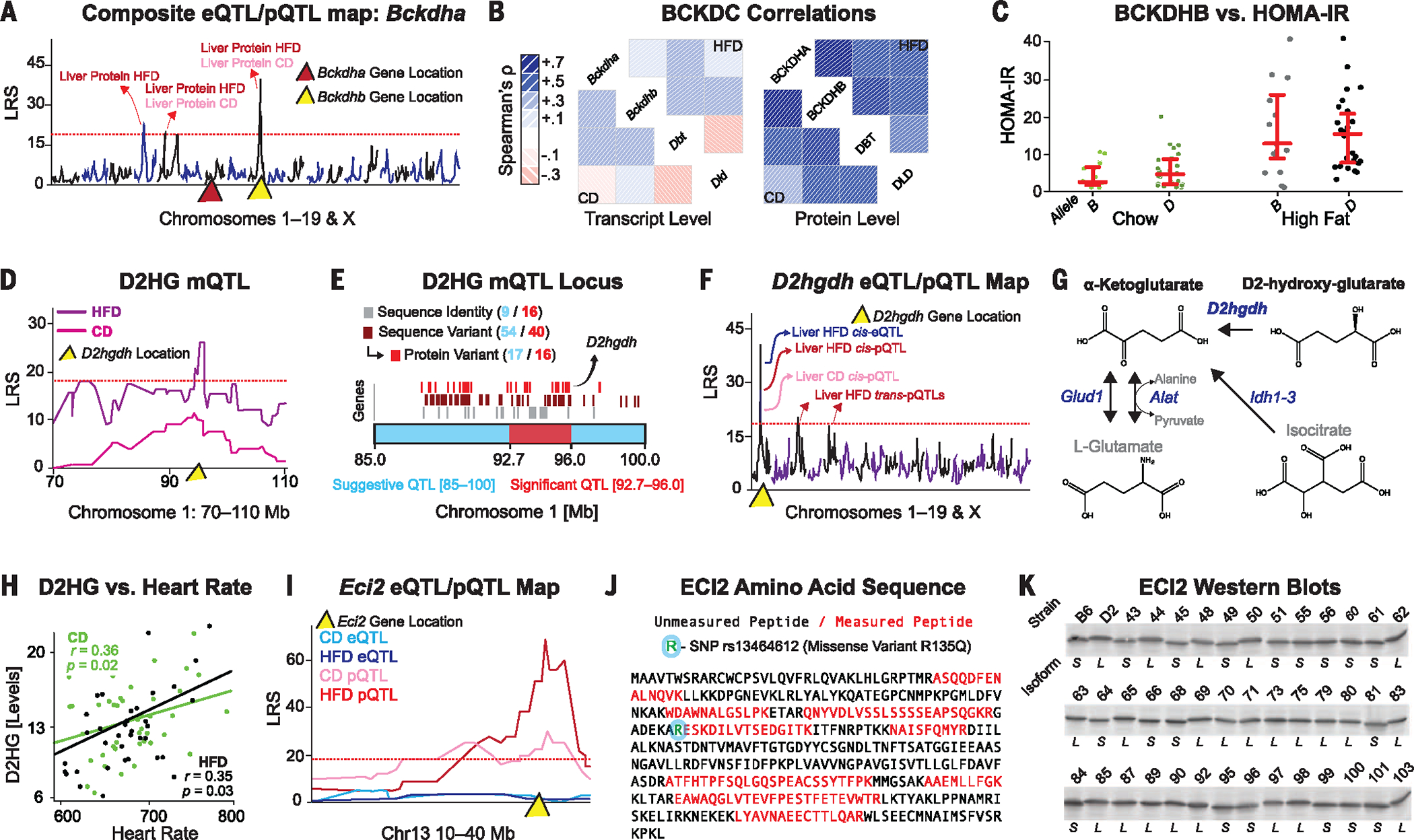
(A) Combined QTL map of Bckdha transcript and protein in both diets. Significant trans-pQTLs map to Bckdhb (yellow triangle) on chromosome 9, whereas no cis-QTLs map to Bckdha on chromosome 7 (red triangle). (B) Spearman correlation matrices of the four subunits of the BCKDC at the transcript or protein level in both diets. (C) Homeostatic model assessment for insulin resistance (HOMA-IR) is significantly increased in HFD cohorts compared with CD (P = 2 × 10−6, Welch’s t test), but no association is seen between Bckdhb allele and HOMA-IR in either dietary cohort. (D) D2HG maps significantly to chromosome 1 in the HFD cohort. (E) This locus contains 56 genes, of which 16 have a major genetic variants variable, including D2hgdh. (F) Composite eQTL and pQTL map for D2hgdh. The protein maps as a cis-pQTL in both diets to the same chromosome 1 locus, whereas only the HFD transcript levels map to a cis-eQTL. (G) D2hgdh drives one of several pathways generating α-ketoglutarate. (H) D2HG is positively associated with heart rate in both diets in a Pearson correlation. (I) Eci2 exhibits no cis-eQTLs, but yields significant cis-pQTLs in both diets. (J) Peptide sequence analysis of ECI2, with the nine measured peptides and the single missense mutation highlighted. (K) ECI2 Western blots show two distinct molecular weight bands depending on the BXD genotype.
We then aimed to identify candidate QTGs behind any of the 302 distinct mQTLs using orthogonal pQTL data. All 856 genes with cis-pQTLs were compared against these mQTLs, with the hypothesis that genes with cis-pQTLs are more likely to be causal for other QTLs mapping to their locus (28). This process highlighted several potential pQTL/mQTL links. In particular, D2HG maps to a locus on chromosome 1 containing 56 genes (Fig. 3, D and E). Among these 56 genes is D-2-hydroxyglutarate dehydrogenase (D2hgdh), which maps consistently to cis-pQTLs (Fig. 3F) and correlates negatively with the upstream metabolite D2HG (ρ = −0.37 and ρ = −0.48 in CD and HFD, respectively). Although the other 55 genes in this locus could contribute to this mQTL, D2hgdh is known to convert D2HG to α-ketoglutarate in the mitochondria (29) (Fig. 3G). In humans, variants in D2HGDH have been linked to severe disease traits such as cardiomyopathy and motor difficulties (20). In the BXDs, we observe moderate but consistent connections between D2HG and cardiovascular phenotypes such as heart rate (Fig. 3H) and exercise capacity (moderate negative correlation), indicating that some mild aspects of the disease phenotype may manifest in this population, even under nonstressed conditions.
The SWATH proteomics analysis is also able to identify nonsynonymous sequence variants across the BXDs, which are detected as peptide-specific cis-pQTLs [cis-peptide(pep)QTLs]. To demonstrate this, we highlight enoyl-CoA delta isomerase 2 (Eci2), a mitochondrial enzyme involved in fatty acid oxidation. Nine distinct peptides were quantified for ECI2, of which one displayed a striking cis-pepQTL in both diets. Interestingly, there are no cis-eQTLs for ECI2 at the gene or exon level (Fig. 3I). Variant analysis revealed a nonsynonymous change (R135Q, rs13464612) adjacent to this peptide that is predicted to abolish the trypsin cleavage site (Fig. 3J). Furthermore, this cis-mapping variant tracks with a small change ECI2 migration in SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 3K), indicating a change in the protein that is not observed at the transcript level. Notably, these analyses highlight the ability to detect putative protein isoforms—an additional source of molecular variation underlying complex phenotypes.
High-dimensional metabolic networks
As shown above, large, multilayered quantitative omics data sets can be used to map and solve QTLs at high throughput. Perhaps the more unique characteristic of more comprehensive measurement techniques, however, is that the resulting data may be used to model extended pathways or functional networks with dozens of proteins and metabolites acting in tandem. To examine this possibility in the BXDs, we performed ontology enrichment analysis on 226 KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (30) to identify which other metabolic pathways are best covered by the proteomic and metabolomics data and may benefit from a transomic approach. Among the most enriched pathways are those involved in fatty acid metabolism and storage, such as the peroxisome proliferator-activated receptor (PPAR) pathway. These sets of genes and pathways are furthermore known to be variable in the BXDs and lead to overt differences in fatty liver and metabolic changes in the liver, including metabolic signatures of liver stress such as increased alanine transaminase (ALAT) and disease phenotypes such as fatty liver disease (21). Of the 41 genes in this pathway, 25 were measured at the protein level, along with a handful of relevant metabolites [e.g., FPP, ALAT, and high-density lipoprotein (HDL)]. Interestingly, although the transcripts and proteins for any single gene did not strikingly correlate (Fig. 4A), significant enrichment of correlations between transcripts and proteins was observed in other parts of the pathway (Fig. 4A, dashed circle), indicating interactions between these two layers and between related metabolic pathways. In turn, the variations in these transcripts and proteins contributed to proximal metabolic changes [e.g., low-density lipoprotein (LDL), HDL, and FPP levels] and to related global phenotypes such as total fat and liver mass. This network is also highly responsive to diet, with clear differences between CD and HFD cohorts in key genes and metabolites (Fig. 4B). While these findings are expected (21), they further emphasize the reliability of cross-layer omics analysis and furthermore provide better networks than those expected using random gene sets (Fig. 4C).
Fig. 4. Network analysis.
