

Abstract
Background
Inflammatory bowel diseases (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), affect millions of people worldwide with increasing incidence.
Objectives
Several studies have shown a link between gut microbiota composition and IBD, but results are often limited by small sample sizes. We aimed to re‐analyze publicly available fecal microbiota data from IBD patients.
Methods
We extracted original fecal 16S rRNA amplicon sequencing data from 45 cohorts of IBD patients and healthy individuals using the BioProject database at the National Center for Biotechnology Information. Unlike previous meta‐analyses, we merged all study cohorts into a single dataset, including sex, age, geography, and disease information, based on which microbiota signatures were analyzed, while accounting for varying technical platforms.
Results
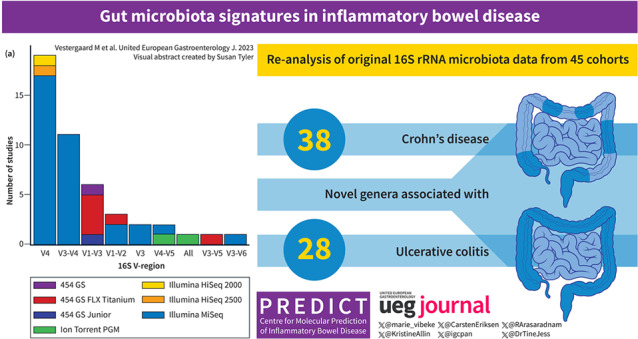
Among 2518 individuals in the combined dataset, we discovered a hitherto unseen number of genera associated with IBD. A total of 77 genera associated with CD, of which 38 were novel associations, and a total of 64 genera associated with UC, of which 28 represented novel associations. Signatures were robust across different technical platforms and geographic locations. Reduced alpha diversity in IBD compared to healthy individuals, in CD compared to UC, and altered microbiota composition (beta diversity) in UC and especially in CD as compared to healthy individuals were found.
Conclusions
Combining original microbiota data from 45 cohorts, we identified a hitherto unseen large number of genera associated with IBD. Identification of microbiota features robustly associated with CD and UC may pave the way for the identification of new treatment targets.
Keywords: 16S rRNA amplicon sequencing, Crohn's disease, Faecalibacterium, IBD, Lachnospiraceae NK4A136 group, microbiome, microbiota, Roseburia, Turicibacter, ulcerative colitis
Key summary.
Summarize the established knowledge on this subject
The gut microbiota associates with inflammatory bowel disease.
Identified associated bacteria vary across studies.
Microbiota studies are influenced by differences in the technical pipeline.
What are the significant and/or new findings of this study
We combined data from 45 different cohorts.
A hitherto unseen large number of genera associated with inflammatory bowel disease was detected.
The associations were consistent across technical pipelines and geographic locations.
INTRODUCTION
The inflammatory bowel diseases (IBDs), ulcerative colitis (UC) 1 and Crohn's disease (CD), 2 are chronic inflammatory diseases of the gastrointestinal tract affecting an increasing number of individuals worldwide. 3 The pathophysiology of IBD is only partially understood. In addition to genetic 4 and environmental 5 risk factors for IBD, IBD is associated with decreased diversity 6 and altered relative abundance of certain bacterial genera and species 7 of the gut microbiota.
Most microbiota studies are based on amplicon sequencing of the bacterial and archeal 16S rRNA gene. 8 However, the Microbiome Quality Control project 9 has shown that variation in each step of the laboratory and bioinformatics pipeline introduces different biases in the resulting microbiota datasets. Pipeline challenges include sample storage, DNA extraction method, primer choice, sequencing depth, and bioinformatics quality control procedures. Also, the choice of statistical model may influence results, 10 and today no gold standard for the conduct of microbiota studies exists.
Previous meta‐analyses of gut microbiota in IBD have primarily summarized published results, 11 , 12 , 13 , 14 without combining original 16S rRNA amplicon sequencing data from existing datasets to ensure that bioinformatic processing of sequencing reads and the following statistical analysis are performed consistently across studies. A large, combined sample size increases the chance of identifying robust and consistent IBD signatures across geographic locations, enables adjustment for individual technical choices, and increases the statistical power to detect new genera associated with IBD.
Therefore, the aim of the present study was to identify, combine, and re‐analyze original 16S rRNA amplicon sequencing data from internationally available cohorts of patients with IBD and healthy individuals, while taking technical variation between studies into account, to provide well‐powered and robust microbiota signatures for IBD.
METHODS
Collection of publicly available original data
To identify available original fecal 16S rRNA amplicon sequencing datasets from IBD patients and healthy individuals worldwide, we searched the BioProject database of the National Center for Biotechnology Information (NCBI) using the search string “(16S OR microbiome OR microbiota OR bacteria) AND (colitis OR ibd OR Crohn's OR uc OR cd OR inflammatory)”. Of the resulting 2270 matches, we included relevant datasets that were based on at least five stool samples and only contained samples from humans, excluding samples from infants, reducing the number of eligible studies to 61. We were unable to match 12 with a publication (Table S1), and they were excluded. For the remaining 49 datasets, if no cohort information was linked to the sequencing reads data, we sent a request for access to the additional information to the corresponding authors. If more than one sample were available per subject, the baseline sample was selected. Of the 49 original datasets, four were excluded due to no information on sample type (fecal or biopsy, accession number PRJNA436359), non‐overlapping paired‐end sequencing reads (accession number PRJNA761255), or forward and reverse reads being inconsistent with each other (accession numbers: PRJNA606913 and PRJNA313074). Non‐overlapping reads were removed as a quality filter of the data, while the inconsistency was not possible to surpass due to the replacement of the fastq IDs with SRA IDs in the files. Finally, 45 datasets were eligible for this study, of which we were able to obtain individual‐level disease status information on 38 and further information on sex and age on 17 (Table 1, Table S2). All 45 cohorts were included when evaluating technical covariates, but analyses of IBD status were limited to the 17 cohorts, including sex and age information.
TABLE 1.
