

Abstract
The alcohol metabolite acetaldehyde is a potent human carcinogen linked to esophageal squamous cell carcinoma (ESCC) initiation and development. Aldehyde dehydrogenase 2 (ALDH2) is the primary enzyme that detoxifies acetaldehyde in the mitochondria. Acetaldehyde accumulation causes genotoxic stress in cells expressing the dysfunctional ALDH2E487K dominant negative mutant protein linked to ALDH2*2, the single nucleotide polymorphism highly prevalent among East Asians. Heterozygous ALDH2*2 increases the risk for the development of ESCC and other alcohol-related cancers. Despite its prevalence and link to malignant transformation, how ALDH2 dysfunction influences ESCC pathobiology is incompletely understood. Herein, we characterize how ESCC and preneoplastic cells respond to alcohol exposure using cell lines, three-dimensional organoids and xenograft models. We find that alcohol exposure and ALDH2*2 cooperate to increase putative ESCC cancer stem cells with high CD44 expression (CD44H cells) linked to tumor initiation, repopulation and therapy resistance. Concurrently, ALHD2*2 augmented alcohol-induced reactive oxygen species and DNA damage to promote apoptosis in the non-CD44H cell population. Pharmacological activation of ALDH2 by Alda-1 inhibits this phenotype, suggesting that acetaldehyde is the primary driver of these changes. Additionally, we find that Aldh2 dysfunction affects the response to cisplatin, a chemotherapeutic commonly used for the treatment of ESCC. Aldh2 dysfunction facilitated enrichment of CD44H cells following cisplatin-induced oxidative stress and cell death in murine organoids, highlighting a potential mechanism driving cisplatin resistance. Together, these data provide evidence that ALDH2 dysfunction accelerates ESCC pathogenesis through enrichment of CD44H cells in response to genotoxic stressors such as environmental carcinogens and chemotherapeutic agents.
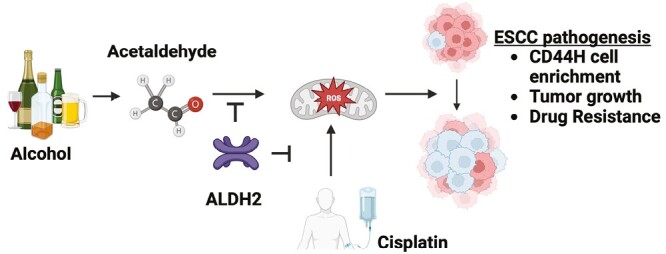
The carcinogenic alcohol metabolite acetaldehyde drives ESCC. Acetaldehyde is detoxified by ALDH2 which is suppressed by its common polymorphism. Here, we demonstrate that ALDH2 dysfunction and alcohol exposure cooperate in ESCC progression through cancer stem cell enrichment.
Graphical Abstract
Graphical Abstract.
Introduction
Alcohol consumption is a leading risk factor for esophageal squamous cell carcinoma (ESCC), a deadly cancer common worldwide (1,2). Alcohol is primarily absorbed in the stomach and metabolized in the liver. Alcohol consumption also exposes the esophageal mucosal surface to high concentrations of ethanol (EtOH) and acetaldehyde, the chief metabolite of EtOH (3,4). After alcohol drinking, acetaldehyde levels in saliva are 10 times higher than in blood (5). Normal human esophageal epithelial cells and ESCC cells can directly metabolize EtOH to produce acetaldehyde via alcohol dehydrogenase ADH1B and CYP2E1 (6,7). Additionally, EtOH or acetaldehyde in circulation promotes ESCC xenograft tumor growth in alcohol-fed mice (6). Understanding the effects of alcohol exposure during the initiation of carcinogenesis and the later stage of progression is equally important.
Acetaldehyde is a highly reactive compound that can damage both mitochondria and DNA (8–10). Acetaldehyde is detoxified by the mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2) which catalyzes the oxidation of acetaldehyde into acetic acid (11). The ALDH2 rs671 (ALDH2*2) is a single nucleotide polymorphism carried by 35–45% of East Asians and is the most common ALDH2 polymorphisms worldwide. The dominant negative mutant ALDH2*2 allele encodes for the E487K change in the ALDH2 protein, which is associated with a dramatic decrease in enzymatic activity compared with wild-type ALDH2*1 (12). Thus, acetaldehyde clearance is delayed in carriers of ALDH2*2. The ALDH2*2 allele confers a higher risk of ESCC, and in particular, excessive alcohol consumption in heterozygous carriers of the ALDH2*2 allele increases the risk for ESCC by ~14-fold, as well as worse prognosis compared with those who develop the disease without ALDH2*1 (13–15). How ALDH2*2 influences the ESCC pathogenesis remains elusive.
Among driving ESCC pathobiology are putative cancer stem cells (CSC) characterized by high expression of CD44 (CD44H cells) that are highly proliferative, capable of initiating tumors, metastatic and therapy resistant (16–22). Within ESCC tumors, CD44H cells display self-renewal and repopulation capabilities (i.e. maintenance of CD44H cells and generation of less-tumorigenic cells with low CD44 expression or CD44L cells) (16,20). Alcohol exposure leads to the enrichment of preexisting CD44H cells that tolerate excessive levels of alcohol-derived reactive oxygen species (ROS) (6). How ALDH2 status influences CD44H cells remains elusive. We hypothesize that ALDH2 dysfunction underlies the observed CD44H cell enrichment.
To address this hypothesis, we leveraged our single cell-derived three-dimensional (3D) esophageal organoid platform that capture the functional, morphological and molecular features of the original tissue (21,23–26). Key to this study, 3D organoids harbor a distinguishable population of CD44H cells that more readily form organoids compared with CD44L cells (16,21,27) and resist chemotherapy (21). We demonstrate that alcohol exposure leads to CD44H cell enrichment to augment tumor growth in an ALDH2*2-dependent manner. These deleterious effects are inhibited by restoring ALDH2 function by the extensively characterized pharmacological ALDH2 activator Alda-1 (28–31). Moreover, we show that alcohol exposure generates ROS and DNA damage to kill CD44L cells, which results in the enrichment of surviving CD44H cells. We further demonstrate that the Aldh2*2 polymorphism modulates response to cisplatin, a commonly used chemotherapeutic for the treatment of ESCC. Together, our data unravel ALDH2 dysfunction as a key axis driving the malignancy of a common and deadly disease.
Materials and methods
Aldh2*2 mutant mice and chemical carcinogenesis
Aldh2*2 (Aldh2E487K) mutant mice carrying the Aldh2*2 knock-in homozygous alleles (Che-Hong Chen and Daria Mochly-Rosen) (30) were crossed with parental C57BL/6 strain carrying wild-type Aldh2*1 homozygous alleles (Aldh2*1 wild-type control mice) to generate mice carrying both mutant and wild-type Aldh2 alleles in heterozygosity, hereafter designated as Aldh2*2 mutant mice. Aldh2*1 and Aldh2*2 mice were subjected to treatment with 100 mg/ml 4-nitroquinline-1-oxide (4NQO) (TCI NO250, Tokyo, Japan) in drinking water for 16 weeks to induce ESCC and intraepithelial neoplasia (IEN), the latter representing a histologic precursor lesion of ESCC (24,32). Following 4NQO withdrawal, mice were killed and dissected at the end of a 10-week observation period to collect esophagi for histopathological examination (Andres Klein-Szanto) and organoid culture. All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Columbia University.
3D mouse-derived organoid culture, treatments and analyses
Mouse-derived organoid (MDO) were generated from mouse esophagi according to detailed protocols published by us (16,24). In brief, esophageal epithelial sheets were isolated from 4NQO-treated mouse esophagi and subjected to mechanical and enzymatic dissociation to prepare single-cell suspensions in advanced DMEM/F12 (ThermoFisher 12634028)-based murine organoid medium (MOM) (24). 2 × 104 single cells were embedded in Matrigel® Basement Membrane Extract (Corning 354234) in a 24 well plate (ThermoFisher 12-556-006) and grown in 500 µl MOM media added into each well and grown for 7–11 days.
Bright-field images of resulting MDOs were captured by the Keyence Fluorescence Microscope BZ-X800 equipped with Keyence software to determine the number and size of organoids, defined as spherical structures ≥5000 µm2 in size. Organoid formation rate (OFR) was determined as the percentage of the number of organoids formed at day 11 per the total number of cells seeded at day 0. Organoid morphology was also determined via phase contrast imaging using the Evos FL Cell Imaging System. Mature organoids were passaged or collected for subsequent analyses as described (6,21).