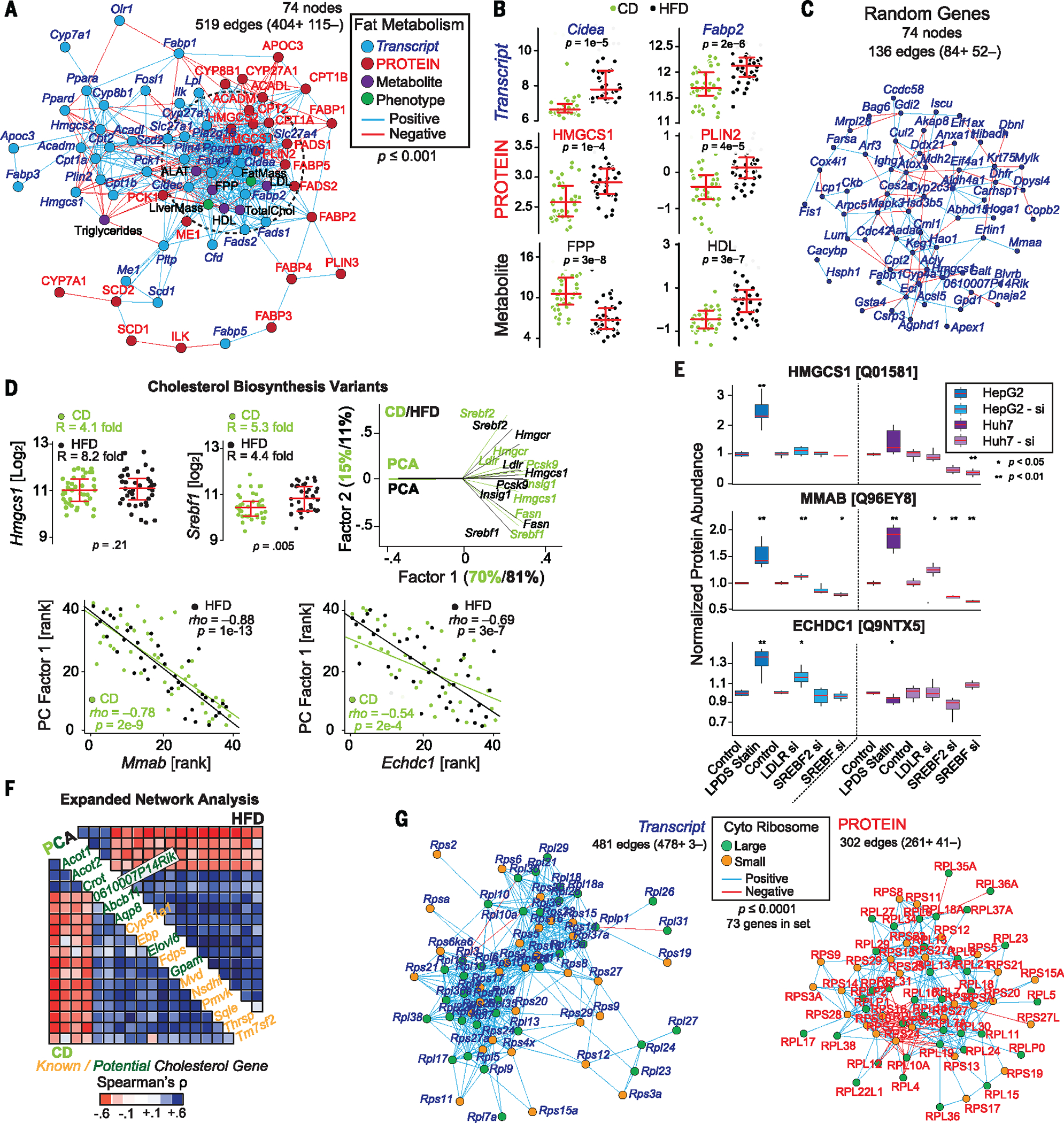
(A) Spearman correlation network showing mixed genes involved in fat metabolism at the transcript and protein level, along with key metabolites and phenotypes. The dashed circle represents the core enriched gene set. Edges are significant at P < 0.001 for a positive (blue) or negative (red) correlation. (B) Diet-dependent expression of key genes and metabolites involved in fat metabolism; P values are for Welch’s t test. (C) A Spearman correlation network of 74 transcripts taken at random from the list of 2600 genes measured at the transcript and protein level, using the same network analysis. Edge counts correspond to the level expected from noise. (D) (Left) Hmgcs1 and Srebf1, as well as other transcripts and proteins in the cholesterol biosynthesis pathway, are highly variable in the BXDs. (Right) PCA of a set of eight cholesterol biosynthesis genes shows that their variances are highly explained by a single factor. (Bottom) Two candidate cholesterol genes, Mmab and Echdc1, which correlate with PC1 in both diets. (E) In vitro validation of HMGCS1, along with two proteins not known to be involved in cholesterol metabolism—MMAB and ECHDC1—which clustered with known cholesterol genes. MMAB and ECHDC1 both respond like HMGCS1 to lipid-deficient serum and statin treatment or to knockdown of LDLR, SREBF2, or SREBF1/2, suggesting that they are indeed involved in cholesterol metabolism. (F) Unbiased Spearman correlation matrices of the first PC in CD (bottom left) and HFD (top right) conditions with other genes turned up many known cholesterol-regulatory genes (orange) as well as new candidates (green). (G) Transcript and protein networks for the 73 genes with paired transcript-protein data in the cytosolic ribosome complex. Both were highly enriched, although with tighter coregulation at the transcriptional level. Edges represent Spearman correlations with P < 0.0001.
Within the fat metabolism gene set, we observed particularly high levels of variability in key genes in cholesterol biosynthesis such as Srebf1 and Hmgcs1 (Fig. 4D, left). Furthermore, key Srebf target genes, including Hmgcr, Pcsk9, Insig1, and Fasn, all clustered tightly in principal components analysis (PCA), with the first principal component of eight core genes explaining 70 and 81% of the total variation in CD and HFD, respectively (Fig. 4D, right). Correlation analyses of the first principal components across the transcriptome and proteome data sets yielded several hits that correlate strongly and independently in both CD and HFD cohorts. These genes included several not known to be involved in cholesterol biosynthesis, such as Mmab and Echdc1 (Fig. 4D, bottom), as well as a number of genes involved in cholesterol that were not included in input, such as Fdps, Mvd, and Dhcr7. We selected two hits, Mmab and Echdc1, which are not well reported in cholesterol literature, for in vitro validation. In HepG2 and Huh7 cell lines, we examined the effect of cells in lipoprotein-deficient serum (LPDS) treated with statins and the effect of Ldlr, Srebf2, or Srebf1/2 small interfering RNA (siRNA) knockdown—conditions that modulate different aspects of the cholesterol biosynthesis pathway (31–33). We observed that MMAB and ECHDC1 proteins are modulated strongly by LPDS + statin treatment and behave similarly (although not identically) to HMGCS1, an established Srebp2 target protein and a key regulatory gene in cholesterol synthesis. Similar genome-wide analyses indicated additional candidate cholesterol-related genes, such as Aqp8, 0610007P14Rik, and Gpam (Fig. 4F). Some of these genes have been identified in previous cholesterol genome-wide association studies, such as for Mmab with blood HDL levels (34, 35), whereas other candidates are likely involved in tangential pathways (e.g., Acot1 and Acot2 are involved in lipid metabolism).
We next examined gene sets that form large and cohesive protein structures, such as the ribosome. Pairwise covariance analysis of transcripts coding for ribosomal proteins have previously been shown to form a tightly connected network (36). To extend this analysis to the protein level, we systematically generated Pearson correlation networks for the 73 genes in the ribosome family that were measured at both the transcript and protein level in all samples. As for the transcripts, the proteins clustered into enriched correlation networks (Fig. 4G). That the ribosomal genes are coregulated is expected, but it illustrates that the data are reliable enough to reveal components of functional modules and thus to support systems analyses.
Following these proofs of concept, we determined which metabolic pathways were most comprehensively covered in the multilayered data sets and triaged them for further analysis, with particular focus on mitochondria-related sets. Among the groups with the most complete protein coverage was the oxidative phosphorylation gene set (oxphos, hsa00190). This gene set is defined by 133 genes, of which 70 were quantified at the protein level. Of these, 67 were also quantified at the transcript level—all except the mitochondrial-encoded ND4, ND5, and ATP8. We performed network analyses on these data and observed highly significant positive correlation networks (Fig. 5A), with the protein network somewhat more enriched than the transcript network. This layer-specific difference in network structure is perhaps logical, given that the proteins are bound together in stoichiometry in their functional complexes, whereas the equivalent transcripts are not. The oxphos network was not strongly affected by diet, with only four proteins and 16 transcripts being variable at the permissive cutoff of P < 0.01. (Zero proteins and four transcripts—Ndufb5, Cox7a2, Atp5b, and Ndufa7—are significantly influenced at adjusted P < 0.05.) Furthermore, observed dietary differences at the transcript level did not reliably indicate any similar change in protein levels (Fig. 5B, outermost green bands.)
Fig. 5. Variable mitochondrial phenotypes in the BXDs.