Publicly available data from microbiota studies of IBD patients.
| Study | Country | UC/CD/HI | % Males | Age span | Status |
|---|---|---|---|---|---|
| Papa et al. (2012) 15 | USA | 43/23/24 | 48.9 | 3–24 | 1 |
| Pérez‐Brocal et al. (2013) 16 | Spain | 0/11/8 | ‐ | ‐ | |
| Gevers et al. (2014) 17 | USA | 0/233/71 | 58.7 | Mean: 12.3 | |
| Norman et al. (2015) 18 | USA, United Kingdom | 59/39/55 | 43.1 | Mean: 41.5 | 4 |
| Pérez‐Brocal et al. (2015) 19 | Spain | 0/12/12 | 37.5 | 18–71 | |
| Duranti et al. (2016) 20 | Italy | 14/0/14 | 39.3 | 25–45 | 2 |
| Jacobs et al. (2016) 21 | USA | 10/26/54 | 55.6 | Mean: 14.0 | 3 |
| Tedjo et al. (2016) 22 | The Netherlands | 0/71/0 | 46.5 | 18–70 | 2 |
| Bajer et al. (2017) 23 | Czech Republic | 32/0/31 | 47.6 | 20–72 | 2 |
| Ijaz et al. (2017) 24 | Scotland | 0/19/31 | 60.0 | IQR: 11–50 | |
| Jacob et al. (2017) 25 | USA | 0/20/2 | 60.0 | 23–71 | 2 |
| Iwasawa et al. (2017) 26 | Japan | 16/0/23 | 43.6 | 3–23 | |
| Santoru et al. (2017) 27 | Italy | 82/50/51 | 54.1 | Mean: 45.9 | 1 |
| Doherty et al. (2018) 28 | USA | 0/306/0 | 38.2 | Mean: 39.0 | |
| Forbes et al. (2018) 29 | Canada | 19/20/23 | 38.7 | Mean: 43.8 | |
| Goyal et al. (2018) 30 | USA | 12/7/0 | 57.1 | 8–21 | |
| Kennedy et al. (2018) 31 | United Kingdom | 0/37/54 | 47.3 | 43–65 | 1 |
| Kump et al. (2018) 32 | Austria | 27/0/0 | 63.0 | Mean: 41.0 | 2 |
| Mcdonald et al. (2018) 33 | Many | 112/82/1770 | 53.3 | 0–89 | |
| Schäffler et al. (2018) 34 | Germany | 0/7/10 | 64.7 | Mean: 33.1 | 2 |
| Shutkever et al. (2018) 35 | United Kingdom | 120/150/0 | 44.1 | Mean: 50.1 | 1 |
| Smolinska et al. (2018) 36 | The Netherlands | 0/68/0 | 44.0 | 18–70 | 2 |
| Zhou et al. (2018) 37 | China | 51/72/71 | 56.1 | Mean: 33.7 | 2 |
| Braun et al. (2019) 38 | Israel | 0/45/22 | 59.7 | Mean: 35.6 | 3 |
| Cold et al. (2019) 39 | Denmark | 7/0/0 | 71.4 | 27–50 | 2, 5 |
| Li et al. (2019) 40 | China | 0/9/9 | 55.1 | Mean: 35.8 | 2 |
| Presti et al. (2019) 41 | Italy | 31/7/47 | 56.5 | 21–74 | |
| Weng et al. (2019) 42 | China | 107/173/42 | 65.8 | IQR: 21–53 | 1 |
| Alam et al. (2020) 43 | United Kingdom | 11/10/9 | ‐ | ‐ | 2, 5 |
| Ambrozkiewicz et al. (2020) 44 | Poland | 0/15/9 | 66.7 a | 20–65 a | 2 |
| Ayelén et al. (2020) 45 | Argentina | 20/0/0 | ‐ | ‐ | |
| Diederen et al. (2020) 46 | The Netherlands | 0/27/15 | 52.0 a | IQR: 12–15 a | 2, 5 |
| MacHiels et al. (2020) 47 | Belgium | 0/54/0 | 47.5 | Mean: 51.4 | 2 |
| Metwaly et al. (2020) 48 | Spain | 0/15/0 | 27.6 | ‐ | 2 |
| Schierová et al. (2020) 49 | Czech Republic | 16/0/0 | 50.0 | 28–66 | 3, 5 |
| Sokol et al. (2020) 50 | France | 0/17/0 | 52.9 | IQR: 29–37 | |
| Wang et al. (2020) 51 | China | 58/0/72 | 51.5 | Mean: 43.9 | 2 |
| Clooney et al. (2021) 52 | Canada, Ireland | 228/303/161 | 54.3 | Mean: 50.3 | 1, 5 |
| Cortez et al. (2021) 53 | Brazil | 12/0/23 | 68.6 | 2–21 | |
| Dai et al. (2021) 54 | China | 24/0/14 | 55.3 | Mean: 34.9 | 2 |
| Frau et al. (2021) 55 | United Kingdom | 20/23/20 | 50.8 | Mean: 44.8 | 1 |
| Imai et al. (2021) 56 | Japan | 42/16/45 | 59.0 | Mean: 29.5 | 4 |
| Maldonado‐Arriaga et al. (2021) 57 | Mexico | 18/0/15 | 57.6 | Mean: 36.9 | 1 |
| Schierova et al. (2021) 58 | Czech Republic | 10/17/37 | 34.4 | IQR: 26–44 | |
| Zakerska‐Banaszak et al. (2021) 59 | Poland | 10/0/10 | 45.0 | Mean: 46.3 | 2, 5 |
Note: A total of 45 studies were eligible for this project. Information on sex and age was collected from each publication for both ulcerative colitis (UC) and Crohn's disease (CD) patients as well as healthy individuals (HI). Status 1 = publicly available data missed individual level information on disease status, age, and sex. Status 2 = publicly available data missed individual level information on age and sex. Status 3 = publicly available data missed individual level information on either age or sex. Status 4 = subject names in publication do not match subject names of sequencing data. Status 5 = Additional individual level data was received from authors. ‐: Not reported in publication.
Abbreviations: CD, Crohn's disease; HI, healthy individuals; IBD, inflammatory bowel diseases; IQR, inter‐quartile range; UC, ulcerative colitis.
Only provided for patients.
Microbiota analysis
All analyses were conducted in R (version 4.1.1). We used the DADA2 pipeline (package version 1.22.0) to infer amplicon sequencing variants (ASVs) from the sequencing reads. 60 Each project was analyzed separately to allow for different error profile estimations and different primers. Taxonomy was assigned to ASVs using the SILVA database version 138.1.
The American Gut project (PRJEB11419) included data from individuals with many different diseases. We selected IBD patients and healthy individuals with no reported diseases and no current pregnancy. Other projects were limited to covering IBD and healthy controls and perhaps one other disease. In this case, we excluded individuals with additional diseases from the dataset. We performed quality control based on the depth of the reads that passed the DADA2 pipeline individually for each project (Figure S1), with the lowest accepted read depth of 2000. To combine all datasets into a single dataset, the microbiota data were pooled at the genus level (Figure S2).
Statistical analysis of diversity measures
We calculated two alpha diversity measures, Chao1 richness and Shannon diversity, based on ASV count data from each project (“estimate_richness”, phyloseq‐package version 1.36.0). Chao1 richness and read depth were transformed using the log‐transformation to approach normality (Figure S3). Alpha diversity measures more than three standard deviations (SD) away from the mean were removed as outliers. This removed 42 samples from the Chao1 dataset and 34 from the Shannon dataset out of a total of 5381 samples. The association between alpha diversity and different variables was analyzed using linear models (“lm”, stats‐package version 4.1.1), and type II ANOVA models (“Anova”, car‐package version 3.0.13) to generate results for numerical and categorical variables, respectively.