Organoids were exposed to EtOH at the indicated final concentration along with or without 20 µM Alda-1 (Tocris 4005) and incubated for the indicated time periods. One hundred percent EtOH (Decon Labs) was added directly into the media to a final concentration of 1 or 2%. To prevent the evaporation of EtOH, plates were sealed with PARAFILM® M (Sigma-Aldrich). To control for any off-target effects of sealing plates, control wells (0% EtOH) were sealed as well. Organoids were treated with cisplatin (Santa Cruz sc-200896) for 72 h. Organoids were converted to monolayer culture by seeding single cells suspended in MOM into 35 mm BioLite Cell Culture Treated Dishes (Thermo 130180).
ESCC cell lines and organoid culture
ESCC cell lines TE11 and TE14 carrying a mutant ALDH2*2 allele and genetically modified derivatives TE11-RFP and T14-RFP (6) were grown in monolayer culture for MitoSOX assays as well as 3D organoids for all flow cytometry and organoid formation assays utilizing RPMI-1640 supplemented with 10% fetal bovine serum as described previously (6,21). TE11 and TE14 organoids were grown for 11 days with media changes every 48 h and subjected to EtOH exposure experiments as described above for MDOs.
Cell line authentication
TE11 (Cellosaurus Expasy CVCL_1761) and TE14 cells (CVCL_3336) (33) were obtained in June 1993 as a gift of Dr Tetsuro Nishihira, Tohoku University School of Medicine, Sendai, Miyagi, Japan. Both cell lines were authenticated by short tandem repeat (STR) profiling (ATCC) prior to use in experiments shown in this work. Upon completion of these studies, both lines were re-authenticated by STR profiling (TE11 in January 2023 and TE14 in March 2023) to ensure that no contamination had occurred during these experiments. All murine lines were derived for the purposes of this study. A board-certified pathologist (AKS) reviewed organoid histology (Supplementary Figure 1, available at Carcinogenesis Online) and diagnosed these lines as having features consistent with preneoplastic IEN.
Immunohistochemistry and H&E staining
Mouse esophageal tissues and organoids were subjected to paraffin embedding and formalin fixation immediately following harvesting as described (24,34). In brief, organoids were treated as indicated, collected, centrifuged for 5–10 s at 2000× g and resuspended in 4% formaldehyde overnight at 4°C. Organoids were then embedded in 2% Bacto-agar (BD 214010) and 2.5% gelatin (ThermoFisher 35050061) prior to being processed for routine paraffin embedding as described previously (17). All samples were stained with hematoxylin and eosin (H&E) as described (17). For immunohistochemistry (IHC), organoids slides were incubated with anti-phospho-histone H2Ax (Ser139) (Cell Signaling 9718) at 1:250. All H&E and IHC data were evaluated by a board-certified pathologist (AJK).
Flow cytometry
Flow cytometric analysis of organoids was performed as described previously (6). Briefly, dissociated organoid cells were washed with PBS and resuspended in PBS containing 1% bovine serum albumin (Sigma-Aldrich A7906). TE11 and TE14 cells were stained with phycoerythrin-conjugated CD44 antibody (1:10, BD Biosciences 555479 Clone G44-26) in dark on ice. Murine cells were stained with an allophycocyanin-conjugated CD44 antibody (BD Pharmingen 559250). Apoptosis was assessed via the FITC-conjugated Annexin V Apoptosis Detection Kit (ThermoFisher) according to the manufacturer's instructions. Dead cells were detected by staining for DAPI (ThermoFisher) or propidium iodide (Biolegend 640914), the latter used in Annexin V assays. Data were analyzed using FlowJo software v10.7.1 (Tree Star). CD44H and CD44L cells were respectively defined as the top 10% and bottom 10% of cells with CD44 intensity within untreated organoids. The CD44 gate was set using these values and applied to all experimental groups to determine CD44L and CD44H cell content following the indicated treatment.
Mitochondrial superoxide assays
TE11 and TE14 cells were grown in monolayer culture as described (10), subjected to the indicated treatments and then incubated with 5 µM MitoSOX™ (M36008; Thermo Fisher Scientific) and 20 µM Hoechst 33342 (ENZ-52401, Enzo Life Sciences) for 30 min at 37°C in a humidified incubator. MitoSOX and Hoechst fluorescence intensity was assessed by imaging with a Keyence microscope and analyzed with ImageJ2 software (Version 2.9.0/1.53t). MitoSOX fluorescent intensity was corrected for the background fluorescence and then normalized to the number of nuclei in the sample. Aldh2*2 and Aldh2*1 MDOs were subjected to the indicated treatments, collected and dissociated into single cells. Single cells were incubated with MitoSOX as above and 4ʹ,6-diamindino-2-phenylindole (DAPI) to detect dead cells. MitoSOX intensity was obtained via flow cytometry and calculated as the average fluorescent intensity in live cells.
Xenograft transplantation
Xenografts were performed as described (6) with the approval of the IACUC at Columbia University. TE11-RFP and TE14-RFP cells were grown in monolayer culture, collected via trypsinization and suspended in 50% Matrigel®. The cell suspension was implanted subcutaneously into the dorsal flanks of 8-week-old athymic nu/nu mice (Taconic Biosciences, Hudson, NY). Ten percent EtOH was given via the drinking water ad libitum from week 2 to week 6 following transplantation. Tumor volume was measured weekly with a digital caliper and calculated as follows: Tumor volume (mm3) = [width (mm)]2 × [length (mm)]2 × 0.5. During the EtOH treatment period, Alda-1 (20 mg/kg/day) or vehicle control (40% polyethylene glycol/10% dimethyl sulfoxide/50% water) was intraperitoneally injected two times per day. At week 6, mice were killed and collected tumors were dissociated into single cells and analyzed by flow cytometry for CD44.
Statistical analyses
Data presented in Figures 1–4 were analyzed/graphed with GraphPad Prism 8.0 software using the Student’s t-test. Data presented in Figure 5 were analyzed/graphed with the following: inkscape (1.2.1), matplotlib (3.4.3), numpy (1.22.4), python (3.8.8), pandas (1.4.3), scipy (1.9.0), seaborn (0.12.1) and statsannotations (0.4.4).
Figure 1.
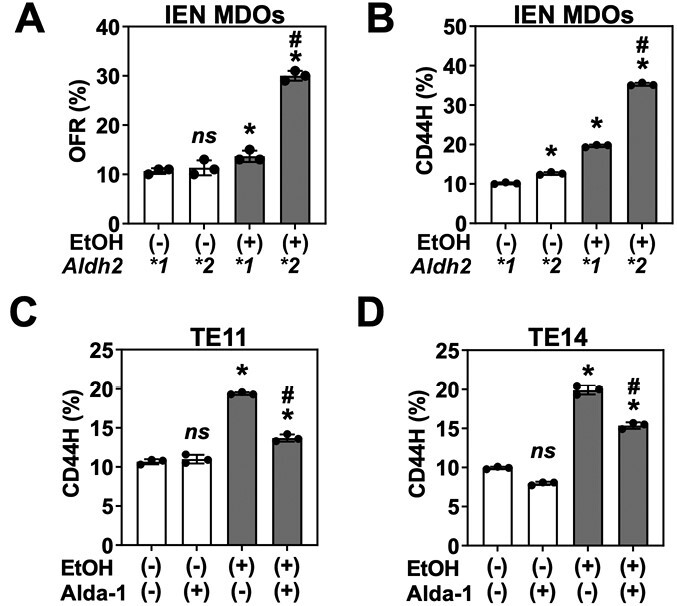
ALDH2 dysfunction cooperates with alcohol in CD44H cell enrichment. IEN MDOs from the indicated genotype were treated with 1% EtOH from day 7 to day 11 of culture. Organoids were collected, dissociated into single cells and then assessed for (A) secondary organoid formation by bright-field microscopy, (B) CD44H cell content by flow cytometry. (C and D) Organoids from two ALDH2*2 human ESCC lines (TE11 or TE14) were treated with 1% EtOH ± 20 µM of the ALDH2 activator Alda-1 from day 7 to day 11 of culture, collected, dissociated into single cells and then subjected to flow cytometry for detection of CD44H cell content. ns, not significant. *P < 0.05 relative to untreated control. #P < 0.05 relative to EtOH-treated Aldh2*1 control (A and B) or EtOH-treated, Alda-1-untreated cells (C and D). Circles represent technical replicates (n = 3).