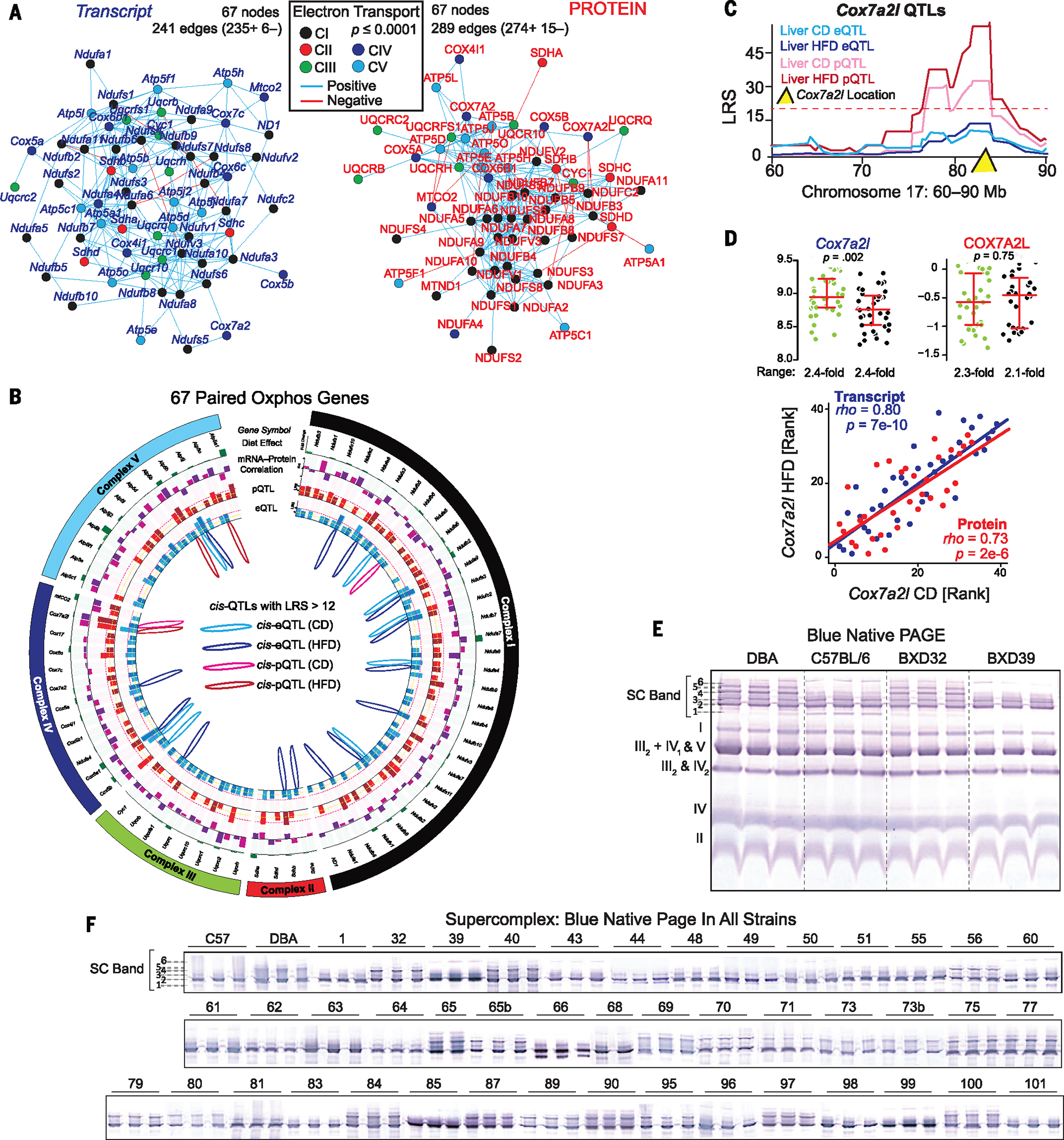
(A) The oxphos protein Spearman correlation network is somewhat more tightly coregulated than the transcript network. In particular, CI proteins cluster more tightly than CI transcripts (black nodes). (B) Circos plot of 67 ETC genes. Green bar ring: effect of diet, relative change between medians. Light green: transcript; dark green: protein. Purple bar ring: correlation between transcript and protein in CD (light purple) or HFD (dark purple). Red bar ring: LRS of peak pQTLs in CD (light red) and HFD (dark red). Blue bar ring: LRS of peak eQTLs in CD (light blue) and HFD (dark blue). Inside: drawing of significant cis-QTLs (LRS ≥ 12). Significant trans-QTLs (LRS ≥ 18) are not drawn. (C) Diet-consistent cis-pQTLs were observed only for COX7A2L, which does not map to significant cis-eQTLs. (D) (Top) The Cox7a2l transcript is affected by diet, whereas both transcript and protein are highly variable across genotype. (Bottom) Expression is consistent across diets within the transcript and protein level, despite the presence of dietary effect in mRNA and its absence in protein. (E) BN-PAGE for four strains with three biological replicates. Individual complexes are labeled. Several distinct upper SC bands are observed, labeled initially as 1 through 6. (F) Upper SCs for all CD cohorts (several independent gels are aligned and spliced together). SCs were quantified in binary fashion by presence (+1) or absence (0) of a particular band.
Consequences of oxphos variation
Given the relatively small effect of diet on transcript and protein levels, surprisingly few QTLs were observed consistently across diet: only six transcripts and one protein (Fig. 5B). For the only such protein, COX7A2L, no corresponding cis-eQTLs were observed (Fig. 5C). Interestingly, the Cox7a2l transcript exhibited significantly different expression in response to diet, whereas the protein levels were unaffected (Fig. 5D, top). The transcript and protein levels were highly correlated across dietary cohorts, suggesting a strong genetic influence on both Cox7a2l and COX7A2L levels, independent of the observed dietary influence on the transcript (Fig. 5D, bottom). Because mitochondrial transcript and protein networks are highly variable and coregulated in the BXDs (Fig. 5A), we hypothesized that these may be associated with clear differences in mitochondrial structures and phenotypes. To broadly test this idea, we performed blue native (BN)–PAGE analysis on isolated mitochondria from three biological replicates across all strains, using the same liver samples as before. Mitochondrial complex levels and formations varied across the BXDs (Fig. 5E), with particularly striking differences in SC formations (Fig. 5F, all strains). The differences in SC patterns across strains, and the consistency within strains, indicate that complex and multi-factorial genetic interactions are driving the mitochondrial effects, at least in part by determining the modularity of supermolecular functional units. To uncover these factors, we assigned the SC bands as quantitative traits, with all bands quantified as “on” (+1) or “off” (0). These traits were then mapped for QTLs. For bands 4 and 5, the data indicated that they are driven by a locus on chromosome 17 (Fig. 6A), containing 35 genes. Notably, this region includes Cox7a2l, and overlaps with its cis-pQTLs (Fig. 5C). Cox7a2l has been recently indicated as causal for certain types of SC formation between different inbred mouse lines (37), although this effect has been debated (38). We thus sought to examine how this locus can affect specific SC formation and whether Cox7a2l is indeed causal.
Fig. 6. Tissue variance in SC formation.
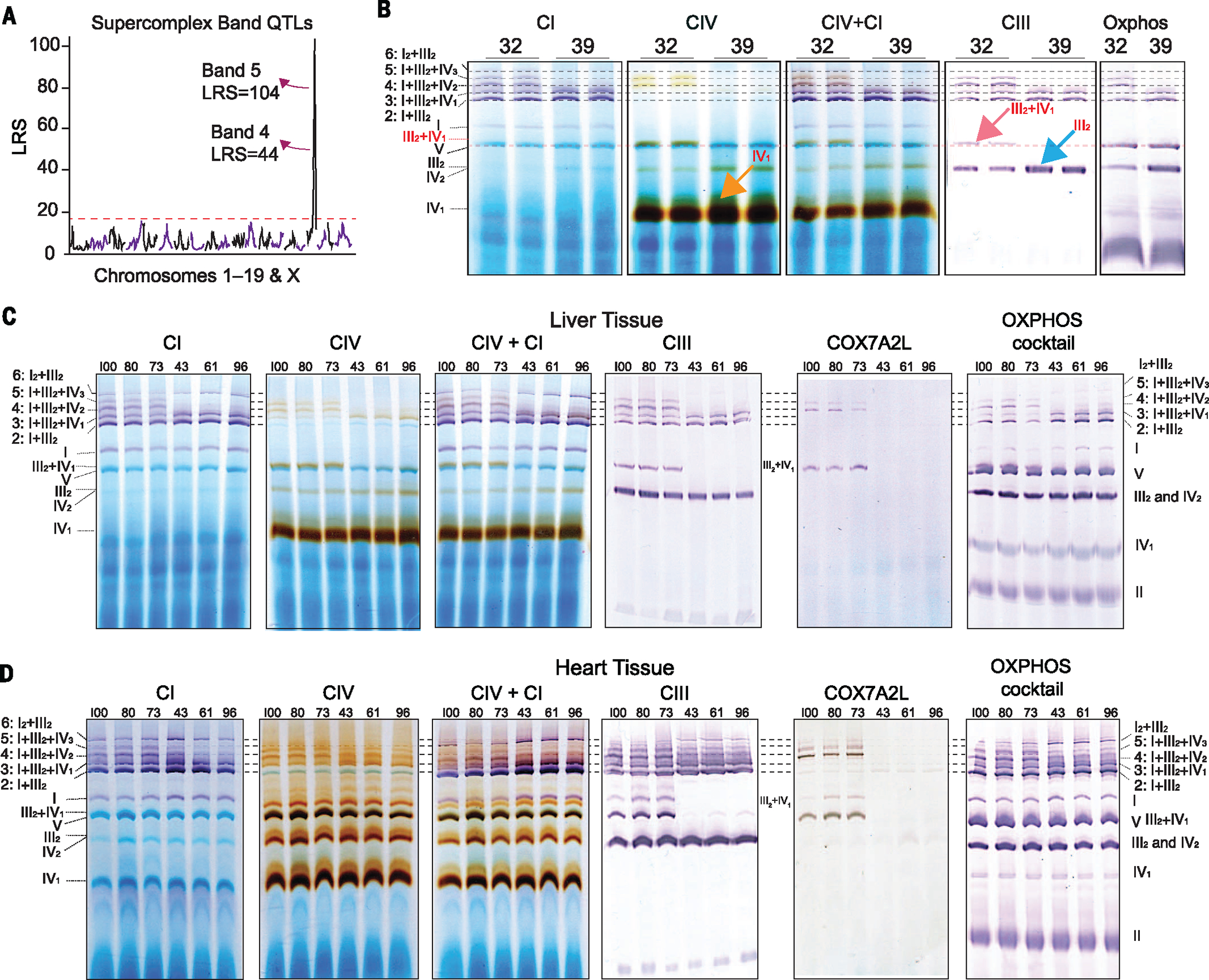
(A) SC bands 4 and 5 mapped significantly as cQTLs to a locus on chromosome 17. (B) In-gel activity assays were performed in the liver tissues to determine SC’s composition and relation to COX7A2L. Bands 2 to 5 could be identified confidently as CI + CIII2 + variable numbers of CIV (0 to 3). (C) In-gel activity assays from livers of six additional BXD strains—three with the B6 allele of Cox7a2l (BXD73, BXD80, and BXD100) and three with the D2 allele (BXD43, BXD61, and BXD96). COX7A2L is present in bands 4 and 5 for strains with the D2 allele. (D) In-gel activity assays from hearts of the same individuals as above. COX7A2L is absent in bands 4 and 5 and III2+IV1 in strains with the B6 allele and present in strains with the D2 allele. Unlike liver, bands 4 and 5 are observed in all strains, albeit at lower levels in strains with the B6 allele of Cox7a2l, indicating tissue-specific differences in SC formation.