We calculated Bray‐Curtis and Jaccard dissimilarities as beta diversity measures based on relative abundance data at the genus level (“distance”, phyloseq‐package version 1.36.0). We conducted analyses of beta diversity using PERMANOVA models (“adonis2”, vegan‐package version 2.5.7). They included a permutation design with permutation within projects, but not between, when analyzing age, sex, body mass index (BMI), smoking status, and the disease status (CD, UC, and healthy individuals). Technical features and geographical location are constant within a project and were analyzed without restrictions to the permutation design. They were not included as covariates in models including the project‐based permutation design. 999 permutations were used, and the marginal effects were reported.
We evaluated all available technical and biological variables for association with alpha and beta diversity to select important confounding variables. The effects of technical features (V‐region, sequencing instrument, sequencing depth, and paired‐end/single‐end layout) were analyzed by including all in the same model. Important technical features (p‐value < 0.05) were included as possible confounders in the following models. Biological features (sex, age, BMI, and smoking status) and geographical location were individually tested for association with alpha and beta diversity due to differences in metadata sparsity in some cohorts. This resulted in 5206, 3202, 2641, 1788 and 1835 samples in the analysis of geographical location, sex, age, BMI, and smoking status, respectively. Potential biological confounders were likewise selected based on p‐values < 0.05. In the final model investigating the association between microbiota features and disease status, technical confounders and the geographical continent of the subjects were made into a single variable to avoid collinearity by merging the variables.
Statistical analysis of individual genera versus disease status
We analyzed associations between disease status and microbiota composition at the genus level. The number of genera to be tested was reduced from 1345 to 144 by excluding all genera present in less than 10% of the samples as advised by Nearing et al. 10 As the choice of statistical method influences the final results, Nearing et al. recommended the use of multiple models when analyzing microbiota data to reduce this bias. 10 We included three different and commonly used models 61 that all enable adjustment for confounders, and we only report results consistent across all three methods in regard to statistical significance (adjusted p‐value < 0.05) and direction of association. We employed a zero‐inflated negative binomial (ZINB) model 62 (“zeroinfl”, pscl‐package version: 1.5.5) to model raw microbiota data without any normalization and transformation. ANCOM‐BC 63 (“ancombc”, ANCOMBC‐package version: 1.4.0) and MaAsLin2 64 (“Maaslin2”, Maaslin2‐package version: 1.8.0) were selected based on a recommendation from Nearing et al. Further information on the three models is available in Table S3. The ZINB model is a mixture model and thus provides both a result for the negative binomial and zero‐inflated part of the model. Thus, only one of these results needed to agree with ANCOM‐BC and MaAsLin2 when evaluating the results. p‐values were adjusted using the Benjamini‐Hochberg method.
The combined variable of technical effects and geographical continent, as well as sex and age of the participants were included as covariates. Further, the log of the sequencing depth was included as an offset in the ZINB model. Eleven of the 144 tested genera failed to fit the ZINB model and were thus only tested in the two other models.
Finally, we constructed a permutation test to evaluate whether the microbiota profile of CD was more similar to UC than expected if overlaps were random. This was validated by permuting the genera 999 times and re‐calculating the number of overlaps between CD and UC findings.
RESULTS
This study included original data from 16S rRNA gene amplicon sequencing of stool samples from 45 IBD cohorts across the world (Table 1, Table S2). The final combined dataset consisted of 5381 stool samples from 36 different countries on five different continents, harboring 1345 different genera. Each sample consisted of at least 2000 summarized genus counts. Information on disease status, sex, and age was available for 2518 samples, of which 934 were patients with IBD (CD, N = 691 and UC, N = 243), and 1584 were healthy individuals (Figure 1). IBD patients consisted of a heterogeneous population of IBD patients including both patients in remission, with active disease, in treatment, naïve to treatment, etc.
FIGURE 1.
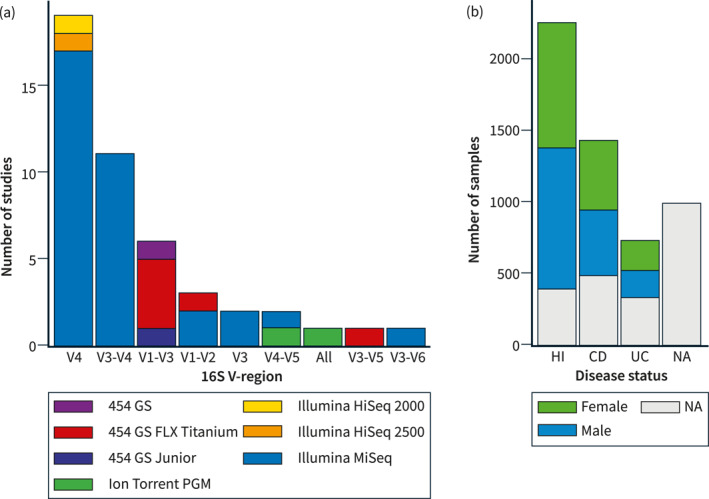
Publicly available microbiota studies on IBD patients and healthy individuals. Forty‐five studies had publicly available data usable for this project. (a) Bar plot summarizing the number of studies available for each 16S V‐region. The studies are further colored according to the sequencing instrument used. (b) Bar plot summarizing the number of samples available for each disease status. The samples are colored by the sex of the subject. CD, Crohn's disease; HI, healthy individuals; IBD, inflammatory bowel diseases; NA, not available; UC, ulcerative colitis.
Potential technical and biological confounders
Technical (sequencing instrument, sequencing layout, analyzed V‐region, and read depth) and biological (sex, age, BMI, and smoking status) variables as well as geographical location were evaluated for association with alpha and beta diversity to assess possible confounding. Most of the technical and biological variables were associated with alpha and beta diversity (Table S4). The combined technical variables obtained an R 2 value of 0.063 when analyzing Bray‐Curtis dissimilarity and 0.074 when analyzing Jaccard dissimilarity. Hence, all the following analyses were corrected for all technical and biological variables, except for BMI and smoking status, which were adjusted only for in a sensitivity analysis to maximize sample size in the main analysis.
Microbiota diversity in IBD versus healthy individuals
Alpha diversity varied across studies (Figure 2a) and was consistently reduced for both UC and CD as compared to healthy individuals (Figure 2b). For CD and UC, respectively, the estimated difference in log‐transformed Chao1 richness to healthy individuals was −0.41 (standard error [SE] = 0.025, p‐value < 2e–16) and −0.27 (SE = 0.032, p‐value < 2e–16). Likewise, the estimated difference in Shannon diversity was −0.34 (SE = 0.047, p‐value = 4e–13) for CD and −0.28 for UC (SE = 0.061, p‐value = 5e–6) as compared to healthy individuals. Alpha diversity was lower in CD patients compared to UC patients using the log‐transformed Chao1 richness for comparison (estimate = −0.14, SE = 0.036, p‐value = 1e–4), but not for the Shannon diversity index (estimate = 0.062, SE = 0.069, p‐value = 0.36).
FIGURE 2.