Figure 4.
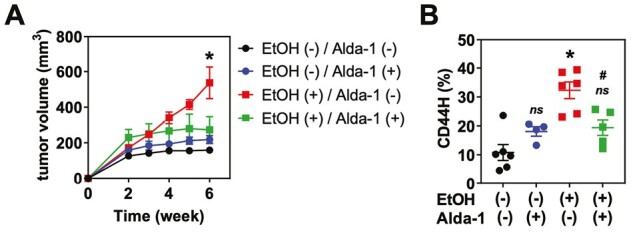
ALDH2 dysfunction accelerates ethanol-stimulated ESCC tumor growth and CD44H cell enrichment. TE14-RFP cells were injected into the flanks of nu/nu mice. 10% EtOH was included in the drinking water ad libitum from week 2 to week 6 following transplantation. During the EtOH treatment period, Alda-1 (20 mg/kg) or vehicle control was intraperitoneally injected twice daily. (A) Tumor volume was measured weekly. (B) At week 6, tumors were dissociated into single cells and analyzed by flow cytometry for CD44. ns, not significant. *P < 0.05 compared with EtOH (−)/Alda-1 (−). #P < 0.05 compared with EtOH (+)/Alda-1 (−). Circles and squares represent technical replicates (n ≥ 4) for both (A) and (B).
Figure 5.
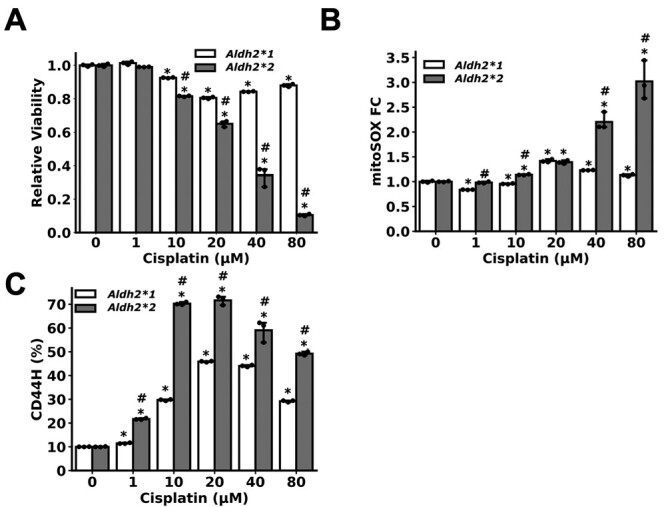
Aldh2 dysfunction potentiates cisplatin-induced CD44H cell enrichment. IEN MDOs from the indicated genotype were treated with the indicated concentration of cisplatin for 72 h, starting from day 7. Organoids were collected on day 10, dissociated into single cells and subjected to flow cytometry to assess for (A) viability by measuring DAPI exclusion, (B) mitochondrial ROS through the mitoSOX assay and (C) CD44H cell content. *P < 0.05 relative to untreated control. #P < 0.05 relative to Aldh2*1 sample treated with identical treatment of cisplatin. Circles = technical replicates (n = 3).
Results
ALDH2 dysfunction and EtOH cooperate to enrich CD44H cells within established preneoplasia and ESCC lesions
Aldh2 dysfunction promotes nitrosamine-induced liver carcinogenesis in Aldh2*2 mice (35). However, Aldh2*2 mice and other rodent strains rarely, if any, develop ESCC or liver cancer in response to alcohol exposure alone (35–37). To evaluate how Aldh2*2 modulates ESCC pathobiology, we treated isogenic Aldh2*2 (mutant) and Aldh2*1 (wild-type) control mice with a potent esophageal carcinogen 4NQO (16). Following 4NQO treatment, we collected esophagi from both Aldh2*2 and Aldh2*1 mice and evaluated the histological progression of ESCC. We observed no statistically significant difference in ESCC progression in Aldh2*2 mice (n = 5) compared with Aldh2*1 (n = 7) (Supplementary Figure 1A, available at Carcinogenesis Online). We then used the remaining tissue to generate MDOs which retain the morphological features of their tissue of origin (Supplementary Figure 1B, available at Carcinogenesis Online). We observed a statistically significant increase in organoids with high-grade neoplastic structures displaying moderate to severe nuclear atypia in the Aldh2*2 condition compared with Aldh2*1 organoids (Supplementary Figure 1C, available at Carcinogenesis Online), suggesting that the single cell-derived organoid system has a high sensitivity to detect neoplastic cells with a higher proliferative capacity that may predominate over normal and less abnormal neoplastic cells ex vivo, in the absence of other constraining factors in vivo such as tumor immunity. In line with the preneoplastic nature, these MDOs did not form xenograft tumors when subcutaneously transplanted in immunodeficient mice (data not shown). We next evaluated Aldh2 expression in these MDOs. Importantly, Aldh2 levels are significantly lower in Aldh2*2 compared with Aldh2*1 MDOs (Supplementary Figure 1D, available at Carcinogenesis Online), which is consistent with reports that this Aldh2 variant has a significantly decreased stability compared with wild-type Aldh2 (30,35,38). Together, we established a tractable organoid model of ESCC pathogenesis in the context of Aldh2 dysfunction.
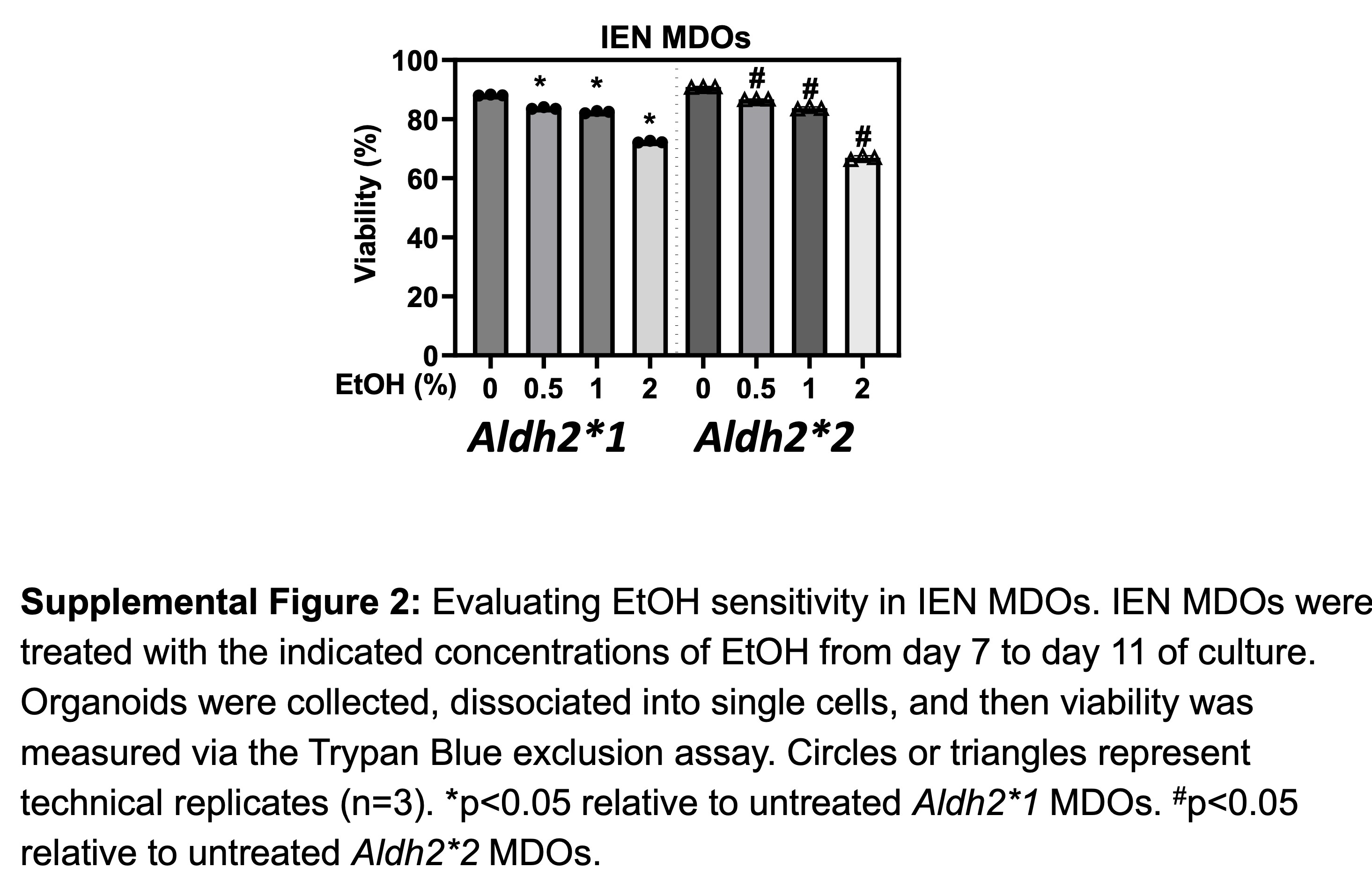
Osei-Sarfo et al. reported that alcohol advanced the 4NQO-induced preneoplastic IEN (dysplasia) lesions in C57BL/6 mice (36). We next leveraged this 3D organoid system to determine how Aldh2*2 influences the ESCC pathogenesis in concert with alcohol exposure. To capture the salient changes that occur during ESCC initiation, we used Aldh2*1 and Aldh2*2 MDOs with histological features consistent with IEN, the ESCC precursor lesion. Using this model, we first determined how Aldh2*2 may alter secondary organoid formation (OFR), a surrogate readout of tumor-initiating capability (6). To do so, we exposed these Aldh2*1 and Aldh2*2 MDOs representing IEN lesions to 1% EtOH for 96 h, which we have determined previously to be a physiologically relevant treatment strategy that mimics EtOH exposure in vivo (6,7). Furthermore, we confirmed that 1% EtOH treatment only modestly decreases IEN MDO viability, which enhances the number of live cells required for the organoid formation assay (Supplementary Figure 2, available at Carcinogenesis Online). We then dissociated these organoids into single cells, passaged these cells and measured their ability to form organoids. We found that EtOH induced a nearly 3-fold increase in OFR in Aldh2*2 organoids compared with a modest ~1.2-fold change in Aldh2*1 organoids (Figure 1A). We next considered whether these changes in OFR were due to the enrichment of the CD44H cell population within each MDO line. To address this possibility, we treated organoids with EtOH as before, collected single cells and performed flow cytometry for CD44H content. Consistent with our OFR results, we observed a nearly 4-fold increase in CD44H cells in the Aldh2*2 organoids following EtOH exposure, compared with a 2-fold increase in the Aldh2*1 organoids (Figure 1B). Together, these results indicate that EtOH exposure results in the enrichment of CD44H cells in the context of Aldh2 dysfunction.