In mammals, SCs are formed by different stoichiometric combinations of three of the five individual complexes in the electron transport chain—complexes I, III, and IV—although it is poorly understood how they are formed or how different complexes influence overall mitochondrial function (39). To determine the stoichiometry of the observed bands, we performed in-gel activity assays for complex I (CI), CIV, CI+IV, and CIII using eight strains: four with the B6 allele of COX7A2L (e.g., BXD39) and four with the D2 allele (e.g., BXD32) (Fig. 6, B and C). We observed that SC formations with multiple copies of CIV—bands 4 (I+III2+IV2) and 5 (I+III2+IV3)—are inhibited in strains with B6-type Cox7a2l. Furthermore, a large increase in free/unconjugated CIII (blue arrow) and CIV (orange arrow) was observed in those strains with B6-type Cox7a2l, indicating indeed a lack of the assembly of these complexes into SCs. This analysis also indicated other variable SC formations at lower molecular weights, particularly complex III2+IV1 (dashed red line and pink arrow, Fig. 6B). Western blot analysis for COX7A2L shows its presence in SC bands 4 and 5 in D2-type strains (along with band III2+IV1; this complex was “hidden” under CV in the total BN-PAGE) and its complete absence in B6-type strains (Fig. 6C). This effect of COX7A2L on SC formation is noted broadly across strains with B6 or D2 alleles of the gene (e.g., Fig. 5F). However, the variant COX7A2L isoform does not seem to influence the formation of SC bands 2 (I+III2) or 3 (I+III2+IV1).
We next examined the possibility that the Cox7a2l may not be the driving factor for SC variation in the BXDs and that the nearby leucine-rich pentatricopeptide (PPR) motif–containing gene (Lrpprc) may be causal (38). Lrpprc is 1.2 Mb downstream from Cox7a2l, and four BXD strains examined have recombinations between this interval: BXD56 has the D2 allele of Cox7a2l and the B6 allele of Lrpprc, whereas BXD44, BXD49, and BXD99 have the opposite. For these strains, SC bands 4 and 5 are absent in BXD44, 49, and 99 and present in BXD56 (Fig. 5F), as expected if Cox7a2l is causal. Furthermore, neither the transcript nor protein measurements of Lrpprc yield QTLs in the BXD livers. This finding does not preclude the possibility that LRPPRC is involved in SC formation. However, it is not the causal gene for the variable SC patterns observed in the BXDs. To further investigate the effects of COX7A2L on SC formation, we extracted mitochondria from the hearts of the same individuals as the liver. Again, SC patterns were strikingly different depending on genotype (Fig. 6D), yet the SC bands representing III2+IV1, I+III2+IV2, and I+III2+IV3 are present in the hearts of strains with the B6 isoform of Cox7a2l, albeit at diminished levels compared with their D2-type counter-parts. Taken together, these data provide substantial evidence to show that COX7A2L is involved in the formation of many CIV-containing SC formations, yet that its influence varies between tissues. Additionally, these data provide a conceptual advancement in the current knowledge of SC formations in B6 by showing that I+III2+IV1 is in fact present [previously reported as absent (37)] and that variants in COX7A2L are causal for many of the differences between B6 and other common inbred strains, particularly D2.
Discussion
We have examined genetically and environmentally variant cohorts of the murine BXD GRP to determine how changes in the genome and environment interact to influence cellular processes and overall variation in complex metabolic traits. To model the molecular factors underlying phenotypic differences across the BXD population, we have applied an in-depth, multilayered approach including genetic, transcriptomic, proteomic, and metabolomic measurements. Systems-level technologies now permit the multilayered measurements of thousands of molecules associated with many physiological processes, at high throughput and with a high degree of quantitative accuracy and reproducibility. We show the first application of SWATH-MS in a diverse mammalian population by quantifying 2622 proteins measured in all 80 cohorts. As in earlier smaller-scale studies (40), genes’ transcript levels are only moderate predictors for the protein levels. The predictive value depends strongly on how variable the transcript (or protein) is. Studies that induce massive transcriptional changes with gain- or loss-of-function techniques can rely on the fact that the resulting mRNA change will almost invariably be reflected in the corresponding protein’s level. In contrast, it cannot be assumed that relatively subtle expression changes in a particular transcript will manifest at its protein level. This latter situation is particularly critical for in vivo population studies, where the top leads identified through microarray or RNA sequencing (RNA-seq) analyses frequently have far more modest differences than findings from in vitro studies. Likewise, genetic variants driving differential transcript expression (e.g., cis-eQTLs) are furthermore only infrequently mirrored at the protein level (e.g., matched cis-pQTLs) and vice versa. Measurement of both transcriptomics and proteomics in tandem appears essential because each measurement level unveils different aspects of the cellular state and regulatory mechanisms.
This greater scope of data analysis allows the identification of hundreds of causal genetic factors that regulate individual transcript and protein levels (i.e., QTLs), as well as for metabolites and phenotypes. This protein analysis allowed novel identification of variants of ECI2 not predicted by genome or transcript data, as well as further delineation of variants affecting the expression of the four proteins in the BCKDC—effects not visible at the transcript level—which lead to a mild form of MSUD in the BXDs. In another, we could readily identify the causal factors driving variance in the metabolite D2HG to the protein D2HGDH. Moreover, however, the increased scope of these data facilitates the modeling and analysis of entire pathways. The PPAR and cholesterol biosynthesis pathways are highly variable in the BXDs due to both genetic and environmental factors and are known to influence the development of metabolic diseases, including fatty liver. Furthermore, we were able to use network analysis to identify Mmab and Echdc1 as likely cholesterol-related genes, which we confirmed through in vitro analysis. For the oxphos gene network, the BXDs display strong levels of variation in both gene expression and the overall mitochondrial assembly of complexes in the ETC. Using the proteomic data, we identified COX7A2L as causal of major variants in SC organization—particularly, a lack of three specific SC bands (III2+IV2, I+III2+IV2, and I+III2+IV3)—and a consequent increase of the unconjugated levels of complexes IV1, III2, and IV2. However, the mechanism of assembly appears strongly tissue dependent: These SCs can be formed in the heart, even in the absence of COX7A2L. Notably, the patterns of mitochondrial complexes are consistent across biological replicates, indicating that the many differences across strains and tissue are due primarily to differential regulation of mitochondria by the nuclear genome. In each of these pathways, the proteomic and metabolomic data extend gene-phenotype links that were previously identified at the transcript level but that were incomplete. To move forward in the analysis of mitochondria and associated disorders, it is hence necessary to analyze the protein levels of all regulators, as well as genetic, environment, and tissue-specific variants. Such implementations of new omics layers will not supersede the now-standard genomic and transcriptomic data sets. Rather, a combined transomic approach can fill in blind spots and assist in defining more detailed metabolic pathways.