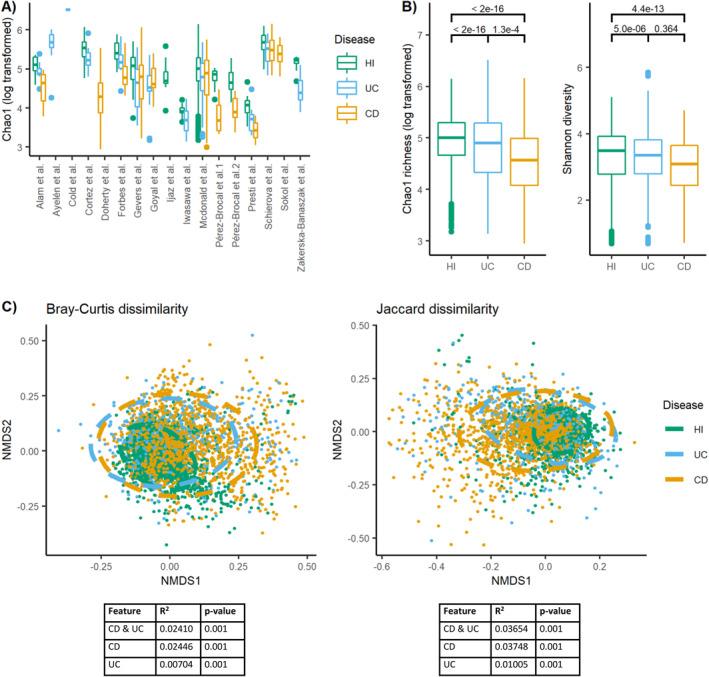
Microbiota diversity is altered in IBD patients versus healthy individuals. (a) The values of Chao1 richness (log transformed) according to IBD disease status within the 17 different study cohorts with available meta‐data visualized using boxplots. If two projects had the same author's name, they were also numbered based on the order presented in Table 1. (b) The values of Chao1 richness (log transformed) and Shannon diversity across all study cohorts as visualized using boxplots and separated by disease status. Numbers above the boxplots display the p‐values of the estimated association. (c) NMDS plots visualizing Bray‐Curtis and Jaccard dissimilarity. Stress Bray‐Curtis = 0.205. Stress Jaccard = 0.196. Ellipses in the NMDS plots are fitted assuming a multivariate t‐distribution. Values below plots represent summary statistics of PERMANOVA analyses of the effect of disease status on the beta diversity measures. Both the combined effect of disease (CD, UC, and healthy individuals) and the separate effect of CD and UC versus healthy individuals are presented. CD, Crohn's disease; HI, healthy individuals; IBD, inflammatory bowel diseases; NMDS, non‐metric multidimensional scaling; UC, ulcerative colitis.
The beta diversity measures, Bray‐Curtis and Jaccard dissimilarity (Figure 2c), revealed that CD explained 2.41%–3.75% of the microbiota variation between individuals, whereas UC only explained 0.70%–1.01% of this variation. Combined, disease status explained 2.41%–3.65% of the variation in the dataset. Healthy individuals were more homogeneous in microbiota composition than IBD patients across cohorts (Figure 2c).
Results were similar upon inclusion of BMI and smoking status in the models (N = 1,602, Table S5).
IBD‐associated genera
A total of 94 genera were identified to be differentially abundant between patients with IBD and healthy individuals (Table 2, Table S6A, and Figure S4). Of these, 55 showed lower abundance in IBD. A ranking of the 94 identified genera based on absolute effect sizes is presented in Table S7 and S8 for CD and UC, respectively. We found that 77 genera were statistically significantly associated with CD, and 64 genera were associated with UC upon adjustment for multiple testing.
TABLE 2.
Overview of genera associated with IBD.
| Higher abundance in both CD and UC | Higher abundance only in CD | Higher abundance only in UC |
|---|---|---|
|
(Clostridium) innocuum group (Eubacterium) brachy group (Ruminococcus) gnavus group Actinomyces Eggerthella Enterococcus Erysipelatoclostridium Faecalitalea Flavonifractor Gemella Haemophilus Intestinibacter Lactococcus Sellimonas Streptococcus Veillonella |
(Eubacterium) fissicatena group Anaerotruncus Atopobium Candidatus Soleaferrea Dialister Dielma Eisenbergiella Fusobacterium Hungatella Lachnoclostridium Lachnospiraceae UCG‐010 Lactobacillus Megasphaera Mogibacterium Ruminococcaceae UBA1819 Solobacterium Tuzzerella Tyzzerella |
Bifidobacterium Gordonibacter Lacticaseibacillus Ruminococcaceae DTU089 Turicibacter |
| Lower abundance in both CD and UC | Lower abundance only in CD | Lower abundance only in UC |
|---|---|---|
|
(Eubacterium) ruminantium group (Eubacterium) siraeum group (Eubacterium) ventriosum group (Eubacterium) xylanophilum group Agathobacter Akkermansia Alistipes Barnesiella Christensenellaceae R‐7 group Coprococcus Fenollaria Fusicatenibacter Intestinimonas Lachnospira Lachnospiraceae CAG‐56 Lachnospiraceae ND3007 group Lachnospiraceae NK4A136 group Lachnospiraceae UCG‐001 Oscillospiraceae NK4A214 group Oscillospiraceae UCG‐002 Oscillospiraceae UCG‐003 Oscillospiraceae UCG‐005 Parabacteroides Paraprevotella Phascolarctobacterium Prevotella Roseburia Ruminococcaceae CAG‐352 Ruminococcus Subdoligranulum |
(Eubacterium) eligens group (Eubacterium) hallii group Colidextribacter Collinsella Corynebacterium Faecalibacterium Holdemanella Lachnospiraceae FCS020 group Marvinbryantia Monoglobus Pseudomonas Romboutsia Turicibacter |
Alloprevotella Angelakisella Bacteroides Bilophila Butyricimonas Coprobacter Desulfovibrio Family XIII UCG‐001 Lachnospiraceae AC2044 group Odoribacter Oxalobacter Prevotella_9 Victivallis |
Note: All listed genera had an adjusted p‐value < 0.05 in all three statistical models and same direction of association. The genera are divided according to association with CD, UC or both CD and UC, and according to whether their abundance in IBD was higher or lower than in healthy individuals. They are named by genus name and sorted alphabetically. Further information is available in Table S6A and Figure S4. The names are colored based on whether the genus previously has been associated with IBD. Green: The same direction of association has been reported in literature. Orange: An opposite direction of association has been reported in literature. Black: The genus has not been associated with IBD previously.
Abbreviations: CD, Crohn's disease; IBD, inflammatory bowel diseases; UC, ulcerative colitis.
A total of 46 genera had the same direction of association in CD and UC. An overlap of 46 similar associations for CD and UC was significantly more than expected from random association (permutation test: p‐value < 0.001). Turicibacter was the only genus showing opposite direction of association between CD and UC.