We next addressed whether our findings translated to established human ESCC tumors. We used ESCC cell lines TE11 and TE14, both carrying ALDH2*2. To isolate the effect of ALDH2 dysfunction using these models, we restored ALDH2 function with the extensively characterized pharmacological activator Alda-1 (28–31). Binding to the AlDH2*2-encoded mutant ALDH2 enzyme (i.e. ALDH2E487K), Alda-1 restores its activity by acting as a structural chaperone (29); however, Alda-1 has no effect on the activity of other cytoplasmic or mitochondrial ALDHs (ALDH1, ALDH3A1, ALDH4A1, ALDH5A1 and ALDH7A1) or alcohol dehydrogenase ADH1 (28,30).
We treated organoids made with TE11 and TE14 cells with 1% EtOH for 96 h in the presence or absence of Alda-1. We then dissociated the organoids into single cells and performed flow cytometry for CD44. Consistent with our results in MDOs, short-term EtOH exposure increased CD44H cells within organoids as determined by flow cytometry (Figure 1C and D). Alda-1 partially inhibited CD44H enrichment, suggesting that ALDH2 dysfunction potentiates the tumor-promoting effects of EtOH on human ESCC pathobiology (Figure 1C and D).
ALDH2 dysfunction cooperates with alcohol to promote CD44L cell apoptosis and enrich surviving CD44H cells
We evaluated the mechanisms underlying CD44H cell enrichment. We hypothesized that CD44H cells may be more resistant to elevated EtOH-induced genotoxic stress that occurs in the context of Aldh2 dysfunction. To determine if such a mechanism was driving CD44H cell enrichment in this model, we first confirmed that EtOH exposure results in increased apoptosis in Aldh2*2 cells. To maximize the number of apoptotic cells for this assay, we increased the dose of EtOH from 1 to 2% (Supplementary Figure 2, available at Carcinogenesis Online). Importantly, 2% is still within physiologically relevant range for mimicking EtOH exposure in vitro (6,7). We grew Aldh2*1 and Aldh2*2 cells in monolayer culture, treated with 2% EtOH for 72 h, collected single cells and performed flow cytometry for Annexin V and Propidium Iodide (PI). We observed a significant increase in Annexin V positive/PI negative early apoptotic cells in the Aldh2*2 organoids exposed to EtOH compared with Aldh2*1 organoids (Figure 2A and B). To confirm that this change was driving the observed CD44H cell enrichment (Figure 1B), we repeated this experiment and measured early apoptosis in CD44H and CD44L cells. We found that EtOH exposure resulted in a significant increase in early apoptotic CD44L cells compared with CD44H cells (Figure 2C). This effect was most pronounced in Aldh2*2 cells. Together, these data indicate EtOH exposure promotes CD44H cell enrichment in the context of Aldh2 dysfunction by promoting apoptosis in CD44L cell populations.
Figure 2.
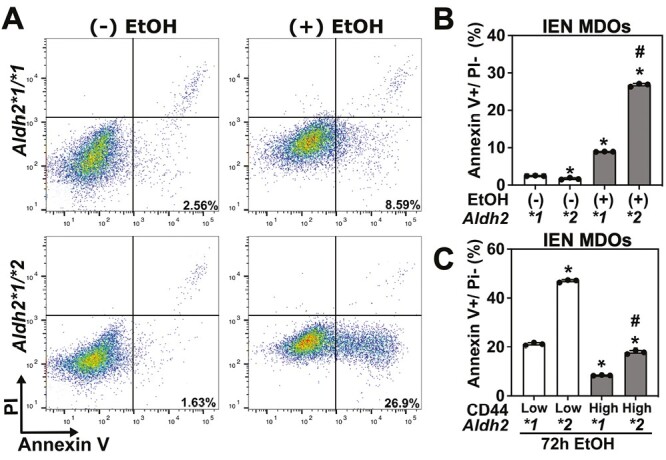
ALDH2 dysfunction cooperates with alcohol in CD44H cell enrichment by promoting CD44L cell apoptosis. Cells dissociated from IEN MDOs from the indicated genotype were grown in monolayer culture, treated with 2% EtOH for 72 h, collected and assessed for apoptosis by flow cytometry for Annexin V/PI. (A) representative dot plot of Annexin V and PI staining, (B) quantification of (A). (C) Apoptosis was measured in CD44H and CD44L cells by flow cytometry. *P < 0.05 compared with Aldh2*1 controls (B) or with Aldh2*1 CD44L cells (C) #P < 0.05 compared with Aldh2*2 control (A) or with Aldh2*2 CD44L cells (C). Circles represent technical replicates (n = 3).
We then addressed whether Aldh1 is involved in EtOH-mediated CD44H cell enrichment. To do so, we evaluated whether 4-diethylaminobenzaldehyde (DEAB), an Aldh1 inhibitor, mediates alcohol-induced CD44L cell apoptosis and CD44H cell enrichment. We treated IEN MDOs with 2% EtOH in the presence or absence of 1.5 µM DEAB, collected single cells and measured Annexin V, PI and CD44 by flow cytometry. We observed a moderate increase in Annexin V positive/PI negative cells in both Aldh2*1 and Aldh2*2 IEN MDOs treated with EtOH (Supplementary Figure 3A, available at Carcinogenesis Online). This increase in early apoptotic cells appear to originate from CD44L cells: DEAB increased Annexin V positive/PI negative population of CD44L, but not in CD44H cells (Supplementary Figure 3B, available at Carcinogenesis Online). Importantly, this increase in apoptosis in DEAB-treated organoids is modest. In contrast, Aldh2 dysfunction generates a dramatic difference in this phenotype (Figure 2A and B), indicating that Aldh2 dysfunction is the primary driver of this phenomenon.