Methods
Population handling
BXD strains were sourced from the University of Tennessee Health Science Center (Memphis, TN, USA) and bred at the École Polytechnique Fédérale de Lausanne (EPFL) animal facility for more than two generations before incorporation into the study. We examined 80 cohorts of the BXD population from 41 strains—41 on CD, 39 on HFD—with male mice from each strain separated into two groups of about five mice for each diet (two strains on HFD were lost before tissue collection). We started with 201 CD and 185 HFD mice, and a total of 183 CD and 168 HFD mice survived until they were killed at 29 weeks of age, with all cohorts having three or more individuals surviving to the end except BXD56 HFD, which had two. Strains were entered into the phenotyping program randomly and had staggered entry, typically by 2 weeks. Most strains entered with both dietary cohorts at the same time, with the exception of BXD50, 68, 69, 71, 84, 85, 89, 95, 96, and 101, where CD cohorts entered before HFD cohorts. All cohorts consisted of littermates. HFD feeding started at 8 weeks of age. Cohorts were communally housed by strain and diet from birth until 23 weeks of age and were then individually housed until they were killed at 29 weeks of age. CD is Harlan 2018 (6% kCal of fat, 20% kCal of protein, and 74% kCal of carbohydrates), and HFD is Harlan 06414 (60% kCal of fat, 20% kCal of protein, and 20% kCal of carbohydrates). All mice were housed under 12 hours of light alternated with 12 hours of dark, with ad libitum access to food and water at all times, except before they were killed, when mice were fasted overnight. All mice were housed in isolator cages with individual air filtration, except during the activity wheel test (10 days) when mice were in open-air cages in a room reserved for that test, after which, mice were returned to the filtered isolator cages. Body weight was measured weekly from 8 weeks of age until killing. Killing took place from 9:00 a.m. until 10:30 a.m., with isoflurane anesthesia followed by a complete blood draw (~1 mL) from the vena cava, followed by perfusion with phosphate-buffered saline. Half of the blood was placed into lithium-heparin (LiHep)–coated tubes and the other half in EDTA-coated tubes; then both were shaken and stored on ice, followed immediately by collection of the liver. The LiHep blood taken for plasma analysis was also centrifuged at 4500 revolutions per minute (rpm) for 10 min at 4°C before being flash-frozen in liquid nitrogen. Whole blood taken for cellular analysis was processed immediately after the killing (i.e., after ~1 to 2 hours on ice). Gallbladders were removed, and the livers were cut into small pieces before freezing in liquid nitrogen until preparation into mRNA, protein, or metabolite samples. Liver and blood serum were then stored at −80°C until analysis. All research was approved by the Swiss cantonal veterinary authorities of Vaud under licenses 2257 and 2257.1.
Phenotyping experiments
A visual summary of the phenotyping program is also included in Fig. 1B. At 16 weeks of age, after 8 weeks of dietary treatment, the cohorts underwent their first phenotyping test: 48 hours of respiration measurements in individual metabolic cages (Oxymax/CLAMS, Columbus Instruments). The first 24 hours were considered adaptation, and the second 24 hours were used for data analysis, including analysis of movement, the volume of oxygen inhaled, the volume of carbon dioxide exhaled, and derived parameters of these two, such as the respiratory exchange ratio (RER). One week later, all cohorts underwent an oral glucose tolerance test. Mice were fasted overnight before the test, and fasted glucose was tested with a glucometer at the tail vein. All individuals were then weighed and given an oral gavage of 20% glucose solution at 10 mL per kg of weight. Glucometer strips were used at 15, 30, 45, 60, 90, 120, 150, and 180 min after the gavage to examine glucose response over time. Blood was also collected at 0 (pregavage), 15, and 30 min to examine insulin levels. Two weeks later, at 19 weeks of age, we performed a noninvasive blood pressure measurement using a tail-cuff system (BP-2000 Blood Pressure Analysis System, Series II, Visitech Systems) over 4 days. The first 2 days were considered as adaptation to the apparatus, and the second 2 days were used for data analysis, and all measurements (systolic blood pressure, diastolic blood pressure, and heart rate) were averaged across both days. Outliers on a per-measurement basis were removed, but outlier mice were retained. Two weeks later, at 21 weeks of age, we performed a cold response test. The basal body temperatures of mice were examined rectally, after which mice were placed individually in prechilled cages in a room at 4°C. The cages were the standard housing cages but with only simple woodchip bedding, without supplement (e.g., tissue paper). Body temperature was checked every hour for 6 hours, after which the mice were returned to their normal housing cage. Two weeks later, at 23 weeks of age, the mice were placed individually in regular housing cages for basal activity recording. The housing cages were then placed in laser detection grids developed by TSE Systems (Bad Homburg, Germany). Within the cages, woodchip bedding was retained, but tissue bedding was removed (as it interferes with the laser detection). Food and water were as normal throughout the standard housing, both of which require rearing to reach. The detection grid has two layers: one for detecting X-Y movement (“ambulation”) the other for Z movement (“rearing”). Both measurements are technically independent, although the measurements of movement are strongly correlated (r ~ 0.70). Mice were housed individually for the 48-hour experiment starting at about 10 a.m., with the night cycles (7 p.m. to 7 a.m., with 30 min of both dawn and dusk) used for movement calculations. We have recently published more interpretation and examination of the results from this experiment (17). After this 2-day experiment, all mice performed a VO2max treadmill experiment using the Metabolic Modular Treadmill (Columbus Instruments). For the first 15 min in the machine for each individual, the treadmill was off while basal respiratory parameters were calculated. The last 2 minutes of data before the treadmill turned on are considered basal levels (most mice spend the first few minutes exploring the device). The treadmill then started at a pace of 4.8 m per minute (m/min), followed by a gradual increase over 60 s to 9 m/min, then 4 min at that pace before increasing to 12 m/min over 60 s, then four min at that pace before increasing to 15 m/min over 60 s, then 4 min at that pace, then the speed increased continuously by 0.015 m per second (or +0.9 m/min) thereafter until the end of the experiment at 63.5 min, 1354.5 m, or when the mouse is exhausted. CD cohorts ran against a 10° incline, whereas HFD cohorts were set at 0°. For this test, no mice reached the maximum distance recorded by the machine—all were taken out when exhausted. The distance, maximum VO2, and maximum RER were recorded. Maximums must be consistent across multiple measurements, and not single-measurement spikes, which were removed. Immediately after the treadmill experiment, mice were placed in individual open-air cages with ad libitum access to activity running wheels (Bioseb BIO-ACTIVW-M, Vitrolles, France). The final 24 hours of activity wheel access were recorded for all strains. For certain strains, all 10 days of activity wheel usage was recorded (depending on the availability of the recording system). After the 10th day, at ~25 weeks of age, mice underwent an identical treadmill experiment to that described above at 23 weeks of age. At this point, with the 10 days of voluntary training, three mice “completed” the experiment—two DBA/2Js on HFD and one BXD81 on CD. As before, the test was stopped for all other mice when they had reached exhaustion (considered as falling off the treadmill and inability to recover and continue running). After this experiment, mice were returned to their standard housing cages—individually—for 4 weeks. Mice were fasted overnight before they were killed. Details about killing are described in the previous section. In addition to the body weight measurements taken each week and before each phenotyping experiment, body composition was recorded at 16, 23, and 25 weeks of age—before respiration and the two VO2max experiments. To do so, each mouse was placed briefly in an EchoMRI (magnetic resonance imaging) machine (the 3-in-1, Echo Medical Systems), where lean and fat mass are recorded, along with total body weight, taking ~1 min per individual. Lean mass is used as a corrective factor for respiratory calculations from the Comprehensive Lab Animal Monitoring System (CLAMS). All other tests are normalized to total body weight in our analyses.
Genomics
The parental lines of C57BL/6J and DBA/2J have been previously sequenced (13). Earlier genotype data—~8000 single-nucleotide polymorphisms (SNPs) per line—have been published previously (42). We have made use of a newer build of the genotype, using ~500,000 SNPs per line (unpublished), which helped refine recombination break-points, such that ~99.99% of the genotype of all BXD strains could be inferred. Full sequence data on the parental lines was published separately (18). The lower density (3806 markers) is available on GeneNetwork as well: www.GeneNetwork.org/genotypes/BXD.geno.
BXD sample preparation and analysis
For mRNA, 100-mg pieces of liver tissue were suspended in TRIzol (Invitrogen) and homogenized with stainless steel beads using a Tissue-Lyser II (Qiagen) at 30 Hz for 2 min, followed by a standard phase separation extraction using chloroform and precipitated by isopropanol. mRNA concentration was measured for all samples and then pooled equally for each cohort (i.e., five biological replicates for BXD103 CD became one mixed pool of BXD103 CD). Pooled RNA was cleaned up using RNEasy (Qiagen). The mRNA of all cohorts was prepared in direct series over a ~2-week period. Seventy-six of the 77 cohorts had high-quality mRNA based on RNA integrity numbers ≥ 8.0, indicating that they are suitable for amplification and subsequent microarray analysis. Arrays were run for all cohorts in direct series over a 3-week period using the Affymetrix MouseGene 1.0 ST array at the Molecular Resource Center of Excellence in the University of Tennessee Health Science Center. Data were normalized using the robust multiarray average method (43), then analyzed in GeneNetwork and R.
For liver protein, the ~100-mg liver sample was homogenized with 4-mL radioimmune precipitation assay-modified buffer (1% Nonidet P-40, 0.1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 50 mM tris, pH 7.5, protease inhibitors EDTA-free, 10 mM NaF, 10 mM sodium pyrophosphate, 5 mM 2-glycerophosphate) in a glass-glass tight Dounce homogenizer (Wheaton Science Products) at 4°C. After the homogenates were centrifuged (20,000 g at 4°C for 15 min), the supernatant was collected and kept at 4°C. The pellets were resuspended with urea-tris buffer (50 mM tris, pH 8.1, 75 mM NaCl, 8 M urea, EDTA-free protease inhibitors, 10 mM NaF, 10 mM sodium pyrophosphate, 5 mM 2-glycerophosphate) and sonicated for 5 min, then centrifuged at 20,000 g for 15 min at 4°C. The supernatants from the two steps were combined, and protein concentrations were determined with the bicinchoninic acid protein assay (Thermo Fisher Scientific). For the precipitation and digestion of proteins in each sample, 200 μg of protein was precipitated with six volumes of ice-cooled acetone and kept 16 hours at −20°C. Then proteins were resuspended in 8 M urea/0.1 M NH4HCO3 buffer, reduced with 12 mM dithiotreitol for 30 min at 37°C, then alkylated with 40 mM iodoacetamide for 45 min at 25°C, in the dark. Samples were diluted with 0.1-M NH4HCO3 to a final concentration of 1.5-M urea, and sequencing grade porcine trypsin (Promega) was added to a final enzyme:substrate ratio of 1:100 and incubated for 16 hours at 37°C. Peptide mixtures were cleaned by Sep-Pak tC18 cartridges (Waters, Milford, MA, USA) and eluted with 40% acetonitrile. The resulting peptide samples were evaporated on a vacuum centrifuge until dry, then resolubilized in 2% acetonitrile/0.1% formic acid to 1μg/μL concentration.