When adjusting for smoking status and BMI, including only datasets where these variables were available, 22 genera were associated with CD and 32 with UC. Of these, 21 and 28, were similar to the genera detected in the main analysis (Figure S5 and Table S6B). An additional sensitivity analysis limited to samples with information on smoking status and BMI, but not including these as covariates in the model, produced similar results (Figure S6).
Distinguishing the gut microbiota in CD and UC
When comparing the microbiota composition in CD versus UC, we found 31 discriminative genera (Table 3, Figure S7, and Table S6C), of which 17 were increased and 14 were decreased in relative abundance in CD patients compared to UC patients.
TABLE 3.
Overview of genera associated with CD versus UC.
| Higher abundance in CD | Higher abundance in UC |
|---|---|
|
|
Note: All listed genera had an adjusted p‐value < 0.05 in all three statistical models and same direction of association. The genera are divided according whether their abundance was higher in CD or in UC. They are named by genus name and sorted alphabetically. Further information is available in Table S6C and Figure S7.
Abbreviations: CD, Crohn's disease; UC, ulcerative colitis.
DISCUSSION
In this study analyzing original 16S fecal microbiota datasets from 45 cohorts across the world, encompassing 934 IBD patients and 1584 healthy individuals, we confirmed reduced alpha diversity in patients with IBD and a microbiota composition that not only differs between IBD and healthy individuals, but also between CD and UC. Through analyses of 144 different genera present in at least 10% of study participants, we discovered the hitherto largest number of genera associated with CD (n = 77) and UC (n = 64). Of these, 46 overlapped across CD and UC. Our analysis was uniquely powered to correct for technical differences that normally introduce bias, was based on coinciding findings from three statistical models, and therefore provides a comprehensive, robust, and generalizable microbiota signature of IBD.
The observed reduced alpha diversity in patients with IBD compared to healthy individuals and in patients with CD compared to UC were in accordance with former studies of smaller sample size. 13 , 65 Also, in analyses of beta diversity, we found altered microbiota composition in UC and especially in CD as compared to healthy individuals. 52 , 65
For the first time, we identified as many as 77 genera associated with CD and 64 associated with UC. When comparing these genera with results from (1) the 45 individual study cohorts eligible for the current study, (2) the four excluded studies, and (3) four previous meta‐analyses of IBD microbiota signatures, 13 , 14 , 66 , 67 we found that 38 genera in CD and 28 genera in UC represented novel associations. These novel findings appear to be robust, since (1) the beta diversity plot did not show clear batch effects, (2) we adjusted for technical differences across studies, and (3) we detected a large overlap with previous studies at the genus level, which was consistent using three different analytical methods.
When examining the ranking of absolute effect sizes of genera found to be associated with IBD in this project, we identified a clear pattern for UC, where genera detected previously in other studies for association with IBD on average showed larger effect sizes than the new discoveries. This supports the notion that sample size is the main explanation for the ability of the present study to detect novel associations. This is further supported by the fact that genera reported in smaller studies with associations opposite to what we observe were ranking lowest in the present study. Thus, a small absolute effect size is more likely to result in an inverse association. We also observed a large variance in ranking across the three different statistical methods (NBZI, ANCOM‐BC and MaAsLin2), which is likely a contributing factor to differences in results across former microbiota studies. It is a strength of this study, that we used three different methods and only reported overlapping findings since we then reduced bias related to statistical methods applied in individual studies and related to individual approaches of normalization and transformation of data. This approach was chosen to overcome the general limitation of microbiota studies, where the choice of statistical model influences the outcome.
Of the four previously mentioned meta‐analyses, the one by Mancabelli et al. 67 may appear partly similar to ours, as the authors re‐analyzed data from 2177 IBD patients and healthy individuals using a common bioinformatic pipeline. However, for statistical analysis at the genus level, each project was analyzed separately, without correcting for technical and biological covariates. Therefore, the authors did not obtain the power obtained in the present study, and the study only detected 18 genera associated with CD and 13 genera associated with UC.
The primary strength and novelty of this study was the combination and re‐analysis of publicly available data from all cohort studies on gut microbiota signatures in IBD, resulting in a hitherto unseen large sample size of IBD patients and healthy individuals. Thus, the bioinformatical pipeline and the statistical method were unified across study data, which eliminated bias and enabled controlling for different techniques. Further, our large sample size enabled detection of a wide range of new genera with smaller effect sizes.
Combining different studies will be influenced by a batch effect caused by technical heterogeneity, which must be accounted for in subsequent analyses. Reducing the complexity of the dataset from the ASV level to the genus level reduced the influence of batch effect. We estimated the remaining batch effect using the available technical information and adjusted for these in all analyses in this paper. We further ran a sensitivity analysis testing whether the results of alpha‐diversity analyses would be influenced by also including a random intercept with the study ID to further correct for batch effect. The sensitivity analysis produced similar results (results not shown). A similar sensitivity analysis was already included in the permutation design of beta‐diversity analyses and was unfortunately not possible in the single genus analysis since all three analysis tools did not allow inclusion of random effects. However, since it did not affect the analyses of diversity, we concluded that the batch effect and technical heterogeneity were sufficiently accounted for in the analysis by including technical and geographical covariates.
The present study also has limitations that need consideration. First, despite access to and combination of original microbiota data from all identified studies, we were unable to obtain complete information on disease status, sex, age, BMI, and smoking status across all study cohorts. This limited the sample size used in analyses. We conducted a sensitivity analysis adjusting for BMI and smoking status. However, the results of the sensitivity analysis were similar to results from analyses just reducing the sample size based on the availability of BMI and smoking status, hence suggesting that differences between main and sensitivity analyses were mainly driven by differences in sample size and not by adjusting for BMI and smoking status in addition to the other covariates.
We corrected for technical covariates available to us (V‐region, sequencing instrument, library layout, and sequencing depth); however, other technical choices might also affect the resulting dataset, including sample storage, DNA extraction method, and primer choice. Furthermore, variation in gut microbiota between IBD subtypes may reflect differences in medication use, disease course, disease location, and disease severity, which was not captured consistently by available data resources either. Not considering this cohort heterogeneity in analyses is a potential limitation to the study. However, the detection of numerous significant associations after correction for multiple testing, consistent across three different statistical models, suggests that results are robust across the diverse IBD populations. Quality control of the sequencing depth of samples was performed differently for each project, and the average sequencing depth varied markedly across projects. We used a sequencing depth of >2000 as the lowest threshold for inclusion. We could have also chosen to exclude projects that ended up being outliers in terms of sequencing depth. However, as an example, samples with more extreme sequencing depth than mean ± 3SD of the log‐transformed sequencing depth only accounted for 8 out of 2518 samples and would thus not change the outcomes of analyses. Lastly, a world‐wide examination of gut microbiota signatures in IBD, based on publicly available studies, will always be prone to selection bias. 16S IBD projects are more likely to be conducted in countries with higher research investments, whereas national data protection rules, on the other hand, may decrease the likelihood of data being publicly available.