ALDH2 dysfunction potentiates ethanol-derived ROS accumulation and DNA damage
We considered how alcohol may promote CD44H cell enrichment in the context of Aldh2 dysfunction. We hypothesized that Aldh2 dysfunction potentiates EtOH-induced ROS accumulation, driving death of CD44L cells while enriching for CD44H cells (Figure 2C). We therefore assessed whether ROS accumulates in Aldh2*2 organoids. We treated organoids with 1% EtOH for 96 h, dissociated organoids into single cells and then measured mitochondrial ROS using the mitochondrial super oxide assay. We found that EtOH exposure results in a significant increase in mitochondrial ROS in Aldh2*2 organoids compared with EtOH-treated Aldh2*1 organoids (Figure 3A). Furthermore, untreated Aldh2*2 organoids exhibit a modest increase in mitochondrial ROS compared with Aldh2*1 controls, highlighting the potentially deleterious effects of Aldh2 dysfunction independent of EtOH exposure. Together, these results indicate that Aldh2 dysfunction augments ROS production following EtOH exposure.
Figure 3.
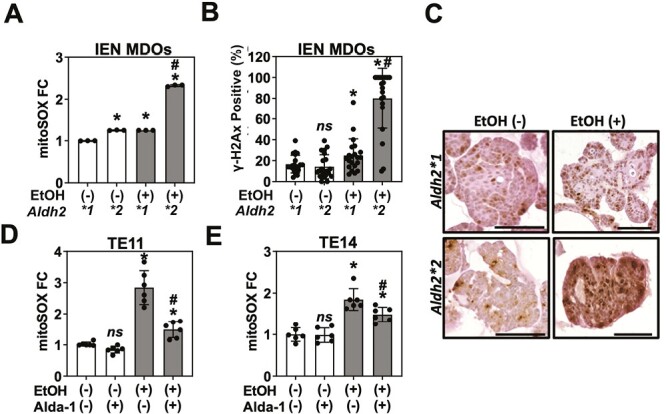
ALDH2 dysfunction potentiates ethanol-derived ROS accumulation and DNA damage. IEN MDOs from the indicated genotype were treated with 1% EtOH from day 7 to day 11 of culture. Organoids were collected and (A) dissociated into single cells to assess mitochondrial ROS via the mitoSOX assay or (B and C) stained for yH2Ax by immunohistochemistry. TE11 or TE14 cells were grown in monolayer culture and then treated with 1% EtOH ±20 µM of the ALDH2 activator Alda-1 for 24 h. Mitochondrial ROS levels were assessed via fluorescence microscopy. ns, not significant. *P < 0.05 relative to untreated control. #P < 0.05 relative to EtOH-treated Aldh2*1 control (A and B), EtOH-treated, Alda-1-untreated cells (D and E). Circles represent technical replicates (n ≥ 3). Scale bar = 100 µm.
We next evaluated the effect of increased ROS accumulation on genome integrity in preneoplastic IEN MDOs. To do so, we performed IHC for the DNA double-strand break marker y-H2AX in IEN MDOs treated with 1% EtOH for 96 h. Consistent with the results of our mitochondrial superoxide assay, we found that EtOH exposure results in a significant increase in the percentage of y-H2AX positive cells in the context of Aldh2 deficiency, with ~80% of cells positive for y-H2AX in Aldh2*2 organoids treated with EtOH compared with <20% rate in untreated controls (Figure 3B and C). Furthermore, we observed a modest significant increase in y-H2AX positivity in Aldh2*1 cells, indicating that EtOH exposure influences ROS and DNA damage in the context of Aldh2 competency, albeit to a lesser extent than in the Aldh2*2 organoids. Together, these data support our hypothesis that ALDH2 limits the accumulation of CD44H cell-enriching ROS following EtOH exposure.
We then assessed whether EtOH exposure results in a similar level of ROS induction in human Aldh2*2 ESCC cell lines. We treated TE11 and TE14 cell lines grown in monolayer with 1% EtOH and 20 µM Alda-1 for 24 h and measured mitochondrial ROS via the mitochondrial superoxide assay. We observed a significant increase in ROS in these ALDH2-dysfunctional cell lines following EtOH exposure, which was reversed following ALDH2 activation by Alda-1 (Figure 3D). These results are consistent with our above-described findings in IEN MDOs. Together, these data corroborate our findings in IEN MDOs that EtOH exposure promotes CD44H cell enrichment and ROS accumulation during ESCC pathogenesis.
ALDH2 dysfunction accelerates ethanol-stimulated ESCC tumor growth and CD44H cell enrichment
We addressed the translational significance of our findings. How the Aldh2*2 polymorphism influences the pathobiology of established tumors following alcohol consumption is undefined. We previously demonstrated that alcohol consumption promotes the growth of xenograft tumors in mice bearing the ESCC cell line TE14 (6). We therefore evaluated whether ALDH2 dysfunction is underlying this phenomenon by subcutaneously transplanting TE14 into the flanks of athymic nu/nu mice. We treated mice with drinking water containing 10% EtOH and 20 mg/kg/day Alda-1 for 6 weeks. We observed a statistically significant increase in tumor size in the EtOH-treated mice compared with vehicle controls (Figure 4A). We next harvested these tumors and performed flow cytometric analysis of their CD44H cell content. We observed a statistically significant increase in the percentage of CD44H cells in EtOH-exposed mice, which was inhibited following treatment with Alda-1 (Figure 4B). Together, our work demonstrates that ethanol exposure promotes tumor growth and CD44H cell enrichment in the context of ALDH2 dysfunction.
Aldh2 dysfunction potentiates cisplatin-induced CD44H cell enrichment
We assessed how Aldh2 status may influence response to other genotoxic agents. Cisplatin is a commonly used chemotherapy for the treatment of ESCC. Since both acetaldehyde and cisplatin induce both ROS and DNA interstrand crosslinks (39,40), we hypothesized that Aldh2*2 cells may respond to cisplatin in a similar manner as they respond to EtOH. To address this hypothesis, we evaluated cisplatin sensitivity in Aldh2*1 and Aldh2*2 MDOs. We collected cisplatin-treated organoids, dissociated into single cells and measured cell viability, CD44 levels and mitochondrial ROS by flow cytometry. We found that the Aldh2*2 organoids were more sensitive to cisplatin, with ~90% of cells dying following treatment with 80 µM cisplatin compared with ~12% of Aldh2*1 cells (Figure 5A). Moreover, cisplatin increased ROS in Aldh2*2, but not Aldh2*1, MDOs in a dose-dependent manner (Figure 5B). Finally, we evaluated whether Aldh2 dysfunction promoted CD44 cell enrichment as observed upon ethanol exposure. We observed a significant increase in CD44H cell levels in Aldh2*2 MDOs following exposure to any dose of cisplatin (Figure 5C). A modest increase in CD44H cells also occurred among cisplatin-surviving cells within Aldh2*1 MDOs. Together, these data suggest that Aldh2*2 may increase cisplatin toxicity to ESCC cells while permitting CD44H cell enrichment to promote therapy resistance, thus indicating that Aldh2 status influences response to commonly used chemotherapy for the treatment of ESCC.
Discussion
Ethanol-related ESCC is a common and deadly disease, particularly in patients heterozygous for ALDH2*2 (15,41). Understanding how ethanol influences ESCC pathogenesis in the context of ALDH2 dysfunction is therefore paramount to identifying the factors underlying the poor prognosis of these patients and identifying new therapeutic strategies to ameliorate the burden of this disease. Here, we demonstrate that ALDH2 dysfunction cooperate with alcohol to augment tumor growth through the enrichment of CD44H cells despite ROS accumulation and DNA damage that promote apoptosis of CD44L cells. These changes are probably driven by the accumulation of acetaldehyde, the EtOH metabolite that is detoxified by ALDH2. Moreover, we find that ALDH2 dysfunction modulates the response to a commonly used chemotherapy, cisplatin. Together, these data indicate that the common ALDH2*2 single nucleotide polymorphism accelerates tumor growth of alcohol-related ESCC and influences response to cisplatin therapy via a similar mechanism permitting CD44H cell enrichment.