For liver metabolites, the ~100-mg liver pieces were homogenized in 1 mL of 70% ethanol at −20°C. Metabolites were extracted by adding 7 mL of 70% ethanol at 75°C for 2 min. Extracts were centrifuged for 10 min at 4000 rpm at 4°C. Clean metabolite extracts were dried in a vacuum centrifuge and resuspended in double-distilled H2O, with volume according to the weight of the extracted liver piece. Quantification of metabolites was performed on an Agilent 6550 quadrupole orthogonal acceleration–time-of-flight (Q-TOF) instrument by flow injection analysis time-of-flight mass spectrometry (24). All samples were injected in duplicates. Ions were annotated based on their accurate mass and the Human Metabolome Database reference list (44), allowing a tolerance of 0.001 Da. Unknown ions and those annotated as adducts were discarded. Theoretical m/z ratios—beyond the significant digits from the measurement sensitivity—are used as the unique index in the data files and online on GeneNetwork. For example, deprotonated fumarate corresponds to 115.0036897_MZ, malate to 133.0142794_MZ, a-ketoglutarate to 145.0141831_MZ, and D2HG to 147.0298102_MZ.
For blood serum analysis, samples were frozen in liquid nitrogen until large “batches” were ready, which were run in multiples of 16 samples. Samples were then thawed on ice, diluted 1:1 in NaCl solution, and then processed on a Dade Behring Dimension Xpand Clinical Chemistry System. Sixteen metabolites were measured based on standard reagent-reaction spectrophotometry. Due to the long period of time for this study, two chemical batch effects were noted for HDL, free fatty acids, aspartate transaminase, lactate dehydrogenase, and creatinine measurements. These metabolite measurements separated distinctly into two batches based on the time of measurement and a change in the batch of reagent used. To account for this, the two batches for these five metabolites were Z-score normalized and then combined, losing information about absolute values but retaining utility for correlation analyses.
BN-PAGE and in-gel activity
For SC analysis, mitochondria were isolated, protein was extracted, and these extracts were prepared and run on BN-PAGE, described in detail in a separate methods paper (45). In brief, ~30 mg of tissue was homogenized and taken for mitochondrial isolation. For BN-PAGE, 50 or 35 μg of mitochondria from liver and heart, respectively, was solubilized in digitonin and sample buffer (Invitrogen, BN 2008). For the liver, these samples were the same tissues used for omics analysis, using all CD cohorts with three biological replicates per cohort. For the heart, these were the same mice as for the liver. Digitonin/protein ratio of 4 g/g was used for liver and 8 g/g was used for heart (for better band resolution, because heart contains more SCs than liver). Electrophoresis was performed using Native PAGE Novex Bis-Tris Gel System (3 to 12%), as per manufacturer’s instructions with minor modification. Gel transfer was performed using Invitrogen iBlot gel transfer system. For detection of the complexes, anti-oxphos cocktail (Invitrogen, 457999) and WesternBreeze Chromogenic Western Blot Immunodetection Kit (Invitogen, WB7103) were used. In the final detection step, incubation of the membrane with the chromogenic substrate was for 8 min for all the gels. Membranes were dried, scanned, and each visible SC band was independently scored from 1 to 5. All samples were then run across several gels, and we observed nearly complete biological reproducibility (heritability) for band presence or absence. Contrast across gels varied significantly; thus, bands were categorized in a binary manner as “present” or “not present” for QTL analysis.
For in-gel activity assays, electrophoresis was performed for 3 hours (30 min at 150 V and 2.5 hours at 250 V). Complex I activity was performed by incubating the gels for 15 to 30 min in the substrate composed of 2 mM tris-HCl pH 7.4; 0.1 mg/mL NADH, and 2.5 mg/mL nitrotetrazolium blue. CIV activity was performed by incubating the gels for 30 to 40 min in the substrate composed of 25 mg of 3,3′-diamidobenzidine tetra-hydrochloride; 50 mg cytochrome c; 45 mL of 50 mM phosphate buffer pH 7.4, and 5 mL water. CIV+CI activity was performed by subsequently incubating the gels in the substrate for CIV followed by incubation in CI. All reactions were stopped with 10% acetic acid.
BN-PAGE was run as well for six cohorts in the HFD state. We observed no clear difference across diets, and no difference related to the COX7A2L-dependent bands. In-gel activity assays were run for CI, CIV, CIV+CI, and CIII for eight strains (four with the B6 allele of Cox7a2l and four for the D2 allele).
Proteomics: Peptide library development
To develop peptide libraries, we chose 58 cohorts and used 100 μg of protein lysate each (digested as described above). The resulting peptides were mixed and loaded for off-gel electrophoresis fractionation as previously described (46). The 24 fractions were combined into 10 fractions and cleaned up with C18 column. Each fraction was analyzed with classical shotgun data acquisition with a AB Sciex TripleTOF 5600 mass spectrometer interfaced to an Eksigent NanoLC Ultra 2D Plus high-performance liquid chromatography system. Samples were loaded on to a PicoFrit emitter coupled with an analytical column (75 μm diameter) with buffer A (2% acetonitrile, 0.1% formic acid) and eluted with a 135-min linear gradient of 2 to 35% buffer B (90% acetonitrile, 0.1% formic acid) with a flow rate of 300 nL/min. The 20 most intense precursors with charge states 2 to 5 were selected for fragmentation, and the MS2 spectra were acquired in the range 50 to 2000 m/z for 100 ms, and precursor ions were excluded from reselection for 15 s.
Profile mode wiff files from shotgun data acquisition were transformed to centroid mode and converted to mzML files using AB Sciex Data Converter, and then converted to mzXML files using FileConverter. The mzXML files were searched against the canonical UniProt complete proteome database for mouse using the Trans-Proteomic Pipeline (47). A decoy database was generated by reversing the amino acid sequences and appended to the target database. Cysteine carboxy-amido-methylation was set as the static modification, and methionine oxidation was set as the variable modification. Peptides with up to one missed cleavage site were allowed. Mass tolerance was set to 25 parts per million for precursor ions and 0.4 Da for fragment ions. The pepXML files were combined using iProphet (48), and the integrated pepXML file was used to generate the redundant spectra library containing all peptide spectra matches using SpectraST (49). Retention time of peptide identification was transformed to indexed retention time (iRT) values based on the linear regression calibrated for each shotgun run using the information of the spiked iRT peptides. The median of iRT values of each peptide were calculated using in-house script, and the consensus library was constructed using SpectraST. We then selected the top five most abundant b and y fragment ions of each peptide to generate the assays for SWATH-MS targeted extraction. The target assay library contains the precursor and fragment m/z values and the relative intensities of the fragment ions, as well as the average iRT value of each precursor. Decoy assays were appended to the target assay library for error rate estimation.
Proteomics: SWATH mass spectrometry and targeted data extraction
SWATH-MS represents the next generation in large-scale quantitative proteomics measurement techniques and provides a substantial leap in both scope and quality over the most commonly used untargeted proteomics technique today, discovery proteomics (also known as shotgun proteomics). Although discovery proteomics achieves high proteome coverage, the identification and quantification are biased toward those proteins with higher abundance in the sample, and it suffers from inherently poor reproducibility when large sample cohorts are being analyzed. This hurdle has limited the implementation of this approach in large population studies. Recently, targeted proteomics methods have been developed to increase the reproducibility of proteome measurement, such as selected reaction monitoring [used in our previous study (19)]. Due to lower throughput, however, studies using these alternative techniques inevitably measure fewer proteins than studies using shotgun proteomics. Recently, we have developed SWATH, which has demonstrated the ability to quantify thousands of proteins with good reproducibility and quantification accuracy across large sample cohorts (3). Consequently, SWATH provides considerable improvements in both proteome coverage and measurement reproducibility.
The SWATH-MS was performed with the 5600 TripleTOF mass spectrometer, as previously described (3). The chromatographic parameters were as described above. For SWATH-MS–based experiments, the mass spectrometer was operated in a looped product ion mode and specifically tuned to allow a quadrupole resolution of 25 Da/mass selection. Using an isolation width of 26 Da (containing 1 Da for the window overlap), a set of 32 overlapping windows was consecutively constructed covering the 400 to 1200 m/z precursor range (3). The collision energy (CE) for each window was determined based on appropriate collision energy for a charge 2+ ion centered upon the window with a spread of 15 eV. An accumulation time (dwell time) of 100 ms was used for all fragment ion scans in high-sensitivity mode. The survey scans were acquired in high-resolution mode at the beginning of each SWATH-MS cycle, resulting in a duty cycle of 3.4 s.
The SWATH-MS results were first converted to profile mzXML files using ProteoWizard (50). The SWATH-MS targeted data extraction was performed using OpenSWATH workflow (51), which applies a target-decoy scoring model to estimate the false discovery rate (FDR) by the mProphet algorithm (52). Retention time alignment between SWATH maps was performed based on the clustering of reference peptides using a nonlinear alignment algorithm (53). Fragment ion chromatograms were extracted according to the target-decoy assay library with a width of 0.05 m/z, and peak groups were scored based on the elution profile of fragment ions, similarity of elution time and relative intensities with the assay library, and the properties of the tandem MS spectrum extracted at the chromatographic peak apex. Finally, peptide FDR was estimated according to the score distribution of target and decoy assays.