CONCLUSION
This comprehensive re‐analysis of data on 2518 IBD patients and healthy individuals from 45 cohorts identified 38 novel genera associated with CD and 28 with UC, in addition to confirming an altered alpha and beta diversity in IBD. Genera detected by the single genus analysis may potentially represent novel targets for IBD prediction and therapy. Especially, genera found to be reduced in the microbiota of IBD patients, such as Lachnospiraceae NK4A136 group, Faecalibacterium, Roseburia, Alistipes and Parabacteroides, may be interesting candidates for future invention strategies. The number of novel findings is surprising considering the large heterogeneity within IBD patients. Future studies would benefit from selecting a more homogenous subgroup or collecting comprehensive characterization of the IBD patients to account for the heterogeneity. Re‐analyzing original gut microbiota data while taking technical issues into account may also pave the way for the identification of robust signals of value for the implementation of microbial‐based treatments in other disease areas.
AUTHOR CONTRIBUTIONS
Conceptualization: Marie Vibeke Vestergaard, Kristine H. Allin, Tine Jess. Data curation: Marie Vibeke Vestergaard. Formal Analysis: Marie Vibeke Vestergaard. Funding acquisition: Tine Jess. Investigation: Oliwia Zakerska‐Banaszak, Ramesh P. Arasaradnam, Mohammad T. Alam. Methodology: Marie Vibeke Vestergaard, Carsten Eriksen, Kristine H. Allin, Susanne Brix, Karsten Kristiansen. Project administration: Marie Vibeke Vestergaard, Tine Jess. Resources: Tine Jess. Software: Marie Vibeke Vestergaard. Supervision: Carsten Eriksen, Kristine H. Allin, Susanne Brix, Karsten Kristiansen, Tine Jess. Visualization: Marie Vibeke Vestergaard. Writing – original draft: Marie Vibeke Vestergaard. Writing – review and editing: All authors.
CONFLICT OF INTEREST STATEMENT
The authors have nothing to disclose.
Supporting information
supportingInformation S1
Table S6
ACKNOWLEDGMENTS
This study was supported by a grant from the Danish National Research Foundation (Grant no. DNRF148).
Vestergaard MV, Allin KH, Eriksen C, Zakerska‐Banaszak O, Arasaradnam RP, Alam MT, et al. Gut microbiota signatures in inflammatory bowel disease. United European Gastroenterol J. 2024;12(1):22–33. 10.1002/ueg2.12485
DATA AVAILABILITY STATEMENT
All data used in this study are already publicly available at the BioProject database of the National Center for Biotechnology Information. All source codes used in this study are available at GitHub: https://github.com/marievibeke/16S_IBD_project.
REFERENCES
- 1. Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017;389(10080):1756–1770. 10.1016/s0140-6736(16)32126-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torres J, Mehandru S, Colombel JF, Peyrin‐Biroulet L. Crohn’s disease. Lancet. 2017;389(10080):1741–1755. 10.1016/s0140-6736(16)31711-1 [DOI] [PubMed] [Google Scholar]
- 3. Alatab S, Sepanlou SG, Ikuta K, Vahedi H, Bisignano C, Safiri S, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(1):17–30. 10.1016/s2468-1253(19)30333-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jostins L, Ripke S, Weersma R, Duerr R, McGovern D, Hui K, et al. Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loftus E. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504–1517. 10.1053/j.gastro.2004.01.063 [DOI] [PubMed] [Google Scholar]
- 6. Walker A, Sanderson J, Churcher C, Parkes G, Hudspith B, Rayment N, et al. High‐throughput clone library analysis of the mucosa‐associated microbiota reveals dysbiosis and differences between inflamed and non‐inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11(1):7. 10.1186/1471-2180-11-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pittayanon R, Lau JT, Leontiadis GI, Tse F, Yuan Y, Surette M, et al. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: a systematic review. Gastroenterology. 2020;158(4):930–946.e1. 10.1053/j.gastro.2019.11.294 [DOI] [PubMed] [Google Scholar]
- 8. Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral J.‐P, Raoult D, et al. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 2000;38(10):3623–3630. 10.1128/jcm.38.10.3623-3630.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sinha R, Abnet CC, White O, Knight R, Huttenhower C. The microbiome quality control project: baseline study design and future directions. Genome Biol. 2015;16(1):1–6. 10.1186/s13059-015-0841-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nearing JT, Douglas GM, Hayes MG, MacDonald J, Desai DK, Allward N, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13(1):1–16. 10.1038/s41467-022-28034-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Metwaly A, Reitmeier S, Haller D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nat Rev Gastroenterol Hepatol. 2022;19(6):1–15. 10.1038/s41575-022-00581-2 [DOI] [PubMed] [Google Scholar]
- 12. Prosberg M, Bendtsen F, Vind I, Petersen AM, Gluud LL. The association between the gut microbiota and the inflammatory bowel disease activity: a systematic review and meta‐analysis. Scand J Gastroenterol. 2016;51(12):1407–1415. 10.1080/00365521.2016.1216587 [DOI] [PubMed] [Google Scholar]
- 13. Sankarasubramanian J, Ahmad R, Avuthu N, Singh AB, Guda C. Gut microbiota and metabolic specificity in ulcerative colitis and Crohn’s disease. Front Med. 2020;7:606298. 10.3389/fmed.2020.606298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang P, Wu S, Luo Q, Zhao X, Chen W.‐H. Metagenomic analysis of common intestinal diseases reveals relationships among microbial signatures and powers multi‐disease diagnostic models. medRxiv. 2021;6(3):19013136. 10.1128/msystems.00112-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papa E, Docktor M, Smillie C, Weber S, Preheim SP, Gevers D, et al. Non‐invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease. PLoS One. 2012;7(6):e39242. 10.1371/journal.pone.0039242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pérez‐Brocal V, García‐López R, Vázquez‐Castellanos JF, Nos P, Beltrán B, Latorre A, et al. Study of the viral and microbial communities associated with Crohn’s disease: a metagenomic approach. Clin Transl Gastroenterol. 2013;4(6):e36. 10.1038/ctg.2013.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gevers D, Kugathasan S, Denson LA, Vázquez‐Baeza Y, Van Treuren W, Ren B, et al. The treatment‐naive microbiome in new‐onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. 10.1016/j.chom.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease‐specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160(3):447–460. 10.1016/j.cell.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pérez‐Brocal V, García‐López R, Nos P, Beltrán B, Moret I, Moya A. Metagenomic analysis of Crohn’s disease patients identifies changes in the virome and microbiome related to disease status and therapy, and detects potential interactions and biomarkers. Inflamm Bowel Dis. 2015;21(11):2515–2532. 10.1097/mib.0000000000000549 [DOI] [PubMed] [Google Scholar]