Our data further the understanding of how the Aldh2*2 polymorphism promotes malignant transformation following ethanol exposure. The preponderance of studies focus on how ethanol exposure promotes malignant transformation by inducing DNA damage, genome instability and changes in gene expression (36,42). In this model, ethanol-induced activation of oncogenes or inactivation of tumor suppressors drives ESCC tumorigenesis. We augment this understanding by demonstrating that brief exposure to ethanol (24–96 h in culture or 6 weeks in xenograft models) induces a dramatic change in the composition of a tumor or pre-malignant lesion. We demonstrate that ethanol enriches CD44H cells in preneoplastic IEN MDOs, ESCC organoids and xenograft tumors (Figures 1 and 4). We confirm that cells positive for the CSC marker CD44H recapitulate the hallmark features of CSCs. In this study, organoid/cell populations with increased CD44H content form larger xenograft tumors (Figure 4), form organoids at a higher rate (Figure 1) and are enriched following treatment with chemotherapeutic agents (Figure 5) than cell populations with low levels of CD44 (i.e. non-CSCs). These findings are all consistent with known features of CSCs, which can form more aggressive and faster-growing tumors that are resistant to a variety of chemotherapies.
CSC enrichment occurs in alcohol-exposed preneoplastic IEN MDOs. While CD44H cells are appreciated to emerge during the pre-malignant stages of ESCC pathogenesis (43), this work is the first to demonstrate that this enrichment occurs in response to ethanol exposure in the context of ALDH2 dysfunction. Furthermore, we find that treatment of IEN MDOs with chemotherapy enriches for CSCs (Figure 5), which tolerate higher levels of cisplatin in other cancer contexts and drive resistance to the drug (44–46). The presence of this aggressive and putatively drug-resistant subset of cells in a pre-malignant lesion (Figure 1B) highlights the need for earlier detection of ESCC pathogenesis. While CD44 is a well-characterized CSC marker, it should be noted that CSCs are heterogeneous, thus warranting to explore the role of EtOH and ALDH2 dysfunction in cell populations defined by other CSC markers such as CD90 and CD133.
Apart from EtOH-induced CD44H cell enrichment, untreated Aldh2*2 MDOs contained a modest but significantly more CD44H cells than Aldh2*1 MDOs (Figure 1B). These data indicate that ALDH2 dysfunction may have both EtOH-dependent and EtOH-independent roles in esophageal carcinogenesis. Our gene expression profiling data comparing CD44H and CD44L cells of transformed human esophageal cell line EPC2T (GSE37993) indicates that there is no significant difference in the expression of CYP2E1, ADH1B and most ALDH family members including all ALDH1 isoforms. In response to EtOH exposure, esophageal epithelial cells undergo mitochondrial dysfunction and ROS production via CYP2E1 (7). CYP2E1 has a pivotal role in esophageal carcinogenesis through the production of acetaldehyde as well as ROS (47–49). CYP2E1 induction and ALDH2 dysfunction are both implicated in DNA adducts formation and aberrant proliferation in the esophageal epithelium in response to chronic alcohol exposure (8,47). In the absence of EtOH exposure, Aldh2*2 MDO displayed slightly more mitochondrial superoxide (ROS) production than Aldh2*1 MDO (Figure 3A). We suspect that Aldh2*2 cells may have a higher level of endogenous aldehydes (50) related to active DNA replication or cell metabolism related to aberrant proliferation.
We further determined that Aldh1 and its isoform Aldh1a1 are disrupted in Aldh2*2 mice: Aldh1a1 protein levels are downregulated relative to Aldh2*1 controls (Supplementary Figure 1D, available at Carcinogenesis Online). The functionality of this downregulation is unclear. Treatment with the Aldh1 inhibitor DEAB produces a significant but modest change in apoptosis in IEN MDOs (Supplementary Figure 3, available at Carcinogenesis Online). In contrast, Aldh2 dysfunction dramatically alters apoptosis relative to Aldh2 wild-type IEN MDOs (Supplementary Figure 3, available at Carcinogenesis Online). Moreover, treatment with Alda-1, an Aldh2-specific activator that has no effect on Aldh1 (28,30), can rescue EtOH-induced CD44H cell enrichment (Figure 1C and D), mitochondrial ROS (Figure 3C and D), tumor growth and tumor CD44H cell content (Figure 4A and B) in ALDH2*2 ESCC cell or organoid lines. Together, these data support our conclusions that ALDH2 dysfunction is the primary driver of EtOH-mediated ESCC progression in samples harboring the ALDH2 polymorphism.
We previously confirmed that the Aldh2*2 mice have disrupted acetaldehyde metabolism: Aldh2 protein levels and enzymatic activity are reduced in liver homogenate of Aldh2*2 mice compared with Aldh2*1 mice (30). In addition, alcohol exposure results in increased blood acetaldehyde levels in Aldh2*2 mice compared with Aldh2*1 mice, highlighting the specificity of this mouse model to disrupted ethanol metabolism. Furthermore, Alda-1 is an extensively characterized pharmacological activator of ALDH2 (28–31). Alda-1 rescues the deleterious effects of alcohol exposure in the context of Aldh2 dysfunction: in ALDH2*2 knock-in mice, Alda-1 reduces EtOH-induced esophageal DNA damage and DNA adduct formation with concurrent restoration of hepatic mitochondrial Aldh2 (31). However, despite this compelling evidence characterizing Aldh2 activity in a variety of tissues and our included data demonstrating that Aldh2 is expressed in esophageal IEN MDOs, we are unable to directly measure Aldh2 activity using our organoids. This is a known challenge: we previously reported that we were unable to measure ALDH2 enzyme activity in esophageal tissues and keratinocytes using modern ALDH2 activity assays (8). We purified the mitochondrial fraction of the mouse liver to measure the ALDH2 activity (0.9 ± 0.22 nmol/min/mg protein), but not in the mouse esophagus and human esophageal keratinocytes, owing to very low ALDH2 expression compared with the liver. This inability to directly quantify changes in acetaldehyde metabolism constitutes a central limitation to our study. Nevertheless, given our extensive indirect evidence demonstrating that this ALDH2 polymorphic variant is dysfunctional when expressed throughout the organism, we are confident that the phenotypes reported in this manuscript are due to ALDH2 dysfunction.
Ultimately, the work herein augments our current understanding of how ethanol and Aldh2*2 influence ESCC pathogenesis. We demonstrate that these factors interact to alter the fundamental makeup of these tumors by supporting the enrichment of CSCs associated with poor patient prognosis and response to chemotherapy.
We hypothesize that the ethanol- and cisplatin-induced ROS accumulation and genotoxic stress represent selective pressures that enrich existing CSCs in the tumor or pre-malignant microenvironments. We have previously demonstrated that ROS or genotoxic stress mediates CD44H enrichment in a variety of cancer contexts (6,16,18). Moreover, cisplatin is known to cause CSC enrichment in other cancer contexts (46) as well as ROS accumulation in the context of Aldh2 dysfunction (51). Supporting our hypothesis, ethanol exposure results in apoptosis in CD44L, but not CD44H cells (Figure 2C and D). How CSCs survive elevated ROS/genotoxic stress is unclear. We have previously demonstrated that CD44H cells activate autophagy to clear ROS that arises from various sources, including alcohol (6,17). In other contexts, CD44H cells survive otherwise cytotoxic levels of ROS by upregulating antioxidants such as SOD2 (18) and NRF2 (52) and other redox regulatory mechanisms (22,53). Alternatively, CD44L cells may be converted to CD44H cells via epithelial-to-mesenchymal transition under oxidative stress (16). Precisely how CSCs survive elevated genotoxic stress induced by alcohol or cisplatin is unclear and represents a limitation of this study.
That ethanol exposure results in increased ROS in the context of ALDH2 dysfunction is well documented. ALDH2*2 carriers cannot efficiently metabolize acetaldehyde, allowing its accumulation following ethanol exposure. Acetaldehyde can directly induce ROS (54). However, how Aldh2 dysfunction results in increased ROS accumulation following cisplatin exposure is not known. One potential explanation is that ALDH2*2 carriers harbor basal levels of mitochondrial damage arising from the inability to metabolize endogenous aldehydes. This increased basal level of ROS may render cells susceptible to increased cisplatin-mediated ROS. Our data support this hypothesis. We observe a modest but significant increase in mitochondrial ROS in untreated Aldh2*2 organoids compared with Aldh2*1 organoids (Figure 3A). This increase may further explain the elevated CD44H cell content that we observed in untreated Aldh2*2 organoids compared with Aldh2*1 organoids (Figure 1B).