Proteomics: SWATH protein classification and quality control
A key in all protein measurement techniques, including antibody-based approaches, is that they must choose and quantify a specific, small subset of the protein’s overall amino acid sequence to analyze. ProteinProphet analysis on the data from OpenSWATH ensures that the peptides identified are proteotypic (54). The majority of the resulting quantified peptides—20,718 of 22,208—are uniquely attributable to a single protein. The remaining 1510 match common regions of up to nine distinct proteins; these peptides were discarded from analysis in this study. All 22,208 peptides are recorded and available in the raw file download on GeneNetwork.org. Peptide quantities were calculated with imsbInfer, an R package (https://github.com/wolski/imsbInfer). We furthermore analyzed several peptide sequences known to target amino acid sequences with missense mutations in the BXD (e.g., the peptide SAVYPT-SAVQMEAALR for the gene Mrsa has a M to L variant at the highlighted amino acid). In this case, we observed striking differences in the alleles that were not observed for other peptides measuring the same gene that do not have the missense mutation. Thus, 100% amino acid matches are necessary for reliable detection, indicating that our unique peptides were accurately assigned. These missense mutations can lead to false-positive cis-pQTLs; thus, in cases where only one peptide mapped to a cis-pQTL, we controlled for sequence variants, often highlighting missense mutations.
After performing all of these controls, we were typically left with multiple peptide measurements corresponding to a single protein (a bit over seven peptides per protein on average, although this number is highly variable). To reduce multiple testing in subsequent multilayered analyses, we thus assigned the “best” peptide to represent the overall gene level. This was done through several sequential criteria: (i) peptides mapping as cis-pQTLs, (ii) peptides with at least nominally significant correlation to transcript (P < 0.05), and (iii) peptides correlating with known controls in independent layers of data [e.g., HMGCS1 should fluctuate with mevalonate (55) (Fig. 2E)]. A total of 632 proteins were assigned based on criteria i, and a further 824 were assigned based on criteria ii, whereas only a single protein was assigned based on criteria iii. The remaining 1165 proteins were assigned to peptides based on intensity—a standard selection criterion in MS analyses (56). cis-pQTL and mRNA-peptide correlations were performed at a per-peptide level. Due to the increased multiple testing issue of taking all individual peptides, network analyses were performed with only one peptide per gene (i.e., per protein), as were trans-pQTL analyses and all other transomic analyses. It is worth noting that this obstacle of multiple peptides per protein is analogous to pioneering quantitative polymerase chain reaction experiments and microarray design, where there were also considerable difficulties and trial-and-error methods for choosing the “correct” primers (or probe sets or RNA-seq reads) that most correctly measure overall transcript levels (57). Although nearly two decades of transcriptome research has led to the fairly reliable establishment of which RNA fragments correspond properly to the overall transcript, no similar database is yet available for proteins. However, as we generally observe strong networks of associated genes (e.g., Fig. 4), we can determine that the peptides, by and large, accurately represent the protein levels. In time, with many studies such as this one, we expect that it will become feasible to determine guidelines and databases for how to best determine overall protein level from individual peptides.
Cholesterol validation
Huh7 and HepG2 cells were grown in Dulbecco’s minimum essential medium (DMEM) or MEM, respectively, and supplemented with 10% fetal bovine serum. Cells were treated for 48 hours (drugs) or 72 hours (siRNA knockdown) before harvesting and preparation of peptides for mass spectrometric analysis. Control conditions for drug experiments consist of untreated and 0.1% dimethyl sulfoxide–treated cells; control conditions for siRNA-knockdown experiments consist of cells that were untreated, mock treated (only lipofectamine RNAiMAX transfection reagent), or cells treated with negative-control siRNA (anti-sense strand: CUACGAUAGACCGGUCGUAtt). Silencer Select siRNAs were used in a concentration of 5 nM. Knockdown of proteins was performed for LDLR (low-density lipoprotein receptor) with siRNAs s224006 and s224007; for SREBF2 with siRNA s27 and s28; for SREBF (sterol regulatory element binding transcription factor) with a mix of s27, s28, s129, and s130, targeting both SREBF1 and SREBF2. For the LPDS + statin condition, cells were incubated in medium containing 10% LPDS and 1 or 5 μM atorvastatin. Protein signal was quantified for three peptides for MMAB and ECHDC1 and five peptides for HMGCS1, and signal was normalized to control conditions. Experiments were performed for all conditions in two or three biological replicates for siRNA or drug experiments, respectively. The box plots consist, therefore, of two (SREBF si), four (SREBF2 si, LDLR si), or six (drug controls, siRNA controls, and LPDS statin) data points.
Metabolomics in liver: Metabolite classification and quality control
We identified 979 unique metabolite features based on m/z using flow-injection TOF-MS. Of these features, 699 could be attributed to a single metabolite, including in cases where of the two “possible” enantiomers, one is more predominant than the other (e.g., L versus D amino acids). The remaining 280 metabolites were “clusters” with no clear predominant feature—for example, the “glucose” metabolite measurements could not be separated from fructose, galactose, or mannose measurements, as all share the same m/z. The “main” metabolite and all possible alternatives are listed with the data on GeneNetwork.org for the raw file download (press the “INFO” button next to the data set on the main search page and download the data set and supplemental data files).
Normality and significance
Outliers were Windsorized for QTL mapping, which was performed using R/qtl (58). The normality of data was checked by the Shapiro-Wilk test in R, with W ≥ 0.90 considered normal. Student’s t test was used for two-group comparisons in normal data, and Welch’s t test was used otherwise. Bonferroni’s correction was used for multiple testing for tests of correlation. FDR calculations were used for peptide scoring. cis-QTLs used a LRS = 12 as the significance threshold, whereas trans-QTLs used a LRS = 18, as cis-QTLs do not need to correct for multiple locus testing across the genome. Raw P values and corrected P values are both reported when applied.
Figure preparation
Network graphs (Figs. 1I; 4, A, C, and G; and 5A) were performed using Spearman correlation, keeping all edges with P values less than or equal to the reported cutoff in the panel legend. These panels were made in R using the custom package imsbInfer, currently on Github (https://github.com/wolski/imsbInfer) but in the process of being added to Bioconductor. The Circos plot (Fig. 5B) was generated using Circos (http://www.circos.ca). Spearman correlation matrices (Figs. 3B and 4F) were generated in R using the corrgram package. Metabolite structures were drawn with ChemBioDraw (Fig. 3G). QTL plots were drawn in R/qtl (e.g., Fig. 3, A, D, and I). Most other figures (e.g., Fig. 1, C to H and J) were generated using standard R plotting packages included in gplots or ggplot2—e.g., stripchart, plotCI, and barplot2. Final figures were all prepared with Adobe Illustrator. Western blots and BN-PAGE gels were transferred and scanned, then cropped for the figure preparation without other rearrangement or editing, except for Fig. 5F, where biological triplets were “cut” and rearranged based on numerical order of the BXDs. For this panel, contrast was edited individually to obtain an even tone across the full complement of strains. Because this is across several independent gels, the relative intensity across strains is unreliable, hence the binary assignment of SC presence rather than quantity.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Lamy, C. Cartoni, and the Center of Phenogenomics (CPG, EPFL) for their help on the phenotyping and L. Rose at the Molecular Resource Center of Excellence at the University of Tennessee Health Science Center for processing the microarrays. J.A. is the Nestlé Chair in Energy Metabolism. Research was further supported by the EPFL, ETHZ, ERC (AdG-670821 and Proteomics v3.0; AdG-233226), the AgingX program of the Swiss Initiative for Systems Biology (51RTP0–151019, 2013/134, and 2013/153), SNSF (31003A-140780, 31003A-143914, and CSRII3–136201), and the NIH (R01AG043930). Raw transcriptome data are published on GEO: GSE60149. Raw MS data for SWATH proteomics are available on PRIDE (41): PXD003266. Raw MS data for metabolomics are available on MassIVE: MSV000079411. Genotype information is available at www.GeneNetwork.org/genotypes/BXD.geno. Full sequence data on the parental lines were published separately (18). All data from this study can also be found and queried on the www.GeneNetwork.org resource. On the main search page, set Species to “Mouse” and Group to “BXD.” Change “Type” to “Phenotype” and search for LISP3 to find phenotype data. Transcriptome data can be downloaded on GeneNetwork by changing Type to “Liver mRNA” and Data Set to any of the four EPFL/LISP entries. Pressing the INFO button to the right will lead to a page with links to download the raw data sets in the upper right. The same process can be performed to download the metabolome (change Type to “Liver Metabolome”) or proteome data (change Type to “Liver Proteome”). The semiprocessed data files used for each omics layer can also be found in the supplementary materials online.