- 20. Duranti S, Gaiani F, Mancabelli L, Milani C, Grandi A, Bolchi A, et al. Elucidating the gut microbiome of ulcerative colitis: bifidobacteria as novel microbial biomarkers. FEMS Microbiol Ecol. 2016;92(12):fiw191. 10.1093/femsec/fiw191 [DOI] [PubMed] [Google Scholar]
- 21. Jacobs JP, Goudarzi M, Singh N, Tong M, McHardy IH, Ruegger P, et al. A disease‐associated microbial and metabolomics state in relatives of pediatric inflammatory bowel disease patients. Cell Mol Gastroenterol Hepatol. 2016;2(6):750–766. 10.1016/j.jcmgh.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tedjo DI, Smolinska A, Savelkoul PH, Masclee AA, Van Schooten FJ, Pierik MJ, et al. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Sci Rep. 2016;6(1):1–10. 10.1038/srep35216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z, et al. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J Gastroenterol. 2017;23(25):4548–4558. 10.3748/wjg.v23.i25.4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ijaz UZ, Quince C, Hanske L, Loman N, Calus ST, Bertz M, et al. The distinct features of microbial ‘dysbiosis’ of Crohn’s disease do not occur to the same extent in their unaffected, genetically‐linked kindred. PLoS One. 2017;12(2):e0172605. 10.1371/journal.pone.0172605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacob V, Crawford C, Cohen‐Mekelburg S, Viladomiu M, Putzel GG, Schneider Y, et al. Single delivery of high‐diversity fecal microbiota preparation by colonoscopy is safe and effective in increasing microbial diversity in active ulcerative colitis. Inflamm Bowel Dis. 2017;23(6):903–911. 10.1097/mib.0000000000001132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwasawa K, Suda W, Tsunoda T, Oikawa‐Kawamoto M, Umetsu S, Inui A, et al. Characterisation of the faecal microbiota in Japanese patients with paediatric‐onset primary sclerosing cholangitis. Gut. 2017;66(7):1344–1346. 10.1136/gutjnl-2016-312533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Santoru ML, Piras C, Murgia A, Palmas V, Camboni T, Liggi S, et al. Cross sectional evaluation of the gut‐microbiome metabolome axis in an Italian cohort of IBD patients. Sci Rep. 2017;7(1):9523. 10.1038/s41598-017-10034-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doherty MK, Ding T, Koumpouras C, Telesco SE, Monast C, Das A, et al. Fecal microbiota signatures are associated with response to ustekinumab therapy among Crohn’s disease patients. mBio. 2018;9(2). 10.1128/mbio.02120-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forbes JD, Chen CY, Knox NC, Marrie RA, El‐Gabalawy H, De Kievit T, et al. A comparative study of the gut microbiota in immune‐mediated inflammatory diseases—does a common dysbiosis exist? Microbiome. 2018;6(1):221. 10.1186/s40168-018-0603-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goyal A, Yeh A, Bush BR, Firek BA, Siebold LM, Rogers MB, et al. Safety, clinical response, and microbiome findings following fecal microbiota transplant in children with inflammatory bowel disease. Inflamm Bowel Dis. 2018;24(2):410–421. 10.1093/ibd/izx035 [DOI] [PubMed] [Google Scholar]
- 31. Kennedy NA, Lamb CA, Berry SH, Walker AW, Mansfield J, Parkes M, et al. The impact of NOD2 variants on fecal microbiota in Crohn’s disease and controls without gastrointestinal disease. Inflamm Bowel Dis. 2018;24(3):583–592. 10.1093/ibd/izx061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kump P, Wurm P, Gröchenig HP, Wenzl H, Petritsch W, Halwachs B, et al. The taxonomic composition of the donor intestinal microbiota is a major factor influencing the efficacy of faecal microbiota transplantation in therapy refractory ulcerative colitis. Aliment Pharmacol Ther. 2018;47(1):67–77. 10.1111/apt.14387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, et al. American gut: an open platform for citizen science microbiome research. mSystems. 2018;3(3). https://journals.asm.org/doi/abs/10.1128/mSystems.00031‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schäffler H, Herlemann DPR, Klinitzke P, Berlin P, Kreikemeyer B, Jaster R, et al. Vitamin D administration leads to a shift of the intestinal bacterial composition in Crohn’s disease patients, but not in healthy controls. J Dig Dis. 2018;19(4):225–234. https://onlinelibrary.wiley.com/doi/full/10.1111/1751‐2980.12591 [DOI] [PubMed] [Google Scholar]
- 35. Shutkever O, Gracie DJ, Young C, Wood HM, Taylor M, Hamlin PJ, et al. No significant association between the fecal microbiome and the presence of irritable bowel syndrome‐type symptoms in patients with quiescent inflammatory bowel disease. Inflamm Bowel Dis. 2018;24(7):1597–1605. 10.1093/ibd/izy052 [DOI] [PubMed] [Google Scholar]
- 36. Smolinska A, Tedjo DI, Blanchet L, Bodelier A, Pierik MJ, Masclee AAM, et al. Volatile metabolites in breath strongly correlate with gut microbiome in CD patients. Anal Chim Acta. 2018;1025:1–11. 10.1016/j.aca.2018.03.046 [DOI] [PubMed] [Google Scholar]
- 37. Zhou Y, Xu ZZ, He Y, Yang Y, Liu L, Lin Q, et al. Gut microbiota offers universal biomarkers across ethnicity in inflammatory bowel disease diagnosis and infliximab response prediction. mSystems. 2018;3(1):188–205. 10.1128/msystems.00188-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Braun T, Di Segni A, Benshoshan M, Neuman S, Levhar N, Bubis M, et al. Individualized dynamics in the gut microbiota precede Crohn’s disease flares. Am J Gastroenterol. 2019;114(7):1142–1151. 10.14309/ajg.0000000000000136 [DOI] [PubMed] [Google Scholar]
- 39. Cold F, Browne PD, Günther S, Halkjaer SI, Petersen AM, Al‐Gibouri Z, et al. Multidonor FMT capsules improve symptoms and decrease fecal calprotectin in ulcerative colitis patients while treated‐an open‐label pilot study Multidonor FMT capsules improve symptoms and decrease fecal calprotectin in ulcerative colitis patients while treated‐an open‐label pilot study. Scand J Gastroenterol. 2021. https://www.tandfonline.com/action/journalInformation?journalCode=igas20 [DOI] [PubMed] [Google Scholar]
- 40. Li P, Zhang T, Xiao Y, Tian L, Cui B, Ji G, et al. Timing for the second fecal microbiota transplantation to maintain the long‐term benefit from the first treatment for Crohn’s disease. Appl Microbiol Biotechnol. 2019;103(1):349–360. 10.1007/s00253-018-9447-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lo PA, Zorzi F, Chierico FD, Altomare A, Cocca S, Avola A, et al. Fecal and mucosal microbiota profiling in irritable bowel syndrome and inflammatory bowel disease. Front Microbiol. 2019;10(JULY):1655. 10.3389/fmicb.2019.01655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weng YJ, Gan HY, Li X, Huang Y, Li ZC, Deng HM, et al. Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J Dig Dis. 2019;20(9):447–459. https://onlinelibrary.wiley.com/doi/full/10.1111/1751‐2980.12795 [DOI] [PubMed] [Google Scholar]