These findings may be applicable to other cancer types in addition to ESCC. ESCC and head and neck squamous cell carcinomas (HNSCC) may develop concurrently or independently in chronic alcohol users heterozygous for ALDH2*2 (55). Furthermore, HNSCC Aldh2*2 patients who consume alcohol have a significantly worse prognosis than Aldh2*1 patients (56). Moreover, 4NQO and EtOH treatment resulted in increased risk for head and neck squamous cell carcinoma pathogenesis which is accompanied by elevated oxidative stress and reduced Aldh2 expression (57,58). We therefore predict that ALDH2 dysfunction will accelerate HNSCC pathogenesis in habitual alcohol users through similar mechanisms related to this paper. Supporting this hypothesis, we have recently demonstrated that CSCs are enriched following alcohol exposure in HNSCC PDOs (6). Additionally, the ALDH2*2 polymorphism is associated with increased risk of colorectal cancer, gastric cancer and breast cancer (59). Although ethanol consumption has been demonstrated to enrich CSCs in these cancers, how ALDH2 status influences these phenomena is unclear (60). Finally, Aldh2 dysfunction promotes hepatocarcinogenesis in mice following ethanol exposure (35,61).
Our findings may be translationally relevant. We provide proof-of-concept that pharmacological ALDH2 activation can reverse alcohol-enhanced tumor growth (Figure 4). Therefore, these activators may have efficacy as single agents. Of note, a next-generation ALDH2 activator, FP-045, is currently in phase 2 clinical trials to treat patients with Fanconi Anemia (FA) (Trial Identifier: NCT04522375), a rare genetic disorder featuring bone marrow failure, leukemia and young-onset HNSCC and ESCC (62). Given that Alda-1 was effective to reduce tumor growth and CD44H cell enrichment in mice drinking alcohol (Figure 4), Alda-1 and FP-045 may be useful to treat ESCC patients who fail to abstain from drinking alcohol against medical advice.
In conclusion, we demonstrate that a common SNP accelerates ESCC pathogenesis in response to a common risk factor of the disease. ALDH2 dysfunction promotes CSC enrichment and tumor growth following short-term alcohol exposure. This phenomenon emerges in pre-malignant cells, highlighting its importance during disease progression. Furthermore, ALDH2 dysfunction modulates response to a commonly used frontline chemotherapy, cisplatin. Both cisplatin and alcohol-mediated CSC enrichment occur concomitantly with ROS accumulation, a well-established driver of this process. Treatment with a pharmacological activator of ALDH2 inhibits these deleterious phenotypes, demonstrating the therapeutic potential of our findings. Together, these data establish a link between ALDH2 dysfunction, CSC enrichment and the progression of ESCC.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We thank Drs. Anil K. Rustgi and Kwok-Kin Wong (NCI P01 Mechanisms of Esophageal Carcinogenesis) and members of the Rustgi and Nakagawa lab for helpful discussions.
Conflict of Interest Statement: Daria Mochly-Rosen and Che-Hong Chen have patents related to Alda-1.
Glossary
Abbreviations:
- ALDH2
aldehyde dehydrogenase 2
- CSCs
cancer stem cells
- DAPI
4ʹ,6-diamindino-2-phenylindole
- ESCC
esophageal squamous cell carcinoma
- H&E
hematoxylin and eosin
- IEN
intraepithelial neoplasia
- IHC
immunohistochemistry
- MDOs
mouse-derived organoids
- MOM
murine organoid medium
- ROS
reactive oxygen species
- STR
short tandem repeat
Contributor Information
Samuel Flashner, Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, Columbia University, New York, NY 10032, USA.
Masataka Shimonosono, Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, Columbia University, New York, NY 10032, USA.
Yasuto Tomita, Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, Columbia University, New York, NY 10032, USA.
Norihiro Matsuura, Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, Columbia University, New York, NY 10032, USA.
Shinya Ohashi, Department of Therapeutic Oncology, Graduate School of Medicine, Kyoto University, Shogoin, Kyoto 606-8507, Japan.
Manabu Muto, Department of Therapeutic Oncology, Graduate School of Medicine, Kyoto University, Shogoin, Kyoto 606-8507, Japan.
Andres J Klein-Szanto, Histopathology Facility, Fox Chase Cancer Center, Philadelphia, PA 19111, USA.
J Alan Diehl, Case Comprehensive Cancer Center, Department of Biochemistry, School of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA.
Che-Hong Chen, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Daria Mochly-Rosen, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Kenneth I Weinberg, Division of Stem Cell Biology and Regenerative Medicine, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA 94305, USA.
Hiroshi Nakagawa, Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, Columbia University, New York, NY 10032, USA; Division of Digestive and Liver Diseases, Department of Medicine, Columbia University Irving Medical Center, Columbia University, New York, NY 10032, USA.
Funding
This work was supported by National Institutes of Health (P01CA098101 to H.N. and J.A.D., R01DK114436 to H.N., R01AA026297 to H.N., L30CA264714 to S.F., 1T32CA265828-01A1 to S.F.) and the Fanconi Anemia Research Fund grant to H.N. and K.W. Furthermore, these studies used the resources of the Herbert Irving Comprehensive Cancer Center Flow Cytometry as well as the Molecular Pathology Shared Resources funded in part through Center Grant P30CA013696 and Organoid & Cell Culture Core of the Columbia University Digestive and Liver Diseases Research Center funded in part through Center Grant P30DK132710.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
- 1. Morgan, E., et al. (2022) The global landscape of esophageal squamous cell carcinoma and esophageal adenocarcinoma incidence and mortality in 2020 and projections to 2040: new estimates from GLOBOCAN 2020. Gastroenterology, 163, 649–658.e2. [DOI] [PubMed] [Google Scholar]
- 2. Rustgi, A.K., et al. (2014) Esophageal carcinoma. N. Engl. J. Med., 371, 2499–2509. [DOI] [PubMed] [Google Scholar]
- 3. Fisher, R.S., et al. (1982) Effect of bolus composition on esophageal transit: concise communication. J. Nucl. Med., 23, 878–882. [PubMed] [Google Scholar]
- 4. Jones, A.W., et al. (1992) Kinetics of ethanol and methanol in alcoholics during detoxification. Alcohol Alcohol., 27, 641–647. [PubMed] [Google Scholar]
- 5. Yokoyama, A., et al. (2008) Salivary acetaldehyde concentration according to alcoholic beverage consumed and aldehyde dehydrogenase-2 genotype. Alcohol. Clin. Exp. Res., 32, 1607–1614. [DOI] [PubMed] [Google Scholar]
- 6. Shimonosono, M., et al. (2021) Alcohol metabolism enriches squamous cell carcinoma cancer stem cells that survive oxidative stress via autophagy. Biomolecules, 11, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chandramouleeswaran, P.M., et al. (2020) Autophagy mitigates ethanol-induced mitochondrial dysfunction and oxidative stress in esophageal keratinocytes. PLoS One, 15, e0239625–e0239629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amanuma, Y., et al. (2015) Protective role of ALDH2 against acetaldehyde-derived DNA damage in oesophageal squamous epithelium. Sci. Rep., 5, 14142. 10.1038/srep14142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanaka, K., et al. (2016) ALDH2 modulates autophagy flux to regulate acetaldehyde-mediated toxicity thresholds. Am. J. Cancer Res., 6, 781–796. [PMC free article] [PubMed] [Google Scholar]
- 10. Peake, J.D., et al. (2021) FANCD2 limits acetaldehyde-induced genomic instability during DNA replication in esophageal keratinocytes. Mol. Oncol., 15, 3109–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klyosov, A.A., et al. (1996) Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry, 35, 4445–4456. [DOI] [PubMed] [Google Scholar]
- 12. Crabb, D.W., et al. (1989) Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH22 allele is dominant. J. Clin. Invest., 83, 314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen, C.H., et al. (2022) Alcohol consumption, ALDH2 polymorphism as risk factors for upper aerodigestive tract cancer progression and prognosis. Life (Basel, Switzerland), 12, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yokoyama, A., et al. (2001) Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis, 22, 433–439. [DOI] [PubMed] [Google Scholar]