REFERENCES AND NOTES
- 1.Lander ES, Initial impact of the sequencing of the human genome. Nature 470, 187–197 (2011). doi: 10.1038/nature09792 [DOI] [PubMed] [Google Scholar]
- 2.Sévin DC, Kuehne A, Zamboni N, Sauer U, Biological insights through nontargeted metabolomics. Curr. Opin. Biotechnol. 34, 1–8 (2015). doi: 10.1016/j.copbio.2014.10.001 [DOI] [PubMed] [Google Scholar]
- 3.Gillet LC et al. , Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717 (2012). doi: 10.1074/mcp.O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selevsek N et al. , Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Mol. Cell. Proteomics 14, 739–749 (2015). doi: 10.1074/mcp.M113.035550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kingsmore SF, Lindquist IE, Mudge J, Beavis WD, Genome-wide association studies: Progress in identifying genetic biomarkers in common, complex diseases. Biomark. Insights 2, 283–292 (2007) [PMC free article] [PubMed] [Google Scholar]
- 6.Mayr M, Madhu B, Xu Q, Proteomics and metabolomics combined in cardiovascular research. Trends Cardiovasc. Med. 17, 43–48 (2007). doi: 10.1016/j.tcm.2006.11.004 [DOI] [PubMed] [Google Scholar]
- 7.Joyce AR, Palsson BO, The model organism as a system: Integrating ‘omics’ data sets. Nat. Rev. Mol. Cell Biol. 7, 198–210 (2006). doi: 10.1038/nrm1857; [DOI] [PubMed] [Google Scholar]
- 8.Franzosa EA et al. , Sequencing and beyond: Integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372 (2015). doi: 10.1038/nrmicro3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emmett MR et al. , Integrative biological analysis for neuropsychopharmacology. Neuropsychopharmacology 39, 5–23 (2014). doi: 10.1038/npp.2013.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero R et al. , The use of high-dimensional biology (genomics, transcriptomics, proteomics, and metabolomics) to understand the preterm parturition syndrome. BJOG 113 (suppl. 3), 118–135 (2006). doi: 10.1111/j.1471-0528.2006.01150.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X et al. , Joint mouse-human phenome-wide association to test gene function and disease risk. Nat. Commun. 7, 10464 (2016). doi: 10.1038/ncomms10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altshuler DM et al. , Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010). doi: 10.1038/nature09298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong N, Morahan G, Stathopoulos M, Proietto J, Andrikopoulos S, A novel mechanism regulating insulin secretion involving Herpud1 in mice. Diabetologia 56, 1569–1576 (2013). doi: 10.1007/s00125-013-2908-y [DOI] [PubMed] [Google Scholar]
- 14.Andreux PA et al. , Systems genetics of metabolism: The use of the BXD murine reference panel for multiscalar integration of traits. Cell 150, 1287–1299 (2012). doi: 10.1016/j.cell.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lush IE, The genetics of tasting in mice. VI. Saccharin, acesulfame, dulcin and sucrose. Genet. Res. 53, 95–99 (1989). doi: 10.1017/S0016672300027968 [DOI] [PubMed] [Google Scholar]
- 16.Dalton TP et al. , Identification of mouse SLC39A8 as the transporter responsible for cadmium-induced toxicity in the testis. Proc. Natl. Acad. Sci. U.S.A. 102, 3401–3406 (2005). doi: 10.1073/pnas.0406085102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams EG et al. , An evolutionarily conserved role for the aryl hydrocarbon receptor in the regulation of movement. PLOS Genet. 10, e1004673 (2014). doi: 10.1371/journal.pgen.1004673; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keane TM et al. , Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011). doi: 10.1038/nature10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y et al. , Multilayered genetics and omics dissection of mitochondrial activity in a mouse reference population. Cell 158, 1415–1430 (2014). doi: 10.1016/j.cell.2014.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Der Knaap MS et al. , D-2-Hydroxyglutaric aciduria: Biochemical marker or clinical disease entity? Ann. Neurol. 45, 111–119 (1999). doi: [DOI] [PubMed] [Google Scholar]
- 21.Hasenfuss SC et al. , Regulation of steatohepatitis and PPARγ signaling by distinct AP-1 dimers. Cell Metab. 19, 84–95 (2014). doi: 10.1016/j.cmet.2013.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue M et al. , Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun. 336, 215–222 (2005). doi: 10.1016/j.bbrc.2005.08.070 [DOI] [PubMed] [Google Scholar]
- 23.Ghazalpour A et al. , Comparative analysis of proteome and transcriptome variation in mouse. PLOS Genet. 7, e1001393 (2011). doi: 10.1371/journal.pgen.1001393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuhrer T, Heer D, Begemann B, Zamboni N, High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 83, 7074–7080 (2011). doi: 10.1021/ac201267k [DOI] [PubMed] [Google Scholar]
- 25.Li-Hawkins J et al. , Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Invest. 110, 1191–1200 (2002). doi: 10.1172/JCI0216309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strauss KA, Puffenberger EG, Morton DH, in GeneReviews RA Pagon et al. , Eds. (Seattle, WA, 2013), http://www.ncbi.nlm.nih.gov/books/NBK1319. [Google Scholar]
- 27.Newgard CB et al. , A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326 (2009). doi: 10.1016/j.cmet.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aylor DL, Not just another trait: Methods for the genetic analysis of gene expression (ProQuest Information and Learning Company, North Carolina State University, 2008); http://www.lib.ncsu.edu/resolver/1840.16/3804.
- 29.Achouri Y et al. , Identification of a dehydrogenase acting on D-2-hydroxyglutarate. Biochem. J. 381, 35–42 (2004). doi: 10.1042/BJ20031933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanehisa M et al. , Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014). doi: 10.1093/nar/gkt1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Izem L et al. , Effect of reduced low-density lipoprotein receptor level on HepG2 cell cholesterol metabolism. Biochem. J. 329, 81–89 (1998). doi: 10.1042/bj3290081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharnagl H et al. , Effect of atorvastatin, simvastatin, and lovastatin on the metabolism of cholesterol and triacylglycerides in HepG2 cells. Biochem. Pharmacol. 62, 1545–1555 (2001). doi: 10.1016/S0006-2952(01)00790-0; [DOI] [PubMed] [Google Scholar]
- 33.Goldstein JL, Brown MS, A century of cholesterol and coronaries: From plaques to genes to statins. Cell 161, 161–172 (2015). doi: 10.1016/j.cell.2015.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teslovich TM et al. , Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010). doi: 10.1038/nature09270; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willer CJ et al. , Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013). doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonke M et al. , Transcriptional networks controlling the cell cycle. Genes Genomes Genetics 3, 75–90 (2013). doi: 10.1534/g3.112.004283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lapuente-Brun E et al. , Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 (2013). doi: 10.1126/science.1230381 [DOI] [PubMed] [Google Scholar]
- 38.Mourier A, Matic S, Ruzzenente B, Larsson NG, Milenkovic D, The respiratory chain supercomplex organization is independent of COX7a2l isoforms. Cell Metab. 20, 1069–1075 (2014). doi: 10.1016/j.cmet.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schon EA, Dencher NA, Heavy breathing: Energy conversion by mitochondrial respiratory supercomplexes. Cell Metab. 9, 1–3 (2009). doi: 10.1016/j.cmet.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 40.Gygi SP, Rochon Y, Franza BR, Aebersold R, Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730 (1999). doi: 10.1128/MCB.19.3.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vizcaíno JA et al. , ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014). doi: 10.1038/nbt.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peirce JL, Lu L, Gu J, Silver LM, Williams RW, A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 5, 7 (2004). doi: 10.1186/1471-2156-5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Irizarry RA et al. , Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003). doi: 10.1093/biostatistics/4.2.249; pmid: [DOI] [PubMed] [Google Scholar]
- 44.Wishart DS et al. , HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 41, D801–D807 (2013). doi: 10.1093/nar/gks1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jha P, Wang X, Auwerx J, Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 6, 1–14 (2016). doi: 10.1002/9780470942390.mo150182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R, Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 (2009). doi: 10.1016/j.cell.2009.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keller A, Eng J, Zhang N, Li XJ, Aebersold R, A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 (2005). doi: 10.1038/msb4100024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shteynberg D et al. , iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10, M111.007690 (2011). doi: 10.1074/mcp.M111.007690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lam H et al. , Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 (2007). doi: 10.1002/pmic.200600625 [DOI] [PubMed] [Google Scholar]
- 50.Kessner D, Chambers M, Burke R, Agus D, Mallick P, ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008). doi: 10.1093/bioinformatics/btn323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Röst HL et al. , OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 (2014). doi: 10.1038/nbt.2841 [DOI] [PubMed] [Google Scholar]
- 52.Reiter L et al. , mProphet: Automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 8, 430–435 (2011). doi: 10.1038/nmeth.1584 [DOI] [PubMed] [Google Scholar]
- 53.Weisser H et al. , An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 12, 1628–1644 (2013). doi: 10.1021/pr300992u; [DOI] [PubMed] [Google Scholar]
- 54.Nesvizhskii AI, Keller A, Kolker E, Aebersold R, A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 (2003). doi: 10.1021/ac0341261 [DOI] [PubMed] [Google Scholar]
- 55.Brown MS, Goldstein JL, The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997). doi: 10.1016/S0092-8674(00)80213-5 [DOI] [PubMed] [Google Scholar]
- 56.Elias JE, Gibbons FD, King OD, Roth FP, Gygi SP, Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nat. Biotechnol. 22, 214–219 (2004). doi: 10.1038/nbt930 [DOI] [PubMed] [Google Scholar]
- 57.Stalteri MA, Harrison AP, Interpretation of multiple probe sets mapping to the same gene in Affymetrix GeneChips. BMC Bioinformatics 8, 13 (2007). doi: 10.1186/1471-2105-8-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arends D, Prins P, Jansen RC, Broman KW, R/qtl: High-throughput multiple QTL mapping. Bioinformatics 26, 2990–2992 (2010). doi: 10.1093/bioinformatics/btq565 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.