- 43. Alam MT, Amos GCA, Murphy ARJ, Murch S, Wellington EMH, Arasaradnam RP. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020;12(1):1. 10.1186/s13099-019-0341-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ambrozkiewicz F, Karczmarski J, Kulecka M, Paziewska A, Niemira M, Zeber‐Lubecka N, et al. In search for interplay between stool microRNAs, microbiota and short chain fatty acids in Crohn’s disease ‐ a preliminary study. BMC Gastroenterol. 2020;20(1):307. 10.1186/s12876-020-01444-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ayelén R, Pablo A, Sofía Q, Jimena C, Renata S, Carolina C, et al. New insights in Ulcerative Colitis Associated Gut Microbiota in South American Population: Akkermansia and Collinsella, two distinctive genera found in Argentine subjects. medRxiv. 2020;7:2020. https://www.medrxiv.org/content/10.1101/2020.07.29.20164764v2 [Google Scholar]
- 46. Diederen K, Li JV, Donachie GE, de Meij TG, de Waart DR, Hakvoort TBM, et al. Exclusive enteral nutrition mediates gut microbial and metabolic changes that are associated with remission in children with Crohn’s disease. Sci Rep. 2020;10(1):1–17. 10.1038/s41598-020-75306-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. MacHiels K, Del Río MP, De La Torre AM, Xie Z, Andreu VP, Sabino J, et al. Early postoperative endoscopic recurrence in Crohn’s disease is characterised by distinct microbiota recolonisation. J Crohns Colitis. 2020;14(11):1535–1546. 10.1093/ecco-jcc/jjaa081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Metwaly A, Dunkel A, Waldschmitt N, Raj ACD, Lagkouvardos I, Corraliza AM, et al. Integrated microbiota and metabolite profiles link Crohn’s disease to sulfur metabolism. Nat Commun. 2020;11(1):1–15. 10.1038/s41467-020-17956-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schierová D, Březina J, Mrázek J, Fliegerová KO, Kvasnová S, Bajer L, et al. Gut microbiome changes in patients with active left‐sided ulcerative colitis after fecal microbiome transplantation and topical 5‐aminosalicylic acid therapy. Cells. 2020;9(10):2283. 10.3390/cells9102283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sokol H, Landman C, Seksik P, Berard L, Montil M, Nion‐Larmurier I, et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: a pilot randomized controlled study. Microbiome. 2020;8(1):12. 10.1186/s40168-020-0792-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang L, Tang L, Feng Y, Zhao S, Han M, Zhang C, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8+ T cells in mice. Gut. 2020;69(11):1988–1997. 10.1136/gutjnl-2019-320105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clooney AG, Eckenberger J, Laserna‐Mendieta E, Sexton KA, Bernstein MT, Vagianos K, et al. Ranking microbiome variance in inflammatory bowel disease: a large longitudinal intercontinental study. Gut. 2021;70(3):499–510. 10.1136/gutjnl-2020-321106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cortez RV, Moreira LN, Padilha M, Bibas MD, Toma RK, Porta G, et al. Gut microbiome of children and adolescents with primary sclerosing cholangitis in association with ulcerative colitis. Front Immunol. 2021;11:3790. 10.3389/fimmu.2020.598152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dai L, Tang Y, Zhou W, Dang Y, Sun Q, Tang Z, et al. Gut microbiota and related metabolites were disturbed in ulcerative colitis and partly restored after mesalamine treatment. Front Pharmacol. 2021;11. 10.3389/fphar.2020.620724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frau A, Ijaz UZ, Slater R, Jonkers D, Penders J, Campbell BJ, et al. Inter‐kingdom relationships in Crohn’s disease explored using a multi‐omics approach. Gut Microb. 2021;13(1). https://www.tandfonline.com/doi/abs/10.1080/19490976.2021.1930871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Imai J, Suzuki H, Kamada N, Golob JL, Kaneko M, Nagata J, et al. A potential pathogenic association between periodontal disease and Crohn’s disease. JCI Insight. 2021;6(23):e1. 10.1172/jci.insight.148543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maldonado‐Arriaga B, Sandoval‐Jiménez S, Rodríguez‐Silverio J, Lizeth Alcaráz‐Estrada S, Cortés‐Espinosa T, Pérez‐Cabeza de Vaca R, et al. Gut dysbiosis and clinical phases of pancolitis in patients with ulcerative colitis. Microbiologyopen. 2021;10(2):e1181. https://onlinelibrary.wiley.com/doi/full/10.1002/mbo3.1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schierova D, Roubalova R, Kolar M, Stehlikova Z, Rob F, Jackova Z, et al. Fecal microbiome changes and specific anti‐bacterial response in patients with IBD during anti‐TNF therapy. Cells. 2021;10(11):3188. 10.3390/cells10113188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zakerska‐Banaszak O, Tomczak H, Gabryel M, Baturo A, Wolko L, Michalak M, et al. Dysbiosis of gut microbiota in Polish patients with ulcerative colitis: a pilot study. Sci Rep. 2021;11(1):1–13. 10.1038/s41598-021-81628-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prodan A, Tremaroli V, Brolin H, Zwinderman AH, Nieuwdorp M, Levin E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One. 2020;15(1):e0227434. 10.1371/journal.pone.0227434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kleine Bardenhorst S, Berger T, Klawonn F, Vital M, Karch A, Rübsamen N. Data analysis strategies for microbiome studies in human populations—a systematic review of current practice. mSystems. 2021;6(1). https://journals.asm.org/doi/full/10.1128/mSystems.01154‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeileis A, Kleiber C, Jackman S. Regression models for count data in R. J Stat Softw. 2008;27(8):1–25. 10.18637/jss.v027.i08 [DOI] [Google Scholar]
- 63. Lin H, Das PS. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11(1):1–11. https://www.nature.com/articles/s41467‐020‐17041‐7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, et al. Multivariable association discovery in population‐scale meta‐omics studies. PLoS Comput Biol. 2021;17(11):e1009442. 10.1371/journal.pcbi.1009442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serrano‐Gómez G, Mayorga L, Oyarzun I, Roca J, Borruel N, Casellas F, et al. Dysbiosis and relapse‐related microbiome in inflammatory bowel disease: a shotgun metagenomic approach. Comput Struct Biotechnol J. 2021;19:6481–6489. 10.1016/j.csbj.2021.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Walters WA, Xu Z, Knight R. Meta‐analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588(22):4223–4233. 10.1016/j.febslet.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mancabelli L, Milani C, Lugli GA, Turroni F, Cocconi D, van Sinderen D, et al. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta‐analysis. FEMS Microbiol Ecol. 2017;93(12):153. 10.1093/femsec/fix153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supportingInformation S1
Table S6
Data Availability Statement
All data used in this study are already publicly available at the BioProject database of the National Center for Biotechnology Information. All source codes used in this study are available at GitHub: https://github.com/marievibeke/16S_IBD_project.