- 15. Liu, P., et al. (2018) Correlations of ALDH2 rs671 and C12orf30 rs4767364 polymorphisms with increased risk and prognosis of esophageal squamous cell carcinoma in the Kazak and Han populations in Xinjiang province. J. Clin. Lab. Anal., 32, e22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Natsuizaka, M., et al. (2017) Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun., 8, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whelan, K.A., et al. (2017) Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene, 36, 4843–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kinugasa, H., et al. (2015) Mitochondrial SOD2 regulates epithelial–mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene, 34, 5229–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato, F., et al. (2015) EGFR inhibitors prevent induction of cancer stem-like cells in esophageal squamous cell carcinoma by suppressing epithelial-mesenchymal transition. Cancer Biol. Ther., 16, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao, J.S., et al. (2011) Tumor initiating cells in esophageal squamous cell carcinomas express high levels of CD44. PLoS One, 6, e21419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kijima, T., et al. (2019) Three-dimensional organoids reveal therapy resistance of esophageal and oropharyngeal squamous cell carcinoma cells. Cmgh, 7, 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Natsuizaka, M., et al. (2014) IGFBP3 promotes esophageal cancer growth by suppressing oxidative stress in hypoxic tumor microenvironment. Am. J. Cancer Res., 4, 29–41. [PMC free article] [PubMed] [Google Scholar]
- 23. Flashner, S., et al. (2021) 3D organoids: an untapped platform for studying host–microbiome interactions in esophageal cancers. Microorganisms, 9, 2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flashner, S., et al. (2022) Modeling oral-esophageal squamous cell carcinoma in 3D organoids. J. Vis. Exp., 190, e64676. 10.3791/64676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sachdeva, U.M., et al. (2021) Understanding the cellular origin and progression of esophageal cancer using esophageal organoids. Cancer Lett., 509, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parikh, A.S., et al. (2023) Patient-derived three-dimensional culture techniques model tumor heterogeneity in head and neck cancer. Oral Oncol., 138, 106330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karakasheva, T.A., et al. (2021) Patient-derived organoids as a platform for modeling a patient’s response to chemoradiotherapy in esophageal cancer. Sci. Rep., 11, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen, C.H., et al. (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science, 321, 1493–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez-Miller, S., et al. (2010) Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol., 17, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zambelli, V.O., et al. (2014) Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci. Transl. Med., 6, 251ra118. https://doi.org10.1126/SCITRANSLMED.3009539/SUPPL_FILE/6-251RA118_SM.PDF [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hirohashi, K., et al. (2020) Protective effects of Alda-1, an ALDH2 activator, on alcohol-derived DNA damage in the esophagus of human ALDH2*2 (Glu504Lys) knock-in mice. Carcinogenesis, 41, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang, X.-H., et al. (2004) Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin. Cancer Res., 10, 301–313. [DOI] [PubMed] [Google Scholar]
- 33. Nishihira, T., et al. (1993) Molecular and cellular features of esophageal cancer cells. J. Cancer Res. Clin. Oncol., 119, 441–449. [DOI] [PubMed] [Google Scholar]
- 34. Karakasheva, T.A., et al. (2020) Generation and characterization of patient-derived head and neck, oral, and esophageal cancer organoids. Curr. Protoc. Stem Cell Biol., 53, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin, S., et al. (2015) ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA, 112, 9088–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Osei-Sarfo, K., et al. (2015) Initiation of esophageal squamous cell carcinoma (ESCC) in a murine 4-nitroquinoline-1-oxide and alcohol carcinogenesis model. Oncotarget, 6, 6040–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soffritti, M., et al. (2002) Results of long-term experimental studies on the carcinogenicity of methyl alcohol and ethyl alcohol in rats. Ann. N. Y. Acad. Sci., 982, 46–69. [DOI] [PubMed] [Google Scholar]
- 38. Xiao, Q., et al. (1996) The mutation in the mitochondrial aldehyde dehydrogenase (ALDH2) gene responsible for alcohol-induced flushing increases turnover of the enzyme tetramers in a dominant fashion. J. Clin. Invest., 98, 2027–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kleih, M., et al. (2019) Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis., 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberts, J.J., et al. (1987) Quantitative estimation of cisplatin-induced DNA interstrand cross-links and their repair in mammalian cells: relationship to toxicity. Pharmacol. Ther., 34, 215–246. [DOI] [PubMed] [Google Scholar]
- 41. Cui, R., et al. (2009) Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology, 137, 1768–1775. [DOI] [PubMed] [Google Scholar]
- 42. Sawada, G., et al. (2016) Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology, 150, 1171–1182. [DOI] [PubMed] [Google Scholar]
- 43. Roye’, G.D., et al. (1996) CD44 expression in dysplastic epithelium and squamous-cell carcinoma of the esophagus. J. Cancer (Pred Oncol), 69, 254–258. [DOI] [PubMed] [Google Scholar]
- 44. Abubaker, K., et al. (2013) Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer, 12, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang, H., et al. (2016) BRCA1/FANCD2/BRG1-driven DNA repair stabilizes the differentiation state of human mammary epithelial cells. Mol. Cell, 63, 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee, J.H., et al. (2020) Tonicity-responsive enhancer-binding protein promotes stemness of liver cancer and cisplatin resistance. EBioMedicine, 58, 102926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Millonig, G., et al. (2011) Ethanol-mediated carcinogenesis in the human esophagus implicates CYP2E1 induction and the generation of carcinogenic DNA-lesions. Int. J. Cancer, 128, 533–540. [DOI] [PubMed] [Google Scholar]
- 48. Seitz, H.K., et al. (2007) Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer, 7, 599–612. [DOI] [PubMed] [Google Scholar]
- 49. Seitz, H.K., et al. (2013) The role of Cytochrome P450 2E1 in ethanol-mediated carcinogenesis. Subcell. Biochem., 67, 131–143. [DOI] [PubMed] [Google Scholar]
- 50. Voulgaridou, G.P., et al. (2011) DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat. Res., 711, 13–27. [DOI] [PubMed] [Google Scholar]
- 51. Kim, J., et al. (2017) Aldehyde dehydrogenase 2*2 knock-in mice show increased reactive oxygen species production in response to cisplatin treatment. J. Biomed. Sci., 24, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ryoo, I., et al. (2018) High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol., 17, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ishimoto, T., et al. (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell, 19, 387–400. [DOI] [PubMed] [Google Scholar]
- 54. Li, S.Y., et al. (2004) Overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene prevents acetaldehyde-induced cell injury in human umbilical vein endothelial cells: role of ERK and p38 mitogen-activated protein kinase. J. Biol. Chem., 279, 11244–11252. [DOI] [PubMed] [Google Scholar]
- 55. Katada, C., et al. (2016) Alcohol consumption and multiple dysplastic lesions increase risk of squamous cell carcinoma in the esophagus, head, and neck. Gastroenterology, 151, 860–869.e7. [DOI] [PubMed] [Google Scholar]
- 56. Shinomiya, H., et al. (2017) Prognostic value of ALDH2 polymorphism for patients with oropharyngeal cancer in a Japanese population. PLoS One, 12, e0187992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Osei-Sarfo, K., et al. (2013) The molecular features of tongue epithelium treated with the carcinogen 4-nitroquinoline-1-oxide and alcohol as a model for HNSCC. Carcinogenesis, 34, 2673–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Urvalek, A.M., et al. (2015) Identification of ethanol and 4-Nitroquinoline-1-oxide induced epigenetic and oxidative stress markers during oral cavity carcinogenesis. Alcohol. Clin. Exp. Res., 39, 1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang, H., et al. (2021) The role of ALDH2 in tumorigenesis and tumor progression: targeting ALDH2 as a potential cancer treatment. Acta Pharm. Sin B, 11, 1400–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu, M., et al. (2016) ErbB2 and p38γ MAPK mediate alcohol-induced increase in breast cancer stem cells and metastasis. Mol. Cancer, 15, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seo, W., et al. (2019) ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J. Hepatol., 71, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Velleuer, E., et al. (2014) Fanconi anemia: young patients at high risk for squamous cell carcinoma. Mol. Cell Pediatr., 1, 9. 10.1186/S40348-014-0009-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.